

Abstract
The US Food and Drug Administration's Center for Drug Evaluation and Research (CDER) developed an investigational Public Health Assessment via Structural Evaluation (PHASE) methodology to provide a structure‐based evaluation of a newly identified opioid's risk to public safety. PHASE utilizes molecular structure to predict biological function. First, a similarity metric quantifies the structural similarity of a new drug relative to drugs currently controlled in the Controlled Substances Act (CSA). Next, software predictions provide the primary and secondary biological targets of the new drug. Finally, molecular docking estimates the binding affinity at the identified biological targets. The multicomponent computational approach coupled with expert review provides a rapid, systematic evaluation of a new drug in the absence of in vitro or in vivo data. The information provided by PHASE has the potential to inform law enforcement agencies with vital information regarding newly emerging illicit opioids.
The US Food and Drug Administration (FDA) is one of many federal agencies involved in efforts to improve the safe use of prescription opioid analgesics and combat misuse and abuse of prescription opioids, as well as new designer street‐drugs. On February 4, 2016, FDA leadership presented the FDA's Opioid Action Plan aimed at reversing the growing opioid abuse epidemic, while assuring appropriate access to pain medications. These efforts include expanding access to abuse‐deterrent formulations for opioid drug products, approving better measures for treating opioid use disorder and preventing deaths from overdose, and taking actions to reduce excess opioids available for abuse.1 The FDA's Action Plan is a critical component of the five‐part Opioid Strategy described by the Department of Health and Human Services in April of 2017.2
Emerging Synthetic Opioids Present A Significant Risk to Public Safety
Chemical analogs are molecules that are structurally similar to a parent compound and typically exhibit a similar biological profile to the parent compound. Therefore, derivatizing a promising drug candidate is a central premise in drug design and optimization in the pharmaceutical industry. The same premise is exploited by clandestine laboratories that generate new opioid analogs, including fentanyl analogs that have been introduced to the street‐drug market and have contributed to the sharp increase in opioid overdose deaths.3 Indeed, fentanyl analogs account for 80% of the emerging synthetic opioids,4 and the rate of drug overdose deaths involving synthetic opioids has increased by 88% per year from 2013 to 2016.5, 6 Furthermore, the variability in potency among fentanyl analogs presents a major risk to public health as some derivatives (e.g., carfentanil) are estimated to be 10,000 times more potent than morphine.7, 8
The fentanyl analog influx is exacerbated by simple modification of the fentanyl core (4‐anilidopiperidine, Figure 1 ). Numerous modifications of the fentanyl core do not alter the binding interaction or activation of the mu opioid receptor (MOR), which can lead to the same analgesic and euphoric effects associated with opioids.9 However, these analogs can evade prosecution. The Federal Analogue Act, modifying the Controlled Substances Act (CSA) at 21 U.S.C. §813, was designed to facilitate the prosecution of chemicals that are structurally “substantially similar” to the parent compound controlled in schedules I or II of the CSA (21 U.S.C. §802(32)(A)), but the definition of structurally “substantially similar” is ambiguous and compounds not “intended” for human consumption are not covered under the Act. Therefore, prosecuting simple modifications to a parent compound has created an undue burden on law enforcement as the degree of similarity and intent must be proven each time a drug is identified.
Figure 1.
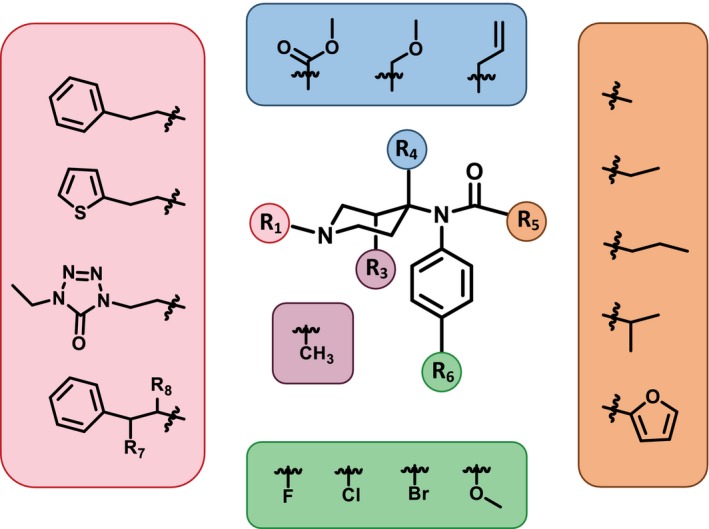
Fentanyl analogs. Commonly modified positions along the 4‐anilidopipeidine core of fentanyl.
The Drug Enforcement Administration (DEA) can also control a drug to protect the public from substances that have high abuse potential. Substances with a high potential for abuse and no accepted medical use are placed into schedule I, whereas substances with an accepted medical use and decreasing degrees of abuse potential are controlled in schedules II−V, respectively. In order to control a newly identified drug, at the request of the DEA, the Department of Health and Human Services provides a comprehensive scientific and medical evaluation using an eight‐factor analysis, which includes evaluation of the substance's pharmacology, mechanism of action, and risks to public health. This mechanism allows for a scientific evaluation of a substance that would either be in support of, or in opposition to, drug control (21 U.S.C. §811).10 Although the eight‐factor analysis thoroughly assesses the chemical properties of newly identified compounds, it can take considerable time to complete. The human resource requirements to perform an eight‐factor analysis, coupled with the vast number of possible fentanyl analogs, is prohibitively resource intensive, which has generated the need for an evaluation protocol of newly identified fentanyl analogs that provides real‐time insight into the risk a new substance may pose to public safety. Rapidly predicting and evaluating the potency for a broad range of newly identified opioids is critical given the diverse, region‐specific synthetic opioid markets.
Therefore, the FDA's Center for Drug Evaluation and Research (CDER) has developed a multipronged computational approach, Public Health Assessment via Structural Evaluation (PHASE; Figure 2 ), for evaluating the similarity of a newly identified drug of abuse to controlled substances and predicting its pharmacology at relevant biological targets. Specifically, PHASE is an investigational approach that uses chemical structure to:
Figure 2.
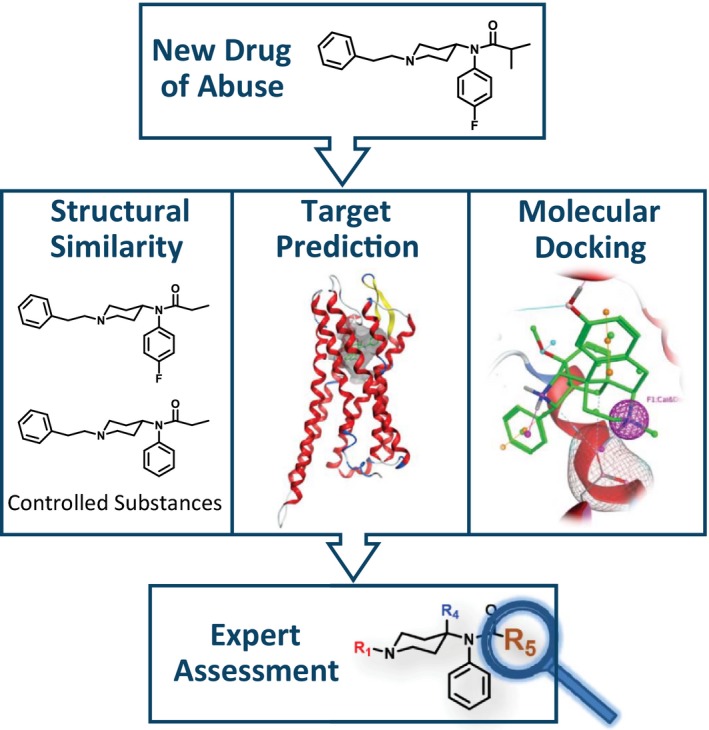
Public Health Assessment via Structural Evaluation (PHASE). When a new drug of abuse is identified, PHASE uses chemical structure to assess the new drug's risk to public safety.
Quantify the new drug's structural similarity with drugs previously controlled in the CSA
Identify probable biological targets
Predict binding affinity at the identified biological targets11
Expert review of the similarity and binding predictions is performed, including consideration of the context of existing literature, to ensure the risk a new substance may pose to public safety is balanced with maintaining appropriate access to pain treatment. PHASE is intended to help inform the public health response related to newly identified, potentially dangerous substances being encountered in the context of illicit drug use.
Characterization of Para‐Fluoroisobutyryl Fentanyl Using Phase
The following section describes and applies each component of the PHASE protocol to para‐fluoroisobutyryl fentanyl (FIBF), a synthetic opioid linked to at least 62 overdose deaths in Maryland alone.12 The DEA issued a temporary scheduling order to place FIBF into schedule I of the CSA on May 3, 2017.13
Structural Similarity Assessment
The structural difference between fentanyl and FIBF is an additional methyl and fluoro group at the R5 and R6 position, respectively (Figure 1 ). The Analogue Act allows a chemical that is “substantially similar” to a controlled substance in schedules I or II of the CSA to be treated, for the purposes of any Federal law, as a controlled substance in schedule I. Considering fentanyl analogs as “substantially similar” to controlled fentanyl substances could involve visual inspection of the drug's core structure and degree of derivatization. PHASE provides a systematic and reproducible approach for quantifying the structural similarity of the new drug with respect to currently scheduled drug substances.
The FDA constructed a controlled substance database containing currently scheduled drugs (I−V), which is linked to a manually curated chemical structure. Linking the controlled substance database to chemical structure instead of naming conventions is particularly powerful when dealing with street‐drugs as many have multiple names that may vary regionally and do not follow standard naming conventions, such as those defined by the International Union of Pure and Applied Chemistry (IUPAC). Therefore, when a new drug is identified, a rapid determination can be made as to whether the drug has ever been evaluated, whether it is being used under a different name, and how similar it is to other controlled substances. Furthermore, the controlled substance database is readily expandable as new, scheduled therapeutics reach the market and newly identified drugs with no medical utility are classified as schedule I by the DEA.
The structural similarity of FIBF to currently scheduled drugs was assessed using a combination of fingerprints (Molecular ACCess System (MACCS) 166 keys) from Molecular Operating Environment14 and the Tanimoto similarity index.15 The Tanimoto coefficient quantifies the similarity between two compounds as the ratio of shared chemical features divided by the union of chemical features for each pair and can be interpreted as a percent similarity. Tanimoto coefficients range from 0−1 (0−100% similar), where compounds with a high degree of structural similarity have a Tanimoto coefficient near 1, whereas structurally dissimilar compounds have a score near 0. To demonstrate the utility of maintaining an updated structure‐linked database, the structural similarity analysis of FIBF is performed using both the 2016 and 2018 controlled substance databases.
The structural similarity assessment identifies four controlled fentanyl analogs as the nearest structural neighbors of FIBF in both the 2016 and 2018 controlled substance databases (Figure 3 a,b). Para‐Fluorofentanyl (T c = 0.92, 92% similar to FIBF), the most structurally similar analog in the 2016 database, is an opioid analgesic that differs by a single methyl group on the R5 position and was placed under international control in 1990. Acetylfentanyl (T c = 0.85) and acetyl‐α‐methylfentanyl (T c = 0.84), both schedule I narcotics, are the next most similar scheduled drugs. Finally, fentanyl was the fourth most structurally similar scheduled substance. Perhaps not surprisingly, given the urgency of the opioid crisis, FIBF and other fentanyl analogs have been scheduled since 2016. The four most structurally similar controlled substances with respect to FIBF in the 2018 database still include p‐fluorofentanyl; however, the three remaining nearest neighbors differ. In addition to scheduling FIBF, p‐chloroisobutyryl fentanyl (T c = 0.96), p‐fluorobutyrylfentanyl (T c = 0.92), and o‐fluorofentanyl (T c = 0.88) have all been scheduled. This analysis highlights the need to compare the structural similarity of a newly identified substance with controlled substances instead of the parent compound, as many analogs may have more structurally related compounds.
Figure 3.
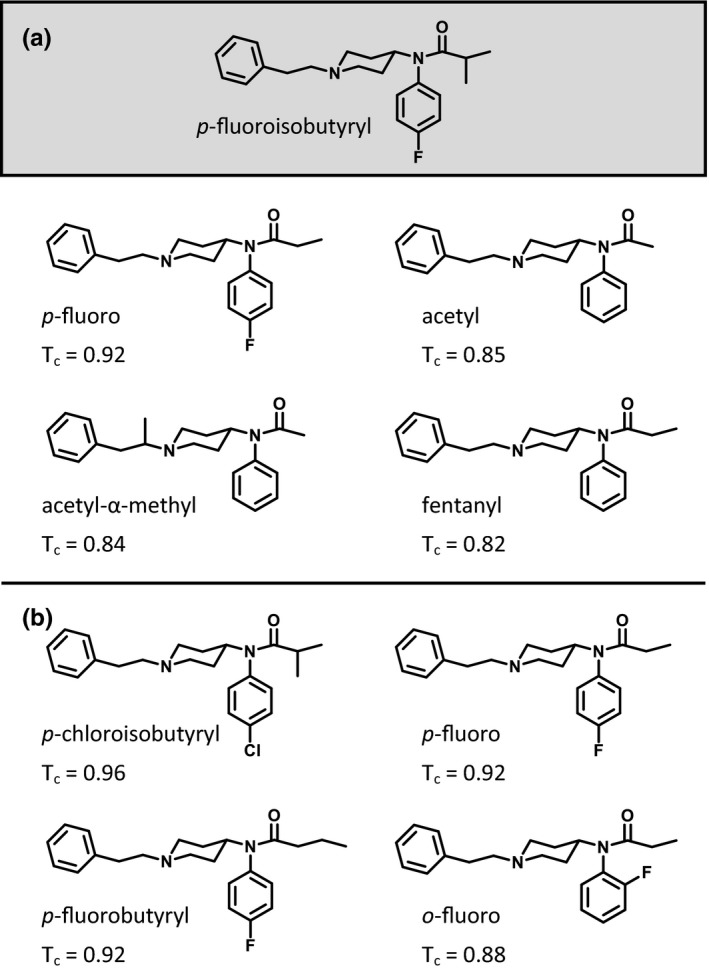
Structural similarity assessment. (a) The most structurally similar controlled substances with respect to para‐fluoroisobutyryl fentanyl (FIBF) in the 2016 database are para‐fluorofentanyl, acetylfentanyl, acetyl‐α‐methylfentanyl, and fentanyl. (b) The most structurally similar controlled substances with respect to FIBF in the 2018 database are p‐chloroisbutyrylfentanyl, p‐fluorofentanyl, p‐fluorobutyrylfentanyl, and o‐fluorofentanyl. Tanimoto coefficient (Tc) ranges from 0−1 (0–100% similar), where one indicates the highest degree of structural similarity.
The structural similarity assessment using Molecular ACCess System keys and the Tanimoto coefficient provides a reproducible, quantitative measure of structural similarity between a newly identified drug and existing DEA controlled substances (schedules I–V). Two compounds that have a large number of overlapping features (T c near 1) and identical chemical scaffolds are more likely to have similar pharmacological profiles than compounds with disparate chemical scaffolds and nonoverlapping chemical features. However, molecular fingerprinting techniques, such as the one described, do not guarantee that two compounds with a high structural similarity score will have the same clinical effect. For example, the (S)‐stereoisomer of thalidomide is teratogenic, but the (R)‐stereoisomer is not.16
Biological Target Prediction
Next, the in silico biological profiles of FIBF were predicted using Chemotargets Clarity17 and SEAware18, 19 (Table 1 ). Chemotargets Clarity is a statistical software tool that uses six independent approaches to identify potential biological targets and mode(s) of action of small molecules by screening over 2,000 mechanisms of action associated with therapeutic activity and safety liabilities. These approaches utilize a variety of structural and target information to predict biological targets, mechanism of action, and binding affinity and include pharmacophore clusters, quantitative structure‐activity relationships, and machine learning techniques, such as random forest, support vector machine, and artificial neural networks. The pharmacology models are trained with a curated dataset of over 1 million compounds derived from the analysis of patents, journal articles, public databases, and data from research collaborations. A ranked list of potential mechanisms of action for the query structure is provided along with structurally similar training set analogs.
Table 1.
Drug target and activity prediction by Chemotargets Clarity and SEAWare
| Drug targets | p‐Fluoroisobutyryl Fentanyl | |
|---|---|---|
| Clarity prediction (activity) | SEAWare prediction | |
| Opioid mu receptor | Binder (agonist) | Binder |
| Opioid kappa receptor | Binder (agonist) | Binder |
| Opioid delta receptor | Binder (agonist) | Binder |
| Serotonin 5‐HT2C receptor | Binder (agonist) | Binder |
| Dopamine D1A receptor | Binder (antagonist) | Binder |
| Dopamine D2 receptor | Binder (antagonist) | Binder |
| Serotonin 5‐HT2A receptor | Binder (antagonist) | Binder |
| Serotonin transporter | Binder (inhibitor) | Binder |
| hERG potassium channel | Binder (inhibitor) | Binder |
| Cannabinoid receptor 1 | Binder (ligand) | Non binder |
| Monoamine oxidase | Non binder | Binder |
hERG, human ether‐a‐go‐go.
Binding activities at the respective drug targets as predicted by Chemotargets Clarity (left column), and binding prediction by SEAWare (right column). Targets not predicted to bind to a particular target by either software platform are reported as non binders.
SEAWare is a software package that analyzes the query structure and predicts receptor binding for ~ 2,300 biological targets based upon structural similarity to target specific training sets. SEAWare uses data from ChEMBL version 2220to group drugs into training sets based on their known targets. SEAWare then applies the Similarity Ensemble Approach,18, 19 which uses Tanimoto coefficients to compare structural similarity between and across the training sets. Predictions are generated by comparing the structural similarity of the query molecule to each target's known ligands, and a P value is calculated to determine the likelihood of a predicted target's ligands occurring by chance. The SEAware P value was used as a binary classifier where a value of < 1e−5 was considered a positive prediction for binding. A ranked list of potential targets for the query structure is provided, along with a similarity score to the most structurally similar training set analog (Tanimoto coefficient) and significance of the prediction (SEAWare P value). Target predictions can be used to support findings from behavioral pharmacology studies as well as field observations by relating structural information to animal and human pharmacology.
The FIBF binding profile (set of predicted biological targets) indicates potential adverse cardiovascular and neuropsychiatric responses, along with more serious and possibly fatal consequences. Notably, FIBF is most strongly predicted to be an MOR agonist, which is associated with analgesia and euphoria and many other serious adverse reactions, including respiratory depression, constipation, and seizures.21 Moreover, activation of the MOR may lead to physical dependence and addiction. The kappa and delta opioid receptors, although generally thought to have fewer serious side effects compared to the MOR, may still be responsible for depression, dysphoria, seizures, and catalepsy.22, 23
In addition to opioid receptors, FIBF is predicted to bind to serotonin receptors and transporter, dopamine receptors, and the human ether‐a‐go‐go potassium channel. Binding to these transporters and receptors can cause a variety of cardiovascular and neuropsychiatric responses. Serotonin receptor and transporter binding is linked to antidepressant and anxiolytic effects; however, negative effects, such as serotonin syndrome and impaired cognition, have also been reported.24, 25 Additionally, many dopamine receptor antagonists are useful in treating schizophrenia, bipolar disorder, and nausea, but they may also be associated with extrapyramidal symptoms.26, 27, 28 Finally, inhibition of the human ether‐a‐go‐go potassium channel is associated with QT prolongation, which may lead to a rare but potentially fatal form of ventricular arrhythmia, Torsade de Pointes.29 Importantly, the in silico binding profiles of newly identified substances can be used to direct which biological targets the compounds should initially be tested against in vitro. The binding profiles may contain false‐positive predictions; however, they are still informative for guiding the further evaluation of potential risks of newly identified drugs of abuse.
Predicting Binding Affinity Using Molecular Docking
Finally, molecular docking is used to predict the binding affinity of FIBF at the MOR. Molecular docking is a type of virtual screen that evaluates the intermolecular interactions between a compound and the active site of a biological target. Initially, thousands of potential binding poses between the compound and biological target are generated by a guided placement procedure to create compound–target complexes. Then, the interactions between each compound–target complex are evaluated with a scoring function to provide a binding energy (score).30, 31, 32 Then, the complex with the most favorable compound–target interactions (lowest score) can be used to predict experimental observables, such as the binding affinity. In brief, the docking model for predicting opioid binding affinity at the MOR was generated by docking and scoring a series of opioids with experimentally determined binding affinities to the MOR crystal structure (PDBID: 51CM33). The strong correlation between the opioid binding scores and the experimentally determined binding affinities allows for the prediction of a newly identified opioid's binding affinity at the MOR. Complete details of the molecular docking model, the associated regression model for the binding affinity prediction, and validation experiments with negative controls have been previously published and are publicly available.11
Building predictive binding models using molecular docking is particularly challenging given the wide range of published in vitro drug‐target binding affinities reported within the literature. A 2011 literature survey of opioid binding affinities with the MOR demonstrated that common opioids have dramatically different values depending on a number of experimental conditions, including the choice of radioligand, tissue source, and species (Figure 4 a).34 Strikingly, fentanyl binding affinity measurements at the MOR span almost five orders of magnitude (0.007–214 nM).35, 36 In light of the diversity of assays for data collection, confounding the comparability of reported data in the literature, the FDA standardized a binding assay and quantified the binding affinity of an array of opioids with the human MOR (blue diamonds, Figure 4 a).34
Figure 4.
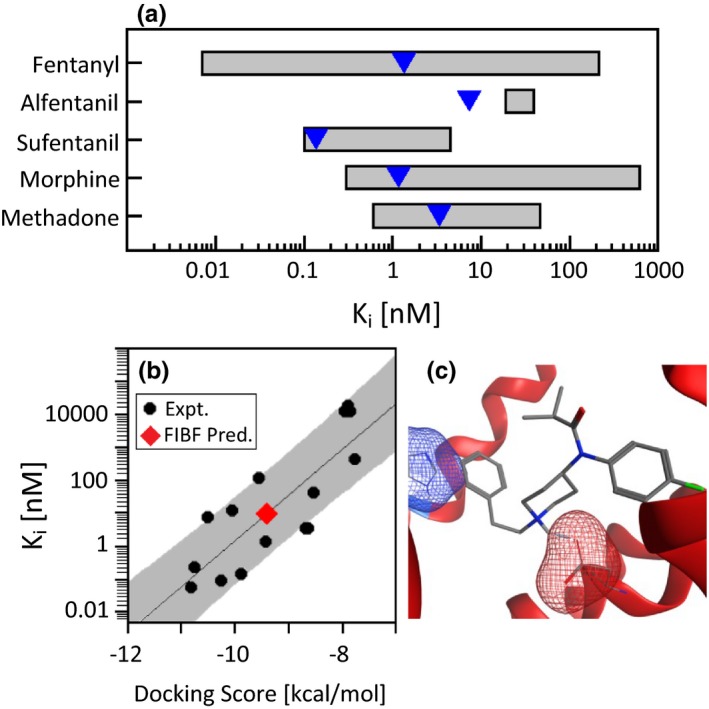
Binding affinity (Ki) using molecular docking. (a) Reported Ki ranges for common opioids with the mu opioid receptor (MOR). The blue triangles represent the measurement performed by the standardized US Food and Drug Administration procedure. (b) Correlation of the docking score with the experimentally determined binding affinity (black circles, r 2 = 0.68) and the predicted binding affinity of para‐fluoroisobutyryl fentanyl (FIBF; red diamond). The solid line is a regression line and the Y‐axis is logarithmic. The gray boundaries present the regression model's predictivity. (c) FIBF docked into the MOR.
The standardized set of binding data allowed for the construction of a computational molecular docking model that accurately classifies opioid binding affinity.11 The docking model distinguished between key structural features of fentanyl derivatives that either increase or decrease the binding affinity. Specifically, compounds with chemical modifications at the four‐position on the piperidine ring increase binding affinity, removal of the N‐phenethyl group significantly diminishes binding affinity, and exchange of a different aromatic group at the N‐phenethyl group does not significantly alter the binding score. Importantly, this allows binding affinity prediction of a new opioid at the MOR receptor, which is critical for assessing the risk of a new fentanyl analog.
The FIBF average docking score is −9.4 kcal/mol (red diamond, Figure 4 b), which equates to a predicted binding affinity between 0.5 and 170 nM. The FIBF average docking score and predicted binding affinity is equivalent to fentanyl. It is not surprising that FIBF is predicted to have a similar binding affinity to fentanyl considering the key binding interactions are maintained, including the salt bridge between the positively charged fentanyl amine and the negatively charged aspartic acid (red mesh) of the receptor, as well as aromatic stacking with the phenethyl (R1) and the histidine within the receptor (blue mesh, Figure 4 c).
In summary, the multicomponent evaluation of FIBF indicates that FIBF presents a risk to public safety. The structural similarity assessment of FIBF identified many relevant fentanyl analogs. The target identification software predicts FIBF is an MOR agonist, and the molecular docking predicts FIBF has a binding affinity with the MOR similar to fentanyl. Finally, literature review of fentanyl structure‐activity relationships does not suggest that any chemical features of FIBF mitigate the risk of MOR agonism.37
Rapid Characterization of Emerging Opioids Can Inform Regulatory Response
The opioid crisis has been severely affected by the widespread use of fentanyl and its analogs being “cut” into heroin, cocaine, and other illicit drugs of abuse. Although fentanyl is significantly already more potent than morphine, simple modification to the fentanyl core can dramatically increase potency to 10,000 times more than morphine.7, 8 The large range in the potency of fentanyl analogs creates unique, region‐specific challenges. The three computational methodologies incorporated into PHASE provide complementary information that can assist with drug scheduling actions of newly identified drugs of abuse that pose a risk to public safety.
First, PHASE quantifies the chemical similarity of a newly identified drug substance to all previously controlled substances contained within a structure‐linked database. Comparing the similarity of a new drug with controlled substances is preferential to only considering the parent drug (e.g., fentanyl), because it provides strong comparators for assessing the new drug's potential risk to public health. Importantly, the controlled substance database is readily expandable as new, scheduled therapeutics reach the market, and newly identified drugs with no approved medical utility are classified as schedule I by the DEA.
Next, PHASE uses multiple target identification programs to develop an in silico binding profile, which contains the predicted biological targets of the new drug. Incorporating multiple prediction platforms increases the confidence in predictions that a drug will bind to a particular target. Both Chemotargets Clarity and SEAware rank the MOR as the primary target of FIBF. Additionally, Chemotargets Clarity predicts FIBF to be an MOR agonist. Functionality prediction is important for determining the abuse potential of fentanyl analogs as MOR agonists are associated with the euphoric effects sought by drug users, whereas MOR antagonists provide potential for reversing opioid overdose. Of note, however, is that fentanyl analogs almost exclusively function as MOR agonists.
Finally, molecular docking models predict the binding affinity of the new drug at the predicted biological targets. Understanding the strength of the binding interaction between a newly identified drug of abuse and its binding target provides critical information for evaluating its risk to public safety. Furthermore, molecular docking models provide an atomically detailed picture of interactions between the drug and the biological target. A primary challenge when building molecular docking models for binding affinity prediction is locating quality experimental datasets generated from the same laboratory. These data are necessary for validating molecular docking models for different drug classes at specific biological targets. Fortunately, external research facilities have measured and published the binding affinities of a range of stimulants (e.g., methamphetamines, 3,4‐methylenedioxymethamphetamine, cocaine, etc.) at the dopamine, serotonin, and norepinephrine transporter receptors.38, 39
PHASE harnesses structure–activity relationships, biological target prediction software, and molecular docking to provide a rapid evaluation of a newly identified drug that lacks in vitro and/or in vivo data. The application of the investigational PHASE approach to the temporarily scheduled FIBF readily identifies the risk FIBF poses to public safety. Specifically, FIBF is most structurally similar to scheduled fentanyl analogs and not the parent drug, fentanyl. The target prediction platforms predict that FIBF binds to the MOR, and finally, molecular docking estimates that FIBF has similar binding affinity to the MOR as fentanyl. Although the PHASE protocol was developed in the context of opioids and fentanyl derivatives, it is generalizable to all drug classes and biological targets. Finally, the current implementation of PHASE does not encompass all drug characteristics, such as lipid solubility, blood–brain barrier transmission, and drug clearance. However, as these predictive models are developed they can be added into the computational workflow.
The opioid crisis presents a critical challenge for protecting public health, and the Department of Health and Human Services is developing several approaches for addressing the issue. One challenge presented by the opioid crisis is that many new drugs of abuse lack proper regulatory control and small modifications to a controlled substance may evade prosecution. PHASE can proactively inform law enforcement and the public with vital information regarding the new substance by providing a mechanism for characterizing the structural similarity of newly identified drugs with currently scheduled drugs and predicting the impact of structural modifications on likely biological targets.
Funding Information
The work was internally funded by the US Food and Drug Administration (FDA).
Conflict of Interest
Rebecca Racz is the Principal Investigator on a Research Collaboration Agreements (RCA) between the FDA’s Center for Drug Evaluation and Research, and Chemotargets.
Disclaimer
This article reflects the views of the authors and should not be construed to represent the FDA's views or policies. The mention of commercial products, their sources, or their use in connection with material reported herein is not to be construed as either an actual or implied endorsement of such products by the Department of Health and Human Services.
Acknowledgment
The authors would like to acknowledge Dr Keith Burkhart for helpful discussions.
References
- 1. Throckmorton, D.C. , Gottlieb, S. & Woodcock, J . The FDA and the next wave of drug abuse – proactive pharmacovigilance. N. Engl. J. Med. 379, 205–207 (2018). [DOI] [PubMed] [Google Scholar]
- 2. Price, T.E. Secretary price announces HHS strategy for fighting opioid crisis. U.S. Department of Health & Human Services. <https://www.hhs.gov/about/leadership/secretary/speeches/2017-speeches/secretary-price-announces-hhs-strategy-for-fighting-opioid-crisis/index.html> (2017).
- 3. Frank, R.G. & Pollack, H.A. Addressing the fentanyl threat to public health. N. Engl. J. Med. 376, 605–607 (2017). [DOI] [PubMed] [Google Scholar]
- 4. Drug Enforcement Administration . Annual enforcement threat report. <https://ndews.umd.edu/sites/ndews.umd.edu/files/dea-emerging-threat-report-2017-annual.pdf> (2017).
- 5. Rudd, R.A. , Seth, P. , David, F. & Scholl, L. Increases in drug and opioid‐involved overdose deaths – United States, 2010–2015. MMWR Morb. Mortal. Wkly. Rep. 65, 1445–1452 (2016). [DOI] [PubMed] [Google Scholar]
- 6. Hedegaard, H. , Warner, M. & Minino, A.M. Drug overdose deaths in the United States, 1999–2016. NCHS Data Brief, 1‐8 (2017). [PubMed]
- 7. United Nations Office on Drugs and Crime . Fentanyl and its analogues – 50 years on. Global Smart Update (2017).
- 8. Janssen, P.A. Potent, new analgesics, tailor‐made for different purposes. Acta Anaesthesiol. Scand. 26, 262–268 (1982). [DOI] [PubMed] [Google Scholar]
- 9. Vuckovic, S. et al Fentanyl analogs: structure‐activity‐relationship study. Curr. Med. Chem. 16, 2468–2474 (2009). [DOI] [PubMed] [Google Scholar]
- 10. Calderon, S.N. & Klein, M. A regulatory perspective on the abuse potential evaluation of novel stimulant drugs in the United States. Neuropharmacology 87, 97–103 (2014). [DOI] [PubMed] [Google Scholar]
- 11. Ellis, C.R. , Kruhlak, N.L. , Kim, M.T. , Hawkins, E.G. & Stavitskaya, L. Predicting opioid receptor binding affinity of pharmacologically unclassified designer substances using molecular docking. PLoS One 13, e0197734 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. United Nations Office on Drugs and Crime . United States: Drug Enforcement Administration to issue a temporary order on para‐fluoroisobutyrfentanyl after fatal intoxications. <https://www.unodc.org/LSS/Announcement/Details/034f817d-9ee6-4854-8910-1e9c8d87bf4a> (2017).
- 13. Drug Enforcement Administration . Schedules of controlled substances: temporary placement of 4‐fluoroisobutyryl fentanyl into schedule I. Fed. Reg. 82, 20544 (2017). [PubMed] [Google Scholar]
- 14. Molecular Operating Environment (MOE) (Chemical Computing Group Inc, Montreal, QC, Canada, 2017).
- 15. Willett, P. Chemical similarity search. J. Chem. Inf. Comput. Sci. 38, 983–996 (1998). [Google Scholar]
- 16. Smith, S.W. Chiral toxicology: it's the same thing…only different. Toxicol. Sci. 110, 4–30 (2009). [DOI] [PubMed] [Google Scholar]
- 17. Garcia‐Serna, R. , Vidal, D. , Remez, N. & Mestres, J. Large‐scale predictive drug safety: from structural alerts to biological mechanisms. Chem. Res. Toxicol. 28, 1875–1887 (2015). [DOI] [PubMed] [Google Scholar]
- 18. Keiser, M.J. , Roth, B.L. , Armbruster, B.N. , Ernsberger, P. , Irwin, J.J. & Shoichet, B.K. Relating protein pharmacology by ligand chemistry. Nat. Biotechnol. 25, 197–206 (2007). [DOI] [PubMed] [Google Scholar]
- 19. Keiser, M.J. et al Predicting new molecular targets for known drugs. Nature 462, 175–181 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Gaulton, A. et al The ChEMBL database in 2017. Nucleic Acids Res. 45, D945–D954 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kieffer, B.L. Opioids: first lessons from knockout mice. Trends Pharmacol. Sci. 20, 19–26 (1999). [DOI] [PubMed] [Google Scholar]
- 22. Lalanne, L. , Ayranci, G. , Kieffer, B.L. & Lutz, P.E. The kappa opioid receptor: from addiction to depression, and back. Front. Psychiatry 5, 170 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Vicente‐Sanchez, A. , Segura, L. & Pradhan, A.A. The delta opioid receptor tool box. Neuroscience 338, 145–159 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Volpi‐Abadie, J. , Kaye, A.M. & Kaye, A.D. Serotonin syndrome. Ochsner J. 13, 533–540 (2013). [PMC free article] [PubMed] [Google Scholar]
- 25. Carhart‐Harris, R.L. & Nutt, D.J. Serotonin and brain function: a tale of two receptors. J. Psychopharmacol. 31, 1091–1120 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Berman, B.D. Neuroleptic malignant syndrome: a review for neurohospitalists. Neurohospitalist 1, 41–47 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Ashok, A.H. et al The dopamine hypothesis of bipolar affective disorder: the state of the art and implications for treatment. Mol. Psychiatry 22, 666–679 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Li, P. , Snyder, G.L. & Vanover, K.E. Dopamine targeting drugs for the treatment of schizophrenia: past, present and future. Curr. Top. Med. Chem. 16, 3385–3403 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Sanguinetti, M.C. & Tristani‐Firouzi, M. hERG potassium channels and cardiac arrhythmia. Nature 440, 463–469 (2006). [DOI] [PubMed] [Google Scholar]
- 30. Jain, A.N. Scoring functions for protein‐ligand docking. Curr. Protein Pept. Sci. 7, 407–420 (2006). [DOI] [PubMed] [Google Scholar]
- 31. Meng, X.Y. , Zhang, H.X. , Mezei, M. & Cui, M. Molecular docking: a powerful approach for structure‐based drug discovery. Curr. Comput. Aided Drug Des. 7, 146–157 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Morris, G.M. & Lim‐Wilby, M. Molecular docking In Molecular Modeling of Proteins (ed. Kukol A.) 365–382 (Humana Press, Totowa, NJ, 2008). [DOI] [PubMed] [Google Scholar]
- 33. Huang, W. et al Structural insights into micro‐opioid receptor activation. Nature 524, 315–321 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Volpe, D.A. et al Uniform assessment and ranking of opioid mu receptor binding constants for selected opioid drugs. Regul. Toxicol. Pharmacol. 59, 385–390 (2011). [DOI] [PubMed] [Google Scholar]
- 35. Chen, J.C. , Smith, E.R. , Cahill, M. , Cohen, R. & Fishman, J.B. The opioid receptor binding of dezocine, morphine, fentanyl, butorphanol and nalbuphine. Life Sci. 52, 389–396 (1993). [DOI] [PubMed] [Google Scholar]
- 36. Traynor, J.R. & Nahorski, S.R. Modulation by mu‐opioid agonists of guanosine‐5’‐O‐(3‐[35S]thio)triphosphate binding to membranes from human neuroblastoma SH‐SY5Y cells. Mol. Pharmacol. 47, 848–854 (1995). [PubMed] [Google Scholar]
- 37. Vardanyan, R.S. & Hruby, V.J. Fentanyl‐related compounds and derivatives: current status and future prospects for pharmaceutical applications. Future Med. Chem. 6, 385–412 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Eshleman, A.J. , Wolfrum, K.M. , Hatfield, M.G. , Johnson, R.A. , Murphy, K.V. & Janowsky, A. Substituted methcathinones differ in transporter and receptor interactions. Biochem. Pharmacol. 85, 1803–1815 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Eshleman, A.J. et al Structure‐activity relationships of substituted cathinones, with transporter binding, uptake, and release. J. Pharmacol. Exp. Ther. 360, 33–47 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]