

Abstract
Colorimetric aptamer-based sensors offer a simple means of on-site or point-of-care analyte detection. However, these sensors are largely incapable of achieving naked-eye detection, because of the poor performance of the target-recognition and signal-reporting elements employed. To address this problem, we report a generalizable strategy for engineering novel multimodule split DNA constructs termed “CBSAzymes” that utilize a cooperative binding split aptamer (CBSA) as a highly target-responsive bioreceptor and a new, highly active split DNAzyme as an efficient signal reporter. CBSAzymes consist of two fragments that remain separate in the absence of target, but effectively assemble in the presence of the target to form a complex that catalyzes the oxidation of 2,2′-azino-bis(3-ethylbenzthiazoline)-6-sulfonic acid, developing a dark green color within 5 min. Such assay enables rapid, sensitive, and visual detection of small molecules, which has not been achieved with any previously reported split-aptamer-DNAzyme conjugates. In an initial demonstration, we generate a cocaine-binding CBSAzyme that enables naked-eye detection of cocaine at concentrations as low as 10 μM. Notably, CBSAzyme engineering is straightforward and generalizable. We demonstrate this by developing a methylenedioxypyrovalerone (MDPV)-binding CBSAzyme for visual detection of MDPV and 10 other synthetic cathinones at low micromolar concentrations, even in biological samples. Given that CBSAzyme-based assays are simple, label-free, rapid, robust, and instrument-free, we believe that such assays should be readily applicable for on-site visual detection of various important small molecules such as illicit drugs, medical biomarkers, and toxins in various sample matrices.
Graphical Abstarct
On-site or point-of-care detection of small molecules is highly desirable for applications such as medical diagnostics,1 environmental monitoring,2 and forensic science.3 Ease of use, cost-effectiveness, and rapid turnaround time are essential features for such assays, alongside typical considerations such as sensitivity and specificity.1 Aptamers have emerged as a promising sensing element for small-molecule detection.4 They are nucleic-acid-based bioaffinity elements isolated from randomized oligonucleotide libraries through an in vitro process known as systematic evolution of ligands by exponential enrichment (SELEX)5 to bind specific targets.6 In addition to exhibiting affinity and specificity that can rival antibodies, aptamers have excellent chemical stability,7 and can be synthesized in an inexpensive and reproducible manner.
Among the numerous aptamer-based sensing platforms described to date, colorimetric assays are especially suitable for on-site detection, because they can be directly interpreted by the naked eye. Gold nanoparticles (AuNPs) have been widely employed with aptamers as sensitive signal reporters for visual small-molecule detection.8–11 However, aptamer-modified AuNPs have several drawbacks. In particular, the production of modified AuNPs is costly, laborious, and time-consuming, and the process is often plagued by batch-to-batch variation.12 Moreover, such AuNPs have limited shelf lives, because of the lability of the thiol–gold bonds that conjugate the aptamer and AuNP.13 Alternatively, aptamer-based colorimetric assays utilizing unmodified AuNPs have been developed,9,10 but these have poor specificity, because the AuNPs are highly unstable and easily undergo nonspecific salt-induced aggregation.14
G-quadruplex-structured, single-stranded DNA enzymes (DNAzymes) are alternative signal reporters for colorimetric aptamer-based assays.15 These DNAzymes mimic the activity of horseradish peroxidase, and they are capable of binding hemin and performing H2O2-mediated oxidation of colorless substrates into colored products.16–18 DNAzymes have several desirable characteristics relative to DNA-AuNP conjugates, including their signal-amplifying ability, chemical stability, ease of mass production without batch-to-batch variation, and the simplicity of incorporating them with different sensing elements.15 DNAzymes16 can be divided into two segments19,20 that can be grafted to the ends of split aptamer fragments to form split aptamer-DNAzyme conjugates.21 The presence of the target promotes the assembly of the split aptamer fragments, bringing the split DNAzymes into close proximity to form an active G-quadruplex structure that can generate a colorimetric readout. This strategy has enabled label-free colorimetric detection of small molecules such as cocaine21 and ATP.22 However, these assays cannot achieve naked-eye detection, because the resulting absorbance changes can only be detected by instruments.21,23–25 This is probably due to the low target responsiveness of the split aptamers, combined with the modest catalytic activity of the split DNAzymes.
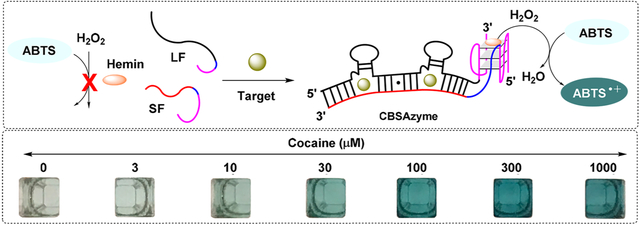
We recently developed new aptamer constructs, termed “cooperative-binding split-aptamers” (CBSAs), which contain two binding domains that bind targets cooperatively.26 CBSAs are far more target-responsive than split aptamers with a single binding domain,26 and we hypothesized that pairing CBSAs with split DNAzymes could enable sensitive visual small-molecule detection. In this work, for the very first time, we have engineered a highly active split DNAzyme sequence and combined it with highly target-responsive CBSAs to create novel “CBSAzyme” constructs. This recognition and signaling element overcomes both the poor target responsiveness observed with split aptamers and the low activity inherent to split G-quadruplex DNAzymes to allow rapid, label-free, visual detection of small molecules. CBSAzymes consist of a pair of DNAzyme-linked aptamer fragments with two tandem target-binding domains. The first target-binding event facilitates binding of the second target through cooperative binding, thus bringing the two fragments together and reconstituting the DNAzyme segments. The assembled DNAzyme subunit can perform peroxide-mediated oxidation of 2,2′-azino-bis(3-ethylbenzthiazoline)-6-sulfonic acid (ABTS), generating a dark green color visible to the naked eye within a few minutes.
We have demonstrated the feasibility of this sensing strategy by developing and optimizing a broadly applicable CBSAzyme template in which the target-binding domain can be substituted with that of any small-molecule binding aptamer to enable colorimetric detection of its respective target. We first rationally engineered a new split DNAzyme from a previously described sequence,19 improving its catalytic activity by 2-fold. We then conjugated this split DNAzyme to a cocaine-binding CBSA and optimized the CBSA stem length, DNAzyme split mode, and the length of the linker between the CBSA and DNAzyme modules. The resulting cocaine-binding CBSAzyme achieved label-free, visual cocaine detection with a detection limit of 10 μM within 5 min. We then demonstrated the generality of our approach by developing a CBSAzyme for the detection of methylenedioxypyrovalerone (MDPV), which is a designer drug of the synthetic cathinone family. This MDPV-binding CBSAzyme was able to achieve naked-eye detection of MDPV, as well as 10 other synthetic cathinones, at a concentration of 30 μM within 5 min. Impressively, the CBSAzyme did not respond to several structurally similar interferents, including other illicit drugs and common cutting agents, even at far higher concentrations. The high sensitivity and specificity of CBSAzymes allow for visual detection of illicit drugs in seized substances as well as in biosamples at low micromolar concentrations. Given the apparent generality of CBSAzyme engineering strategy and the resultant capacity to achieve rapid and sensitive visual small-molecule detection, we believe that this sensor framework serves as a valuable tool for the on-site detection of other small-molecule targets of medical, environmental, and forensic interest.
EXPERIMENTAL SECTION
CBSAzyme-Based Colorimetric Detection of Cocaine.
Detection was performed by incubating cocaine (final concentrations: 0, 0.1, 0.3, 1, 3, 10, 30, 100, 300, or 1000 μM) with 1 μM each of COC–CBSAzyme-SF and COC–CBSAzyme-LF in 40 mM HEPES (pH 7.0) with 1 μM hemin, 1 mM KCl, 30 mM NaCl, 0.05% Triton X-100, and 1% DMSO for 15 min at room temperature. The sample was then loaded into a well of a 384-well microplate, and the enzymatic reaction was initiated by the addition of 2 mM H2O2 and 1.5 mM ABTS (final concentrations). The absorbance at 418 nm was monitored every minute using a Tecan Infinite M1000 PRO microplate reader. The signal gain was calculated by A/A0, where A and A0 are the absorbance values of CBSAzyme mixture with and without target, respectively. For all calibration curves, error bars show standard deviation of signal gain from three measurements at each concentration.
CBSAzyme-Based Colorimetric Detection of MDPV.
Briefly, 1 μM each of MDPV-CBSAzyme-SF and MDPV-CBSAzyme-LF were incubated with MDPV (final concentrations: 0, 0.1, 0.3, 1, 3, 10, 30, 100, 300, or 1000 μM) in 40 mM HEPES (pH 7.0) with 1 μM hemin, 7 mM KCl, 77 mM NaCl, 0.05% Triton X-100, and 1% DMSO. This mixture was incubated for 15 min at room temperature. The sample was then loaded into a well of a 384-well microplate and the enzymatic reaction was initiated by the addition of 2 mM H2O2 and 1.5 mM ABTS (final concentrations). The absorbance at 418 nm was monitored every minute using a Tecan Infinite M1000 PRO microplate reader. The signal gain was calculated by A/A0, where A and A0 are the absorbance values of CBSAzyme mixture with and without target, respectively. For all calibration curves, error bars show standard deviation of signal gain from three measurements at each concentration.
RESULTS AND DISCUSSION
Engineering a New Split DNAzyme with Improved Catalytic Activity.
Peroxidase-mimicking DNAzymes are G-quadruplex-structured oligonucleotides that can perform a catalytic reaction similar to that of horseradish peroxidase.18 These DNAzymes can be split into two fragments as signal reporters.19,20 For example, Willner et al. have split a single-stranded DNAzyme into two symmetrical segments, with each strand containing two GGG repeats.19 This is known as the 2:2 split mode. The split DNAzyme segments can be reconstituted with hemin when they are brought into close proximity19 to form a layered G-quadruplex complex that exhibits peroxidase-like activity, catalyzing the oxidation of colorless ABTS into dark green ABTS• + using H2O2.27 However, the catalytic activity of this split DNAzyme is low.21,25 We believed that its activity could be improved by altering the spacers between the guanine triplets.
To test this hypothesis, we designed pairs of duplex DNA–DNAzyme conjugates with each fragment containing one segment of the 2:2 split DNAzyme (Figure 1A). We systematically mutated the original split DNAzyme and tested the activity of the resulting constructs (Figure 1B, Gq1, Gq2, Gq3, and Gq4) using the initial rate of ABTS•+ production as a benchmark. We first observed that the reaction rate for the native split DNAzyme (Gq1 (A0-B0); see Table S1 in the Supporting Information (SI)) was 14.1 nM/s (Figure 1B). By removing the thymine at position X (Gq2 (A1-B0); see Table S1), we observed a higher reaction rate of 19.0 nM/s (see Figure 1B, as well as Figure S1A in the SI), indicating that the thymine bulge originally present within the GGG repeat may be disruptive to the assembly of the split DNAzyme. We further replaced the adenine at position Y with the less bulky cytosine (Gq3 (A2-B0); see Table S1), and we found that the reaction rate further increased to 20.9 nM/s (see Figure 1B, as well as Figure S1A). This improvement in activity may be attributed to the lower steric hindrance of cytosine relative to adenine, which provides better flexibility for DNAzyme assembly. Finally, we removed the adenine at position Z from the adenine-cytosine dinucleotide spacer (Gq4 (A2-B1); see Table S1) and found that the split DNAzyme displayed even greater activity (27.3 nM/s) (see Figure 1B, as well as Figure S1B in the SI), essentially 2-fold higher than the original split DNAzyme. These mutation studies indicated that shorter spacers promoted the formation of a more compact G-quadruplex structure that boosts the catalytic activity of the split DNAzyme.
Figure 1.

Optimizing the activity of a split G-quadruplex-structured DNAzyme using a duplex DNA template. (A) Structures of hemin, ABTS, and the duplex DNA–DNAzyme conjugate formed by strands A and B and the scheme for DNAzyme-mediated oxidation of ABTS to produce the green-colored ABTS• +. X, Y, and Z indicate selected modification sites within the spacers between GGG repeats. (B) Structures of the assembled split DNAzyme modules (Gq1, Gq2, Gq3, and Gq4). (C) Reaction rates of each DNAzyme in terms of nanomolar ABTS• + produced per second.
Engineering Cocaine-Binding CBSA-DNAzyme Conjugates (CBSAzymes).
We have previously reported a cocaine-binding CBSA (COC-5335) with high target responsiveness that enabled one-step fluorescence detection of cocaine within 15 min.26 We demonstrated that the addition of a second binding pocket introduces cooperativity and significantly increases target binding affinity, in comparison with the corresponding parent split aptamer with a single binding pocket. In particular, the K1/2 (which represents the target concentration producing half occupancy of the receptor) of this CBSA (36 μM) is lower than that of the KD value of the parent split cocaine-binding aptamer with only a single binding site (~1 mM).26 To achieve visual detection of cocaine, we set out to develop a CBSA-DNAzyme conjugate using COC-5335 and the optimized split DNAzyme studied above. Specifically, we attached one split DNAzyme segment from Gq4 (5′-GGGCGGGT-3′) to the 3′ terminus of the long fragment of COC-5335 via an AT dinucleotide linker, and then we attached the other Gq4 segment (5′-AGGGCGGG-3′) to the 5′ terminus of the short fragment of COC-5335 via an AA dinucleotide linker (see Figure S2 in the SI). We termed this construct “CBSAzyme-5335–22”, wherein the first four numbers (5335) refers to the CBSA module and the last two numbers (22) refers to the split mode of the DNAzyme. We expected that these two CBSAzyme fragments are separated in the absence of cocaine and the split DNAzyme remains unassembled and incapable of oxidizing ABTS; thus, the solution remains clear. In the presence of cocaine, both fragments assemble, bringing the two DNAzyme segments into close proximity. The assembled DNAzyme module accommodates hemin within its G-quadruplex, allowing for H2O2-mediated oxidation of ABTS to ABTS• +, thereby rapidly turning the solution from clear to dark green.
Effect of CBSA Stem Length on CBSAzyme Performance.
We mixed the short and long fragments of CBSAzyme-5335–22 in reaction buffer in the presence or absence of cocaine. As expected, we observed that the absorption of ABTS• + at 418 nm greatly increased in the presence of cocaine. In contrast, the absorbance intensity only slightly increased in the absence of cocaine, most likely due to nonspecific background assembly of the CBSAzyme (see Figure S3A in the SI). To evaluate whether reducing thermostability can mitigate background assembly of the CBSAzyme and improve target response, we synthesized derivatives of CBSAzyme-5335–22 by truncating one or two base pairs from the termini of the CBSA fragments closest to the DNAzyme segments to generate CBSAzyme-5334–22 and CBSAzyme-5333–22, respectively. We then tested the performance of these derivatives under their optimized reaction conditions. CBSAzyme-5334–22 achieved lower background assembly while maintaining excellent catalytic activity (see Figure S3B in the SI), producing a higher target response than CBSAzyme-5335–22. CBSAzyme-5333–22 (Figure S3C in the SI) showed slightly decreased activity while having the same background signal as CBSAzyme-5334–22, demonstrating that further decreases in the thermostability of the CBSAzyme are deleterious for assay performance.
Effects of the DNAzyme Split Mode on CBSAzyme Performance.
We then set out to further improve the assay performance of CBSAzyme-5334–22 by destabilizing the DNAzyme module as a means of reducing the background signal. Studies have shown that symmetrically split DNAzymes can more easily self-assemble than those that are asymmetrically split.28 Therefore, we split our DNAzyme in a 1:3 mode, with one segment having three GGG repeats and the other only having one, and joined these fragments to our CBSA. Specifically, we coupled the three-GGG-repeat segment to the short fragment of the CBSA (COC-5334-SF), and the single-GGG-repeat segment onto the long fragment (COC-5334-LF), thereby generating CBSAzyme-5334–13 (Figure 2A). We compared the performance of CBSAzyme-5334–13 and CBSAzyme-5334–22 in the absence and presence of cocaine, and observed different levels of activity and background (Figures 2B and 2C). CBSAzyme-5334–13 demonstrated much lower background assembly, relative to CBSAzyme-5334–22 in the absence of cocaine, most likely due to the low stability of 1:3 split DNAzymes. The 1:3 split CBSAzyme also had a higher level of activity in the presence of cocaine, relative to the 2:2 split CBSAzyme. We further determined the Michaelis–Menten constant (KM) and turnover number (kcat) for both CBSAzymes. In the absence of cocaine, CBSAzyme-5334–22 had a slightly lower KM and higher kcat than the 1:3 split CBSAzyme, which explains its higher background signal. In the presence of cocaine, CBSAzyme-5334–13 had a much lower KM and higher kcat than the 2:2 split CBSAzyme (see Figure S4 in the SI), consistent with the observed higher catalytic activity.
Figure 2.

Effect of DNAzyme split mode on catalytic activity. (A) Constructing the 1:3 mode split DNAzyme and subsequent conjugation to COC-5334 via dinucleotide linkers to generate CBSAzyme-5334–13. Structures and time-course absorbance measurements for (B) CBSAzyme-5334–13 and (C) CBSAzyme-5334–22 in the absence and presence of 250 μM cocaine. [Each fragment] = 1 μM.
We then characterized the target-induced conformational change of CBSAzyme-5334–13 using circular dichroism (see Figure S5 in the SI). We observed that the CBSAzyme alone produced two distinct peaks: a negative peak at 245 nm and a broad positive peak spanning from ~260 nm to 280 nm. We believe the broad positive peak possibly encompasses two merged peaks, wherein one peak at 260 nm arises from a parallel G-quadruplex and another at ~270 nm originates from a duplex DNA structure.29 Furthermore, both the G-quadruplex and duplex DNA structures may contribute to the presence of the negative peak at 245 nm.29 When hemin was added, no measurable change in the shape or size of these peaks occurred, indicating that hemin itself cannot further assemble the CBSA or DNAzyme. The ellipticity increased at 270 nm when only cocaine was added, demonstrating target-induced CBSA assembly. Further addition of hemin induced a peak shift from 270 nm to 265 nm with an accompanying increase in the ellipticity of the peak, which likely corresponded to full assembly of the CBSA and DNAzyme modules.29 This shows that although cocaine can assemble the CBSA module, hemin is required to stabilize the DNAzyme. As a control, we monitored the conformational changes of CBSAzyme-5334–22 (see Figure S6 in the SI). The circular dichroism spectrum for this CBSAzyme resembled that of CBSAzyme-5334–13. Hemin alone again induced no observable change in the size or shape of the spectra. In the presence of only cocaine, the ellipticity of the peaks at 263 nm and 270 nm notably increased, but the subsequent addition of hemin did not cause any meaningful further change in the spectra. These results indicate that the split DNAzyme segments can readily assemble even without hemin, supporting the notion that the 2:2 split DNAzyme has a strong tendency to self-assemble. This likely explains the improvement in background signal that was observed when using the 1:3 split mode.
Optimization of the CBSAzyme Linker.
To further optimize the signal gain produced by our CBSAzyme, we investigated whether the linker length between the CBSA and DNAzyme modules had any impact on target-induced assembly and catalytic response. We first synthesized a variant of the long fragment of CBSAzyme-5334–13 in which we shortened the linker from AT to A and then performed our assay with the corresponding short fragment in the presence and absence of cocaine. In comparison to the original construct, which achieved a signal gain of 4.50 (see Figure S7A in the SI), shortening the linker of the long fragment yielded improved CBSAzyme performance with a higher signal gain of 5.27 (see Figure S7B in the SI). This is presumptively due to the fact that a shorter linker favors the assembly of two DNAzyme segments. We then modified the linker of the short fragment of CBSAzyme-5334–13 by replacing the original AA linker with a single A, and tested this variant’s performance in conjunction with the optimized long fragment. Shortening this linker resulted in a high background signal and a lower signal gain of 2.83 (see Figure S7C in the SI). Therefore, we retained the original AA linker of the short fragment. We believe that the optimized A/AA linker combination favors assembly of both the CBSA and the split DNAzyme. Circular dichroism analysis of this optimized CBSAzyme indicated that the assembled split DNAzyme retained a parallel G-quadruplex structure in the presence of hemin and cocaine (see Figure S8 in the SI), corresponding to its high DNAzyme activity.30,31
Colorimetric Detection of Cocaine with the Optimized CBSAzyme.
We then employed the optimized construct, hereon termed COC–CBSAzyme, for the visual detection of cocaine. A series of control experiments verified that signal is only produced in the presence of cocaine (Figure 3A). We observed no signal with H2O2 and ABTS alone. When hemin was added, ABTS was slowly oxidized by H2O2 to produce a minor increase in absorbance at 418 nm. Further addition of the long fragment did not produce any signal change, but addition of the short fragment with hemin in the absence of long fragment produced low levels of background. This is probably because the short fragment contains three GGG repeats, which may form intramolecular or intermolecular G-quadruplexes32 that can accommodate hemin to catalyze the oxidation of ABTS. When both fragments were included in the same mixture without cocaine, we observed low levels of background signal that were identical to that obtained with the short fragment alone, indicating that the fragments only underwent minimal assembly in the absence of target. The addition of cocaine promoted assembly of the CBSAzyme, yielding a large change in absorbance (Figure 3A) and rapid development of a dark green color within 15 min (see Figure S9 in the SI). To determine the optimum concentrations of short and long fragment, we performed the COC–CBSAzyme-based assay with varying concentrations of short fragment (0.2 to 2 μM) while keeping the concentration of the long fragment at 1 μM and vice versa. We found that 1 μM of each fragment produced the highest signal gain (see Figure S10 in the SI). To demonstrate the importance of cooperative target binding for sensitive target detection, we compared the performance of our CBSAzyme to a DNAzyme-linked split aptamer with a single binding site. We engineered this variant by truncating the 5-bp double-stranded duplex and the adjacent binding site from the 5′ end of the long fragment of COC–CBSAzyme to form SAzyme-334 (see Figure S11A in the SI). We observed a clear color change with COC–CBSAzyme at cocaine concentrations as low as 10 μM after only 2 min; however, no color change was obtained with SAzyme-334 at cocaine concentrations lower than 300 μM, even after 15 min (see Figures S11B and S11C in the SI). This demonstrates the far lower target responsiveness of the SAzyme-334, relative to COC–CBSAzyme.
Figure 3.

Utilizing COC–CBSAzyme for the visual detection of cocaine. (A) Time-dependent absorbance change at 418 nm with (1) reaction buffer alone, (2) hemin alone, (3) short fragment with hemin, (4) long fragment with hemin, and both fragments plus hemin in the (5) absence or (6) presence of 250 μM cocaine. (B) Calibration curve generated using 0–1000 μM cocaine. Inset shows the linear range from 0 to 10 μM cocaine. (C) Photographs of samples containing 0–1000 μM cocaine after 15 min of reaction. [Each fragment] = 1 μM. For the calibration curve, error bars show standard deviation of absorbance from three measurements at each concentration.
We finally determined the performance of COC–CBSAzyme for the visual detection of cocaine. In the absence of cocaine, the color of the solution only slightly changed after 30 min of reaction. However, in the presence of cocaine, the absorbance of ABTS• + gradually increased and the solution rapidly developed a dark green color. We observed that the color intensity of the solution was proportional to the concentration of cocaine and we were able to detect as low as 10 μM cocaine with the naked eye within only 5 min (see Figure S12 in the SI). We used a microplate reader to generate a calibration curve using the absorbance of ABTS• + at 418 nm after 15 min of reaction and obtained a measurable limit of detection of 1 μM with a linear range of 0 to 100 μM (see Figures 3B and 3C). We further tested the specificity of our assay against 250 μM concentrations of various common cutting agent and adulterants found in seized drug samples, including chlorpromazine, diphenhydramine, promazine, scopolamine, caffeine, levamisole, lidocaine, and sucrose (see Figure S13A in the SI). We found no cross-reactivity to scopolamine and sucrose, and only minimal cross-reactivity to caffeine, chlorpromazine, promazine, and levamisole at this concentration. However, moderate cross-reactivity was observed for diphenhydramine and lidocaine (see Figure S13B and S13C in the SI). This was expected, given that the cocainebinding aptamer has previously reported cross-reactivity to these compounds,33,34 as well as steroids,35 quinine/quinineanalogs,36,37 and aminoglycosides.38 Nevertheless, the successful performance of COC–CBSAzyme for rapid visual cocaine detection highlights the promise of this strategy for on-site drug detection in seized substances.
Demonstrating the Generality of the CBSAzyme Assay Format for the Detection of Other Small Molecules.
MDPV is a member of the family of designer drugs known as synthetic cathinones39—a class of drugs for which assay development lags well behind the emergence of new molecules into the market.40 To demonstrate the generality of our CBSAzyme approach, we first engineered an MDPV-binding CBSA based on our recently isolated three-way-junction-structured DNA aptamer (Figure 4A, I) that binds to MDPV with KD = 6 μM.41 We used the principles reported by Heemstra42 and ourselves26 to design the MDPV-binding CBSA. Briefly, an incision is made at the loop of one of the stem loops, the stems are shortened, and two of these constructs are linked together at their main stems. Per Heemstra, splitting at the other stem loop should not affect binding affinity as long as such regions are not involved directly in target binding. Specifically, we derived two different parent split aptamer pairs (Figure 4A, II) with a single binding pocket from the MDPV-binding aptamer, in which the GAA loop from stem 3 was removed and the number of base pairs in all stems was decreased. We then connected stem 1 of one parent split aptamer to stem 3 of the second via a single thymine linker on each strand to form a MDPV-binding CBSA, MDPV-6335 (see Figure 4A, III). To determine the binding affinity and cooperativity of MDPV-6335, we modified the short fragment with a 5′ IowaBlack RQ quencher and a 3′ Cy5 fluorophore (Figure 4B). In the absence of MDPV, the two fragments remain separate, with the fluorophore–quencher pair of the short fragment remaining in close proximity to each other and thus yielding no fluorescence signal (Figure 4B, left). When the target is added, the CBSA assembles to form a rigid target-CBSA complex, separating the fluorophore–quencher pair and producing a large fluorescence signal (Figure 4B, right). We used this fluorophore–quencher-modified version of MDPV-6335 to generate a binding curve for MDPV concentrations ranging from 0 to 3000 μM, and fit the resulting curve with the Hill equation43 (Figure 4C). We determined that MDPV-6335 had a binding affinity (K1/2) of 140.6 μM with a cooperativity (nH) of 1.8,43 which shows the high degree of target binding cooperativity.
Figure 4.

Engineering and characterization of an MDPV-binding CBSA. (A) A previously isolated MDPV-binding aptamer (I) was used to generate a pair of split aptamers with destabilized stems (II), which were then merged to create MDPV-6335 (III). (B) Scheme of a MDPV fluorescence assay using a fluorophore–quencher-modified version of MDPV-CBSA. (C) Binding curve of MDPV-6335 generated using 0–3000 μM MDPV and the calculated values for K1/2 and nH, using the Hill equation. Error bars show standard deviation of signal gain from three measurements at each concentration.
We then generated a MDPV-binding CBSAzyme by incorporating the optimized DNAzyme and linker sequences described above into the short and long fragments of MDPV-6335 to form MDPV–CBSAzyme (see Figure S14A in the SI). We tested the performance of this CBSAzyme and obtained a high signal gain in the presence of MDPV after 15 min (Figure S14B in the SI). We also used circular dichroism to confirm the structure and target-induced conformational changes of MDPV–CBSAzyme (see Figure S15 in the SI). For the CBSAzyme alone, two peaks were observed: a negative peak at 241 nm and a broad positive peak with a maximum at 275 nm and a shoulder at 260 nm. The peaks at 260 and 275 nm correspond to background assembly of the G-quadruplex-structured DNAzyme and duplex-structured CBSA, respectively, while the peak at 241 nm represents assembly of both modules.29 With the addition of hemin, the spectra did not change, indicating that hemin was unable to assemble either the CBSA or the split DNAzyme. With the addition of MDPV but no hemin, we observed a slight increase in the 275 nm positive peak, indicating low levels of target-induced CBSA assembly. When both hemin and MDPV were added, the positive peaks at 260 and 275 nm increased, confirming that both hemin and MDPV are required for efficient assembly.
We finally evaluated the performance of MDPV–CBSAzyme for target detection. Control experiments confirmed that the CBSAzyme produces a target-specific signal. We observed no background signal with hemin alone, or with the long fragment and hemin. As with our cocaine assay, we saw a very small absorbance signal with the mixture of short fragment and hemin. We observed similar low levels of absorbance signal when both long and short fragments were combined with hemin in the absence of target, but no visible color change was observed after 15 min of reaction (see Figure S16 in the SI). When MDPV was added, the absorbance of ABTS• + increased and the solution rapidly developed a dark green color after 15 min (see Figure S17A in the SI). We then generated a calibration curve using the absorbance ABTS• + at 418 nm after 15 min and obtained a detection limit of 3 μM MDPV with a linear range from 0 to 100 μM (see Figures S17B and S17C in the SI). Importantly, we were able to detect MDPV concentrations as low as 30 μM by the naked eye within 5 min (see Figure S18 in the SI). Notably, the CBSAzyme-based assay is robust and performs well in complex matrices. As a demonstration, we used MDPV–CBSAzyme to detect MDPV at different concentrations in biological samples such as saliva and urine. We found that the color of the MDPV-containing samples progressively changed over time, while the MDPV-free samples showed no obvious color changes (see Figures S19A and S20A in the SI). A limit of detection of 3 μM MDPV for both 50% saliva and 50% urine samples was obtained, based on the calibration curve generated after 5 min of reaction (see Figure S19B and S20B in the SI).
The isolated MDPV-binding aptamer has been reported to cross-react to many synthetic cathinones that share the same beta-keto phenethylamine core structure as MDPV, while having no binding affinity to common cutting agents.44 Therefore, we tested whether our MDPV–CBSAzyme retained this valuable feature of its parent aptamer, which would allow us to use a single assay to visually detect a variety of synthetic cathinones. We challenged our CBSAzyme-based assay against 250 μM concentrations of 11 different synthetic cathinones, including MDPV, methylone, pentylone, 3,4-methylenedioxy-α-pyrrolidinobutiophenone (MDPBP), mephedrone, 4′-methyl-α-pyrrolidinobutiophenone (MPBP), 4′-methyl-α-pyrrolidinohexanophenone (MPHP), naphyrone, methedrone, ethylone, and butylone. As expected, we observed a dark green color (see Figure 5A) and a high signal gain after 15 min for all tested synthetic cathinones (see Figure S21 in the SI), indicating that MDPV–CBSAzyme retains excellent cross-reactivity to structurally similar synthetic cathinone analogues. In addition, our CBSAzyme-based assay retained excellent specificity against structurally similar interferents, as we observed no measurable signal with 250 μM concentrations of common cutting agents and illicit drugs such as caffeine, benzocaine, lidocaine, sucrose, and methamphetamine (see Figure 5B and Figure S22 in the SI). Given the excellent sensitivity, cross-reactivity, and specificity of MDPV–CBSAzyme, we foresee that such construct can be employed for on-site visual detection of synthetic cathinones in seized substances and biosamples. Furthermore, based on the success of our CBSAzyme-based assays for colorimetric cocaine and synthetic cathinone detection, we believe that our CBSAzyme constructs can be generalized to detect any small molecule in a simple, label- and instrument-free manner.
Figure 5.

Cross-reactivity and specificity of the colorimetric MDPV-CBSAzyme-based assay. Photographs of samples containing (A) buffer-only control sample or 11 synthetic cathinones and (B) MDPV or 5 interferents after 15 min of reaction. All drugs or interferents were present at 250 μM. [Each fragment] = 1 μM.
CONCLUSION
In this work, we present a broadly applicable strategy for developing new CBSAzyme constructs that combine cooperative-target-binding and signal-reporting functionalities to enable rapid, visual small-molecule detection. CBSAzymes consist of two oligonucleotide fragments, each with a CBSA region containing two tandem target-binding domains and a split horseradish-peroxidase-mimicking DNAzyme module. CBSAzymes are ideal sensing elements for small-molecule detection, because they inherit the high target responsiveness of CBSAs26 and the catalytic activity of DNAzymes, which enables label-free, amplified naked-eye detection.
Using our optimized cocaine-binding CBSAzyme, we were able to observe a visible color change after just 5 min of reaction in the presence of 10 μM cocaine, with a measurable limit of detection of 1 μM using a microplate reader. Compared to other aptamer-based assays for colorimetric cocaine detection (see Table S2 in the SI), our assay is rapid, free of instruments, labels, and enzymes, and sufficiently sensitive for on-site drug screening in seized substances, where the concentration of the drugs is typically above 100 μM. A control experiment using a split aptamer with a single target-binding site conjugated to the same DNAzyme produced only a slight color change in the presence of 300 μM cocaine after 15 min, highlighting the importance of cooperative target-binding feature of CBSAzyme. Finally, we demonstrated the generality of the CBSAzyme engineering strategy by developing a new MDPV binding CBSAzyme for the detection of MDPV and other synthetic cathinones. This CBSAzyme enabled naked-eye detection of MDPV at concentrations as low as 30 μM after only 5 min, with a measurable limit of detection of 3 μM. The CBSAzyme also enabled robust MDPV detection in saliva and urine with a similar detection limit. Notably, this is the first reported assay capable of visual synthetic-cathinone detection in biological samples. Importantly, MDPV-CBSAzyme cross-reacted to 11 different synthetic cathinones while yielding no response to five interferents at a concentration of 250 μM, demonstrating the potential for a broadly applicable assay to detect the family of synthetic cathinones in seized substances.
Notably, this is the first work utilizing split-aptamer–DNAzyme conjugates to achieve sensitive, naked-eye small-molecule detection. The engineering strategy described herein offers a powerful means for on-site detection of small molecules for several reasons. First, CBSAzymes are simple to design, and we have developed a general model in which the target-binding domain of the CBSA module can be replaced with that of any other three-way-junction-structured aptamer, enabling the detection of a variety of small molecules. Such aptamers can be readily isolated via SELEX using three-way-junction structured libraries.41,45 Second, since CBSAzymes are nucleic acids, with no chemical modifications required, they can be easily synthesized in a cost-effective and reproducible manner, and the CBSAzymes themselves are chemically stable and resistant to harsh environmental conditions. Third, CBSAzyme-based assays require no instrumentation, and are simple to perform, requiring only sample-reagent mixing and visual interpretation. Fourth, such assays are rapid, as naked-eye detection can be achieved within just a few minutes. Therefore, we believe that CBSAzymes will prove broadly useful for the rapid and streamlined development of on-site colorimetric assays for other small-molecule targets including illicit drugs, toxins, disease biomarkers, and pharmaceuticals for applications such as law enforcement, environmental monitoring, and clinical diagnostics.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by the National Institutes of Health—National Institute on Drug Abuse [No. R15DA036821], the State Scholarship Fund of China [No. 201607060006], and the National Institute of Justice—Office of Justice Programs—U.S. Department of Justice Award [No. 2016-DN-BX-0167]. O. A. acknowledges the Presidential Fellowship awarded by the University Graduate School of Florida International University. The authors thank Yu Han for her help on protocol development of DNAzyme experiments.
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.analchem.9b00507.
Optimization of the catalytic activity of duplex DNA–split DNAzyme conjugates; strategy for engineering the cocaine-binding CBSAzyme from a cocaine-binding CBSA; effect of CBSA stem length on the performance of the cocaine-detecting CBSAzyme; determination of the enzyme kinetics of CBSAzyme-5334–22 and CBSAzyme-5334–13; circular dichroism spectra of CBSAzyme-5334–13, CBSAzyme-5334–22, and COC–CBSAzyme; optimization of the linker of CBSAzyme-5334–13; utilizing COC–CBSAzyme for the naked-eye detection of cocaine; optimization of the concentrations of the long and short fragments of COC–CBSAzyme in the CBSAzyme-based assay; comparison of the target responsiveness of COC–CBSAzyme and SAzyme-334; visual detection of cocaine using COC–CBSAzyme; specificity of the COC–CBSAzyme-based assay for cocaine detection; design and performance of an MDPV-binding CBSAzyme; circular dichroism spectra of MDPV–CBSAzyme; utilizing MDPV–CBSAzyme for the naked-eye detection of MDPV; the time-course visual detection of MDPV in buffer, 50% saliva and 50% urine using MDPV–CBSAzyme; target cross-reactivity and specificity of the MDPV–CBSAzyme-based assay; oligonucleotide sequences used in this work; comparison of this work to other aptamer-based assays for visual cocaine detection (PDF)
The authors declare no competing financial interest.
REFERENCES
- (1).Srinivasan B; Tung SJ Lab. Autom 2015, 20, 365–389. [DOI] [PubMed] [Google Scholar]
- (2).Long F; Zhu A; Shi H Sensors 2013, 13, 13928–13948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Harper L; Powell J; Pijl EM Harm Reduct. J 2017, 14, 52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Pfeiffer F; Mayer G Front. Chem 2016, 4, 25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Tuerk C; Gold L Science 1990, 249, 505–510. [DOI] [PubMed] [Google Scholar]
- (6).Lee JF; Hesselberth JR; Meyers LA; Ellington AD Nucleic Acids Res 2004, 32, D95–D100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Pendergrast PS; Marsh HN; Grate D; Healy JM; Stanton MJ Biomol. Technol 2005, 16, 224–234. [PMC free article] [PubMed] [Google Scholar]
- (8).Liu J; Lu Y Angew. Chem., Int. Ed 2006, 45, 90–94. [Google Scholar]
- (9).Zhang J; Wang L; Pan D; Song S; Boey FYC; Zhang H; Fan C Small 2008, 4, 1196–1200. [DOI] [PubMed] [Google Scholar]
- (10).Xia F; Zuo X; Yang R; Xiao Y; Kang D; Vallée-Bélisle A; Gong X; Yuen JD; Hsu BBY; Heeger AJ; Plaxco KW. Proc. Natl. Acad. Sci. U. S. A 2010, 107, 10837–10841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Yu H; Canoura J; Guntupalli B; Alkhamis O; Xiao Y Anal. Chem 2018, 90, 1748–1758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Liang P; Canoura J; Yu H; Alkhamis O; Xiao Y ACS Appl. Mater. Interfaces 2018, 10, 4233–4242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Li Z; Jin R; Mirkin CA; Letsinger RL Nucleic Acids Res 2002, 30, 1558–1562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Enüstün BV; Turkevich JJ Am. Chem. Soc 1963, 85, 3317–3328. [Google Scholar]
- (15).Ruttkay-Nedecky B; Kudr J; Nejdl L; Maskova D; Kizek R; Adam V Molecules 2013, 18, 14760–14779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Travascio P; Li Y; Sen D Chem. Biol 1998, 5, 505–517. [DOI] [PubMed] [Google Scholar]
- (17).Li Y; Sen D Biochemistry 1997, 36, 5589–5599. [DOI] [PubMed] [Google Scholar]
- (18).Travascio P; Bennet AJ; Wang DY; Sen D Chem. Biol 1999, 6, 779–787. [DOI] [PubMed] [Google Scholar]
- (19).Xiao Y; Pavlov V; Gill R; Bourenko T; Willner I ChemBioChem 2004, 5, 374–379. [DOI] [PubMed] [Google Scholar]
- (20).Deng M; Zhang D; Zhou Y; Zhou XJ Am. Chem. Soc 2008, 130, 13095–13102. [DOI] [PubMed] [Google Scholar]
- (21).Elbaz J; Moshe M; Shlyahovsky B; Willner I Chem. - Eur. J 2009, 15, 3411–3418. [DOI] [PubMed] [Google Scholar]
- (22).Freeman R; Liu X; Willner IJ Am. Chem. Soc 2011, 133, 11597–11604. [DOI] [PubMed] [Google Scholar]
- (23).Kong DM; Wang N; Guo XX; Shen HX Analyst 2010, 135, 545–549. [DOI] [PubMed] [Google Scholar]
- (24).Hao Y; Guo Q; Wu H; Guo L; Zhong L; Wang J; Lin T; Fu F; Chen G Biosens. Bioelectron 2014, 52, 261–264. [DOI] [PubMed] [Google Scholar]
- (25).Li T; Shi L; Wang E; Dong S Chem. - Eur. J 2009, 15, 3347–3350. [DOI] [PubMed] [Google Scholar]
- (26).Yu H; Canoura J; Guntupalli B; Lou X; Xiao Y Chem. Sci 2017, 8, 131–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Li T; Dong S; Wang E Chem. Commun 2007, 0, 4209–4211. [DOI] [PubMed] [Google Scholar]
- (28).Ren J; Wang J; Wang J; Luedtke NW; Wang E Biosens. Bioelectron 2012, 31, 316–322. [DOI] [PubMed] [Google Scholar]
- (29).Kypr J; Kejnovská I; Renčiuk D; Vorlíčková M NucleicAcids Res 2009, 37, 1713–1725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Cheng X; Liu X; Bing T; Cao Z; Shangguan D Biochemistry 2009, 48, 7817–7823. [DOI] [PubMed] [Google Scholar]
- (31).Kong DM; Cai LL; Guo JH; Wu J; Shen HX Biopolymers 2009, 91, 331–339. [DOI] [PubMed] [Google Scholar]
- (32).Víglaský V; Bauer L; Tlučková K Biochemistry 2010, 49, 2110–2120. [DOI] [PubMed] [Google Scholar]
- (33).Wang Z; Yu H; Canoura J; Liu Y; Alkhamis O; Fu F; Xiao Y Nucleic Acids Res 2018, 46, No. e81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Sharma AK; Heemstra JM J. Am. Chem. Soc 2011, 133, 12426–12429. [DOI] [PubMed] [Google Scholar]
- (35).Reinstein O; Neves MAD; Saad M; Boodram SN; Lombardo S; Beckham SA; Brouwer J; Audette GF; Groves P; Wilce MCJ; Johnson PE Biochemistry 2011, 50, 9368–9376. [DOI] [PubMed] [Google Scholar]
- (36).Slavkovic S; Altunisik M; Reinstein O; Johnson PE Bioorg. Med. Chem 2015, 23, 2593–2597. [DOI] [PubMed] [Google Scholar]
- (37).Reinstein O; Yoo M; Han C; Palmo T; Beckham SA; Wilce MCJ; Johnson PE Biochemistry 2013, 52, 8652–8662. [DOI] [PubMed] [Google Scholar]
- (38).Sachan A; Ilgu M; Kempema A; Kraus GA; Nilsen-Hamilton M Anal. Chem 2016, 88, 7715–7723. [DOI] [PubMed] [Google Scholar]
- (39).Banks ML; Worst TJ; Rusyniak DE; Sprague JE J. Emerg. Med 2014, 46, 632–642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Ellefsen KN; Anizan S; Castaneto MS; Desrosiers NA; Martin TM; Klette KL; Huestis MA Drug Test. Anal 2014, 6, 728–738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Yu H; Yang W; Alkhamis O; Canoura J; Yang K-A; Xiao Y Nucleic Acids Res 2018, 46, No. e43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Kent AD; Spiropulos NG; Heemstra JM Anal. Chem 2013, 85, 9916–9923. [DOI] [PubMed] [Google Scholar]
- (43).Hill AV J. Physiol 1910, 40, iv–vii. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Cole C; Jones L; McVeigh J; Kicman A; Syed Q; Bellis M Drug Test. Anal 2011, 3, 89–96. [DOI] [PubMed] [Google Scholar]
- (45).Yang K-A; Pei R; Stefanovic D; Stojanovic MN J. Am. Chem. Soc 2012, 134, 1642–1647. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.