

Abstract
γ-Secretase is a membrane-embedded protease complex, with presenilin as the catalytic component containing two transmembrane aspartates in the active site. With more than 90 known substrates, the γ-secretase complex is considered “the proteasome of the membrane”, with central roles in biology and medicine. The protease carries out hydrolysis within the lipid bilayer to cleave the transmembrane domain of the substrate multiple times before releasing secreted products. For many years, elucidation of γ-secretase structure and function largely relied on small-molecule probes and mutagenesis. Recently, however, advances in cryo-electron microscopy have led to the first detailed structures of the protease complex. Two new reports of structures of γ-secretase bound to membrane protein substrates provide great insight into the nature of substrate recognition and how Alzheimer’s disease-causing mutations in presenilin might alter substrate binding and processing. These new structures offer a powerful platform for elucidating enzyme mechanisms, deciphering effects of disease-causing mutations, and advancing Alzheimer’s disease drug discovery.
Graphical Abstract
The γ-secretase complex is a founding member of a family of intramembrane-cleaving proteases (I-CLiPs) that carry out hydrolysis of substrates within the hydrophobic environment of the lipid bilayer.1 These proteases contain membrane-embedded active sites that converged evolutionarily on the same basic mechanisms found in soluble proteases and include site 2 protease (S2P) metalloproteases, presenilin-type aspartyl proteases, and the rhomboid serine proteases.2 Found in virtually all forms of life, I-CLiPs cut within the transmembrane domain (TMD) of their substrates and play a broad range of critical roles in biology.3–6
γ-Secretase was initially defined as an activity that cleaved the TMD of the amyloid precursor protein (APP) to produce the amyloid β-peptide (Aβ) that is deposited as cerebral plaques in Alzheimer’s disease.7 Missense mutations in the small Aβ region of APP cause familial Alzheimer’s disease (FAD), altering the production or properties of Aβ to strengthen its tendency to aggregate.8,9 In 1995, other FAD missense mutations were found in presenilin-1 and −2 (PSEN1 and PSEN2, respectively),10,11 multipass membrane proteins that were then only known to be distantly related to an obscure gene in the roundworm Caenorhabditis elegans involved in spermatogenesis.12
Presenilin FAD mutations were soon found to alter Aβ production at the level of γ-secretase,13–18 and knockout of PSEN1 dramatically reduced the level of Aβ formation by γ-secretase.19,20 Meanwhile, pharmacological evidence suggested that γ-secretase is an aspartyl protease.21 Together, these observations led to the discovery of two completely conserved TMD aspartates in presenilin that are essential for γ-secretase activity22 and the identification of presenilin as the target of inhibitors of γ-secretase directed to its active site.23,24 Thus, presenilin was identified as a novel membrane-embedded protease, an aspartyl I-CLiP.
Concurrently, genetic studies in C. elegans and mice connected presenilins to the Notch family of cell-surface receptors.25–27 Signaling from Notch is essential for the determination of cell fate in embryogenesis in all metazoans,28 and knockout of presenilins leads to embryonic-lethal phenotypes similar to that seen upon knockout of Notch1.26,27 Signaling from Notch1 was found to be dependent on the release of its intracellular domain through cleavage within the TMD of the receptor.29 These findings converged with those for APP proteolysis, leading to the discovery that the presenilin-dependent γ-secretase is the same protease that cleaves Notch and that γ-secretase is an essential component of the Notch signaling pathway.30,31
Follow-up reports quickly confirmed that the two conserved TMD aspartates in presenilin are essential for its endoproteolysis to the N-terminal fragment (NTF) and C-terminal fragment (CTF) and for γ-secretase activity.32–34 Nevertheless, lingering doubts about the proteolytic function of presenilin remained, because the protein could not be demonstrated to have this activity on its own. These concerns were partially allayed by the discovery of another polytopic membrane protein as an aspartyl protease, bacterial type 4 prepilin peptidase (TFPP).35 Although TFPPs have catalytic aspartates outside of the membrane and are evolutionarily unrelated to presenilins, they do share a GxGD motif containing one of the catalytic aspartates.36 This motif is important not only for protease activity but also for substrate selectivity in presenilins (e.g., Notch vs APP).36 Complete acceptance of presenilin as a protease came with the discovery of a family of presenilin homologues with catalytic activity on their own, exemplified by signal peptide peptidase.37,38
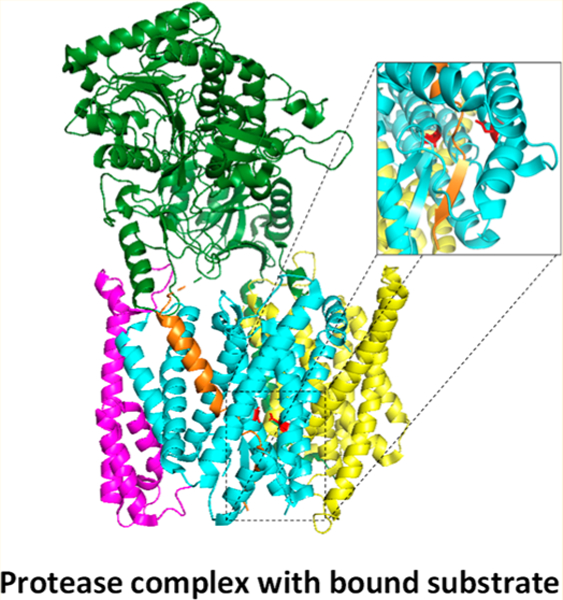
Although presenilin was identified as the catalytic component of γ-secretase, evidence clearly pointed to presenilin being part of a larger complex. Presenilin alone did not show γ-secretase activity, and presenilin expressed in the endoplasmic reticulum (ER) is cleaved into a tightly regulated and metabolically stable NTF and CTF in the Golgi apparatus before moving further along the secretory pathway and to the cell surface.39–43 Moreover, presenilin NTFs and CTFs remain associated and enter into a high-molecular weight complex.43–46 Ultimately, through extensive genetic and biochemical investigations, three other membrane protein components were identified: nicastrin,47 anterior pharynx-defective 1 (Aph-1),48–50 and presenilin enhancer 2 (Pen-2)51,52 (Figure 1). Expression of these three proteins along with presenilin reconstituted γ-secretase activity in mammalian cells53,54 and yeast,55 and all four proteins, with presenilin as NTF and CTF, co-purified using an immobilized activity-based probe.54
Figure 1.

Intramembrane proteolysis by the γ-secretase complex. γ-Secretase cleaves >90 different type I integral membrane proteins after ectodomain release by membrane-tethered sheddases. The protease complex carries out proteolysis in the transmembrane domain of these substrates to secrete N-terminal cleavage products into the extracellular milieu and release C-terminal cleavage products into the cytoplasm. The γ-secretase complex is composed of four different multipass membrane proteins, with presenilin as the catalytic component containing two transmembrane aspartates in the active site. Upon assembly with the other subunits (nicastrin, Aph-1, and Pen-2), presenilin undergoes autoproteolysis into an N-terminal fragment (NTF) and C-terminal fragment (CTF) to form active γ-secretase.
The γ-secretase complex is now known to process more than 90 different type I integral membrane proteins after sheddase-mediated removal of their ectodomains.56–58 Although in a few cases the release of substrate intracellular domain leads to cell signaling analogous to that of Notch,59,60 cleavage of most substrates by γ-secretase is thought to be a means of clearing these protein stubs from the membrane. For this reason, γ-secretase has been dubbed “the proteasome of the membrane”.61 Moreover, substrate processing by γ-secretase is now known to be quite complicated, with an initial cut occurring in the substrate TMD near the membrane–cytosol interface followed by C-terminal trimming of the membrane-bound product to smaller secreted peptides.
Processing of APP is the most studied. An initial endoproteolytic cut leads to the formation of a 48-or 49-residue Aβ peptide (Aβ48 or Aβ49, respectively) and corresponding APP intracellular domain (AICD) fragments.62–66 Aβ48 or Aβ49 is then processed through a carboxypeptidase activity of γ-secretase,67,68 trimming generally every three amino acids.69–71 Thus, the production of Aβ occurs along two pathways: Aβ49 → Aβ46 → Aβ43 → Aβ40 and Aβ48 → Aβ45 → Aβ42 → Aβ38 (Figure 2). Interestingly, presenilin endoproteolysis also involves processive tripeptide trimming,72 and Notch1 is likely processed similarly by γ-secretase.73 As the γ-secretase complex contains only one of each component,74 the protease has only one active site that is apparently responsible for all of these substrate TMD processing events.
Figure 2.
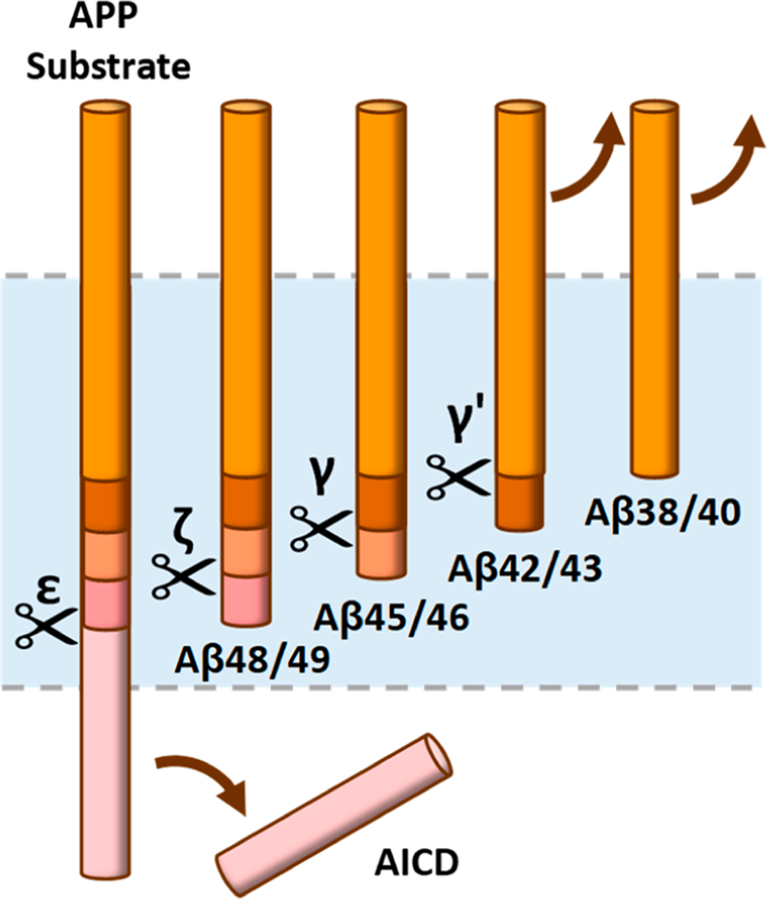
Processive proteolysis by γ-secretase. The protease first carries out endoproteolysis (ε) near the cytosolic end of the TMD of the APP substrate, with release of the intracellular domain (AICD). This is followed by carboxypeptidase cleavages (ζ, γ, and γ′) of the remaining long Aβ peptides, in intervals of roughly three amino acids, to secreted peptides that are 38–43 residues in length. There are two general pathways for Aβ generation: Aβ49 → Aβ46 → Aβ43 → Aβ40 and Aβ48 → Aβ45 → Aβ42 → Aβ38.
STRUCTURE AND FUNCTION: MOLECULAR PROBES AND MUTAGENESIS
Early studies of γ-secretase structure and function relied largely on peptidomimetic inhibitors and mutagenesis of substrates or the enzyme. The first designed inhibitors were substrate-based transition-state analogue (TSA) inhibitors, difluoroketone peptidomimetics based on the APP TMD.21,75 Hydration of the difluoroketone results in a geminal diol that closely resembles an intermediate formed in aspartyl protease catalysis when water attacks the carbonyl carbon of the scissile amide bond.76 These and related peptidomimetic TSA inhibitors suggested that γ-secretase is an aspartyl protease.21 This finding, along with the requirement of presenilin for γ-secretase processing of APP in cells,19 led to the discovery of presenilin as the catalytic component of the enzyme through mutagenesis of two conserved TMD aspartates.22 These aspartates were also found to be absolutely required for the processing of presenilin into NTF and CTF, suggesting that presenilin is a zymogen that undergoes autoproteolysis.22 Identification of the other components of the enzyme demonstrated that this autoproteolysis is triggered only after assembly of all four members into the complex.53–55
Variations of these early peptidomimetic TSA inhibitors were used as probes for substrate binding pockets in the enzyme active site. The protease was found to have relatively loose specificity,21 a finding consistent with the later discovery of many other TMD substrates with little or no sequence conservation.56–58 TSA probes also suggested large pockets –S1, S1′, and S3′–in the active site that accommodate substrate residues P1, P1′, and P3′, while the S2′ pocket is smaller.77,78 Moreover, TSA residues P1′, P2′, and P3′ were essential for inhibitory potency, but adding a P4′ residue had essentially no effect on potency.78 Thus, the enzyme apparently has only three corresponding S′ pockets, a feature later revealed to be an important determinant of the carboxypeptidase trimming activity of γ-secretase.79 TSA inhibitors were also converted into affinity labeling reagents that were covalently attached to presenilin NTF and CTF.23,24 These observations suggested that the active site was at the interface between these two presenilin subunits, consistent with the fact that NTF and CTF each contain one of the conserved aspartates essential for γ-secretase activity.
Other types of substrate-based probe that helped understand γ-secretase structure and function are helical peptide inhibitors (HPIs). A series of peptides based on the APP TMD were designed, installing the helix-inducing α-aminoisobutyric acid (Aib) residue.80 Certain APP TMD residues were swapped with Aibs, spacing them apart so that Aib residues would array along one face of the helix, presenting APP residues along the rest of the TMD for interaction with the protease. The idea was to mimic the helical conformation of the APP TMD as it would be upon initial interaction with γ-secretase. The designed HPIs could be remarkably potent inhibitors of γ-secretase activity, both in cells and in isolated enzyme assays.80,81 Conversion of potent HPIs to affinity labeling reagents led to covalent binding to presenilin NTF and CTF,82 as seen with TSA probes. However, TSA inhibitors could not compete with 10-residue HPI affinity probes, and 10-residue HPIs could not compete with TSA affinity probes, indicating different binding sites for the two types of probes. The labeling of PSEN1 NTF and CTF by HPI and TSA probes is consistent with a recent tour de force report of systematic mutagenesis of the recombinant APP substrate with an unnatural photo-cross-linkable amino acid.83 Very little cross-linking to other γ-secretase components was observed. Intriguingly, repeating these experiments with two different FAD mutant PSEN1-containing γ-secretase complexes resulted in altered patterns of photolabeling to PSEN1 NTF and CTF, suggesting that these disease-causing mutations alter the positioning of the substrate for proteolysis.
The findings with HPI-and TSA-based affinity probes were consistent with the observation that the endogenous APP substrate could be co-purified with γ-secretase using an immobilized TSA inhibitor,84 demonstrating that the substrate could bind to the protease even when the active site was occupied by an inhibitor. This co-purification suggested the existence of an initial substrate docking site distinct from the active site, part of a lateral gating mechanism for entry of the substrate into the active site. The water-containing active site was expected to be inside presenilin, sequestered from the hydrophobic environment of the lipid bilayer, with substrate TMD first interacting with presenilin on its lipid-exposed surface. The 10-residue HPI probes apparently bind to this docking exosite. Interestingly, potent 13-residue HPI probes could compete with both 10-residue HPI and TSA,82 suggesting that the docking site and active sites are proximal, within the distance spanned by three amino acids.
The finding that mimics of the APP TMD in a helical conformation could bind tightly to the enzyme was also consistent with an earlier phenylalanine scanning mutagenesis experiment of the APP TMD in transfected cells.85 Systematic Phe mutation in the APP TMD resulted in effects on the major secreted Aβ peptides (Aβ40 and Aβ42), with changes in the Aβ42/Aβ40 ratio that followed a repeating pattern every three or four residues (approximately the turn of an α-helix). However, this Phe scanning approach was revisited recently,79 with the understanding of the carboxypeptidase activity and two pathways by which Aβ40 and Aβ42 are produced (see Figure 2). The new Phe scanning study was also undertaken with the information gained from TSA inhibitor probes that the S2′ pocket is smaller than S1, S1′, and S3′ and cannot accommodate a Phe residue.78 Phe mutation affected the Aβ42/Aβ40 ratio in a manner completely predictable with processive proteolysis along the two pathways and with cleavage blocked wherever Phe is in the P2′ position in the APP TMD. Moreover, placing Phe in both positions 50 and 51 (Aβ numbering) virtually blocked ε cleavage and all Aβ production, but the mutant substrate could nevertheless co-immunoprecipitate with the γ-secretase complex. Taken together, the results suggest a model in which substrate TMD interacts as an α-helix with the enzyme before unwinding in part into the active site and filling pockets S1′–S3′. After ε cleavage, this unwinding is repeated, with trimming generally every three amino acids dictated by these three pockets (Figure 3). The stability of the interaction between longer Aβ peptide intermediates and the protease likely determines the likelihood of C-terminal trimming versus release as products, and FAD mutations can apparently decrease the affinity of the enzyme for Aβ intermediates.86,87
Figure 3.

General mechanism of substrate recognition and processing by γ-secretase. Helical substrate TMD initially binds to presenilin at a docking exosite (where the helix is bound in ES1). This is followed by movement of the substrate in whole or in part (as shown) into the active site, with unwinding of the substrate TMD to set up the transition state (ES1*) for ε cleavage. After release of the intracellular domain, the remaining enzyme-bound product (ES2) again unwinds into the active site for carboxypeptidase cleavage (ES2*). Three pockets in the enzyme active site dictate trimming every three amino acids. Successive carboxypeptidase trimming occurs until short peptide products are released. The inset shows pockets S1′ and S3′ are relatively large and can accommodate bulky aromatic amino acids such as Phe, while S2′ is smaller and cannot accommodate Phe.
Cysteine mutagenesis in PSEN1 coupled with cross-linking via disulfide bond formation has also proven to be a valuable approach to deciphering the structure and function of presenilin within the active γ-secretase complex. PSEN1 contains five native cysteine residues, and all of these can be replaced with serine to form a cysteine-less PSEN1 that retains the ability to assemble into an active γ-secretase complex.88,89 Partial replacement of native cysteines along with oxidation with copper and 1,10-phenanthroline led to the finding that Cys92 in TMD1 could cross-link to either Cys410 or Cys419 in TMD8, observed as cross-linking of PSEN1 NTF and CTF by sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS–PAGE).90 Thus, TMD1 and TMD8 in PSEN1 are apparently directly adjacent, a finding later borne out by cryo-electron microscopy (cryo-EM) structure determination.91 Cysteine mutagenesis of the catalytic aspartates in TMD6 and TMD7 in an otherwise cysteine-less PSEN1 results in assembly with other complex members, but this mutant PSEN1 is unable to undergo autoproteolysis and is completely inactive.89 Nevertheless, oxidative conditions led to the shifting of the PSEN1 holoprotein by SDS–PAGE, suggesting that the two cysteines replacing the catalytic aspartates are immediately proximal as expected.
Systematic replacement of PSEN1 residues with Cys in the otherwise Cys-less mutant was also coupled with use of biotinylated thiol-containing probes, an approach called the substituted cysteine accessibility method (SCAM).92 Using SCAM, the water accessibility of specific residues could be determined. Cross-linking using whole cells could identify water-accessible residues on the extracellular side, while cross-linking using proteoliposomes could also reveal water-accessible residues on the intracellular side. In this way, TMD6 and TMD7 were found to be exposed to a hydrophilic environment, with the two catalytic aspartates more limited in their water accessibility,88,89 findings confirmed by the first detailed cryo-EM structures of γ-secretase. The SCAM method could also identify key residues not initially observed by cryo-EM. A portion of hydrophilic loop 1 (L1) was labeled with a pattern consistent with a short α-helical conformation, and this region was thought to be involved in substrate recognition.93 These findings were recently confirmed upon cryo-EM elucidation of substrate-bound γ-secretase.94,95 Double Cys labeling and use of cross-linking reagents could additionally reveal proximal residues in NTF and CTF (i.e., by generating PSEN1 NTF/CTF that co-migrated with holoprotein).96 SCAM labeling could also be conducted in the presence of small-molecule γ-secretase inhibitors and modulators. Blocking of specific SCAM labeling sites thereby suggested sites of compound binding. In this way, the binding of HPI and TSA probes to distinct sites was confirmed and tentatively identified residues involved in the docking site and active site.97
Aside from SCAM analysis of PSEN1, the presenilins have been extensively mutated in attempts to understand structure and function. While a full review of these studies is beyond the scope of this Perspective, three critical motifs in PSEN1 identified through mutagenesis are worth mentioning here: the GxGD motif, discussed previously as being critical for proteolytic function and substrate selectivity,36,98–100 a PAL motif located in TMD9, and the hydrophilic loop 1 (L1)/TMD2 region. The PAL motif is essential for PSEN1 endoproteolysis and γ-secretase activity. Nonconservative mutations lead to complete loss of proteolytic function101 and alter the binding of active site-directed TSA probes.102 These findings were later confirmed by cryo-EM elucidation of the γ-secretase structure, revealing that the PAL motif is part of the active site.91 Mutagenesis of L1/TMD2 of PSEN1 suggested that this region is critical for PSEN1 autoproteolysis and coordination between the substrate docking site and the catalytic core.103 This notion is likewise supported by recent cryo-EM structures of γ-secretase bound to substrates.94,95
While small-molecule probes and mutagenesis helped to improve our understanding of the structure and function of presenilin in γ-secretase, other approaches were required to begin elucidating how the subunits of the protease complex interact with each other. Partial dissociation of the isolated complex using the detergent dodecyl d-maltoside revealed that PSEN1 NTF interacts with Pen-2 and nicastrin interacts with Aph-1.104 The PSEN1 CTF was found in two other partial complexes, one with PSEN1 NTF and Pen-2 and the other with Aph-1 and nicastrin. These findings of subunit interactions were confirmed using cross-linking reagents105 and were consistent with evidence that the apparent order of assembly of the complex involves nicastrin and Aph-1 interaction,49 then addition of PSEN1 holoprotein to form a tripartite complex,53,106,107 and finally addition of Pen-2, which triggers PSEN1 autoproteolysis53 (Figure 4). TMD swapping experiments with PSEN1 identified TMD4 as being essential for interaction with Pen-2, and more focused mutagenesis revealed a YNF motif in TMD4 as being specifically critical.108,109 All of these interactions were later confirmed by the cryo-EM structures of γ-secretase. This confirmation works both ways. Cryo-EM reconstructions should be consistent with results from biochemical experiments to mitigate concerns about the validity of the cryo-EM model.
Figure 4.

Assembly and activation of the γ-secretase complex. Nicastrin and Aph-1 form a stable subcomplex (step 1), followed by addition of presenilin (step 2). Interaction of Pen-2 with TMD4 of presenilin (step 3) triggers autoproteolysis of presenilin into NTF and CTF subunits (step 4) to form γ-secretase capable of cleaving substrates.
Biochemical experiments also helped to improve our understanding of the role of nicastrin in substrate recognition by the γ-secretase complex. The nicastrin ectodomain was found to have a sequence similar to those of certain aminopeptidases,110 although all conserved catalytic residues are not present (i.e., nicastrin does not have aminopeptidase activity111). Co-immunoprecipitation of Notch and APP substrates of γ-secretase with the nicastrin ectodomain led to the suggestion that nicastrin interacts with the N-terminus of γ-secretase substrates, thereby guiding the substrate to the active site on presenilin.112 However, these findings were inconsistent with other studies,113 the most notable being a recent report114 revealing that (1) peptides based on the N-terminus of substrate do not inhibit substrate processing, (2) mutagenesis of the N-terminal residue of substrate has no effect on the kinetics of substrate processing, and (3) acetylation of the substrate N-terminus also did not affect the substrate processing rate. Instead, the length of the extracellular ectodomain of the substrate was found to be the critical factor. Longer substrates are cleaved more poorly,114–116 and only the substrate TMD is required for high-affinity binding and processing by γ-secretase.114 These observations suggested that the nicastrin ectodomain functions as a gatekeeper for substrates, sterically blocking the approach of substrates with ectodomains that are too long.114 Consistent with this model, reduction of the disulfide bonds in nicastrin resulted in conformational changes (i.e., partial unfolding) and increased access of longer substrates for processing by γ-secretase. This topic will be revisited later in light of detailed cryo-EM structure determination of the protease complex.
STRUCTURE AND FUNCTION: CRYO-EM IMAGES
Recent advances in cryo-EM–particularly the development of direct electron detection in combination with computational single-particle analysis, image sorting, and three-dimensional (3D) reconstruction–have revolutionized structural biology, providing atomic-resolution structures of previously inaccessible protein complexes.117 The size, complexity, hydrophobicity, and heterogeneous glycosylation of γ-secretase make it highly challenging to crystallize for X-ray diffraction. Other I-CLiP family members have proven to be amenable to crystallography,118–121 but the rhomboid serine proteases, the S2P metalloproteases, and the presenilin homologue aspartyl proteases are much smaller and single polypeptides, with chemically and thermally stable orthologs found in the microbial world. The crystal structure of an archaeal presenilin homologue confirmed the nine-TMD topology of PSEN1 and showed the two catalytic aspartates in TMD6 and TMD7 adjacent to each other.121 However, these aspartates were not close enough or oriented properly to coordinate with and activate a water molecule for use in hydrolysis.
The first reported cryo-EM images of the γ-secretase complex provided poor resolution, ranging from 12 to 48 Å.122–125 Although rather amorphous, these 3D reconstructions provided rough dimensions of the complex, suggested the location of the nicastrin ectodomain and a membrane-embedded hydrophobic belt, and indicated the presence of cavities for entry of water into the active site. Subsequent structures suggested flexibility in the nicastrin ectodomain, with three conformational states captured, and a more compact structure of the complex in the presence of inhibitors.126,127 Nevertheless, the inability to resolve amino acids or even TMDs severely limited a more specific understanding of structure and function.
In 2014, the first detailed structure of the γ-secretase complex, determined by cryo-EM single-particle analysis with 4.5 Å overall resolution, was reported from the laboratories of Y. Shi at Tsinghua University in Beijing and S. Scheres at the MRC Laboratory of Molecular Biology in Cambridge, U.K.128 In addition to the advances in cryo-EM technology, the ability to express large quantities of the complex proved to be critical. The development of a tetracistronic construct allowed expression of all four components of γ-secretase in roughly equal levels upon transient transfection into human embryonic kidney (HEK) 293 cells in a suspension culture. This first detailed structure of the protease complex revealed the TMDs in a horseshoe-shaped arrangement. A lack of side-chain features for the TMDs prevented assignment of the four γ-secretase subunits. However, the crystal structure of an archaeal presenilin homologue121 led to a speculative assignment for PSEN1 and tentative location of the active site on the concave side of the horseshoe-shaped TMD arrangement. Hovering over the active site was the aminopeptidase-like domain of nicastrin, seemingly positioned perfectly to interact with the substrate N-terminus and guide substrate TMD into the active site. Crystallization of a microbial eukaryotic nicastrin homologue129 validated the 3D reconstruction of this subunit within the γ-secretase complex as elucidated by cryo-EM, and indeed, the ectodomain is structurally similar to a large family of peptidases.
The following year, however, a new report emerged from the Shi–Scheres collaboration of γ-secretase at 3.4 Å overall resolution, with clear assignment of the TMDs.91 The revised structure revealed that the active site actually resides on the convex side of the horseshoe-shaped TMD arrangement (Figure 5). The new arrangement had immediate implications for the role of nicastrin, validating a concurrent report (mentioned earlier here) that nicastrin serves as a gatekeeper to sterically block potential substrates with long extracellular ectodomains from approaching the active site114 (Figure 6). The aminopeptidase-like domain of nicastrin was now positioned on the opposite side from approaching the substrate, in a way that would require extreme conformational contortions to bind the substrate N-terminus in a productive manner.
Figure 5.

First detailed structure of the γ-secretase complex determined by cryo-EM, single-particle analysis, and image reconstruction, with the revised assignment of the presenilin TMDs: green for nicastrin, yellow for Aph-1, magenta for Pen-2, cyan (NTF) and aquamarine (CTF) for PS1, TMDs numbered, and red for catalytic aspartates. TMD2 was not resolved but was modeled using the crystal structure of an archaeal presenilin homologue. This TMD (“2”) is depicted as the dashed outlined cylinder. Looking only at the 19 TMDs of the complex from the cytosolic side (right) illustrates the horseshoe-shaped arrangement, with the active site that can be approached by the substrate from the convex side and flexible TMD2 as the apparent gate. Protein Data Bank entry 5A63.
Figure 6.

Nicastrin serves as a gatekeeper of the γ-secretase complex. The large ectodomain of nicastrin juts out over the entryway to the active site, sterically preventing access of substrates with ectodomains that are >20 Å long. Conformationally flexible TMD2 of presenilin is believed to be the gate, allowing substrate entry within the membrane. Protein Data Bank entry 5A63.
The new structure, now correctly identifying presenilin-1 and its TMDs, was also much more consistent with the previously reported crystal structure of an archaeal presenilin homologue.121 Interestingly, TMD2 of PSEN1 was poorly resolved, implying a mobility that suggested that this TMD could function as the gate for the lateral approach of substrate into the active site. Nevertheless, an atomic model for this TMD was generated on the basis of the crystal structure of the archaeal presenilin homologue. In both cases, the catalytic PSEN1 aspartates in TMD6 and TMD7 were positioned ∼10 Å apart (from Cα to Cα), relatively close but farther away than in active aspartyl proteases. Meanwhile, the PAL motif in PSEN1 TMD9 was proximal to these aspartates and appears to be part of the active site, as had been previously suggested.101,102 These observations led to the speculation that substrate binding initiates conformational changes that bring the aspartates closer, allowing them to coordinate with water and activate it for proteolysis.
The arrangement of the subunits within the complex was consistent with previous biochemical studies104,105 and revealed further details. The single nicastrin TMD interacts with TMD1, TMD5, and TMD7 of Aph-1, while TMD2 and TMD4 of Aph-1 associate with TMD8 and TMD9 of PSEN1 (as the CTF subunit). The three most C-terminal residues of the PSEN1 CTF (FYI) are inserted into a hydrophobic pocket in Aph-1 on the extracellular side. The Pen-2 topology was revealed as three TMDs, with two of these being short and together dipping in and out of the predicted boundary of the membrane on the intracellular side. Pen-2 was bound to TMD4 of the PSEN1 NTF, specifically to a YN motif, as suggested from mutagenesis experiments.108,109
The Scheres–Shi collaboration further developed their cryo-EM single-particle analysis of γ-secretase, using image classification along with masked refinement to focus on conformationally flexible parts of the protease complex, PSEN1 in particular.130 In this way, three different classes of enzyme conformation were identified, with an overall resolution of 3.5 Å. Major differences were seen among the three classes, particularly with respect to PSEN1 TMD2 and TMD6. TMD2 was visible in only class 1 (Figure 7A), while TMD6 adopted a different conformation in all three classes. An unidentified rod-shaped density was observed in class 1 and class 2, filling in a cavity created by PSEN1 TMD2, TMD3, and TMD5. Side chains of this rod-shaped density could not be observed, but the peptide backbone adopted a clear helical conformation, becoming random coiled and then invisible as it approached the catalytic aspartates. This peptide was suggested to be a composite of various γ-secretase substrates that each co-purified with separate enzyme complexes. The conformational flexibility of TMD2 again suggested a role in the lateral gating mechanism for entry of TMD substrates into the complex. Interestingly, Pen-2 also undergoes a conformational change in class 3 relative to class 1 and class 2, along with changes in PSEN1, consistent with strong evidence that interaction of Pen-2 with TMD4 of PSEN1 is essential for protease function.
Figure 7.

Structures of γ-secretase determined using image classification and masked refinement. (A) Class 1 structure of the apoenzyme, with resolution of TMD2 of presenilin and the presence of an unidentified helical density (orange) in presenilin, thought to be a composite of multiple substrates co-purified with the protease complex. This density appears to unwind and disappear as it approaches the active site (catalytic aspartates colored red). TMDs of presenilin are numbered, and loop 1 (L1) is indicated. Protein Data Bank entry 5FN3. (B) Structure of the enzyme bound to small peptide inhibitor DAPT. The protease complex assumes a conformation similar to the class 1 structure but without the unidentified density. A cavity formed where this density would be is indicated by the large purple arrow. Other parts of presenilin become visible or rearrange, most notably the cytoplasmic side of TMD6, which becomes kinked and extended. DAPT, which was only partly resolved binding near the active site, is not shown for the sake of clarity. Protein Data Bank entry 5FN2.
The enzyme complex was also analyzed by cryo-EM in the presence of an inhibitor called DAPT {N-[N-(3,5-difluor-ophenacetyl)-l-alanyl]-S-phenylglycine tert-butyl ester}, resulting in a structure of a single class determined at 4.2 Å overall resolution, with no general unknown helical density surrounded by PSEN1 TMDs130 (Figure 7B). While many side chains were unresolved, backbone features were clearly seen. Previous studies provided evidence that this tripeptide-like compound binds proximal to the enzyme active site,131,132 and indeed, a density attributed to the inhibitor was observed close to the catalytic aspartates. Parts of PSEN1 that had been unresolved in the apo structures now became clearly defined in the inhibitor-bound structure. These PSEN1 regions include TMD2 and its flanking loops L1 and L2 as well as the cytosolic sides of TMD3 and TMD6. Also resolved is part of large loop L6, which undergoes autoproteolysis during the maturation and activation of the γ-secretase complex. Specifically, the conserved hydrophobic region of L6 (E277–Y288), which represents the C-terminal end of the PSEN1 NTF subunit, appears to contribute to the enzyme active site, along with the now resolved cytosolic region of TMD6 (P263–Q276). The rest of L6, which is not conserved and comprises the N-terminus of the PSEN1 CTF, remains invisible. L1 is also relatively large and contains a conserved hydrophobic stretch. This loop dips in and out of the transmembrane region to interact with TMD2, TMD3, and TMD5.
Notably, the DAPT-bound structure shows a cavity bound by TMD2, TMD3, and TMD5–TMD7130 (see arrow in Figure 7B). In the class 1 and class 2 reconstructions of the apoenzyme, this cavity is occupied by the unaccounted for rodlike density attributed to bound, co-purified cellular substrates. Lining this cavity are many sites of FAD-causing PSEN1 missense mutations. Taken together, the DAPT-bound structure of γ-secretase suggests this inhibitor induces PSEN1 into a conformation similar to what it assumes upon substrate binding. Thus, this compound apparently inhibits the protease by trapping this conformation and precluding substrate binding.
Two new reports from the Shi laboratory provide the first detailed structures of the γ-secretase complex bound to APP and Notch substrates.94,95 Two modifications were needed to stably trap the substrate on the enzyme. The first was mutation of one of the active site aspartates to alanine, to prevent proteolysis of the bound substrate. The second was cysteine mutation of both the substrate and presenilin to allow disulfide cross-linking. The sites of Cys mutation were in the N-terminal extracellular region of the substrate and in L1 of PSEN1. Thus, the enzyme was catalytically inactive, and the substrate was covalently bound to the enzyme in a manner that increases the likelihood of artifacts. Nevertheless, the resulting enzyme–substrate complexes, with a bound substrate structure determined at 2.7 and 2.6 Å resolution for Notch-and APP-derived substrates, respectively, provide considerable insights into the nature of substrate recognition by γ-secretase that are consistent with previous studies, discussed earlier here, and extend these findings with specific substrates.
In both cases, the bound substrate is located in the cavity observed in the DAPT-bound complex and filled by the unaccounted for rodlike density in class 1 and class 2 structures of the apoenzyme (Figure 8). Substrate TMD assumes a helical conformation for the first 14 or 15 residues beginning at the extracellular side, and this helical region is surrounded by TMD2, TMD3, and TMD5. The conserved hydrophobic region of L1 that dips into the transmembrane region also appears to approach the substrate TMD, although this part of L1 was largely mutated to polyalanine, perhaps because this provided better cryo-EM images or better disulfide cross-linking between the substrate and enzyme. Examination of this region of L1 in the class 1 and class 2 apoenzyme structures, which ostensibly contain the bound unidentified substrate, appears to validate the idea that this part of L1 is directly involved in substrate binding. The structures of bound APP and Notch substrates differ from that of the co-purified generic substrate on the extracellular side. For the former, the substrate turns into the complex, as it is forced to do through disulfide cross-linking, while the latter kinks away from the complex. As the generic substrate is not artificially cross-linked, the conformation of the extracellular region may be closer to reality.
Figure 8.

Structures of γ-secretase bound to substrates. (A) γ-Secretase bound to Notch1-derived substrate. Protein Data Bank entry 6IDF. (B) γ-Secretase bound to the APP-derived substrate. Protein Data Bank entry 6IYC. In both structures, the substrate is located inside PSEN1 with the same basic arrangement as that seen in the class 1 apoenzyme with the unidentified density (Figure 7A). Both substrates are now resolved as they unwind and enter the catalytically disabled active site (PSEN1 TMD7 Asp mutated to Ala). Insets show the substrate TMD assumes a β-strand conformation near the cytoplasmic side, stabilized by a β-strand in PSEN1 TMD7, which is in turn stabilized by a short β-strand at the C-terminus of PSEN1 NTF.
Toward the cytosolic side of Notch and APP substrate TMD, the helical conformation ends just before entry into the active site, becoming first partially unwound and then fully extended into a β-strand. The β-strand of the substrate interacts with an antiparallel β-strand in the cytosolic region of TMD7, which in turn interacts with a β-strand in the conserved hydrophobic region at the C-terminus of PSEN1 NTF (part of what had been L6 before autoproteolysis) (see the insets in Figure 8). The β-strand of substrate TMD also interacts on its other side with an extended region of TMD9. Thus, the helical region of substrate TMD interacts with the PSEN1 NTF subunit, the β-strand region of substrate TMD interacts with the PS1 CTF subunit, and the region of the substrate in between, in the process of being unwound, interacts with both PSEN1 NTF and CTF. The unwinding of the substrate TMD helix in the active site and extension into a β-strand is consistent with biochemical expectations, as the scissile amide bond is not accessible when the substrate is in a helical conformation. These findings are also consistent with what is known about interactions of the substrate with soluble proteases. Substrates are generally bound to the active site in an extended conformation, although exceptions exist.133
The new structures with a bound substrate also revealed that many hot spots for Alzheimer’s disease-causing mutations in PSEN1 (positions with two or more different amino acid mutations134) directly interact with the substrate, for example, those in TMD2, TMD3, and TMD5 that surround the helical region of substrate TMD (Figure 9). In the DAPT-bound structure, these residues line the cavity filled by the substrate in the new co-structures. Other mutations are found elsewhere in the PSEN1 structure and may exert their effects indirectly, through allosteric mechanisms. Whether these mutations would be expected to affect substrate binding, helix unwinding, proteolysis in general, or specific proteolytic events is not clear solely on the basis of the structure of the inactive but otherwise wild-type enzyme with the cross-linked substrate. No Alzheimer’s disease presenilin mutation has yet been found to cause a complete loss of proteolytic function. The closest case is L435F in PSEN1.135 This leucine, part of the PAL motif, is found immediately proximal to both catalytic aspartates in the substrate–enzyme co-structure, in a way that makes it easy to envision that mutation to phenylalanine could disrupt the ability of these aspartates to work together to activate a water molecule. However, L435F PSEN1 shows a clear ability to produce Aβ43,136,137 and this specific Aβ peptide was found deposited in the post-mortem brains of two human carriers of this mutation.137 Such observations run counter to the presenilin loss-of-function hypothesis.138
Figure 9.

Familial Alzheimer’s disease (FAD) hot spots in PSEN1. (A) Sixty-one residues that are each mutated to two or more different amino acids in FAD are colored purple. Only PSEN1 and the APP substrate are shown for the sake of clarity. Note that many of these mutation sites appear to face or interact with the substrate. (B) Mutation sites that interact with the helical region of the substrate. (C) Mutation sites that interact, directly or indirectly, with the substrate in the active site and with the substrate β-strand. Protein Data Bank entry 6IYC.
UNANSWERED QUESTIONS
The new cryo-EM structures of the γ-secretase complex with bound Notch and APP substrates provide substantial insight into the nature of substrate recognition. In both cases, the substrate fills a cavity formed upon binding of DAPT, implying that DAPT inhibits the protease by trapping it in the conformation the protease otherwise assumes upon substrate binding. The co-structures of γ-secretase with substrate are also consistent with previous findings using chemical probes and mutagenesis, discussed above, with the bound substrate TMD in a helical conformation until unwinding and extension occur in the active site. The fascinating visualization of the details of bound substrate now raises important follow-up questions about substrate recognition and enzyme mechanism.
The first question is how γ-secretase activates water within the confines of the lipid bilayer for use in hydrolysis. So far, the catalytic aspartates have been not quite close enough or not resolved clearly enough to offer an answer. In the case of the new structures with the bound substrate, the active site has been disabled by mutation of one of the catalytic aspartates to alanine. Without both aspartates, coordination with a water molecule is not possible, and the bound substrates in the new structures are not poised to undergo proteolysis. The cross-linking of the substrate to presenilin in the complex also increases the potential for artifacts, with substrate adjusting itself for interaction with the enzyme in ways compatible with the artificial covalent attachment. The means of trapping the substrate to the enzyme without cross-linking or inactivation of the protease are needed to gain a more physiological picture of substrate recognition and intramembrane proteolysis.
The presence of both bound substrates surrounded by presenilin raises a second critical question. How does the substrate gain access to the interior of presenilin? These substrate-bound structures reveal neither the location of the docking site nor the path of lateral entry. Mobile TMD2 is likely part of the lateral gating mechanism, but this remains to be tested. Lateral gating of substrates into the active site of a rhomboid serine proteases was explored through extensive mutagenesis to identify point mutations that increase the level of proteolytic cleavage, by loosening the contact between rhomboid TMD2 and TMD5.139 Double-cysteine mutagenesis in both TMD2 and TMD5 allowed control of gate closing and opening through oxidation and reduction, respectively. Similar approaches could be taken to determine the substrate lateral gating pathway for γ-secretase. Regardless of the specific pathway for lateral gating, however, large loop L1 would appear to hinder substrate progress. This loop may swing upward toward the extracellular region to make way for substrate entry, followed by swinging back inward to clamp the substrate.
Unwinding of the substrate is apparently a critical step in preparing the scissile amide bond for attack by activated water. How this unwinding occurs is the third critical question regarding substrate recognition and processing. Perhaps a network of hydrogen bond donors and acceptors on presenilin work to disrupt the helical conformation of substrate TMD, replacing the intramolecular hydrogen bonding arrangement with intermolecular interactions. Backbone amide bond NH and C═O in PSEN1 would be good candidates, as these stabilize the extended conformation of the substrate in the active site. These regions of presenilin likely undergo major conformational changes themselves in the process of unwinding substrate TMD. The cytoplasmic side of TMD7 and the C-terminus of the PSEN1 NTF subunit are invisible in the absence of the substrate but become ordered and extended upon interacting with the extended region of the substrate.
A fourth major question is how γ-secretase carries out processive proteolysis after endoproteolytic cleavage. Some-how the substrate must be repositioned to trim the initially formed N-terminal cleavage product in intervals of three amino acids. The presence of three S′ pockets apparently dictates carboxypeptidase cleavage every three residues, but does the substrate reposition itself through random motions to refill these pockets? Or does the enzyme actively participate in further substrate unwinding? Doing so may involve opening up the presenilin subunit to carry out successive unwinding steps. Such conformational “breathing” may occur not only for the cytoplasmic side of TMD7 and the C-terminus of PSEN1 NTF but also for TMD2 and loop L1.
Beyond the mechanistic questions, there is also great interest in the implications of the new structures for the design of Alzheimer’s disease therapeutics. γ-Secretase inhibitors have failed in clinical trials for Alzheimer’s disease,140,141 in part due to a lack of selectivity for APP over Notch processing. Interference with proper Notch signaling results in immuno-suppression, gastrointestinal toxicity, and skin lesions. The new structures could provide the basis for the computer-aided design of inhibitors that are selective for blocking APP proteolysis over Notch. While some inhibitors apparently have such selectivity (so-called “Notch-sparing” γ-secretase inhibitors), one such compound also displayed toxicities connected to Notch signaling deficiencies in clinical trials,141 suggesting that selectivity was not sufficient to decrease the level of Aβ production in the brain without inhibiting Notch signaling in the periphery. In any event, the clinical failures of γ-secretase inhibitors are not only due to blocking Notch processing and signaling. Cognitive function actually worsened in Alzheimer’s disease subjects treated with inhibitors in these trials. Evidence from animal models suggests that cognitive worsening may be due to blocking of APP proteolysis and the resulting increase in levels of the APP-derived membrane-bound substrates.142
Rather than inhibition, modulation of γ-secretase activity would be preferred for the potential treatment of Alzheimer’s disease. Specifically, the goal would be to stimulate the carboxypeptidase trimming of long Aβ peptides to shorter versions, as this particular proteolytic function of γ-secretase is decreased in PSEN1 Alzheimer’s disease mutant γ-secretase complexes.86,87,143,144 Such modulators would neither increase substrate levels nor block processing of Notch and other critical γ-secretase substrates. Interestingly, such modulators were first identified in 2001 as agents that lower Aβ42 levels while increasing Aβ38 levels.145 It is now clear that these compounds specifically stimulate the γ-secretase-mediated trimming of Aβ42 to Aβ38,86 and potent next-generation modulators are apparently advancing into clinical trials.146–148 The success of such agents, however, depends on Aβ42 being the trigger of Alzheimer’s disease pathogenesis and the ability to therapeutically intervene early enough. Neither of these critical issues has been resolved, emphasizing the need for more basic biomedical research before pushing through therapeutic candidates. Toward this end, the discovery of γ-secretase modulators that affect other specific Aβ trimming steps would offer valuable chemical tools for addressing critical questions about the molecular basis of Alzheimer’s disease. Such chemical tools may also be therapeutic prototypes that provide in vivo proof of principle and essential target validation. The new advances in understanding the structure and mechanism of γ-secretase should greatly facilitate the design and discovery of such new chemical tools and drug prototypes.
The γ-secretase complex is a central enzyme in biology and medicine, with the ability to carry out processive proteolysis in the membrane. As such, the enzyme continues to fascinate on many levels. Our collective understanding of the structure and mechanism of this membrane-embedded proteolytic machine has advanced dramatically in recent years with the application of cutting-edge cryo-EM technology. The consistency of the new detailed structures with findings using small-molecule probes and mutagenesis is gratifying and reassuring. Further work is essential to elucidate critical open questions regarding structure and mechanism, how Alzheimer’s disease-causing mutations alter structure and function, and how new understanding can be leveraged for drug discovery. The new structures provide a solid platform for future advances on all of these important fronts.
Funding
M.S.W. is supported for this work through Grant GM122894 from the National Institutes of Health.
Footnotes
The author declares no competing financial interest.
REFERENCES
- (1).Wolfe MS, De Los Angeles J, Miller DD, Xia W, and Selkoe DJ (1999) Are presenilins intramembrane-cleaving proteases? Implications for the molecular mechanism of Alzheimer’s disease. Biochemistry 38, 11223–11230. [DOI] [PubMed] [Google Scholar]
- (2).Wolfe MS, and Kopan R (2004) Intramembrane proteolysis: theme and variations. Science 305, 1119–1123. [DOI] [PubMed] [Google Scholar]
- (3).Hooper NM, and Lendeckel U, Eds. (2007) Intramembrane-Cleaving Proteases (I-CLiPs), Vol. 6, Springer, Dordrecht, The Netherlands. [Google Scholar]
- (4).Freeman M (2014) The rhomboid-like superfamily: molecular mechanisms and biological roles. Annu. Rev. Cell Dev. Biol 30, 235–254. [DOI] [PubMed] [Google Scholar]
- (5).Rawson RB (2013) The site-2 protease. Biochim. Biophys. Acta, Biomembr 1828, 2801–2807. [DOI] [PubMed] [Google Scholar]
- (6).Schneider JS, and Glickman MS (2013) Function of site-2 proteases in bacteria and bacterial pathogens. Biochim. Biophys. Acta, Biomembr 1828, 2808–2814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Selkoe DJ (1994) Cell biology of the amyloid β-protein precursor and the mechanism of Alzheimer’s disease. Annu. Rev. Cell Biol 10, 373–403. [DOI] [PubMed] [Google Scholar]
- (8).Chartier-Harlin MC, Crawford F, Houlden H, Warren A, Hughes D, Fidani L, Goate A, Rossor M, Roques P, Hardy J, et al. (1991) Early-onset Alzheimer’s disease caused by mutations at codon 717 of the β-amyloid precursor protein gene. Nature 353, 844–846. [DOI] [PubMed] [Google Scholar]
- (9).Goate A, Chartier-Harlin MC, Mullan M, Brown J, Crawford F, Fidani L, Giuffra L, Haynes A, Irving N, James L, et al. (1991) Segregation of a missense mutation in the amyloid precursor protein gene with familial Alzheimer’s disease. Nature 349, 704–706. [DOI] [PubMed] [Google Scholar]
- (10).Sherrington R, Rogaev EI, Liang Y, Rogaeva EA, Levesque G, Ikeda M, Chi H, Lin C, Li G, Holman K, Tsuda T, Mar L, Foncin JF, Bruni AC, Montesi MP, Sorbi S, Rainero I, Pinessi L, Nee L, Chumakov I, Pollen D, Brookes A, Sanseau P, Polinsky RJ, Wasco W, Da Silva HA, Haines JL, Pericak-Vance MA, Tanzi RE, Roses AD, Fraser PE, Rommens JM, and St George-Hyslop PH (1995) Cloning of a gene bearing missense mutations in early-onset familial Alzheimer’s disease. Nature 375, 754–760. [DOI] [PubMed] [Google Scholar]
- (11).Rogaev EI, Sherrington R, Rogaeva EA, Levesque G, Ikeda M, Liang Y, Chi H, Lin C, Holman K, Tsuda T, Mar L, Sorbi S, Nacmias B, Piacentini S, Amaducci L, Chumakov I, Cohen D, Lannfelt L, Fraser PE, Rommens JM, and St George-Hyslop PH (1995) Familial Alzheimer’s disease in kindreds with missense mutations in a gene on chromosome 1 related to the Alzheimer’s disease type 3 gene. Nature 376, 775–778. [DOI] [PubMed] [Google Scholar]
- (12).L’Hernault SW, and Arduengo PM (1992) Mutation of a putative sperm membrane protein in Caenorhabditis elegans prevents sperm differentiation but not its associated meiotic divisions. J. Cell Biol 119, 55–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Borchelt DR, Thinakaran G, Eckman CB, Lee MK, Davenport F, Ratovitsky T, Prada CM, Kim G, Seekins S, Yager D, Slunt HH, Wang R, Seeger M, Levey AI, Gandy SE, Copeland NG, Jenkins NA, Price DL, Younkin SG, and Sisodia SS (1996) Familial Alzheimer’s disease-linked presenilin 1 variants elevate A β1–42/1–40 ratio in vitro and in vivo. Neuron 17, 1005–1013. [DOI] [PubMed] [Google Scholar]
- (14).Citron M, Westaway D, Xia W, Carlson G, Diehl T, Levesque G, Johnson-Wood K, Lee M, Seubert P, Davis A, Kholodenko D, Motter R, Sherrington R, Perry B, Yao H, Strome R, Lieberburg I, Rommens J, Kim S, Schenk D, Fraser P, St George Hyslop P, and Selkoe DJ (1997) Mutant presenilins of Alzheimer’s disease increase production of 42-residue amyloid β-protein in both transfected cells and transgenic mice. Nat. Med 3, 67–72. [DOI] [PubMed] [Google Scholar]
- (15).Tomita T, Maruyama K, Saido TC, Kume H, Shinozaki K, Tokuhiro S, Capell A, Walter J, Grunberg J, Haass C, Iwatsubo T, and Obata K (1997) The presenilin 2 mutation (N141I) linked to familial Alzheimer disease (Volga German families) increases the secretion of amyloid β protein ending at the 42nd (or 43rd) residue. Proc. Natl. Acad. Sci. U. S. A 94, 2025–2030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Xia W, Zhang J, Kholodenko D, Citron M, Podlisny MB, Teplow DB, Haass C, Seubert P, Koo EH, and Selkoe DJ (1997) Enhanced production and oligomerization of the 42-residue amyloid β-protein by Chinese hamster ovary cells stably expressing mutant presenilins. J. Biol. Chem 272, 7977–7982. [DOI] [PubMed] [Google Scholar]
- (17).Duff K, Eckman C, Zehr C, Yu X, Prada CM, Perez-Tur J, Hutton M, Buee L, Harigaya Y, Yager D, Morgan D, Gordon MN, Holcomb L, Refolo L, Zenk B, Hardy J, and Younkin S (1996) Increased amyloid-β42(43) in brains of mice expressing mutant presenilin 1. Nature 383, 710–713. [DOI] [PubMed] [Google Scholar]
- (18).Scheuner D, Eckman C, Jensen M, Song X, Citron M, Suzuki N, Bird TD, Hardy J, Hutton M, Kukull W, Larson E, Levy-Lahad E, Viitanen M, Peskind E, Poorkaj P, Schellenberg G, Tanzi R, Wasco W, Lannfelt L, Selkoe D, and Younkin S (1996) Secreted amyloid β-protein similar to that in the senile plaques of Alzheimer’s disease is increased in vivo by the presenilin 1 and 2 and APP mutations linked to familial Alzheimer’s disease. Nat. Med 2, 864–870. [DOI] [PubMed] [Google Scholar]
- (19).De Strooper B, Saftig P, Craessaerts K, Vanderstichele H, Guhde G, Annaert W, Von Figura K, and Van Leuven F (1998) Deficiency of presenilin-1 inhibits the normal cleavage of amyloid precursor protein. Nature 391, 387–390. [DOI] [PubMed] [Google Scholar]
- (20).Naruse S, Thinakaran G, Luo JJ, Kusiak JW, Tomita T, Iwatsubo T, Qian X, Ginty DD, Price DL, Borchelt DR, Wong PC, and Sisodia SS (1998) Effects of PS1 deficiency on membrane protein trafficking in neurons. Neuron 21, 1213–1221. [DOI] [PubMed] [Google Scholar]
- (21).Wolfe MS, Xia W, Moore CL, Leatherwood DD, Ostaszewski B, Rahmati T, Donkor IO, and Selkoe DJ (1999) Peptidomimetic probes and molecular modeling suggest Alzheimer’s γ-secretases are intramembrane-cleaving aspartyl proteases. Biochemistry 38, 4720–4727. [DOI] [PubMed] [Google Scholar]
- (22).Wolfe MS, Xia W, Ostaszewski BL, Diehl TS, Kimberly WT, and Selkoe DJ (1999) Two transmembrane aspartates in presenilin-1 required for presenilin endoproteolysis and γ-secretase activity. Nature 398, 513–517. [DOI] [PubMed] [Google Scholar]
- (23).Li YM, Xu M, Lai MT, Huang Q, Castro JL, DiMuzio-Mower J, Harrison T, Lellis C, Nadin A, Neduvelil JG, Register RB, Sardana MK, Shearman MS, Smith AL, Shi XP, Yin KC, Shafer JA, and Gardell SJ (2000) Photoactivated γ-secretase inhibitors directed to the active site covalently label presenilin 1. Nature 405, 689–694. [DOI] [PubMed] [Google Scholar]
- (24).Esler WP, Kimberly WT, Ostaszewski BL, Diehl TS, Moore CL, Tsai J-Y, Rahmati T, Xia W, Selkoe DJ, and Wolfe MS (2000) Transition-state analogue inhibitors of γ-secretase bind directly to presenilin-1. Nat. Cell Biol 2, 428–434. [DOI] [PubMed] [Google Scholar]
- (25).Levitan D, and Greenwald I (1995) Facilitation of lin-12-mediated signalling by sel-12, a Caenorhabditis elegans S182 Alzheimer’s disease gene. Nature 377, 351–354. [DOI] [PubMed] [Google Scholar]
- (26).Shen J, Bronson RT, Chen DF, Xia W, Selkoe DJ, and Tonegawa S (1997) Skeletal and CNS defects in Presenilin-1-deficient mice. Cell 89, 629–639. [DOI] [PubMed] [Google Scholar]
- (27).Wong PC, Zheng H, Chen H, Becher MW, Sirinathsinghji DJ, Trumbauer ME, Chen HY, Price DL, Van der Ploeg LH, and Sisodia SS (1997) Presenilin 1 is required for Notch1 and DII1 expression in the paraxial mesoderm. Nature 387, 288–292. [DOI] [PubMed] [Google Scholar]
- (28).Kopan R, and Ilagan MX (2009) The canonical Notch signaling pathway: unfolding the activation mechanism. Cell 137, 216–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Schroeter EH, Kisslinger JA, and Kopan R (1998) Notch-1 signalling requires ligand-induced proteolytic release of intracellular domain. Nature 393, 382–386. [DOI] [PubMed] [Google Scholar]
- (30).De Strooper B, Annaert W, Cupers P, Saftig P, Craessaerts K, Mumm JS, Schroeter EH, Schrijvers V, Wolfe MS, Ray WJ, Goate A, and Kopan R (1999) A presenilin-1-dependent γ-secretase-like protease mediates release of Notch intracellular domain. Nature 398, 518–522. [DOI] [PubMed] [Google Scholar]
- (31).Struhl G, and Greenwald I (1999) Presenilin is required for activity and nuclear access of Notch in Drosophila. Nature 398, 522–525. [DOI] [PubMed] [Google Scholar]
- (32).Steiner H, Duff K, Capell A, Romig H, Grim MG, Lincoln S, Hardy J, Yu X, Picciano M, Fechteler K, Citron M, Kopan R, Pesold B, Keck S, Baader M, Tomita T, Iwatsubo T, Baumeister R, and Haass C (1999) A loss of function mutation of presenilin-2 interferes with amyloid β-peptide production and notch signaling. J. Biol. Chem 274, 28669–28673. [DOI] [PubMed] [Google Scholar]
- (33).Kimberly WT, Xia W, Rahmati T, Wolfe MS, and Selkoe DJ (2000) The transmembrane aspartates in presenilin 1 and 2 are obligatory for γ-secretase activity and amyloid β-protein generation. J. Biol. Chem 275, 3173–3178. [DOI] [PubMed] [Google Scholar]
- (34).Leimer U, Lun K, Romig H, Walter J, Grunberg J, Brand M, and Haass C (1999) Zebrafish (Danio rerio) presenilin promotes aberrant amyloid β-peptide production and requires a critical aspartate residue for its function in amyloidogenesis. Biochemistry 38, 13602–13609. [DOI] [PubMed] [Google Scholar]
- (35).LaPointe CF, and Taylor RK (2000) The type 4 prepilin peptidases comprise a novel family of aspartic acid proteases. J. Biol. Chem 275, 1502–1510. [DOI] [PubMed] [Google Scholar]
- (36).Steiner H, Kostka M, Romig H, Basset G, Pesold B, Hardy J, Capell A, Meyn L, Grim ML, Baumeister R, Fechteler K, and Haass C (2000) Glycine 384 is required for presenilin-1 function and is conserved in bacterial polytopic aspartyl proteases. Nat. Cell Biol 2, 848–851. [DOI] [PubMed] [Google Scholar]
- (37).Weihofen A, Binns K, Lemberg MK, Ashman K, and Martoglio B (2002) Identification of signal peptide peptidase, a presenilin-type aspartic protease. Science 296, 2215–2218. [DOI] [PubMed] [Google Scholar]
- (38).Ponting CP, Hutton M, Nyborg A, Baker M, Jansen K, and Golde TE (2002) Identification of a novel family of presenilin homologues. Hum. Mol. Genet 11, 1037–1044. [DOI] [PubMed] [Google Scholar]
- (39).Thinakaran G, Borchelt DR, Lee MK, Slunt HH, Spitzer L, Kim G, Ratovitsky T, Davenport F, Nordstedt C, Seeger M, Hardy J, Levey AI, Gandy SE, Jenkins NA, Copeland NG, Price DL, and Sisodia SS (1996) Endoproteolysis of presenilin 1 and accumulation of processed derivatives in vivo. Neuron 17, 181–190. [DOI] [PubMed] [Google Scholar]
- (40).Ratovitski T, Slunt HH, Thinakaran G, Price DL, Sisodia SS, and Borchelt DR (1997) Endoproteolytic processing and stabilization of wild-type and mutant presenilin. J. Biol. Chem 272, 24536–24541. [DOI] [PubMed] [Google Scholar]
- (41).Podlisny MB, Citron M, Amarante P, Sherrington R, Xia W, Zhang J, Diehl T, Levesque G, Fraser P, Haass C, Koo EH, Seubert P, George-Hyslop P St., Teplow DB, and Selkoe DJ (1997) Presenilin proteins undergo heterogeneous endoproteolysis between Thr291 and Ala299 and occur as stable N-and C-terminal fragments in normal and Alzheimer brain tissue. Neurobiol. Dis 3, 325–337. [DOI] [PubMed] [Google Scholar]
- (42).Thinakaran G, Harris CL, Ratovitski T, Davenport F, Slunt HH, Price DL, Borchelt DR, and Sisodia SS (1997) Evidence that levels of presenilins (PS1 and PS2) are coordinately regulated by competition for limiting cellular factors. J. Biol. Chem 272, 28415–28422. [DOI] [PubMed] [Google Scholar]
- (43).Steiner H, Capell A, Pesold B, Citron M, Kloetzel PM, Selkoe DJ, Romig H, Mendla K, and Haass C (1998) Expression of Alzheimer’s disease-associated presenilin-1 is controlled by proteolytic degradation and complex formation. J. Biol. Chem 273, 32322–32331. [DOI] [PubMed] [Google Scholar]
- (44).Seeger M, Nordstedt C, Petanceska S, Kovacs DM, Gouras GK, Hahne S, Fraser P, Levesque L, Czernik AJ, George-Hyslop PS, Sisodia SS, Thinakaran G, Tanzi RE, Greengard P, and Gandy S (1997) Evidence for phosphorylation and oligomeric assembly of presenilin 1. Proc. Natl. Acad. Sci. U. S. A 94, 5090–5094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Capell A, Grunberg J, Pesold B, Diehlmann A, Citron M, Nixon R, Beyreuther K, Selkoe DJ, and Haass C (1998) The proteolytic fragments of the Alzheimer’s disease-associated presenilin-1 form heterodimers and occur as a 100–150-kDa molecular mass complex. J. Biol. Chem 273, 3205–3211. [DOI] [PubMed] [Google Scholar]
- (46).Yu G, Chen F, Levesque G, Nishimura M, Zhang DM, Levesque L, Rogaeva E, Xu D, Liang Y, Duthie M, George-Hyslop PH St., and Fraser PE (1998) The presenilin 1 protein is a component of a high molecular weight intracellular complex that contains β-catenin. J. Biol. Chem 273, 16470–16475. [DOI] [PubMed] [Google Scholar]
- (47).Yu G, Nishimura M, Arawaka S, Levitan D, Zhang L, Tandon A, Song YQ, Rogaeva E, Chen F, Kawarai T, Supala A, Levesque L, Yu H, Yang DS, Holmes E, Milman P, Liang Y, Zhang DM, Xu DH, Sato C, Rogaev E, Smith M, Janus C, Zhang Y, Aebersold R, Farrer LS, Sorbi S, Bruni A, Fraser P, and St George-Hyslop P (2000) Nicastrin modulates presenilin-mediated notch/glp-1 signal transduction and betaAPP processing. Nature 407, 48–54. [DOI] [PubMed] [Google Scholar]
- (48).Goutte C, Tsunozaki M, Hale VA, and Priess JR (2002) APH-1 is a multipass membrane protein essential for the Notch signaling pathway in Caenorhabditis elegans embryos. Proc. Natl. Acad. Sci. U. S. A 99, 775–779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Gu Y, Chen F, Sanjo N, Kawarai T, Hasegawa H, Duthie M, Li W, Ruan X, Luthra A, Mount HT, Tandon A, Fraser PE, and St George-Hyslop P (2003) APH-1 interacts with mature and immature forms of presenilins and nicastrin and may play a role in maturation of presenilin-nicastrin complexes. J. Biol. Chem 278, 7374–7380. [DOI] [PubMed] [Google Scholar]
- (50).Lee SF, Shah S, Li H, Yu C, Han W, and Yu G (2002) Mammalian APH-1 interacts with presenilin and nicastrin and is required for intramembrane proteolysis of amyloid-β precursor protein and Notch. J. Biol. Chem 277, 45013–45019. [DOI] [PubMed] [Google Scholar]
- (51).Francis R, McGrath G, Zhang J, Ruddy DA, Sym M, Apfeld J, Nicoll M, Maxwell M, Hai B, Ellis MC, Parks AL, Xu W, Li J, Gurney M, Myers RL, Himes CS, Hiebsch R, Ruble C, Nye JS, and Curtis D (2002) aph-1 and pen-2 are required for Notch pathway signaling, γ-secretase cleavage of betaAPP, and presenilin protein accumulation. Dev. Cell 3, 85–97. [DOI] [PubMed] [Google Scholar]
- (52).Steiner H, Winkler E, Edbauer D, Prokop S, Basset G, Yamasaki A, Kostka M, and Haass C (2002) PEN-2 is an integral component of the γ-secretase complex required for coordinated expression of presenilin and nicastrin. J. Biol. Chem 277, 39062–39065. [DOI] [PubMed] [Google Scholar]
- (53).Takasugi N, Tomita T, Hayashi I, Tsuruoka M, Niimura M, Takahashi Y, Thinakaran G, and Iwatsubo T (2003) The role of presenilin cofactors in the γ-secretase complex. Nature 422, 438–441. [DOI] [PubMed] [Google Scholar]
- (54).Kimberly WT, LaVoie MJ, Ostaszewski BL, Ye W, Wolfe MS, and Selkoe DJ (2003) γ-Secretase is a membrane protein complex comprised of presenilin, nicastrin, aph-1, and pen-2. Proc. Natl. Acad. Sci. U. S. A 100, 6382–6387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Edbauer D, Winkler E, Regula JT, Pesold B, Steiner H, and Haass C (2003) Reconstitution of γ-secretase activity. Nat. Cell Biol 5, 486–488. [DOI] [PubMed] [Google Scholar]
- (56).Beel AJ, and Sanders CR (2008) Substrate specificity of γ-secretase and other intramembrane proteases. Cell. Mol. Life Sci 65, 1311–1334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (57).Haapasalo A, and Kovacs DM (2011) The many substrates of presenilin/γ-secretase. J. Alzheimer’s Dis 25, 3–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (58).Hemming ML, Elias JE, Gygi SP, and Selkoe DJ (2008) Proteomic profiling of γ-secretase substrates and mapping of substrate requirements. PLoS Biol 6, No. e257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (59).Sardi SP, Murtie J, Koirala S, Patten BA, and Corfas G (2006) Presenilin-dependent ErbB4 nuclear signaling regulates the timing of astrogenesis in the developing brain. Cell 127, 185–197. [DOI] [PubMed] [Google Scholar]
- (60).Marambaud P, Wen PH, Dutt A, Shioi J, Takashima A, Siman R, and Robakis NK (2003) A CBP binding transcriptional repressor produced by the PS1/epsilon-cleavage of N-cadherin is inhibited by PS1 FAD mutations. Cell 114, 635–645. [DOI] [PubMed] [Google Scholar]
- (61).Kopan R, and Ilagan MX (2004) γ-secretase: proteasome of the membrane? Nat. Rev. Mol. Cell Biol 5, 499–504. [DOI] [PubMed] [Google Scholar]
- (62).Gu Y, Misonou H, Sato T, Dohmae N, Takio K, and Ihara Y (2001) Distinct intramembrane cleavage of the β-amyloid precursor protein family resembling γ-secretase-like cleavage of Notch. J. Biol. Chem 276, 35235–35238. [DOI] [PubMed] [Google Scholar]
- (63).Yu C, Kim SH, Ikeuchi T, Xu H, Gasparini L, Wang R, and Sisodia SS (2001) Characterization of a presenilin-mediated amyloid precursor protein carboxyl-terminal fragment γ. Evidence for distinct mechanisms involved in γ-secretase processing of the APP and Notch1 transmembrane domains. J. Biol. Chem 276, 43756–43760. [DOI] [PubMed] [Google Scholar]
- (64).Weidemann A, Eggert S, Reinhard FB, Vogel M, Paliga K, Baier G, Masters CL, Beyreuther K, and Evin G (2002) A novel var epsilon-cleavage within the transmembrane domain of the Alzheimer amyloid precursor protein demonstrates homology with Notch processing. Biochemistry 41, 2825–2835. [DOI] [PubMed] [Google Scholar]
- (65).Sato T, Dohmae N, Qi Y, Kakuda N, Misonou H, Mitsumori R, Maruyama H, Koo EH, Haass C, Takio K, Morishima-Kawashima M, Ishiura S, and Ihara Y (2003) Potential link between amyloid β-protein 42 and C-terminal fragment γ 49–99 of β-amyloid precursor protein. J. Biol. Chem 278, 24294–24301. [DOI] [PubMed] [Google Scholar]
- (66).Sastre M, Steiner H, Fuchs K, Capell A, Multhaup G, Condron MM, Teplow DB, and Haass C (2001) Presenilin-dependent γ-secretase processing of β-amyloid precursor protein at a site corresponding to the S3 cleavage of Notch. EMBO Rep 2, 835–841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (67).Funamoto S, Morishima-Kawashima M, Tanimura Y, Hirotani N, Saido TC, and Ihara Y (2004) Truncated carboxyl-terminal fragments of β-amyloid precursor protein are processed to amyloid β-proteins 40 and 42. Biochemistry 43, 13532–13540. [DOI] [PubMed] [Google Scholar]
- (68).Fernandez MA, Klutkowski JA, Freret T, and Wolfe MS (2014) Alzheimer Presenilin-1 Mutations Dramatically Reduce Trimming of Long Amyloid β-Peptides (Aβ) by γ-Secretase to Increase 42-to-40-Residue Aβ. J. Biol. Chem 289, 31043–31052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (69).Qi-Takahara Y, Morishima-Kawashima M, Tanimura Y, Dolios G, Hirotani N, Horikoshi Y, Kametani F, Maeda M, Saido TC, Wang R, and Ihara Y (2005) Longer forms of amyloid β protein: implications for the mechanism of intramembrane cleavage by γ-secretase. J. Neurosci 25, 436–445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (70).Yagishita S, Morishima-Kawashima M, Ishiura S, and Ihara Y (2008) Aβ46 is processed to Aβ40 and Aβ43, but not to Aβ42, in the low density membrane domains. J. Biol. Chem 283, 733–738. [DOI] [PubMed] [Google Scholar]
- (71).Takami M, Nagashima Y, Sano Y, Ishihara S, Morishima-Kawashima M, Funamoto S, and Ihara Y (2009) γ-Secretase: successive tripeptide and tetrapeptide release from the transmembrane domain of β-carboxyl terminal fragment. J. Neurosci 29, 13042–13052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (72).Fukumori A, Fluhrer R, Steiner H, and Haass C (2010) Three-amino acid spacing of presenilin endoproteolysis suggests a general stepwise cleavage of γ-secretase-mediated intramembrane proteolysis. J. Neurosci 30, 7853–7862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (73).Okochi M, Steiner H, Fukumori A, Tanii H, Tomita T, Tanaka T, Iwatsubo T, Kudo T, Takeda M, and Haass C (2002) Presenilins mediate a dual intramembranous γ-secretase cleavage of Notch-1. EMBO J 21, 5408–5416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (74).Sato T, Diehl TS, Narayanan S, Funamoto S, Ihara Y, De Strooper B, Steiner H, Haass C, and Wolfe MS (2007) Active γ-secretase complexes contain only one of each component. J. Biol. Chem 282, 33985–33993. [DOI] [PubMed] [Google Scholar]
- (75).Wolfe MS, Citron M, Diehl TS, Xia W, Donkor IO, and Selkoe DJ (1998) A substrate-based difluoro ketone selectively inhibits Alzheimer’s γ-secretase activity. J. Med. Chem 41, 6–9. [DOI] [PubMed] [Google Scholar]
- (76).Thaisrivongs S, Pals DT, Kati WM, Turner SR, Thomasco LM, and Watt W (1986) Design and synthesis of potent and specific renin inhibitors containing difluorostatine, difluorostatone, and related analogues. J. Med. Chem 29, 2080–2087. [DOI] [PubMed] [Google Scholar]
- (77).Moore CL, Leatherwood DD, Diehl TS, Selkoe DJ, and Wolfe MS (2000) Difluoro Ketone Peptidomimetics Suggest a Large S1 Pocket for Alzheimer’s γ-Secretase: Implications for Inhibitor Design. J. Med. Chem 43, 3434–3442. [DOI] [PubMed] [Google Scholar]
- (78).Esler WP, Das C, and Wolfe MS (2004) Probing pockets S2-S4′ of the γ-secretase active site with (hydroxyethyl)urea peptidomimetics. Bioorg. Med. Chem. Lett 14, 1935–1938. [DOI] [PubMed] [Google Scholar]
- (79).Bolduc DM, Montagna DR, Seghers MC, Wolfe MS, and Selkoe DJ (2016) The amyloid-β forming tripeptide cleavage mechanism of γ-secretase. eLife 5, No. e17578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (80).Das C, Berezovska O, Diehl TS, Genet C, Buldyrev I, Tsai JY, Hyman BT, and Wolfe MS (2003) Designed helical peptides inhibit an intramembrane protease. J. Am. Chem. Soc 125, 11794–11795. [DOI] [PubMed] [Google Scholar]
- (81).Bihel F, Das C, Bowman MJ, and Wolfe MS (2004) Discovery of a subnanomolar helical D-tridecapeptide inhibitor of γ-secretase. J. Med. Chem 47, 3931–3933. [DOI] [PubMed] [Google Scholar]
- (82).Kornilova AY, Bihel F, Das C, and Wolfe MS (2005) The initial substrate-binding site of γ-secretase is located on presenilin near the active site. Proc. Natl. Acad. Sci. U. S. A 102, 3230–3235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (83).Fukumori A, and Steiner H (2016) Substrate recruitment of γ-secretase and mechanism of clinical presenilin mutations revealed by photoaffinity mapping. EMBO J 35, 1628–1643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (84).Esler WP, Kimberly WT, Ostaszewski BL, Ye W, Diehl TS, Selkoe DJ, and Wolfe MS (2002) Activity-dependent isolation of the presenilin/γ-secretase complex reveals nicastrin and a γ substrate. Proc. Natl. Acad. Sci. U. S. A 99, 2720–2725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (85).Lichtenthaler SF, Wang R, Grimm H, Uljon SN, Masters CL, and Beyreuther K (1999) Mechanism of the cleavage specificity of Alzheimer’s disease γ-secretase identified by phenyl-alanine-scanning mutagenesis of the transmembrane domain of the amyloid precursor protein. Proc. Natl. Acad. Sci. U. S. A 96, 3053–3058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (86).Okochi M, Tagami S, Yanagida K, Takami M, Kodama TS, Mori K, Nakayama T, Ihara Y, and Takeda M (2013) γ-secretase modulators and presenilin 1 mutants act differently on presenilin/γ-secretase function to cleave Aβ42 and Aβ43. Cell Rep 3, 42–51. [DOI] [PubMed] [Google Scholar]
- (87).Szaruga M, Munteanu B, Lismont S, Veugelen S, Horre K, Mercken M, Saido TC, Ryan NS, De Vos T, Savvides SN, Gallardo R, Schymkowitz J, Rousseau F, Fox NC, Hopf C, De Strooper B, and Chavez-Gutierrez L (2017) Alzheimer’s-causing mutations shift Aβ length by destabilizing γ-secretase-Aβn interactions. Cell 170, 443–456. [DOI] [PubMed] [Google Scholar]
- (88).Sato C, Morohashi Y, Tomita T, and Iwatsubo T (2006) Structure of the catalytic pore of γ-secretase probed by the accessibility of substituted cysteines. J. Neurosci 26, 12081–12088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (89).Tolia A, Chavez-Gutierrez L, and De Strooper B (2006) Contribution of presenilin transmembrane domains 6 and 7 to a water-containing cavity in the γ-secretase complex. J. Biol. Chem 281, 27633–27642. [DOI] [PubMed] [Google Scholar]
- (90).Kornilova AY, Kim J, Laudon H, and Wolfe MS (2006) Deducing the transmembrane domain organization of presenilin-1 in γ-secretase by cysteine disulfide cross-linking. Biochemistry 45, 7598–7604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (91).Bai XC, Yan C, Yang G, Lu P, Ma D, Sun L, Zhou R, Scheres SH, and Shi Y (2015) An atomic structure of human γ-secretase. Nature 525, 212–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (92).Tomita T (2017) Probing the structure and function relationships of presenilin by substituted-cysteine accessibility method. Methods Enzymol 584, 185–205. [DOI] [PubMed] [Google Scholar]
- (93).Takagi-Niidome S, Sasaki T, Osawa S, Sato T, Morishima K, Cai T, Iwatsubo T, and Tomita T (2015) Cooperative roles of hydrophilic loop 1 and the C-terminus of presenilin 1 in the substrate-gating mechanism of γ-secretase. J. Neurosci 35, 2646–2656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (94).Yang G, Zhou R, Zhou Q, Guo X, Yan C, Ke M, Lei J, and Shi Y (2019) Structural basis of Notch recognition by human γ-secretase. Nature 565, 192–197. [DOI] [PubMed] [Google Scholar]
- (95).Zhou R, Yang G, Guo X, Zhou Q, Lei J, and Shi Y (2019) Recognition of the amyloid precursor protein by human γ-secretase. Science 363, No. eaaw0930. [DOI] [PubMed] [Google Scholar]
- (96).Takeo K, Watanabe N, Tomita T, and Iwatsubo T (2012) Contribution of the γ-secretase subunits to the formation of catalytic pore of presenilin 1 protein. J. Biol. Chem 287, 25834–25843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (97).Sato C, Takagi S, Tomita T, and Iwatsubo T (2008) The C-terminal PAL motif and transmembrane domain 9 of presenilin 1 are involved in the formation of the catalytic pore of the γ-secretase. J. Neurosci 28, 6264–6271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (98).Yamasaki A, Eimer S, Okochi M, Smialowska A, Kaether C, Baumeister R, Haass C, and Steiner H (2006) The GxGD motif of presenilin contributes to catalytic function and substrate identification of γ-secretase. J. Neurosci 26, 3821–3828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (99).Perez-Revuelta BI, Fukumori A, Lammich S, Yamasaki A, Haass C, and Steiner H (2010) Requirement for small side chain residues within the GxGD-motif of presenilin for γ-secretase substrate cleavage. J. Neurochem 112, 940–950. [DOI] [PubMed] [Google Scholar]
- (100).Kretner B, Fukumori A, Kuhn PH, Perez-Revuelta BI, Lichtenthaler SF, Haass C, and Steiner H (2013) Important functional role of residue x of the presenilin GxGD protease active site motif for APP substrate cleavage specificity and substrate selectivity of γ-secretase. J. Neurochem 125, 144–156. [DOI] [PubMed] [Google Scholar]
- (101).Wang J, Brunkan AL, Hecimovic S, Walker E, and Goate A (2004) Conserved “PAL” sequence in presenilins is essential for γ-secretase activity, but not required for formation or stabilization of γ-secretase complexes. Neurobiol. Dis 15, 654–666. [DOI] [PubMed] [Google Scholar]
- (102).Wang J, Beher D, Nyborg AC, Shearman MS, Golde TE, and Goate A (2006) C-terminal PAL motif of presenilin and presenilin homologues required for normal active site conformation. J. Neurochem 96, 218–227. [DOI] [PubMed] [Google Scholar]
- (103).Gong P, Vetrivel KS, Nguyen PD, Meckler X, Cheng H, Kounnas MZ, Wagner SL, Parent AT, and Thinakaran G (2010) Mutation analysis of the presenilin 1 N-terminal domain reveals a broad spectrum of γ-secretase activity toward amyloid precursor protein and other substrates. J. Biol. Chem 285, 38042–38052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (104).Fraering PC, LaVoie MJ, Ye W, Ostaszewski BL, Kimberly WT, Selkoe DJ, and Wolfe MS (2004) Detergent-dependent dissociation of active γ-secretase reveals an interaction between Pen-2 and PS1-NTF and offers a model for subunit organization within the complex. Biochemistry 43, 323–333. [DOI] [PubMed] [Google Scholar]
- (105).Steiner H, Winkler E, and Haass C (2008) Chemical crosslinking provides a model of the γ-secretase complex subunit architecture and evidence for close proximity of the C-terminal fragment of presenilin with APH-1. J. Biol. Chem 283, 34677–34686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (106).LaVoie MJ, Fraering PC, Ostaszewski BL, Ye W, Kimberly WT, Wolfe MS, and Selkoe DJ (2003) Assembly of the γ-secretase complex involves early formation of an intermediate subcomplex of Aph-1 and nicastrin. J. Biol. Chem 278, 37213–37222. [DOI] [PubMed] [Google Scholar]
- (107).Luo WJ, Wang H, Li H, Kim BS, Shah S, Lee HJ, Thinakaran G, Kim TW, Yu G, and Xu H (2003) PEN-2 and APH-1 coordinately regulate proteolytic processing of presenilin 1. J. Biol. Chem 278, 7850–7854. [DOI] [PubMed] [Google Scholar]
- (108).Kim SH, and Sisodia SS (2005) Evidence that the “NF” motif in transmembrane domain 4 of presenilin 1 is critical for binding with PEN-2. J. Biol. Chem 280, 41953–41966. [DOI] [PubMed] [Google Scholar]
- (109).Watanabe N, Tomita T, Sato C, Kitamura T, Morohashi Y, and Iwatsubo T (2005) Pen-2 is incorporated into the γ-secretase complex through binding to transmembrane domain 4 of presenilin 1. J. Biol. Chem 280, 41967–41975. [DOI] [PubMed] [Google Scholar]
- (110).Fagan R, Swindells M, Overington J, and Weir M (2001) Nicastrin, a presenilin-interacting protein, contains an amino-peptidase/transferrin receptor superfamily domain. Trends Biochem. Sci 26, 213–214. [DOI] [PubMed] [Google Scholar]
- (111).Fergani A, Yu G, George-Hyslop P St., and Checler F (2001) Wild-type and mutated nicastrins do not display amino-peptidase M-and B-like activities. Biochem. Biophys. Res. Commun 289, 678–680. [DOI] [PubMed] [Google Scholar]
- (112).Shah S, Lee SF, Tabuchi K, Hao YH, Yu C, LaPlant Q, Ball H, Dann CE 3rd, Sudhof T, and Yu G (2005) Nicastrin functions as a γ-secretase-substrate receptor. Cell 122, 435–447. [DOI] [PubMed] [Google Scholar]
- (113).Chavez-Gutierrez L, Tolia A, Maes E, Li T, Wong PC, and de Strooper B (2008) Glu 332 in the Nicastrin ectodomain is essential for γ-secretase complex maturation but not for its activity. J. Biol. Chem 283, 20096–20105. [DOI] [PubMed] [Google Scholar]
- (114).Bolduc DM, Montagna DR, Gu Y, Selkoe DJ, and Wolfe MS (2016) Nicastrin functions to sterically hinder γ-secretase-substrate interactions driven by substrate transmembrane domain. Proc. Natl. Acad. Sci. U. S. A 113, E509–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (115).Struhl G, and Adachi A (2000) Requirements for presenilin-dependent cleavage of notch and other transmembrane proteins. Mol. Cell 6, 625–636. [DOI] [PubMed] [Google Scholar]
- (116).Funamoto S, Sasaki T, Ishihara S, Nobuhara M, Nakano M, Watanabe-Takahashi M, Saito T, Kakuda N, Miyasaka T, Nishikawa K, Saido TC, and Ihara Y (2013) Substrate ectodomain is critical for substrate preference and inhibition of γ-secretase. Nat. Commun 4, 2529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (117).Quentin D, and Raunser S (2018) Electron cryomicroscopy as a powerful tool in biomedical research. J. Mol. Med 96, 483–493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (118).Wang Y, Zhang Y, and Ha Y (2006) Crystal structure of a rhomboid family intramembrane protease. Nature 444, 179–180. [DOI] [PubMed] [Google Scholar]
- (119).Wu Z, Yan N, Feng L, Oberstein A, Yan H, Baker RP, Gu L, Jeffrey PD, Urban S, and Shi Y (2006) Structural analysis of a rhomboid family intramembrane protease reveals a gating mechanism for substrate entry. Nat. Struct. Mol. Biol 13, 1084–1091. [DOI] [PubMed] [Google Scholar]
- (120).Feng L, Yan H, Wu Z, Yan N, Wang Z, Jeffrey PD, and Shi Y (2007) Structure of a site-2 protease family intramembrane metalloprotease. Science 318, 1608–1612. [DOI] [PubMed] [Google Scholar]
- (121).Li X, Dang S, Yan C, Gong X, Wang J, and Shi Y (2013) Structure of a presenilin family intramembrane aspartate protease. Nature 493, 56–61. [DOI] [PubMed] [Google Scholar]
- (122).Lazarov VK, Fraering PC, Ye W, Wolfe MS, Selkoe DJ, and Li H (2006) Electron microscopic structure of purified, active γ-secretase reveals an aqueous intramembrane chamber and two pores. Proc. Natl. Acad. Sci. U. S. A 103, 6889–6894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (123).Osenkowski P, Li H, Ye W, Li D, Aeschbach L, Fraering PC, Wolfe MS, Selkoe DJ, and Li H (2009) Cryoelectron microscopy structure of purified γ-secretase at 12 Å resolution. J. Mol. Biol 385, 642–652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (124).Renzi F, Zhang X, Rice WJ, Torres-Arancivia C, Gomez-Llorente Y, Diaz R, Ahn K, Yu C, Li YM, Sisodia SS, and Ubarretxena-Belandia I (2011) Structure of γ-secretase and its trimeric pre-activation intermediate by single-particle electron microscopy. J. Biol. Chem 286, 21440–21449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (125).Ogura T, Mio K, Hayashi I, Miyashita H, Fukuda R, Kopan R, Kodama T, Hamakubo T, Iwastubo T, Tomita T, and Sato C (2006) Three-dimensional structure of the γ-secretase complex. Biochem. Biophys. Res. Commun 343, 525–534. [DOI] [PubMed] [Google Scholar]
- (126).Li Y, Lu SH, Tsai CJ, Bohm C, Qamar S, Dodd RB, Meadows W, Jeon A, McLeod A, Chen F, Arimon M, Berezovska O, Hyman BT, Tomita T, Iwatsubo T, Johnson CM, Farrer LA, Schmitt-Ulms G, Fraser PE, and St George-Hyslop PH (2014) Structural interactions between inhibitor and substrate docking sites give insight into mechanisms of human PS1 complexes. Structure 22, 125–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (127).Elad N, De Strooper B, Lismont S, Hagen W, Veugelen S, Arimon M, Horre K, Berezovska O, Sachse C, and Chavez-Gutierrez L (2015) The dynamic conformational landscape of γ-secretase. J. Cell Sci 128, 589–598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (128).Lu P, Bai XC, Ma D, Xie T, Yan C, Sun L, Yang G, Zhao Y, Zhou R, Scheres SH, and Shi Y (2014) Three-dimensional structure of human γ-secretase. Nature 512, 166–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (129).Xie T, Yan C, Zhou R, Zhao Y, Sun L, Yang G, Lu P, Ma D, and Shi Y (2014) Crystal structure of the γ-secretase component nicastrin. Proc. Natl. Acad. Sci. U. S. A 111, 13349–13354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (130).Bai XC, Rajendra E, Yang G, Shi Y, and Scheres SH (2015) Sampling the conformational space of the catalytic subunit of human γ-secretase. eLife 4 (pii), No. e11182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (131).Kornilova AY, Das C, and Wolfe MS (2003) Differential effects of inhibitors on the γ-secretase complex. Mechanistic implications. J. Biol. Chem 278, 16470–16473. [DOI] [PubMed] [Google Scholar]
- (132).Morohashi Y, Kan T, Tominari Y, Fuwa H, Okamura Y, Watanabe N, Sato C, Natsugari H, Fukuyama T, Iwatsubo T, and Tomita T (2006) C-terminal fragment of presenilin is the molecular target of a dipeptidic γ-secretase-specific inhibitor DAPT (N-[N-(3,5-difluorophenacetyl)-L-alanyl]-S-phenylglycine t-butyl ester). J. Biol. Chem 281, 14670–14676. [DOI] [PubMed] [Google Scholar]
- (133).Timmer JC, Zhu W, Pop C, Regan T, Snipas SJ, Eroshkin AM, Riedl SJ, and Salvesen GS (2009) Structural and kinetic determinants of protease substrates. Nat. Struct. Mol. Biol 16, 1101–1108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (134).http://www.alzforum.org/mutations.
- (135).Xia D, Watanabe H, Wu B, Lee SH, Li Y, Tsvetkov E, Bolshakov VY, Shen J, and Kelleher RJ 3rd. (2015) Presenilin-1 knockin mice reveal loss-of-function mechanism for familial Alzheimer’s disease. Neuron 85, 967–981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (136).Veugelen S, Saito T, Saido TC, Chavez-Gutierrez L, and De Strooper B (2016) Familial Alzheimer’s disease mutations in presenilin generate amyloidogenic Aβ peptide seeds. Neuron 90, 410–416. [DOI] [PubMed] [Google Scholar]
- (137).Kretner B, Trambauer J, Fukumori A, Mielke J, Kuhn PH, Kremmer E, Giese A, Lichtenthaler SF, Haass C, Arzberger T, and Steiner H (2016) Generation and deposition of Aβ43 by the virtually inactive presenilin-1 L435F mutant contradicts the presenilin loss-of-function hypothesis of Alzheimer’s disease. EMBO Mol. Med 8, 458–465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (138).Kelleher RJ 3rd, and Shen J (2017) Presenilin-1 mutations and Alzheimer’s disease. Proc. Natl. Acad. Sci. U. S. A 114, 629–631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (139).Baker RP, Young K, Feng L, Shi Y, and Urban S (2007) Enzymatic analysis of a rhomboid intramembrane protease implicates transmembrane helix 5 as the lateral substrate gate. Proc. Natl. Acad. Sci. U. S. A 104, 8257–8262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (140).Doody RS, Raman R, Farlow M, Iwatsubo T, Vellas B, Joffe S, Kieburtz K, He F, Sun X, Thomas RG, Aisen PS, Siemers E, Sethuraman G, and Mohs R (2013) A phase 3 trial of semagacestat for treatment of Alzheimer’s disease. N. Engl. J. Med 369, 341–350. [DOI] [PubMed] [Google Scholar]
- (141).Coric V, Salloway S, van Dyck CH, Dubois B, Andreasen N, Brody M, Curtis C, Soininen H, Thein S, Shiovitz T, Pilcher G, Ferris S, Colby S, Kerselaers W, Dockens R, Soares H, Kaplita S, Luo F, Pachai C, Bracoud L, Mintun M, Grill JD, Marek K, Seibyl J, Cedarbaum JM, Albright C, Feldman HH, and Berman RM (2015) Targeting prodromal Alzheimer disease with avagacestat: a randomized clinical trial. JAMA Neurol 72, 1324–1333. [DOI] [PubMed] [Google Scholar]
- (142).Mitani Y, Yarimizu J, Saita K, Uchino H, Akashiba H, Shitaka Y, Ni K, and Matsuoka N (2012) Differential effects between γ-secretase inhibitors and modulators on cognitive function in amyloid precursor protein-transgenic and nontransgenic mice. J. Neurosci 32, 2037–2050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (143).Quintero-Monzon O, Martin MM, Fernandez MA, Cappello CA, Krzysiak AJ, Osenkowski P, and Wolfe MS (2011) Dissociation between the processivity and total activity of γ-secretase: implications for the mechanism of Alzheimer’s disease-causing presenilin mutations. Biochemistry 50, 9023–9035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (144).Fernandez MA, Klutkowski JA, Freret T, and Wolfe MS (2014) Alzheimer presenilin-1 mutations dramatically reduce trimming of long amyloid β-peptides (Aβ) by γ-secretase to increase 42-to-40-residue Aβ. J. Biol. Chem 289, 31043–31052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (145).Weggen S, Eriksen JL, Das P, Sagi SA, Wang R, Pietrzik CU, Findlay KA, Smith TE, Murphy MP, Bulter T, Kang DE, Marquez-Sterling N, Golde TE, and Koo EH (2001) A subset of NSAIDs lower amyloidogenic Aβ42 independently of cyclooxygenase activity. Nature 414, 212–216. [DOI] [PubMed] [Google Scholar]
- (146).Bursavich MG, Harrison BA, and Blain JF (2016) γ-secretase modulators: new Alzheimer’s drugs on the horizon? J. Med. Chem 59, 7389–7409. [DOI] [PubMed] [Google Scholar]
- (147).Boy KM, Guernon JM, Zuev DS, Xu L, Zhang Y, Shi J, Marcin LR, Higgins MA, Wu YJ, Krishnananthan S, Li J, Trehan A, Smith D, Toyn JH, Meredith JE, Burton CR, Kimura SR, Zvyaga T, Zhuo X, Lentz KA, Grace JE, Denton R, Morrison JS, Mathur A, Albright CF, Ahlijanian MK, Olson RE, Thompson LA, and Macor JE (2019) Identification and preclinical evaluation of the bicyclic pyrimidine γ-secretase modulator BMS-932481. ACS Med. Chem. Lett 10, 312–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (148).Wagner SL, Rynearson KD, Duddy SK, Zhang C, Nguyen PD, Becker A, Vo U, Masliah D, Monte L, Klee JB, Echmalian CM, Xia W, Quinti L, Johnson G, Lin JH, Kim DY, Mobley WC, Rissman RA, and Tanzi RE (2017) Pharmacological and toxicological properties of the potent oral γ-secretase modulator BPN-15606. J. Pharmacol. Exp. Ther 362, 31–44. [DOI] [PMC free article] [PubMed] [Google Scholar]