

Abstract
Cell death is a natural process for the turnover of aged cells, but it can also arise as a result of pathological conditions. Cell death is recognized as a key feature in both acute and chronic hepatobiliary diseases caused by drug, alcohol, and fat uptake; by viral infection; or after surgical intervention. In the case of chronic disease, cell death can lead to (chronic) secondary inflammation, cirrhosis, and the progression to liver cancer. In liver transplantation, graft preservation and ischemia/reperfusion injury are associated with acute cell death. In both cases, so‐called programmed cell death modalities are involved. Several distinct types of programmed cell death have been described of which apoptosis and necroptosis are the most well known. Parenchymal liver cells, including hepatocytes and cholangiocytes, are susceptible to both apoptosis and necroptosis, which are triggered by distinct signal transduction pathways. Apoptosis is dependent on a proteolytic cascade of caspase enzymes, whereas necroptosis induction is caspase‐independent. Moreover, different from the “silent” apoptotic cell death, necroptosis can cause a secondary inflammatory cascade, so‐called necroinflammation, triggered by the release of various damage‐associated molecular patterns (DAMPs). These DAMPs activate the innate immune system, leading to both local and systemic inflammatory responses, which can even cause remote organ failure. Therapeutic targeting of necroptosis by pharmacological inhibitors, such as necrostatin‐1, shows variable effects in different disease models.
Abbreviations
- AIH
autoimmune hepatitis
- APAP
acetaminophen
- ATP
adenosine triphosphate
- CCA
cholangiocarcinoma
- cIAP
cellular inhibitor of apoptosis protein
- ConA
concanavalin A
- CYLD
cylindromatosis
- CYP2E1
cytochrome P450 2E1
- DAMP
damage‐associated molecular pattern
- DC
dendritic cell
- Drp1
dynamin‐related protein 1
- FADD
Fas‐associated protein with death domain
- FLIP
FLICE‐inhibitory protein
- GalN
D‐galactosamine
- HBV
hepatitis B virus
- HCC
hepatocellular carcinoma
- HCV
hepatitis C virus
- HFD
high‐fat diet
- HMGB1
high‐mobility group box 1
- IDO
indoleamine 2,3‐dioxygenase
- IFNγ
interferon γ
- IKK‐α
inhibitor of nuclear factor kappa B kinase subunit beta
- IKK‐β
inhibitor of nuclear factor kappa B kinase subunit beta
- IL
interleukin
- IP
intraperitoneally
- IRI
ischemia/reperfusion injury
- IV
intravenously
- JNK
c‐Jun N‐terminal kinase
- KC
Kupffer cell
- LO2
human fetal hepatocyte cell line
- LPS
lipopolysaccharide
- LUBAC
linear ubiquitin chain assembly complex
- MCD
methionine‐choline‐deficient
- miRNA
microRNA
- MLKL
mixed‐lineage kinase domain‐like
- mRNA
messenger RNA
- NAFLD
nonalcoholic fatty liver disease
- NASH
nonalcoholic steatohepatitis
- Nec
necrostatin
- NEMO
nuclear factor kappa B essential modulator
- NET
neutrophil
- NF‐κB
nuclear factor kappa B
- NKT
natural killer T cell
- NLRP3
nucleotide‐binding oligomerization domain–like receptor protein 3
- NPC
nonparenchymal cell
- PARP‐1
poly(adenosine diphosphate ribose) polymerase
- PGAM5
phosphoglycerate mutase 5
- PLC
parenchymal liver cell
- PMH
primary mouse hepatocyte
- pMLKL
pseudokinase mixed‐lineage kinase domain‐like
- po
per os
- RIPK
receptor‐interacting serine/threonine‐protein kinase
- ROA
retro‐orbital administration
- ROS
reactive oxygen species
- TAB1
transforming growth factor β–activated kinase 1 binding protein‐1
- TAK1
transforming growth factor β–activated kinase 1
- TLR
toll‐like receptor
- TNF‐α
tumor necrosis factor α
- TNFR1
tumor necrosis factor receptor 1
- TRADD
tumor necrosis factor 1–associated death domain
- TRAF2
tumor necrosis factor receptor–associated factor 2
- TRAIL
tumor necrosis factor–related apoptosis‐inducing ligand
In this review, we will discuss the mechanisms of necroptosis, and we will focus on liver transplantation and liver diseases, such as acute liver failure, fatty liver diseases, cholestatic liver diseases, chronic viral hepatitis, and primary liver cancer. Furthermore, we will review the clinical relevance of necroptotic cell death and its therapeutic potential by targeting cell death in liver diseases.
Cell death is a fundamental process that is essential in embryonic and (neo)natal development and homeostasis in all organs, including the liver. Cell death is a means of removing aged and damaged cells that otherwise might play a role in organ dysfunction and cancer development. For instance, if transformed hepatocytes with genetic aberrations become resistant to cell death, this may lead to cancer initiation and tumorigenesis.1 In response to the overwhelming cellular stress, hepatocytes can die through active suicide, termed “apoptosis.” Another type of cell death, termed “necrosis,” is a more passive killing of cells. Apoptosis is characterized by a cascade of specific intracellular events leading to so‐called programmed cell death, whereas necrosis occurs as a consequence of extracellular events leading to physical damage and nonregulated (nonprogrammed) cell death.2 In addition to apoptosis and necrosis, a new form of cell death that shared both proporties of apoptosis and necrosis was identified approximately a decade ago. This form of programmed necrosis has been termed “necroptosis.” The molecular events involved in necrosis, programmed apoptotic, and necroptotic cell death are summarized in Fig. 1.
Figure 1.
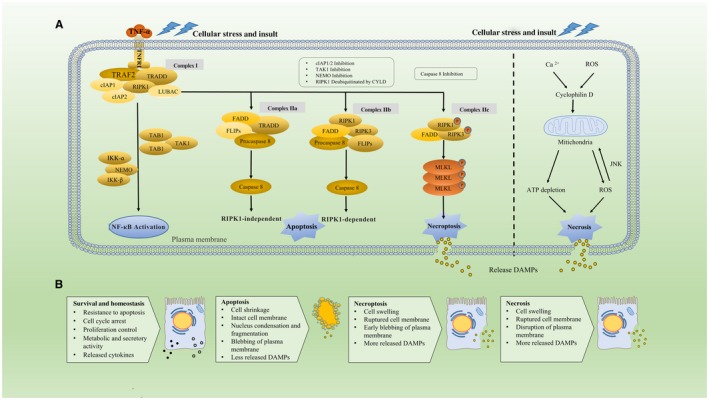
Distinct molecular and morphologic features of apoptotic, necroptotic, and necrotic cell death. (A) Molecular pathways of cell death in PLCs. The binding of TNF‐α and TNFR1 recruits TRADD, TRAF2, RIPK1, cIAP1/2, and LUBAC and forms the complex I leading to the activation of the NF‐κB signaling and a prosurvival pathway. Following the dissociation from TNFR1, complex I is transformed into complex IIa, which includes TRADD, FADD, FLIPs, and procaspase 8, and contributes to the activation of caspase 8 and subsequent RIPK1‐independent apoptosis. Hyperactivation of cylindromatosis (CYLD) deubiquitinates RIPK1 and thus destabilizes complex I and promotes the formation of complex IIb, which is involved in RIPK1‐dependent apoptosis. Complex IIb consists of RIPK1, RIPK3, FADD, FLIPs, and caspase 8, and it can be promoted by inhibition of NEMO, cIAPs, or TAK1. Nevertheless, once caspase 8 is inhibited, RIPK3 is activated to interact with RIPK1 and binds to MLKL, forming the complex IIc (necrosome) by which necroptosis is promoted. RIPK3 phosphorylates MLKL in the complex IIc and thereby triggers oligomerization of MLKL, driving the permeabilization step. Nonprogrammed cell death by necrosis is characterized by mitochondrial impairment with resulting ATP depletion and triggering of the ROS‐JNK loop. After the cell membrane ruptures in necrotic or necroptotic cells, intracellular DAMPs are released and act as activators and amplifiers of necroinflammation. Conversely, release of a lower amount of DAMPs from apoptotic cells leads to much milder necroinflammation. (B) Summary of hallmark events and characteristics of cell survival and cell death by apoptosis, necroptosis, or necrosis.
Necroptosis is characterized as an active and well‐regulated form of necrosis that is morphologically and biochemically distinct from apoptosis. A special feature of necroptosis is the loss of integrity of the plasma membrane and subsequent release of damage‐associated molecular patterns (DAMPs), triggering inflammation and exacerbating tissue damage, ie, so‐called necroinflammation.3 Necroptosis is one of the main contributors to necroinflammation. Inflammation and the subsequent immunological response play an important role in many liver diseases as well as in ischemia/reperfusion injury (IRI) or rejection after liver transplantation. Necroptosis is increasingly considered to play a role in the pathophysiology of these processes, and insight into necroptosis offers promising therapeutic intervention methods to treat liver diseases by targeting necroptosis. For instance, in hepatocellular carcinoma (HCC) or biliary cancer, resistance to apoptosis severely hampers the efficacy of chemotherapy (due to anticancer drug resistance). In this field, the pharmacological switch of cell death toward necroptosis may lead to therapeutic applications in drug‐resistant cancers. This review focuses on necroptosis and summarizes not only related regulatory mechanisms but also the clinical relevance and bench‐to‐bedside translational potential in liver diseases and liver transplantation.
Mechanisms of Necroptosis
Necroptosis can be initiated by a range of factors, such as IRI, release of reactive oxygen species (ROS), antineoplastic events, and calcium overload.4 Furthermore, intracellular factors, such as tumor necrosis factor α (TNF‐α), Fas ligand, tumor necrosis factor–related apoptosis‐inducing ligand (TRAIL), interferon γ (IFNγ), double‐stranded RNA, and adenosine triphosphate (ATP) depletion, are also known to be involved in inducing necroptosis.5 The binding of TNF‐α to tumor necrosis factor receptor 1 (TNFR1) is one of the most extensively studied signaling pathways promoting not only necroptosis but also apoptosis and activation of the nuclear factor kappa B (NF‐κB) pathway (Fig. 1A, left side).6 Generally, TNF‐α can trigger the formation of prosurvival‐ and proinflammatory‐related complexes and caspase‐dependent apoptosis.7 Specifically, TNFR1 recruits TNFR1‐associated death domain (TRADD) protein, tumor necrosis factor receptor–associated factor 2 (TRAF2), receptor‐interacting serine/threonine‐protein kinase (RIPK) 1, cellular inhibitor of apoptosis protein (cIAP) 1/2, and the linear ubiquitin chain assembly complex (LUBAC) to form the complex I, which contributes to the activation of the NF‐κB signaling pathway.8 After dissociation from TNFR1, complex I will be transformed into complex IIa (comprising TRADD, Fas‐associated protein with death domain [FADD], FLICE‐inhibitory protein [FLIP], and procaspase 8), leading to activation of caspase 8 and rendering RIPK1‐independent apoptosis.9 Conversely, the formation of complex IIb, consisting of RIPK1, RIPK3, FADD, FLIPs, and caspase 8, can be promoted by knockdown of the nuclear factor kappa B essential modulator (NEMO), blockage of cIAPs, or transforming growth factor β–activated kinase 1 (TAK1).10 Hyperactivation of cylindromatosis deubiquitinates RIPK1 and, thus, destabilizes complex I, promoting the formation of complex IIb, which is involved in RIPK1‐dependent apoptosis.11 However, when caspase 8 is down‐regulated or inhibited, RIPK3 is activated to interact with RIPK1, which binds to mixed lineage kinase domain‐like (MLKL). Together, this forms the cytosolic complex IIc (necrosome) by which necroptosis is initiated (Fig. 1A, right side).12 Necrostatin (Nec)–1 is a well‐investigated inhibitor of necroptosis by targeting the catalytic and allosteric functions of RIPK1,13 and with that, preventing the formation of the necrosome.
From the above, it is clear that RIPK1 and RIPK3 do not play an exclusive role in the modulation of cell death. Moreover, RIPK1 is the switch node between the prosurvival pathway and apoptosis and/or necroptosis.14 As the ultimate execution step, RIPK3 phosphorylates MLKL in the complex IIc and thereby triggers oligomerization of MLKL, which is indispensable for its translocation to the plasma membrane.15 These oligomers can destabilize the plasma membrane through a pore‐forming complex or by incapacitating Ca2+ or Na+ channels indirectly.16 The late permeabilization step, characterized by an increase of intracellular osmotic pressure and the opening of membrane pores, represents one of the hallmarks of necroptotic cell death.17
Necroinflammation is a form of sterile inflammation triggered by DAMPs that are released from necrotic cells through the ruptured membrane. Necroptosis is one of the most important initiators. DAMPs act as activators and amplifiers of the inflammation response18 and can be categorized into 2 groups:
Molecules with no inflammatory activity in normal cells but which, upon release, exhibit immune activity (ie, heat shock proteins and extracellular ATP).
Alarmins that exhibit specific cytokines initiating an inflammatory response once released (ie, interleukin [IL] 1α and IL33).
DAMPs are recognized by a series of receptors called “pattern recognition receptors,” such as toll‐like receptors (TLRs) and nucleotide oligomerization domain‐like receptors, which activate the innate immunity and thereby evoke the release of cytokines that, in turn, induce more necrosis and trigger an inflammatory cascade reaction.19 This vicious inflammatory circle is strongly associated with the development of chronic liver disease and liver fibrosis3 and is involved in acute liver injury, graft injury, and rejection after liver transplantation.20
Necroptosis in Liver Diseases
Drug‐Induced Liver Injury
Drug‐induced liver injury is mostly caused by acetaminophen (APAP) toxicity.21 It has been reported that necrosis, independent of caspase and the TNF receptor, is mostly involved in APAP‐induced liver injury22 and that the role of apoptosis is limited as was demonstrated by the insensitivity to caspase inhibitors (Table 1).32 In a murine model, both genetic blockage and chemical inhibition of RIPK1 could ameliorate APAP‐induced liver injury.27 In contrast, Li et al.31 reported that neither Nec‐1 nor RIPK1 silencing was able to protect human hepatocytes from cell death, suggesting a differential role of RIPK1 in mice or humans undergoing APAP‐induced liver injury. Likewise, Ramachandran et al.25 demonstrated that RIPK3 is an early mediator of drug‐induced liver injury. Knockout of RIPK3 protected mice from APAP toxicity but only during a very short time frame (no more than 24 hours in vivo and 48 hours in vitro). One potential explanation for this is that the abrogation of RIPK3 cannot alleviate the secondary injury that occurs after APAP overdose.33 Deutsch et al.28 also reported increased expression of RIPK3 in liver tissue of patients with hepatic failure caused by APAP overdose. Silencing or chemical inhibition of RIPK3 protects human hepatocytes from drug‐induced liver injury.31 However, a study by Dara et al.23 shows that knockout of RIPK3 did not alleviate liver injury and the activation of RIPK3 is not observed in the murine APAP‐induced liver injury model. Furthermore, they also reported that the MLKL messenger RNA (mRNA) level is elevated quickly after APAP treatment and knockout of MLKL cannot rescue mice from APAP‐induced liver injury,23 demonstrating that the up‐regulation of MLKL is not the mediator but the consequence of APAP toxicity. Therefore, the involvement of necroptosis in APAP‐induced liver injury is still questionable. The role of RIPK1 independent of RIPK3 and MLKL in such injury needs to be clarified. Moreover, further investigation should focus not only on animal models but should also include clinical human samples because there might be species‐specific differences within these processes.
Table 1.
Necroptosis in APAP‐Induced Liver Injury
| Researchers | Subject (Mice) | APAP Treatment | Findings |
|---|---|---|---|
| Dara et al.23 | Male C57BL/6n | 300 mg/kg IP |
|
| An et al.24 | Male C57BL/6 | 300 mg/kg IP |
|
| Ramachandran et al.25 | Male C57Bl/6J | 200 mg/kg IP |
|
| Takemoto et al.26 | Male C57BL/6 | 800 mg/kg IP |
|
| Zhang et al.27 | Male C57Bl/6J | 300 mg/kg IP |
|
| Deutsch et al.28 | Male C57BL/6 | 500 mg/kg IP |
|
| Yan et al.29 | Male C57BL/6 | 300 mg/kg IP |
|
| Lee et al.30 | C3H/He | 400 mg/kg po |
|
| Li et al.31 | Male C57Bl/6J | 300 mg/kg IP |
|
Immune‐Mediated Liver Injury
Immune‐mediated liver injury plays a critical role in liver diseases through innate and adaptive immune responses. Specifically, autoimmune hepatitis (AIH) is a severe necroinflammatory liver disease that progressively contributes to liver failure and mortality.34 Administration of the lectin concanavalin A (ConA), a T cell mitogen, to mice is the most used model to study immune‐mediated liver injury. Typically, in this model, early stage apoptosis is followed by massive necrosis at later stages. The kinase activity of RIPK1 is elevated when stimulated with ConA and cell death is caspase‐independent.35 However, knockout of RIPK1 in parenchymal liver cells (PLCs) could not rescue liver damage because of exacerbated TNF‐α–mediated and caspase‐dependent apoptosis (Table 2).28 Actually, under steady‐state conditions, RIPK1 also functions as a scaffold protecting hepatocytes from apoptosis through NF‐κB activation or NF‐κB‐independent pathways.40 This means that RIPK1 plays a dual role in the ConA‐induced liver injury. Deutsch et al.28 reported the elevated expression of RIPK3 in liver tissue of AIH patients, but genetic silence of RIPK3 in a murine model could not relieve ConA hepatitis. Conversely, Kang et al.38 demonstrated that genetic silencing of RIPK3 reduced the ConA‐induced elevation of serum aminotransferase concentrations as well as inflammatory markers such as IFNγ and TNF protein. This discrepancy may arise from using different gene modification strategies in these studies. Actually, as reported by Kang et al.,38 the expression of RIPK3 in natural killer T cells (NKTs) is much higher compared with hepatocytes. RIPK3 is involved in the function of NKTs, a crucial step in ConA hepatitis, through activation of RIPK3–phosphoglycerate mutase 5 (PGAM5)–dynamin‐related protein 1 (Drp1)/nuclear factor of activated T cell signaling. This is in accordance with results of He et al.41 that showed that PGAM5‐Drp1 axis–mediated mitochondrial fission drives the hepatic necrosis in ConA hepatitis. In addition, pseudokinase mixed‐lineage kinase domain‐like (pMLKL) has been reported to be elevated in human AIH biopsies, and in murine models, it has been proven that ConA hepatitis is driven by an MLKL‐dependent pathway that occurs independent of RIPK3.42 Taken together, the actual pathway of programmed necrosis during ConA hepatitis is still not clear. Although the involvement of necroptotic mediators, such as RIPK1, RIPK3, and MLKL, has been reported, they are more likely to exhibit independent roles in diverse signaling pathways that drive programmed necrosis. Moreover, as demonstrated by Liedtke et al.,36 simultaneous blockage of RIPK1 and caspase 8 during ConA hepatitis completely inhibited liver injury by suppressing necrosis as well as apoptosis and might be a potential therapy for immune‐mediated liver injury.
Table 2.
Necroptosis in ConA‐Induced Liver Injury
| Researchers | Subject (Mice) | Con A Treatment | Findings |
|---|---|---|---|
| Liedtke et al.36 | Unclear | 25 mg/kg IV |
|
| Jouan‐Lanhouet et al.37 | Female C57Bl/6 | 20 mg/kg ROA |
|
| Kang et al.38 | C57Bl/6 | 20 mg/kg IP |
|
| Deutsch et al.28 | Male C57BL/6 | 20 mg/kg IV |
|
| Filliol et al.35 | Male C57BL/6 | 20 mg/kg IV |
|
| Le Cann et al.39 | C57Bl/6 | 12 mg/kg IV |
|
| Filliol et al.40 | Alfp‐Cre transgenic mice | 12 mg/kg IP |
|
| He et al.41 | Unclear | 25 mg/kg IV |
|
Fatty Liver Diseases
Nonalcoholic fatty liver disease (NAFLD) is a common pathological condition associated with obesity, diabetes, and metabolic syndrome, resulting in fat accumulation in the liver without the history of alcohol abuse. NAFLD can develop into nonalcoholic steatohepatitis (NASH), where fat accumulation triggers inflammation (hepatitis).43 NAFLD/NASH is currently the most rapidly expanding indication for liver transplantation in developed countries. Progressive steatohepatitis is associated with extensive apoptosis in hepatocytes induced by free fatty acids.44 Several novel agents inhibiting apoptosis have been applied in clinical trials, but no significant protective effect was observed so far.45, 46 In addition, inhibition of apoptosis by blocking caspase 8 in a murine model for alcohol‐induced liver injury could not mitigate hepatic cell death, which might imply a possible switch from apoptosis to necroptosis.47 This is in line with results of Gautheron et al.48 that showed that deletion of caspase 8 renders mice more susceptible to methionine‐choline‐deficient (MCD) diet–induced liver steatosis, including extensively increased RIPK3 expression and subsequent massive liver injury and fibrosis. In concordance with these mouse models, significant elevation of RIPK3 in liver biopsies from NASH and NAFLD patients has also been reported.48, 49 Circulating RIPK3, pMLKL, and necrosis markers were also found to be increased in the serum of NAFLD patients.49 Deficiency of RIPK3 attenuates murine MCD‐induced liver injury, steatosis, inflammation, fibrosis, and oxidative stress.49 However, in mice that were fed a high‐fat diet (HFD) to induce NAFLD, the absence of RIPK3 exacerbated liver injury with increased inflammation and hepatocyte apoptosis as well as early fibrotic responses.50 The discrepancy between these findings may arise from the difference between the MCD and HFD models. Generally, mice treated with HFD exhibit glucose intolerance and insulin resistance similar to NAFLD patients, whereas MCD‐fed mice do not show these features and might be a more appropriate model for NASH.51 This also reveals the potential different role of necroptosis in NASH and NAFLD patients. In further studies on NASH/NAFLD as well as other disease models, the differences between the various types of cell death and also a careful evaluation of using the right model should obviously be taken into consideration.
Cholestatic Diseases
Cholestatic diseases are defined by a reduction in bile flow caused by impaired secretion by hepatocytes or cessation in bile flow through intrahepatic or extrahepatic bile ducts. Cholestasis often occurs as a result of chronic liver and biliary diseases, such as hepatitis, primary sclerosing cholangitis, primary biliary cirrhosis, and graft‐versus‐host disease.52 Accumulation of toxic bile acids can induce apoptosis of both hepatocytes and cholangiocytes and is strongly associated with hepatocarcinogenesis as was shown in a rodent model.53 Unfortunately, potent caspase inhibitors could only moderately alleviate liver injury after bile duct ligation, which implies that apoptosis is not the only type of cell death in cholestasis. As also described for NAFLD/NASH, a switch to necroptosis may occur in cholestatic diseases.54 Afonso et al.55 reported high expression levels of RIPK3 and MLKL in primary biliary cirrhosis patients, demonstrating the occurrence of necroptosis in this disease. They also found that RIPK3 deficiency could inhibit necroinflammation that occurred during bile duct ligation. Actually, in the case of cholestasis, caspase 8–dependent apoptosis was found to drive compensatory proliferation in hepatocytes and nonparenchymal cells (NPCs) through c‐Jun N‐terminal kinase (JNK) activation. However, when the necroptotic pathway is triggered, activation of RIPK3 inhibits caspase 8–dependent activation of JNK in both PLCs and non‐PLCs. This, in turn, limits the immune response and compensatory proliferation of PLCs. Subsequently, the development of jaundice and cholestasis is promoted, and inflammatory hepatocarcinogenesis is impeded.56 Another study also showed that inactivated phosphorylation of RIPK1 in PLCs inhibits the compensatory proliferation of both hepatocytes and intrahepatic biliary cells, which promotes cholestasis.57 This indicates the possible independent role of RIPK1 and RIPK3 in cholestatic diseases, which needs to be clarified in further studies.
Viral Hepatitis
Chronic viral hepatitis (hepatitis B virus [HBV] and hepatitis C virus [HCV]) is a major cause of chronic liver disease worldwide. HBV/HCV–related liver injury constitutes a major risk factor for cirrhosis and HCC.58 Necroinflammation in viral hepatitis is strongly associated with liver fibrosis progression and hepatocarcinogenesis.58, 59 However, the mechanism of both necroptosis and necroinflammation in chronic viral hepatitis remains unclear due to a limited number of studies. Afonso et al.49 demonstrated high expression of RIPK3 in liver tissue from HBV and HCV patients by immunostaining, which indicates the potential involvement of necroptosis in chronic viral hepatitis. HCV has been proven to influence the death receptor–mediated pathway and the apoptotic pathway. Consequently, caspase inhibitors, such as IDN‐6556 (Emricasan, a caspase inhibitor), were reported to be used as a potential therapy,60 but again, the possible necroptotic switch should also be taken into account here. Lim et al.61 found that the HCV‐induced cell death of human hepatoma cells could be rescued not only using a pancaspase inhibitor but also by Nec‐1. However, the researchers did not provide evidence for the type of cell death involved. In addition, HBV‐related hepatoma cell lines have been described to express high levels of apoptosis inhibitors, which may explain the resistance of HCC‐HBV to apoptosis induction therapy.62 The HBV X protein–induced microRNA (miRNA) 21 could suppresses cell apoptosis in HCC by targeting IL12.63 In addition, HBV core protein could also inhibit Fas‐mediated apoptosis in HCC by regulating membrane‐bound Fas/Fas ligand and soluble Fas expression.64 Taken together, this implies that targeting necroptosis might be further explored as a potentially interesting therapy for HBV‐related HCC‐HBV.
Liver Cancer
HCC and cholangiocarcinoma (CCA) are the most common primary malignancies in the liver. They differ markedly in their morphology, metastatic potential, and responses to therapy. It has been suggested that both forms may arise from liver progenitor cells and have potentially overlapping pathways of oncogenesis.1 The regulatory molecules and tissue context that commit transformed hepatic cells toward HCC or CCA are still largely unknown, but a recent mouse study showed that hepatocytes with aberrantly activated oncogenes give rise to CCA when embedded in a necroptosis‐dominated hepatic microenvironment.65 DAMPs released by necroptotic hepatocytes can activate immune cells to secrete various specific cytokines to form a robust inflammatory environment, determining the outgrowth of CCA from transformed hepatocytes. In contrast, hepatocytes that harbor the same oncogenic driver will give rise to HCC if it is not adjacent to necroptotic hepatocytes. Notably, different from hepatocytes, transformed cholangiocytes can only develop into CCA and not HCC.66 This finding may explain why CCA exhibits resistance to apoptosis, which is possibly due to higher endogenic expression of myeloid cell leukemia 1.61 Further work is warranted to unravel the extracellular and intracellular mechanisms and interactions between cytokines and transformed hepatocytes in detail, which, in turn, will be helpful to find new targeted therapies against HCC and CCA.
Necroptosis in Liver Transplantation
Liver transplantation is widely accepted as the only effective intervention for patients with end‐stage liver disease. During liver transplantation, IRI is inevitable, although ischemia‐free transplantations are feasible as described recently.67 IRI is a detrimental process that not only damages the liver graft but is also associated with distant organ damage, such as acute kidney injury after liver transplantation.68 Apoptosis and necrosis are the 2 most important types of cell death related to IRI. With strict morphological criteria, Gujral et al.69 demonstrated that only a small subset of sinusoidal endothelial cells and hepatocytes underwent apoptosis after warm ischemia followed by reperfusion. Necrosis appears to be the dominant type of cell death in IRI, especially during reperfusion injury, accounting for more than 90% of total cell death and, consequently, caspase inhibitors cannot prevent IRI‐related cell death. A schematic overview of necroptosis in liver transplantation is shown in Fig. 2. Haga et al.70 demonstrated that hypoxia/reoxygenation leads to an increased expression of necroptosis mediators in murine liver cell lines, which subsequently could be inhibited by Nec‐1. Several studies also interpret the role of necroptosis in IRI using a mouse hepatic IRI model. Hong et al.71 reported that RIPK1 and RIPK3 expression is dramatically increased and is accompanied by the formation of the RIPK1/RIPK3 complex after IRI. Treatment with Nec‐1 not only mitigated necroptosis but also decreased the serum levels of TNF‐α and IL6. Nevertheless, 2 other studies done using similar animal models reveal the converse conclusion that necroptosis is not involved in the IRI process,72, 73 demonstrating controversial findings resulting in unclear and contradicting conclusions in this field. Also, there is no protective effect observed after administration of Nec‐1 or cyclosporine A, which is an inhibitor of mitochondrial permeability transition. This indicates that neither necroptosis nor mitochondrial permeability transition contributes to necrosis in hepatic IRI. This contradictory conclusion may arise from variations during the establishment of the model and needs more careful analysis. Administration of Nec‐1 before ischemia occurs or before reperfusion is done might also explain this discrepancy in results because the injury type and signaling are differential in various stages during transplantation surgery.69 Furthermore, Liss et al.74 found that hepatic IRI in the setting of steatosis led to increased expression of RIPK1, RIPK3, and MLKL, which might indicate the potential involvement of necroptosis in IRI in steatotic liver grafts.
Figure 2.
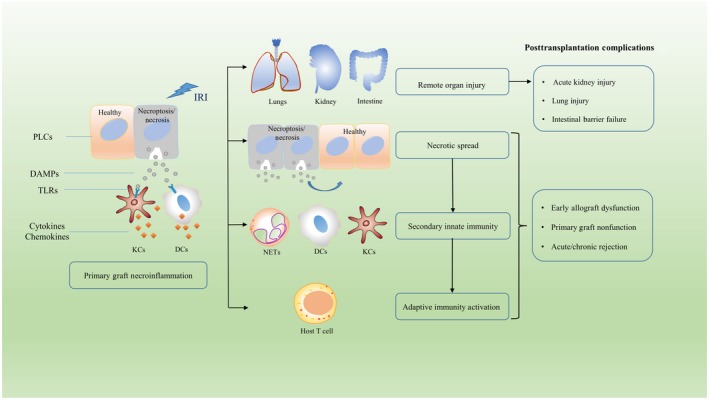
Schematic overview of necrosis and necroinflammation during liver transplantation. During ischemia and reperfusion injury, both necroptosis and necrosis of PLCs can occur. Rupture of the cell membrane facilitates the release of intracellular DAMPs and subsequent inflammatory responses. TLRs on both KCs and DCs are activated that promote the production and release of cytokines and chemokines. This will trigger migration of innate immune cells to the liver graft but also give rise to necrotic spread by further induction of necroptosis in surrounding cells. This necrotic spread could cause early allograft dysfunction or total graft failure causing primary nonfunction. Furthermore, this necrotic spread and necroinflammation can lead to remote organ injury outside the graft. Robust innate immunity can also active host T cells and evoke adaptive immune response that is associated with acute and chronic rejection after transplantation.
Necroinflammation resulting from IRI has proven to determine the fate of the liver graft and the outcome of patients undergoing liver transplantation (Fig. 2). Necroptosis is one of the critical triggers of necroinflammation, though its role in hepatic IRI is still unclear. Following hepatic IRI, DAMPs are released from necrotic cells and exacerbate inflammation and liver injury.75 Activation of TLRs by DAMPs also recruits innate immune cells to the graft and ultimately contributes to graft rejection.76 High‐mobility group box 1 (HMGB1), a nuclear protein regulating transcription, is one of the most‐investigated DAMPs, which binds to TLR4 and induces the generation of proinflammatory cytokines and subsequent graft liver damage.77 Increased plasma levels of HMGB1 are a marker of hepatocellular injury in recipients.78 Down‐regulation of nuclear HMGB1 by small interfering RNA in a mouse IRI model protected the liver from IRI and alleviated inflammation response.79 Moreover, a clinical trial demonstrated that a TLR4 single‐nucleotide polymorphism, leading to diminished binding with HMGB1, reduced the risk of graft loss after liver transplantation.80 In general, necroinflammation is a promising new therapeutic target for liver transplantation. Furthermore, the booming development of ex vivo normothermic machine perfusion should also be addressed because this can provide an appropriate model for the investigation on necroptosis and necroinflammation in human liver grafts.
Pharmacological Strategies Targeting Necroptosis
Although apoptosis inhibitors have been used in a clinical trial against apoptosis‐mediated liver injury, the protective effect of these inhibitors remains questionable and might be dependent on whether or not the switch from apoptosis to necroptosis is made.81 Therefore, the clinical application of necroptosis inhibitors should also be considered. Nec‐1 is the first and extensively used compound identified as an inhibitor of necroptosis acting on the kinase activity of RIPK1.82 Because of its striking specificity for necroptosis, it is also widely interpreted as indicating proof of necroptosis in various conditions. However, even though Nec‐1 was described more than a decade ago, it has only been used in preclinical trials to treat amyotrophic lateral sclerosis.83 Low potency and the short half‐life of Nec‐1 in vivo restrains its clinical application. Moreover, Nec‐1 is not exclusively inhibiting for necroptosis because it also acts on the indoleamine 2,3‐dioxygenase (IDO) enzyme that regulates the innate and adaptive immune system.84 Instead, the development of Nec‐1s, which is more stable, has great potential in vivo. Nec‐1s lacks IDO inhibitory activity and possesses a longer half‐life in vivo than Nec‐1, which makes Nec‐1s more promising for bench‐to‐bed translation.85 Administration of radio immunoprecipitation assay 56, another RIPK1 inhibitor, is also a potential therapy against necroptosis that has been validated in murine systemic inflammatory response syndrome.86
Considering the crosstalk of RIPK1 in apoptosis, necroptosis, and NF‐κB pathways, inhibition of RIPK3 kinase activity seems to be a more specific and therefore more potential therapeutic option targeting necroptosis. RIPK3 inhibitors can also be applied in some RIPK1‐independent pathologic conditions, such as viral infection and pancreatitis.87 Compounds including low concentrations of GSKʹ840, GSKʹ843, GSKʹ872, and GW392 have been verified in vivo and in vitro to inhibit necroptosis.82, 88 However, RIPK3 inhibitors used in high concentrations might induce apoptosis by activating caspase 8.89 Furthermore, necrosulfonamide, which targets MLKL, is found to inhibit necroptosis in vitro. More studies are needed to investigate the efficiency and safety of necrosulfonamide in vivo.
Different from the above inhibitors, some drugs that are already used in the clinic also inhibit necroptosis activity. Their safety in patients has already been validated. For instance, dabrafenib is a US Food and Drug Administration–approved drug for metastatic melanoma treatment,90 and Li et al.31 demonstrated that dabrafenib could inhibit RIPK3 and alleviate APAP‐induced liver injury in vitro and in vivo. Sorafenib is a clinically used drug to treat HCC and acute myeloid leukemia,91 and it can inhibit both RIPK1 and RIPK3 kinase activity and thus protects against TNF‐induced systemic inflammatory response syndrome and kidney IRI.92 Likewise, ponatinib and pazopanib, 2 anticancer agents, were also found to be inhibitors of necroptosis in human cells.93 Phenytoin, a clinically used anticonvulsant drug, can also block necroptosis in systemic inflammatory response syndrome and was shown to be efficient in a kidney IRI model. In addition, melatonin is synthesized endogenously by the pineal gland and functions as an indirect antioxidant. It has been demonstrated to attenuate carbon tetrachloride–induced liver injury and to prevent fibrosis by inhibiting necroptosis‐associated inflammatory signaling.94
Conclusion
In this review, we strived to illuminate the role of necroptosis in acute and chronic liver diseases as well as in liver transplantation. Although our primary view was on necroptosis, the crosstalk between apoptosis and necroptosis should not be ignored. In most cases, these 2 types of cell death are intermixed in the pathogenesis of liver diseases, although the role of necroptosis in some liver diseases is still unconfirmed. The switch between apoptosis and necroptosis should be noted when using either caspase inhibitors or necroptosis inhibitors. For instance, caspase inhibitors can prevent apoptosis but may induce necroptosis in turn, which gives rise to detrimental effects, ranging from less effectiveness to massive secondary inflammation response. Insight into necroptosis also offers a convincing explanation for the limited clinical benefits of caspase inhibitors. Actually, necroptosis mediators, such as RIPK1 and RIPK3, can also be involved in other signaling pathways, mitochondrial dysfunction, and apoptosis. This highlights the difficulty to target cell death in pathological situations. Further studies should be focused on the investigation of the potential crosstalk between various cell‐survival and cell‐death signaling pathways in liver diseases. It is also notable that RIPK1, RIPK3, and MLKL could function independently in some liver diseases, such as acute liver injury and cholestatic diseases, through a nonnecroptosis pathway. What would be important to help the research field forward is consensus on the precise definition of necroptosis and its morphology and signaling hallmarks. In addition, the results of some recent studies using experimental mouse models exhibit controversial results, which could be caused by the differences in genetic background of the mouse strains and the different experimental conditions used in the studies. For instance, in studies focusing on fatty liver disease, diverse treatment may very well lead to inconsistent results when choosing either MCD or HFD models. Related studies involving humans are still restricted to clinical liver biopsies. It is also a promising direction to use in vitro experimental models derived from patients, such as organoids,95, 96 which represent individual or patient‐specific pathological features. In addition, as a severe consequence of necroptosis, necroinflammation plays a crucial role in both acute and chronic liver diseases. Several of these DAMPs could serve as potential biomarkers for the evaluation of liver injury, for instance, during normothermic graft preservation by machine perfusion for liver transplantation. In summary, necroptosis is a promising therapeutic target for treatment of liver diseases and during graft preservation and should be further explored both to deepen our understanding of how liver cells die and clarify the clinical perspective for translation of the knowledge in medical practice.
Acknowledgment
We thank Petra de Ruiter for critical reading of the manuscript.
This work was supported by the China Scholarship Council (Shaojun Shi, number 201706230252).
Potential conflict of interest: Nothing to report.
References
- 1. Sia D, Villanueva A, Friedman SL, Llovet JM. Liver cancer cell of origin, molecular class, and effects on patient prognosis. Gastroenterology 2017;152:745‐761. [DOI] [PubMed] [Google Scholar]
- 2. Schwabe RF, Luedde T. Apoptosis and necroptosis in the liver: a matter of life and death. Nat Rev Gastroenterol Hepatol 2018;15:738‐752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Luedde T, Kaplowitz N, Schwabe RF. Cell death and cell death responses in liver disease: mechanisms and clinical relevance. Gastroenterology 2014;147:765‐783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Fulda S. The mechanism of necroptosis in normal and cancer cells. Cancer Biol Ther 2013;14:999‐1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Golstein P, Kroemer G. Cell death by necrosis: towards a molecular definition. Trends Biochem Sci 2007;32:37‐43. [DOI] [PubMed] [Google Scholar]
- 6. Galluzzi L, Kepp O, Chan FK, Kroemer G. Necroptosis: mechanisms and relevance to disease. Annu Rev Pathol 2017;12:103‐130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Wang L, Du F, Wang X. TNF‐alpha induces two distinct caspase‐8 activation pathways. Cell 2008;133:693‐703. [DOI] [PubMed] [Google Scholar]
- 8. Dara L. The receptor interacting protein kinases in the liver. Semin Liver Dis 2018;38:73‐86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Suda J, Dara L, Yang L, Aghajan M, Song Y, Kaplowitz N, Liu ZX. Knockdown of RIPK1 markedly exacerbates murine immune‐mediated liver injury through massive apoptosis of hepatocytes, independent of necroptosis and inhibition of NF‐κB. J Immunol 2016;197:3120‐3129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Geng J, Ito Y, Shi L, Amin P, Chu J, Ouchida AT, et al. Regulation of RIPK1 activation by TAK1‐mediated phosphorylation dictates apoptosis and necroptosis. Nat Commun 2017;8:359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Dondelinger Y, Aguileta MA, Goossens V, Dubuisson C, Grootjans S, Dejardin E, et al. RIPK3 contributes to TNFR1‐mediated RIPK1 kinase‐dependent apoptosis in conditions of cIAP1/2 depletion or TAK1 kinase inhibition. Cell Death Differ 2013;20:1381‐1392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Moriwaki K, Chan FK. RIP3: a molecular switch for necrosis and inflammation. Genes Dev 2013;27:1640‐1649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Degterev A, Maki JL, Yuan J. Activity and specificity of necrostatin‐1, small‐molecule inhibitor of RIP1 kinase. Cell Death Differ 2013;20:366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Blériot C, Lecuit M. RIPK1, a key survival factor for hepatocytes. J Hepatol 2017;66:1118‐1119. [DOI] [PubMed] [Google Scholar]
- 15. Wang H, Sun L, Su L, Rizo J, Liu L, Wang LF, et al. Mixed lineage kinase domain‐like protein MLKL causes necrotic membrane disruption upon phosphorylation by RIP3. Mol Cell 2014;54:133‐146. [DOI] [PubMed] [Google Scholar]
- 16. Zhao J, Jitkaew S, Cai Z, Choksi S, Li Q, Luo J, Liu ZG. Mixed lineage kinase domain‐like is a key receptor interacting protein 3 downstream component of TNF‐induced necrosis. Proc Natl Acad Sci U S A 2012;109:5322‐5327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ros U, Peña‐Blanco A, Hänggi K, Kunzendorf U, Krautwald S, Wong WW, García‐Sáez AJ. Necroptosis execution is mediated by plasma membrane nanopores independent of calcium. Cell Rep 2017;19:175‐187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Land WG, Agostinis P, Gasser S, Garg AD, Linkermann A. Transplantation and damage‐associated molecular patterns (DAMPs). Am J Transplant 2016;16:3338‐3361. [DOI] [PubMed] [Google Scholar]
- 19. Guicciardi ME, Malhi H, Mott JL, Gores GJ. Apoptosis and necrosis in the liver. Compr Physiol 2013;3:977‐1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Land WG, Agostinis P, Gasser S, Garg AD, Linkermann A. DAMP‐induced allograft and tumor rejection: the circle is closing. Am J Transplant 2016;16:3322‐3337. [DOI] [PubMed] [Google Scholar]
- 21. Jaeschke H. Acetaminophen: dose‐dependent drug hepatotoxicity and acute liver failure in patients. Dig Dis 2015;33:464‐471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Xie Y, McGill MR, Dorko K, Kumer SC, Schmitt TM, Forster J, Jaeschke H. Mechanisms of acetaminophen‐induced cell death in primary human hepatocytes. Toxicol Appl Pharmacol 2014;279:266‐274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Dara L, Johnson H, Suda J, Win S, Gaarde W, Han D, Kaplowitz N. Receptor interacting protein kinase 1 mediates murine acetaminophen toxicity independent of the necrosome and not through necroptosis. Hepatology 2015;62:1847‐1857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. An J, Mehrhof F, Harms C, Lättig‐Tünnemann G, Lee SL, Endres M, et al. ARC is a novel therapeutic approach against acetaminophen‐induced hepatocellular necrosis. J Hepatol 2013;58:297‐305. [DOI] [PubMed] [Google Scholar]
- 25. Ramachandran A, McGill MR, Xie Y, Ni HM, Ding WX, Jaeschke H. Receptor interacting protein kinase 3 is a critical early mediator of acetaminophen‐induced hepatocyte necrosis in mice. Hepatology 2013;58:2099‐2108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Takemoto K, Hatano E, Iwaisako K, Takeiri M, Noma N, Ohmae S, et al. Necrostatin‐1 protects against reactive oxygen species (ROS)‐induced hepatotoxicity in acetaminophen‐induced acute liver failure. FEBS Open Bio 2014;4:777‐787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Zhang YF, He W, Zhang C, Liu XJ, Lu Y, Wang H, et al. Role of receptor interacting protein (RIP)1 on apoptosis‐inducing factor‐mediated necroptosis during acetaminophen‐evoked acute liver failure in mice. Toxicol Lett 2014;225:445‐453. [DOI] [PubMed] [Google Scholar]
- 28. Deutsch M, Graffeo CS, Rokosh R, Pansari M, Ochi A, Levie EM, et al. Divergent effects of RIP1 or RIP3 blockade in murine models of acute liver injury. Cell Death Dis 2015;6:e1759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Yan T, Wang H, Zhao M, Yagai T, Chai Y, Krausz KW, et al. Glycyrrhizin protects against acetaminophen‐induced acute liver injury via alleviating tumor necrosis factor α‐mediated apoptosis. Drug Metab Dispos 2016;44:720‐731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Lee BW, Jeon BS, Yoon BI. Exogenous recombinant human thioredoxin‐1 prevents acetaminophen‐induced liver injury by scavenging oxidative stressors, restoring the thioredoxin‐1 system and inhibiting receptor interacting protein‐3 overexpression. J Appl Toxicol 2018;38:1008‐1017. [DOI] [PubMed] [Google Scholar]
- 31. Li JX, Feng JM, Wang Y, Li XH, Chen XX, Su Y, et al. The B‐Raf(V600E) inhibitor dabrafenib selectively inhibits RIP3 and alleviates acetaminophen‐induced liver injury. Cell Death Dis 2014;5:e1278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Jaeschke H, Cover C, Bajt ML. Role of caspases in acetaminophen‐induced liver injury. Life Sci 2006;78:1670‐1676. [DOI] [PubMed] [Google Scholar]
- 33. Gardner CR, Laskin JD, Dambach DM, Sacco M, Durham SK, Bruno MK, et al. Reduced hepatotoxicity of acetaminophen in mice lacking inducible nitric oxide synthase: potential role of tumor necrosis factor‐α and interleukin‐10. Toxicol Appl Pharmacol 2002;184:27‐36. [PubMed] [Google Scholar]
- 34. Krawitt EL. Autoimmune hepatitis. N Engl J Med 2006;354:54‐66. [DOI] [PubMed] [Google Scholar]
- 35. Filliol A, Piquet‐Pellorce C, Le Seyec J, Farooq M, Genet V, Lucas‐Clerc C, et al. RIPK1 protects from TNF‐α‐mediated liver damage during hepatitis. Cell Death Dis 2016;7:e2462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Liedtke C, Bangen JM, Freimuth J, Beraza N, Lambertz D, Cubero FJ, et al. Loss of caspase‐8 protects mice against inflammation‐related hepatocarcinogenesis but induces non‐apoptotic liver injury. Gastroenterology 2011;141:2176‐2187. [DOI] [PubMed] [Google Scholar]
- 37. Jouan‐Lanhouet S, Arshad MI, Piquet‐Pellorce C, Martin‐Chouly C, Le Moigne‐Muller G, Van Herreweghe F, et al. TRAIL induces necroptosis involving RIPK1/RIPK3‐dependent PARP‐1 activation. Cell Death Differ 2012;19:2003‐2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Kang YJ, Bang BR, Han KH, Hong L, Shim EJ, Ma J, et al. Regulation of NKT cell‐mediated immune responses to tumours and liver inflammation by mitochondrial PGAM5‐Drp1 signalling. Nat Commun 2015;6:8371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Le Cann F, Delehouzé C, Leverrier‐Penna S, Filliol A, Comte A, Delalande O, et al. Sibiriline, a new small chemical inhibitor of receptor‐interacting protein kinase 1, prevents immune‐dependent hepatitis. FEBS J 2017;284:3050‐3068. [DOI] [PubMed] [Google Scholar]
- 40. Filliol A, Farooq M, Piquet‐Pellorce C, Genet V, Dimanche‐Boitrel MT, Vandenabeele P, et al. RIPK1 protects hepatocytes from death in Fas‐induced hepatitis. Sci Rep 2017;7:9205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. He GW, Günther C, Kremer AE, Thonn V, Amann K, Poremba C, et al. PGAM5‐mediated programmed necrosis of hepatocytes drives acute liver injury. Gut 2017;66:716‐723. [DOI] [PubMed] [Google Scholar]
- 42. Günther C, He GW, Kremer AE, Murphy JM, Petrie EJ, Amann K, et al. The pseudokinase MLKL mediates programmed hepatocellular necrosis independently of RIPK3 during hepatitis. J Clin Invest 2016;126:4346‐4360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Farrell GC, Haczeyni F, Chitturi S. Pathogenesis of NASH: how metabolic complications of overnutrition favour lipotoxicity and pro‐inflammatory fatty liver disease. Adv Exp Med Biol 2018;1061:19‐44. [DOI] [PubMed] [Google Scholar]
- 44. Wang Y, Ausman LM, Russell RM, Greenberg AS, Wang XD. Increased apoptosis in high‐fat diet‐induced nonalcoholic steatohepatitis in rats is associated with c‐Jun NH2‐terminal kinase activation and elevated proapoptotic Bax. J Nutr 2008;138:1866‐1871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Ratziu V, Sheikh MY, Sanyal AJ, Lim JK, Conjeevaram H, Chalasani N, et al. A phase 2, randomized, double‐blind, placebo‐controlled study of GS‐9450 in subjects with nonalcoholic steatohepatitis. Hepatology 2012;55:419‐428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Bouziana SD, Tziomalos K. Inhibition of apoptosis in the management of nonalcoholic fatty liver disease. World J Gastrointest Pharmacol Ther 2013;4:4‐8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Hao F, Cubero FJ, Ramadori P, Liao L, Haas U, Lambertz D, et al. Inhibition of caspase‐8 does not protect from alcohol‐induced liver apoptosis but alleviates alcoholic hepatic steatosis in mice. Cell Death Dis 2017;8:e3152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Gautheron J, Vucur M, Reisinger F, Cardenas DV, Roderburg C, Koppe C, et al. A positive feedback loop between RIP3 and JNK controls non‐alcoholic steatohepatitis. EMBO Mol Med 2014;6:1062‐1074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Afonso MB, Rodrigues PM, Carvalho T, Caridade M, Borralho P, Cortez‐Pinto H, et al. Necroptosis is a key pathogenic event in human and experimental murine models of non‐alcoholic steatohepatitis. Clin Sci (Lond) 2015;129:721‐739. [DOI] [PubMed] [Google Scholar]
- 50. Roychowdhury S, McCullough RL, Sanz‐Garcia C, Saikia P, Alkhouri N, Matloob A, et al. Receptor interacting protein 3 protects mice from high‐fat diet‐induced liver injury. Hepatology 2016;64:1518‐1533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Gautheron J, Vucur M, Luedde T. Necroptosis in nonalcoholic steatohepatitis. Cell Mol Gastroenterol Hepatol 2015;1:264‐265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Santiago P, Scheinberg AR, Levy C. Cholestatic liver diseases: new targets, new therapies. Therap Adv Gastroenterol 2018;11:1756284818787400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Canbay A, Guicciardi ME, Higuchi H, Feldstein A, Bronk SF, Rydzewski R, et al. Cathepsin B inactivation attenuates hepatic injury and fibrosis during cholestasis. J Clin Invest 2003;112:152‐159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Gujral JS, Liu J, Farhood A, Jaeschke H. Reduced oncotic necrosis in Fas receptor‐deficient C57BL/6J‐lpr mice after bile duct ligation. Hepatology 2004;40:998‐1007. [DOI] [PubMed] [Google Scholar]
- 55. Afonso MB, Rodrigues PM, Simão AL, Ofengeim D, Carvalho T, Amaral JD, et al. Activation of necroptosis in human and experimental cholestasis. Cell Death Dis 2016;7:e2390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Vucur M, Reisinger F, Gautheron J, Janssen J, Roderburg C, Cardenas DV, et al. RIP3 inhibits inflammatory hepatocarcinogenesis but promotes cholestasis by controlling caspase‐8‐ and JNK‐dependent compensatory cell proliferation. Cell Rep 2013;4:776‐790. [DOI] [PubMed] [Google Scholar]
- 57. Koppe C, Verheugd P, Gautheron J, Reisinger F, Kreggenwinkel K, Roderburg C, et al. IκB kinaseα/β control biliary homeostasis and hepatocarcinogenesis in mice by phosphorylating the cell‐death mediator receptor‐interacting protein kinase 1. Hepatology 2016;64:1217‐1231. [DOI] [PubMed] [Google Scholar]
- 58. Saeed U, Waheed Y, Ashraf M. Hepatitis B and hepatitis C viruses: a review of viral genomes, viral induced host immune responses, genotypic distributions and worldwide epidemiology. Asian Pac J Trop Dis 2014;4:88‐96. [Google Scholar]
- 59. Pawlotsky JM, Negro F, Aghemo A, Berenguer M, Dalgard O, Dusheiko G, et al.; for European Association for the Study of the Liver . EASL recommendations on treatment of hepatitis C 2018. J Hepatol 2018;69:461‐511. [DOI] [PubMed] [Google Scholar]
- 60. Masuoka HC, Guicciardi ME, Gores GJ. Caspase inhibitors for the treatment of hepatitis C. Clin Liver Dis 2009;13:467‐475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Lim EJ, El Khobar K, Chin R, Earnest‐Silveira L, Angus PW, Bock CT, et al. Hepatitis C virus‐induced hepatocyte cell death and protection by inhibition of apoptosis. J Gen Virol 2014;95:2204‐2215. [DOI] [PubMed] [Google Scholar]
- 62. Lu X, Lee M, Tran T, Block T. High level expression of apoptosis inhibitor in hepatoma cell line expressing hepatitis B virus. Int J Med Sci 2005;2:30‐35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Yin D, Wang Y, Sai W, Zhang L, Miao Y, Cao L, et al. HBx‐induced miR‐21 suppresses cell apoptosis in hepatocellular carcinoma by targeting interleukin‐12. Oncol Rep 2016;36:2305‐2312. [DOI] [PubMed] [Google Scholar]
- 64. Liu W, Lin YT, Yan XL, Ding YL, Wu YL, Chen WN, Lin X. Hepatitis B virus core protein inhibits Fas‐mediated apoptosis of hepatoma cells via regulation of mFas/FasL and sFas expression. FASEB J 2015;29:1113‐1123. [DOI] [PubMed] [Google Scholar]
- 65. Seehawer M, Heinzmann F, D'Artista L, Harbig J, Roux PF, Hoenicke L, et al. Necroptosis microenvironment directs lineage commitment in liver cancer. Nature 2018;562:69‐75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Guest RV, Boulter L, Kendall TJ, Minnis‐Lyons SE, Walker R, Wigmore SJ, et al. Cell lineage tracing reveals a biliary origin of intrahepatic cholangiocarcinoma. Cancer Res 2014;74:1005‐1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. He X, Guo Z, Zhao Q, Ju W, Wang D, Wu L, et al. The first case of ischemia‐free organ transplantation in humans: a proof of concept. Am J Transplant 2018;18:737‐744. [DOI] [PubMed] [Google Scholar]
- 68. Klune JR, Tsung A. Molecular biology of liver ischemia/reperfusion injury: established mechanisms and recent advancements. Surg Clin North Am 2010;90:665‐677. [DOI] [PubMed] [Google Scholar]
- 69. Gujral JS, Bucci TJ, Farhood A, Jaeschke H. Mechanism of cell death during warm hepatic ischemia‐reperfusion in rats: apoptosis or necrosis? Hepatology 2001;33:397‐405. [DOI] [PubMed] [Google Scholar]
- 70. Haga S, Kanno A, Ozawa T, Morita N, Asano M, Ozaki M. Detection of necroptosis in ligand‐mediated and hypoxia‐induced injury of hepatocytes using a novel optic probe detecting receptor‐interacting protein (RIP)1/RIP3 binding. Oncol Res 2018;26:503‐513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Hong JM, Kim SJ, Lee SM. Role of necroptosis in autophagy signaling during hepatic ischemia and reperfusion. Toxicol Appl Pharmacol 2016;308:1‐10. [DOI] [PubMed] [Google Scholar]
- 72. Rosentreter D, Funken D, Reifart J, Mende K, Rentsch M, Khandoga A. RIP1‐dependent programmed necrosis is negatively regulated by caspases during hepatic ischemia‐reperfusion. Shock 2015;44:72‐76. [DOI] [PubMed] [Google Scholar]
- 73. Saeed WK, Jun DW, Jang K, Chae YJ, Lee JS, Kang HT. Does necroptosis have a crucial role in hepatic ischemia‐reperfusion injury? PLoS One 2017;3:e0184752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Liss KHH, McCommis KS, Chambers KT, Pietka TA, Schweitzer GG, Park SL, et al. The impact of diet‐induced hepatic steatosis in a murine model of hepatic ischemia/reperfusion injury. Liver Transpl 2018;24:908‐921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Scaffidi P, Misteli T, Bianchi ME. Release of chromatin protein HMGB1 by necrotic cells triggers inflammation. Nature 2002;418:191‐195. [DOI] [PubMed] [Google Scholar]
- 76. Braza F, Brouard S, Chadban S, Goldstein DR. Role of TLRs and DAMPs in allograft inflammation and transplant outcomes. Nat Rev Nephrol 2016;12:281‐290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Tsung A, Sahai R, Tanaka H, Nakao A, Fink MP, Lotze MT, et al. The nuclear factor HMGB1 mediates hepatic injury after murine liver ischemia‐reperfusion. J Exp Med 2005;201:1135‐1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Ilmakunnas M, Tukiainen EM, Rouhiainen A, Rauvala H, Arola J, Nordin A, et al. High mobility group box 1 protein as a marker of hepatocellular injury in human liver transplantation. Liver Transpl 2008;14:1517‐1525. [DOI] [PubMed] [Google Scholar]
- 79. Zhao G, Fu C, Wang L, Zhu L, Yan Y, Xiang Y, et al. Down‐regulation of nuclear HMGB1 reduces ischemia‐induced HMGB1 translocation and release and protects against liver ischemia‐reperfusion injury. Sci Rep 2017;7:46272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Dhillon N, Walsh L, Krüger B, Ward SC, Godbold JH, Radwan M, et al. A single nucleotide polymorphism of Toll‐like receptor 4 identifies the risk of developing graft failure after liver transplantation. J Hepatol 2010;53:67‐72. [DOI] [PubMed] [Google Scholar]
- 81. Ni HM, McGill MR, Chao X, Woolbright BL, Jaeschke H, Ding WX. Caspase inhibition prevents tumor necrosis factor‐α–induced apoptosis and promotes necrotic cell death in mouse hepatocytes in vivo and in vitro. Am J Pathol 2016;186:2623‐2636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Conrad M, Angeli JP, Vandenabeele P, Stockwell BR. Regulated necrosis: disease relevance and therapeutic opportunities. Nat Rev Drug Discov 2016;15:348‐366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Ito Y, Ofengeim D, Najafov A, Das S, Saberi S, Li Y, et al. RIPK1 mediates axonal degeneration by promoting inflammation and necroptosis in ALS. Science 2016;353:603‐608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Vandenabeele P, Grootjans S, Callewaert N, Takahashi N. Necrostatin‐1 blocks both RIPK1 and IDO: consequences for the study of cell death in experimental disease models. Cell Death Differ 2013;20:185‐187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Martens S, Goossens V, Devisscher L, Hofmans S, Claeys P, Vuylsteke M, et al. RIPK1‐dependent cell death: a novel target of the Aurora kinase inhibitor Tozasertib (VX‐680). Cell Death Dis 2018;9:211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Ren Y, Su Y, Sun L, He S, Meng L, Liao D, et al. Discovery of a highly potent, selective, and metabolically stable inhibitor of receptor‐interacting protein 1 (RIP1) for the treatment of Systemic Inflammatory Response Syndrome. J Med Chem 2017;60:972‐986. [DOI] [PubMed] [Google Scholar]
- 87. Arora D, Sharma PK, Siddiqui MH, Shukla Y. Necroptosis: modules and molecular switches with therapeutic implications. Biochimie 2017;137:35‐45. [DOI] [PubMed] [Google Scholar]
- 88. Rodriguez DA, Weinlich R, Brown S, Guy C, Fitzgerald P, Dillon CP, et al. Characterization of RIPK3‐mediated phosphorylation of the activation loop of MLKL during necroptosis. Cell Death Differ 2016;23:76‐88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Mandal P, Berger SB, Pillay S, Moriwaki K, Huang C, Guo H, et al. RIP3 induces apoptosis independent of pronecrotic kinase activity. Mol Cell 2014;56:481‐495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Fiskus W, Mitsiades N. B‐Raf inhibition in the clinic: present and future. Annu Rev Med 2016;67:29‐43. [DOI] [PubMed] [Google Scholar]
- 91. Wilhelm S, Carter C, Lynch M, Lowinger T, Dumas J, Smith RA, et al. Discovery and development of sorafenib: a multikinase inhibitor for treating cancer. Nat Rev Drug Discov 2006;5:835‐844. [DOI] [PubMed] [Google Scholar]
- 92. Martens S, Jeong M, Tonnus W, Feldmann F, Hofmans S, Goossens V, et al. Sorafenib tosylate inhibits directly necrosome complex formation and protects in mouse models of inflammation and tissue injury. Cell Death Dis 2017;8:e2904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Fauster A, Rebsamen M, Huber KV, Bigenzahn JW, Stukalov A, Lardeau CH, et al. A cellular screen identifies ponatinib and pazopanib as inhibitors of necroptosis. Cell Death Dis 2015;6:e1767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Choi HS, Kang JW, Lee SM. Melatonin attenuates carbon tetrachloride‐induced liver fibrosis via inhibition of necroptosis. Transl Res 2015;166:292‐303. [DOI] [PubMed] [Google Scholar]
- 95. Huch M, Gehart H, van Boxtel R, Hamer K, Blokzijl F, Verstegen MM, et al. Long‐term culture of genome‐stable bipotent stem cells from adult human liver. Cell 2015;160:299‐312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Nantasanti S, de Bruin A, Rothuizen J, Penning LC, Schotanus BA. Concise review: organoids are a powerful tool for the study of liver disease and personalized treatment design in humans and animals. Stem Cells Transl Med 2016;5:325‐330. [DOI] [PMC free article] [PubMed] [Google Scholar]