

Abstract
Ca2+/calmodulin-dependent protein kinase II (CAMK2) is a key player in synaptic plasticity and memory formation. Mutations in Camk2a or Camk2b cause intellectual disability in humans, and severe plasticity and learning deficits in mice, indicating unique functions for each isoform. However, considering the high homology between CAMK2A and CAMK2B, it is conceivable that for critical functions, one isoform compensates for the absence of the other, and that the full functional spectrum of neuronal CAMK2 remains to be revealed.
Here we show that germline as well as adult deletion of both CAMK2 isoforms in male or female mice is lethal. Moreover, Ca2+-dependent activity as well as autonomous activity of CAMK2 is essential for survival. Loss of both CAMK2 isoforms abolished LTP, whereas synaptic transmission remained intact. The double-mutants showed no gross morphological changes of the brain, and in contrast to the long-considered role for CAMK2 in the structural organization of the postsynaptic density (PSD), deletion of both CAMK2 isoforms did not affect the biochemical composition of the PSD. Together, these results reveal an essential role for CAMK2 signaling in early postnatal development as well as the mature brain, and indicate that the full spectrum of CAMK2 requirements cannot be revealed in the single mutants because of partial overlapping functions of CAMK2A and CAMK2B.
SIGNIFICANCE STATEMENT CAMK2A and CAMK2B have been studied for over 30 years for their role in neuronal functioning. However, most studies were performed using single knock-out mice. Because the two isoforms show high homology with respect to structure and function, it is likely that some redundancy exists between the two isoforms, meaning that for critical functions CAMK2B compensates for the absence of CAMK2A and vice versa, leaving these functions to uncover. In this study, we generated Camk2a/Camk2b double-mutant mice, and observed that loss of CAMK2, as well as the loss of Ca2+-dependent and Ca2+-independent activity of CAMK2 is lethal. These results indicate that despite 30 years of research the full spectrum of CAMK2 functioning in neurons remains to be unraveled.
Keywords: CAMK2, hippocampus, survival, synaptic plasticity
Introduction
Since the discovery of the Ca2+/calmodulin-dependent protein kinase II (CAMK2) protein family in the 1970s, >2000 papers have been published in which the function of CAMK2A or CAMK2B, the most abundant CAMK2 isoforms in the brain, has been studied. The generation of different Camk2a mutants (of which the knock-out was already published >25 years ago (Silva et al., 1992a,b)) and Camk2b mutants, greatly contributed to the understanding of the role of these two isoforms in neuronal functioning, learning, and plasticity in mice (Mayford et al., 1995; Giese et al., 1998; Elgersma et al., 2002; Borgesius et al., 2011; Achterberg et al., 2014; Kool et al., 2016). Very recently, the importance of CAMK2A and CAMK2B for normal human neurodevelopment was shown (Küry et al., 2017; Stephenson et al., 2017; Akita et al., 2018; Chia et al., 2018).
CAMK2A and CAMK2B are estimated to have evolved from a common ancestral CAMK2 gene ∼1 billion years ago (Ryan and Grant, 2009) and are highly homologous, consisting both of a catalytic, regulatory, variable, and association domain. The catalytic and regulatory domain show an 89–93% sequence homology in rats (Tobimatsu and Fujisawa, 1989), whereas the differences lie within the variable domain, where CAMK2B but not CAMK2A contains an F-actin binding domain.
CAMK2 forms a holoenzyme of ∼12 subunits, which can consist of both CAMK2A and CAMK2B subunits. This CAMK2 holoenzyme is able to convert a short high-frequency signal into a long-term change in synaptic strength (for review, see Lisman et al., 2002; Hell, 2014). With the difference in binding affinity for Calmodulin, which is ∼8-fold higher for CAMK2B homomers than for CAMK2A homomers (half-maximum autophosphorylation of CAMK2 is achieved at 15 vs 130 nm calmodulin, respectively; Brocke et al., 1999), the subunit composition of the CAMK2 holoenzyme determines the sensitivity for fluctuating calcium levels (Thiagarajan et al., 2002). Upon calcium influx Ca2+/calmodulin binds CAMK2 in the regulatory domain (Vallano, 1989), allowing the release of a pseudosubstrate region of the protein from the catalytic domain. When two adjacent subunits within the holoenzyme are activated by Ca2+/calmodulin, one subunit can phosphorylate the neighboring subunit on Thr286 (CAMK2A) or Thr287 (CAMK2B) leaving this subunit autonomously active (Ca2+-independent activity) when calcium levels drop to baseline (Miller and Kennedy, 1986; Hanson et al., 1994). However, upon detachment of Ca2+/calmodulin from CAMK2, Thr305/Thr306 (CAMK2A) or Thr306/Thr307 (CAMK2B) within the calmodulin binding region can be phosphorylated thereby preventing future binding of Ca2+/calmodulin (thus Ca2+-dependent activity). The importance of the autophosphorylation events for CAMK2 function, was shown by generating Camk2a point mutants, in which the Thr286 or Thr305/Thr306 were mutated to either phosphomimic residues (e.g., Thr305Asp), or phosphodead residues (e.g., Thr286Ala). All of these mutations resulted in learning and plasticity phenotypes (Mayford et al., 1995; Giese et al., 1998; Elgersma et al., 2002).
In addition to an important enzymatic function, there are also studies showing that CAMK2A and CAMK2B play important and unique structural roles, using either Camk2a or Camk2b knock-out mice. For example, CAMK2A has been shown to play an important structural role in the presynapse in short-term plasticity (Hojjati et al., 2007) and CAMK2B plays an important structural role in determining the localization of CAMK2A during hippocampal plasticity, through its F-actin binding domain (Borgesius et al., 2011). Thus, the unique functions of CAMK2A and CAMK2B in neuronal functioning are well established. However, CAMK2A and CAMK2B are highly homologous, thus it is conceivable that there is substantial redundancy in function, and that these functions of CAMK2 are missed when studying the Camk2a or Camk2b single mutants.
In this study we aimed to reveal novel CAMK2 functions by studying different Camk2a/Camk2b double-mutants, showing that despite the enormous wealth of literature on CAMK2 functions, its full spectrum is still not uncovered and that the role of CAMK2 signaling in neurons is much more important than what was previously thought.
Materials and Methods
Animals.
In this study the following mice were used: Camk2a−/− (Camk2atm3Sva, MGI:2389262) and Camk2b−/− mice to generate Camk2a+/+;Camk2b+/+ (WT mice); Camk2a+/−;Camk2b−/− (mice heterozygous for Camk2a and knock-out for Camk2b); Camk2a−/−;Camk2b+/− (mice knock-out for Camk2a and heterozygous for Camk2b); Camk2a−/−;Camk2b−/− (Camk2a and Camk2b double knock-out mice); Camk2aT286A/T286A (Camk2atm2Sva, MGI:2158733) and Camk2bT287A/T287A mice to generate Camk2a+/T286A;Camk2bT287A/T287A (mice heterozygous for a T286A knock-in mutation in Camk2a and homozygous for a T287A knock-in mutation in Camk2b); Camk2aT286A/T286A;Camk2b+/T287A (mice homozygous for a T286A knock-in mutation in Camk2a and heterozygous for a T287A knock-in mutation in Camk2b); Camk2aT286A/T286A;Camk2bT287A/T287A (homozygous for T286A and T287A knock-in mutations in Camk2a and Camk2b, respectively); Camk2aT305D/T305D (Camk2atm5Sva, MGI: 2389272) and Camk2bA303R/A303R (Camk2btm2.1Yelg, MGI:5285573) mice to generate Camk2a+/T305D;Camk2bA303R/A303R (mice heterozygous for a T305D knock-in mutation in Camk2a and homozygous for a A303R knock-in mutation in Camk2b); Camk2aT305D/T305D;Camk2b+/A303R (mice homozygous for a T305D knock-in mutation in Camk2a and heterozygous for a A303R knock-in mutation in Camk2b); Camk2aT305D/T305D;Camk2bA303R/A303R (homozygous for T305D and A303R knock-in mutations in Camk2a and Camk2b, respectively); Camk2af/f;Camk2bf/f (homozygous floxed Camk2a (Camk2atm1.1Yelg, MGI:5662417) and Camk2b mice with no Cre expression; controls); Camk2af/f;Camk2bf/f;CAG-CreESR (homozygous floxed Camk2a and Camk2b mice with transgenic Cre expression throughout the body after injection with tamoxifen (Tg(CAG-cre/Esr1*)5Amc; MGI:2182767), and Camk2af/f;Camk2bf/f;CA3-Cre (knock-out mutants for Camk2a and Camk2b specifically in the CA3 region of the hippocampus (Grik4tm1.1(cre)Slab; MGI: 4398684, kindly provided by Ralf Schoepfer, Laboratory for Molecular Pharmacology, NPP, University College London, and York Rudhard, In Vitro Pharmacology, Evotec AG, Manfred Eigen Campus; Filosa et al., 2009). All mice were backcrossed >16 times in a C57BL/6J background and were group-housed in IVC cages (Sealsafe 1145 T, Tecniplast) with bedding material (Lignocel BK 8/15, Rettenmayer) on a 12 h light/dark cycle in 21°C (±1°C), humidity at 40–70% and with chow (No. 1 maintenance autoclave pellets, Special Diets Services) and water available ad libitum. Experimenters were blind to all genotypes throughout experiments and data analysis. Mice (males and females) were genotyped when they were 7-d-old, and re-genotyped after the mice were killed. Genotyping records were obtained and kept by a technician not involved in the experimental design, performance, and analysis. All experiments were done during the light phase, with animals between 2 and 4 months of age. All experiments were done with approval of the local Dutch Animal Ethical Committee for animal research and were in accordance with the European Communities Council Directive (86/609/EEC).
Generation of mouse mutants.
The generation of both the floxed and knock-out Camk2a (Elgersma et al., 2002; Achterberg et al., 2014) and Camk2b (Borgesius et al., 2011; Kool et al., 2016) mouse mutants have been described previously. All knock-in mutants used in this study have been published before as well: Camk2aT286A (Giese et al., 1998); Camk2aT305D (Elgersma et al., 2002); Camk2bT287A (Kool et al., 2016); and Camk2aA303R (Borgesius et al., 2011). To generate a CA3-specific deletion of the Camk2a and Camk2b genes we crossed Camk2af/f;Camk2bf/f mice with Grik4-Cre-Neo (in this study referred to as CA3-Cre mice). CA3-Cre expression starts as early as P5 and is predominantly restricted to the CA3 area of the hippocampus. To make sure that full deletion of the gene-of-interest had taken place, experiments were started at a minimum age of 8 weeks.
Tamoxifen injections.
Adult Camk2af/f;Camk2bf/f and Camk2af/f;Camk2bf/f;CAG-CreESR mice (8–10 weeks of age) were injected intraperitoneally with tamoxifen (Sigma-Aldrich; 0.1 mg/g bodyweight) for 8 consecutive days. To keep the levels of tamoxifen constant throughout injection days we kept a tight injection scheme, injecting mice 24 +/− 1 h after the previous injection. Tamoxifen was dissolved in sunflower oil (20 mg/ml). For electrophysiological experiments we killed adult mice (12–16 weeks old) 25 d after the first tamoxifen injection. Though tamoxifen is not known to have an effect on emotional reactivity, neurological functioning, or learning (Vogt et al., 2008) we injected both Camk2af/f;Camk2bf/f and Camk2af/f;Camk2bf/f;CAG-CreESR mice to control for any possible effects of tamoxifen.
Mass spectrometry.
Cortical tissue was isolated from adult Camk2af/f;Camk2bf/f and Camk2af/f;Camk2bf/f;CAG-CreESR mice 21 d after tamoxifen injection. Cell lysis was performed in 50 mm Tris-HCl, pH 8.2, with 0.5% sodium deoxycholate. Briefly, cells were incubated with the buffer and then boiled and sonicated for 10 min using a Bioruptor (Diagenode). Protein quantitation was performed using the colorimetric absorbance BCA protein assay kit (ThermoFisher Scientific). Proteins were reduced using 5 mm 1,4-dithiothreitol for 30 min at 50°C and subsequently alkylated using 10 mm iodoacetamide for 15 min in the dark. Proteins were first digested for 4 h with Lys-C (Wako Pure Chemicals; 1:200 enzyme–substrate ratio) and then overnight with trypsin (ThermoFisher Scientific; 1:50 enzyme–substrate ratio) at 30°C. The detergent was then removed by adding trifluoroacetic acid to 0.5% and precipitated detergent was spun down at 10,000 × g for 10 min. Extracted proteolytic peptides were labeled with TMT 6-plex labeling reagents (ThermoFisher Scientific) allowing for peptide quantitation. Peptides were mixed at the 6-plex level and further fractionated into six fractions by HILIC chromatography. Fractions were collected and analyzed by nanoflow LC-MS/MS. nLC-MS/MS was performed on EASY-nLC 1000 coupled to an Orbitrap Fusion Tribid mass spectrometer (ThermoFisher Scientific) operating in positive mode and equipped with a nanospray source. Peptides were separated on a ReproSil C18 reversed phase column (Dr. Maisch GmbH; column dimensions 15 cm × 50 μm, packed in-house) using a linear gradient from 0 to 80% B [A = 0.1% formic acid; B = 80% (v/v) acetonitrile, 0.1% formic acid] in 70 min and at a constant flow rate of 200 nl/min using a splitter. The column eluent was directly sprayed into the electrospray ionization source of the mass spectrometer. Mass spectra were acquired in continuum mode; fragmentation of the peptides was performed in data-dependent mode using the multinotch SPS MS3 reporter ion-based quantification method.
PSD fraction isolation.
Cortical tissue was isolated from adult Camk2af/f;Camk2bf/f and Camk2af/f;Camk2bf/f;CAG-CreESR mice 21 d after tamoxifen injection (Group 1) or in the days preceding death (4–5 weeks after the first tamoxifen injection; Group 2), and placed on ice until further processing. The lysates were prepared in homogenization buffer containing 0.32 m Sucrose, 1 mm NaHCO3, 1 mm MgCl2, 10 mm HEPES, pH 7.4, and protease and phosphatase inhibitors cocktails (P8340, P5726, and P0044, Sigma-Aldrich). This extract was immediately processed for the isolation of synaptosomes as described by Carlin et al. (1980). Protein concentration of the synaptosome fraction was determined using the BCA protein assay kit (Pierce) and adjusted to 1 mg/ml. Postsynaptic densities (PSD) were obtained from 100 μg of synaptosomes by adding 1% (v/v) Triton X-100 and HEPES, standing on ice for 15 min and centrifugation for 30 min. The pellet (PSD fraction) was dissolved in Laemmli sample buffer (1×). Samples were then used for subsequent Western blotting.
Western blot.
Mice were anesthetized using isoflurane and killed by decapitation. Brain samples (or acute hippocampal slices in the case of the Western blots after the electrophysiology experiments) were taken out quickly and stored in liquid nitrogen. Lysates were then first prepared and brain samples were homogenized in lysis buffer (10 mm Tris-HCl 6.8, 2.5% SDS, 2 mm EDTA). Protein concentration in the samples was determined and lysate concentrations were adjusted to 1 mg/ml. Western blots were probed with primary antibodies against either CAMK2A (clone 6G9, 1:20,000; Millipore, catalog #MAB8699; RRID:AB_2067919), CAMK2B (clone CB-β1, 1:10.000; ThermoFisher Scientific, catalog #13-9800; RRID:AB_2533045), PSD95 (1:1000; Proteintech Group, catalog #20665-1-AP; RRID:AB_2687961), GRIA2 (1:1000; Proteintech Group, catalog #11994-1-AP; RRID:AB_2113725), GRIN2B (1:2000; Proteintech Group, catalog #21920-1-AP; RRID:AB_11232223), and actin (1:20,000; Millipore, catalog #MAB1501R; RRID:AB_2223041) and secondary antibodies (goat anti-mouse and/or goat anti-rabbit, both 1:3000; Jackson ImmunoResearch, catalog #115-007-003; RRID:AB_2338476; and catalog #111-007-003; RRID:AB_2337925). Blots were stained either with Enhanced Chemiluminescence (ECL; 32106, Pierce) or stained and quantified using LI-COR Odyssey Scanner and Odyssey 3.0 software (Odyssey CLx; RRID:SCR_014579). Quantification of Western blot in ECL was done using ImageJ (Fiji; RRID:SCR_002285).
Immunohistochemistry and immunofluorescence.
Mice were anesthetized with pentobarbital and perfused transcardially with PBS followed by freshly prepared 4% paraformaldehyde (PFA) solution (Sigma-Aldrich). Brains were taken out after perfusion, postfixed for 1.5 h in PFA, and afterward kept in 30% sucrose solution overnight. Free-floating 40-μm-thick frozen sections were made and for immunohistochemistry, a standard avidin-biotin-immunoperoxidase complex method (ABC, Vector Laboratories) with CAMK2A (clone 6G9, 1:10,000; Millipore, catalog #MAB8699; RRID:AB_2067919) as the primary antibody and diaminobenzidine (0.05%) as the chromogen was used. For immunofluorescence, free-floating 40-μm-thick sections were washed in PBS once and afterward primary antibody was added (anti-CAMK2B, 1:1000; Abcam, catalog #ab34703; RRID:AB_2275072) diluted in PBS containing 2% NHS, 0.5% Triton X-100, and 150 mm bovine serum albumin (BSA) and kept at 4°C overnight for 48 h. Two days later sections were washed three times with PBS and secondary antibodies were added (Cy3 rabbit, 1:200 for immunofluorescence; Jackson ImmunoResearch, catalog #711-165-152; RRID:AB_2307443) diluted in PBS containing 2% NHS, 0.5% Triton X-100, and 150 mm BSA. After 1–2 h incubation of the secondary antibody at room temperature sections were washed four times in PB (0.05 m) and mounted on slides using chrome(3) potassium sulfatedodecahydrate and left to dry. Finally, for immunofluorescence, sections were covered using Mowiol (Sigma-Aldrich). For immunohistochemistry, the slices were, after drying, dehydrated in alcohol, cleared with xylene and covered using Permount (Fisher Scientific).
Half-life calculations.
For the protein degradation curves, mice received tamoxifen injections and were killed 4, 8, 10, 12, 15, 18, 21, and 24 d after the start of the experiment (n = 2 for each time point). Camk2af/f;Camk2bf/f mice without Cre were taken along for baseline levels. Protein levels were measured using Western blot and data were plotted using Prism data analysis software (GraphPad Prism; RRID:SCR_002798).
Local field potential surgery.
Mice were anesthetized with a mixture of isoflurane and oxygen (5% for induction and <2% for maintenance) and body temperature was kept constant at 37° during the entire surgical procedure. Temgesic (0.3 mg/ml) and lidocaine (Xylocaine, 100 mg/ml) were used for general and local analgesia. After fixation in a custom-designed stereotaxic apparatus, the scalp was opened to expose the skull. The membranous tissue underneath was cleared and the bone was surgically prepared with Optibond prime and adhesive (Kerr). The placement of the recording electrodes (Bear Lab Chronic Microelectrodes, 30070, FHC) was determined using a digital x-y manipulator according to the following coordinates: for the somatosensory cortex from the bregma AP: −1.94 mm, DL: −3.00 mm, DV: 0.6 mm; for the motor cortex from the bregma AP: +1.42, DL: + 1.75, DV: 0.5 mm. A reference electrode (silver wire) was placed on top of the vermis in the cerebellum. A small brass pedestal was attached to the skull with Charisma (Heraeus Kulzer) to ensure the fixation of the mice to the head bar during recording.
Local field potential recordings.
Two days after the surgical procedure, mice were head-fixed to a brass bar suspended over a cylindrical treadmill to allow anesthesia-free recording sessions and placed in a light-isolated Faraday cage. Mice were allowed to habituate to the setup before proceeding to the recording. Local field potential (LFP) signals were acquired every 2 d in sessions of 20 min each until the days preceding death, using the Open Ephys platform with a sampling rate of 3 kS/s and a bandpass filter between 0.1 and 200 Hz. Mice were observed daily and humanely killed when showing signs of behavioral discomfort (not before day 32 postinjection).
Electrophysiology.
Mice were killed after being anesthetized with isoflurane (Nicholas Piramal) and the brain was taken out quickly and submerged in ice-cold oxygenated (95%) and carbonated (5%) artificial CSF (ACSF; <4.0°) containing the following (in mm): 120 NaCl, 3.5 KCl, 2.5 CaCl2, 1.3 MgSO4, 1.25 NaH2PO4, 26 NaHCO3, and 10 d-glucose. Using a vibratome 400-μm-thick sagittal slices were made for CA3–CA1 experiments and 400-μm-thick coronal slices for CA3–CA3 experiments. Hippocampal sections were dissected out afterward and maintained at room temperature for at least 1.5 h in an oxygenated and carbonated bath to recover before experiments were initiated. At the onset of experiments hippocampal slices were placed in a submerged recording chamber and perfused continuously at a rate of 2 ml/min with ACSF equilibrated with 95% O2, 5% CO2 at 30°C. Extracellular recording of field EPSPs (fEPSPs) and stimulation were done using bipolar platinum (Pt)/iridium (Ir) electrodes (Frederick Haer). Stimulus duration of 100 μs for all experiments was used. In CA3–CA1 measurements, the stimulating electrode and recording electrode were placed on the CA3–CA1 Schaffer collateral afferents and apical dendrites of CA1 pyramidal cells (both 150–200 μm from stratum pyramidale), respectively. In CA3–CA3 measurements both the stimulating electrode and recording electrode were placed on the stratum radiatum of the CA3 area. The stratum lucidum was carefully avoided. Upon placement of the electrodes slices were given 20–30 min to rest before continuing measurements. All paired-pulse facilitation (PPF) experiments were stimulated at one-third of slice maximum. Varying intervals were used in PPF: 10, 25, 50, 100, 200, and 400 ms. CAMK2-dependent LTP was evoked using four different tetani: (1) 100 Hz (1 train of 1 s at 100 Hz, stimulated at one-third of slice maximum), (2) 200 Hz (4 trains of 0.5 s at 200 Hz, spaced by 5 s, stimulated at one-third of slice maximum), (3) theta burst (2 trains of 4 stimuli at 100 Hz, 200 ms apart, stimulated at two-thirds of slice maximum), and (4) CA3–CA3 LTP (2 trains of 1 s at 100 Hz 10 s apart, stimulated at one-third of slice maximum). A possible caveat in fEPSP measurements in the CA3 area is distinguishing between the mossy fiber pathway and the commissural (CA3–CA3) pathway. Therefore, we took several measures to make sure we recorded from the commissural pathway. First, with respect to the mossy fibers we used antidromic stimulation in the CA3 area. Second, we made use of the electrophysiological parameters PPF and 1 Hz frequency facilitation that differ between these two pathways. Mossy fiber transmission shows very strong facilitation (± 215% for PPF and ± 250% for 1 Hz; Scanziani et al., 1997), hence, we chose an upper limit of 180% for PPF and 130% for 1 Hz, and excluded all slices exceeding those limits. Finally, at the end of all experiments we used a pharmacological approach using DCG-IV (3 μm) to distinguish between both pathways. DCG-IV (3 μm) is known to reduce mossy fiber transmission by 80% (Kirschstein et al., 2004). This way, we felt confident that we only included data from experiments where we specifically stimulated CA3–CA3 synapses. Chemical LTD was induced using a 5 min wash-in of DHPG (100 μm; Tocris Biosciences) 20 min after establishing a stable baseline. For PKA-dependent LTP a similar baseline was established before chemical induction. We added picrotoxin (50 μm) to the ACSF throughout the experiment and LTP was induced chemically (cLTP) by bath application of picrotoxin (50 μm), forskolin (50 μm), and rolipram (0.1 μm) for 15 min, after which bath circulation was returned to ACSF with only picrotoxin (50 μm). During LTP slices were stimulated once per minute. Potentiation was measured as the normalized increase of the mean fEPSP slope for the duration of the baseline. During induction of chemical LTP slices were stimulated at half of slice maximum. Only stable recordings were included and this judgment was made blind to genotype. Average LTP was defined as the mean last 10 min of the normalized fEPSP slope.
Data analysis and statistics.
Statistical tests were performed using a two-way repeated-measures ANOVA or Student's t test to determine the effect of genotype in the experiments. In LTP experiments, the last 10 data points were used for comparison. The mass spectrometry data were analyzed with Proteome Discoverer 2.1 (RRID:SCR_014477). Peak lists were automatically created from raw data files using the Mascot Distiller software v2.3; Matrix Science; RRID:SCR_000307). The Mascot search algorithm (version 2.3.2, Matrix Science; RRID:SCR_000307) was used for searching against the UniProt database (taxonomy: Mus musculus, version December 2015; RRID:SCR_002380). The peptide tolerance was typically set to 10 ppm and the fragment ion tolerance was set to 0.8 Da. The reporter ion tolerance was set to 0.003 Da. A maximum number of 2 missed cleavages by trypsin were allowed and carbamidomethylated cysteine and oxidized methionine were set as fixed and variable modifications, respectively. Typical contaminants were omitted from the output tables. Protein ratios were calculated from the scaled normalized abundances of the reporter ions over the six quantitation channels. Gene ontology analysis was performed on the statistically different proteins with >20% difference in abundance ratio identified through mass spectrometry using the PANTHER Overrepresentation Test (GO Ontology database, Release date 2019-01-01, PANTHER; RRID:SCR_004869). The full list of proteins identified in the mass spectrometry analysis was used as reference dataset. A binomial test with Bonferroni correction was used for the statistical analysis. Statistical difference of PSD-associated proteins was assessed using a two-tailed Student's t test. For the analysis of the Western blots for the PSD associated protein levels, unpaired one-tailed or two-tailed Student's t test was used. For the LFP analysis, the average power density spectrum of the last 3 d of recording was obtained using MATLAB software (MathWorks; RRID:SCR_001622). The mean relative power was calculated over four frequency bands relative to the total power: delta (2–4 Hz), theta (5–8 Hz), β (13–30 Hz), and gamma (30–50 Hz). After determining normality of the distribution using the Wilk–Shapiro test, we determined statistical significance using an unpaired two-tailed Student's t test to assess the effect of genotype across each band frequency. For all statistical analyses α was set at 0.05. Values are represented as average ± SEM. Group sizes can be found in the figure legends. All values are based on number of slices measured. Each experimental group contained at minimum three different mice. All statistical tests were performed either using GraphPad Prism (RRID:SCR_002798) or SPSS Statistics v22.0 (RRID:SCR_002865).
Results
Loss of both CAMK2A and CAMK2B results in neonatal death
To unravel the full spectrum of CAMK2 functions, the Camk2a/Camk2b double-heterozygous mice (Camk2a−/+;Camk2b−/+) were intercrossed to obtain F2 Camk2a/Camk2b double knock-out mice (Camk2a−/−;Camk2b−/−; see Materials and Methods). Genotyping performed at day 7 on 222 pups, revealed 0 Camk2a−/−;Camk2b−/− double-mutants, whereas 14 pups were expected based on a Mendelian distribution (Fig. 1a), indicating that the double-mutant might be lethal. Moreover, all of the Camk2a+/−;Camk2b−/− mice died within 36 d after birth, whereas only 28% of the Camk2a−/−;Camk2b+/− died within the same period, indicating that complete loss of CAMK2B is less tolerated than complete loss of CAMK2A. Importantly, other genotypes obtained by this breeding (such as Camk2a+/−;Camk2b+/− mice, data not shown) appeared just as vital as wild-type mice. To understand whether the lethality of Camk2a−/−;Camk2b−/− mice was prenatal or postnatal, mice were monitored immediately from birth on. We observed that a small number of the born pups died within the first day after birth, which all appeared to be Camk2a−/−;Camk2b−/− upon genotyping. Taken these pups into account, we found that 6 of 100 pups were Camk2a−/−;Camk2b−/− double-mutants, which is the expected number of double-mutants, indicating that the Camk2a−/−;Camk2b−/− mutants are born at normal frequency (χ2: 9.4, p = 0.31), but die within 1 d after birth. When observing the pups directly after birth it was not possible to predict which pup would die, because the Camk2a−/−;Camk2b−/− pups did not show notable growth retardation or morphological changes, and a milk spot was visible in the abdomen, indicating that it was not lack of food intake that killed the pups. Additionally, immunohistochemistry showed no gross morphological changes in brains of pups on P0 (data not shown). Together, this shows that simultaneous loss of both CAMK2A and CAMK2B results in neonatal death, indicating a critical role of CAMK2 during this period, which cannot be revealed by studying the CAMK2 isoforms in isolation.
Figure 1.

Multiple Camk2a mutants crossed with Camk2b mutants and their survival in percentage of their total group size. a, Double knock-out mice for both Camk2a and Camk2b (Camk2a−/−;Camk2b−/−) die on P0. Homozygosity for Camk2b (with 1 functioning allele of Camk2a: Camk2a+/−;Camk2b−/−, n = 18) has a more severe impact on survival than homozygosity for Camk2a (and 1 functioning allele of Camk2b: Camk2a−/−;Camk2b+/−, n = 25). Camk2a+/+;Camk2b+/+ were used as controls (n = 16). b, Homozygous loss of Ca2+-dependent activity of both CAMK2A and CAMK2B (Camk2aT305D/T305D;Camk2b;A303R/A303R, n = 2) results in early death. Homozygosity for a A303R knock-in mutation in Camk2b and a heterozygous T305D knock-in mutation for Camk2a (Camk2a+/T305D;Camk2bA303R/A303R, n = 18) has a more severe impact on survival than a homozygous knock-in mutation for Camk2a and a heterozygous A303R knock-in mutation for Camk2b (Camk2aT305D/T305D;Camk2b+/A303R, n = 15). Camk2a+/+;Camk2b+/+ were used as controls (n = 4). c, Homozygous loss of autonomous activity of both CAMK2A and CAMK2B (Camk2aT286A/T286A;Camk2bT287A/T287A, n = 12) results in early death. Again, homozygosity for a T287A knock-in mutation in Camk2b (and a heterozygous T286A knock-in mutation for Camk2a: Camk2a+/T286A;Camk2bT287A/T287A, n = 34) has a more severe impact on survival than a homozygous knock-in mutation for Camk2a (and a heterozygous T287A knock-in mutation for Camk2b: Camk2aT286A/T286A;Camk2b+/T287A, n = 20). Camk2a+/+;Camk2b+/+ were used as controls (n = 14). With the exception of one mouse in the first experiment (a), all Camk2a+/+;Camk2b+/+ mice survived a minimum of up to 50 d postnatally. d, Model showing the effect of the different mutations used on the activity of the holoenzyme for the survival experiments. Green, CAMK2A; red, CAMK2B; yellow, calcium/calmodulin.
Loss of Ca2+-independent and Ca2+-dependent activity of CAMK2 results in neonatal death
Activity of CAMK2 is governed by multiple phosphorylation sites, of which the Thr286 (Thr287 in CAMK2B) is important for Ca2+-independent activity (Fig. 1d, bottom) and Thr305/Thr306 (Thr306/Thr307 in CAMK2B) for the Ca2+-dependent activity (Fig. 1d, middle), because they are located within the Ca2+/calmodulin binding site on CAMK2. Considering the phenotypes of the Camk2a and Camk2b single-mutants, we know that the CAMK2A phosphomimic mutation at Thr305 (CAMK2A-T305D), which blocks Ca2+/calmodulin binding and keeps CAMK2 in its inactivated state, is more detrimental than not having CAMK2A at all (Elgersma et al., 2002). This is also the case for a similar mutation in CAMK2B (CAMK2B-A303R) with respect to locomotion (Kool et al., 2016), although this is not the case for hippocampal learning (Borgesius et al., 2011). Hence, we expected that a double-mutant of CAMK2A-T305D and CAMK2B-A303R might be lethal as well. Indeed, when intercrossing Camk2a+/T305D;Camk2b+/A303R double-mutant mice we found that upon P7, only two Camk2aT305D/T305D;Camk2bA303R/A303R mutants were found in a total of 126 pups (number expected was 8). The two pups that survived until P7 still died a premature death within 16–23 d after birth (Fig. 1b). Similar to the Camk2a/Camk2b double-mutants, homozygous mutations in the Ca2+/calmodulin binding of CAMK2B were less tolerated than comparable mutations in CAMK2A (Fig. 1b).
We then tested whether Ca2+-independent activity (also known as autonomous activity) was essential for life. To that end, we intercrossed Camk2a+/T286A;Camk2b+/T287A double-mutant mice, such that autophosphorylation of both CAMK2A and CAMK2B at the Thr286/287 site is prevented (Giese et al., 1998; Kool et al., 2016). Surprisingly, despite the fact that these mice still have Ca2+-dependent activity, we found that the Camk2aT286A/T286A;Camk2bT287A/T287A mice started dying from P11 onward and that all had died by P27 (Fig. 1c). Again, homozygous mutations in CAMK2B were less tolerated than comparable mutations in CAMK2A. This indicates that CAMK2 autonomous activity is essential for survival.
Adult deletion of CAMK2A and CAMK2B is lethal
The premature death observed in the various Camk2a/Camk2b double-mutants described in the previous section, indicate a crucial role for CAMK2-dependent signaling during development. Using inducible Camk2a and Camk2b knock-out mice, we have recently shown that CAMK2-dependent signaling is also important after brain development (Achterberg et al., 2014; Kool et al., 2016). Notably, the phenotypes observed when deleting the Camk2a gene in adult mice are as severe as when deleting the gene at germline (Achterberg et al., 2014). Therefore, we postulated that deletion of both CAMK2A and CAMK2B could potentially also be lethal in adult mice. We generated inducible Camk2af/f;Camk2bf/f;CAG-CreESR mice, which were injected daily for 4 consecutive days with tamoxifen at 8 weeks of age to induce deletion of both Camk2a and Camk2b. Up until 4 d after the onset of gene deletion (first tamoxifen injection), the protein levels remained the same, but after that time point the levels reduced exponentially. Both CAMK2A and CAMK2B showed similar half-lives and decay constants in the cortex (CAMK2A, half-life: 3.5 d; CAMK2B, half-life: 2.8 d) as well as in the hippocampus (CAMK2A, half-life: 5.3 d; CAMK2B, half-life: 4.4 d; Fig. 2a). Immunohistochemical stainings at 21 d after onset of gene deletion showed that despite a few CAMK2A- or CAMK2B-positive cells, most brain areas were devoid of CAMK2 staining (Fig. 2b). Around 15–19 d (depending on the brain region) after onset of gene deletion, the levels of CAMK2A and CAMK2B dropped <10%, after which the Camk2af/f;Camk2bf/f;CAG-CreESR mice started to die. All injected Camk2af/f;Camk2bf/f;CAG-CreESR mice died within 24–53 d, with a median survival of 37 d (Fig. 2c). These mice did not show any obvious alterations in behavior until their last 24 h, during which they would stop moving, eating, and drinking. Importantly, Camk2af/f;Camk2bf/f mice without CAG-CreESR (control group) all survived.
Figure 2.

Adult loss of CAMK2A and CAMK2B is lethal. a, Western blot of cortical (top) and hippocampal (bottom) lysates using antibodies targeted against CAMK2A and CAMK2B. Actin was used as loading control. Days after first injection are indicated above the blots. Cre- mice were killed 4 d after the first tamoxifen injection. Bottom left graph, Nonlinear regression curve showing protein degradation in cortex, showing no difference in protein degradation rate of both CAMK2A and CAMK2B (n = 2 for each time point). Bottom right graph, Nonlinear regression curve showing protein degradation in hippocampus, where CAMK2B degradation is faster than CAMK2A degradation. Comparing both graphs, protein degradation of both CAMK2A and CAMK2B is faster in the cortex than in the hippocampus. b, Immunohistological stainings showing effective loss after tamoxifen injections of CAMK2A (top) and CAMK2B (bottom) in Camk2af/f;Camk2bf/f;CAG-CreESR mice 21 d after onset of gene deletion. c, Loss of both CAMK2A and CAMK2B (Camk2af/f;Camk2bf/f;CAG-CreESR) in adulthood is lethal. Both groups of mice [Camk2af/f;Camk2bf/f;CAG-CreESR (n = 9) and Camk2af/f;Camk2bf/f (n = 8)] received tamoxifen injections (see Materials and Methods).
CAMK2A and CAMK2B deletion does not result in overt changes in PSD composition
Analysis of the double-mutant brains did not reveal any gross morphology changes (Fig. 2b, and data not shown). Because both CAMK2A and CAMK2B have been shown to play an important structural role during plasticity and, upon activation, CAMK2 becomes highly enriched in PSD (Shen and Meyer, 1999), we hypothesized that the acute loss of both isoforms might interfere with the PSD protein composition and/or its stability. To evaluate this, we performed a proteomics analysis on cortical tissue from Camk2af/f;Camk2bf/f;CAG-CreESR mice compared with Camk2af/f;Camk2bf/f controls 21 d after onset of gene deletion. Surprisingly, in addition to a reduction of CAMK2A and CAMK2B, we found little changes in the key PSD-associated proteins (Sheng and Kim, 2011) in the Camk2af/f;Camk2bf/f;CAG-CreESR samples. Only SynGAP, GRIP1, the NMDA receptor subunits and the GRIA2 subunit of the AMPA receptor revealed a small (<10–20%) but significant increase in expression level in the Camk2af/f;Camk2bf/f;CAG-CreESR compared with Camk2af/f;Camk2bf/f control samples (Table 1; p values are two-tailed unpaired Student's t tests; for the raw values, see Table 1-1). The full list of proteins revealed some more proteins showing a significant difference of 20% or more in abundance ratio (upregulated or downregulated) between the Camk2af/f;Camk2bf/f;CAG-CreESR and Camk2af/f;Camk2bf/f control samples (Table 1-1). Subsequent GO-analysis did not reveal any overrepresented or underrepresented GO term in this dataset (PANTHER GO-analysis; Thomas et al., 2003).
Table 1.
Protein ratio abundances of key PSD enriched proteins
| Protein name | Gene | Uniprot ID | No. of Peptides | Abundance ratio: (wt)/(mut) | p |
|---|---|---|---|---|---|
| Calcium/calmodulin-dependent protein kinase type II subunit alpha | Camk2a | P11798 | 20 | 8.1 | <0.0001 |
| Calcium/calmodulin-dependent protein kinase type II subunit beta | Camk2b | Q5SVJ0 | 23 | 5.2 | <0.0001 |
| Glutamate receptor interacting protein 2 | Grip2 | G3XA20 | 2 | 1.3 | 0.394 |
| SH3 and multiple ankyrin repeat domains protein 2 | Shank2 | Q80Z38-3 | 31 | 1.3 | 0.317 |
| SH3 and multiple ankyrin repeat domains protein 3 | Shank3 | Q4ACU6-9 | 19 | 1.1 | 0.599 |
| Synaptic functional regulator FMR1 | Fmr1 | P35922 | 13 | 1.1 | 0.267 |
| Septin-7 | Sept7 | E9Q1G8 | 25 | 1.1 | 0.234 |
| alpha-actinin 4 | Actn4 | P57780 | 41 | 1.0 | 0.085 |
| MAGUK p55 subfamily member 5 | Mpp5 | B2RRY4 | 3 | 1.0 | 0.674 |
| Cadherin-2 | Cdh2 | P15116 | 11 | 1.0 | 0.255 |
| alpha-actinin 1a | Actn1 | A1BN54 | 42 | 1.0 | 0.273 |
| Src substrate cortactin | Cttn | Q60598 | 18 | 1.0 | 0.026 |
| Cortactin-binding protein 2 | Cttnbp2 | B9EJA2 | 20 | 1.0 | 0.161 |
| Catenin beta-1 | Ctnnb1 | Q02248 | 28 | 1.0 | 0.463 |
| PRKCA-binding protein | Pick1 | E9PUZ5 | 7 | 1.0 | 0.691 |
| Homer protein homolog 1 | Homer1 | Q9Z2Y3 | 20 | 1.0 | 0.401 |
| MAGUK p55 subfamily member 3 | Mpp3 | Q6XE40 | 6 | 1.0 | 0.88 |
| Neuroligin-3 | Nlgn3 | A2AGI2 | 6 | 1.0 | 0.878 |
| Metabotropic glutamate receptor 3 | Grm3 | Q9QYS2 | 15 | 1.0 | 0.879 |
| Neuroligin-4 like | Nlgn-4l | B0F2B4 | 7 | 1.0 | 0.961 |
| Neuroligin-2 | Nlgn2 | Q69ZK9 | 10 | 1.0 | 0.916 |
| Metabotropic glutamate receptor 5 | Grm5 | Q3UVX5 | 16 | 1.0 | 0.907 |
| MAGUK p55 subfamily member 2 | Mpp2 | Q9WV34-2 | 23 | 1.0 | 0.739 |
| Cytoplasmic FMR1-interacting protein 2 | Cyfip2 | Q5SQX6 | 36 | 1.0 | 0.802 |
| Glutamate receptor 3 | Gria3 | Q9Z2W9 | 22 | 1.0 | 0.868 |
| MAGUK p55 subfamily member 6 | Mpp6 | Q9JLB0-2 | 20 | 1.0 | 0.486 |
| SH3 and multiple ankyrin repeat domains protein 3 | Shank3 | A0A0A0MQD5 | 32 | 1.0 | 0.663 |
| Homer protein homolog 2 | Homer2 | Q9QWW1 | 4 | 1.0 | 0.807 |
| SH3 and multiple ankyrin repeat domains protein 1 | Shank1 | D3YZU1 | 36 | 1.0 | 0.604 |
| SH3 and multiple ankyrin repeat domains protein 2 | Shank2 | D3Z5K8 | 33 | 1.0 | 0.527 |
| Cytoplasmic FMR1-interacting protein 1 | Cyfip1 | Q7TMB8 | 23 | 1.0 | 0.633 |
| alpha-actinin 2 | Actn2 | Q9JI91 | 27 | 1.0 | 0.613 |
| Kalirin | Kalrn | A2CG49-7 | 29 | 1.0 | 0.228 |
| Glutamate receptor ionotropic, NMDA 2B | Grin2b | G3X9V4 | 11 | 1.0 | 0.099 |
| Glutamate receptor 2 | Gria2 | P23819-4 | 30 | 0.9 | 0.046 |
| Glutamate receptor 3 | Gria3 | B0QZW1 | 22 | 0.9 | 0.267 |
| Metabotropic glutamate receptor 2 | Grm2 | Q14BI2 | 16 | 0.9 | 0.336 |
| Metabotropic glutamate receptor 1 | Grm1 | P97772 | 4 | 0.9 | 0.134 |
| Glutamate receptor 1 | Gria1 | P23818 | 17 | 0.9 | 0.128 |
| Glutamate receptor-interacting protein 1 | Grip1 | Q925T6 | 4 | 0.9 | 0.043 |
| Glutamate receptor ionotropic, NMDA 1 | Grin1 | A2AI21 | 15 | 0.9 | 0.009 |
| Glutamate receptor ionotropic, NMDA 2A | Grin2a | P35436 | 13 | 0.9 | 0.022 |
| Neuroligin-1 | Nlgn1 | Q99K10 | 7 | 0.9 | 0.229 |
| Ras/Rap GTPase-activating protein SynGAP | Syngap1 | F6SEU4 | 36 | 0.9 | <0.001 |
| Glutamate receptor 4 | Gria4 | Q9Z2W8 | 8 | 0.8 | 0.465 |
| Glutamate receptor ionotropic, NMDA 2D | Grin2d | Q03391 | 1 | 0.8 | 0.0059 |
Selection of PSD-enriched proteins detected with mass spectrometry performed on Camk2af/f;Camk2bf/f;CAG-CreESR (n = 3) and Camk2af/f;Camk2bf/f control mice (n = 3) killed 21 d after onset of gene deletion. Protein ratios were calculated from the scaled normalized abundances of the reporter ions over the six quantitation channels. A Student's t test analysis was performed over the scaled normalized abundances to evaluate significance. For the full list of proteins analyzed using mass spectrometry, see Table 1-1. p-values lower then 0.05 are in put in bold.
List of significantly different protein abundance ratios as analyzed by mass spectrometry. Peak lists were automatically created from raw data files using the Mascot Distiller software (version 2.3; MatrixScience). The Mascot search algorithm (version 2.3.2, MatrixScience) was used for searching against the Uniprot database (taxonomy: Mus musculus, version December 2015). The peptide tolerance was typically set to 10 ppm and the fragment ion tolerance was set to 0.8 Da. The reporter ion tolerance was set to 0.003 Da. A maximum number of 2 missed cleavages by trypsin were allowed and carbamidomethylated cysteine and oxidized methionine were set as fixed and variable modifications, respectively. Typical contaminants were omitted from the output tables. Protein ratios were calculated from the scaled normalized abundances of the reporter ions over the six quantitation channels. Download Table 1-1, XLSX file (156.6KB, xlsx)
To zoom in further on the PSD itself, we isolated the synaptosomes from cortical tissue of Camk2af/f;Camk2bf/f;CAG-CreESR mice and Camk2af/f;Camk2bf/f control mice. Group 1 was killed 21 d after onset of gene deletion and the Group 2 was killed 1 d before they would die (assessed by observation, between 35 and 42 d after onset of gene deletion). We focused on the major PSD proteins PSD95, the NR2B subunit of the NMDA receptor (the subunit to which CAMK2A binds), and the GluA2 subunit of the AMPA receptor. Because the proteomics results showed only minor changes in the expression of PSD-related proteins from total lysates, equal amounts of synaptosomes were used as a starting point for the PSD fraction enrichment for both groups as shown in Figure 3, b and d (Group 1: PSD95, t(10) = 0.4, p = 0.7; NR2B, t(10) = 0.11, p = 0.91; GluR2, t(10) = 0.03, p = 0.98; Group 2: PSD95, t(14) = 0.68, p = 0.51; NR2B, t(14) = 0.32, p = 0.75; GluR2, t(14) = 0.35, p = 0.74; two-tailed unpaired t test). Despite the successful deletion of both CAMK2 isoforms as seen in the synaptosome fraction and in the PSD-enriched fraction (synaptosome fraction Group 1: CAMK2A, t(10) = 13.55, p < 0.0001; CAMK2B, t(10) = 7.45, p < 0.0001; PSD fraction Group 1: CAMK2A, t(10) = 10.82, p < 0.0001; CAMK2B, t(10) = 3.79, p < 0.005; synaptosome fraction Group 2: CAMK2A, t(14) = 7.69, p < 0.0001; CAMK2B, t(14) = 7.40, p < 0.0001; PSD fraction Group 2: CAMK2A, t(13) = 8.37, p < 0.0001; CAMK2B, t(13) = 3.59, p < 0.005; one-tailed unpaired t test; Fig. 3b,d), no significant differences in the protein levels of any of the PSD associated proteins were found, neither at 21 d after onset of gene deletion (PSD95: t(10) = 1.58, p = 0.15; NR2B: t(10) = 1.93, p = 0.08; GluR2: t(10) = 0.33, p = 0.75; two-tailed unpaired t test) nor close to death (PSD95: t(13) = 0.83, p = 0.42; NR2B: t(12) = 0.37, p = 0.72; GluR2: t(10) = 0.003, p = 0.1; two-tailed unpaired t test) in the Camk2af/f;Camk2bf/f;CAG-CreESR group compared with the Camk2af/f;Camk2bf/f control group. Together, this suggests that, in contrast to what could be expected, acute deletion of both CAMK2 isoforms does not lead to major alterations of the PSD composition.
Figure 3.

Adult loss of both CAMK2A and CAMK2B does not cause alterations of the postsynaptic density. a, b, Western blot examples of synaptosomes, soluble fraction and PSD-enriched fraction probed with common PSD proteins antibodies (PSD95, NR2B, and GluR2) of cortical lysates from Camk2af/f;Camk2bf/f;CAG-CreESR (a, n = 6; b, n = 6) and Camk2af/f;Camk2bf/f control mice (a, n = 6; b, n = 8) killed at 21 d after gene deletion (Group 1; a) or just before death (Group 2; b). Actin was used as a control to show efficient enrichment in the PSD fraction of solely PSD proteins. c, d, Quantification of protein levels in the synaptosomes fraction (top) and in the PSD fraction (bottom). Synaptosomes show equal starting levels for both Camk2af/f;Camk2bf/f;CAG-CreESR and control mice for all PSD proteins. Quantification of the PSD associated fraction of PSD95, NR2B, GluR2 shows no difference between the control mice and Camk2af/f;Camk2bf/f;CAG-CreESR mice at either time points of analysis. Error bars indicate SEM. **p < 0.005; ***p < 0.0001.
Adult loss of CAMK2A and CAMK2B does not cause changes in brain activity
Because it is known that downregulation of CAMK2A results in increased neuronal excitability and seizures (Butler et al., 1995), we assessed whether epilepsy could be the cause of death in these mice. Even though we did not observe any seizures in the Camk2af/f;Camk2bf/f;CAG-CreESR mice, we performed continuous EEG recordings on a subset of the mice, to monitor epileptic activity more carefully. None of the tested mice showed epileptic activity in their EEG recordings (data not shown).
To assess whether there is any decline of brain activity upon simultaneous deletion of CAMK2A and CAMK2B, the LFP was measured starting from 23 d after onset of gene deletion until 35 d after onset of gene deletion and detailed analysis of the power spectrum was performed. Power spectrum analysis on the last days of recording revealed no difference in the total power between Camk2af/f;Camk2bf/f;CAG-CreESR and Camk2af/f;Camk2bf/f control mice in [Somatosensory cortex (SScx), Total power: t(11) = 0.25, p = 0.81; Motor cortex (M1), Total power: t(11) = 0.01, p = 0.99; Fig. 4]. Also the contribution of specific frequency bands to the total power (normalized against the total power), did not reveal any differences (SScx: delta, t(5.5) = 0.59, p = 0.58; theta, t(11) = 1.81, p = 0.10; beta, t(4.19) = 1.66, p = 0.17; gamma, t(4.12) = 1.46, p = 0.22; M1: delta, t(11) = 0.60, p = 0.56; theta, t(11) = 0.24, p = 0.82; beta, t(11) = 0.13, p = 0.90; gamma, t(11) = 0.38, p = 0.71; Fig. 4a,b). These results indicate that there is no decline in brain activity upon simultaneous deletion of CAMK2A and CAMK2B.
Figure 4.

Power spectra analysis reveal no changes in brain activity upon deletion of both CAMK2A and CAMK2B. a, Example traces of LFP recordings obtained from somatosensory cortex of Camk2af/f;Camk2bf/f control mice (top trace; n = 8) and Camk2af/f;Camk2bf/f;CAG-CreESR (bottom trace; n = 5) during the last day of recording (35 d after onset of gene deletion). Bar graphs depict calculated total power across the last 3 d of recording (bottom left) and percentage of relative power normalized against the total power across four different frequency bands: delta (2–4 Hz), theta (5–10 Hz), beta (13–30 Hz), and gamma (30–50 Hz) (bottom right). b, Example traces of LFP recordings obtained from motor cortex of Camk2af/f;Camk2bf/f control mice (top trace; n = 8) and Camk2af/f;Camk2bf/f;CAG-CreESR (bottom trace; n = 5) during the last day of recording (35 d after gene deletion). Bar graphs depict averaged total power across the last 3 d of recording (bottom left) and percentage of relative power normalized against the total power across four different frequency bands: delta (2–4 Hz), theta (5–10 Hz), beta (13–30 Hz), and gamma (30–50 Hz) (bottom right). No differences were observed in the days preceding death in either total power or specific frequency bands. Error bars depict the SEM.
Loss of CAMK2A and CAMK2B completely abolishes LTP
We next tested the effect of the combined loss of CAMK2A and CAMK2B on basal synaptic transmission and LTP. Thus far, previous reports on conventional and inducible Camk2a and Camk2b single knock-out mice showed an impairment of LTP upon Camk2 gene deletion with ∼50% of residual LTP left compared with wild-type levels (Hinds et al., 1998; Elgersma et al., 2002; Borgesius et al., 2011; Achterberg et al., 2014). It is likely that the remaining fraction of LTP present in these mutant mice is provided by the remaining isoform present (CAMK2B in the case of Camk2a mutant mice and vice versa). To test this, we injected 8-week-old mice and chose 25 d after onset of gene deletion as the moment of kill and electrophysiological testing, corresponding to the moment when CAMK2 levels have dropped to a minimum but well before most of these mice start dying, to keep confounding effects of dying on the LTP measurements to a minimum (Fig. 5a). We measured basal synaptic transmission, PPF, LTP, and DHPG-induced LTD in the well studied CA3–CA1 Schaffer collateral pathway in acute hippocampal slices. In agreement with the lack of gross brain morphology changes, Camk2af/f;Camk2bf/f;CAG-CreESR mice still showed normal basal synaptic transmission as fiber volley amplitude, fEPSP slope, and their ratio did not differ significantly between both Camk2af/f;Camk2bf/f;CAG-CreESR and Camk2af/f;Camk2bf/f mice (effect of genotype: fiber volley: F(1,57) = 0,53, p = 0.47; fEPSP slope: F(1,90) = 0.74, p = 0.39; repeated-measures ANOVA; Fig. 5b). Subsequently PPF was not impaired in Camk2af/f;Camk2bf/f;CAG-CreESR mice (effect of genotype: PPF: F(1,89) = 0.34, p = 0.56; repeated-measures ANOVA; Fig. 5c). We then tested LTP by giving a 100 Hz tetanus and found a complete abolishment of LTP 50 min after induction in Camk2af/f;Camk2bf/f;CAG-CreESR mice (effect of genotype: 100 Hz LTP: F(1,35) = 19.86, p < 0.001; repeated-measures ANOVA; Fig. 5d). We then tested a much stronger LTP induction protocol (4 trains of 200 Hz for 0.5 s, spaced 5 s apart) known to activate different pools of CAMK2 in the spines (Lee et al., 2009). This LTP induction protocol yields normal LTP in Camk2b−/− mice and only partially reduces LTP in Camk2a−/− mice (Borgesius et al., 2011). However, like in the 100 Hz LTP protocol, Camk2af/f;Camk2bf/f;CAG-CreESR mutants showed complete absence of LTP in the 4 × 200 Hz protocol (effect of genotype: 200 Hz LTP: F(1,18) = 27.19, p < 0.001; repeated-measures ANOVA; Fig. 5e). To investigate whether other LTP inducing pathways were similarly affected, we tested PKA-dependent plasticity, using a 15 min wash-in of forskolin (50 μm) and rolipram (0.1 μm) in the presence of picrotoxin (50 μm) to induce cLTP. We found that also this LTP pathway was affected in the Camk2af/f;Camk2bf/f;CAG-CreESR mice, although considerable potentiation was still observed (effect of genotype: PKA LTP: F(1,55) = 9.75, p < 0.01; repeated-measures ANOVA; Fig. 5f). Finally, we found no involvement of CAMK2A and CAMK2B in DHPG-induced LTD (effect of genotype: DHPG LTD: F(1,20) = 1.05, p = 0.32; repeated-measures ANOVA; Fig. 5g). As a control for the efficiency of gene deletion, we performed Western blot analysis on the acute hippocampal slices used in these experiments. As expected, the slices of Camk2af/f;Camk2bf/f;CAG-CreESR mice showed a clear absence of CAMK2A and CAMK2B (Fig. 5h).
Figure 5.
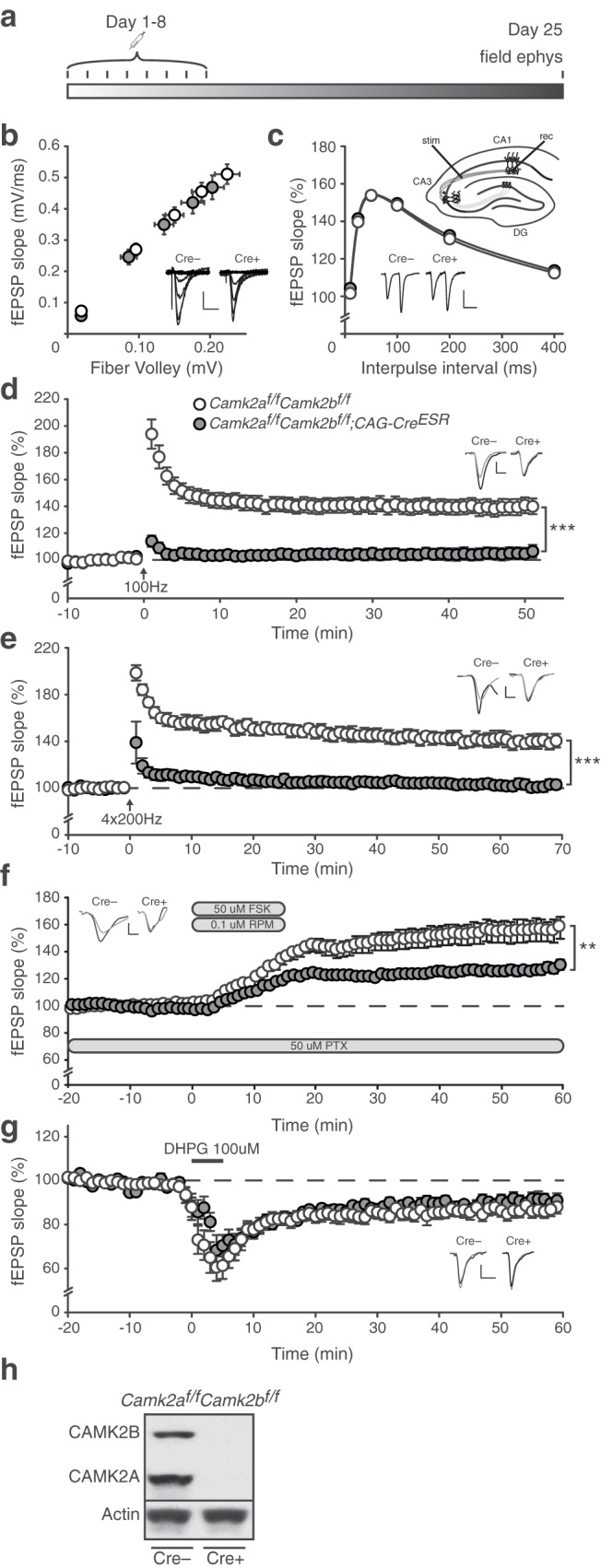
CAMK2A and CAMK2B are essential for CA3–CA1 LTP. a, Timeline showing the loss of CAMK2A and CAMK2B upon induction of genomic deletion with Tamoxifen injections (see Materials and Methods). Mice were killed 25 d after the first injection to conduct electrophysiological experiments. b, Camk2af/f;Camk2bf/f;CAG-CreESR mice [fiber volley: (n = 30 from 11 mice), fEPSP slope: (n = 42 from 11 mice)] show normal basal synaptic transmission compared with Camk2af/f;Camk2bf/f mice [fiber volley: (n = 29 from 15 mice), fEPSP slope: (n = 50 from 15 mice)]. c, Inset, Schematic overview of LTP induction in the CA3–CA1 pathway (see Materials and Methods). stim, Stimulating electrode; rec, recording electrode; DG, dentate gyrus. Camk2af/f;Camk2bf/f;CAG-CreESR mice (n = 40 from 11 mice) show normal PPF compared with Camk2af/f;Camk2bf/f mice (n = 51 from 15 mice). d, Camk2af/f;Camk2bf/f;CAG-CreESR mice (n = 16 from 6 mice) show a complete loss of 100 Hz LTP compared with Camk2af/f;Camk2bf/f mice (n = 21 from 9 mice). e, Camk2af/f;Camk2bf/f;CAG-CreESR mice (n = 11 from 4 mice) show a complete loss of 200 Hz LTP compared with Camk2af/f;Camk2bf/f mice (n = 9 from 5 mice). f, Camk2af/f;Camk2bf/f;CAG-CreESR mice (n = 28 from 7 mice) show impaired forskolin/rolipram-induced (50 μm/0.1 μm) LTP compared with Camk2af/f;Camk2bf/f mice (n = 29 from 7 mice). FSK, Forskolin; RPM, rolipram; PTX, picrotoxin. g, Camk2af/f;Camk2bf/f;CAG-CreESR mice (n = 9 from 5 mice) show normal DHPG-induced (100 μm) LTD compared with Camk2af/f;Camk2bf/f mice (n = 13 from 6 mice). h, Western blot showing efficient loss of both CAMK2A and CAMK2B in the acute hippocampal slices of Camk2af/f;Camk2bf/f;CAG-CreESR mice with normal CAMK2A and CAMK2B expression in Camk2af/f;Camk2bf/f mice. Actin levels are shown as loading control. Error bars indicate SEM. Electrophysiological example traces can be found within the figures. Scale bars: y, 0.2 mV; x, 10 ms. **p < 0.005; ***p < 0.0001.
Presynaptic CAMK2 is indispensable for CA3–CA1 LTP
CAMK2 was originally found as a presynaptic protein, involved in the phosphorylation of Synapsin I (DeLorenzo et al., 1979; Kennedy and Greengard, 1981; Kennedy et al., 1983). Additionally, more recent literature shows involvement of CAMK2A in vesicle release and short-term presynaptic plasticity as well as a role for presynaptic CAMK2 in LTP in culture conditions (Llinás et al., 1985; Nichols et al., 1990; Chapman et al., 1995; Hinds et al., 2003; Ninan and Arancio, 2004; Lu and Hawkins, 2006; Hojjati et al., 2007; Jiang et al., 2008; Pang et al., 2010; Achterberg et al., 2014). Therefore, it is likely that loss of both presynaptic as well as postsynaptic CAMK2 contributes to the LTP deficits described above. To investigate the requirement of presynaptic CAMK2 for LTP induction, we deleted Camk2 in the CA3 region of the hippocampus, without affecting CAMK2 expression in the other hippocampal regions, by crossing Camk2af/f;Camk2bf/f mice with a Cre-line in which Cre is under the control of the GRIK4 promotor (glutamate ionotropic receptor kainate type subunit 4). This gene is highly expressed in CA3 neurons, but absent in CA1 neurons (Filosa et al., 2009). We confirmed the specificity of this cre-line in Camk2af/f;CA3-Cre and Camk2bf/f;CA3-Cre mice. These mice showed specific deletion of CAMK2A and CAMK2B in the pyramidal cells of the CA3 region of the hippocampus at the age of 8 weeks, with no deletion in other parts of the hippocampus (Fig. 6a). Note that the mossy fibers coming from the dentate gyrus still express CAMK2A and can now be readily observed crossing through the CA3 area. We used immunofluorescence to confirm the deletion of CAMK2B in these mice. Although confirmation of the deletion of CAMK2B is complicated by the residual expression of CAMK2B in oligodendrocytes (Waggener et al., 2013) and interneurons (Lamsa et al., 2007), we could still observe a decrease in fluorescence in the stratum pyramidale of the CA3 region of the hippocampus (Fig. 6a).
Figure 6.

Presynaptic CAMK2A and CAMK2B are not necessary for CA3–CA1 LTP. a, Specific deletion of CAMK2A (Top) and CAMK2B (middle and bottom) in the CA3 area of the hippocampus. Immunohistochemical stainings showing deletion of CAMK2A in the CA3 area in Camk2af/f;CA3-Cre mice (top right). Note the absence of CAMK2A staining in all layers of the CA3 area except for mossy fibers coming from the dentate gyrus that still express CAMK2A. Immunofluorescent images showing deletion of CAMK2B in the CA3 area in Camk2bf/f;CA3-Cre mice (bottom right). Note that interneurons, oligodendrocytes, and mossy fibers coming from the dentate gyrus still express CAMK2B. The middle and bottom right pictures are enlarged images of boxed areas. Arrowheads show CA3 pyramidal cell somas in Camk2bf/f mice (middle right) and absence of fluorescence in cell somas in Camk2bf/f;CA3-Cre mice (bottom right). b, Camk2af/f;Camk2bf/f;CA3-Cre mice [fiber volley: (n = 19 from 6 mice), fEPSP slope: (n = 19 from 6 mice)] show normal basal synaptic transmission compared with Camk2af/f;Camk2bf/f mice [fiber volley: (n = 22 from 6 mice), fEPSP slope: (n = 22 from 6 mice)]. c, Inset, Schematic overview of LTP induction in the CA3–CA1 pathway (see Materials and Methods). stim, Stimulating electrode; rec, recording electrode; DG, dentate gyrus. Camk2af/f;Camk2bf/f;CA3-Cre mice (n = 41 from 6 mice) show normal PPF compared with Camk2af/f;Camk2bf/f mice (n = 44 from 6 mice). d, Camk2af/f;Camk2bf/f;CA3-Cre mice (n = 20 from 6 mice) show normal 100 Hz LTP compared with Camk2af/f;Camk2bf/f mice (n = 14 from 6 mice). e, Camk2af/f;Camk2bf/f;CA3-Cre mice (n = 9 from 4 mice) show normal theta burst LTP compared with Camk2af/f;Camk2bf/f mice (n = 10 from 4 mice). Error bars indicate SEM. Electrophysiological example traces can be found within the figures. Scale bars: y, 0.2 mV; x, 10 ms.
As we observed in the Camk2af/f;Camk2bf/f;CAG-CreESR mice, synaptic transmission and PPF were not affected in the absence of presynaptic CAMK2A and CAMK2B (effect of genotype: fiber volley: F(1,39) = 0.15, p = 0.70; fEPSP: F(1,39) = 0.89, p = 0.35; PPF: F(1,83) = 0.89, p = 0.35; repeated-measures ANOVA; Fig. 6b,c). As different LTP induction protocols rely on different molecular pathways (Grover and Teyler, 1990; Cavuş and Teyler, 1996; Raymond and Redman, 2002) and are reported to differ in their dependence on presynaptic CAMK2A (Lu and Hawkins, 2006), we tested 100 Hz as well as theta burst (2 Theta; see Materials and Methods) LTP-induction protocols. Surprisingly, both the 100 Hz and theta burst LTP-inducing protocols resulted in normal LTP in Camk2af/f;Camk2bf/f;CA3-Cre mice (effect of genotype: 100 Hz LTP: F(1,32) = 0.002, p = 0.96; Theta burst LTP: F(1,17) = 0.001, p = 0.98; repeated-measures ANOVA; Fig. 6d,e), indicating that presynaptic CAMK2A and CAMK2B are not required for LTP at the CA3–CA1 synapse of the hippocampus. Thus the complete loss of high-frequency stimulation (HFS) induced LTP in Camk2af/f;Camk2bf/f;CAG-CreESR mice is completely caused by loss of postsynaptic CAMK2.
Role of CAMK2A and CAMK2B in plasticity at the CA3–CA3 synapse
Control over gene deletion in the CA3 area of the hippocampus does not only provide a great tool for investigating a presynaptic role in the well studied CA3–CA1 synapse, it can also be used to investigate the role of CAMK2A and CAMK2B both presynaptically and postsynaptically in the associational/commissural pathway (CA3–CA3 synapse). The CA3–CA3 synapse in the hippocampus has been widely proposed to play a pivotal role in spatial processing and previous studies have shown that LTP at this synapse is NMDA dependent (Debanne et al., 1998). Therefore, we examined the role of CAMK2A and CAMK2B in the properties of the CA3–CA3 synapse in our Camk2af/f;Camk2bf/f;CA3-Cre mutant (Fig. 7a).
Figure 7.

Redundancy of CAMK2A and CAMK2B in CA3–CA3 LTP. a, Schematic overview of LTP induction in the CA3–CA3 pathway (see Materials and Methods). stim, Stimulating electrode; rec, recording electrode; DG, dentate gyrus. b, Camk2af/f;Camk2bf/f;CA3-Cre mice [fiber volley: (n = 27 from 4 mice), fEPSP slope: (n = 23 from 4 mice)] mice show normal basal synaptic transmission compared with Camk2af/f;Camk2bf/f mice [fiber volley: (n = 24 from 4 mice) fEPSP slope: (n = 24 from 4 mice)]. c, Camk2af/f;Camk2bf/f;CA3-Cre (n = 17 from 4 mice) mice show normal PPF compared with Camk2af/f;Camk2bf/f (n = 21 from 4 mice) mice. d, Camk2af/f;Camk2bf/f;CA3-Cre (n = 16 from 4 mice) show reduced 100 Hz LTP compared with Camk2af/f;Camk2bf/f (n = 18 from 4 mice) mice. e, DCG-IV only minimally blocks the fEPSP signal in both Camk2af/f;Camk2bf/f;CA3-Cre (n = 21 from 4 mice) and Camk2af/f;Camk2bf/f (n = 21 from 4 mice) mice. f, Camk2af/f;CA3-Cre (n = 29 from 8 mice) show normal 100 Hz LTP compared with Camk2a+/+ (n = 16 from 5 mice) mice. g, Camk2bf/f;CA3-Cre (n = 28 from 8 mice) show normal 100 Hz LTP compared with Camk2bf/f (n = 24 from 8 mice) mice. Error bars indicate SEM. Electrophysiological example traces can be found within the figures. Scale bars: y, 0.1 mV; x, 10 ms. *p = 0.01
Although CA3–CA3 basal synaptic transmission and PPF were not affected in the Camk2af/f;Camk2bf/f;CA3-Cre mice (effect of genotype: fiber volley: F(1,45) = 0.18, p = 0.68; fEPSP slope: F(1,49) = 0.03, p = 0.87; PPF: F(1,36) = 0.92, p = 0.34, repeated-measures ANOVA; Fig. 7b,c), deletion of both CAMK2A and CAMK2B in the CA3 region of the hippocampus resulted in a significant impairment of LTP at the CA3–CA3 synapse (effect of genotype: F(1,32) = 6.89, p = 0.01; repeated-measures ANOVA; Fig. 7d). Pyramidal neurons in the CA3 region of the hippocampus receive inputs from different pathways. The major source of inputs originates from the CA3 itself through the commissural pathway (CA3–CA3 synapse), but also the mossy fibers coming from the dentate gyrus (DG–CA3 synapse) form an important source. To distinguish between these different pathways, we used three different approaches: (1) antidromic stimulation (Fig. 5a), (2) differences in physiological characteristics of 1 Hz facilitation and PPF, and (3) differences in sensitivity to DCG-IV (see Materials and Methods). Importantly, 1 Hz stimulation did not show any facilitation [1 Hz facilitation (10th stimulus): Camk2af/f;Camk2bf/f;CA3-Cre 106.8 ± 2.1 vs Camk2af/f;Camk2bf/f 103.8 ± 2.3; 1 Hz Facilitation (10 stimuli, data not shown): effect of genotype: F(1,40) = 1.56, p = 0.22, repeated-measures ANOVA]. Furthermore, 3 μm DCG-IV reduced transmission by <20% in our experiments (81.9 ± 2.7% and 78.1 ± 3.3% transmission in the last 10 min for Camk2af/f;Camk2bf/f and Camk2af/f;Camk2bf/f;CA3-Cre, respectively; DCG-IV: t = 1.12, p = 0.27; unpaired two tailed t test; Fig. 7e). Therefore, we are confident that we selectively stimulated CA3–CA3 connections.
Notably, the phenotype observed in the Camk2af/f;Camk2bf/f;CA3-Cre mice appeared to be the result of the combined deletion of CAMK2A and CAMK2B and not because of absence of CAMK2A or CAMK2B alone, as the same parameters were not affected in the Camk2af/f;CA3-Cre or Camk2bf/f;CA3-Cre mice, in which only CAMK2A or CAMK2B is deleted from the CA3 region of the hippocampus (effect of genotype: Camk2af/f;CA3-Cre LTP: F(1,43) = 0.43, p = 0.52; Camk2bf/f;CA3-Cre LTP: F(1,50) = 0.004, p = 0.95, repeated-measures ANOVA; Fig. 7f,g). Because we did not observe an effect in the expression level of CAMK2A in Camk2af/f animals without Cre in comparison with wild-type mice (data not shown), we used the wild-type littermate Camk2a+/+ mice as the control group for one of these experiments (Fig. 7f). Importantly, basal synaptic transmission and PPF were both not significantly different and the control experiments for mossy fiber stimulation 1 Hz facilitation and DCG-IV wash-in were all within normal range and not significantly different for both Camk2af/f;CA3-Cre and Camk2bf/f;CA3-Cre mice (data not shown). Together, these data show that either CAMK2A or CAMK2B needs to be expressed at the CA3–CA3 synapse for normal CA3–CA3 LTP. Hence, this confirms the notion that some functions of CAMK2 cannot be uncovered using Camk2a or Camk2b single-mutants.
Discussion
CAMK2A and CAMK2B are the most abundant proteins in the brain and are shown to be crucial for learning, memory, and plasticity in mice and for normal neurodevelopment in humans (Silva et al., 1992a,b; van Woerden et al., 2009; Borgesius et al., 2011; Li et al., 2013; Küry et al., 2017; Stephenson et al., 2017). Because of the high homology in their structure, we contemplated that the full spectrum of CAMK2 functions has yet to be revealed, because crucial functions can potentially be masked due to compensation by the non-deleted isoform. In this study we confirm this hypothesis with the following findings. (1) Loss of CAMK2A and CAMK2B simultaneously is lethal both during development as well as in adult mice. (2) Loss of the combined CAMK2A and CAMK2B Ca2+-dependent or Ca2+-independent activity is lethal. (3) Simultaneous loss of CAMK2A and CAMK2B results in complete absence of LTP at the CA3–CA1 synapse in the hippocampus. (4) Simultaneous loss of CAMK2A and CAMK2B but not of CAMK2A or CAMK2B alone results in LTP deficits at the CA3–CA3 synapse. Together these results show that there is quite some redundancy in the CAMK2A and CAMK2B isoform function, and that crucial functions of CAMK2 remain to be uncovered.
It remains to be investigated why absence or dysregulation of CAMK2 function results in lethality. CAMK2 is critical for NMDA receptor-dependent signaling, and it is known that absence of the NR1 subunit of the NMDA receptor results in neonatal death due to respiratory failure in these mice (Forrest et al., 1994). CAMK2 activity mediates the emergence and maintenance of synchronous activity in the pre-Bötzinger complex, a center in the brainstem important for respiratory motor output (Mironov, 2013). Thus respiratory distress could underlie the lethality seen in the Camk2a−/−;Camk2b−/− mutants. Also NR2B-null mutants are not viable (Kutsuwada et al., 1996), which is because of a defect in the suckling response. Indeed lethality was rescued when the pups were hand-fed (Kutsuwada et al., 1996). We do not think that a similar mechanism underlies the lethality seen in our mice, because all pups had milk in their stomach just before they died. Moreover, when looking at the catalytically and autonomously inactive mutants (Camk2aT305D/T305D;Camk2bA303R/A303R and Camk2aT286A/T286A;Camk2bT287A/T287A, respectively) it seems unlikely that either feeding or breathing deficits are the cause of death, because some of these mice survive until P17 or later. These findings also indicate that enzymatic activity of CAMK2 is dispensable in the first 2 weeks after birth and that the survival of these mice depends solely on the presence of CAMK2, pointing out an essential structural role of CAMK2. Interestingly, during the early postnatal period, LTP is dependent on PKA, but not on CAMK2 and this switches ∼P9 (Yasuda et al., 2003), further supporting the lack of need of the enzymatic activity of CAMK2 during this early postnatal period. Our results further indicate that loss of CAMK2B is less tolerated than loss of CAMK2A, because the mice that were heterozygous for CAMK2A (knock-out or point mutant) and homozygous for CAMK2B died earlier than mice carrying a homozygous CAMK2A mutation and heterozygous CAMK2B mutation. This could be caused by the earlier onset of expression of CAMK2B, ∼E12.5, compared with CAMK2A, which starts to be expressed ∼P1 (Bayer et al., 1999).
Although homozygous loss of CAMK2A and CAMK2B resulted in death, it is surprising how little biochemical, morphological and electrophysiological phenotypes we observed in these double-mutants: (1) we observed no gross brain morphology differences. This is similar to the NR1- or the NR2B-null mutants (Forrest et al., 1994; Kutsuwada et al., 1996), and is consistent with the finding that the anatomical development of the brain does not need CAMK2-NMDA receptor-dependent plasticity or functional synapses (Sando et al., 2017; Sigler et al., 2017). (2) Although proteomic analysis indicated that simultaneous deletion of CAMK2A and CAMK2B did result in the upregulation or downregulation of a few PSD-associated proteins, we did not observe significant changes in the biochemical composition of the PSD. These findings indicate that presence of CAMK2 is not necessary for maintaining the PSD composition, which is a surprising finding given that it is generally believed that CAMK2 plays a critical role in the structural organization of the PSD (for review, see Hell, 2014). Future experiments are needed to investigate the structural integrity of the PSD in these mutants. (3) Total brain activity as measured with LFP recordings, remained unchanged, even close to death. This indicates that CAMK2 does not play a role in the background oscillatory activity present in the brain. (4) We did not observe changes in basal synaptic transmission, which we will discuss further.
The complete loss of HFS-induced LTP upon deletion of both CAMK2A and CAMK2B is not surprising considering the impairments both Camk2a−/− (Hinds et al., 1998; Elgersma et al., 2002) and Camk2b−/− (Borgesius et al., 2011) mice show in this kind of LTP. The complete loss of LTP in Camk2af/f;Camk2bf/f;CAG-CreESR mice suggests that the residual LTP left in the single knock-out mice is mediated by the remaining isoform. Indeed a recent study confirms the lack of a CAMK2-independent form of NMDA receptor-dependent LTP (Incontro et al., 2018). This is further strengthened by the observation that single amino acid mutations in Camk2a mutant mice such as Camk2aT286A (Giese et al., 1998) and Camk2aT305D (Elgersma et al., 2002) have more detrimental effects on LTP than the loss of the Camk2a isoform as a whole. This could well be explained by the dominant-negative effects of these Camk2a point mutations on the proper functioning of the heteromeric CAMK2A-CAMK2B holoenzyme.
In contrast to impaired LTP, basal synaptic transmission and PPF in the CA3–CA1 area were unaffected in absence of both CAMK2A and CAMK2B. Given the abundance of CAMK2 in synapses both presynaptically and postsynaptically and its major role in AMPA phosphorylation, GluN2B binding and AMPA insertion (for review, see Lisman et al., 2012), these findings were unexpected. However, these findings are in line with the observation that the PSD composition seems to be largely unaffected by the absence of CAMK2. Hence, synaptic strength in the CA3–CA1 synapse can be maintained independently of the presence of CAMK2A and CAMK2B. These findings are in contrast to a recent study that found a role for CAMK2A, but not CAMK2B, in basal synaptic strength in a subset of neurons (Incontro et al., 2018). Using CRISPR/Cas9 in slices and in utero electroporation to induce sparse deletion of CAMK2, these authors found that basal AMPA receptor- and NMDA receptor-dependent synaptic transmission requires CAMK2A, but not CAMK2B, that CAMK2A signaling is critically dependent on its binding to the NMDA receptor. Possibly, the discrepancy between our findings and theirs is explained by the different approaches. Targeting a subset of neurons, as done by Incontro et al. (2018), could induce a transcellular competitive process, which is absent in our global knock-out mutants.
Forskolin-induced LTP has been shown to be primarily PKA-dependent (Sokolova et al., 2006), but also NMDA-dependent components have been described (Otmakhov et al., 2004). Thus the impairment seen in cLTP in our Camk2af/f;Camk2bf/f;CAG-CreESR could be caused by the selective loss of the NMDA (CAMK2)-dependent pathway while leaving the PKA-dependent pathway intact.
The finding that presynaptic CAMK2 is dispensable for LTP at the CA3–CA1 synapse is surprising, considering that CAMK2 was first discovered as a presynaptic protein (DeLorenzo et al., 1979; Kennedy and Greengard, 1981; Kennedy et al., 1983). Indeed, CAMK2A has long been shown to play a role in presynaptic plasticity and regulation of neurotransmitter release at the CA3–CA1 synapse in brain slices (Llinás et al., 1985; Nichols et al., 1990; Chapman et al., 1995; Hinds et al., 2003; Hojjati et al., 2007; Jiang et al., 2008; Pang et al., 2010; Achterberg et al., 2014). Additionally, a previous study reported a role for presynaptic CAMK2 in LTP in dissociated hippocampal neurons (Ninan and Arancio, 2004). In this study, the role of presynaptic CAMK2 was assessed using different CAMK2 inhibitors, which when applied resulted in reduced LTP. Thus, in light of these studies, we expected to find a requirement for presynaptic CAMK2A and CAMK2B in synaptic transmission and LTP at the CA3–CA1 synapse. However, we found that presynaptic CAMK2 is completely indispensable for LTP at the Schaffer collateral pathway. There are a few reasons to explain the discordance between literature and our findings. First, in our model CAMK2 is already absent from as early as P5. We cannot exclude that the lack of effect in our experiments is because of presynaptic compensatory mechanisms during this phase of development. Second, to study the role of presynaptic CAMK2 in LTP in hippocampal cultures, CAMK2 inhibitors were used to block presynaptic CAMK2 activity. These are general CAMK2 blockers, blocking not only CAMK2A or CAMK2B, but also CAMK2D and CAMK2G. It could be that in our experiments CAMK2D and CAMK2G are compensating for the absence of CAMK2A and CAMK2B. Indeed, evidence for a neuronal role for CAMK2G is emerging (Ma et al., 2014; Cohen et al., 2018; Proietti Onori et al., 2018). Together, it is clear that complete understanding of the precise role of presynaptic CAMK2 at the CA3–CA1 Schaffer collateral synapse is still lacking.
CA3–CA3 LTP shares many characteristics with Schaffer collateral (CA3–CA1) LTP. Most notably, both CA3–CA3 and CA3–CA1 LTP depend on NMDA receptor activation and a postsynaptic rise in Ca2+ (Bradler and Barrionuevo, 1990; Zalutsky and Nicoll, 1990; Debanne et al., 1998, 1999; Bains et al., 1999; Smith and Swann, 1999; Pavlidis et al., 2000; Kakegawa et al., 2004). We show here that CA3–CA3 LTP is impaired when deleting both CAMK2A and CAMK2B, but not upon deletion of either CAMK2A or CAMK2B, indicating that one of the two CAMK2 isoforms needs to be present to support normal LTP. These results are in agreement with a previous study using a pharmacological approach, which described a presynaptic and postsynaptic role for CAMK2 in LTP at the CA3–CA3 synapse (Lu and Hawkins, 2006). Using a CAMK2 synthetic peptide (281–309) CAMK2 inhibitor, on organotypic slice cultures the authors saw a decrease of 50% in theta burst LTP when CAMK2 was blocked presynaptically or postsynaptically. This inhibitor does not distinguish between the four different CAMK2 isoforms, leaving open the question which CAMK2 isoform is responsible for the LTP deficit. Here we showed that the loss of either CAMK2A or CAMK2B isoform does not affect LTP, but that only the simultaneous absence of both CAMK2A and CAMK2B causes a CA3–CA3 LTP deficit. Interestingly, whereas CA3–CA1 LTP is completely abolished in absence of CAMK2, CA3–CA3 LTP is only reduced indicating that there are some fundamental differences underlying CA3–CA1 LTP compared with CA3–CA3 LTP.
Together, our results show that despite our vast knowledge about CAMK2, there are many aspects of CAMK2 function that remain to be uncovered.
Footnotes
This work was supported by a NWO-ALW Veni Grant (863.12.017 to G.M.v.W.), NWO-ZonMW-Vici Grant (918.866.10 to Y.E.).We thank Erika Goedknegt and Marcel de Brito van Velze for technical support, and Ralf Schoepfer and York Rudhard for kindly providing the Grik4tm1.1(cre)Slab mice.
The authors declare no competing financial interests.
References
- Achterberg KG, Buitendijk GH, Kool MJ, Goorden SM, Post L, Slump DE, Silva AJ, van Woerden GM, Kushner SA, Elgersma Y (2014) Temporal and region-specific requirements of αCaMKII in spatial and contextual learning. J Neurosci 34:11180–11187. 10.1523/JNEUROSCI.0640-14.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akita T, Aoto K, Kato M, Shiina M, Mutoh H, Nakashima M, Kuki I, Okazaki S, Magara S, Shiihara T, Yokochi K, Aiba K, Tohyama J, Ohba C, Miyatake S, Miyake N, Ogata K, Fukuda A, Matsumoto N, Saitsu H (2018) De novo variants in CAMK2A and CAMK2B cause neurodevelopmental disorders. Ann Clin Transl Neurol 5:280–296. 10.1002/acn3.528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bains JS, Longacher JM, Staley KJ (1999) Reciprocal interactions between CA3 network activity and strength of recurrent collateral synapses. Nat Neurosci 2:720–726. 10.1038/11184 [DOI] [PubMed] [Google Scholar]
- Bayer KU, Löhler J, Schulman H, Harbers K (1999) Developmental expression of the CaM kinase II isoforms: ubiquitous gamma- and delta-CaM kinase II are the early isoforms and most abundant in the developing nervous system. Brain Res Mol Brain Res 70:147–154. 10.1016/S0169-328X(99)00131-X [DOI] [PubMed] [Google Scholar]
- Borgesius NZ, van Woerden GM, Buitendijk GH, Keijzer N, Jaarsma D, Hoogenraad CC, Elgersma Y (2011) βCaMKII plays a nonenzymatic role in hippocampal synaptic plasticity and learning by targeting αCaMKII to synapses. J Neurosci 31:10141–10148. 10.1523/JNEUROSCI.5105-10.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradler JE, Barrionuevo G (1990) Heterosynaptic correlates of long-term potentiation induction in hippocampal CA3 neurons. Neuroscience 35:265–271. 10.1016/0306-4522(90)90080-N [DOI] [PubMed] [Google Scholar]
- Brocke L, Chiang LW, Wagner PD, Schulman H (1999) Functional implications of the subunit composition of neuronal CaM kinase II. J Biol Chem 274:22713–22722. 10.1074/jbc.274.32.22713 [DOI] [PubMed] [Google Scholar]
- Butler LS, Silva AJ, Abeliovich A, Watanabe Y, Tonegawa S, McNamara JO (1995) Limbic epilepsy in transgenic mice carrying a Ca2+/calmodulin-dependent kinase II alpha-subunit mutation. Proc Natl Acad Sci U S A 92:6852–6855. 10.1073/pnas.92.15.6852 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlin RK, Grab DJ, Cohen RS, Siekevitz P (1980) Isolation and characterization of postsynaptic densities from various brain regions: enrichment of different types of postsynaptic densities. J Cell Biol 86:831–845. 10.1083/jcb.86.3.831 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavuş I, Teyler T (1996) Two forms of long-term potentiation in area CA1 activate different signal transduction cascades. J Neurophysiol 76:3038–3047. 10.1152/jn.1996.76.5.3038 [DOI] [PubMed] [Google Scholar]
- Chapman PF, Frenguelli BG, Smith A, Chen CM, Silva AJ (1995) The alpha-Ca2+/calmodulin kinase II: a bidirectional modulator of presynaptic plasticity. Neuron 14:591–597. 10.1016/0896-6273(95)90315-1 [DOI] [PubMed] [Google Scholar]
- Chia PH, Zhong FL, Niwa S, Bonnard C, Utami KH, Zeng R, Lee H, Eskin A, Nelson SF, Xie WH, Al-Tawalbeh S, El-Khateeb M, Shboul M, Pouladi MA, Al-Raqad M, Reversade B (2018) A homozygous loss-of-function CAMK2A mutation causes growth delay, frequent seizures and severe intellectual disability. ELife 7: e32451. 10.7554/eLife.32451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen SM, Suutari B, He X, Wang Y, Sanchez S, Tirko NN, Mandelberg NJ, Mullins C, Zhou G, Wang S, Kats I, Salah A, Tsien RW, Ma H (2018) Calmodulin shuttling mediates cytonuclear signaling to trigger experience-dependent transcription and memory. Nat Commun 9:2451. 10.1038/s41467-018-04705-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Debanne D, Gähwiler BH, Thompson SM (1998) Long-term synaptic plasticity between pairs of individual CA3 pyramidal cells in rat hippocampal slice cultures. J Physiol 507:237–247. 10.1111/j.1469-7793.1998.237bu.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Debanne D, Gähwiler BH, Thompson SM (1999) Heterogeneity of synaptic plasticity at unitary CA3–CA1 and CA3–CA3 connections in rat hippocampal slice cultures. J Neurosci 19:10664–10671. 10.1523/JNEUROSCI.19-24-10664.1999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeLorenzo RJ, Freedman SD, Yohe WB, Maurer SC (1979) Stimulation of Ca2+-dependent neurotransmitter release and presynaptic nerve terminal protein phosphorylation by calmodulin and a calmodulin-like protein isolated from synaptic vesicles. Proc Natl Acad Sci U S A 76:1838–1842. 10.1073/pnas.76.4.1838 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elgersma Y, Fedorov NB, Ikonen S, Choi ES, Elgersma M, Carvalho OM, Giese KP, Silva AJ (2002) Inhibitory autophosphorylation of CaMKII controls PSD association, plasticity, and learning. Neuron 36:493–505. 10.1016/S0896-6273(02)01007-3 [DOI] [PubMed] [Google Scholar]
- Filosa A, Paixão S, Honsek SD, Carmona MA, Becker L, Feddersen B, Gaitanos L, Rudhard Y, Schoepfer R, Klopstock T, Kullander K, Rose CR, Pasquale EB, Klein R (2009) Neuron-glia communication via EphA4/ephrin-A3 modulates LTP through glial glutamate transport. Nat Neurosci 12:1285–1292. 10.1038/nn.2394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forrest D, Yuzaki M, Soares HD, Ng L, Luk DC, Sheng M, Stewart CL, Morgan JI, Connor JA, Curran T (1994) Targeted disruption of NMDA receptor 1 gene abolishes NMDA response and results in neonatal death. Neuron 13:325–338. 10.1016/0896-6273(94)90350-6 [DOI] [PubMed] [Google Scholar]
- Giese KP, Fedorov NB, Filipkowski RK, Silva AJ (1998) Autophosphorylation at Thr286 of the alpha calcium-calmodulin kinase II in LTP and learning. Science 279:870–873. 10.1126/science.279.5352.870 [DOI] [PubMed] [Google Scholar]
- Grover LM, Teyler TJ (1990) Two components of long-term potentiation induced by different patterns of afferent activation. Nature 347:477–479. 10.1038/347477a0 [DOI] [PubMed] [Google Scholar]
- Hanson PI, Meyer T, Stryer L, Schulman H (1994) Dual role of calmodulin in autophosphorylation of multifunctional CaM kinase may underlie decoding of calcium signals. Neuron 12:943–956. 10.1016/0896-6273(94)90306-9 [DOI] [PubMed] [Google Scholar]
- Hell JW. (2014) CaMKII: claiming center stage in postsynaptic function and organization. Neuron 81:249–265. 10.1016/j.neuron.2013.12.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinds HL, Tonegawa S, Malinow R (1998) CA1 long-term potentiation is diminished but present in hippocampal slices from alpha-CaMKII mutant mice. Learn Mem 5:344–354. [PMC free article] [PubMed] [Google Scholar]
- Hinds HL, Goussakov I, Nakazawa K, Tonegawa S, Bolshakov VY (2003) Essential function of -calcium/calmodulin-dependent protein kinase II in neurotransmitter release at a glutamatergic central synapse. Proc Natl Acad Sci U S A 100:4275–4280. 10.1073/pnas.0530202100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hojjati MR, van Woerden GM, Tyler WJ, Giese KP, Silva AJ, Pozzo-Miller L, Elgersma Y (2007) Kinase activity is not required for alphaCaMKII-dependent presynaptic plasticity at CA3–CA1 synapses. Nat Neurosci 10:1125–1127. 10.1038/nn1946 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Incontro S, Díaz-Alonso J, Iafrati J, Vieira M, Asensio CS, Sohal VS, Roche KW, Bender KJ, Nicoll RA (2018) The CaMKII/NMDA receptor complex controls hippocampal synaptic transmission by kinase-dependent and independent mechanisms. Nat Commun 9:2069. 10.1038/s41467-018-04439-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang X, Lautermilch NJ, Watari H, Westenbroek RE, Scheuer T, Catterall WA (2008) Modulation of CaV2.1 channels by Ca2+/calmodulin-dependent protein kinase II bound to the C-terminal domain. Proc Natl Acad Sci U S A 105:341–346. 10.1073/pnas.0710213105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kakegawa W, Tsuzuki K, Yoshida Y, Kameyama K, Ozawa S (2004) Input- and subunit-specific AMPA receptor trafficking underlying long-term potentiation at hippocampal CA3 synapses. Eur J Neurosci 20:101–110. 10.1111/j.1460-9568.2004.03461.x [DOI] [PubMed] [Google Scholar]
- Kennedy MB, Greengard P (1981) Two calcium/calmodulin-dependent protein kinases, which are highly concentrated in brain, phosphorylate protein I at distinct sites. Proc Natl Acad Sci U S A 78:1293–1297. 10.1073/pnas.78.2.1293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kennedy MB, Bennett MK, Erondu NE (1983) Biochemical and immunochemical evidence that the “major postsynaptic density protein” is a subunit of a calmodulin-dependent protein kinase. Proc Natl Acad Sci U S A 80:7357–7361. 10.1073/pnas.80.23.7357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirschstein T, von der Brelie C, Steinhäuser M, Vinçon A, Beck H, Dietrich D (2004) L-CCG-I activates group III metabotropic glutamate receptors in the hippocampal CA3 region. Neuropharmacology 47:157–162. 10.1016/j.neuropharm.2004.04.004 [DOI] [PubMed] [Google Scholar]
- Kool MJ, van de Bree JE, Bodde HE, Elgersma Y, van Woerden GM (2016) The molecular, temporal and region-specific requirements of the beta isoform of calcium/calmodulin-dependent protein kinase type 2 (CAMK2B) in mouse locomotion. Sci Rep 6:26989. 10.1038/srep26989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Küry S, van Woerden GM, Besnard T, Proietti Onori M, Latypova X, Towne MC, Cho MT, Prescott TE, Ploeg MA, Sanders S, Stessman HAF, Pujol A, Distel B, Robak LA, Bernstein JA, Denommé-Pichon AS, Lesca G, Sellars EA, Berg J, Carré W, et al. (2017) De novo mutations in protein kinase genes CAMK2A and CAMK2B cause intellectual disability. Am J Hum Genet 101:768–788. 10.1016/j.ajhg.2017.10.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kutsuwada T, Sakimura K, Manabe T, Takayama C, Katakura N, Kushiya E, Natsume R, Watanabe M, Inoue Y, Yagi T, Aizawa S, Arakawa M, Takahashi T, Nakamura Y, Mori H, Mishina M (1996) Impairment of suckling response, trigeminal neuronal pattern formation, and hippocampal LTD in NMDA receptor epsilon 2 subunit mutant mice. Neuron 16:333–344. 10.1016/S0896-6273(00)80051-3 [DOI] [PubMed] [Google Scholar]
- Lamsa KP, Heeroma JH, Somogyi P, Rusakov DA, Kullmann DM (2007) Anti-hebbian long-term potentiation in the hippocampal feedback inhibitory circuit. Science 315:1262–1266. 10.1126/science.1137450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SJ, Escobedo-Lozoya Y, Szatmari EM, Yasuda R (2009) Activation of CaMKII in single dendritic spines during long-term potentiation. Nature 458:299–304. 10.1038/nature07842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li K, Zhou T, Liao L, Yang Z, Wong C, Henn F, Malinow R, Yates JR 3rd, Hu H (2013) βCaMKII in lateral habenula mediates core symptoms of depression. Science 341:1016–1020. 10.1126/science.1240729 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lisman J, Schulman H, Cline H (2002) The molecular basis of CaMKII function in synaptic and behavioural memory. Nat Rev Neurosci 3:175–190. 10.1038/nrn753 [DOI] [PubMed] [Google Scholar]
- Lisman J, Yasuda R, Raghavachari S (2012) Mechanisms of CaMKII action in long-term potentiation. Nat Rev Neurosci 13:169–182. 10.1038/nrn3192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Llinás R, McGuinness TL, Leonard CS, Sugimori M, Greengard P (1985) Intraterminal injection of synapsin I or calcium/calmodulin-dependent protein kinase II alters neurotransmitter release at the squid giant synapse. Proc Natl Acad Sci U S A 82:3035–3039. 10.1073/pnas.82.9.3035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu FM, Hawkins RD (2006) Presynaptic and postsynaptic Ca2+ and CaMKII contribute to long-term potentiation at synapses between individual CA3 neurons. Proc Natl Acad Sci U S A 103:4264–4269. 10.1073/pnas.0508162103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma H, Groth RD, Cohen SM, Emery JF, Li B, Hoedt E, Zhang G, Neubert TA, Tsien RW (2014) γCaMKII shuttles Ca2+/CaM to the nucleus to trigger CREB phosphorylation and gene expression. Cell 159:281–294. 10.1016/j.cell.2014.09.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayford M, Wang J, Kandel ER, O'Dell TJ (1995) CaMKII regulates the frequency-response function of hippocampal synapses for the production of both LTD and LTP. Cell 81:891–904. 10.1016/0092-8674(95)90009-8 [DOI] [PubMed] [Google Scholar]
- Miller SG, Kennedy MB (1986) Regulation of brain type II Ca2+/calmodulin-dependent protein kinase by autophosphorylation: a Ca2+-triggered molecular switch. Cell 44:861–870. 10.1016/0092-8674(86)90008-5 [DOI] [PubMed] [Google Scholar]
- Mironov SL. (2013) Calmodulin and calmodulin kinase II mediate emergent bursting activity in the brainstem respiratory network (preBötzinger complex). J Physiol 591:1613–1630. 10.1113/jphysiol.2012.237362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nichols RA, Sihra TS, Czernik AJ, Nairn AC, Greengard P (1990) Calcium/calmodulin-dependent protein kinase II increases glutamate and noradrenaline release from synaptosomes. Nature 343:647–651. 10.1038/343647a0 [DOI] [PubMed] [Google Scholar]
- Ninan I, Arancio O (2004) Presynaptic CaMKII is necessary for synaptic plasticity in cultured hippocampal neurons. Neuron 42:129–141. 10.1016/S0896-6273(04)00143-6 [DOI] [PubMed] [Google Scholar]
- Otmakhov N, Khibnik L, Otmakhova N, Carpenter S, Riahi S, Asrican B, Lisman J (2004) Forskolin-induced LTP in the CA1 hippocampal region is NMDA receptor dependent. J Neurophysiol 91:1955–1962. 10.1152/jn.00941.2003 [DOI] [PubMed] [Google Scholar]
- Pang ZP, Cao P, Xu W, Südhof TC (2010) Calmodulin controls synaptic strength via presynaptic activation of calmodulin kinase II. J Neurosci 30:4132–4142. 10.1523/JNEUROSCI.3129-09.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavlidis P, Montgomery J, Madison DV (2000) Presynaptic protein kinase activity supports long-term potentiation at synapses between individual hippocampal neurons. J Neurosci 20:4497–4505. 10.1523/JNEUROSCI.20-12-04497.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Proietti Onori M, Koopal B, Everman DB, Worthington JD, Jones JR, Ploeg MA, Mientjes E, van Bon BW, Kleefstra T, Schulman H, Kushner SA, Küry S, Elgersma Y, van Woerden GM (2018) The intellectual disability-associated CAMK2G p.Arg292Pro mutation acts as a pathogenic gain-of-function. Hum Mutat 39:2008–2024. 10.1002/humu.23647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raymond CR, Redman SJ (2002) Different calcium sources are narrowly tuned to the induction of different forms of LTP. J Neurophysiol 88:249–255. 10.1152/jn.2002.88.1.249 [DOI] [PubMed] [Google Scholar]
- Ryan TJ, Grant SG (2009) The origin and evolution of synapses. Nat Rev Neurosci 10:701–712. 10.1038/nrn2717 [DOI] [PubMed] [Google Scholar]
- Sando R, Bushong E, Zhu Y, Huang M, Considine C, Phan S, Ju S, Uytiepo M, Ellisman M, Maximov A (2017) Assembly of excitatory synapses in the absence of glutamatergic neurotransmission. Neuron 94:312–321.e3. 10.1016/j.neuron.2017.03.047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scanziani M, Salin PA, Vogt KE, Malenka RC, Nicoll RA (1997) Use-dependent increases in glutamate concentration activate presynaptic metabotropic glutamate receptors. Nature 385:630–634. 10.1038/385630a0 [DOI] [PubMed] [Google Scholar]
- Shen K, Meyer T (1999) Dynamic control of CaMKII translocation and localization in hippocampal neurons by NMDA receptor stimulation. Science 284:162–166. 10.1126/science.284.5411.162 [DOI] [PubMed] [Google Scholar]
- Sheng M, Kim E (2011) The postsynaptic organization of synapses. Cold Spring Harb Perspect Biol 3:a005678. 10.1101/cshperspect.a005678 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sigler A, Oh WC, Imig C, Altas B, Kawabe H, Cooper BH, Kwon HB, Rhee JS, Brose N (2017) Formation and maintenance of functional spines in the absence of presynaptic glutamate release. Neuron 94:304–311.e4. 10.1016/j.neuron.2017.03.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silva AJ, Stevens CF, Tonegawa S, Wang Y (1992a) Deficient hippocampal long-term potentiation in alpha-calcium-calmodulin kinase II mutant mice. Science 257:201–206. 10.1126/science.1378648 [DOI] [PubMed] [Google Scholar]
- Silva AJ, Paylor R, Wehner JM, Tonegawa S (1992b) Impaired spatial learning in alpha-calcium-calmodulin kinase II mutant mice. Science 257:206–211. 10.1126/science.1321493 [DOI] [PubMed] [Google Scholar]
- Smith KL, Swann JW (1999) Long-term depression of perforant path excitatory postsynaptic potentials following synchronous network bursting in area CA3 of immature hippocampus. Neuroscience 89:625–630. 10.1016/S0306-4522(98)00651-4 [DOI] [PubMed] [Google Scholar]
- Sokolova IV, Lester HA, Davidson N (2006) Postsynaptic mechanisms are essential for forskolin-induced potentiation of synaptic transmission. J Neurophysiol 95:2570–2579. 10.1152/jn.00617.2005 [DOI] [PubMed] [Google Scholar]
- Stephenson JR, Wang X, Perfitt TL, Parrish WP, Shonesy BC, Marks CR, Mortlock DP, Nakagawa T, Sutcliffe JS, Colbran RJ (2017) A novel human CAMK2A mutation disrupts dendritic morphology and synaptic transmission, and causes ASD-related behaviors. J Neurosci 37:2216–2233. 10.1523/JNEUROSCI.2068-16.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thiagarajan TC, Piedras-Renteria ES, Tsien RW (2002) α- and βCaMKII. inverse regulation by neuronal activity and opposing effects on synaptic strength. Neuron 36:1103–1114. 10.1016/S0896-6273(02)01049-8 [DOI] [PubMed] [Google Scholar]
- Thomas PD, Campbell MJ, Kejariwal A, Mi H, Karlak B, Daverman R, Diemer K, Muruganujan A, Narechania A (2003) PANTHER: a library of protein families and subfamilies indexed by function. Genome Res 13:2129–2141. 10.1101/gr.772403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tobimatsu T, Fujisawa H (1989) Tissue-specific expression of four types of rat calmodulin-dependent protein kinase II mRNAs. J Biol Chem 264:17907–17912. [PubMed] [Google Scholar]
- Vallano ML. (1989) Separation of isozymic forms of type II calcium/calmodulin-dependent protein kinase using cation-exchange chromatography. J Neurosci Methods 30:1–9. 10.1016/0165-0270(89)90067-8 [DOI] [PubMed] [Google Scholar]
- van Woerden GM, Hoebeek FE, Gao Z, Nagaraja RY, Hoogenraad CC, Kushner SA, Hansel C, De Zeeuw CI, Elgersma Y (2009) betaCaMKII controls the direction of plasticity at parallel fiber-purkinje cell synapses. Nat Neurosci 12:823–825. 10.1038/nn.2329 [DOI] [PubMed] [Google Scholar]
- Vogt MA, Chourbaji S, Brandwein C, Dormann C, Sprengel R, Gass P (2008) Suitability of tamoxifen-induced mutagenesis for behavioral phenotyping. Exp Neurol 211:25–33. 10.1016/j.expneurol.2007.12.012 [DOI] [PubMed] [Google Scholar]
- Waggener CT, Dupree JL, Elgersma Y, Fuss B (2013) CaMKII regulates oligodendrocyte maturation and CNS myelination. J Neurosci 33:10453–10458. 10.1523/JNEUROSCI.5875-12.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yasuda H, Barth AL, Stellwagen D, Malenka RC (2003) A developmental switch in the signaling cascades for LTP induction. Nat Neurosci 6:15–16. 10.1038/nn985 [DOI] [PubMed] [Google Scholar]
- Zalutsky RA, Nicoll RA (1990) Comparison of two forms of long-term potentiation in single hippocampal neurons. Science 248:1619–1624. 10.1126/science.2114039 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
List of significantly different protein abundance ratios as analyzed by mass spectrometry. Peak lists were automatically created from raw data files using the Mascot Distiller software (version 2.3; MatrixScience). The Mascot search algorithm (version 2.3.2, MatrixScience) was used for searching against the Uniprot database (taxonomy: Mus musculus, version December 2015). The peptide tolerance was typically set to 10 ppm and the fragment ion tolerance was set to 0.8 Da. The reporter ion tolerance was set to 0.003 Da. A maximum number of 2 missed cleavages by trypsin were allowed and carbamidomethylated cysteine and oxidized methionine were set as fixed and variable modifications, respectively. Typical contaminants were omitted from the output tables. Protein ratios were calculated from the scaled normalized abundances of the reporter ions over the six quantitation channels. Download Table 1-1, XLSX file (156.6KB, xlsx)