

Abstract
Background
Over 96% of high‐grade ovarian carcinomas and 50% of all cancers are characterized by alterations in the p53 gene. Therapeutic strategies to restore and/or reactivate the p53 pathway have been challenging. By contrast, p63, which shares many of the downstream targets and functions of p53, is rarely mutated in cancer.
Methods
A novel strategy is presented for circumventing alterations in p53 by inducing the tumor‐suppressor isoform TAp63 (transactivation domain of tumor protein p63) through its direct downstream target, microRNA‐130b (miR‐130b), which is epigenetically silenced and/or downregulated in chemoresistant ovarian cancer.
Results
Treatment with miR‐130b resulted in: 1) decreased migration/invasion in HEYA8 cells (p53 wild‐type) and disruption of multicellular spheroids in OVCAR8 cells (p53‐mutant) in vitro, 2) sensitization of HEYA8 and OVCAR8 cells to cisplatin (CDDP) in vitro and in vivo, and 3) transcriptional activation of TAp63 and the B‐cell lymphoma (Bcl)‐inhibitor B‐cell lymphoma 2‐like protein 11 (BIM). Overexpression of TAp63 was sufficient to decrease cell viability, suggesting that it is a critical downstream effector of miR‐130b. In vivo, combined miR‐130b plus CDDP exhibited greater therapeutic efficacy than miR‐130b or CDDP alone. Mice that carried OVCAR8 xenograft tumors and were injected with miR‐130b in 1,2‐dioleoyl‐sn‐glycero‐3‐phosphatidylcholine (DOPC) liposomes had a significant decrease in tumor burden at rates similar to those observed in CDDP‐treated mice, and 20% of DOPC–miR‐130b plus CDDP‐treated mice were living tumor free. Systemic injections of scL–miR‐130b plus CDDP in a clinically tested, tumor‐targeted nanocomplex (scL) improved survival in 60% and complete remissions in 40% of mice that carried HEYA8 xenografts.
Conclusions
The miR‐130b/TAp63 axis is proposed as a new druggable pathway that has the potential to uncover broad‐spectrum therapeutic options for the majority of p53‐altered cancers.
Keywords: 3‐dimensional (3D) spheroids, B‐cell lymphoma 2‐like protein 11 (BIM), chemosensitization, cisplatin, leoyl‐sn‐glycero‐3‐phosphatidylcholine (DOPC), microRNA 130b (miR‐130b), ovarian cancer, transactivation (TA) and N‐terminally truncated (ΔN) isoforms of the p63 protein (TAp63/ΔNp63), tumor protein p53, tumor‐targeted nanocomplex (scL)
Short abstract
This study uncovered a new feed‐forward loop that involves tumor protein p53 family member TAp63 and its downstream target microRNA 130b (miR‐130b). Injection of miR‐130b using 1,2‐dioleoyl‐sn‐glycero‐3‐phosphocholine liposomes (nontargeted) and a clinically tested, tumor‐targeted nanocomplex (scL) resulted in significantly decreased tumor burden and improved overall survival, suggesting that the miR‐130b/TAp63 tumor‐suppressor axis could open up novel therapeutic options for many cancers with alterations in the p53 gene and/or pathway.
Introduction
Ovarian cancer (OVCA) is the most lethal gynecologic malignancy because there is a lack of effective screening tools and OVCA has the ability to acquire resistance to chemotherapy.1 Cisplatin (CDDP) is a first‐line chemotherapeutic agent used to treat OVCA.2, 3 The 5‐year survival rate for patients with OVCA is 92% if it is diagnosed when the cancer is localized, 72% when it has spread to lymph nodes, 27% when it has metastasized to distant sites, and 22% when it is unstaged (Surveillance, Epidemiology, and End Results statistics). Over 75% of women present with advanced‐stage disease, >60% experience disease recurrence from chemoresistant tumors, and from 70% to 90% die of their disease.1
The p53 pathway was impaired in 96% of 489 high‐grade serous ovarian cancers that were studied.4 Reactivation of p53 has been difficult to implement therapeutically, because from 75% to 80% of p53 mutations are missense mutations that can acquire dominant negative activity to inhibit wild‐type p535, 6, 7 and induce chemoresistance.8, 9 Therefore, we hypothesized that p63, which shares a high degree of conservation with p53 in the DNA binding and transactivation domains, would be a powerful target for cancer therapy.10, 11, 12, 13 Activating p63 triggers cell‐cycle arrest, apoptosis, and senescence, even in the absence of p53.14, 15 It has been observed that p63 compensates for the loss of p53 in cancer cells under genotoxic stress, and it is rarely mutated in cancer.16
We selected microRNA 130b (miR‐130b), a direct downstream target that is transcriptionally activated by the transactivation (TA) domain of tumor protein p63 (TAp63), as a candidate tumor‐suppressor microRNA that has the potential to overcome mutations in p53 by activating the TAp63 pathway. Furthermore, miR‐130b is epigenetically silenced and/or downregulated in chemoresistant OVCA and is negatively correlated with tumor stage, suggesting that it can sensitize tumors to chemotherapy.3, 17, 18, 19, 20, 21
We tested the in vivo impact of miR‐130b using a nontargeted nanoliposome (1,2‐dioleoyl‐sn‐glycero‐3‐phosphatidylcholine [DOPC]) and a tumor‐targeted nanocomplex (scL) as delivery vehicles. DOPC is in phase 1 clinical trials for testing ephrin type‐A receptor 2 precursor (EphA2) small interfering RNA (siRNA) in OVCA.22, 23 Prototypes of scL have completed phase 1a and 1b clinical trials for testing p5324 or retinoblastoma 94 (RB94)25 gene therapy in patients with advanced solid tumors. Antitumor effects were produced by scL‐p53, and it was well tolerated at therapeutic doses in phase 2. scL is a cationic liposome, the surface of which is decorated with an antitransferrin receptor antibody fragment as the targeting moiety,24, 26, 27, 28 which, when injected systemically, specifically and efficiently delivers the payload to primary and metastatic tumor cells throughout the body by binding to the transferrin receptor, the level of which is elevated in most tumor types.29
We observed that the treatment of mice carrying HEYA8 and OVCAR8 xenografts with the miR‐130b plus CDDP combination significantly increased apoptosis and long‐term survival in mice compared with CDDP treatment alone. Molecular studies have demonstrated that miR‐130b, which is a direct downstream target of TAp63,30 can establish a feed‐forward loop to transcriptionally activate TAp63 and significantly induce TAp63 to target the B‐cell lymphoma 2 (BCL2)‐inhibitor BCL2‐like protein 11 (BIM). We also demonstrated that, in HEYA8 mice, miR‐130b can downregulate N‐terminally truncated (ΔN) p63 (ΔNp63), the oncogenic isoform, which is a competitive inhibitor of TAp63 and p53. We also established that TAp63 is a critical downstream effector of miR‐130b–mediated tumor suppression. We propose that targeting the miR‐130b/TAp63 axis is a clinically attractive therapeutic strategy not only for the treatment of OVCA but also for the treatment of many of the 80% of all cancers that carry mutations in p53.
Materials and Methods
miRNA Transfection and Drug Treatments
Cells (6 × 104 per well) were seeded in 6‐well plates for 24 hours. Then, 20 nM of microRNA mimics and negative controls (NCs) (Ambion Inc) and 2 μL Lipofectamine 2000 (Invitrogen) per transfection were transfected according to the manufacturer’s instructions. Twenty‐four hours after transduction, media were replaced with media containing 5 μg/mL CDDP (Sigma).
3‐Dimensional Culture Assay
To form 3‐dimensional (3D) structures, cells (with or without miRNA transfection) were incubated overnight with magnetic gold‐polymer–iron‐oxide hydrogel (Nanoshuttle‐PL; n3D Biosciences, Inc) to allow attachment to the cells. The cells were magnetically levitated for 4 hours to form a 3D structure with the extracellular matrix; then, they were mechanically disrupted with a pipette, transferred to a 96‐well plate, and the culture was printed into a dot shape by placing a magnet drive underneath the plate for 15 minutes. The 3D culture area was automatically imaged over time by using the IncuCyte ZOOM live‐cell imaging system (Essen BioScience Inc), plotted for each well, and statistical significance was determined using a 1‐way analysis of variance (R software; R Foundation for Statistical Computing).
Western Blot Analysis
Total proteins were extracted from cellular pellets using a radioimmunoprecipitation assay buffer (Sigma Corporation) with protease inhibitors (Complete‐Mini; Roche Diagnostics). Thirty micrograms of protein was used to perform Western blot analyses, as previously described.17 Primary antibodies were added at the concentrations suggested by each manufacturer (P53 OP29 from CalBiochem; TAp63, ΔNp63, p21‐12D1, BIM‐C34C5, and cleaved caspase 3‐D175 from Cell Signaling; and actin‐F3165 from Sigma).
In Vivo Treatment With DOPC–miR‐130b and CDDP
miR‐130b and NC miRNAs (Life Technologies) were conjugated with DOPC as previously described.31 The animal experiments were performed in accordance with protocols approved by the Institutional Animal Care and Use Committee of The University of Texas MD Anderson Cancer Center. Female athymic nude mice were injected into the peritoneal cavity with 1 × 106 OVCAR8 cells that stably expressed the luciferase gene. Mice were divided into 4 groups (n = 5 per group) as follows: 1) DOPC‐NC, 2) DOPC–miR‐130b, 3) DOPC‐NC plus CDDP, and 4) DOPC–miR‐130b plus CDDP. One week after injection, each miRNA was administered twice weekly at a dose of 200 μg/kg body weight. Treatment was continued for 6 weeks with 11 injections. Mouse weight and luciferase intensity were recorded weekly using an in vivo bioluminescent imaging system.
Preparation of scL Nanocomplexes
The nanocomplexes were prepared in a manner similar to that previously described using siRNA.24 Briefly, the cationic liposome was combined with the targeting antitransferrin receptor single‐chain antibody fragment (TfRscFv) and then was mixed with the appropriate amount of miRNA to form the scL–miR‐130b nanocomplex. Before use, the size of the complex was measured using the Malvern Zetasizer nano‐ZS version 6.01 and software according to the manufacturer’s instructions (Malvern Instruments). For further details of the cell lines and assays, see the Supporting Materials and Methods.
In Vivo Imaging for the scL‐miRNA Mouse Model
For in vivo tumor‐targeting studies, mice bearing HEYA8 tumors were injected once into the tail vein with 25 μg of scL‐NC labeled with indocarbocyanine (cy5) dye. Image acquisition and analyses were performed using the Maestro in vivo fluorescence imaging system (Caliper Life Sciences) with the cy5 detection filter. Identical parameters were used to image the tumors and normal organs. SigmaPlot 11.0 (Systat Software, Inc) was used to graph the data and to compute statistical significance using the Student t test.
Animal Model and Treatment With scL‐miR
Athymic nude mice aged 4 to 6 weeks were obtained from Harlan Sprague Dawley Inc and housed as a group in the Division of Comparative Medicine at Georgetown University School of Medicine. All animal experiments were performed in accordance with, and under protocols approved by the Georgetown University Animal Care and Use Committee. Mice were injected intraperitoneally with 2.0 × 106 HEYA8‐luc cells or OVCAR8‐luc cells in 200 μL phosphate‐buffered saline. Seven days after tumor cell inoculation, mice received an intraperitoneal injection twice weekly for a total of 5 weeks with 50 μg scL–miR‐130b, with 60 to 75 μg CDDP, or with scL–miR‐130b plus CDDP. At the appropriate time, or when the animals were becoming moribund, they were killed by inhalation of isoflurane according to institutional guidelines.
Results
miR‐130b Inhibits Migration, Invasion, and Multicellular Spheroid Formation in 3D Culture Models of OVCA Cells
Scratch‐wound assays revealed that treatment with miR‐130b modestly impaired migration in both HEYA8 and OVCAR8 cells. In HEYA8 cells, the relative wound density (RWD) at 14 hours was 23% lower (P = 6.1E‐13) (Fig. 1A); whereas, in OVCAR8 cells, the RWD was 20% lower at 24 hours compared with untreated (NT) cells (P = 8.3E‐13) or cells treated with NC (P = 2.8E‐10) (Fig. 1B). Overexpression of miR‐130b also reduced the ability of the cells to invade a Matrigel matrix. HEYA8 cells had a 45% reduction in RWD at 60 hours (P = 5.4E‐13) (Fig. 1C). The effect was less pronounced in OVCAR8 cells, with a 17% reduction of RWD in cells treated with miR‐130b compared with those treated with NC (P = 9.1E‐3) (Fig. 1D). The migration and invasion assays for the cell lines SKOV3, OVCAR3, OVCAR4, and OVCAR5 did not differ significantly between miR‐130b treatment and NC treatment. By using 3D cell culture models, which are considered better representatives of tumors in vivo, using the magnetic levitation system,32 we observed that NT and NC‐treated HEYA8 and OVCAR8 cells formed tight, multicellular spheroids by 72 hours after levitation. By contrast, HEYA8 and OVCAR8 cells transiently transfected with miR‐130b were unable to form compact spheroids and exhibited considerable cell death (Fig. 1E) (Supporting Fig. 1). The p53‐null SKOV3 spheroids treated with miR‐130b were only marginally disrupted, and the impact of miR‐130b on p53‐mutant OVCAR3, OVCAR4, and OVCAR5 spheroids ranged from moderate to significant (Fig. 1E, Supporting Fig. 1). Collectively, these results suggest that miR‐130b is likely to impair metastatic progression in p53‐mutant OVCA cell lines by disrupting spheroids, which are the primary agents of metastatic spread. The findings also suggest that the effects of miR‐130b overexpression on migration, invasion, and spheroid formation phenotypes are sensitive to the genetic background of the cancer cell.
Figure 1.
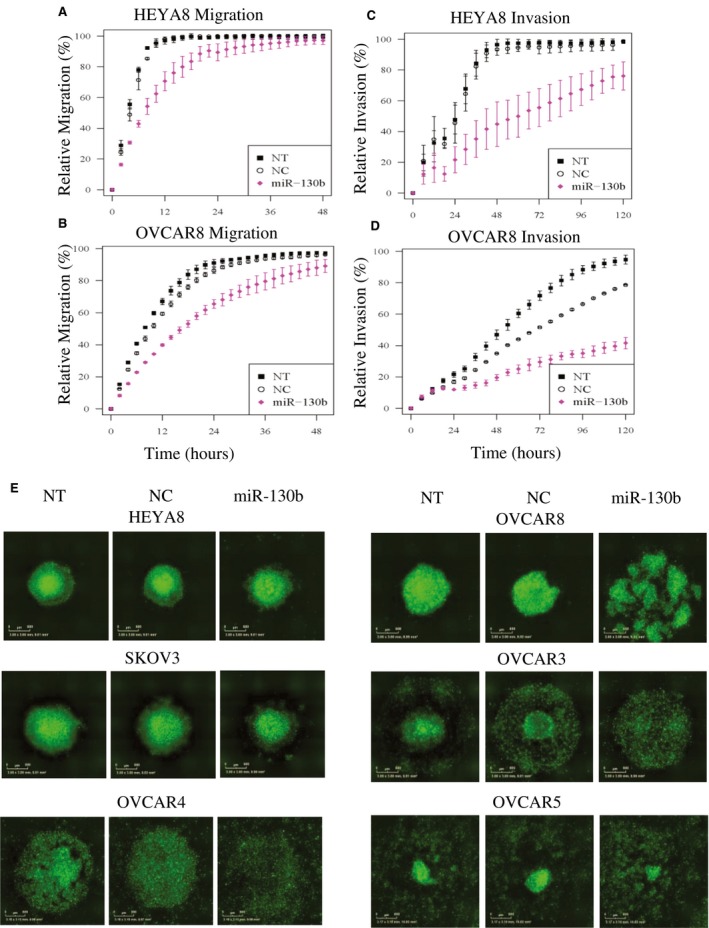
Migration/invasion assays were performed on HEYA8 and OVCAR8 cells and on 3‐dimensional (3D) spheroid formation in 6 ovarian cancer cell lines treated with microRNA 130b (miR‐130b). Migration of (A) HEYA8 and (B) OVCAR8 cells and invasion of (C) HEYA8 and (D) OVCAR8 cells are illustrated. Error bars indicate standard deviations of 3 experiments. The time courses started 72 hours after transfection. (E) Green fluorescent 3D spheroid images of HEYA8, OVCAR8, SKOV3, OVCAR3, OVCAR4, and OVCAR5 cells 72 hours after transfection are shown. NC indicates treated with 20 nM scrambled negative control; NT, untreated; miR‐130b: treated with 20 nM miR‐130b mimics.
The Impact of miR‐130b on the p53 and p63 Pathways
Because miR‐130b is a direct target of p63, a p53 family member, and a tumor suppressor in multiple cancers,33, 34 we analyzed the effects of miR‐130b overexpression on selected genes from the p53 and p63 pathways. Transient transfection with miR‐130b mimics resulted in miR‐130b levels that were increased >800‐fold in HEYA8 cells and >2500‐fold in OVCAR8 cells 72 hours after transfection (Supporting Fig. 2A). After miR‐130b transfection, p53, p21, mouse double minute 2 homolog (MDM2), p63, phosphatase and tensin homolog (PTEN), and retinoblastoma 1 (RB1) transcripts were upregulated in HEYA8 cells (Supporting Fig. 2B). In contrast, only p63 transcripts were significantly induced, and MDM2 and PTEN levels were moderately increased in OVCAR8 cells (Supporting Fig. 2B). When we used p53 siRNA, the up‐regulation of MDM2 by miR‐130b depended on the expression of p53 in HEYA8 cells (Supporting Fig. 2C) and was unaffected in OVCAR8 cells. The upregulation of p63 was completely abrogated by p53 siRNA in OVCAR8 cells (Supporting Fig. 2D) and was unaffected in HEYA8 cells.
To investigate the effect of miR‐130b on different isoforms of p63 and BIM, we used Western blot analyses and observed that TAp63, which is not expressed in HEYA8 or OVCAR8 cells, was induced 3.89‐fold in HEYA8 cells and 3.06‐fold in OVCAR8 cells compared with NC (Fig. 2); whereas ΔNp63, which is expressed at high levels in both cell lines, was downregulated 0.5‐fold in HEYA8 cells and upregulated 1.24‐fold in OVCAR8 cells. The BCL2‐inhibitor BIM was induced 4.47‐fold in HEYA8 cells and 9.44‐fold in OVCAR8 cells (Fig. 2). In addition, miR‐130b upregulated caspase‐3, p21, p27, and cyclin‐dependent kinase 6 (CDK6) and downregulated CDK4 and PUMA (p53 upregulated modulator of apoptosis) in HEYA8 cells. In OVCAR8 cells, miR‐130b upregulated mutant p53 and CDK6 and downregulated CDK4 and p21 (Supporting Fig. 2E).
Figure 2.
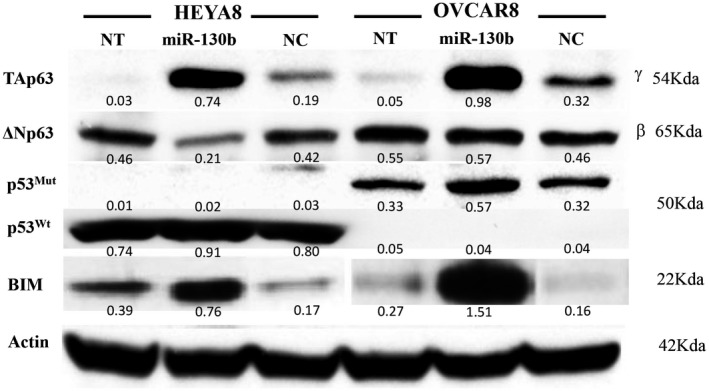
The effect of microRNA 130b (miR‐130b) on the p63 transactivation (Tap63) and N‐terminally truncated (ΔNp63) axis protein levels. Representative blots from duplicate Western blot analyses after transfection with miR‐130b in HEYA8 and OVCAR8 cells are shown here, with quantification indicated under each blot. BIM indicates B‐cell lymphoma 2‐like protein 11; NC, negative control; NT, no treatment; p53mut, mutant p53; p53wt, wild‐type p53.
p63 Is a Critical Downstream Effector of miR‐130b
In head and neck cancers, p63 is regulated by miR‐130b.35 To determine whether it is a direct target of miR‐130b in ovarian cancer cells, we cloned the 3′‐untranslated region of p63, which contains a predicted miR‐130b binding site according to TargetScan 6.0 software, and we performed a luciferase reporter assay. We observed that the miR‐130b–transfected cells decreased luciferase activity by 50% for HEYA8 cells and by 30% for OVCAR8 cells (Supporting Fig. 3A). To determine whether the induction of TAp63 is necessary and sufficient for miR‐130b–driven tumor suppression, we transfected cells with an expression vector carrying TAp63 or ΔNp63. Cell proliferation measured through an MTS (3‐[4,5‐dimethylthiazol‐2‐yl]‐5‐[3‐carboxymethoxyphenyl]‐2‐[4‐sulfophenyl]‐2H‐tetrazolium) assay revealed that TAp63 overexpression led to a decrease in cell viability by 80% in HEYA8 cells at 48 hours after transfection and by 60% in OVCAR8 cells at 72 hours after transfection (Fig. 3A). In contrast, overexpression of the other isoform, ΔNp63, had less impact on cell viability compared with TAp63 (Supporting Fig. 3B). To assess the clinical significance of our findings, we integrated miRNAseq and RNAseq data from The Cancer Genome Atlas Ovarian Cancer Project, in which p63 is altered (mostly amplified) in 25% of patients who have data available on clinical outcomes (Fig. 3B). We observed that patients who had tumors that expressed higher levels of miR‐130b had better overall survival (P = .034), those who had higher levels of TAp63 trended toward better survival (P = .099), and those who had higher levels of ΔNp63 trended toward worse survival (P = .018) (Fig. 3C).
Figure 3.
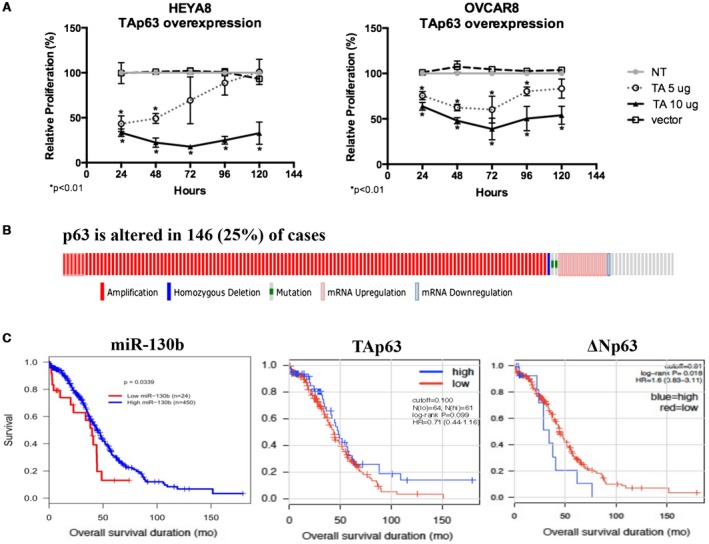
p63 is a critical downstream effector of microRNA 130b (miR‐130b). (A) The relative proliferation of HEYA8 and OVCAR8 cells after overexpression of the transactivation (TA) domain of tumor protein p63 (TAp63). Error bars indicate the standard deviation of 3 biologic replicates. Asterisks indicate P < .01. NT indicates no treatment; vector, pCDNA3.1 empty vector. (B) Oncoprints generated from the cBioPortal Cancer Genomics (available at: http://www.cbioportal.org/, Accessed March 3, 2019) indicate that p63 is altered in 146 patients who had 603 samples (25%). (C) This is a Kaplan‐Meier survival curve of miR‐130b, TAp63, and the N‐terminally truncated (ΔN) isoform of p63 (ΔNp63) from The Cancer Genome Atlas. HR indicates hazard ratio.
In Vivo Sensitization of OVCAR8 Xenograft Tumors to CDDP by DOPC–miR‐130b
Mice were inoculated intraperitoneally with OVCAR8 cells carrying the stably transfected luciferase gene to induce tumors (day 0). Seven days after inoculation, mice (5 per group) were treated intraperitoneally with 10 μg DOPC–miR‐130b/NC, either alone or in combination with CDDP, twice weekly for a total of 11 treatments. The dose of CDDP was 60 μg per mouse per injection for the first 5 treatments and 40 μg per mouse per injection for the last 6 treatments. Mice were imaged every week using an in vivo bioluminescent imaging system to assess tumor response. Compared with NC mice, mice treated with CDDP or with DOPC–miR‐130b had tumor regression. In addition, mice that received DOPC–miR‐130b plus CDDP had the lowest signal intensity, indicating major tumor growth inhibition/reduction (Fig. 4A,B). The untreated animals died or were humanely killed because of tumor burden by 95 days after tumor cell inoculation. Treatment with either DOPC–miR‐130b or CDDP extended survival, and all mice in these single‐agent treatment groups died from their disease by day 111. Treatment with the DOPC–miR‐130b plus CDDP considerably increased the lifespan (P < .03). One hundred percent of the combination‐treated mice survived until day 88, and 20% survived for >200 days (Fig. 4C).
Figure 4.
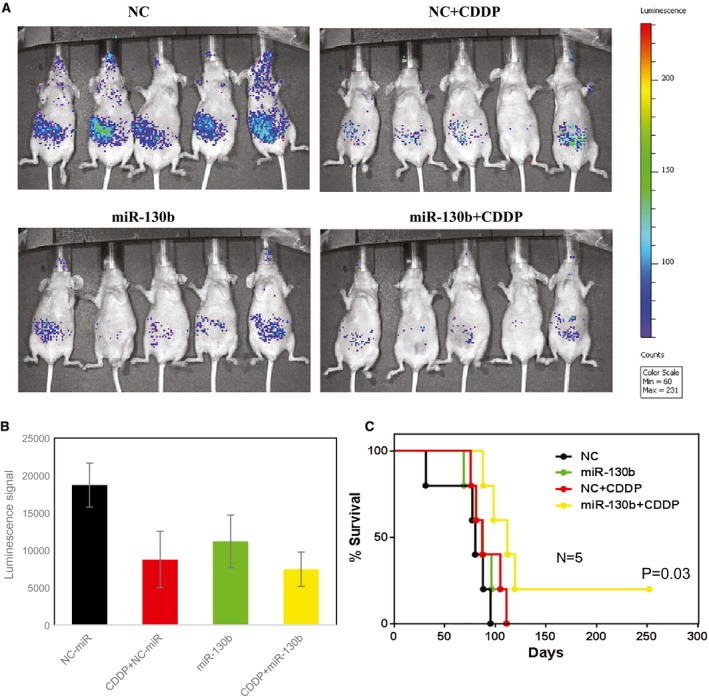
In vivo chemosensitization of ovarian tumors by 1,2‐dioleoyl‐sn‐glycero‐3‐phosphatidylcholine (DOPC)–microRNA 130b (miR‐130b) is illustrated in an orthotopic mouse model of ovarian cancer. Athymic nude mice bearing OVCAR8 xenograft tumors were treated with DOPC–miR‐130b and/or cisplatin (CDDP) (miR‐130b + CDDP) twice weekly for 5 weeks. Normal control (NC) mice treated with DOPC–miR were used as controls. (A) In these images (from week 3), green corresponds to the highest signal intensity (and, thus, to tumor burden), and blue corresponds to the lowest signal intensity. Bar charts illustrate (B) the quantification of luminescence signal intensities and (C) Kaplan‐Meier curves for the chemosensitization study.
In Vitro Sensitization of HEYA8 and OVCAR8 Cells to CDDP by scL–miR‐130b
The DOPC delivery platform has been extensively characterized and is now in early stage clinical trials. Because delivery is such a major component of noncoding RNA therapeutics, we tested an additional platform scL, which also has been tested clinically and achieved tumor targeting, to demonstrate the consistency of biologic effects. OVCA cells transfected with cy5‐labeled scL–miR‐130b/NC had high levels of fluorescence after transfection, demonstrating efficient uptake. A dose‐dependent cell death response was observed when cells were transfected with 100 nM of scL–miR‐130b, with an even greater effect using 200 nM. In contrast, HEYA8 and OVCAR8 cells transfected with scL‐NC exhibited cell death only at 200 nM, likely because of nonspecific cytotoxicity (Fig. 5A). To test whether miR‐130b could sensitize chemoresistant OVCA cells to CDDP, HEYA8 and OVCAR8 cells were treated with increasing doses of CDDP, and the 50% inhibitory concentration (IC50) value (the drug dose resulting in 50% cell death) was determined by an 2,3‐bis‐(2‐methoxy‐4‐nitro‐5‐sulfophenyl)‐2H‐tetrazolium‐5‐carboxanilide (XTT) cell‐survival assay. Both OVCA lines had marked increases in sensitization to CDDP when treated with scL–miR‐130b, with IC50 values of 3 and 0.8 μM for HEYA8 and OVCAR8 cells, respectively (Fig. 5B). In contrast, at the maximum dose of CDDP tested, controls (liposome only, CDDP only, and scL‐NC) did not reach an IC50 in either cell line. Furthermore, nonspecific cytotoxicity from the liposome was ruled out because of a lack of significant cell death in the liposome‐only treated controls. Collectively, these experiments demonstrate that, in vitro, miR‐130b‐scL is able to overcome the resistance of both p53‐wild type and p53‐mutant OVCA cell lines to CDDP.
Figure 5.
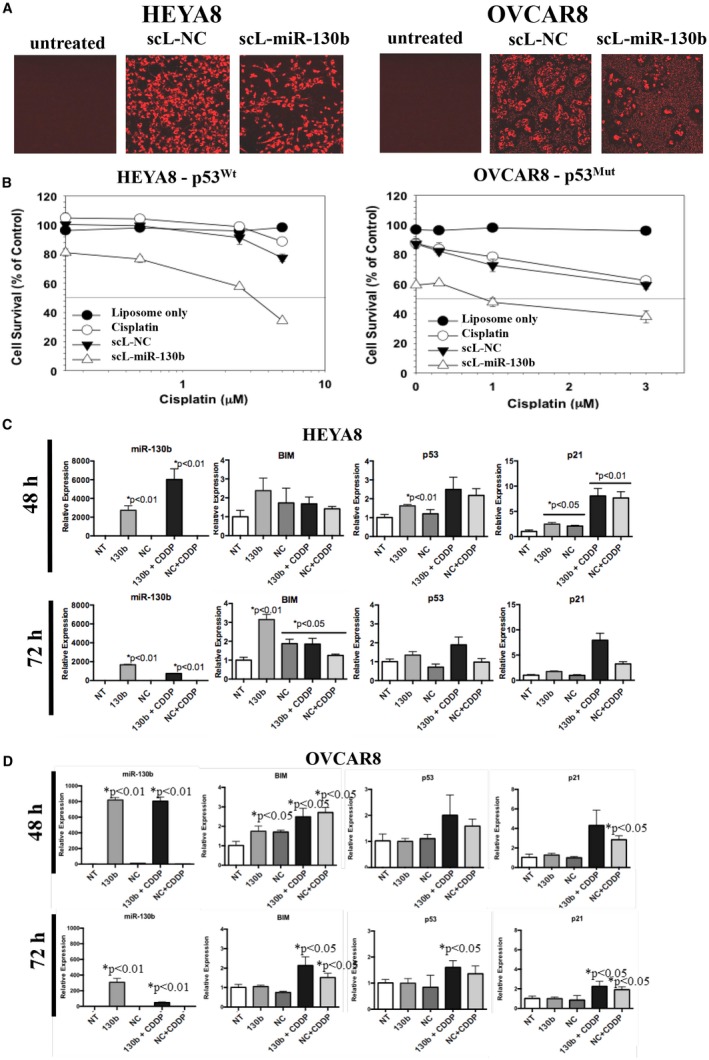
In vitro chemosensitization of ovarian cancer cells was achieved by tumor‐targeted nanocomplex (scL)‐delivered microRNA 130b (miR‐130b). (A) HEYA8 and OVCAR8 human ovarian cancer cells were seeded at a density of 4.0 × 103 cells per well in 96‐well plates. Twenty‐four hours later, the cells were transfected with indocarbocyanine (cy5)‐labeled scL–miR‐130b or scL‐normal control (NC) microRNA at 200 nM miRNA per well. After a 5‐hour incubation, 10% serum was added, and the cells were incubated for an additional 67 hours then imaged without fixing. p53Mut indicates mutant p53; p53Wt, wild‐type p53. (B) HEYA8 and OVCAR8 cells were transfected with 35 nM scL–miR‐130b or scL‐NC. Five hours after transfection, increasing doses of cisplatin (CDDP) were added. Seventy‐two hours later, the level of cell survival was assessed using a 2,3‐bis‐(2‐methoxy‐4‐nitro‐5‐sulfophenyl)‐2H‐tetrazolium‐5‐carboxanilide (XTT) assay, and the results were plotted. Expression levels of selected miR‐130b–responsive genes in are illustrated in (C) HEYA8 and (D) OVCAR8 cells that were transfected with scL–miR‐130b at 48 and 72 hours after transfection. BIM indicates B‐cell lymphoma 2‐like protein 11; NT, not treated.
To assess the impact on downstream targets, RNA was harvested from cells treated with 60 nM of either scL–miR‐130b or scL–miR‐130b plus CDDP (3 μM) at 48 and 72 hours after treatment. At 48 hours after treatment with scL–miR‐130b and scL–miR‐130b plus CDDP, the levels of miR‐130b increased approximately 3000‐fold and 6000‐fold, respectively, in HEYA8 cells (Fig. 5C) and approximately 800‐fold in OVCAR8 cells (Fig. 5D). In HEYA8 cells, scL–miR‐130b treatment resulted in increases >3‐fold in BIM and approximately 1.5‐fold to 2.5‐fold increases in p53 and p21, and scL–miR‐130b plus CDDP treatment resulted in increases of approximately 2‐fold to 3‐fold in p53 and approximately 7.5‐fold to 8‐fold in p21 (Fig. 5C). In OVCAR8 cells, no significant increase was observed in BIM, p53, or p21 with scL–miR‐130b treatment, whereas treatment with scL–miR‐130b plus CDDP led to modest increases of 2.2‐fold in BIM and approximately 2.5‐fold in p21 72 hours after treatment (Fig. 5D).
In Vivo Sensitization and Long‐Term Survival of HEYA8 Ovarian Xenograft Tumors to scL–miR‐130b Plus CDDP
To assess whether scL injected systemically can bypass normal cells and preferentially deliver miRNAs to both primary and metastatic tumors in vivo, mice bearing HEYA8 subcutaneous xenograft tumors were injected intravenously into the tail vein with 25 μg of cy5‐labeled scL‐NC. Two individual tumors from the same animal and normal organs (liver, kidneys, lungs, and spleen) were harvested 40 hours postinjection and imaged. High levels of red fluorescence were evident in both tumors (Fig. 6A). More significantly, the fluorescence was evident not only on the dorsal and ventral surfaces of the tumor but also in the tumor cross‐sections, demonstrating the successful delivery and penetrance of the scL nanocomplex to the interior of the tumor (Fig. 6A). In contrast, no fluorescence was evident in any of the normal organs examined except the kidneys. At this time point of 40 hours postinjection, the signal in the kidneys is likely caused by the excretion of either cy5‐labeled miR‐130b or, more likely, only the excretion of cy5 as a degradation product. The dark area in the center of the cross‐sectional tumor images in Figure 6 is because of necrosis in the center of these relatively large (>600 mm3) tumors. These results demonstrate both the tumor‐targeting specificity of the systemically administered scL delivery system as well as the efficient uptake of scL–miR‐130b by ovarian tumors in vivo.
Figure 6.
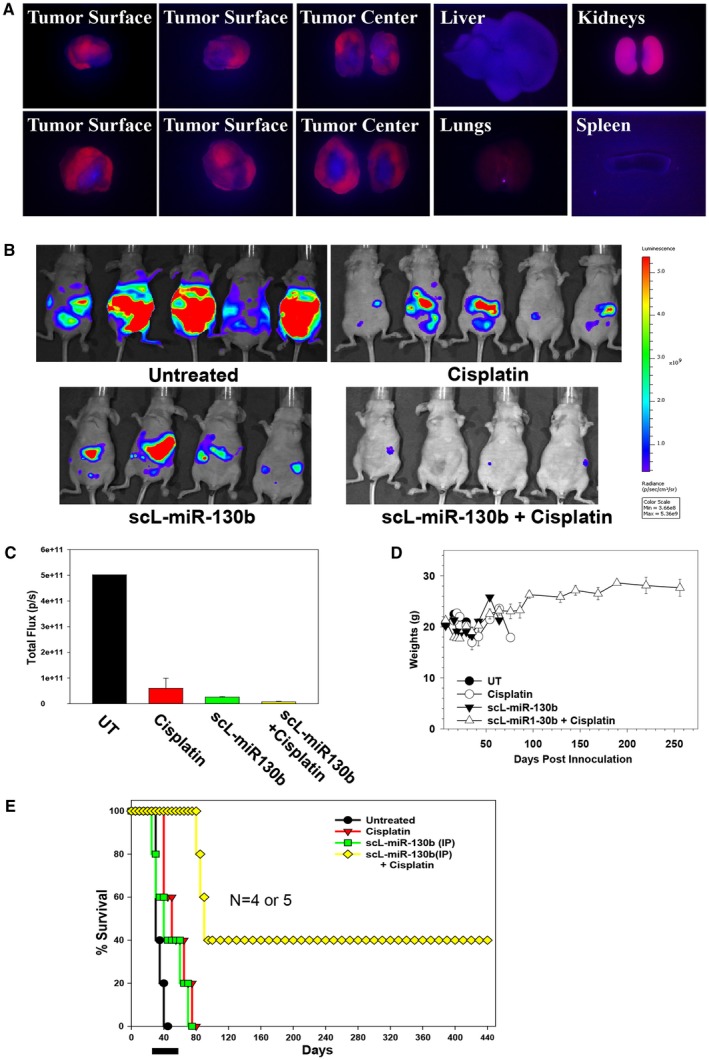
In vivo tumor targeting was achieved by systemically administered, tumor‐targeted nanocomplex (scL)‐microRNA and the chemosensitization of ovarian tumors by scL–microRNA 130b (miR‐130b) in an orthotopic mouse model of ovarian cancer. (A) Athymic nude mice bearing HEYA8 subcutaneous xenograft tumors were injected once into the tail vein with 25 μg indocarbocyanine (cy5)‐labeled, tumor‐targeted nanocomplex. Forty hours after injection, the dorsal and ventral sides and the central portion (cross‐section) of 2 individual tumors (liver, lungs, kidneys, and spleen) were imaged using an in vivo fluorescence imaging system with a cy5 detection filter. (B) Athymic nude mice bearing HEYA8 subcutaneous xenograft tumors were treated with scL–miR‐130b and/or cisplatin twice weekly for 5 weeks. Untreated animals were used as controls. The animals were imaged on day 42. Red corresponds to the highest signal intensity (maximum [max]) and, thus, tumor burden; and purple corresponds to the lowest signal intensity (minimum [min]). (C) Quantification of Xenogen signal intensities for the images were obtained 42 days post‐tumor cell inoculation. (D) The weights of the mice were monitored on a regular basis. (E) Kaplan‐Meier curves were generated from the in vivo chemosensitization study. The black bar indicates the duration of treatment. IP indicates intraperitoneal; UT, untreated.
Mice were injected intraperitoneally with HEYA8 cells carrying the stably transfected luciferase gene to induce tumors (day 0). Seven days after inoculation, groups of mice (4‐5 mice per group) were injected intraperitoneally with scL–miR‐130b (50 μg miR‐130b per mouse per injection), either alone or in combination with CDDP, twice weekly for a total of 10 treatments. Control groups of mice were untreated or received CDDP only. The dose of CDDP was 60 μg per mouse per injection for the first 7 treatments and 75 μg per mouse per injection for the last 3 treatments. Four weeks after HEYA8 cell inoculation, the untreated mice had developed significant tumor burden and ascites. The animals were imaged using the Xenogen in vivo imaging system to assess tumor response. A high level of luciferase expression was observed in the untreated mice, indicating a considerable amount of tumor burden in the abdomen (Fig. 6B). Although treatment with CDDP resulted in some tumor growth inhibition, tumor burden was still apparent, suggesting the development of CDDP resistance. A similar tumor response was observed in scL–miR‐130b–treated mice, indicating that the antitumor efficacy of miR‐130b alone is similar to that of CDDP (Fig. 6B). In contrast, mice that received scL–miR‐130b plus CDDP treatment were nearly free of tumor (Fig. 6B). On day 42 (the last day of treatment), a lower tumor burden was evident in all treatment groups (P < .01) compared with untreated controls, and mice that were treated with scL–miR‐130b plus CDDP displayed the lowest signal intensity, indicating major tumor growth inhibition/reduction and complete tumor elimination (Fig. 6C). The mice maintained a healthy weight before, during, and long after treatment had ended, indicating a lack of major toxicities associated with the scL–miR‐130b treatment regimen (Fig. 6D).
In our long‐term survival studies, untreated animals died or were humanely killed because of tumor burden by day 42 after tumor cell inoculation. Although the single‐agent treatment groups had extended survival, all of the mice died of their disease by day 80 (Fig. 6E). In contrast, treatment with combined scL–miR‐130b plus CDDP considerably increased the lifespan, with 100% of the combination‐treated mice surviving until day 83 and 40% surviving beyond day 440 (Fig. 6E). Necropsy results after their eventual death revealed no indication of tumor recurrence. The significant antitumor efficacy and long‐term survival, combined with the lack of toxicity from scL–miR‐130b plus CDDP treatments, demonstrate that scL–miR‐130b not only targets and efficiently delivers miR‐130b to tumors in an orthotopic mouse model of human OVCA, but also can enhance the response of ovarian tumors to CDDP, a first‐line therapeutic agent.
Reverse‐Phase Protein Array Analysis of miR‐130b–Treated OVCA
Reverse‐phase protein arrays conducted to assess the global impact of miR‐130b demonstrated that miR‐130b treatments downregulated proto‐oncogene tyrosine protein kinase Src (SRC), postmeiotic segregation increased, Saccharomyces cerevisiae 2 (PMS2), and v‐myc avian myelocytomatosis viral oncogene homolog (MYC) in HEYA8 cells and downregulated SRC, PMS2, and poly(adenine diphosphate‐ribose) polymerase 1 (PARP1) in OVCAR8 cells (Fig. 7A, Supporting Table 1). Olaparib (AZD2281), the first PARP inhibitor to be tested in OVCA, demonstrated clinical benefit in recurrent ovarian tumors carrying mutations in BRAC1/BRCA2. We propose that, by downregulating PARP1 and PMS2, drugs targeting the miR‐130b/TAp63 axis can act as synthetic lethal agents to sensitize patients with OVCA who carry mutations in BRCA1/BRCA2 and DNA‐repair pathway genes, including ataxia‐telangiectasia mutated (ATM), serine/threonine protein kinase ATR (ATR), and checkpoint kinase 1 (CHEK1), to clinically approved PARP inhibitors. Our model for how the miR‐130b/TAp63 tumor‐suppressor axis could be developed to sensitize ovarian tumors and other p53‐mutant tumors to CDDP and other clinically approved drugs is shown illustrated in Figure 7B.
Figure 7.
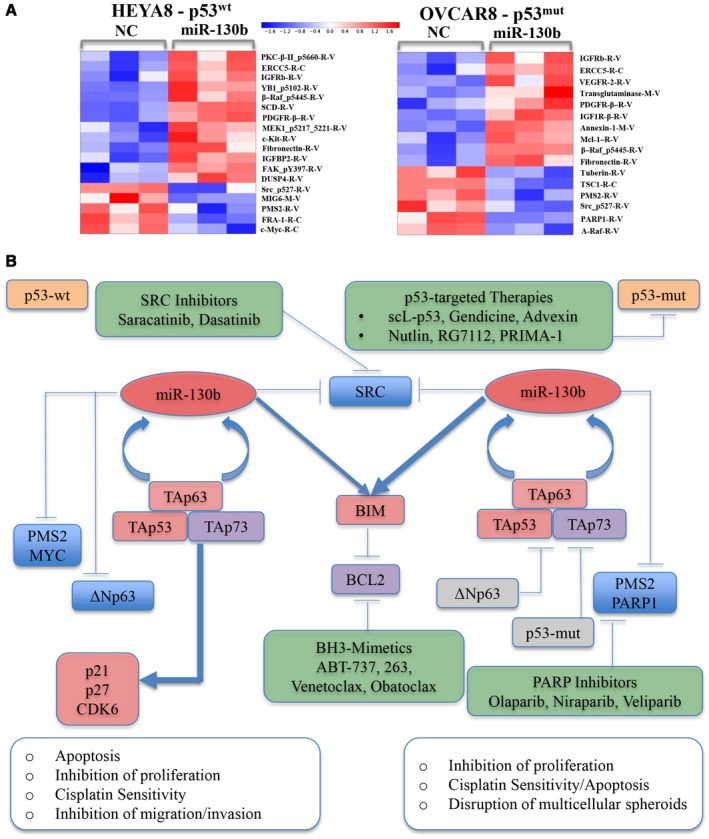
The proteomic footprint of microRNA 130b (miR‐130b) is illustrated in HEYA8 and OVCAR8 xenografts, a model for critical effectors downstream of miR‐130b, and a framework for the therapeutic development of miR‐130b for treating ovarian and other cancers. (A) Heat maps of proteins that are downregulated (blue) and upregulated (red) by >1.25‐fold upon treatment of HEYA8 and OVCAR8 cells with miR‐130b or negative control (NC) are depicted after reverse phase protein array analysis. (B) Genes that were upregulated (red), downregulated (blue), and did not change in response to miR‐130b (gray) treatment are depicted along with genes that have been established as connected (but were not measured) in the current study (purple). Therapeutic agents and/or strategies are indicated that are currently used or are being clinically tested in phase 1 or 2 trials for treating ovarian cancer (OVCA) (green) that target 1 or several direct or indirect downstream targets of miR‐130b. ABT‐737 indicates a small‐molecule B‐cell lymphoma (Bcl‐2) and Bcl‐xL drug; BCL2, B‐cell lymphoma; BIM, B‐cell lymphoma 2‐like protein 11;p53mut, mutant p53; p53wt, wild‐type p53; PARP, poly‐adenosine diphosphatase polymerase; PMS2, postmeiotic segregation increased, Saccharomyces cerevisiae, 2; PRIMA‐1, proline‐rich membrane anchor 1; scL‐p53, scL‐p53 tumor‐targeted nanocomplex; SRC, proto‐oncogene tyrosine protein kinase Src; TAp53, transactivation domain of tumor protein p53; TAp63, transactivation domain of tumor protein p63; TAp73, transactivation domain of tumor protein p73; ΔNp63, N‐terminally truncated (ΔN) isoform of the p63 protein.
Discussion
The treatment of OVCA remains an unmet clinical need despite decades of work. Because >96% of tumors in this study contained mutations in p53, we searched for tumor‐suppressor miRNAs that could circumvent mutations in p53.4 Here, we present work that reveals activating TAp63 as a novel therapeutic option for overcoming chemoresistance not in only OVCA but also in many of the 80% of all cancers that carry mutations in p53, in which TAp63 is an established tumor suppressor.34, 35 We propose that, in the acute phase of treatment, miR‐130b induces its own transcriptional activator (TAp63) to activate a self‐reinforcing, feed‐forward loop that can enhance wild‐type p53 and circumvent mutant p53. Furthermore, miR‐130b functions to represses the oncogenic isoform ΔNp63, which is the only p63 isoform expressed in wild‐type p53 to attenuate the dominant negative impact of ΔNp63 to enable the p53/TAp63/TAp73 tumor‐suppressor complex. These, together with miR‐130b–mediated upregulation of the BCL2‐inhibitor BIM and downregulation of the MYC and SRC oncogenes, lead to increased apoptosis, impaired cell survival, inhibition of migration/invasion, and sensitization to CDDP. It has been reported that mutant p53 promotes epithelial‐mesenchymal transition by antagonizing the miR‐130b–mediated downregulation of ZEB1 (zinc finger E‐box binding homeobox 1); therefore, reactivation of the miR‐130b/TAp63 axis also could attenuate metastatic progression.19, 33, 36
The miR‐130b/TAp63 feed‐forward loop that we present here is clinically attractive because TAp63 is induced by many chemotherapeutic agents and is 1 mechanism of chemoresistance.37, 38 Furthermore, TAp63 is specifically and highly expressed in oocytes, in which it guards the oocytes during the quiescent period before fertilization.39, 40 Therefore, in addition to minimizing side effects from chemotherapy, another very important attribute of TAp63 is its potential for preserving fertility after chemotherapy. Finally, targeted delivery achieved through the TfRscFv‐targeting moiety on the scL nanocomplex is expected to significantly increase treatment efficacy while minimizing side effects on normal tissues, as demonstrated in phase 1 and 2 clinical trials for scL‐p53.25, 27
Funding Support
This work was supported by a High‐Impact High‐Risk Award (RP110355) from the Cancer Prevention and Research Institute of Texas (CPRIT) and by the McNair Foundation (Preethi H. Gunaratne); grants from the CPRIT (RP120124) and the National Cancer Institute (NCI) (1R01CA160394‐01A1) to Elsa R. Flores and Preethi H. Gunaratne; a grant from the CPRIT (RP150094) to Elsa R. Flores, Preethi H. Gunaratne, and Chunru Lin; grants from the National Institute of Diabetes and Digestive and Kidney Diseases (R00DK094981) and the NCI (1R01CA218025‐01) to Chunru Lin; a McCammon Foundation Award to Yinghong Pan; a fellowship form the National Council on Science and Technology in Mexico to Anadulce Hernandez‐Herrera); grants from the NCI (5R01CA132012‐02) and National Foundation for Cancer Research (HU0001) to Esther H. Chang; a research grant from SynerGene Therapeutics, Inc to Kathleen F. Pirollo; a National Institutes of Health (NIH) training grant (5T32CA009686‐19) to Abhi K. Rao; and grants from the National Science Foundation (CAREER DBI‐1254185) and the University of California Cancer Research Coordinating Committee to Abhi K. Rao. These studies were conducted in part using the Preclinical Imaging Research Laboratory and Animal Core Facilities, which are supported by NCI Cancer Center Support grant 2P30‐CA‐51,008, US. Public Health Service grant 1S10RR15768‐01, and an NIH Research Facilities Improvement grant (C06RR14567).
Conflict of Interest Disclosures
Esther H. Chang and Kathleen F. Pirollo are 2 of the inventors of scL, for which several patents are owned by Georgetown University and licensed to SynerGene Therapeutics Inc. Esther H. Chang is an equity stakeholder and serves as a nonpaid consultant to SynerGene Therapeutics Inc. Preethi H. Gunaratne, Ashley L. Benham, and Anadulce Hernandez‐Herrera are 3 of the inventors of microRNA‐130a,b as a tumor suppressor and sensitizing agent for chemotherapy US. Patent No. 9,192,622), for which a patent is owned by the University of Houston. The patent is under consideration for licensing to NEXTmiRNA Technologies, where Preethi H. Gunaratne is the founding scientist and the majority stakeholder. The remaining authors have no disclosures.
Author Contributions
Preethi H. Gunaratne: Contributed to study design; conceived, supervised, and directed the study; provided conceptual advice; and wrote the article. Yinghong Pan : Generated migration and invasion results, carried out the 3D assays, confirmed TP63 to be a direct target of miR‐130b using luciferase reporter assays, led the DOPC‐in vivo study, and wrote the article. Abhi K. Rao: Performed the cell survival assays, performed the confocal microscopy studies, was responsible for the in vivo tumor targeting and in vivo efficacy experiments, analyzed data for these experiments, and wrote the article. Chunru Lin: Led the DOPC‐in vivo study. Anadulce Hernandez‐Herrera: Performed all cell viability and apoptosis assays, generated migration and invasion results, performed all Western blot analyses (other than p63, p53, and caspase 3) and all quantitative polymerase chain reaction analyses, and wrote the article. Ke Liang: Performed the in vivo injections and mouse studies. Antonina S. Rait: Performed the cell survival assays. Avinashnarayan Venkatanarayan: Contributed the p63, p53, and caspase 3 Western blots. Ashley L. Benham: Provided preliminary evidence for miR‐130b as a potential tumor suppressor of ovarian cancer and constructed the luciferase‐expressing HEYA8 cell line used to perform the scL–miR‐130b work. Farwah Rubab: Responsible for the in vivo tumor targeting and in vivo efficacy experiments. SangSoo Kim: Performed the confocal microscopy studies. Clara K. Chan: Generated migration and invasion results. Anil Sood: Contributed the DOPC‐miRNA and DOPC‐NC. Amy C. Rowat: Generated migration and invasion results. Kathleen K. Pirollo: Analyzed data for the experiments and wrote the article. Elsa R. Flores: Contributed the p63, p53, and caspase 3 Western blots. Esther H. Chang: Contributed to study design; conceived, supervised, and directed the study; provided conceptual advice; and analyzed data for the experiments. Lingegowda S. Mangala and Gabriel Lopez‐Berestein: Contributed DOPC‐miRNA preparation. Kimal Rajapakshe and Cristian Coarfa: Contributed to RPPA data analysis.
Supporting information
The first two authors contributed equally to this work.
The third, fourth, and fifth authors contributed equally to this work.
We thank Gordon Robertson of Canada’s Michael Smith Genome Sciences Center at the British Columbia Cancer Agency for generating the Kaplan‐Meier survival curves from The Cancer Genome Atlas ovarian cancer sequence data (Fig. 2C) and for critically reading the article.
Contributor Information
Preethi H. Gunaratne, Email: phgunaratne@uh.edu.
Esther H. Chang, Email: change@georgetown.edu.
References
- 1. Cannistra SA. Cancer of the ovary. N Engl J Med. 2004;351:2519‐2529. [DOI] [PubMed] [Google Scholar]
- 2. Florea AM, Busselberg D. Cisplatin as an anti‐tumor drug: cellular mechanisms of activity, drug resistance and induced side effects. Cancers (Basel). 2011;3:1351‐1371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Sorrentino A, Liu CG, Addario A, Peschle C, Scambia G, Ferlini C. Role of microRNAs in drug‐resistant ovarian cancer cells. Gynecol Oncol. 2008;111:478‐486. [DOI] [PubMed] [Google Scholar]
- 4. Cancer Genome Atlas Research Network . Integrated genomic analyses of ovarian carcinoma. Nature. 2011;474:609‐615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Wang Y, Suh YA, Fuller MY, et al. Restoring expression of wild‐type p53 suppresses tumor growth but does not cause tumor regression in mice with a p53 missense mutation. J Clin Invest. 2011;121:893‐904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Hong B, van den Heuvel AP, Prabhu VV, Zhang S, El‐Deiry WS. Targeting tumor suppressor p53 for cancer therapy: strategies, challenges and opportunities. Curr Drug Targets. 2014;15:80‐89. [DOI] [PubMed] [Google Scholar]
- 7. Brosh R, Rotter V. When mutants gain new powers: news from the mutant p53 field. Nat Rev Cancer. 2009;9:701‐713. [DOI] [PubMed] [Google Scholar]
- 8. Rusch V, Klimstra D, Venkatraman E, et al. Aberrant p53 expression predicts clinical resistance to cisplatin‐based chemotherapy in locally advanced non‐small cell lung cancer. Cancer Res. 1995;55:5038‐5042. [PubMed] [Google Scholar]
- 9. Lin X, Howell SB. DNA mismatch repair and p53 function are major determinants of the rate of development of cisplatin resistance. Mol Cancer Ther. 2006;5:1239‐1247. [DOI] [PubMed] [Google Scholar]
- 10. Fang L, Lee SW, Aaronson SA. Comparative analysis of p73 and p53 regulation and effector functions. J Cell Biol. 1999;147:823‐830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Guo X, Keyes WM, Papazoglu C, et al. TAp63 induces senescence and suppresses tumorigenesis in vivo. Nat Cell Biol. 2009;11:1451‐1457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Jung MS, Yun J, Chae HD, et al. P53 and its homologues, p63 and p73, induce a replicative senescence through inactivation of NF‐Y transcription factor. Oncogene. 2001;20:5818‐5825. [DOI] [PubMed] [Google Scholar]
- 13. Schmale H, Bamberger C. A novel protein with strong homology to the tumor suppressor p53. Oncogene. 1997;15:1363‐1367. [DOI] [PubMed] [Google Scholar]
- 14. Belyi VA, Ak P, Markert E, et al. The origins and evolution of the p53 family of genes [serial online]. Cold Spring Harb Perspect Biol. 2020;2:a001198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Dotsch V, Bernassola F, Coutandin D, Candi E, Melino G. P63 and p73, the ancestors of p53 [serial online]. Cold Spring Harb Perspect Biol. 2010;2:a004887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Yao JY, Chen JK. TAp63 plays compensatory roles in p53‐deficient cancer cells under genotoxic stress. Biochem Biophys Res Commun. 2010;403:310‐315. [DOI] [PubMed] [Google Scholar]
- 17. Creighton CJ, Benham AL, Zhu H, et al. Discovery of novel microRNAs in female reproductive tract using next generation sequencing [serial online]. PLoS One. 2010;5:e9637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Wang L, Zhu MJ, Ren AM, et al. A ten‐microRNA signature identified from a genome‐wide microRNA expression profiling in human epithelial ovarian cancer [serial online]. PLoS One. 2014;9:e96472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Yang C, Cai J, Wang Q, et al. Epigenetic silencing of miR‐130b in ovarian cancer promotes the development of multidrug resistance by targeting colony‐stimulating factor 1. Gynecol Oncol. 2012;124:325‐334. [DOI] [PubMed] [Google Scholar]
- 20. Chen C, Hu Y, Li L. NRP1 is targeted by miR‐130a and miR‐130b, and is associated with multidrug resistance in epithelial ovarian cancer based on integrated gene network analysis. Mol Med Rep. 2016;13:188‐196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Paudel D, Zhou W, Ouyang Y, et al. MicroRNA‐130b functions as a tumor suppressor by regulating RUNX3 in epithelial ovarian cancer. Gene. 2016;586:48‐55. [DOI] [PubMed] [Google Scholar]
- 22. Tseng JH, Bisogna M, Hoang LN, et al. miR‐200c‐driven mesenchymal‐to‐epithelial transition is a therapeutic target in uterine carcinosarcomas [serial online]. Sci Rep. 2017;7:3614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Landen CN Jr, Chavez‐Reyes A, Bucana C, et al. Therapeutic EphA2 gene targeting in vivo using neutral liposomal small interfering RNA delivery. Cancer Res. 2005;65:6910‐6918. [DOI] [PubMed] [Google Scholar]
- 24. Xu L, Huang CC, Huang W, et al. Systemic tumor‐targeted gene delivery by anti‐transferrin receptor scFv‐immunoliposomes. Mol Cancer Ther. 2002;1:337‐346. [PubMed] [Google Scholar]
- 25. Senzer N, Nemunaitis J, Nemunaitis D, et al. Phase I study of a systemically delivered p53 nanoparticle in advanced solid tumors. Mol Ther. 2013;21:1096‐1103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kim M, Kasinski AL, Slack FJ. MicroRNA therapeutics in preclinical cancer models. Lancet Oncol. 2011;12:319‐321. [DOI] [PubMed] [Google Scholar]
- 27. Pirollo KF, Rait A, Zhou Q, et al. Materializing the potential of small interfering RNA via a tumor‐targeting nanodelivery system. Cancer Res. 2007;67:2938‐2943. [DOI] [PubMed] [Google Scholar]
- 28. Rait AS, Pirollo KF, Ulick D, Cullen K, Chang EH. HER‐2‐targeted antisense oligonucleotide results in sensitization of head and neck cancer cells to chemotherapeutic agents. Ann N Y Acad Sci. 2003;1002:78‐89. [DOI] [PubMed] [Google Scholar]
- 29. Daniels TR, Bernabeu E, Rodriguez JA, et al. The transferrin receptor and the targeted delivery of therapeutic agents against cancer. Biochim Biophys Acta. 2012;1820:291‐317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Ventura A, Kirsch DG, McLaughlin ME, et al. Restoration of p53 function leads to tumour regression in vivo. Nature. 2007;445:661‐665. [DOI] [PubMed] [Google Scholar]
- 31. Gutierrez‐Puente Y, Tari AM, Stephens C, Rosenblum M, Guerra RT. Lopez‐Berestein G. Safety, pharmacokinetics, and tissue distribution of liposomal P‐ethoxy antisense oligonucleotides targeted to Bcl‐2. J Pharmacol Exp Ther. 1999;291:865‐869. [PubMed] [Google Scholar]
- 32. Yamada KM, Cukierman E. Modeling tissue morphogenesis and cancer in 3D. Cell. 2007;130:601‐610. [DOI] [PubMed] [Google Scholar]
- 33. Dong P, Karaayvaz M, Jia N, et al. Mutant p53 gain‐of‐function induces epithelial‐mesenchymal transition through modulation of the miR‐130b‐ZEB1 axis. Oncogene. 2013;32:3286‐3295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Su X, Chakravarti D, Cho MS, et al. TAp63 suppresses metastasis through coordinate regulation of Dicer and miRNAs. Nature. 2010;467:986‐990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Melino G. P63 is a suppressor of tumorigenesis and metastasis interacting with mutant p53. Cell Death Differ. 2011;18:1487‐1499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Chan CK, Pan Y, Nyberg K, et al. Tumour‐suppressor microRNAs regulate ovarian cancer cell physical properties and invasive behaviour [serial online]. Open Biol. 2016;6:pii 160275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Lu C, Lu S, Liang W, et al. TAp63alpha mediates chemotherapeutic agent‐induced apoptosis in human bone marrow mesenchymal stem cells. Stem Cells Dev. 2011;20:1319‐1326. [DOI] [PubMed] [Google Scholar]
- 38. Sen T, Sen N, Brait M, et al. DeltaNp63alpha confers tumor cell resistance to cisplatin through the AKT1 transcriptional regulation. Cancer Res. 2011;71:1167‐1176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Gonfloni S, Di Tella L, Caldarola S, et al. Inhibition of the c‐Abl‐TAp63 pathway protects mouse oocytes from chemotherapy‐induced death. Nature Med. 2009;15:1179‐1185. [DOI] [PubMed] [Google Scholar]
- 40. Suh EK, Yang A, Kettenbach A, et al. P63 protects the female germ line during meiotic arrest. Nature. 2006;444:624‐628. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials