

Abstract
The intramolecular [2+2] photocycloaddition of 3‐alkenyl‐2‐cycloalkenones was performed in an enantioselective fashion (nine representative examples, 54–86 % yield, 76–96 % ee) upon irradiation at λ=366 nm in the presence of an AlBr3‐activated oxazaborolidine as the Lewis acid. An extensive screening of proline‐derived oxazaborolidines showed that the enantioface differentiation depends strongly on the nature of the aryl group at the 3‐position of the heterocycle. DFT calculations of the Lewis acid–substrate complex indicate that attractive dispersion forces may be responsible for a change of the binding mode. The catalytic [2+2] photocycloaddition was shown to proceed on the triplet hypersurface with a quantum yield of 0.05. The positive effect of Lewis acids on the outcome of a given intramolecular [2+2] photocycloaddition was illustrated by optimizing the key step in a concise total synthesis of the sesquiterpene (±)‐italicene.
Keywords: cycloaddition, enantioselectivity, Lewis acids, photochemistry, total synthesis
Introduction
The intramolecular [2+2] photocycloaddition1 of appropriately substituted 2‐cycloalkenones is an enormously powerful transformation which has been extensively used in the total synthesis of natural products.2 The reaction can be performed by direct irradiation, typically at a wavelength (λ) of 300–370 nm. Since intersystem crossing in enones is fast (k ISC≅1011 s−1),3 the reaction proceeds via the first excited triplet state (T1) which has π–π* character. Compared to intermolecular reactions, there is an improved regioselectivity as the internal olefin is enforced to approach the photoexcited enone via an initial cyclization to a 1,4‐diradical.4 Five‐membered ring formation is preferred where possible and dictates the regioselectivity of the reaction.5
Historically, the reaction belongs to one of the first photochemical transformations known to organic chemists. In 1908, Ciamician and Silber reported on the formation of carvonecamphor (2) upon exposure of carvone (1) to sunlight (Scheme 1).6 The same observation was made by Sernagiotto a few years later.7 In 1957, Büchi and Goldman isolated product 2—still prepared by sunlight irradiation—and proved its constitution and configuration.8 Meinwald and Schneider optimized the reaction employing an artificial light source with an emission maximum at λ=355 nm and could isolate the desired product with a maximum yield of 35 %.9
Scheme 1.
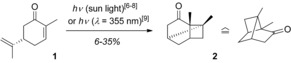
The conversion of carvone (1) into carvonecamphor (2) representing the first intramolecular [2+2] photocycloaddition reaction of a cyclic enone.6, 7, 8, 9
Despite the fact, that the reaction is so powerful, enantioselective variants of the enone [2+2] photocycloaddition have relied, until very recently, on the covalent attachment of a chiral auxiliary.10 In 2013, our group presented the first enantioselective11 enone [2+2] photocycloaddition reaction mediated by chiral Lewis acids.12, 13 The substrates were 5,6‐dihydro‐4‐pyridones14 to which an alkenyl chain was attached at the nitrogen atom. The chiral Lewis acid acts by coordination to the carbonyl carbon atom and lowers the energy difference between the ground state (S0) and the first excited state (S1). The chromophore is activated15 and the allowed π–π* absorption is red‐shifted to absorb at λ ≥360 nm. Although there is a weak n–π* absorption of uncomplexed dihydropyridone at a similar wavelength, the Lewis acid complex has a much higher absorption coefficient and the reaction thus proceeds enantioselectively. Despite the fact that the reaction could be extended to the intramolecular [2+2] photocycloaddition of 3‐alkenyloxy‐2‐cycloalkenones,16 the intermolecular reaction of simple 2‐cycloalkenones, such as 2‐cyclohexenone, remained elusive until very recently.17 In the context of the latter topic, we had performed optimization reactions with 3‐alkenyl‐2‐cycloalkenones of general structure A (Figure 1) for which there has not yet been a report on an enantioselective variant.
Figure 1.
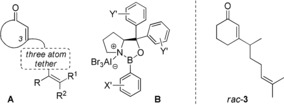
General structure of [2+2] photocycloaddition substrates A and of putative chiral catalysts B; structure of chiral [2+2] photocycloaddition substrate rac‐3.
The reaction was studied with a broad variety of chiral oxazaborolidine Lewis acids18 B (variation of X′ and Y′) and the results of this study are disclosed in this Full Paper. In addition, we could show with chiral substrate rac‐3 that Lewis acid coordination lends a significantly improved selectivity to the reaction. This reaction was used in the total synthesis of italicene and isoitalicene. Mechanistic studies confirm the fact that the reaction proceeds on the triplet surface and computational studies offer an explanation for an unexpected reversal in the enantioselectivity of the process.
Results and Discussion
Synthesis of starting materials and racemic [2+2] photocycloaddition
Our experiments focussed on 2‐cyclopentenones and 2‐cyclohexenones, which carry an alkenyl chain at position C‐3. To the best of our knowledge, the first intramolecular [2+2] photocycloaddition of a 3‐alkenyl‐substituted 2‐cycloalkenone was mentioned in a communication by Corey and Sestanj.19 Since then, this compound class has been extensively used and there are a plethora of examples for their intramolecular [2+2] photocycloaddition.1
The precursors are typically prepared from 3‐ethoxy‐2‐cycloalkenones by addition of the respective alkenyl metal reagent and subsequent hydrolysis.20 The previously unknown compounds 5 c and 5 h were synthesized from the chloromethyl‐substituted olefinic acetal 4 which was obtained by a known procedure21 and which underwent nucleophilic substitution by an allylic alcohol (Scheme 2).
Scheme 2.
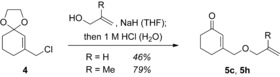
Preparation of photocycloaddition substrates 5 c and 5 h from allylic chloride 4.
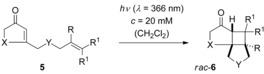
Table 1 lists the substrates 5 and racemic products rac‐6 which were investigated in the present study. Irradiation was performed with fluorescent lamps which exhibit an emission maximum at λ=366 nm.22 Typically, full conversion of 2‐cyclohexenones (5 a–5 d, 5 f–5 i) was achieved after a maximum irradiation time of 8 h. The blue‐shifted absorption of cyclopentenones 5 e and 5 j required for substrate 5 e a longer irradiation time of 47 h while in the case of enone 5 j a short‐wavelength emitter (λ=350 nm) was used to complete the reaction in a reasonable period of time.
Table 1.
Racemic [2+2] photocycloaddition reactions of enones 5: Substitution patterns, reaction times and yields.
| ||||||
|---|---|---|---|---|---|---|
| Substrate[a] | X | Y | R | R1 | t [h] | Yield [%] |
| 5 a | CH2CH2 | CH2 | H | H | 8 | 91 |
| 5 b | CH2CH2 | CMe2 | H | H | 3 | 87 |
| 5 c | CH2CH2 | O | H | H | 5 | 87 |
| 5 d | CMe2CH2 | CH2 | H | H | 5 | 79 |
| 5 e | CH2 | CH2 | H | H | 47 | 56 |
| 5 f | CH2CH2 | CH2 | Me | H | 5 | 68 |
| 5 g | CH2CH2 | CH2 | H | Me | 5.5 | 51[b] |
| 5 h | CH2CH2 | O | Me | H | 8 | 80 |
| 5 i | CMe2CH2 | CH2 | Me | H | 5 | 88 |
| 5 j | CH2 | CH2 | Me | H | 8 | 63[c] |
[a] Unless noted otherwise, the reactions were performed under anhydrous and oxygen‐free conditions at an irradiation wavelength of λ=366 nm (emission maximum of the light source) and at a substrate concentration of 20 mm in CH2Cl2 as the solvent at ambient temperature. [b] Olefinic by‐products were removed by ozonolysis. [c] An irradiation wavelength of λ=350 nm (emission maximum of the light source) was used.
Oxazaborolidine Lewis acids of general formula B (Figure 1) were prepared from the respective amino alcohols23 by oxazaborolidine formation and subsequent complexation with AlBr3 as the activating Lewis acid. Other activators did not match the enantioselectivity achieved with this Lewis acid neither did other amino alcohol skeletons. In the optimization experiments, we thus focussed on amino alcohols derived from proline. Accordingly, the synthesis commenced with known l‐proline methyl ester 7 23 which was converted to amino alcohols 8 by treatment with an excess (2.5 equiv) of the respective Grignard reagent (Scheme 3). The latter in turn was formed from aryl (Ar) bromide by direct magnesiation in the presence of catalytic amounts of iodine. Yields were high (>80 %) both for the formation of the alcohols and for the subsequent hydrogenolysis of the N‐benzyl group to the target compounds (see the Supporting Information for more details).
Scheme 3.

Synthesis of various diaryl‐substituted prolinols 9 from N‐benzyl proline methyl ester (7).
There was some concern regarding a possible racemization at the stereogenic center of proline during the Grignard addition which is the reason we attempted to determine the enantiomeric excess (ee) of the amino alcohols after deprotection. However, a satisfactory separation of the enantiomers by chiral HPLC could not be achieved and an unambiguous ee determination was impossible. Two representative amino alcohols were hence converted into the respective oxazolidinones 10 a and 10 b which were amenable to HPLC separation (Figure 2). Both compounds turned out to be essentially enantiopure (98.8 % ee for 10 a, 99.9 % ee for 10 b). Oxazaborolidine 11 was prepared from aminoalcohol 9 i by condensation with 2,4,6‐trifluorophenyl boronic acid and it was characterized by NMR analysis (1H, 11B, 13C, 19F). The NMR data matched reported data of other previously synthesized oxazaborolidines.24 In the 11B NMR there was a single signal for the oxazaborolidine boron atom at 30.2 ppm24a (for further details, see the Supporting Information). In the catalytic experiments, the oxazaborolidines were freshly prepared but not individually characterized. After thorough removal of water they were directly activated by addition of AlBr3.
Figure 2.
Structures of oxazolidinones 10 and of oxazaborolidine 11.
Enantioselective [2+2] photocycloaddition
As mentioned in the introduction, Lewis acids display a profound influence on the absorption properties of enones.15 Figure 3 shows the absorption spectrum of enone 5 a in dichloromethane solution (c=0.5 mm). The strong π–π* absorption of the compound appears at short wavelength with an absorption maximum at λ max=234 nm (ϵ=17 010 m −1 cm−1). The n–π* absorption that is responsible for the observed photocycloaddition reaction (Table 1) could not be identified due to its low absorbance but it was clearly detectable at higher concentration (c=50 mm, see Supporting Information). Its absorption maximum was found at λ max=324 nm (ϵ=50 m −1 cm−1). Addition of Lewis acids led to a bathochromic shift of the allowed π–π* transition (Figure 3). Assuming full complexation to occur with 20 equivalents of Lewis acid, the respective UV/Vis absorption data are for 5 a⋅EtAlCl2 λ max=281 nm (ϵ=13 750 m −1 cm−1) and for 5 a⋅BCl3 λ max=288 nm (ϵ=17 830 m −1 cm−1). In both cases, there is a significant absorption at longer wavelength that exceeds in terms of quantitative absorbance the n–π* absorption of the uncomplexed substrate 5 a. In the presence of a Lewis acid there is no n–π* transition.15
Figure 3.

UV/Vis spectra of cyclic alkenone 5 a in the absence (black line) and in the presence of 20 equivalents of either EtAlCl2 (yellow) or BCl3 (red) (c=0.5 mm in CH2Cl2).
The above‐mentioned scenario is typical for enones15 and is the prerequisite for the use of chiral Lewis acids in a subsequent intramolecular [2+2] photocycloaddition reaction. The higher absorption coefficient of the Lewis acid‐substrate complex allows this complex to harvest the respective long wavelength photons and to suppress the racemic background reaction that occurs upon excitation of the uncomplexed substrates via their n–π* transition. Indeed, it was found for the reaction of compound 5 a that several AlBr3‐activated oxazaborolidines derived from 2,3,4‐trifluoroboronic acid and amino alcohols 9 promoted an enantioselective intramolecular [2+2] photocycloaddition to product 6 a (Scheme 4).
Scheme 4.

Evaluation of various oxazaborolidines 12 in the enantioselective intramolecular [2+2] photocycloaddition to tricyclic product 6 a.
Upon diastereoselective reduction to the respective secondary alcohol, the absolute configuration of the major enantiomer 6 a was elucidated by Mosher analysis.25 The selection of the aryl boronic acid was based on a preliminary screen performed with the 3,5‐dimethylphenyl‐substituted alcohol 9 i and various boronic acids. In this first set of experiments, the 2,3,4‐trifluorophenyl boronic acid performed the best (see Supporting Information). Surprisingly, we found that the enantioface differentiation in the reaction 5 a→6 a was not consistent but that it depended strongly on the aryl group Ar in catalyst 12. In some cases, there was even a slight preference for the other enantiomer ent‐6 a which is presented in Scheme 4 as a negative enantiomeric excess (ee).
The highest enantioselectivity (78 % ee) was recorded for oxazaborolidine 12 e derived from amino alcohol 9 e (Ar=2,3‐dimethylphenyl). Several other oxazaborolidines 12 with alkyl‐ and methoxy‐substituted aryl groups also led to selectivities >50 % ee (12 d, 12 g–12 l, 12 o). A substitution in para‐position of the aryl group and fluorine substituents led to low or negative enantioselectivities (12 f, 12 m, 12 n, 12 p, 12 r).
When searching for ways to optimize the enantioselectivity of the intramolecular [2+2] photocycloaddition we initially turned towards the irradiation conditions. The emission source that we employed exhibits an emission maximum at λ=366 nm (Figure 4) but has a notable emission in the short wavelength (λ<360 nm) region. It was speculated that this emission might lead to a direct excitation of substrate 5 a and might favor the racemic transformation by direct excitation. We could show that an Fe2(SO4)3 filter solution26 indeed suppresses the short wavelength emission and the effect on the enantioselectivity was clearly notable. Upon direct irradiation at λ=366 nm the reaction with Lewis acid 12 e⋅AlBr3 had provided product 6 a (Table 1) in 62 % yield and with 78 % ee. When the light was filtered by an Fe2(SO4)3 solution (c=600 mg L−1) the yield increased to 80 % and the enantioselectivity increased to 84 % ee.
Figure 4.
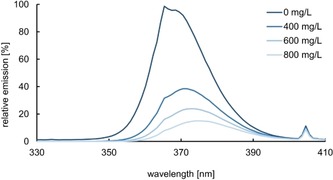
Calculated emission profile of a 366 nm fluorescent lamp tuned by an Fe2(SO4)3 filter solution (in 0.01 m HCl) of varying concentration.
Simultaneously to the emission experiments, we revisited the aryl boronic acids and performed another screening of their influence on the enantioselectivity now employing prolinol 9 e (Ar=2,3‐dimethylphenyl) as the amino alcohol. It was found that 2,4,6‐trifluorophenyl boronic acid led to an improvement that parallels the improvement achieved with the filter solution. When catalyzed by AlBr3‐activated oxazaborolidine 13, the yield for the reaction 5 a→6 a improved as compared to the reaction promoted by catalyst 12 e to 80 % and the enantioselectivity to 83 % ee (Scheme 5). Since direct irradiation at λ=366 nm is operationally easier than irradiation through a filter solution, alkenones 5 a‐5 j were subsequently subjected to the former conditions. Substrate consumption was complete after 24 h and the products were—with a single exception (vide infra)—obtained in good to high yields (54–86 %). More importantly, the enantiocontrol was high and exceeded in eight out of ten examples a level of 80 % ee. Upon Lewis acid‐promoted conversion of enone 5 g, a significant amount of olefinic side products were identified which had to be removed from the product by ozonolysis.
Scheme 5.
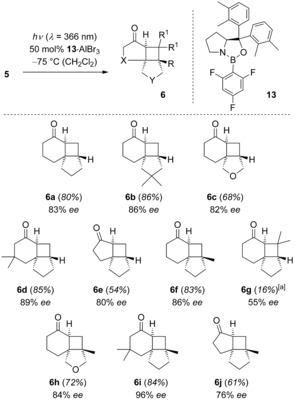
Enantioselective intramolecular [2+2] photocycloaddition of enones 6 in the presence of AlBr3‐activated oxazaborolidine 13.[a]Olefinic side‐products were removed by ozonolysis prior to purification.
This method thus paves—for the first time—an enantioselective route to access typical intramolecular [2+2] photocycloaddition products of 2‐cyclohexenones (6 a–6 d, 6 f, 6 h, 6 i) and 2‐cyclopentenones (6 e, 6 j). The product configuration was assigned in analogy to major enantiomer 6 a which was dextrorotatory ([α]D=+156). Likewise, all other cyclobutane products showed a high positive specific rotation in CH2Cl2 solution ([α]D=+98 to [α]D=+292).
Computational and mechanistic studies
The coordination of enones to acid‐activated oxazaborolidines has been discussed in previous work27, 28 and most commonly a weak non‐classical hydrogen bond between the α‐hydrogen atom of the enone substrate and the oxygen atom of the oxazaborolidine is invoked. This interaction avoids rotation around the strong coordinating bond between the carbonyl group and the boron atom. In order to visualize the reactive complex, we optimized the structure of model substrate 3‐methyl‐2‐cyclohexenone coordinated to Lewis acid 12 i⋅AlBr3 (Figure 5). All computations were carried out with Gaussian 1629 using the B3LYP‐D3BJ functional30 and the cc‐pVTZ basis set.31 In order to match experimental conditions, computations include thermal corrections at −75 °C and a PCM solvation model for CH2Cl2 (for details see the Supporting Information). Si‐C was found to be the most stable conformation of complex C with the above‐mentioned non‐classical hydrogen bond (2.46 Å) clearly visible and the Si face of the β‐carbon atom being exposed for an intramolecular attack. The lower aryl group (Ardown) is slightly turned with one side pointing towards the enone double bond thereby blocking this enantiotopic face. The 2,3,4‐trifluorophenyl group shields the area above the enone α‐carbon atom and is very likely responsible for the fact that olefins with terminal substituents (substrate 5 g) react poorly in the intramolecular [2+2] photocycloaddition. Likewise, it was found in the previously studied intermolecular variant of this reaction17 that tri‐ and tetrasubstituted olefins reacted sluggishly. A major difference between the former and the latter photocycloaddition is the fact that the first C−C bond formation in the former case will occur exclusively in the β‐position (“rule of five”)5 while in the latter case the α‐position is likely the position of initial attack. In addition, the intramolecular reaction is faster and thus outcompetes possible degradation pathways of the photoexcited enone. Indeed, Lewis acids 11⋅AlBr3 and 12 i⋅AlBr3 performed poorly in the intermolecular [2+2] photocycloaddition which was ascribed to the fact that they underwent decomposition by hydrogen abstraction from the enone.
Figure 5.

Optimized structure (right) displaying the preferred conformation within the complex of 3‐methyl‐2‐cyclohexenone and Lewis acid 12 i⋅AlBr3 (Si‐C, left).
The complex of 3‐methyl‐2‐cyclohexenone and Lewis acid 12 n⋅AlBr3 (complex D) was also studied computationally since the latter is the least structurally different from 12 i⋅AlBr3 but nonetheless had led to an inverted enantioselectivity (Scheme 4, −29 % ee). Although the ee shift from 75 % ee (12 i⋅AlBr3) to −29 % ee (12 n⋅AlBr3) corresponds only to a difference in free enthalpies of approx. 4 kJ mol−1 at −75 °C, we were curious whether different conformational properties of the two complexes C and D could be identified that would rationalize an influence of the rather subtle changes in catalyst aryl group substitution. We first explored enone binding to the opposite concave side of the catalyst which has been discussed in the context of cycloaddition reactions27, 32 and obtained the corresponding conformers for both C and D. However, both structures were very similar and no apparent characteristics were identified that would explain the strong influence of the aryl para‐substituent (see Supporting Information for details). In a complimentary approach encouraged by computational studies of complexes without C H‐O contacts but rather π‐interactions with Ardown 33 we turned our attention to a coordination pattern in which the substrate is rotated by 180° around the O−B bond between enone and catalyst. This complex would also lead to product of inverse absolute configuration through shielding of the opposite enantiotopic face and, indeed, we identified conformation Re‐D in which the Re face of the β‐carbon atom is exposed towards an attack.34 In this conformation, Ardown and enone are perfectly parallel to each other (Figure 6) thereby suggesting attractive dispersion interactions between the two entities. Additionally, the aryl para‐substituent and the substrate β‐substituent are brought in close proximity in Re‐D, which might serve as a plausible hint towards the difference between C and D. While this result should not be taken in any way as quantitative (the computed ground state energies for Re‐ and Si‐complexes differ by less than 5 kJ mol−1), it shows qualitatively how cyclic enones can coordinate to certain Lewis acid‐activated oxazaborolidines in a way that explains a reversal in enantioselectivity.
Figure 6.
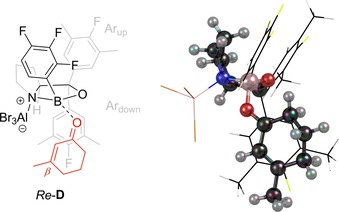
Optimized complex conformer structure (right) of 3‐methyl‐2‐cyclohexenone and Lewis acid 12 n⋅AlBr3 which displays the Re face of the β‐carbon atom towards an attack (Re‐D, left).
Due to an efficient symmetry‐allowed ISC from the n–π* singlet state,3, 35 cyclic enones undergo [2+2] photocycloaddition reactions from the π–π* triplet state. We determined the quantum yield for the racemic reaction 5 a→rac‐6 a at λ=368 nm (LED) and at −80 °C to be Φ=0.38 (±0.02). The high value which compares well with quantum yields previously obtained for this and related [2+2] photocycloaddition reactions36 is a testimony to the high efficiency with which both ISC and ring closure occur. Under the same conditions it was attempted to determine the quantum yield of the Lewis acid‐promoted reaction employing Lewis acid 13⋅AlBr3. The high sensitivity of the Lewis acid towards air and moisture made it difficult to take samples at given time intervals and to monitor the progress of the reaction. Instead, the reaction was stopped after 10 min and the conversion was determined (see the Supporting Information for further details). The measurement was performed in triplicate and delivered a quantum yield of Φ=0.052 (±0.007). Given that ISC occurs in this case from a π–π* singlet the quantum yield is remarkably high. The value seems to support—as earlier suggested by calculations37—the hypothesis that the Lewis acids facilitates the forbidden ISC to the π–π* triplet state. An alternative explanation for the efficiency of the Lewis acid‐catalyzed process would be a rapid cyclization on the singlet hypersurface prior to ISC. In order to distinguish between the two pathways and to substantiate the fact that the catalyzed reaction proceeds indeed on the triplet hypersurface, we turned to a classical experiment that had been earlier performed with substrates 5 k by Becker and co‐workers.36a Upon irradiation at λ>330 nm (uranium glass filter), it had been found that both diastereoisomers cis‐5 k and trans‐5 k gave in separate reactions products rac‐6 k as a cis/trans mixture in a ratio of 50:50. The reaction was not stereospecific and implied the intermediacy of triplet diradical 14 which allows for free rotation around the indicated single bond (Figure 7). Likewise, we studied the two substrates cis‐5 k and trans‐5 k in separate reactions which were performed with the Lewis acid 13⋅AlBr3 under the conditions of Scheme 5. The reaction turned out to be also stereoconvergent and led to a mixture of trans‐6 k and cis‐6 k in an almost identical diastereomeric ratio (d.r.). When starting from cis‐5 k, the d.r. was 83:17 whereas the d.r. was 86:14 with trans‐5 k. In both reactions, the trans compound trans‐6 k prevailed and was formed with 79 % ee in the former and with 86 % ee in the latter case.
Figure 7.
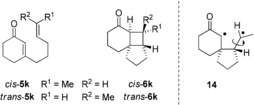
Structures of irradiation precursors cis‐5 k and trans‐5 k, of photocycloaddition products cis‐6 k and trans‐6 k, and of putative triplet intermediate 14.
While the stereoconvergency of the reactions 5 k→6 k supports the intermediacy of 1,4‐diradical 14, the high simple diastereoselectivity is remarkable if compared to the non‐existent diastereoselectivity (d.r.=50:50) observed for the racemic reaction.36a The finding seems to indicate that intermediate 14 remains in the coordination sphere of the catalyst which forces the methyl group in the trans position of the resulting cyclobutane ring. If intermediate 14 had dissociated from the catalyst prior to ring closure38 the d.r. should have been similar to the racemic reaction. The observation parallels with the sensitivity of the Lewis acid‐mediated [2+2] photocycloaddition towards steric hindrance (substrate 5 g).
Total synthesis of (±)‐italicene and (±)‐isoitalicene
The sesquiterpenes italicene and isoitalicene39 are the most prominent natural products which feature the octahydrocyclopenta[1,4]cyclobuta[1,2]benzene skeleton that in turn is built up in the intramolecular [2+2] photocycloaddition of 3‐(pent‐4‐enyl)‐2‐cyclohexenones. Synthetic approaches towards these compounds have been reported40 and to this date there exist two completed total syntheses.39, 41
Following a retrosynthetic [2+2] disconnection, 2‐cyclohexenone rac‐3 seemed to be a reasonable starting material which would allow the implementation of a photochemical key step in the synthesis. Indeed, this reaction had been previously studied by Hoye et al.42 but the results were somewhat sobering (Scheme 6). At ambient temperature, not a single selectivity parameter was satisfactorily controlled and the reaction delivered four isomeric products in relative proportions of almost unity. Optimization of the intramolecular [2+2] photocycloaddition in our study rested on three parameters: choice of the irradiation wavelength, temperature, and Lewis acid. There was a minor improvement in yield at λ=366 nm but the d.r. (rac‐15 a/rac‐15 b) remained low (67:33) and the formation of compounds rac‐16 was not suppressed. As already found by Hoye et al., a decrease of the reaction temperature to −75 °C enhanced the d.r. to 78:22 but olefins rac‐16 remained present. Eventually, we discovered that with AlBr3 as the (achiral) Lewis acid there was a perfect type selectivity and even an increase of the d.r. to 82:18 (Scheme 7).
Scheme 6.
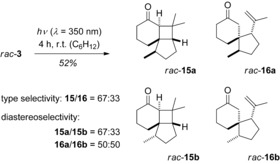
Low‐type selectivity and facial diastereoselectivity in the [2+2] photocycloaddition of substrate rac‐3.42
Scheme 7.
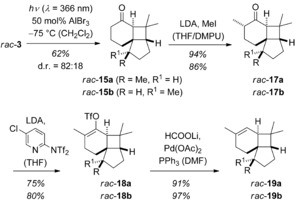
Total synthesis of (±)‐italicene (rac‐19 a) and (±)‐isoitalicene (rac‐19 b) via a selective Lewis‐acid promoted [2+2] photocycloaddition.
The diastereomeric photocycloaddition products rac‐15 were separable and the relative configuration of the major isomer rac‐15 a was confirmed.42 Its preferred formation can be explained by a chair‐like transition state in which the methyl group at the stereogenic center is in an equatorial position.43 While it was initially attempted to perform the α‐methylation and to follow a known route towards italicene and isoitalicene,39 we failed to separate the isomers at the stage of the natural products. In addition, the final dehydration step yielded repeatedly several product isomers apart from the two natural products. As an alternative, we decided to process the diastereoisomers separately and devised an alternative sequence for the introduction of the olefinic double bond. The α‐methylation proceeded smoothly and with high diastereoselectivity for both epimers rac‐15 a and rac‐15 b. The ketones rac‐17 a and rac‐17 b were converted via the respective enolates into unstable triflates44 rac‐18 a and rac‐18 b which had to be purified on deactivated neutral alumina. The reduction was eventually performed with lithium formate employing Pd(OAc)2 (10 mol %) as the catalyst.45 The conditions were found to be superior to other reported procedures which led to the formation of inseparable nonpolar side products.46 Starting from 4‐bromoanisole as precursor for enone rac‐3,42 (±)‐italicene (rac‐19 a) was synthesized in seven steps with an overall yield of 14 %. The Lewis acid was responsible for a significant improvement in the selectivity of the photochemical stey step which accounts for the high overall yield. (±)‐Isoitalicene (rac‐19 b) was obtained via the minor photocycloaddition diastereoisomer rac‐15 b with an overall yield of 3 %.
Given that the starting alkene rac‐3 is doubly substituted at the terminal carbon atom, the chances were low to process the compound in a kinetic photochemical resolution (cf. substrate 5 g). Still, it was attempted to perform the [2+2] photocycloaddition in the presence of Lewis acids 11⋅AlBr3, 12 e⋅AlBr3, 12 i⋅AlBr3, and 13⋅AlBr3 under standard conditions (Scheme 5). The highest enantioselectivity was recorded with the Lewis acid 11⋅AlBr3 if the reaction was stopped after one hour. The major diastereoisomer 15 a that was formed with a d.r. of 84:16 displayed an enantiomeric excess of 42 %. The conversion was low, however, and 71 % of the starting material 3 was recovered (10 % ee).
Conclusion
In summary, this study provided new information about the mode of action of Lewis acid‐mediated [2+2] photocycloaddition reactions. For the enantioselective intramolecular reaction of substrates 5, it was found that the choice of substituents at the chiral oxazaborolidine has a large influence on the degree of enantioselecitivity. According to DFT calculations of complex 12 i⋅AlBr3 with an enone, the lower aryl group (Ardown) at the carbon atom and the aryl group at the boron atom control the accessibility to the substrate. The enone encounters a high enantioface differentiation at the β‐carbon atom at which the first C−C bond formation occurs. Moreover, the aryl group at the boron atom limits the available space in cis‐position of the terminal carbon atom in the alkenyl chain. It was found that a reversal of enantioselectivity is possible by varying the aryl groups Ar of the oxazaborolidine and a different binding mode of the enone to the Lewis acid was identified. This discovery may allow to access in the future opposite enantiomers of a given photoproduct by alteration of the substituents. Regarding the mechanism of the Lewis acid‐catalyzed reaction, a reaction pathway via the π–π* triplet was substantiated. Although the higher absorbance at λ=366 nm of the Lewis acid‐enone complex vs. the free enone facilitates its preferred excitation, the faster ISC rate of the free enone compensates its lower absorbance. As a consequence, the quantum yield of the Lewis acid‐mediated reaction is by a factor of 10 lower which in turn requires a high catalyst loading of 50 mol % to achieve a high degree of enantioselectivity. The effect of Lewis acids on the selectivity of photochemical reactions is not limited to aspects of enantioselectivity. We found an improved type selectivity in the intramolecular [2+2] photocycloaddition of substrate rac‐3. Hydrogen abstraction products rac‐16 were absent if the reaction was performed in the presence of AlBr3 as the Lewis acid and the facial diastereoselectivity in favor of product rac‐15 a increased. A concise and high‐yielding synthesis of (±)‐italicene (rac‐19 a) could thus be achieved.
Experimental Section
General information: All air and moisture sensitive reactions were carried out in heat gun‐dried glassware under an argon atmosphere using standard Schlenk techniques. Room temperature refers to 22–26 °C. Temperatures of 0 °C were obtained using an ice/water bath. Temperatures of −78 °C were obtained using a dry ice/isopropanol bath. For moisture sensitive reactions, tetrahydrofuran (THF), diethyl ether (Et2O) and dichloromethane (CH2Cl2) were dried using a MBSPS 800 MBraun solvent purification system. The following columns were used: Tetrahydrofuran: 2×MB‐KOL‐M type 2 (3 Å molecular sieve); Diethyl ether: 1×MB‐KOL‐A type 2 (aluminum oxide), 1×MB‐KOL‐M type 2 (3 Å molecular sieve); Dichloromethane: 2×MB‐KOL‐A type 2 (aluminum oxide). The following dry solvents are commercially available and were used without further purification: Toluene: Acros Organics, 99.8 % extra dry, over molecular sieves. For photochemical reactions, dry dichloromethane was degassed by three freeze‐pump‐thaw cycles and stored over 4 Å molecular sieves. Technical solvents [pentane (P), diethyl ether (Et2O), dichloromethane (CH2Cl2), methanol (MeOH), n‐hexane (nHex), ethyl acetate (EtOAc), cyclohexane (cHex)] were distilled prior to column chromatography. Commercially available chemicals were purchased from the suppliers ABCR, Acros, Alfa‐Aesar, Sigma–Aldrich (now Merck KGaA), and TCI, and were used without further purification. For isomerizations of the photoproducts, basic alumina (Merck, aluminum oxide 90 active basic, 0.063–0.200 mm) was used.
Analytical methods and equipment: Photochemical experiments were carried out in heat gun‐dried Duran tubes in a positive geometry setup (cylindrical array of 16 fluorescent tubes, 8 W nominal power) with the sample placed in the center of the illumination chamber. Fluorescent tubes of the type Hitachi UV‐A (BI‐B) (λ max=350 nm) and Philips Blue Light (λ max=366 nm) were employed. Enantioselective reactions were carried out at −75 °C using a Duran cooling finger which was attached to a high‐performance cryostat (Huber CC80). Ozone was generated by a FisherTechnology ozone‐generator Type 502. Flash column chromatography was performed with silica 60 (Merck, 230–400 mesh) as the stationary phase with the indicated eluent mixtures. Deactivation of neutral alumina (Merck, aluminum oxide 90 active neutral, 70–230 mesh) was carried out by the addition of 36 wt % water in small portions. Subsequently, the powder was spread in a petri dish and was allowed to dry on air for at least two days. Thin Layer Chromatography (TLC) was performed on silica coated glass plates (Merck, silica 60 F254) with detection by UV‐light (λ=254 nm) and/or by staining with a potassium permanganate solution [KMnO4] or with a cerium ammonium molybdate solution [CAM] followed by heat treatment: KMnO4‐staining solution: potassium permanganate (3.00 g), potassium carbonate (20.0 g) and aqueous sodium hydroxide solution (5 wt %, 5.00 mL) in water (300 mL). CAM‐staining solution: cerium sulfate tetrahydrate (1.00 g), ammonium molybdate (25.0 g) and concentrated sulfuric acid (25.0 mL) in water (250 mL). NMR spectra were recorded at room temperature either on a Bruker AVHD‐300, AVHD‐400, AVHD‐500 or an AV‐500 cryo. 1H NMR spectra were referenced to the residual proton signal of chloroform‐d1 (δ=7.26 ppm), [D4]MeOH (δ=3.31 ppm), [D6]benzene (δ=7.16 ppm) or deuterium oxide (δ=4.79 ppm). 13C NMR spectra were referenced to the 13C‐D triplet of CDCl3 (δ=77.16 ppm), to the 13C‐D septet of CD3OD (δ=49.00 ppm) or to the 13C‐D triplet of C6D6 (δ=128.06 ppm). 19F NMR spectra were referenced to the 19F signal of CCl3F (δ=0 ppm). Apparent multiplets which occur as a result of coupling constant equality between magnetically non‐equivalent protons are marked as virtual (virt.). The following abbreviations for single multiplicities were used: br‐broad, s‐singlet, d‐doublet, t‐triplet, q‐quartet, quint‐quintet, sext‐sextet, sept‐septet. Assignment and multiplicity of the 13C NMR signals were determined by two‐dimensional NMR experiments (COSY, HSQC, HMBC). Protons oriented above the molecular plane are labeled as α and those oriented below as β. Infrared spectra were recorded on a PerkinElmer Frontier IR‐FTR spectrometer by ATR technique. The signal intensity is assigned using the following abbreviations: br (broad), vs. (very strong), s (strong), m (medium), w (weak). Low resolution and high resolution mass spectra were recorded on a Thermo Scientific LTQ‐FT Ultra (ESI) or a Thermo Scientific DFS‐HRMS spectrometer (EI). All melting points were determined using a Büchi M‐565 melting point apparatus, with a range quoted to the nearest integer. UV/Vis spectra were measured on a PerkinElmer Lambda 35 UV/Vis spectrometer. Spectra were recorded using a Hellma precision cell made of quartz SUPRASIL® with a pathway of 1 mm or 1 cm. Solvents and concentrations are given for each spectrum. GC analysis was performed on an Agilent 7890 B gas chromatograph using an Agilent HP‐5 column (30 m×0.32 mm×0.25 μm, SN: 19091J‐413) with a flame ionization detector. The temperature method is given for the corresponding compounds. Chiral GC analysis was performed on an Agilent 7890 B gas chromatograph using an Agilent Cyclosil‐B column (30 m×0.25 mm×0.25 μm, SN: USF620714 H) or a Macherey–Nagel Lipodex E column (25 m×0.25 mm, SN: 23393‐92) with a flame ionization detector. The temperature method is given for the corresponding compounds. Chiral HPLC was performed on a Thermo‐Fisher HPLC system comprising a SR3000 solvent rack, a LPG3400 SD pump, a WPS‐3000 SL autosampler, a TCC‐3000 SD column compartment and a DAD‐3000 UV/Vis detector fitted with the appropriate Daicel column as chiral stationary phase (flow rate: 1.0 mL min−1, Daicel column, time and eluent are given for the corresponding compounds). Optical rotations were recorded on a Bellingham+Stanley ADP440+ polarimeter using a cuvette with a path length of 0.05 dm. All measurements were performed using the sodium D line (λ=589 nm) at room temperature. The specific rotation is reported as follows: [α]D T=100×α/(l×c) [10−1 grad cm2 g−1] (α: optical rotation [deg], l: path length [dm], c: concentration of sample [g 100 cm−3]).
Preparation of starting materials, calculations, and mechanistic studies: The synthesis of the photocycloaddition precursors and of the proline‐derived amino alcohols 9 is described in the Supporting Information, which also contains details on the preparation of the oxazaborolidines and other amino alcohol derivatives, on the preparation of the activated oxazaborolidines, on the DFT calculations, and on the mechanistic studies.
Racemic intramolecular [2+2] photocycloaddition (General Procedure 1): A solution of the respective irradiation precursor (1.00 equiv) in dichloromethane (1–3 mL) was transferred to a Duran phototube. Dichloromethane was added until a concentration of 20 mm was reached. The solution was irradiated at λ=366 nm for the respective amount of time. After complete conversion, the solvent was removed in vacuo. The residue was purified by column chromatography with the given eluent mixture. The obtained cis/trans mixture was equilibrated over basic alumina in a small amount of dichloromethane overnight. The suspension was filtered, washed with small portions of diethyl ether, and the filtrate was concentrated.
Enantioselective intramolecular [2+2] photocycloaddition (General Procedure 2): A solution of the respective irradiation precursor (1.00 equiv) in dichloromethane (1–3 mL) was transferred to a heat‐gun dried Duran phototube and the vessel was washed twice with small portions of dichloromethane. Then, a solution of activated oxazaborolidine catalyst 13⋅AlBr3 (50.0 mol %) in dichloromethane (1–3 mL) was transferred to the reaction mixture and the vessel was washed with small portions of dichloromethane. Dichloromethane was added until a concentration of 20 mm was reached. The solution was cooled to −75 °C within 30 min and was subsequently irradiated at λ=366 nm for 24 h. The reaction mixture was poured into suspended silica in dichloromethane and the solvent was removed in vacuo. The dry‐loaded product was purified by column chromatography with a given eluent mixture. The obtained cis/trans mixture was equilibrated over basic alumina in a small amount of dichloromethane overnight. The suspension was filtered, washed with small portions of diethyl ether, and the filtrate was concentrated.
Photocycloaddition product 6 a: Racemic: Following GP1, enone 5 a (131 mg, 800 μmol, 1.00 equiv) was irradiated in dichloromethane (40 mL) for 8 h. After purification by column chromatography (silica, P/Et2O=4:1), ketone rac‐6 a (119 mg, 725 μmol, 91 %) was obtained as a colorless oil. Enantioselective: Following GP2, enone 5 a (16.4 mg, 100 μmol, 1.00 equiv) was irradiated in dichloromethane (5 mL). After purification by column chromatography (silica, P/Et2O=4:1), ketone 6 a (13.2 mg, 80.4 μmol, 80 %, 83 % ee) was obtained as a colorless oil. R f=0.42 (pentane/Et2O 6:4) [KMnO4]; [α]D 25=+156 (c=1.0 in CH2Cl2); 1H NMR (500 MHz, CDCl3, 25 °C, TMS): δ=1.34 (virt. td, 2 J ≈3 J 1=12.6 Hz, 3 J 2=6.8 Hz, 1 H, HH‐1), 1.50–1.58 (m, 2 H, H‐8), 1.58–1.64 (m, 3 H, HH‐1, H‐3), 1.77–1.93 (m, 3 H, H‐2, HH‐4), 1.93–2.04 (m, 2 H, H‐7), 2.07 (ddd, 2 J=13.1 Hz, 3 J 1=9.7 Hz, 3 J 2=7.0 Hz, 1 H, HH‐4), 2.17 (dddd, 2 J=18.0 Hz, 3 J 1=11.4 Hz, 3 J 2=6.9 Hz, 4 J=1.1 Hz, 1 H, HH‐6), 2.37–2.43 (m, 1 H, H‐3a), 2.48 (virt. ddq, 3 J 1=11.4 Hz, 3 J 2=7.0 Hz, 4 J 1 ≈4 J 2=1.2 Hz, 1 H, H‐4a), 2.57 ppm (virt. dddt, 2 J=18.0 Hz, 3 J 1=4.7 Hz, 3 J 2=3.4 Hz, 4 J 1 ≈4 J 2=1.2 Hz, 1 H, HH‐6); 13C NMR (126 MHz, CDCl3, 27 °C, TMS): δ=21.2 (t, C‐7), 25.1 (t, C‐2), 26.9 (t, C‐4), 32.9 (t, C‐8), 33.1 (t, C‐3), 39.6 (d, C‐3a), 39.6 (t, C‐6), 40.4 (t, C‐1), 47.3 (d, C‐4a), 50.0 (s, C‐8a), 215.7 ppm (s, C‐5); Chiral GC: τ R (major)=157.2 min, τ R (minor)=161.8 min, [60 °C (1 min), 100 °C (30 °C min−1), 100 °C (157 min), 135 °C (3 °C min−1), 200 °C (20 °C min−1), 200 °C (3 min)], Cyclosil‐B. The analytical data obtained matched those reported in the literature.47
Photocycloaddition product 6 b: Racemic: Following GP1, enone 5 b (38.5 mg, 200 μmol, 1.00 equiv) was irradiated in dichloromethane (10 mL) for 3 h. After purification by column chromatography (silica, P/Et2O=4:1), ketone rac‐6 b (33.5 mg, 174 μmol, 87 %) was obtained as a colorless oil. Enantioselective: Following GP2, enone 5 b (38.5 mg, 200 μmol, 1.00 equiv) was irradiated in dichloromethane (10 mL). After purification by column chromatography (silica, P/Et2O=6:1), ketone 6 b (33.0 mg, 172 μmol, 86 %, 86 % ee) was obtained as a colorless oil. R f=0.51 (pentane/Et2O 1:1) [KMnO4]; [α]D 25=+97.7 (c=1.5 in CH2Cl2); 1H NMR (500 MHz, CDCl3, 25 °C, TMS): δ=0.94 (s, 3 H, Me‐2), 1.17 (s, 3 H, Me‐2), 1.49 (dd, 2 J=13.4 Hz, 4 J=1.7 Hz, 1 H, HH‐1), 1.51 (dd, 2 J=13.1 Hz, 3 J=6.2 Hz, 1 H, HH‐3), 1.57 (ddd, 2 J=14.0 Hz, 3 J 1=11.1 Hz, 3 J 2=3.3 Hz, 1 H, HH‐8), 1.70 (d, 2 J=13.4 Hz, 1 H, HH‐1), 1.75 (dddd, 2 J=14.0 Hz, 3 J 1=6.4 Hz, 3 J 2=3.0 Hz, 4 J=1.1 Hz, 1 H, HH‐8), 1.81–1.91 (m, 2 H, HH‐3, HH‐7), 1.94–2.07 (m, 2 H, HH‐4, HH‐7), 2.12–2.18 (m, 1 H, HH‐4), 2.18–2.25 (m, 1 H, HH‐6), 2.44–2.49 (m, 1 H, H‐3a), 2.49–2.56 (m, 1 H, HH‐6), 2.77–2.82 ppm (m, 1 H, H‐4a); 13C NMR (126 MHz, CDCl3, 27 °C, TMS): δ=20.7 (t, C‐7), 27.9 (t, C‐4), 29.6 (q, Me‐2), 30.0 (q, Me‐2), 35.0 (t, C‐8), 38.9 (t, C‐6), 41.8 (d, C‐3a), 43.2 (s, C‐2)*, 49.4 (t, C‐3), 50.9 (d, C‐4a), 51.3 (s, C‐8a)*, 56.6 (t, C‐1), 216.7 ppm (s, C‐5) [*Assignment of signals is interconvertible.]; IR (ATR): ṽ=2927 (s, sp3‐CH), 2863 (m, sp3‐CH), 1696 (vs., C=O), 1462 (m, sp3‐CH), 907 cm−1 (w); MS (EI, 70 eV): m/z (%): 192 (33) [M]+, 177 (23) [M−CH3]+, 164 (17) [M−CO]+, 159 (11), 136 (15), 122 (30) [C8H10O]+, 110 (100) [M−C6H10]+, 107 (58), 93 (20), 83 (23) [C6H11]+, 67 (24), 55 (51) [C4H7]+, 41 (20); HRMS (EI, 70 eV): calcd for C13H20O [M]+: 192.1509; found: 192.1504; calcd for C12 13CH20O [M]+: 193.1542; found: 193.1541; Chiral GC: τ R (major)=173.4 min, τ R (minor)=174.0 min, [60 °C (1 min), 100 °C (30 °C min−1), 100 °C (157 min), 135 °C (3 °C min−1), 200 °C (20 °C min−1), 200 °C (3 min)], Cyclosil‐B.
Photocycloaddition product 6 c: Racemic: Following GP1, enone 5 c (16.6 mg, 100 μmol, 1.00 equiv) was irradiated in dichloromethane (5 mL) for 5 h. After purification by column chromatography (silica, P/EtOAc=1:1), ketone rac‐6 c (14.4 mg, 86.6 μmol, 87 %) was obtained as a colorless oil. Enantioselective: Following GP2, enone 5 c (33.2 mg, 200 μmol, 1.00 equiv) was irradiated in dichloromethane (10 mL). After purification by column chromatography (silica, P/EtOAc=1:1), ketone 6 c (22.6 mg, 136 μmol, 68 %, 82 % ee) was obtained as a colorless oil. R f=0.31 (pentane/Et2O 1:1) [KMnO4]; [α]D 26=+138 (c=1.4 in CH2Cl2); 1H NMR (500 MHz, CDCl3, 25 °C, TMS): δ=1.52 (ddd, 2 J=13.9 Hz, 3 J 1=11.9 Hz, 3 J 2=4.3 Hz, 1 H, HH‐8), 1.70 (dddd, 2 J=13.9 Hz, 3 J 1=4.5 Hz, 3 J 2=3.2 Hz, 4 J=1.5 Hz, 1 H, HH‐8), 1.94–2.13 (m, 4 H, H‐4, H‐7), 2.18 (dddd, 2 J=17.4 Hz, 3 J 1=12.3 Hz, 3 J 2=6.0 Hz, 4 J=1.1 Hz, 1 H, HH‐6), 2.54–2.61 (m, 2 H, H‐3a, HH‐6), 2.71 (dd, 3 J 1=10.7 Hz, 3 J 2=7.2 Hz, 1 H, H‐4a), 3.28 (d, 2 J=9.3 Hz, 1 H, HH‐1), 3.61 (dd, 2 J=9.3 Hz, 3 J=5.2 Hz, 1 H, HH‐3), 3.86 (d, 2 J=9.3 Hz, 1 H, HH‐1), 3.86 ppm (d, 2 J=9.3 Hz, 1 H, HH‐3); 13C NMR (126 MHz, CDCl3, 27 °C, TMS): δ=21.3 (t, C‐7), 26.8 (t, C‐4), 28.6 (t, C‐8), 39.9 (t, C‐6), 40.9 (d, C‐3a), 46.6 (d, C‐4a), 51.1 (s, C‐8a), 74.4 (t, C‐3), 78.9 (t, C‐1), 214.1 ppm (s, C‐5); IR (ATR): ṽ=2938 (m, sp3‐CH), 2843 (m, sp3‐CH), 1697 (vs., C=O), 1107 (s, sp3‐CO), 914 cm−1 (vs., sp3‐CO); MS (EI, 70 eV): m/z (%): 166 (58) [M]+, 137 (27) [M−CO]+, 121 (84) [M−C2H5O]+, 110 (100) [M−C3H4O]+, 96 (82) [M−C4H6O]+, 82 (78) [C5H6O]+, 79 (90), 67 (66), 55 (58) [C4H7]+, 41 (61) [C3H5]+; HRMS (EI, 70 eV): calcd for C10H14O2 [M]+: 166.0988; found: 166.0985; calcd for C9 13CH14O2 [M]+: 167.1022; found: 167.1022; Chiral GC: τ R (minor)=37.5 min, τ R (major)=37.7 min, [60 °C (0 min), 245 °C (3 °C min−1), 245 °C (3 min)], Cyclosil‐B.
Photocycloaddition product 6 d: Racemic: Following GP1, enone 5 d (38.5 mg, 200 μmol, 1.00 equiv) was irradiated in dichloromethane (10 mL) for 5 h. After purification by column chromatography (silica, P/Et2O=6:1), ketone rac‐6 d (30.4 mg, 158 μmol, 79 %) was obtained as a colorless oil. Enantioselective: Following GP2, enone 5 d (38.5 mg, 200 μmol, 1.00 equiv) was irradiated in dichloromethane (10 mL). After purification by column chromatography (silica, P/Et2O=6:1), ketone 6 d (32.6 mg, 170 μmol, 85 %, 89 % ee) was obtained as a colorless oil. R f=0.63 (pentane/Et2O 1:1) [KMnO4]; [α]D 25=+156 (c=1.3 in CH2Cl2); 1H NMR (500 MHz, CDCl3, 25 °C, TMS): δ=0.98 (s, 3 H, Me‐7), 1.06 (s, 3 H, Me‐7), 1.36 (virt. td, 2 J ≈3 J 1=12.3 Hz, 3 J 2=7.2 Hz, 1 H, HH‐1), 1.52 (dd, 2 J=12.6 Hz, 3 J=6.0 Hz, 1 H, HH‐3), 1.54–1.62 (m, 2 H, HH‐3, HH‐8), 1.69 (d, 2 J=14.5 Hz, 1 H, HH‐8), 1.72–1.78 (m, 1 H, HH‐1), 1.78–1.89 (m, 3 H, H‐2, HH‐4), 2.15 (d, 2 J=14.8 Hz, 1 H, HH‐6), 2.19 (ddd, 2 J=13.0 Hz, 3 J 1=9.5 Hz, 3 J 2=6.6 Hz, 1 H, HH‐4), 2.25 (d, 2 J=14.8 Hz, 1 H, HH‐6), 2.36 (dd, 3 J 1=11.6 Hz, 3 J 2=6.6 Hz, 1 H, H‐4a), 2.38–2.43 ppm (m, 1 H, H‐3a); 13C NMR (126 MHz, CDCl3, 27 °C, TMS): δ=24.8 (t, C‐2), 27.3 (t, C‐4), 28.5 (q, Me‐7), 31.0 (q, Me‐7), 33.0 (t, C‐3), 34.6 (s, C‐7), 42.7 (d, C‐3a), 43.1 (t, C‐1), 46.0 (d, C‐4a), 47.7 (t, C‐8), 49.6 (s, C‐8a), 53.8 (t, C‐6), 216.7 ppm (s, C‐5); IR (ATR): ṽ=2941 (s, sp3‐CH), 2895 (m, sp3‐CH), 2868 (m, sp3‐CH), 1700 (vs., C=O), 1467 cm−1 (m, sp3‐CH); MS (EI, 70 eV): m/z (%): 192 (40) [M]+, 177 (18) [M−CH3]+, 149 (41) [M−C3H7]+, 136 (81) [M−C4H8]+, 125 (35) [C8H13O]+, 108 (56) [C7H8O]+, 93 (44), 82 (100) [C6H10]+, 54 (30), 41 (18); HRMS (EI, 70 eV): calcd for C13H20O [M]+: 192.1509; found: 192.1513; Chiral GC: τ R (minor)=169.7 min, τ R (major)=170.3 min, [60 °C (1 min), 100 °C (30 °C min−1), 100 °C (157 min), 135 °C (3 °C min−1), 200 °C (20 °C min−1), 200 °C (3 min)], Cyclosil‐B.
Photocycloaddition product 6 e: Racemic: Following GP1, enone 5 e (30.0 mg, 200 μmol, 1.00 equiv) was irradiated in dichloromethane (10 mL) for 47 h. After purification by column chromatography (silica, P/Et2O=5:1), ketone rac‐6 e (16.6 mg, 111 μmol, 56 %) was obtained as a colorless oil. Enantioselective: Following GP2, enone 5 e (30.0 mg, 200 μmol, 1.00 equiv) was irradiated in dichloromethane (10 mL). After purification by column chromatography (silica, P/Et2O=5:1), ketone 6 e (16.2 mg, 108 μmol, 54 %, 80 % ee) was obtained as a colorless oil. R f=0.26 (pentane/Et2O 5:1) [KMnO4]; [α]D 25=+292 (c=1.1 in CH2Cl2); 1H NMR (500 MHz, CDCl3, 25 °C, TMS): δ=1.42 (ddd, 2 J=13.0 Hz, 3 J 1=10.7 Hz, 3 J 2=9.1 Hz, 1 H, HH‐7), 1.54–1.59 (m, 1 H, HH‐5), 1.59–1.67 (m, 1 H, HH‐5), 1.68–1.74 (m, 1 H, HH‐7), 1.76–1.83 (m, 1 H, HH‐4), 1.83–1.91 (m, 4 H, HH‐6, HH‐4, H‐1), 1.92 (virt. dq, 2 J=4.8 Hz, 3 J 1 ≈3 J 2 ≈3 J 3=2.2 Hz, 1 H, HH‐6), 2.22 (ddd, 3 J 1=10.7 Hz, 3 J 2=4.4 Hz, 4 J=2.0 Hz, 1 H, H‐3a), 2.35 (virt. ddt, 2 J=17.9 Hz, 3 J 1=7.8 Hz, 3 J 2 ≈4 J=2.0 Hz, 1 H, HH‐2), 2.49–2.56 (m, 1 H, H‐4a), 2.78 ppm (virt. dddt, 2 J=17.9 Hz, 3 J 1=12.5 Hz, 3 J 2=9.6 Hz, 4 J 1 ≈4 J 2=0.8 Hz, 1 H, HH‐2); 13C NMR (126 MHz, CDCl3, 27 °C, TMS): δ=25.8 (t, C‐4), 26.0 (t, C‐6), 32.1 (t, C‐1), 33.4 (t, C‐5), 37.4 (t, C‐7), 38.2 (t, C‐2), 40.6 (d, C‐4a), 47.1 (d, C‐3a), 53.0 (s, C‐7a), 222.8 ppm (s, C‐3); Chiral GC: τ R (minor)=82.4 min, τ R (major)=90.8 min, [60 °C (1 min), 100 °C (30 °C min−1), 100 °C (157 min), 135 °C (3 °C min−1), 200 °C (20 °C min−1), 200 °C (3 min)], Cyclosil‐B. The analytical data obtained matched those reported in the literature.48
Photocycloaddition product 6 f: Racemic: Following GP1, enone 5 f (35.7 mg, 200 μmol, 1.00 equiv) was irradiated in dichloromethane (10 mL) for 5 h. After purification by column chromatography (silica, P/Et2O=5:1), ketone rac‐6 f (24.2 mg, 136 μmol, 68 %) was obtained as a colorless oil. Enantioselective: Following GP2, enone 5 f (35.7 mg, 200 μmol, 1.00 equiv) was irradiated in dichloromethane (10 mL). After purification by column chromatography (silica, P/Et2O=5:1), ketone 6 f (29.5 mg, 165 μmol, 83 %, 86 % ee) was obtained as a colorless oil. R f=0.58 (pentane/Et2O 1:1) [KMnO4]; [α]D 25=+155 (c=1.1 in CH2Cl2); 1H NMR (500 MHz, CDCl3, 25 °C, TMS): δ=1.06 (s, 3 H, Me‐3a), 1.29–1.36 (m, 1 H, HH‐3), 1.36–1.42 (m, 1 H, HH‐1), 1.45 (ddd, 2 J=13.7 Hz, 3 J 1=9.1 Hz, 3 J 2=4.2 Hz, 1 H, HH‐8), 1.57–1.63 (m, 1 H, HH‐3), 1.71–1.83 (m, 4 H, HH‐1, H‐2, HH‐8), 1.86 (ddd, 2 J=12.7 Hz, 3 J=7.2 Hz, 4 J=1.4 Hz, 1 H, HH‐4), 1.88–1.98 (m, 2 H, H‐7), 2.01 (dd, 2 J=12.7 Hz, 3 J=11.0 Hz, 1 H, HH‐4), 2.21–2.28 (m, 1 H, HH‐6), 2.37–2.44 ppm (m, 2 H, H‐4a, HH‐6); 13C NMR (126 MHz, CDCl3, 27 °C, TMS): δ=22.0 (t, C‐7), 22.9 (q, Me‐3a), 23.9 (t, C‐2), 29.5 (t, C‐8), 35.1 (t, C‐4), 40.0 (t, C‐6), 41.7 (t, C‐1), 42.0 (t, C‐3), 44.6 (s, C‐3a), 45.3 (d, C‐4a), 51.3 (s, C‐8a), 216.5 ppm (s, C‐5); Chiral GC: τ R (minor)=131.9 min, τ R (major)=136.7 min, [60 °C (1 min), 100 °C (30 °C min−1), 100 °C (157 min), 135 °C (3 °C min−1), 200 °C (20 °C min−1), 200 °C (3 min)], Cyclosil‐B. The analytical data obtained matched those reported in the literature.49
Photocycloaddition product 6 g: Racemic: Following GP1, enone 5 g (38.5 mg, 200 μmol, 1.00 equiv) was irradiated in dichloromethane (10 mL) for 5.5 h. After purification by column chromatography (silica, P/Et2O=6:1), a product mixture was obtained, which contains inseparable impurities. To facilitate purification, the mixture was submitted to ozonolysis which was conducted at −78 °C in dichloromethane (3 mL). Completion of the reaction was indicated by blue coloration during ozone introduction. The blue color was removed by an argon gas flow and dimethyl sulfide (1 mL) was added. Subsequently, the mixture was warmed to room temperature and the solvent was removed in vacuo. The residue was purified by column chromatography (silica, P/Et2O=6:1). After the work‐up process, ketone 6 g (19.7 mg, 102 μmol, 51 %) was obtained as a colorless oil. Enantioselective: Following GP2, enone 5 g (38.5 mg, 200 μmol, 1.00 equiv) was irradiated in dichloromethane (10 mL). After purification by column chromatography (silica, P/Et2O=6:1), a product mixture was obtained, which contains inseparable impurities. To facilitate purification, the mixture was submitted to ozonolysis which was conducted at −78 °C in dichloromethane (3 mL). Completion of the reaction was indicated by blue coloration during ozone introduction. The blue color was removed by an argon gasflow and dimethyl sulfide (1 mL) was added. Subsequently, the mixture was warmed to room temperature and the solvent was removed in vacuo. The residue was purified by column chromatography (silica, P/Et2O=6:1). After the work‐up process, ketone 6 g (6.20 mg, 32.2 μmol, 16 %, 55 % ee) was obtained as a colorless oil. R f=0.66 (pentane/Et2O 1:1) [KMnO4]; [α]D 25=+140 (c=1.1 in CH2Cl2); 1H NMR (500 MHz, CDCl3, 25 °C, TMS): δ=1.03 (s, 3 H, Me‐4), 1.05 (s, 3 H, Me‐4), 1.21–1.30 (m, 1 H, HH‐1), 1.50–1.62 (m, 1 H, HH‐3), 1.68–1.78 (m, 5 H, HH‐1, HH‐2, HH‐3, HH‐7, HH‐8), 1.80–1.91 (m, 2 H, HH‐2, HH‐8), 1.91–1.98 (m, 1 H, HH‐7), 1.98–2.00 (m, 1 H, H‐3a), 2.17 (s, 1 H, H‐4a), 2.18–2.33 ppm (m, 2 H, H‐6); 13C NMR (126 MHz, CDCl3, 27 °C, TMS): δ=21.4 (t, C‐7), 25.1 (q, Me‐4), 26.9 (t, C‐2), 27.6 (q, Me‐4), 28.2 (t, C‐3), 34.5 (t, C‐8), 36.9 (s, C‐4), 40.4 (t, C‐1), 41.1 (t, C‐6), 45.1 (s, C‐8a), 52.0 (d, C‐3a), 57.4 (d, C‐4a), 214.3 ppm (s, C‐5); Chiral GC: τ R (minor)=157.6 min, τ R (major)=161.9 min, [60 °C (1 min), 100 °C (30 °C min−1), 100 °C (157 min), 135 °C (3 °C min−1), 200 °C (20 °C min−1), 200 °C (3 min)], Cyclosil‐B. The analytical data obtained matched those reported in the literature.42
Photocycloaddition product 6 h: Racemic: Following GP1, enone 5 h (18.0 mg, 100 μmol, 1.00 equiv) was irradiated in dichloromethane (5 mL) for 8 h. After purification by column chromatography (silica, P/EtOAc=2:1), ketone rac‐6 h (14.4 mg, 79.9 μmol, 80 %) was obtained as a colorless oil. Enantioselective: Following GP2, enone 5 h (36.1 mg, 200 μmol, 1.00 equiv) was irradiated in dichloromethane (10 mL). After purification by column chromatography (silica, P/EtOAc=2:1), ketone 6 h (25.8 mg, 143 μmol, 72 %, 84 % ee) was obtained as a colorless oil. R f=0.37 (pentane/Et2O 1:1) [KMnO4]; [α]D 26=+142 (c=1.5 in CH2Cl2); 1H NMR (500 MHz, CDCl3, 25 °C, TMS): δ=1.08 (s, 3 H, Me‐3a), 1.45 (ddd, 2 J=14.4 Hz, 3 J 1=8.6 Hz, 3 J 2=5.9 Hz, 1 H, HH‐8), 1.77 (virt. dt, 2 J=14.4 Hz, 3 J 1 ≈3 J 2=5.0 Hz, 1 H, HH‐8), 1.87–2.02 (m, 3 H, HH‐4, H‐7), 2.20–2.31 (m, 2 H, HH‐4, HH‐6), 2.46 (virt. dt, 2 J=16.6 Hz, 3 J 1 ≈3 J 2=5.2 Hz, 1 H, HH‐6), 2.68 (dd, 3 J 1=11.0 Hz, 3 J 2=7.0 Hz, 1 H, H‐4a), 3.26 (d, 2 J=9.1 Hz, 1 H, HH‐3), 3.30 (d, 2 J=9.2 Hz, 1 H, HH‐1), 3.83 (d, 2 J=9.1 Hz, 1 H, HH‐3), 3.95 ppm (d, 2 J=9.2 Hz, 1 H, HH‐1); 13C NMR (126 MHz, CDCl3, 27 °C, TMS): δ=18.1 (q, Me‐3a), 21.9 (t, C‐7), 24.9 (t, C‐8), 34.8 (t, C‐4), 40.2 (t, C‐6), 45.0 (d, C‐4a), 45.5 (s, C‐3a), 51.7 (s, C‐8a), 80.0 (t, C‐1), 81.1 (t, C‐3), 214.6 ppm (s, C‐5); IR (ATR): ṽ=2935 (m, sp3‐CH), 2838 (m, sp3‐CH), 1699 (vs., C=O), 1054 (s, sp3‐CO), 932 cm−1 (s, sp3‐CO); MS (EI, 70 eV): m/z (%): 180 (15) [M]+, 135 (55) [C9H11O]+, 122 (31), 109 (100) [C7H9O]+, 95 (46), 79 (61), 67 (51), 55 (97) [C4H7]+, 41 (45) [C3H5]+; HRMS (EI, 70 eV): calcd for C11H16O2 [M]+: 180.1145; found: 180.1143; calcd for C10 13CH16O2 [M]+: 181.1178; found: 181.1183; Chiral GC: τ R (minor)=42.7 min, τ R (major)=43.3 min, [60 °C (0 min), 130 °C (30 °C min−1), 130 °C (38 min), 160 °C (5 °C min−1), 240 °C (15 °C min−1), 240 °C (2 min)], Cyclosil‐B.
Photocycloaddition product 6 i: Racemic: Following GP1, enone 5 i (41.3 mg, 200 μmol, 1.00 equiv) was irradiated in dichloromethane (10 mL) for 5 h. After purification by column chromatography (silica, P/Et2O=6:1), ketone rac‐6 i (36.4 mg, 176 μmol, 88 %) was obtained as a colorless oil. Enantioselective: Following GP2, enone 5 i (41.3 mg, 200 μmol, 1.00 equiv) was irradiated in dichloromethane (10 mL). After purification by column chromatography (silica, P/Et2O=6:1), ketone 6 i (34.7 mg, 168 μmol, 84 %, 96 % ee) was obtained as a colorless oil. R f=0.62 (pentane/Et2O 1:1) [KMnO4]; [α]D 25=+228 (c=1.3 in CH2Cl2); 1H NMR (500 MHz, CDCl3, 25 °C, TMS): δ=0.90 (s, 3 H, Me‐7), 1.01 (s, 3 H, Me‐3a), 1.04 (s, 3 H, Me‐7), 1.23–1.34 (m, 2 H, HH‐3, HH‐8), 1.39 (virt. td, 2 J ≈3 J 1=12.2 Hz, 3 J 2=7.0 Hz, 1 H, HH‐1), 1.62 (virt. ddt, 2 J=12.7 Hz, 3 J=6.2 Hz, 4 J 1≈4 J 2=1.5 Hz, 1 H, HH‐3), 1.69–1.84 (m, 3 H, H‐2, HH‐4), 1.91 (d, 2 J=14.2 Hz, 1 H, HH‐8), 1.93–1.99 (m, 1 H, HH‐1), 2.02 (dd, 2 J=12.5 Hz, 3 J=11.2 Hz, 1 H, HH‐4), 2.12 (ddd, 2 J=16.1 Hz, 4 J 1=2.5 Hz, 4 J 2=1.3 Hz, 1 H, HH‐6), 2.21 (dd, 2 J=16.1 Hz, 4 J=0.8 Hz, 1 H, HH‐6), 2.36 ppm (ddd, 3 J 1=11.2 Hz, 3 J 2=7.7 Hz, 4 J=1.3 Hz, 1 H, H‐4a); 13C NMR (126 MHz, CDCl3, 27 °C, TMS): δ=24.3 (q, Me‐3a), 24.3 (t, C‐2), 28.0 (q, Me‐7), 31.8 (q, Me‐7), 33.9 (s, C‐7), 34.8 (t, C‐4), 40.8 (t, C‐3), 42.8 (t, C‐8), 43.7 (d, C‐4a), 44.0 (t, C‐1), 45.4 (s, C‐3a), 50.0 (s, C‐8a), 52.7 (t, C‐6), 216.2 ppm (s, C‐5); Chiral GC: τ R (major)=94.3 min, τ R (minor)=95.0 min, [60 °C (0.5 min), 70 °C (10 °C min−1), 114 °C (0.4 °C min−1), 200 °C (10 °C min−1), 200 °C (3 min)], Lipodex E. The analytical data obtained matched those reported in the literature.49
Photocycloaddition product 6 j: Racemic: Following GP1, enone 5 j (32.9 mg, 200 μmol, 1.00 equiv) was irradiated in dichloromethane (10 mL) for 8 h at λ=350 nm. After purification by column chromatography (silica, P/Et2O=5:1), ketone rac‐6 j (20.6 mg, 125 μmol, 63 %) was obtained as a colorless oil. Enantioselective: Following GP2, enone 5 j (32.9 mg, 200 μmol, 1.00 equiv) was irradiated in dichloromethane (10 mL). After purification by column chromatography (silica, P/Et2O=5:1), ketone 6 j (20.2 mg, 123 μmol, 61 %, 76 % ee) was obtained as a colorless oil. R f=0.60 (pentane/Et2O 1:1) [KMnO4]; [α]D 25=+222 (c=1.1 in CH2Cl2); 1H NMR (500 MHz, CDCl3, 25 °C, TMS): δ=1.14 (s, 3 H, Me‐4a), 1.30–1.38 (m, 1 H, HH‐5), 1.44–1.52 (m, 1 H, HH‐7), 1.59–1.70 (m, 3 H, HH‐4, HH‐5, HH‐7), 1.70–1.84 (m, 3 H, HH‐1, H‐6), 2.01–2.09 (m, 2 H, HH‐1, HH‐4), 2.20 (ddd, 3 J 1=11.0 Hz, 3 J 2=4.7 Hz, 4 J=2.0 Hz, 1 H, H‐3a), 2.34 (virt. ddt, 2 J=18.8 Hz, 3 J 1=9.8 Hz, 3 J 2 ≈4 J=2.0 Hz, 1 H, HH‐2), 2.62–2.71 ppm (m, 1 H, HH‐2); 13C NMR (126 MHz, CDCl3, 27 °C, TMS): δ=23.2 (q, Me‐4a), 25.1 (t, C‐6), 27.1 (t, C‐1), 34.5 (t, C‐4), 38.5 (t, C‐7), 38.8 (t, C‐2), 42.3 (t, C‐5), 43.8 (s, C‐4a), 45.6 (d, C‐3a), 53.6 (s, C‐7a), 223.4 ppm (s, C‐3); Chiral GC: τ R (minor)=14.8 min, τ R (major)=14.9 min, [60 °C (0 min), 120 °C (30 °C min−1), 120 °C (10 min), 240 °C (30 °C min−1), 240 °C (2 min)], Cyclosil‐B. The analytical data obtained matched those reported in the literature.48
Photocycloaddition products trans ‐6 k and cis ‐6 k: Racemic: Following GP1, enone cis‐5 k (35.7 mg, 200 μmol, 1.00 equiv) was irradiated in dichloromethane (10 mL) for 8 h at a wavelength of λ=366 nm. After purification by column chromatography (P/Et2O=4:1), a mixture of ketones rac‐trans‐6 k and rac‐cis‐6 k (32.7 mg, 183 μmol, 92 %, d.r.=50:50) was obtained as a colorless oil. Following GP1, enone trans‐5 k (35.7 mg, 200 μmol, 1.00 equiv) was irradiated in dichloromethane (10 mL) for 8 h at a wavelength of λ=366 nm. After purification by column chromatography (P/Et2O=4:1), a mixture of ketones rac‐trans‐6 k and rac‐cis‐6 k (33.0 mg, 185 μmol, 93 %, d.r.=50:50) was obtained as a colorless oil. Enantioselective: Following GP2, enone cis‐5 k (35.7 mg, 200 μmol, 1.00 equiv) was irradiated in dichloromethane (10 mL). After purification by column chromatography (P/Et2O=5:1), a mixture of ketones trans‐6 k (79 % ee) and cis‐6 k (55 % ee) (13.5 mg, 75.7 μmol, 38 %, d.r.=83:17) was obtained as a colorless oil. Following GP2, enone trans‐5 k (35.7 mg, 200 μmol, 1.00 equiv) was irradiated in dichloromethane (10 mL). After purification by column chromatography (P/Et2O=5:1), a mixture of ketones trans‐6 k (86 % ee) and cis‐6 k (74 % ee) (15.7 mg, 88.1 μmol, 44 %, d.r.=86:14) was obtained as a colorless oil. trans‐6 k: R f=0.58 (pentane/Et2O 1:1) [KMnO4]; 1H NMR (500 MHz, CDCl3, 25 °C, TMS): δ=0.94 (d, 3 J=7.5 Hz, 3 H, Me‐4), 1.29–1.39 (m, 1 H, HH‐3), 1.49–1.63 (m, 5 H, H‐1, HH‐3, H‐8), 1.75–1.84 (m, 2 H, H‐2), 1.89–1.98 (m, 2 H, H‐7), 1.98–2.04 (m, 1 H, H‐3a), 2.04–2.14 (m, 2 H, H‐4, HH‐6), 2.40 (virt. dt, 2 J=18.4 Hz, 3 J 1 ≈3 J 2=3.6 Hz, 1 H, HH‐6), 2.52 ppm (d, 3 J=11.2 Hz, 1 H, H‐4a); 13C NMR (126 MHz, CDCl3, 27 °C, TMS): δ=17.3 (q, Me‐4), 21.3 (t, C‐7), 25.2 (t, C‐2), 32.5 (t, C‐8)*, 33.0 (t, C‐1)*, 33.3 (d, C‐4), 40.6 (t, C‐3), 41.1 (t, C‐6), 45.8 (s, C‐8a), 47.9 (d, C‐3a), 51.3 (s, C‐4a), 214.9 ppm (s, C‐5) [*Assignment of signals is interconvertible.]; Chiral GC: τ R (major)=55.4 min, τ R (minor)=58.1 min, [60 °C (0 min), 115 °C (15 °C min−1), 115 °C (50 min), 160 °C (5 °C min−1), 220 °C (30 °C min−1), 220 °C (2 min)], Cyclosil‐B. cis‐6 k: R f=0.58 (pentane/Et2O 1:1) [KMnO4]; 1H NMR (500 MHz, CDCl3, 25 °C, TMS): δ=0.98 (d, 3 J=6.8 Hz, 3 H, Me‐4), 1.24–1.38 (m, 1 H), 1.47–1.63 (m, 3 H), 1.56–1.75 (m, 1 H), 1.68–1.74 (m, 1 H), 1.75–1.86 (m, 2 H), 1.89–1.98 (m, 1 H), 2.03–2.21 (m, 3 H, H‐4a), 2.33–2.46 (m, 2 H, H‐3a, H‐4), 2.57 ppm (virt. dt, 2 J=17.8 Hz, 3 J 1 ≈3 J 2=3.9 Hz, 1 H, HH‐6); 13C NMR (126 MHz, CDCl3, 27 °C, TMS): δ=14.7 (q, Me‐4), 20.9 (t), 26.7 (t), 26.9 (t), 31.3 (d, C‐4), 32.5 (t), 39.0 (t), 39.6 (t), 43.3 (d, C‐3a), 47.4 (s, C‐8a), 56.0 (d, C‐4a), 214.0 ppm (s, C‐5); Chiral GC: τ R (minor)=56.8 min, τ R (major)=57.6 min, [60 °C (0 min), 115 °C (15 °C min−1), 115 °C (50 min), 160 °C (5 °C min−1), 220 °C (30 °C min−1), 220 °C (2 min)], Cyclosil‐B. The analytical data obtained matched those reported in the literature.47
Photocycloaddition products 15 a and 15 b: Racemic: Following GP1, enone rac‐3 (273 mg, 1.32 mmol, 1.00 equiv) was irradiated in dichloromethane (66 mL) for 14 h. Different from GP1, the reaction mixture was treated with triethylamine (1 mL) instead of basic alumina and the solvent was removed in vacuo. The residue was purified by column chromatography (silica, P/Et2O=5:1). Starting material rac‐3 as well as the product mixture, which was submitted to ozonolysis, were isolated. The ozonolysis was conducted at −78 °C in dichloromethane (3 mL). Completion of the reaction was indicated by blue coloration during ozone introduction. The blue color was removed by an argon gasflow and dimethyl sulfide (1 mL) was added. Subsequently, the mixture was warmed to room temperature, the solvent was removed in vacuo and the residue was purified by column chromatography (silica, P/Et2O=5:1). After the work‐up process, ketones rac‐15 a and rac‐15 b (211 mg, 1.02 mmol, 77 %, d.r.=67:33, rac‐15 a/ rac‐15 b) were obtained as a colorless oil and starting material rac‐3 (17.7 mg, 85.8 μmol, 6 %) was recovered. Racemic in the presence of aluminum bromide: In analogy to GP2, enone rac‐3 (41.3 mg, 200 μmol, 1.00 equiv) was irradiated in dichloromethane (10 mL) using aluminum bromide as catalyst for 24 h. After purification by column chromatography (silica, P/Et2O=6:1), ketones rac‐15 a and rac‐15 b (25.5 mg, 124 μmol, 62 %, d.r.=82:18, rac‐15 a/ rac‐15 b) were obtained as a colorless oil. Enantioselective: Following GP2, enone rac‐3 (41.3 mg, 200 μmol, 1.00 equiv) was irradiated in dichloromethane (10 mL) using activated catalyst 11⋅AlBr3 for 1 h. After purification by column chromatography (silica, P/Et2O=6:1), ketones 15 a and 15 b [5.30 mg, 25.7 μmol, 13 %, d.r.=84:16, 15 a (43 % ee)/15 b (21 % ee)] were obtained as a colorless oil and starting material ent‐3 (29.1 mg, 141 μmol, 71 %, 10 % ee) was recovered. Separation of diastereoisomers: A mixture of diastereomers rac‐15 a and rac‐15 b (500 mg) was separated by column chromatography (silica, P/Et2O=30:1) with a conventional column (36 mm diameter, 300 mm length). The collected fractions were analyzed by gas chromatography and were combined to six fractions [(content of rac‐15 b)]: [F1 (≥99.5 %)], [F2 (90<99.5 %)], [F3 (10<90 %)]; [(content of rac‐15 a)]: [F4 (90<99 %)], [F5 (99<99.5)], [F6 (≥99.5 %)]. The fractions F3, F4 and F5 were purified under the same conditions iteratively (subsequently from F3 to F5) until F3 contained less than 15 mg of the product mixture. Finally, F2, F3, F4, F5 were purified subsequently. 15 a: R f=0.66 (pentane/Et2O 1:1) [CAM, KMnO4]; 1H NMR (500 MHz, CDCl3, 25 °C, TMS): δ=0.84 (d, 3 J=7.2 Hz, 3 H, Me‐1), 1.03 (s, 3 H, Me‐4β), 1.07 (s, 3 H, Me‐4α), 1.48–1.55 (m, 1 H, HH‐2), 1.59–1.76 (m, 4 H, H‐3, HH‐7, HH‐8), 1.84–2.07 (m, 5 H, H‐1, HH‐2, H‐3a, HH‐7, HH‐8), 2.15–2.25 (m, 2 H, H‐4a, HH‐6), 2.28–2.36 ppm (m, 1 H, HH‐6); 13C NMR (126 MHz, CDCl3, 27 °C, TMS): δ=16.7 (q, Me‐1), 20.5 (t, C‐7), 25.1 (t, C‐3), 25.7 (q, Me‐4β), 27.6 (q, Me‐4α), 29.0 (t, C‐8), 34.7 (t, C‐2), 37.0 (s, C‐4), 40.9 (t, C‐6), 41.0 (d, C‐1), 47.8 (s, C‐8a), 51.6 (d, C‐3a), 58.4 (d, C‐4a), 214.7 ppm (s, C‐5); Chiral GC: τ R (minor)=25.3 min, τ R (major)=25.9 min, [60 °C (0.5 min), 200 °C (4 °C min−1), 200 °C (5 min)], Lipodex E. 15 b: R f=0.66 (pentane/Et2O 1:1) [CAM, KMnO4]; 1H NMR (500 MHz, CDCl3, 25 °C, TMS): δ=0.89 (d, 3 J=6.7 Hz, 3 H, Me‐1), 1.03 (s, 3 H, Me‐4β), 1.06 (s, 3 H, Me‐4α), 1.42 (virt. qd, 2 J ≈3 J 1 ≈3 J 2=12.3 Hz, 3J3=6.6 Hz, 1 H, HH‐2), 1.48–1.73 (m, 4 H, H‐1, H‐3, HH‐8), 1.73–1.91 (m, 3 H, HH‐2, HH‐7, HH‐8), 1.94–2.04 (m, 1 H, HH‐7), 2.06 (d, 3 J=8.0 Hz, 1 H, H‐3a), 2.11–2.21 (m, 1 H, HH‐6), 2.25–2.35 ppm (m, 2 H, H‐4a, HH‐6); 13C NMR (126 MHz, CDCl3, 27 °C, TMS): δ=12.8 (q, Me‐1), 21.5 (t, C‐7), 24.9 (q, Me‐4β), 26.6 (t, C‐3), 27.3 (q, Me‐4α), 33.0 (t, C‐8), 35.4 (t, C‐2), 36.3 (s, C‐4), 41.1 (t, C‐6), 44.7 (d, C‐1), 46.5 (s, C‐8a), 52.2 (d, C‐4a), 53.1 (d, C‐3a), 214.1 ppm (s, C‐5); Chiral GC: τ R (major)=24.3 min, τ R (minor)=24.6 min, [60 °C (0.5 min), 200 °C (4 °C min−1), 200 °C (5 min)], Lipodex E. The analytical data obtained matched those reported in the literature.42
Ketone rac ‐17 a: A solution of n‐butyllithium (2.50 m in hexane, 3.54 mL, 8.85 mmol, 6.00 equiv) was added to a solution of diisopropylamine (955 mg, 1.33 mL, 9.44 mmol, 6.40 equiv) in tetrahydrofuran (15 mL, 630 mm) at −78 °C. The resulting mixture was stirred for 1 h at −78 °C. A solution of ketone rac‐15 a (304 mg, 1.47 mmol, 1.00 equiv) in tetrahydrofuran (15 mL, 100 mm) was added dropwise to the freshly prepared lithium diisopropylamide solution at −78 °C. After 6 h, DMPU (2.27 g, 2.14 mL, 17.7 mmol, 12.0 equiv) and iodomethane (1.67 g, 735 μL, 11.8 mmol, 8.00 equiv) were added in sequence, during which a colorless precipitate was formed. The suspension was allowed to slowly warm to room temperature over the course of 15 h. The brown reaction mixture was transferred to a silica‐packed column, the reaction vessel was rinsed with dichloromethane and the reaction mixture was filtered with a solvent mixture (P/Et2O=5:1). The product containing fractions were combined and after removal of the solvent in vacuo, the residue was purified by column chromatography (silica, P/Et2O=10:1). Ketone rac‐17 a (305 mg, 1.38 mmol, 94 %, d.r.=90:10) was obtained as a colorless oil. R f=0.77 (pentane/Et2O 1:1) [CAM, KMnO4]; 1H NMR (500 MHz, CDCl3, 25 °C, TMS): δ=0.82 (d, 3 J=7.2 Hz, 3 H, Me‐1), 1.01 (s, 3 H, Me‐4β), 1.09 (s, 3 H, Me‐4α), 1.09 (d, 3 J=7.1 Hz, 3 H, Me‐6), 1.33 (virt. tdd, 2 J ≈3 J 1=13.5 Hz, 3 J 2=11.9 Hz, 3 J 3=2.3 Hz, 1 H, HH‐7), 1.49–1.56 (m, 1 H, HH‐2), 1.56–1.64 (m, 1 H, HH‐3), 1.70 (virt. tt, 2 J ≈3 J 1=13.0 Hz, 3 J 2 ≈3 J 3=7.6 Hz, 1 H, HH‐3), 1.76–1.83 (m, 1 H, HH‐8), 1.84–1.92 (m, 2 H, H‐1, HH‐7), 1.94 (d, 3 J=8.2 Hz, 1 H, H‐3a), 2.01 (virt. tt, 2 J ≈3 J 1=13.1 Hz, 3 J 2 ≈3 J 3=6.9 Hz, 1 H, HH‐2), 2.07–2.15 (m, 2 H, H‐6, HH‐8), 2.19 ppm (s, 1 H, H‐4a); 13C NMR (126 MHz, CDCl3, 27 °C, TMS): δ=16.5 (q, Me‐6), 16.9 (q, Me‐1), 24.9 (t, C‐3), 25.5 (q, Me‐4β), 27.9 (q, Me‐4α), 29.0 (t, C‐8), 29.3 (t, C‐7), 34.6 (t, C‐2), 37.2 (s, C‐4), 39.9 (d, C‐1), 45.8 (d, C‐6), 48.7 (s, C‐8a), 51.9 (d, C‐3a), 57.9 (d, C‐4a), 216.5 ppm (s, C‐5); The analytical data obtained matched those reported in the literature.42
Ketone rac ‐17 b: A solution of n‐butyllithium (2.50 m in hexane, 1.51 mL, 3.76 mmol, 6.00 equiv) was added to a solution of diisopropylamine (406 mg, 566 μL, 4.01 mmol, 6.40 equiv) in tetrahydrofuran (6.27 mL, 640 mm) at −78 °C. The resulting mixture was stirred for 1 h at −78 °C. A solution of ketone rac‐15 b (129 mg, 627 μmol, 1.00 equiv) in tetrahydrofuran (6.27 mL, 100 mm) was added dropwise to the freshly prepared lithium diisopropylamide solution at −78 °C. After 6 h, DMPU (965 mg, 910 μL, 7.53 mmol, 12.0 equiv) and iodomethane (712 mg, 312 μL, 5.02 mmol, 8.00 equiv) were added in sequence, during which a colorless precipitate was formed. The suspension was allowed to slowly warm to room temperature over the course of 15 h. The brown reaction mixture was transferred to a silica‐packed column, the reaction vessel was rinsed with dichloromethane and the reaction mixture was filtered with a solvent mixture (P/Et2O=5:1). The product containing fractions were combined and after removal of the solvent in vacuo, the residue was purified by column chromatography (silica, P/Et2O=10:1). Ketone rac‐17 b (119 mg, 540 μmol, 86 %) was obtained as a colorless oil. R f=0.77 (pentane/Et2O 1:1) [CAM, KMnO4]; 1H NMR (500 MHz, CDCl3, 25 °C, TMS): δ=0.90 (d, 3 J=6.7 Hz, 3 H, Me‐1), 1.00 (s, 3 H, Me‐4β), 1.07 (d, 3 J=7.0 Hz, 3 H, Me‐6), 1.09 (s, 3 H, Me‐4α), 1.41 (virt. qd, 2 J ≈3 J 1 ≈3 J 2=12.3 Hz, 3 J 3=7.0 Hz, 1 H, HH‐2), 1.49–1.67 (m, 4 H, H‐1, H‐3, HH‐7), 1.75 (virt. dt, 2 J=12.4 Hz, 3 J 1 ≈3 J 2=6.2 Hz, 1 H, HH‐2), 1.89 (ddd, 2 J=13.7 Hz, 3 J 1=5.6 Hz, 3 J 2=3.5 Hz, 1 H, HH‐8), 1.94–2.07 (m, 3 H, H‐3a, HH‐7, HH‐8), 2.16–2.26 (m, 1 H, H‐6), 2.31 ppm (s, 1 H, H‐4a); 13C NMR (126 MHz, CDCl3, 27 °C, TMS): δ=13.2 (q, Me‐1), 15.8 (q, Me‐6), 24.9 (q, Me‐4β), 26.3 (t, C‐3), 27.8 (q, Me‐4α), 30.4 (t, C‐7), 33.7 (t, C‐8), 35.7 (t, C‐2), 36.7 (s, C‐4), 45.1 (d, C‐6), 45.9 (d, C‐1), 47.0 (s, C‐8a), 51.8 (d, C‐4a), 54.2 (d, C‐3a), 215.6 ppm (s, C‐5); The analytical data obtained matched those reported in the literature.42
Triflate rac ‐18 a: A solution of n‐butyllithium (2.50 m in hexane, 363 μL, 908 μmol, 4.00 equiv) was added to a solution of diisopropylamine (96.4 mg, 135 μL, 953 μmol, 4.20 equiv) in tetrahydrofuran (2.27 mL, 420 mm) at −78 °C. The resulting mixture was stirred for 30 min. A solution of ketone rac‐17 a (50.0 mg, 227 μmol, 1.00 equiv) in tetrahydrofuran (2.27 mL, 100 mm) was added dropwise to the freshly prepared lithium diisopropylamide solution at −78 °C. After 7.5 h, a solution of Comins reagent (401 mg, 1.02 mmol, 4.50 equiv) in tetrahydrofuran (1.02 mL, 1.00 m) was added dropwise to the enolate solution, during which the solution turned deep brown. After 5 min, the reaction mixture was allowed to warm to room temperature in the course of 30 min. The brown reaction mixture was transferred to a column packed with deactivated, neutral alumina*, the reaction vessel was rinsed with dichloromethane and the reaction mixture was filtered with a solvent mixture (P/CH2Cl2=15:1). The product containing fractions were combined and after removal of the solvent in vacuo, the residue was purified by column chromatography (deactivated neutral alumina, P/CH2Cl2=15:1) three consecutive times in order to remove residual Comins reagent. Triflate rac‐18 a (59.8 mg, 170 μmol, 75 %) was obtained as a colorless oil. [*Deactivation is described in the general information.] R f=0.47 (pentane) [CAM, KMnO4]; 1H NMR (500 MHz, C6D6, 25 °C, TMS): δ=0.58 (d, 3 J=7.2 Hz, 3 H, Me‐1), 1.00 (s, 3 H, Me‐4β), 1.04 (s, 3 H, Me‐4α), 1.24–1.39 (m, 2 H, HH‐2, HH‐8), 1.41–1.51 (m, 3 H, H‐3, HH‐8), 1.51–1.74 (m, 7 H, H‐1, H‐3a, Me‐6, H‐7), 1.85 (virt. dtd, 2 J=13.3 Hz, 3 J 1 ≈3 J 2=9.5 Hz, 3 J 3=6.7 Hz, 1 H, HH‐2), 2.39 ppm (br s, 1 H, H‐4a); 13C NMR (126 MHz, CDCl3, 27 °C, TMS): δ=15.9 (q, Me‐1), 17.3 (q, Me‐6), 24.8 (t, C‐3), 24.9 (q, Me‐4β), 26.8 (q, Me‐4α), 26.8 (t, C‐8), 29.5 (t, C‐7), 34.9 (t, C‐2), 35.6 (s, C‐4), 40.6 (d, C‐1), 48.7 (s, C‐8a), 49.4 (d, C‐4a), 51.1 (d, C‐3a), 119.1 (qs, 1 J CF=320 Hz, CF3), 128.5 (s, C‐6)*, 145.2 ppm (s, C‐5) [*The 13C signal of C‐6 overlaps with the solvent signal of C6D6. However, the signal can be located with the help of a HMBC crosspeak with the proton signal of Me‐6 to assign the 13C signal of C‐6.]; 19F NMR (376 MHz, C6D6, 25 °C, TMS): δ=−75.4 (s, 3 F, CF3); IR (ATR): ṽ=2953 (m, sp3‐CH), 2870 (m, sp3‐CH), 1410 (s, SO), 1201 (vs., sp3‐CF), 1141 (vs., sp3‐CF), 898 cm−1 (vs., SO); MS (EI, 70 eV): m/z (%): 352 (51) [M]+, 308 (14), 281 (74), 266 (100), 252 (14), 220 (17), 205 (68), 187 (64), 159 (56), 145 (73), 109 (39), 82 (55), 55 (42) [C4H7]+; HRMS (EI, 70 eV): calcd for C16H23O3F3 32S [M]+: 352.1315; found: 352.1310; calcd for C15 13CH23O3F3 32S [M]+: 353.1348; found: 353.1344.
Triflate rac ‐18 b: A solution of n‐butyllithium (2.50 m in hexane, 363 μL, 908 μmol, 4.00 equiv) was added to a solution of diisopropylamine (96.4 mg, 135 μL, 953 μmol, 4.20 equiv) in tetrahydrofuran (2.27 mL, 420 mm) at −78 °C. The resulting mixture was stirred for 40 min. A solution of ketone rac‐17 b (50.0 mg, 227 μmol, 1.00 equiv) in tetrahydrofuran (2.27 mL, 100 mm) was added dropwise to the freshly prepared lithium diisopropylamide solution at −78 °C. After 8.5 h, a solution of Comins reagent (401 mg, 1.02 mmol, 4.50 equiv) in tetrahydrofuran (1.02 mL, 1.00 m) was added dropwise to the enolate solution, during which the solution turned deep brown. After 5 min, the reaction mixture was allowed to warm to room temperature in the course of 30 min. The brown reaction mixture was transferred to a column packed with deactivated, neutral alumina*, the reaction vessel was rinsed with dichloromethane and the reaction mixture was filtered with a solvent mixture (P/CH2Cl2=15:1). The product containing fractions were combined and after removal of the solvent in vacuo, the residue was purified by column chromatography (deactivated neutral alumina, P/CH2Cl2=15:1) three consecutive times in order to remove residual Comins reagent. Triflate rac‐18 b (63.8 mg, 181 μmol, 80 %) was obtained as a colorless oil. [*Deactivation is described in the general information.] R f=0.47 (pentane) [CAM, KMnO4]; 1H NMR (500 MHz, C6D6, 25 °C, TMS): δ=0.85 (d, 3 J=5.6 Hz, 3 H, Me‐1), 0.95 (s, 3 H, Me‐4α), 0.97 (s, 3 H, Me‐4β), 1.26–1.36 (m, 4 H, HH‐1, HH‐2, HH‐3, HH‐8), 1.40–1.52 (m, 2 H, HH‐3, HH‐8), 1.55–1.61 (m, 2 H, HH‐2, H‐3a), 1.62 (s, 3 H, Me‐6), 1.72 (virt. dt, 2 J=16.7 Hz, 3 J 1 ≈3 J 2=6.1 Hz, 1 H, HH‐7), 1.90 (virt. dt, 2 J=16.7 Hz, 3 J 1 ≈3 J 2=6.7 Hz, 1 H, HH‐7), 2.49 ppm (br s, 1 H, H‐4a); 13C NMR (126 MHz, CDCl3, 27 °C, TMS): δ=13.0 (q, Me‐1), 17.2 (q, Me‐6), 23.9 (q, Me‐4β), 26.5 (t, C‐3), 26.7 (q, Me‐4α), 30.5 (t, C‐7), 31.0 (t, C‐8), 35.7 (t, C‐2), 35.8 (s, C‐4), 43.2 (d, C‐4a), 44.6 (d, C‐1), 48.1 (s, C‐8a), 52.8 (d, C‐3a), 119.1 (qs, 1 J CF=320 Hz, CF3), 128.6 (s, C‐6)*, 145.5 ppm (s, C‐5) [*The 13C signal of C‐6 overlaps with the solvent signal of C6D6. However, the signal can be located with the help of a HMBC crosspeak with the proton signal of Me‐6 to assign the 13C signal of C‐6.]; 19F NMR (376 MHz, C6D6, 25 °C, TMS): δ=−75.4 (s, 3 F, CF3); IR (ATR): ṽ=2953 (m, sp3‐CH), 2870 (m, sp3‐CH), 1410 (s, SO), 1201 (vs., sp3‐CF), 1141 (vs., sp3‐CF), 898 cm−1 (vs., SO); MS (EI, 70 eV): m/z (%): 352 (51) [M]+, 308 (14), 281 (74), 266 (100), 252 (14), 220 (17), 205 (68), 187 (64), 159 (56), 145 (73), 109 (39), 82 (55), 55 (42) [C4H7]+; HRMS (EI, 70 eV): calcd for C16H23O3F3 32S [M]+: 352.1315; found: 352.1310; calcd for C15 13CH23O3F3 32S [M]+: 353.1348; found: 353.1344.
Italicene (rac‐19 a): Palladium(II) acetate (3.91 mg, 17.0 μmol, 10.0 mol %) was added to a solution of triflate rac‐18 a (59.8 mg, 170 μmol, 1.00 equiv), triphenylphosphine (13.4 mg, 50.9 μmol, 30.0 mol %) and lithium formate monohydrate (59.4 mg, 848 μmol, 5.00 equiv) in dimethylformamide (3.39 mL, 50.0 mm). The resulting mixture was heated to 60 °C. The reaction mixture turned black in 7 min. After stirring for 20 min, the reaction mixture was allowed to cool to room temperature. The suspension was transferred to a column packed with deactivated, neutral alumina*, the reaction vessel was rinsed with dichloromethane and the reaction mixture was filtered with pentane. The product containing fractions were combined and after removal of the solvent in vacuo, the residue was purified by column chromatography (deactivated neutral alumina, pentane) three consecutive times to remove residual triphenylphosphine. In order to remove pentane completely without losing too much of the volatile product rac‐19 a, the vessel was evacuated at room temperature to 100 mbar and loaded with air in sequence five times. The title compound rac‐19 a (31.5 mg, 154 μmol, 91 %) was obtained as a colorless oil. [*Deactivation is described in the general information.] R f=0.71 (pentane) [CAM, KMnO4]; 1H NMR (500 MHz, CDCl3, 25 °C, TMS): δ=0.78 (d, 3 J=7.2 Hz, 3 H, Me‐1), 0.91 (s, 3 H, Me‐4β), 0.96 (s, 3 H, Me‐4α), 1.46 (dd, 2 J=12.4 Hz, 3 J=6.9 Hz, 1 H, HH‐2α), 1.53–1.59 (m, 1 H, HH‐3β), 1.60–1.69 (m, 2 H, HH‐3α, HH‐8), 1.69–1.76 (m, 5 H, H‐1, H‐3a, Me‐6), 1.76–1.81 (m, 2 H, H‐7), 1.84 (virt. dt, 2 J=12.8 Hz, 3 J 1 ≈3 J 2=3.5 Hz, 1 H, HH‐8), 1.88 (br s, 1 H, H‐4a), 2.02 (virt. tt, 2 J ≈3 J 1=12.3 Hz, 3 J 2 ≈3 J 3=7.0 Hz, 1 H, HH‐2β), 5.30–5.34 ppm (m, 1 H, H‐5); 13C NMR (126 MHz, CDCl3, 27 °C, TMS): δ=16.6 (q, Me‐1), 24.5 (q, Me‐6), 24.9 (t, C‐3), 24.9 (q, Me‐4β), 27.2 (q, Me‐4α), 27.8 (t, C‐7), 28.1 (t, C‐8), 34.8 (s, C‐4), 35.0 (t, C‐2), 39.7 (d, C‐1), 45.4 (s, C‐8a), 48.0 (d, C‐4a), 51.5 (d, C‐3a), 121.1 (d, C‐5), 136.2 ppm (s, C‐6); The analytical data obtained matched those reported in the literature.39
Isoitalicene (rac‐19 b): Palladium(II) acetate (4.06 mg, 18.1 μmol, 10.0 mol %) was added to a solution of triflate rac‐18 b (63.8 mg, 181 μmol, 1.00 equiv), triphenylphosphine (14.3 mg, 54.3 μmol, 30.0 mol %) and lithium formate monohydrate (63.3 mg, 905 μmol, 5.00 equiv) in dimethylformamide (3.62 mL, 50.0 mm). The resulting mixture was heated to 60 °C. The reaction mixture turned black in 10 min. After stirring for 20 min, the reaction mixture was allowed to cool to room temperature. The suspension was transferred to a column packed with deactivated, neutral alumina*, the reaction vessel was rinsed with dichloromethane and the reaction mixture was filtered with pentane. The product containing fractions were combined and after removal of the solvent in vacuo, the residue was purified by column chromatography (deactivated neutral alumina, pentane) three consecutive times to remove residual triphenylphosphine. In order to remove pentane completely and avoid any loss of the volatile product rac‐19 b, the vessel was evacuated at room temperature to 100 mbar and loaded with air in sequence five times. The title compound rac‐19 b (35.8 mg, 175 μmol, 97 %) was obtained as a colorless oil. [*Deactivation is described in the general information.] R f=0.71 (pentane) [CAM, KMnO4]; 1H NMR (500 MHz, CDCl3, 25 °C, TMS): δ=0.82 (d, 3 J=6.3 Hz, 3 H, Me‐1), 0.90 (s, 3 H, Me‐4β), 0.91 (s, 3 H, Me‐4α), 1.39–1.67 (m, 5 H, H‐1, HH‐2, H‐3, HH‐8), 1.68–1.77 (m, 5 H, HH‐2, H‐3a, Me‐6), 1.82 (ddd, 2 J=14.9 Hz, 3 J 1=9.5 Hz, 3 J 2=5.5 Hz, 1 H, HH‐8), 1.90 (virt. dt, 2 J=16.5 Hz, 3 J 1 ≈3 J 2=5.8 Hz, 1 H, HH‐7), 1.94–2.02 (m, 2 H, H‐4a, HH‐7), 5.36–5.40 (m, 1 H, H‐5); 13C NMR (126 MHz, CDCl3, 27 °C, TMS): δ=13.5 (q, Me‐1), 24.1 (q, Me‐4β), 24.5 (q, Me‐6), 26.5 (t, C‐3), 27.2 (q, Me‐4α), 28.6 (t, C‐7), 33.1 (t, C‐8), 35.2 (s, C‐4), 36.3 (t, C‐2), 41.0 (d, C‐4a), 43.9 (s, C‐8a), 45.0 (d, C‐1), 53.6 (d, C‐3a), 121.8 (d, C‐5), 135.7 (s, C‐6); The analytical data obtained matched those reported in the literature.39
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
Financial support by the European Research Council under the European Union's Horizon 2020 research and innovation programme (grant agreement No 665951‐ELICOS) and by the Fonds der Chemischen Industrie (Liebig fellowship to G.S.) is gratefully acknowledged. We thank O. Ackermann and J. Kudermann for their help with the HPLC and GLC analyses. Dr. C. Brenninger is acknowledged for his help in the preparation and characterization of products cis‐5 k and trans‐5 k. The authors gratefully acknowledge the computing and data resources provided by the Leibniz Supercomputing Centre (www.lrz.de).
S. Poplata, A. Bauer, G. Storch, T. Bach, Chem. Eur. J. 2019, 25, 8135.
References
- 1.Reviews covering intramolecular [2+2] photocycloaddition reactions:
- 1a. Becker D., Haddad N., Org. Photochem. 1989, 10, 1–162; [Google Scholar]
- 1b. Margaretha P., in Synthetic Organic Photochemistry, Molecular and Supramolecular Photochemistry, Vol. 12 (Eds.: A. G. Griesbeck, J. Mattay), Dekker, New York, 2005, pp. 211–237; [Google Scholar]
- 1c. Poplata S., Tröster A., Zou Y.-Q., Bach T., Chem. Rev. 2016, 116, 9748–9815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Reviews:
- 2a. Iriondo-Alberdi J., Greaney M. F., Eur. J. Org. Chem. 2007, 4801–4815; [Google Scholar]
- 2b. Hoffmann N., Chem. Rev. 2008, 108, 1052–1103; [DOI] [PubMed] [Google Scholar]
- 2c. Bach T., Hehn J. P., Angew. Chem. Int. Ed. 2011, 50, 1000–1045; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2011, 123, 1032–1077. [Google Scholar]
- 3.
- 3a. Schalk O., Schuurman M. S., Wu G., Lang P., Mucke M., Feifel R., Stolow A., J. Phys. Chem. A 2014, 118, 2279–2287; [DOI] [PubMed] [Google Scholar]
- 3b. Riedle E., Bradler M., Wenninger M., Sailer C. F., Pugliesi I., Faraday Discuss. 2013, 163, 139–158. [DOI] [PubMed] [Google Scholar]
- 4. Schuster D. I., in CRC Handbook of Photochemistry and Photobiology (Eds.: W. M. Horspool, F. Lenci), CRC Press, Boca Raton, 2004, pp. 72/1–72/24. [Google Scholar]
- 5. Maradyn D. J., Weedon A. C., J. Am. Chem. Soc. 1995, 117, 5359–5360. [Google Scholar]
- 6. Ciamician G., Silber P., Ber. Dtsch. Chem. Ges. 1908, 41, 1928–1935. [Google Scholar]
- 7.
- 7a. Sernagiotto E., Gazz. Chim. Ital. 1917, 47, 153–159; [Google Scholar]
- 7b. Sernagiotto E., Gazz. Chim. Ital. 1918, 48, 52–61. [Google Scholar]
- 8. Büchi G., Goldman I. M., J. Am. Chem. Soc. 1957, 79, 4741–4748. [Google Scholar]
- 9. Meinwald J., Schneider R. A., J. Am. Chem. Soc. 1965, 87, 5218–5229. [Google Scholar]
- 10.
- 10a. Tolbert L. M., Ali M. B., J. Am. Chem. Soc. 1982, 104, 1742–1744; [Google Scholar]
- 10b. Lange G. L., Decicco C., Tan S. L., Chamberlain G., Tetrahedron Lett. 1985, 26, 4707–4710; [Google Scholar]
- 10c. Herzog H., Koch H., Scharf H.-D., Runsink A. J., Tetrahedron 1986, 42, 3547–3558; [Google Scholar]
- 10d. Inoue Y., Chem. Rev. 1992, 92, 741–770; [Google Scholar]
- 10e. Chen C., Chang V., Cai X., Duesler E., Mariano P. S., J. Am. Chem. Soc. 2001, 123, 6433–6434; [DOI] [PubMed] [Google Scholar]
- 10f. Faure S., Piva-Le-Blanc S., Bertrand C., Pete J.-P., Faure R., Piva O., J. Org. Chem. 2002, 67, 1061–1070; [DOI] [PubMed] [Google Scholar]
- 10g. Inhülsen I., Akiyama N., Tsutsumi K., Nishiyama Y., Kakiuchi K., Tetrahedron 2013, 69, 782–790. [Google Scholar]
- 11.
- 11a. Brimioulle R., Bach T., Science 2013, 342, 840–843; [DOI] [PubMed] [Google Scholar]
- 11b. Brimioulle R., Bauer A., Bach T., J. Am. Chem. Soc. 2015, 137, 5170–5176. [DOI] [PubMed] [Google Scholar]
- 12.For recent reviews covering enantioselective [2+2] photocycloaddition reactions, see:
- 12a. Xu Y., Conner M. L., Brown M. K., Angew. Chem. Int. Ed. 2015, 54, 11918–11928; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 12086–12097; [Google Scholar]
- 12b. Brimioulle R., Lenhart D., Maturi M. M., Bach T., Angew. Chem. Int. Ed. 2015, 54, 3872–3890; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 3944–3963. [Google Scholar]
- 13.For alternative approaches towards enantioselective intramolecular [2+2] photocycloaddition reactions, see:
- 13a. Müller C., Bauer A., Bach T., Angew. Chem. Int. Ed. 2009, 48, 6640–6642; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2009, 121, 6767–6769; [Google Scholar]
- 13b. Müller C., Bauer A., Maturi M. M., Cuquerella M. C., Miranda M. A., Bach T., J. Am. Chem. Soc. 2011, 133, 16689–16697; [DOI] [PubMed] [Google Scholar]
- 13c. Du J., Skubi K. L., Schultz D. M., Yoon T. P., Science 2014, 344, 392–396; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13d. Vallavoju N., Selvakumar S., Jockusch S., Sibi M. P., Sivaguru J., Angew. Chem. Int. Ed. 2014, 53, 5604–5608; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 5710–5714; [Google Scholar]
- 13e. Skubi K. L., Kidd J. B., Jung H., Guzei I. A., Baik M.-H., Yoon T. P., J. Am. Chem. Soc. 2017, 139, 17186–17192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Guerry P., Blanco P., Brodbeck H., Pasteris O., Neier R., Helv. Chim. Acta 1991, 74, 163–178. [Google Scholar]
- 15. Brenninger C., Jolliffe J. D., Bach T., Angew. Chem. Int. Ed. 2018, 57, 14338–14349; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 14536–14547. [Google Scholar]
- 16. Brimioulle R., Bach T., Angew. Chem. Int. Ed. 2014, 53, 12921–12924; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 13135–13138. [Google Scholar]
- 17. Poplata S., Bach T., J. Am. Chem. Soc. 2018, 140, 3228–3231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Review: Corey E. J., Angew. Chem. Int. Ed. 2009, 48, 2100–2117; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2009, 121, 2134–2151. [Google Scholar]
- 19. Dilling W. L., Chem. Rev. 1966, 66, 373–393. [Google Scholar]
- 20. Mattay J., Banning A., Bischof E. W., Heidbreder A., Runsink J., Chem. Ber. 1992, 125, 2119–2127. [Google Scholar]
- 21.
- 21a. Tsunoda T., Suzuki M., Noyori R., Tetrahedron Lett. 1980, 21, 1357–1358; [Google Scholar]
- 21b. Noyori R., Murata S., Suzuki M., Tetrahedron 1981, 37, 3899–3910. [Google Scholar]
- 22.For the reaction set-up, see ref. [17].
- 23. Sparr C., Tanzer E.-M., Bachmann J., Gilmour R., Synthesis 2010, 1394–1397. [Google Scholar]
- 24.
- 24a. Mahender Reddy K., Bhimireddy E., Thirupathi B., Breitler S., Yu S., Corey E. J., J. Am. Chem. Soc. 2016, 138, 2443–2453; [DOI] [PubMed] [Google Scholar]
- 24b. Brimioulle R., Guo H., Bach T., Chem. Eur. J. 2012, 18, 7552–7560; [DOI] [PubMed] [Google Scholar]
- 24c. Conner M. L., Xu Y., Brown M. K., J. Am. Chem. Soc. 2015, 137, 3482–3485. [DOI] [PubMed] [Google Scholar]
- 25.
- 25a. Dale J. A., Mosher H. S., J. Am. Chem. Soc. 1973, 95, 512–519; [Google Scholar]
- 25b. Hoye T. R., Jeffrey C. S., Shao F., Nat. Protoc. 2007, 2, 2451–2458. [DOI] [PubMed] [Google Scholar]
- 26. Pellicori S. F., Appl. Opt. 1964, 3, 361–366. [Google Scholar]
- 27.
- 27a. Ryu D. H., Lee T. W., Corey E. J., J. Am. Chem. Soc. 2002, 124, 9992–9993; [DOI] [PubMed] [Google Scholar]
- 27b. Ryu D. H., Corey E. J., J. Am. Chem. Soc. 2003, 125, 6388–6390; [DOI] [PubMed] [Google Scholar]
- 27c. Wiest J. M., Conner M. L., Brown M. K., J. Am. Chem. Soc. 2018, 140, 15943–15949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.
- 28a. Paddon-Row M. N., Anderson C. D., Houk K. N., J. Org. Chem. 2009, 74, 861–868; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28b. Sakata K., Fujimoto H., J. Org. Chem. 2013, 78, 3095–3103. [DOI] [PubMed] [Google Scholar]
- 29.Gaussian 16, Revision B.01, M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman, D. J. Fox, Gaussian, Inc., Wallingford CT, 2016.
- 30.
- 30a. Stephens P. J., Devlin F. J., Chabalowski C. F., Frisch M. J., J. Phys. Chem. 1994, 98, 11623–11627; [Google Scholar]
- 30b. Becke A. D., J. Chem. Phys. 1993, 98, 5648–5652; [Google Scholar]
- 30c. Lee C., Yang W., Parr R. G., Phys. Rev. B 1988, 37, 785–789; [DOI] [PubMed] [Google Scholar]
- 30d. Grimme S., Ehrlich S., Goerigk L., J. Comput. Chem. 2011, 32, 1456–1465. [DOI] [PubMed] [Google Scholar]
- 31. Kendall R. A., Dunning T. H., Harrison R. J., J. Phys. Chem. 1992, 96, 6796–6806. [Google Scholar]
- 32. Paton R. S., Org. Biomol. Chem. 2014, 12, 1717–1720. [DOI] [PubMed] [Google Scholar]
- 33. Paddon-Row M. N., Kwan L. C. H., Willis A. C., Sherburn M. S., Angew. Chem. Int. Ed. 2008, 47, 7013–7017; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2008, 120, 7121–7125. [Google Scholar]
- 34.This coordination pattern of the enone substrate was observed to occur concomitant to energetically favoring pyrrolidine ring inversion.
- 35.
- 35a. El-Sayed M. A., Acc. Chem. Res. 1968, 1, 8–16; [Google Scholar]
- 35b. Klán P., Wirz J., Photochemistry of Organic Compounds; Wiley, Chichester, 2009, pp. 38–39. [Google Scholar]
- 36.
- 36a. Becker D., Nagler M., Sahali Y., Haddad N., J. Org. Chem. 1991, 56, 4537–4543; [Google Scholar]
- 36b. Gleiter R., Fischer E., Chem. Ber. 1992, 125, 1899–1911. [Google Scholar]
- 37. Wang H., Cao X., Chen X., Fang W., Dolg M., Angew. Chem. Int. Ed. 2015, 54, 14295–14298; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 14503–14506. [Google Scholar]
- 38.Lifetimes of 1,4-diradicals are in the range of 10–100 ns which appears to be a tentative lower limit for the association of enone intermediates to the Lewis acid:
- 38a. Becker D., Haddad N., Sahali Y., Tetrahedron Lett. 1989, 30, 2661–2664; [Google Scholar]
- 38b. Rudolph A., Weedon A. C., Can. J. Chem. 1990, 68, 1590–1597; [Google Scholar]
- 38c. Kaprinidis N. A., Lem G., Courtney S. H., Schuster D. I., J. Am. Chem. Soc. 1993, 115, 3324–3325. [Google Scholar]
- 39. Leimner J., Marschall H., Meier N., Weyerstahl P., Chem. Lett. 1984, 13, 1769–1772. [Google Scholar]
- 40.
- 40a. Ihara M., Ohnishi M., Takano M., Makita K., Taniguchi N., Fukumoto K., J. Am. Chem. Soc. 1992, 114, 4408–4410; [Google Scholar]
- 40b. Ihara M., Taniguchi T., Makita K., Takano M., Ohnishi M., Taniguchi N., Fukumoto K., Kabuto C., J. Am. Chem. Soc. 1993, 115, 8107–8115; [Google Scholar]
- 40c. Weyerstahl P., Marschall H., Christiansen C., Seelmann I., Liebigs Ann. 1996, 1641–1644; [Google Scholar]
- 40d. Harada Y., Maki S., Niwa H., Hirano T., Yamamura S., Synlett 1998, 1313–1314; [Google Scholar]
- 40e. Faure S., Piva O., Tetrahedron Lett. 2001, 42, 255–259. [Google Scholar]
- 41. Honda T., Ueda K., Tsubuki M., Toya T., Kurozumi A., J. Chem. Soc. Perkin Trans. 1 1991, 1749–1754. [Google Scholar]
- 42. Hoye T. R., Martin S. J., Peck D. R., J. Org. Chem. 1982, 47, 331–337. [Google Scholar]
- 43.
- 43a. Birch A. M., Pattenden G., J. Chem. Soc. Chem. Commun. 1980, 1195–1197; [Google Scholar]
- 43b. Oppolzer W., Zutterman F., Bättig K., Helv. Chim. Acta 1983, 66, 522–533. [Google Scholar]
- 44. Comins D. L., Dehghani A., Tetrahedron Lett. 1992, 33, 6299–6302. [Google Scholar]
- 45.Lithium formate was used as reducing agent in order to avoid partial decomposition of triflates rac-18 a and rac-18 b which was observed when using formic acid/trialkylamine salts as reducing agents: Cacchi S., Morera E., Ortar G., Tetrahedron Lett. 1984, 25, 4821–4824. [Google Scholar]
- 46. Fürstner A., Hannen P., Chem. Eur. J. 2006, 12, 3006–3019. [DOI] [PubMed] [Google Scholar]
- 47. Brenninger C., Pöthig A., Bach T., Angew. Chem. Int. Ed. 2017, 56, 4337–4341; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 4401–4405. [Google Scholar]
- 48. Matlin A. R., George C. F., Wolff S., Agosta W. C., J. Am. Chem. Soc. 1986, 108, 3385–3394. [Google Scholar]
- 49. Hörmann F. M., Chung T. S., Rodriguez E., Jakob M., Bach T., Angew. Chem. Int. Ed. 2018, 57, 827–831; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 835–839. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary