

Abstract
Significantly increased production of biobased polymers is a prerequisite to replace petroleum‐based materials towards reaching a circular bioeconomy. However, many renewable building blocks from wood and other plant material are not directly amenable for polymerization, due to their inert backbones and/or lack of functional group compatibility with the desired polymerization type. Based on a retro‐biosynthetic analysis of polyesters, a chemoenzymatic route from (−)‐α‐pinene towards a verbanone‐based lactone, which is further used in ring‐opening polymerization, is presented. Generated pinene‐derived polyesters showed elevated degradation and glass transition temperatures, compared with poly(ϵ‐decalactone), which lacks a ring structure in its backbone. Semirational enzyme engineering of the cyclohexanone monooxygenase from Acinetobacter calcoaceticus enabled the biosynthesis of the key lactone intermediate for the targeted polyester. As a proof of principle, one enzyme variant identified from screening in a microtiter plate was used in biocatalytic upscaling, which afforded the bicyclic lactone in 39 % conversion in shake flask scale reactions.
Keywords: biopolymers, enzymes, lactones, retrosynthesis, terpene
Polymeric materials are of immense relevance for our everyday life and society in application areas that include light‐weight metal replacements, textiles, packaging, adhesives, coatings, electronics, aerospace, and medicine. Approximatively 300 megatons of synthetic polymers are manufactured annually;1 a number that is projected to grow significantly in the near future.1a, 2 At present, only 1 % of all man‐made synthetic materials are generated from renewable resources.1 Hence, current manufacturing of polymers is dependent on exhaustive use of petroleum‐based virgin synthons; a process associated with severe impacts on our environment and climate.
Chemical retrosynthesis,3 which was initially developed by Corey, has revolutionized synthetic chemistry: by disconnecting complex target molecules into readily accessible fragments, which can be reassembled (in the synthetic direction) by C−X bond formations and/or functional group interconversions (FGIs), their production is facilitated. In analogy, chemical retrosynthesis applied to the polymer field readily identifies cyclic ketones as potent building blocks for polyesters through Baeyer–Villiger (BV)4 oxidation and ring‐opening polymerization (ROP; Scheme 1, top).1a, 5 There is tremendous potential to incorporate biocatalysis into retrosynthesis, to expand the available reaction space,6 as initially suggested by Turner and O'Reilly7 and reviewed more recently.8 Herein, we present a retro‐biosynthesis‐based route to generate pinene‐derived polyesters by chemoenzymatic catalysis (Scheme 1). Pinenes and other monoterpenes are accessible from nonarable land and processes that do not compete with food production (e.g., forestry).1b Their commercial availability amounts to 300 000 tons per year globally.9 Furthermore, there is an untapped potential to acquire 3 from pulp and paper processing and the wood industry. Per ton of Nordic bleached sulfate Kraft pulp, about 1–2 kg turpentine are produced. Hence, the amount of turpentine accessible in the Nordic countries alone can be estimated as in the range of 15–20 000 tons annually. Improved recovery of turpentine could increase these numbers significantly. A large portion of turpentine produced is used as an internal fuel.
Scheme 1.
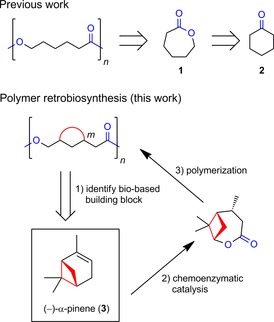
Chemical retrosynthetic analysis applied to polymers (top) is exemplified for poly(ϵ‐caprolactone). Its corresponding monomer fragment, ϵ‐caprolactone (1), can be generated from cyclohexanone (2) by FGI. Polymer retro‐biosynthesis herein (bottom) is investigated by targeting a polyester with a ring (colored in red) incorporated in its backbone. An identified, and potentially suitable, renewable building block harboring a cyclobutane ring (corresponding to m=4) is shown in the box. The envisioned transformation into the key intermediate is indicated. Relevant functional groups are highlighted in blue.
Incorporating ring structures into monomers tailors the biophysical and mechanical properties of the corresponding macromolecules by increasing their rigidity.10 Polymers harboring rings in their backbones (represented by 4–7; Scheme 2 A) are thus a contemporary research area.1b, 11 Potential renewable starting materials for the production of the latter include cellulose, hemicellulose, lignin, and terpenes.1, 12 In particular, terpenes have, in recent years, emerged as an important pool of renewable carbons for biochemicals and ‐polymers.1b, 11j, 12a The multicyclic skeleton of terpene metabolites13 is of specific interest for the generation of biobased polymers with pendant rings, for example, limonene‐derived polycarbonates11e 5 (Scheme 2 A). Activation of inert11j carbocyclic terpenes by oxidation11d, 11e, 14 and FGI11h is often required to generate activated monomers amenable for polymer production. Herein, as a case study, we performed a retro‐biosynthetic analysis for polyesters harboring rings as backbone motifs (Scheme 1). We envisioned that polyesters with rings as backbone motifs could be afforded through activation of bicyclic monoterpene scaffolds into lactones by FGI (Scheme 1, bottom). According to our hypothesis, valorization of 3 would result in the protruding ring remaining intact in the generated biobased polyester. The latter is in contrast to (−)‐menthide,11g and other previously investigated terpene‐based cyclic esters,1a, 11j which give rise to polyester chains that lack a ring structure in the backbone upon ROP.
Scheme 2.
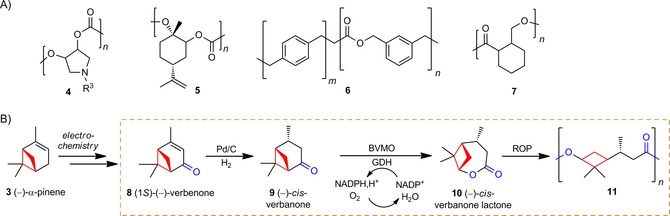
A) Representative polymers with rings incorporated into their backbones.11⋅B) Chemoenzymatic synthesis of (−)‐α‐pinene‐derived lactone 10 and polyester 11 harboring a cyclobutane unit. The focus of this work is shown in the box. BVMO: Baeyer–Villiger monooxygenase, GDH: glucose dehydrogenase. For clarity, only the most substituted (i.e., normal) lactone 10 is shown and substrates for cofactor regeneration by GDH are omitted.
Based on this strategy, we present a chemoenzymatic pathway to upgrade 3 into a verbanone‐based lactone 10 amenable for ROP, to generate polyester 11 with a cyclobutane ring in its backbone (Schemes 1 and 2). This pathway is based on several FGIs and associated oxidations/reductions: 1) allylic oxidation of 3, 2) reduction of the ene group to afford saturated ketone 9, 3) BV oxidation to generate lactone 10, and 4) ROP to yield polyester 11.
Efficient generation of the insecticide 8, starting from 3, has recently been reported through benign chemical synthetic methods.15 It is noted that 9 is readily accessible through the chemical reduction of 8 (see below). For polymer retro‐biosynthesis, it is important to demonstrate that the production of key intermediates through enzyme catalysis is, in principle, possible. P450‐catalyzed oxidation of α‐pinene into 8 has been described in the literature.16 In this investigation, we focused on the enzymatic transformation of ketone 9 into lactone 10. Inspired by the fact that bulky bi‐ and macrocyclic lactones are abundant in nature,17 we reasoned that BVMOs constitute suitable biocatalysts to catalyze the biotransformation of 9 into 10. We turned our attention to the cyclohexanone monooxygenase from Acinetobacter calcoaceticus (CHMOAcineto).18 More specifically, we employed a quadruple mutant of CHMOAcineto with higher stability19 during biocatalysis (abbreviated herein as CHMOAcineto_QM; sequences are given in the Supporting Information). To facilitate the presumably challenging conversion of 9 into the bulky and bicyclic lactone 10, enzyme engineering guided by molecular modeling was utilized. In the absence of an available crystal structure of CHMO from A. calcoaceticus, homology modeling was performed by using the “tight” conformation of the cyclohexanone monooxygenase from Rhodococcus sp. HI‐31 as a template (PDB ID: 4RG320). The tight conformation corresponds to an important structural snapshot along the reaction coordinate for BVMOs21 and has previously been used as a framework to engineer and decipher (regio‐)selectivity for this enzyme family.20, 22 From a basal energy minimization of the homology model, eight amino acids were selected as potential hot spots (orange; Figure 1) based on a side chain pointing towards the protruding methyl groups and their residues being within 6 Å heavy‐atom distance of the modeled lactone product.
Figure 1.
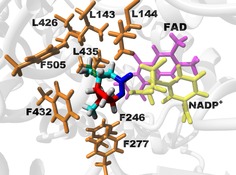
Energy‐minimized homology model of CHMOAcineto_QM (gray), complexed with lactone 10 (shown in enlarged sticks). The cyclobutane ring of the product is highlighted in red and the ester group is shown in blue. Identified hot spot amino acids are in orange sticks. For clarity, only parts of the cofactors (FAD in magenta and nicotinamide adenine dinucleotide phosphate (NADP+) in yellow) are shown.
Mutations of identified residues generated a small enzyme library of 26 variants. Single, double, and triple variants were expressed in Escherichia coli BL21(DE3). Although all variants were expressed in soluble form, F505W, L435F, L426A/L435A, L435A/F505L, L144A/L435A, L143A/L435A, L143A/L426A, L143A/L144A/L435A, L143A/L426A/L435A, and L143A/L435A/F505L showed higher expression than that of CHMOAcineto_QM (Figure S1 in Supporting Information). The extracted cell‐free lysates were tested in terms of reduced nicotinamide adenine dinucleotide phosphate (NADPH) consumption in the presence of the natural substrate, 2 (Table 1), and experimentally screened for the oxidation of 9, in the presence of the GDH cofactor regeneration system.
Table 1.
Screening of BV oxidations of ketones 2 and 9 by the cell lysate of recombinant E. coli containing CHMOAcineto_QM variants.[a]
| Enzyme variant | Volumetric activity on 2 [U mL−1] |
Conversion of 9
[%] |
|---|---|---|
| CHMOAcineto_QM | 1.01±0.07 | – |
| F432A | 0.36±0.01 | – |
| F277A/F432A | 0.05±0.01 | – |
| F505A | 0.99±0.04 | – |
| F277A | 0.19±0.01 | 0.78±0.11 |
| L435A | 2.06±0.28 | 2.29±0.57 |
| L143A | 1.30±0.04 | – |
| F505W | 0.51±0.02 | – |
| L143F | 1.01±0.16 | – |
| L435F | 0.08±0.01 | – |
| F246A | 0.42±0.01 | 1.16±0.05 |
| L426A/L435A | 1.19±0.03 | 2.27±0.87 |
| L144A/F505L | 0.11±0.01 | – |
| L435A/F505L | 0.96±0.04 | – |
| L143A/F505L | 1.70±0.02 | – |
| L144A/L435A | 0.17±0.01 | – |
| L143A/L144A | 0.10±0.01 | – |
| L143A/L435A | 1.17±0.04 | 1.40±0.03 |
| L426A | 1.46±0.04 | 4.92±1.21 |
| L144A | 0.15±0.02 | – |
| F505L | 1.28±0.68 | – |
| L143A/L426A | 0.94±0.57 | – |
| L144A/L426A | 0.08±0.00 | 0.81±0.21 |
| L426A/F505L | 0.94±0.02 | – |
| L143A/L144A/L435A | 0.19±0.01 | – |
| L143A/L426A/L435A | 0.61±0.02 | 0.83±0.03 |
| L143A/L435A/F505L | 0.62±0.03 | 0.75±0.05 |
[a] Volumetric activity is given for 2 and conversion data is given for 9. Standard deviations are shown (n=3).
Interestingly, all variants possessing the side‐chain variation L144A showed lower activity on the natural substrate 2 compared with that of CHMOAcineto_QM. The L144 residue is present in proximity to the flavin adenine dinucleotide (FAD) and NADPH binding pocket. The variation of this amino acid to a smaller residue, such as Ala, could destabilize the optimal fitting of the cofactors, especially in the presence of 2. Similarly, the side‐chain variation F277A caused significant loss of activity towards 2, as shown in both the F277A and F277A/F432A variants. In the latter case, the addition of the variation F432A further reduced the activity, which was in line with the fact that the F432A single variant displayed lower activity than that of CHMOAcineto_QM. It is possible that replacing Phe residues in proximity of FAD/NADPH leads to suboptimal positioning of the cofactors. However, the substitution of L435 to the smaller side chain Ala (L435A) increased the activity of the enzyme twofold towards 2, whereas, in the presence of the bulkier residue Phe (L435F), the activity was dramatically reduced. This substitution could possibly displace the FAD cofactor to reduce the enzymatic activity. The combination of L435A together with the benign substitution L426A did not increase the activity of the variant L426A/L435A on the substrate 2.
To determine if the different activities shown on 2 of the variants could lead to higher activity on the bulkier substrate 9, the variants were tested for the biocatalytic conversion of 9 into 10. Whereas CHMOAcineto_QM showed no conversion (Table 1) under our screening conditions (in a deep‐well plate under suboptimal oxygen supply conditions;23 see the Experimental Section), several variants that readily oxidized 9 into 10 were identified. Specifically, the variant L426A showed a conversion of 5 % of 9 into 10, which was the highest among all 26 variants. We were pleased to find that scaling up of the L426A‐catalyzed reaction to a 2 L baffled conical flask (see the Experimental Section), which allowed a higher availability of oxygen, resulted in a 39 % conversion of 9 in 24 h, corresponding to 25 mg of product formation according to GC analysis (spectra shown in Figure S2). Under the same conditions, the template variant CHMO_QM reached 7 % conversion. NMR spectroscopy analysis of the crude reaction mixture after scaling up of the enzymatic synthesis for the L426A variant (Figure S3) showed a relative abundance of 30 % of the extracted lactone; relating to roughly 24 mg of product. This finding underlines the high potential of optimizing the biocatalytic step further, based on hits from the generated library. The residue L426 is present in the substrate binding pocket and substitution to a smaller alanine at this position would allow the binding of bulkier substrate 9, and its conversion into 10. Although the construction of double and triple mutants carrying this substitution did not lead to higher conversion, all variants were able to convert 9, except variant L143A/L426A. The coexistence of these two substitutions could possibly create an unfavorable spacing in the active site of CHMOAcineto_QM. The activity was restored, although to a smaller extent, by the introduction of the substitution L435A, for which the single substitution also showed conversion (Table 1). The variants containing the substitution of F505 to different hydrophobic residues (Trp, Leu, and Ala) did not show any conversion of 9 into 10, except in the case of the triple variant L143A/L435A/F505L, which contained the two substitutions L143A and L435A that improved the conversion. Residue F505 is present in the substrate‐binding domain, but points away from the cofactor binding pocket. Modification to a leucine side chain probably increased the volume of the active site, which allowed 9 to be converted into 10 in the presence of L143A and L435A, creating enough space for the non‐natural substrate.
In addition to the enzymatic transformation, chemical BV oxidation was performed with m‐chloroperbenzoic acid (mCPBA) under an inert atmosphere in anhydrous dichloromethane. The product was obtained in 37 % yield and could easily be identified by the downfield shift of the epsilon proton (δ=2.6 to 4.3 ppm; Figures S4 and S5). Analysis of 10 was further corroborated by results from HRMS analysis (Figure S6). NMR spectroscopy and MS analysis confirmed the normal lactone 10. Chemically synthesized lactone 10 was then cationically polymerized by ROP with methane sulfonic acid (MSA), a catalyst considered to be environmentally benign,24 to investigate its potential as a monomer. To explore the reactivity of lactone 10 towards other monomers and its ability to form copolymers, it was also copolymerized together with ϵ‐decalactone, by using MSA as a catalyst and benzyl alcohol (BnOH) as an initiator. The monomer conversion was determined by means of 1H NMR spectroscopy of the crude reaction mixture by using the shift of the epsilon protons from δ=4.3 to 4.5 ppm, showing conversions above 90 % for all polymers. 2D NMR spectroscopy was performed to confirm the incorporation of the intact ring into the polymer backbone (Figures S7 and S8). The theoretical molecular weights, M n, were calculated by the ratio between the epsilon protons of the repeating unit (δ=4.5 ppm) and the terminal epsilon protons (δ=3.6 ppm) and corresponded well with M n measured by size exclusion chromatography (SEC; Table 1 and Figures S9 and S10). However, it should be noted that the ratio between the terminal epsilon protons and the benzylic protons (δ=5.2 ppm) indicate that less than 40 % of the chains are actually initiated from the initiator; the remainder are most likely opened by water or hydrolyzed lactones. This can be explained by the nonoptimized reaction conditions, which lead to the presence of residual traces of water that give rise to a higher number of initiated chains than that targeted ([M]/[I]). The incorporation of two different lactones into the copolymer was further analyzed by determining the ratio between the epsilon protons of the repeating units (δ=4.5 ppm for verbanone and δ=4.9 ppm for ϵ‐decalactone) and corresponded well with 49 % ϵ‐decalactone and 51 % 10 to the targeted one to one ratio (Figure S10).
To investigate the impact of the ring on the physiochemical properties, the synthesized polymers were analyzed by means of thermogravimetric analysis (TGA) and differential scanning calorimetry (DSC; Figure 2). TGA analysis showed enhanced thermal stability in the case of PϵVaL, relative to that of PϵDL. DSC analysis showed that PϵDL had a glass transition temperature (T g) of −57 °C, which was in good agreement with previously reported data.25 In the case of PϵVaL, the T g was markedly increased up to 25 °C, as expected. The degradation temperature (T d) and glass transition temperature (T g) of all polymers (defined at 5 % weight loss) are listed in Table 2.
Figure 2.
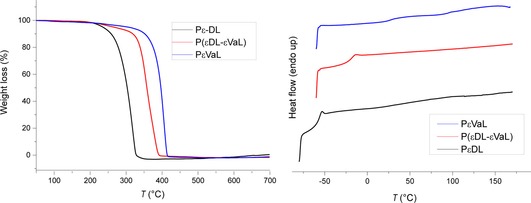
Thermograms of the synthesized polyesters (left: TGA; right: DSC). PϵVaL: poly(ϵ‐verbanone lactone), PϵDL: poly(ϵ‐decalactone), P(ϵDL‐ϵVaL): poly(ϵ‐decalactone–ϵ‐verbanone lactone).
Table 2.
Polymerization parameters and properties of generated biobased materials.
| Polymer | [M]/[BnOH]/ | Conversion | M n [g mol−1] | Ð | T g | T d (5 %) | |
|---|---|---|---|---|---|---|---|
| [MSA] | [%] | NMR | SEC | [°C] | [°C] | ||
| PϵDL | 100:1:2 | 93 | 3500 | 3000 | 1.1 | −57 | 227 |
| P(ϵDL‐ϵVaL) | (50:50):1:2 | 95 | 3400 | 3200 | 1.12 | −21 | 254 |
| PϵVaL | 100:1:2 | 94 | 3000 | 3300 | 1.14 | 26 | 306 |
Towards reaching a more sustainable society, generating biobased polymers from renewable feedstock has gained increasing attention.1, 12a This development is illustrated by the recent generation of biomaterials from pectin26 and limonene,11e which are minor components in apple and orange peels, respectively. According to European Bioplastics, the market for biobased polymers is predicted to grow annually by approximately 4 % in the coming years.27 In particular, polyesters are of high industrial significance1a, 2, 28 and can display favorable properties, in terms of their sustainability,1a if generated from renewable synthons. Rigid, thermally stable, symmetrical, biobased aliphatic molecules suitable for polymers are rare.29 The creation of rigid cycloaliphatic structures in aliphatic polyesters has the potential to significantly improve the thermal and mechanical properties of, for instance, poly(ϵ‐caprolactone)‐based materials for industrial use. We were able to produce a pinene‐derived polyester with enhanced thermal stability and higher glass transition temperatures relative to those of PϵDL. Furthermore, engineering of the enzyme CHMOAcineto_QM, which was a more stable variant than that of wild‐type CHMOAcineto, allowed us to convert the sterically demanding ketone 9 into lactone 10. Aligned with this finding, it has recently been shown that the CHMO from A. calcoaceticus can process bulky α,α‐dialkyl cyclic ketones.30 Further reaction optimization for enhanced oxygen supply,23 in combination with protein engineering, could increase the enzymatic transformation to reach industrially relevant conversion and amounts of the lactone. To the best of our knowledge, bicyclic α‐pinene‐derived lactone 10 has not yet been explored and is currently unknown in terpene metabolic pathways.31 Biocatalytic activation of inert building blocks, either by designed microorganisms or by in vitro approaches, associated with high‐turnover numbers,32 could allow for the generation of these remarkable building blocks on a larger scale. In particular, oxidoreductases have recently been highlighted as promising and emerging biocatalysts33 for the industrial generation of pharmaceuticals, agrochemicals, and platform chemicals. We believe that our concept of polymer retro‐biosynthesis has a high potential to enable the biobased production of a targeted polymer type.
Experimental Section
Chemicals and reagents: All chemicals and reagents used in this work were of analytical grade. Buffer components, FAD, and catalase from bovine liver were purchased from Sigma–Aldrich. Reduced NADPH was purchased from Roche. Dichloromethane was passed through a column packed with Al2O3 (activated, neutral, Brockmann Activity I) and stored over molecular sieves.
Preparation of (−)‐cis‐verbanone (9): Large‐scale synthesis of 9 was performed as follows: 5 % Pd on activated carbon (paste type 395, Johnson Matthey, 0.5 g) was added to 8 (35 mL, 228 mol) in a stirred pressure autoclave. The autoclave was sealed and flushed with hydrogen gas three times, then heated to 80 °C for 2 h. After the reaction mixture was cooled, it was filtered through Celite to give verbanone (31.2 g, 91 %; Figure S4) as a slightly yellow oil.
Small‐scale synthesis: The mixture obtained from large‐scale synthesis was dissolved in CH2Cl2 (0.5 m) followed by the addition of 10 % (w/w) of Pd/C. The round‐bottomed flask was sealed with a septum and stirred under H2 (≈3 bar) for 4 h. Afterwards, the solution was filtered through a p5 glass filter to remove the catalyst. The filtrate was concentrated to afford 9 in quantitative yields. Stereochemistry was evaluated from H–H coupling constants in the 1H NMR spectrum and confirmed by previous reports.34 1H NMR (400 MHz, CDCl3): δ=1.00 (s, 3 H), 1.16 (d, J=7.4 Hz, 3 H), 1.33 (s, 3 H), 1.39 (d, J=10.2 Hz, 1 H), 2.20–2.07 (m, 2 H), 2.42–2.29 (m, 1 H), 2.63–2.50 (m, 2 H), 2.86 ppm (dd, J=20.0, 10.8 Hz, 1 H).
Chemical lactone synthesis: mCPBA (2 equiv) was dissolved in anhydrous CH2Cl2, dried over magnesium sulfate, and concentrated in a round‐bottomed flask equipped with a magnetic stirrer under an inert atmosphere. Anhydrous CH2Cl2 (0.5 m) and 9 (1 equiv) were added to the flask. The reaction mixture was stirred for 18 h under reflux and an inert atmosphere. After 18 h, the reaction was cooled to room temperature and the resulting slurry was filtered. The filtrate was subsequently washed with saturated sodium bisulfite and saturated sodium bicarbonate, dried over magnesium sulfate, and concentrated. The organic phase was purified by means of MPLC (0 % EtOAc in heptane over 2CV then up to 30 % EtOAc in heptane over 8CV) and concentrated to afford the normal, most substituted lactone 10 as a colorless oil. HRMS analysis of 10 (Agilent Technologies gas chromatograph 6890 Series coupled to a GCT time of flight mass spectrometer, Waters) with methane as a reagent gas resulted in a measured mass of m/z 169.1225 (calcd: 169.1229) and the expected formula C10H17O2. 1H NMR (400 MHz, CDCl3): δ=1.14 (s, 3 H), 1.15 (d, 3 H), 1.31 (s, 3 H), 1.70 (dd, 1 H), 2.20–2.32 (m, 2 H), 2.69–2.85 (overlapping m, 2 H), 3.12–3.21 (m, 1 H), 4.3 ppm (dd, 1 H).
Polymer and monomer analysis: 1H (400 MHz) and 13C (100 MHz) NMR spectra were recorded with a Bruker Avance AM 400 spectrometer (USA). The signal of the deuterated solvent, CDCl3 (δ=7.26 ppm), was used as reference. For SEC, a TOSOH EcoSEC HLC‐8320GPC system (Japan), equipped with an EcoSEC RI detector and three PSS PFG 5 μm columns (microguard, 100 Å, and 300 Å; USA), was used. Poly(ethylene glycol) (PEG) standards were used for calibration and toluene was used as an internal standard. DSC was performed by using a Mettler Toledo DSC 820 module. Samples (5–10 mg) were prepared in 100 μL aluminum crucibles. The samples were subjected to heating from 30 to 170 °C (or 160 °C), then cooled to −60 °C (or −80 °C), and then heated again to 170 °C (or 160 °C) at a heating/cooling rate of 10 °C min−1 under a flow of nitrogen (50 mL min−1). The data obtained from the second heating step was used for analyses. For TGA, a Mettler Toledo TGA/DSC1 instrument was used. Samples (5–7 mg) were prepared in 70 μL alumina crucibles and heated from 40 to 700 °C at a heating rate of 10 °C min−1 under a flow of nitrogen (50 mL min−1).
Polymerization reactions: The lactone was azeotropically distilled with toluene. BnOH and MSA were used as received. All glassware was dried at 150 °C for 24 h and additionally dried with a heating gun at 600 °C under reduced pressure. The lactone (2.38 mmol, 100 equiv) was added to the reaction vessel and distilled azeotropically with toluene (2 m final concentration of toluene). MSA (0.045 mmol, 2 equiv) and BnOH (0.023 mmol, 1 equiv) were added under argon and three vacuum–argon cycles were conducted. The reaction was started by placing the sealed vial (equipped with a magnetic stirrer and septum) in a preheated oil bath at 70 °C. After stirring for 24 h, the polymers were precipitated by the dropwise addition of the solution into an excess of MeOH (−78 °C) under stirring. The solution was filtered and the recovered polymers rinsed with MeOH. The polymers were then dried under reduced pressure overnight.
Homology modeling and generation of the energy‐minimized structure of CHMO Acineto_QM complexed with 10: To generate a homology model of CHMOAcineto_QM, the crystal structure of the homologous enzyme from Rhodococcus sp. HI‐31 (PDB ID: 4RG320) in the catalytically relevant tight conformation was used as template.20, 22 Cofactors, the bound lactone product 1, and crystallographic waters were deleted from the available Rhodococcus sp. structure (PDB ID: 4RG320). The resulting protein was uploaded to Swiss‐Model35 as a template together with the CHMOAcineto_QM query sequence (see the Supporting Information). The spatial locations of the cofactors (FAD and NADP+) and lactone product (1) in the homology model were dissected from the Rhodococcus sp. structure following structural superposition. Hydrogen atoms were added by using the software suite YASARA.36 Initial energy minimization was performed by keeping all backbone atoms fixed and by using the AMBER14 force field, as implemented in YASARA (standard settings, periodic boundary condition was used, cutoff for noncovalent interactions was 8 Å, particle mesh Ewald (PME)‐captured long‐range electrostatics). Subsequently, explicit water was added, the pH was set to 8 by the pK a prediction tool in YASARA,37 and energy minimization of the whole structure was performed. Finally, ϵ‐caprolactone (1) was rebuilt into lactone 10. Relevant force‐field parameters were obtained by the AutoSMILES methodology in YASARA.36
Library construction, bacterial strains, plasmids, and media: Based on the generated molecular model in Figure 1, a small library of BVMO mutants was constructed by alteration of identified hot‐spot residues. Site‐directed mutagenesis was performed by GeneArt (Thermo Fisher Scientific), except for the F246A variant, which was generated in‐house through the Phusion DNA polymerase (Thermo Fisher Scientific, USA), under standard conditions (primer sequences are given in the Supporting Information). The in‐house available gene of CHMOAcineto_QM (see the Supporting Information) in a pET28a(+) vector containing an N‐terminal His6 tag, was used as a template. Following gene verification by DNA sequencing, plasmids harboring mutated genes were transformed by heat‐shock into E. coli BL21(DE3). The E. coli strain was grown in 2× YT medium (16 g L−1 tryptone, 10 g L−1 yeast extract, 5 g L−1 sodium chloride) containing 40 μg mL−1 kanamycin sulfate (Sigma–Aldrich). Cell density at OD600 was determined by using a V‐1200 spectrophotometer (VWR, UK) and a plate reader SpectraMax i3x (Molecular Devices, USA).
Recombinant expression of CHMO Acineto _QM and variants: Freshly transformed E. coli BL21(DE3) cells were inoculated in 2× YT medium (1 mL) containing kanamycin sulfate (40 μg mL−1) in 96‐deep‐well plates, and cultivated overnight at 37 °C and 200 rpm. The overnight culture was used to inoculate fresh medium (3 mL) to OD600=0.1 and incubated at 37 °C and 200 rpm until OD600=0.6–0.8 was reached. Induction was performed for 20 h at 25 °C and 180 rpm by using isopropyl β‐d‐1‐thiogalactopyranoside (IPTG; 0.05 mm; Sigma–Aldrich). Cells were harvested by centrifugation at 2276 g for 15 min at 4 °C on a Thermo Scientific Sorvall ST16R centrifuge coupled to a M20 rotor (USA). Cell lysis was performed with B‐PER Complete Bacterial Protein Extraction Reagent (Thermo Fisher Scientific), at 5 mL g−1 to resuspend the cell pellet. The solution was incubated at room temperature for 15 min under mixing (750 rpm) to lyse the cells. The cell debris were removed by centrifugation at 2276 g for 30 min at 4 °C.
For scaling up, a preculture of 2× YT medium (30 mL) containing kanamycin sulfate (40 μg mL−1) was incubated overnight at 37 °C and 200 rpm. The overnight culture was used to incubate fresh medium (200 mL) to OD600=0.1 and incubated at 37 °C and 200 rpm until OD600=0.6–0.8 was reached. Induction and cell harvesting were performed as described above. The wet cell pellet (1 g) was resuspended in Tris buffer (50 mm, 5 mL, pH 8.5) and sonicated for 1 min with 1 s with 2 s pause (total time 3 min), 60 % duty cycle under ice cooling with Misonix sonifier cell disruptor ultrasonic S‐4000 probe (Misonix Inc, USA). Cellular debris were removed by centrifugation at 40 000 g and 4 °C for 20 min.
Protein analysis: The protein concentration was measured by using the procedure reported by Bradford,38 with the Bio‐Rad protein assay kit (Bio‐Rad, USA) and bovine serum albumin (BSA) as the protein standard. SDS‐PAGE was performed according to the method reported by Laemmli,39 with 4 % stacking and 15 % separating gels, purchased from Bio‐Rad (USA). SeeBlue Plus2 prestained protein standard was purchased from Thermo Fisher Scientific and used as a molecular‐weight marker. Proteins were stained with InstantBlue (Expedeon, UK).
BVMO activity determined by NADPH measurements: The measurement of the activity of the E. coli cell lysate containing CHMOAcineto_QM variants was performed spectrophotometrically, with a Spark TECAN plate reader (Switzerland). Cell lysate (20 μL) was mixed with reaction mixture (200 μL; 50 mm Tris⋅HCl pH 8.5, 0.6 mm 2 (60 mm in ethanol), 0.25 mm NADPH). The consumption of reduced NADPH was determined at λ=340 nm for 120 s in 96‐well plates (with an extinction coefficient of ϵ NADPH=6.4 mm −1 cm−1).
Biocatalytic lactone synthesis: Biocatalysis reactions were performed with cell lysate in 96‐deep‐well plates sealed with a breathable seal, at 30 °C and 200 rpm for 24 h. The cell lysate (40 μL) was mixed with 0.2 mm (NADPH, from a 0.1 m stock in 50 mm Tris⋅HCl, pH 8.5) and 2 mm 9 (1 m stock in ethanol), supplemented with FAD (62 μm; 62 mm stock in 50 mm Tris⋅HCl pH 8.5) and catalase from bovine liver (290 U mL−1). The GDH cofactor regeneration system was used by adding GDH (Codexis, USA) at a concentration of 0.09 mg mL−1 and activity 0.03 U μL−1 with d‐glucose (0.2 m; 1 m stock in 50 mm Tris⋅HCl, pH 8.5).
Scaling up was performed in 2 L baffled shake flasks to increase oxygen supply to the reaction. The flasks contained reaction mixture (200 mL) and were sealed with a breathable seal. The cell lysate (16 mL) was added to the reaction, as described above. The reaction was performed at 30 °C and 200 rpm.
Extraction of biocatalysis samples and GC analysis: Biocatalysis reactions were extracted with ethyl acetate (2×500 μL) spiked with decane (2 mm) as an internal standard. Each extraction was followed by vortexing for 5 s and centrifugation at 9000 g for 10 min. The organic phase was analyzed by means of GC/flame ionization detection (FID) and GC/MS on a GCMS‐QP2010 Ultra (Shimadzu, Japan) instrument equipped with a AOC‐20i auto injector (Shimadzu, Japan). Rxi‐5ms capillary columns (30 m×250 μm×0.25 μm, Restek, USA) were used with argon as the carrier gas. A Sky Liner PTV 2010 liner with glass wool was used as an inlet liner and a splitless injection mode (1 μL) was used with the following GC temperatures: injection port, 225 °C; initial column temperature, 70 °C; first temperature ramp, 20 °C min−1; second column temperature, 300 °C; second temperature ramp, 3 °C min−1; final temperature, 340 °C; final hold time, 10 min; total run time 35 min. The MSD source was kept at 200 °C and the interface temperature at 200 °C. The solvent was delayed for 2 min. The scaled up reaction was first filtered to remove insoluble residues. The filtrate was then extracted three times with EtOAc and washed once with deionized water. The organic phase was dried over magnesium sulfate, filtered, and concentrated. The obtained slightly yellow and clear oil was analyzed by means of NMR spectroscopy and GC/FID.
Conflict of interest
The authors declare no conflict of interest.
Biographical Information
Per‐Olof Syrén obtained his PhD in biotechnology from KTH in Sweden in 2011 under the supervision of Prof. Karl Hult. After postdoctoral research in the laboratory of Prof. Bernhard Hauer in Stuttgart as an Alexander von Humboldt Fellow, he returned to KTH to establish his independent research group. His research combines biotechnology, biocatalysis, enzyme design, and polymer chemistry with the goal of contributing to a better environment and health. His research program bridges three departments: Science for Life Laboratory, the Department of Protein Science, and the Department of Fibre and Polymer Technology. He has been an Associate Professor in Chemistry since 2016.

Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
This work was generously funded by a FORMAS young‐research leader grant (#942‐2016‐66). We acknowledge support from the Swedish Research Council (VR, #2016‐06160). The PDC Center for High‐Performance Computing at the Royal Institute of Technology (KTH) is greatly acknowledged. We thank Birger Sjöberg and Ylva Gravenfors at the Science for Life Laboratory, and Tomas Gustafsson at RISE Bioeconomy for support with chemical synthesis and fruitful discussions.
A. Stamm, A. Biundo, B. Schmidt, J. Brücher, S. Lundmark, P. Olsén, L. Fogelström, E. Malmström, U. T. Bornscheuer, P.-O. Syrén, ChemBioChem 2019, 20, 1664.
References
- 1.
- 1a. Zhang X., Fevre M., Jones G. O., Waymouth R. M., Chem. Rev. 2018, 118, 839–885; [DOI] [PubMed] [Google Scholar]
- 1b. Zhu Y., Romain C., Williams C. K., Nature 2016, 540, 354–362. [DOI] [PubMed] [Google Scholar]
- 2. Geyer R., Jambeck J. R., Law K. L., Sci. Adv. 2017, 3, e1700782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.
- 3a. Corey E. J., Chem. Soc. Rev. 1988, 17, 111–133; [Google Scholar]
- 3b. Corey E. J., Angew. Chem. Int. Ed. Engl. 1991, 30, 455–465; [Google Scholar]; Angew. Chem. 1991, 103, 469–479. [Google Scholar]
- 4. Baeyer A., Villiger V., Ber. Dtsch. Chem. Ges. 1899, 32, 3625–3633. [Google Scholar]
- 5. Nuyken O., Pask S. D., Polymer 2013, 5, 361–403. [Google Scholar]
- 6. Birmingham W. R., Starbird C. A., Panosian T. D., Nannemann D. P., Iverson T. M., Bachmann B. O., Nat. Chem. Biol. 2014, 10, 392–399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Turner N. J., O'Reilly E., Nat. Chem. Biol. 2013, 9, 285–288. [DOI] [PubMed] [Google Scholar]
- 8.
- 8a. Hönig M., Sondermann P., Turner N. J., Carreira E. M., Angew. Chem. Int. Ed. 2017, 56, 8942–8973; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 9068–9100; [Google Scholar]
- 8b. de Souza R. O. M. A., Miranda L. S. M., Bornscheuer U. T., Chem. Eur. J. 2017, 23, 12040–12063. [DOI] [PubMed] [Google Scholar]
- 9. Gandini A., Monomers and Macromonomers from Renewable Resources, Wiley-VCH, Weinheim, 2011, pp. 1–33. [Google Scholar]
- 10. Kean Z. S., Niu Z., Hewage G. B., Rheingold A. L., Craig S. L., J. Am. Chem. Soc. 2013, 135, 13598–13604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.
- 11a. Zhu J.-B., Watson E. M., Tang J., Chen E. Y.-X., Science 2018, 360, 398–403; [DOI] [PubMed] [Google Scholar]
- 11b. Nadif S. S., Kubo T., Gonsales S. A., VenkatRamani S., Ghiviriga I., Sumerlin B. S., Veige A. S., J. Am. Chem. Soc. 2016, 138, 6408–6411; [DOI] [PubMed] [Google Scholar]
- 11c. Liu Y., Zhou H., Guo J.-Z., Ren W.-M., Lu X.-B., Angew. Chem. Int. Ed. 2017, 56, 4862–4866; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 4940–4944; [Google Scholar]
- 11d. Miyaji H., Satoh K., Kamigaito M., Angew. Chem. Int. Ed. 2016, 55, 1372–1376; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 1394–1398; [Google Scholar]
- 11e. Hauenstein O., Agarwal S., Greiner A., Nat. Commun. 2016, 7, 11862; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11f. Xie F., Deng X., Kratzer D., Cheng K. C. K., Friedmann C., Qi S., Solorio L., Lahann J., Angew. Chem. Int. Ed. 2017, 56, 203–207; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 209–213; [Google Scholar]
- 11g. Zhang D., Hillmyer M. A., Tolman W. B., Biomacromolecules 2005, 6, 2091–2095; [DOI] [PubMed] [Google Scholar]
- 11h. Winnacker M., Sag J., Chem. Commun. 2018, 54, 841–844; [DOI] [PubMed] [Google Scholar]
- 11i. Winnacker M., Rieger B., ChemSusChem 2015, 8, 2455–2471; [DOI] [PubMed] [Google Scholar]
- 11j. Winnacker M., Angew. Chem. Int. Ed. 2018, 57, 14362–14371; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 14560–14569. [Google Scholar]
- 12.
- 12a. Hillmyer M. A., Science 2017, 358, 868–870; [DOI] [PubMed] [Google Scholar]
- 12b. Sun Z., Fridrich B., de Santi A., Elangovan S., Barta K., Chem. Rev. 2018, 118, 614–678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.
- 13a. Oldfield E., Lin F.-Y., Angew. Chem. Int. Ed. 2012, 51, 1124–1137; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2012, 124, 1150–1163; [Google Scholar]
- 13b. Christianson D. W., Chem. Rev. 2017, 117, 11570–11648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Lowe J. R., Martello M. T., Tolman W. B., Hillmyer M. A., Polym. Chem. 2011, 2, 702–708. [Google Scholar]
- 15. Horn E. J., Rosen B. R., Chen Y., Tang J., Chen K., Eastgate M. D., Baran P. S., Nature 2016, 533, 77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Bell S. G., Chen X., Sowden R. J., Xu F., Williams J. N., Wong L.-L., Rao Z., J. Am. Chem. Soc. 2003, 125, 705–714. [DOI] [PubMed] [Google Scholar]
- 17.
- 17a. Rioz-Martínez A., de Gonzalo G., Torres Pazmiño D. E., Fraaije M. W., Gotor V., Eur. J. Org. Chem. 2009, 2526–2532; [DOI] [PubMed] [Google Scholar]
- 17b. Grogan G., Roberts S., Wan P., Willetts A., Biotechnol. Lett. 1993, 15, 913–918; [Google Scholar]
- 17c. Mihovilovic M. D., Kapitán P., Tetrahedron Lett. 2004, 45, 2751–2754; [Google Scholar]
- 17d. Alphand V., Archelas A., Furstoss R., Tetrahedron Lett. 1989, 30, 3663–3664; [Google Scholar]
- 17e. Snajdrova R., Grogan G., Mihovilovic M. D., Bioorg. Med. Chem. Lett. 2006, 16, 4813–4817; [DOI] [PubMed] [Google Scholar]
- 17f. Koch A. A., Hansen D. A., Shende V. V., Furan L. R., Houk K. N., Jimenez-Oses G., Sherman D. H., J. Am. Chem. Soc. 2017, 139, 13456–13465; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17g. Leisch H., Morley K., Lau P. C. K., Chem. Rev. 2011, 111, 4165–4222. [DOI] [PubMed] [Google Scholar]
- 18.
- 18a. Abril O., Ryerson C. C., Walsh C., Whitesides G. M., Bioorg. Chem. 1989, 17, 41–52; [Google Scholar]
- 18b. Donoghue N. A., Norris D. B., Trudgill P. W., Eur. J. Biochem. 1976, 63, 175–192; [DOI] [PubMed] [Google Scholar]
- 18c. Chen Y. C., Peoples O. P., Walsh C. T., J. Bacteriol. 1988, 170, 781–789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.
- 19a. Balke K., Beier A., Bornscheuer U. T., Biotechnol. Adv. 2018, 36, 247–263; [DOI] [PubMed] [Google Scholar]
- 19b. Opperman D. J., Reetz M. T., ChemBioChem 2010, 11, 2589–2596. [DOI] [PubMed] [Google Scholar]
- 20. Yachnin B. J., McEvoy M. B., MacCuish R. J. D., Morley K. L., Lau P. C. K., Berghuis A. M., ACS Chem. Biol. 2014, 9, 2843–2851. [DOI] [PubMed] [Google Scholar]
- 21.
- 21a. Yachnin B. J., Sprules T., McEvoy M. B., Lau P. C. K., Berghuis A. M., J. Am. Chem. Soc. 2012, 134, 7788–7795; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21b. Ryerson C. C., Ballou D. P., Walsh C., Biochemistry 1982, 21, 2644–2655. [DOI] [PubMed] [Google Scholar]
- 22.
- 22a. Balke K., Schmidt S., Genz M., Bornscheuer U. T., ACS Chem. Biol. 2016, 11, 38–43; [DOI] [PubMed] [Google Scholar]
- 22b. Balke K., Bäumgen M., Bornscheuer U. T., ChemBioChem 2017, 18, 1627–1638. [DOI] [PubMed] [Google Scholar]
- 23. Chapman M. R., Cosgrove S. C., Turner N. J., Kapur N., Blacker A. J., Angew. Chem. Int. Ed. 2018, 57, 10535–10539; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 10695–10699. [Google Scholar]
- 24. Gazeau-Bureau S., Delcroix D., Martín-Vaca B., Bourissou D., Navarro C., Magnet S., Macromolecules 2008, 41, 3782–3784. [Google Scholar]
- 25. Olsén P., Borke T., Odelius K., Albertsson A.-C., Biomacromolecules 2013, 14, 2883–2890. [DOI] [PubMed] [Google Scholar]
- 26. Zhao S., Malfait W. J., Demilecamps A., Zhang Y., Brunner S., Huber L., Tingaut P., Rigacci A., Budtova T., Koebel M. M., Angew. Chem. Int. Ed. 2015, 54, 14282–14286; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 14490–14494. [Google Scholar]
- 27.BIOPLASTICS Facts and Figures, European Bioplastics, 2017.
- 28. Robert C., de Montigny F., Thomas C. M., Nat. Commun. 2011, 2, 586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.
- 29a. Hoppens N. C., Hudnall T. W., Foster A., Booth C. J., J. Polym. Sci. Part A 2004, 42, 3473–3478; [Google Scholar]
- 29b. Kelsey D. R., Scardino B. M., Grebowicz J. S., Chuah H. H., Macromolecules 2000, 33, 5810–5818. [Google Scholar]
- 30. Morrill C., Jensen C., Just-Baringo X., Grogan G., Turner N. J., Procter D. J., Angew. Chem. Int. Ed. 2018, 57, 3692–3696; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 3754–3758. [Google Scholar]
- 31. Can Baser K. H., Buchbauer G., Handbook of Essential Oils: Science Technology, and Applications , 2nd ed., CRC, Boca Raton, 2015. [Google Scholar]
- 32. Tufvesson P., Lima-Ramos J., Nordblad M., Woodley J. M., Org. Process Res. Dev. 2011, 15, 266–274. [Google Scholar]
- 33. Dong J., Fernández-Fueyo E., Hollmann F., Paul C. E., Pesic M., Schmidt S., Wang Y., Younes S., Zhang W., Angew. Chem. Int. Ed. 2018, 57, 9238–9261; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 9380–9404. [Google Scholar]
- 34. Hirata T., Izumi S., Shimoda K., Hayashi M., J. Chem. Soc. Chem. Commun. 1993, 1426–1427. [Google Scholar]
- 35.
- 35a. Bertoni M., Kiefer F., Biasini M., Bordoli L., Schwede T., Sci. Rep. 2017, 7, 10480; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35b. Benkert P., Biasini M., Schwede T., Bioinformatics 2011, 27, 343–350; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35c. Bienert S., Waterhouse A., de Beer T. A. P., Tauriello G., Studer G., Bordoli L., Schwede T., Nucleic Acids Res. 2017, 45, D313–D319; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35d. Guex N., Peitsch M. C., Schwede T., Electrophoresis 2009, 30, S162–S173; [DOI] [PubMed] [Google Scholar]
- 35e. Waterhouse A., Bertoni M., Bienert S., Studer G., Tauriello G., Gumienny R., Heer F. T., de Beer T. A. P., Rempfer C., Bordoli L., Lepore R., Schwede T., Nucleic Acids Res. 2018, 46, W296–W303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Krieger E., Darden T., Nabuurs S. B., Finkelstein A., Vriend G., Proteins Struct. Funct. Bioinf. 2004, 57, 678–683. [DOI] [PubMed] [Google Scholar]
- 37. Krieger E., Nielsen J. E., Spronk C. A. E. M., Vriend G., J. Mol. Graphics Modell. 2006, 25, 481–486. [DOI] [PubMed] [Google Scholar]
- 38. Bradford M. M., Anal. Biochem. 1976, 72, 248–254. [DOI] [PubMed] [Google Scholar]
- 39. Laemmli U. K., Nature 1970, 227, 680. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary