

Abstract
The synthesis of 5‐(cyano)dibenzothiophenium triflate 9, prepared by activation of dibenzo[b,d]thiophene‐5‐oxide with Tf2O and subsequent reaction with TMSCN is reported, and its reactivity as electrophilic cyanation reagent evaluated. The scalable preparation, easy handling and broad substrate scope of the electrophilic cyanation promoted by 9, which includes amines, thiols, silyl enol ethers, alkenes, electron rich (hetero)arenes and polyaromatic hydrocarbons, illustrate the synthetic potential of this reagent. Importantly, Lewis acid activation of the reagent is not required for the transfer process. We additionally report herein biomimetic cyanocyclization cascade reactions, which are not promoted by typical electrophilic cyanation reagents, demonstrating the superior ability of 9 to trigger challenging transformations.
Keywords: electrophilic cyanation, metal-free functionalizations, sulfonium salts, transfer reagents, umpolung
The synthetic versatility of the cyano group, which is a privileged precursor of amines, amides, aldehydes, carboxylic acids, and N‐containing heterocycles,1 as well as the prevalence of nitriles in natural products,2 pharmaceuticals,3 agrochemicals,4 dyes5 and high performance materials and polymers6 has stimulated the development of efficient methodologies for the selective incorporation of CN substituents at specific positions of elaborated organic scaffolds.7 Despite of the sophistication achieved in this area,8 most of the protocols described still use the inherent nucleophilicity of the cyanide anion to promote the C−CN bond‐forming event and hence, they are reminiscent to the classical Sandmeyer9 and Rosenmund von Braun reactions for the preparation of aromatic nitriles,10 or the Kolbe nitrile synthesis for aliphatic ones.11 Disconnection strategies based on the umpolung of the CN moiety are comparatively scarce and their synthetic potential has still not been fully exploited, mainly because of the disadvantages associated with the use of the available electrophilic cyanide sources.12 Namely, the extreme toxicity and volatility of the pseudo halogens 1–3,13 or the considerably lower reactivity of N‐cyano sulfonamides 4, N‐cyanobenzotriazole 5 and N‐cyanobenzoimidazole 6, which require the use of strong C‐nucleophiles such as organometallic reagents or enolates to transfer the formal [CN]+ unit.14 It was not until the introduction of cyanobenziodoxone 7 by Zhdankin and co‐workers,15 and its intensive use by the groups of Waser16 and others17 that metal‐free electrophilic cyanation at unfunctionalized C−H positions of electron‐rich organic substrates could be efficiently achieved (Figure 1). We recently contributed to this area with the introduction of the imidazolium thiocyanate 8, which has a reactivity profile similar to the one of 7, but is not based on thermally unstable hypervalent I(III) platforms.18
Figure 1.
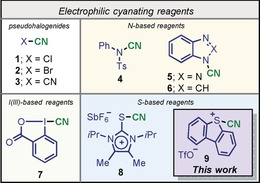
Representative electrophilic cyanation reagents.
Nature often uses carbocationic cascade cyclizations to produce complex molecular architectures from simple precursors, and chemists have been quite successful at transferring this strategy into routine synthesis planning.19 Initiation of such cyclizations is often triggered by elimination of a leaving group, the activation of olefins by π‐acid catalysts, or the reaction of carbon–carbon double bonds with electrophiles.20 However, we are not aware of the use of either 7 or 8 as promoters for the metal‐free cyanofunctionalization of olefins, presumably because of the moderate electrophilic character of the cyano moieties in these two [CN]+ synthons.21
Given the synthetic potential that such cyano functionalization may have, we were encouraged to develop a new electrophilic cyanation reagent to trigger these transformations. We envisioned that the dibenzothiophenium unit, which has been previously used for the preparation of electrophilic trifluoromethylation and alkynylation reagents, might be an adequate platform to achieve a more efficient umpolung of the CN group.22 Herein, we report the synthesis of 5‐(cyano)dibenzothiophenium triflate 9, and preliminarily evaluate its reactivity on the cyanation of typical organic nucleophiles, and the cyanofunctionalization of a variety of indole derivatives.
The desired 5‐(cyano)dibenzothiophenium salt 9 was been obtained in a one‐pot synthesis from the sulfoxide 10 by activation with one equivalent of triflic acid anhydride at −50 °C and quenching of the in situ generated bistriflate with TMSCN. Compound 9 precipitates from the reaction mixture and is isolated as a beige solid. This synthetic route has been scaled up to ten grams with a reproducible 60 % yield (Scheme 1). Diagnostic spectroscopic features of 9 are the 13C NMR signal for the carbon atom of the cyano moiety (δ=103.8 ppm), which is upfield shifted if compared with that of arylsulfonyl cyanides (δ=110–119 ppm.), and the stretching frequency of the C≡N moiety at (CN)=2191.7 cm−1.
Scheme 1.
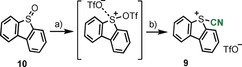
Synthesis of 5‐(cyano)dibenzothiophenium triflate. Reagents and conditions: a) Tf2O (1 equiv), −50 °C, 1 h, (not isolated); c) TMSCN (1 equiv), CH2Cl2, −50 °C, 8 h, 60 %.
X‐ray diffraction analysis of monocrystals of 9 confirmed the expected connectivity (Figure 2). In the cationic part the sulfur atom remains at the plane defined by the dibenzothiophene skeleton. However, the C−S bond distances within the aromatic moiety, 1.7911(8) Å for S1−C2 and 1.7991(8) Å for S1−C13, are significantly longer than in dibenzothiophene (1.740 Å). This difference probably results from the reduction of aromaticity at the thiophene platform after coordination of the cyano moiety. In addition, the central sulfur atom adopts a trigonal‐pyramidal coordination environment, being the sum of the bond angles around S1 286.6°(4), thus, there is a stereochemically relevant electron pair at this atom. Also of particular relevance is the short contact S1−O3 [2.6085(7) Å], which is significantly shorter than the sum of the van der Waals radii of the corresponding elements (3.32 Å). This interaction evidences some Lewis acidity at S1 and suggests that the initial step of the cyano transfer using 9 might imply the approach of the incoming nucleophile to the electron‐poor sulfur atom, followed by a reductive elimination type process. An analogous mechanism has been proposed for cyanation reactions promoted by 7.23
Figure 2.
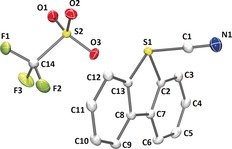
Molecular structure of compound 9 in the solid state. Anisotropic displacement shown at 50 % probability level and hydrogen atoms omitted for clarity. Selected bond lengths [Å] and angles [deg]: S1−C1, 1.7404(9); S1−C2, 1.7911(8); S1−C13, 1.7991(8); S1−O3, 2.6085(7); O3−S1−C1, 176.07(3); C2−S1−C1, 96.06(4); C13−S1−C1, 98.92(4).24
Once 9 was completely characterized, we started our investigation by subjecting benchmark N‐, S‐, and C‐based nucleophiles to its action. Optimization of the reaction conditions using anilines revealed Cs2CO3 as the optimal base (Scheme 2, Method A). Under these reaction conditions the cyanamides 11–23 can be obtained in moderate to excellent yields. Aromatic thiols do not require the addition of base to afford the thiocyanates 24–27 (Method B), while for aliphatic ones Cs2CO3 is again necessary to obtain acceptable results, 28. Silyl enol ethers and terminal olefins, as well as electron‐rich homo‐, hetero‐, and polycyclic aromatics were also transformed into the desired nitriles 29–38 under similar reaction conditions.25 While the profile of 8 and 9 regarding substrate scope is quite similar, all cyano‐transfer reactions shown in Scheme 2 took place at relatively low temperatures (−50 °C→50 °C), and without activation of the transfer reagent with BF3⋅OEt2. This result suggests inherently higher electrophilicity at the CN moiety of 9.
Scheme 2.
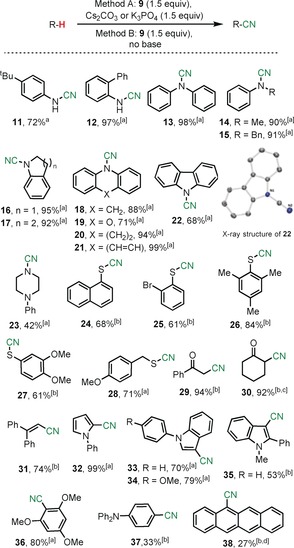
Substrate scope of the electrophilic cyanation using 9. All yields are those of isolated products unless otherwise stated. [a] Method A was applied. [b] Method B was applied. [c] Yield determined by NMR spectroscopy. The identity of 30 was confirmed by its derivatization with TsNH‐NH2 and isolation of the corresponding 3‐aminopyrazole. [d] Two equivalents of 9 were employed.
To further evaluate the utility of 9 in more complex transformations, we tested its reactivity towards the indole 39, in which the most reactive position 3 is blocked with a methyl substituent. A mixture of two products, 40 and 41, was obtained under these reaction conditions. The connectivity of 40, which could be assessed by X‐ray analysis, corresponds to a dimeric structure originating from the initial attack of the [CN]+ moiety at position 3 of 39 and subsequent trapping of the in situ generated iminium salt with a second equivalent of 39. Compound 41 is the product of additional cyanation at the most nucleophilic amino group of 40 (Scheme 3 a). Only the diasteromer coming from the syn attack of both groups to the carbon–carbon double bond was detected. Simultaneous decoration of the indole substrate with two alkyl substituents at positions 2 and 3, as is the case of 42 and 44, quenches the dimerization pathway. In these cases, after the initial electrophilic cyanation step deprotonation of the N takes place, affording the indolenines 43 and 45 (see the Supporting Information for the X‐ray structure of 43).
Scheme 3.
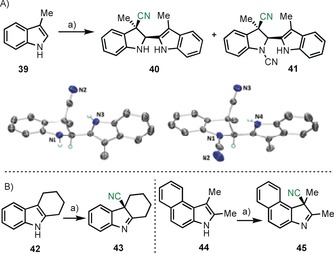
A) Reaction of 9 with 3‐methylindole (39). Reagents, conditions, and yields of isolated products: a) 9 (1.5 equiv), Cs2CO3 (1.0 equiv) 0 °C, DCM, 2 h, 40 (50 %), 41 (30 %). Molecular structures of compounds 40 and 41 in the solid state. Anisotropic displacement shown at 30 % probability level. Only selected hydrogen atoms are shown.24 B) Reaction of 9 with 42 and 44. Reagents, conditions, and yields of isolated products: a) 9 (1.5 equiv), CH3CN, RT, 15 min, 43 (76 %), 45 (86 %).
To explore the generality and synthetic relevance of these cyanation reactions, we first decided to equip a series of indoles with an internal nucleophilic group and explore intramolecular cyanofunctionalizations. Hence, a series of N‐protected tryptamine derivatives were initially subjected to the action of 9. All substrates tested, regardless of the nature of the N‐protecting group, smoothly delivered the corresponding tricyclic pyrroloindolines in good chemical yields 46–55 (Scheme 4). The length of the tether connecting the olefin and amino functionalities explains that only the diasteromer derived from the anti‐cyanoamination is observed. The pyrroloindoline core is found in a number of alkaloids,26 and has classically been synthesized by treatment of N‐protected tryptophan esters with electrophilic reagents such as halonium, selenonium, or even sulfonium ions.27 Promotion of this kind of cyclization by electrophilic cyano moieties is to the best of our knowledge unprecedented, and renders cyano pyrroloindolines of potential synthetic and/or pharmacological interest.
Scheme 4.
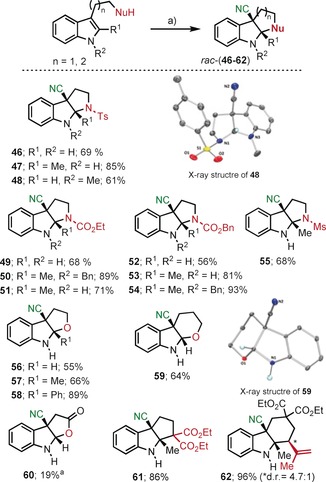
Reaction scope of the cyano cyclization of triptamine and triptophol derivatives. Reagents, conditions: a) 9 (1.5 equiv), RT, CH3CN, 2 h, unless otherwise stated. [a] CH2Cl2 was used as solvent. Molecular structures of 48 and 59 in the solid state. Anisotropic displacement shown at 50 % probability level. Only selected hydrogen atoms are shown.24
The superiority of 9 as a [CN]+ synthon when compared with 7 or 8 can be inferred from these cyanoaminations. Under identical reaction conditions neither 7 nor 8 are capable of promoting the cyanoamination of the simplest substrate of the series, N‐tosyl tryptamine, to afford 46 (see the Supporting Information). Moreover, the scope of this reaction can be likewise extended to indole substrates tethered with O or even C nucleophiles. Except for compound 62, where an additional chiral center is formed, only one diasteromer is observed (Scheme 4).
Finally, having established reaction conditions for the effective cyanation of 2,3‐disubstituted indoles towards 43 and 45, we evaluated the possible involvement of these substrates in a subsequent [4+2] Povarov‐type cyclization. Such cascade process is not easy because it requires the consecutive dearomatization of both rings of the original indole. However, a couple single precedents found in the literature for cousin structures indicated that the transformation might be possible.28 Hence, allyl‐substituted indoles were prepared and submitted to react with 9. To our delight, the desired products 63–68, containing a tetracyclic skeleton, were smoothly delivered with excellent diasteroselectivities (Scheme 5 a). Note that despite four consecutive chiral centers being formed, three of them on quaternary carbon atoms, only the diasteromer shown is formed. The slightly lower chemical yield obtained for 64 is explained by the concurrent direct cyanation at position 2 of the indole core. The depicted transformation also takes place intermolecularly. In this case, liberated from the constrains exerted by the malonate tether, the incoming olefin preferentially attacks the azadiene intermediate from its less sterically hindered face, that is, the one occupied by the cyano substituent (Scheme 5 b). This explains the reverse facial selectivity observed in 69 and 70.
Scheme 5.
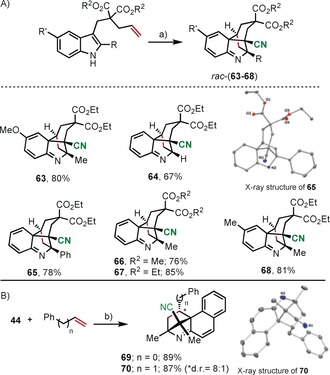
Reaction scope of the electrophilic cyanation‐Povarov cascade. Reagents, conditions: a) 9 (1.0–1.5 equiv), r.t., CH2Cl2, 3–36 h; b) 9 (1.5 equiv), r.t., CH2Cl2, 11–36 h. aDiasteromeric mixture (8:1 ratio); the second diasteromer only differs in the configuration of the carbon marked (*). Molecular structures of compounds 65 and 70 in the solid state. Anisotropic displacement shown at 50 % probability level. Only selected hydrogen atoms are shown.24
In summary, in 5‐(cyano)dibenzothiophenium triflate 9, whose synthesis is described along this paper, the umpolung of the cyano moiety is so efficient that this moiety cannot only be transferred to typical organic nucleophiles, but is also capable of promoting electrophilic cyanocyclization cascades in appropriately designed substrates. The development of asymmetric versions of the reaction sequences herein described, and the applications of structurally related sulfonium salts in other chemical transformations are currently under investigation in our laboratory.
Experimental Section
Synthesis of 5‐(cyano)dibenzothiophenium triflate (9): Tf2O (25 mmol, 1 equiv, 4.205 mL) was added dropwise within 5 minutes to a solution of dibenzo[b,d]thiophene 5‐oxide (1.00 equiv, 25 mmol, 5.007 g) in dry dichloromethane (350 mL) at −50 °C. After stirring the resulting mixture for 1 hour, TMSCN (25 mmol, 1 equiv, 3.353 mL) was added dropwise and the mixture was further stirred at −50 °C for 8 additional hours. Then, the cooling system was removed, and the formed suspension warmed to room temperature. Filtration of the solvents afforded 9 as a white/beige solid, which was further washed with dichloromethane (2×50 mL), and finally dried under vacuum (5.39 g.; 60 %).
Conflict of interest
A patent regarding reagent 9 and its synthetic applications has been filed by the University of Göttingen (application number DE102018211606.7).
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
Support from the European Research Council (ERC CoG 771295), the Deutsche Forschungsgemeinschaft (INST 186/1237‐1), and the University of Göttingen is gratefully acknowledged. We also thank B. Waldecker and F. Kraft for preliminary experimental contributions towards the synthesis of 9 and Dr. M. John for assistance with NMR analysis.
X. Li, C. Golz, M. Alcarazo, Angew. Chem. Int. Ed. 2019, 58, 9496.
Dedicated to Prof. Armin de Meijere on the occasion of his 80th birthday
References
- 1. Fatiadi A. J., Preparation and Synthetic Applications of Cyano Compounds (Eds.: S. Patai, Z. Rappoport), Wiley-VCH, Weinheim, 1983. [Google Scholar]
- 2. Fleming F. F., Nat. Prod. Rep. 1999, 16, 597–606. [Google Scholar]
- 3. Fleming F. F., Yao L., Ravikumar P. C., Funk L., Shook B. C., J. Med. Chem. 2010, 53, 7902–7917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Pollack P., Fine Chemicals: The industry and the Buisness, Wiley, Hoboken, 2007. [Google Scholar]
- 5. Huang S. T., Hsu Y. C., Yen Y. S., Chou H. H., Lin J. T., Chang C. W., Hsu C. P., Tsai C., Yin D. J., J. Phys. Chem. C 2008, 112, 19739–19747. [Google Scholar]
- 6. Zil′berman E. N., Russ. Chem. Rev. 1986, 55, 39–48. [Google Scholar]
- 7.For recent reviews see:
- 7a. Yan G., Zhang Y., Wang J., Adv. Synth. Catal. 2017, 359, 4068–4105. [Google Scholar]
- 8.
- 8a. Zanon J., Klapars A., Buchwald S. L., J. Am. Chem. Soc. 2003, 125, 2890–2891; [DOI] [PubMed] [Google Scholar]
- 8b. Ramnauth J., Bhardwaj N., Renton P., Rakhit S., Maddaford S. P., Synlett 2003, 14, 2237–2239; [Google Scholar]
- 8c. Sundermeier M., Zapf A., Beller M., Angew. Chem. Int. Ed. 2003, 42, 1661–1664; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2003, 115, 1700–1703; [Google Scholar]
- 8d. Sundermeier M., Mutyala S., Zapf A., Spannenberg A., Beller M., J. Organomet. Chem. 2003, 684, 50–55; [Google Scholar]
- 8e. Chidambaram R., Tetrahedron Lett. 2004, 45, 1441–1444; [Google Scholar]
- 8f. Schareina T., Zapf A., Beller M., Chem. Commun. 2004, 1388–1389; [DOI] [PubMed] [Google Scholar]
- 8g. Weissman S. A., Zewge D., Chen C., J. Org. Chem. 2005, 70, 1508–1510; [DOI] [PubMed] [Google Scholar]
- 8h. Schareina T., Zapf A., Beller M., Tetrahedron Lett. 2005, 46, 2585–2588; [Google Scholar]
- 8i. Cristau H.-J., Ouali A., Spindler J.-F., Taillefer M., Chem. Eur. J. 2005, 11, 2483–2492; [DOI] [PubMed] [Google Scholar]
- 8j. Veauthier J. M., Carlson C. N., Collis G. E., Kiplinger J. L., John K. D., Synthesis 2005, 16, 2683–2686; [Google Scholar]
- 8k. Jensen R. S., Gajare A. S., Toyota K., Yoshifuj M., Ozawa F., Tetrahedron Lett. 2005, 46, 8645–8647; [Google Scholar]
- 8l. Grossman O., Gelman D., Org. Lett. 2006, 8, 1189–1191; [DOI] [PubMed] [Google Scholar]
- 8m. Schareina T., Zapf A., Mägerlein W., Müller N., Beller M., Tetrahedron Lett. 2007, 48, 1087–1090; [Google Scholar]
- 8n. Zhu Y.-Z., Cai C., Eur. J. Org. Chem. 2007, 2401–2404; [Google Scholar]
- 8o. Buono F. G., Chidambaram R., Mueller R. H., Waltermire R. E., Org. Lett. 2008, 10, 5325–5328; [DOI] [PubMed] [Google Scholar]
- 8p. Jia X., Yang D., Zhang S., Cheng J., Org. Lett. 2009, 11, 4716–4719; [DOI] [PubMed] [Google Scholar]
- 8q. Yan G., Kuang C., Zhang Y., Wang J., Org. Lett. 2010, 12, 1052–1055; [DOI] [PubMed] [Google Scholar]
- 8r. Ushkov A. V., Grushin V. V., J. Am. Chem. Soc. 2011, 133, 10999–11005; [DOI] [PubMed] [Google Scholar]
- 8s. Schareina T., Zapf A., Cotté A., Gotta M., Beller M., Adv. Synth. Catal. 2011, 353, 777–780; [Google Scholar]
- 8t. Yeung P. Y., So C. M., Lau C. P., Kwong F. Y., Org. Lett. 2011, 13, 648–651; [DOI] [PubMed] [Google Scholar]
- 8u. Chen J., Sun Y., Liu B., Liu D., Cheng J., Chem. Commun. 2012, 48, 449–451. [DOI] [PubMed] [Google Scholar]
- 9.
- 9a. Sandmeyer T., Ber. Dtsch. Chem. Ges. 1884, 17, 1633–1635; [Google Scholar]
- 9b. Sandmeyer T., Ber. Dtsch. Chem. Ges. 1884, 17, 2650–2653; [Google Scholar]
- 9c. Sandmeyer T., Ber. Dtsch. Chem. Ges. 1885, 18, 1492–1496; [Google Scholar]
- 9d. Sandmeyer T., Ber. Dtsch. Chem. Ges. 1885, 18, 1946–1948. [Google Scholar]
- 10. Rosenmund K. W., Struck E., Ber. Dtsch. Chem. Ges. 1919, 52, 1749–1756. [Google Scholar]
- 11. Wang Z., Comprehensive Organic Name Reactions and Reagents, Wiley, Hoboken, 2010, pp. 1661–1663. [Google Scholar]
- 12.
- 12a. Kim J., Kim J. H., Chang S., Angew. Chem. Int. Ed. 2012, 51, 11948–11959; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2012, 124, 12114–12125; [Google Scholar]
- 12b. Schörgenhumer J., Waser M., Org. Chem. Front. 2016, 3, 1535–1540; [Google Scholar]
- 12c. Nauth A. M., Opatz T., Org. Biomol. Chem. 2019, 17, 11–23. [DOI] [PubMed] [Google Scholar]
- 13. Noller C. R., Lehrbuch der Organischen Chemie, Springer, Berlin, 2013. [Google Scholar]
- 14.
- 14a. Hughes T. V., Hammond S. D., Cava M. P., J. Org. Chem. 1998, 63, 401–402; [Google Scholar]
- 14b. Wu Y. Q., Limburg D. C., Wilkinson D. E., Hamilton G. S., Org. Lett. 2000, 2, 795–797; [DOI] [PubMed] [Google Scholar]
- 14c. Anbarasan P., Neumann H., Beller M., Chem. Eur. J. 2010, 16, 4725–4728; [DOI] [PubMed] [Google Scholar]
- 14d. Anbarasan P., Neumann H., Beller M., Chem. Eur. J. 2011, 17, 4217–4222; [DOI] [PubMed] [Google Scholar]
- 14e. Akula R., Xiong Y., Ibrahim H., RSC Adv. 2013, 3, 10731–10735; [Google Scholar]
- 14f. Cui J., Song J., Liu Q., Liu H., Dong Y., Chem. Asian J. 2018, 13, 482–495. [DOI] [PubMed] [Google Scholar]
- 15. Zhdankin V. V., Kuehl C. J., Krasutsky A. P., Bolz J. T., Mismash B., Woodward J. K., Simonsen A. J., Tetrahedron Lett. 1995, 36, 7975–7978. [Google Scholar]
- 16.
- 16a. Frei R., Courant T., Wodrich M. D., Waser J., Chem. Eur. J. 2015, 21, 2662–2668; [DOI] [PubMed] [Google Scholar]
- 16b. Le Vaillant F., Wodrich M. D., Waser J., Chem. Sci. 2017, 8, 1790–1800; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16c. Declas N., Le Vaillant F., Waser J., Org. Lett. 2019, 21, 524–528. [DOI] [PubMed] [Google Scholar]
- 17. Wang X., Studer A., Angew. Chem. Int. Ed. 2018, 57, 11792–11796; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 11966–11970. [Google Scholar]
- 18.
- 18a. Talavera G., Peña J., Alcarazo M., J. Am. Chem. Soc. 2015, 137, 8704–8707; [DOI] [PubMed] [Google Scholar]
- 18b. Barrado A. G., Zieliński A., Goddard R., Alcarazo M., Angew. Chem. Int. Ed. 2017, 56, 13401–13405; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 13586–13590. [Google Scholar]
- 19. Tantillo D. J., Chem. Soc. Rev. 2010, 39, 2847–2854. [DOI] [PubMed] [Google Scholar]
- 20. Ungarean C. N., Southgate E. H., Sarlah D., Org. Biomol. Chem. 2016, 14, 5454–5467. [DOI] [PubMed] [Google Scholar]
- 21.The electrophilic C−H cyanation of alkenes employing an in situ generated polyfluorinated I(III) reagent has been recently reported, see Ref. [17].
- 22.For other transfer reagents based on the dibenzothiophene unit, see:
- 22a. Umemoto T., Ishihara S., J. Am. Chem. Soc. 1993, 115, 2156–2164; [Google Scholar]
- 22b. Waldecker B., Kraft F., Golz C., Alcarazo M., Angew. Chem. Int. Ed. 2018, 57, 12538–12542; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 12718–12722. [Google Scholar]
- 23. Frei R., Courant T., Wodrich M. D., Waser J., Chem. Eur. J. 2015, 21, 2662–2668. [DOI] [PubMed] [Google Scholar]
- 24.CCDC 1904769, 1904770, 1904771, 1904772, 1904773, 1904774, 1904775, 1904776, 1904777, 1904778, 1907655, 1907656, and 1907657 contain the supplementary crystallographic data for this paper. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre.
- 25.Noteworthy, the nitrile formation takes place for these more challenging substrates without the necessity of activation of the cyanation reagent with a Lewis acid catalyst.
- 26. Crich D., Banerjee A., Acc. Chem. Res. 2007, 40, 151–161. [DOI] [PubMed] [Google Scholar]
- 27.Selected references:
- 27a. Marsden S. P., Depew K. M., Danishefsky S. J., J. Am. Chem. Soc. 1994, 116, 11143–11144; [Google Scholar]
- 27b. Silva-López C., Pérez-Balado C., Rodríguez-Graña P., de Lera A. R., Org. Lett. 2008, 10, 77–80; [DOI] [PubMed] [Google Scholar]
- 27c. Tayu M., Hui Y., Takeda S., Higuchi K., Saito N., Kawasaki T., Org. Lett. 2017, 19, 6582–6585; [DOI] [PubMed] [Google Scholar]
- 27d. Jiang X., Zhu W., Yang L., Zheng Z., Yu C., Eur. J. Org. Chem. 2019, 2268–2274. [Google Scholar]
- 28.
- 28a. Hájíček J., Trojánek J., Tetrahedron Lett. 1981, 22, 1823–1826; [Google Scholar]
- 28b. Šoral M., Markus J., Doháňošová J., Šoralová S., Dvoranová D., Chyba A., Moncol J., Berkeš D., Liptaj T., J. Mol. Struct. 2017, 1128, 230–238. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary