

Abstract
Synaptotagmin-1 and -7 constitute the main calcium sensors mediating SNARE-dependent exocytosis in mouse chromaffin cells, but the role of a closely related calcium-binding protein, Doc2b, remains enigmatic. We investigated its role in chromaffin cells using Doc2b knock-out mice and high temporal resolution measurements of exocytosis. We found that the calcium dependence of vesicle priming and release triggering remained unchanged, ruling out an obligatory role for Doc2b in those processes. However, in the absence of Doc2b, release was shifted from the readily releasable pool to the subsequent sustained component. Conversely, upon overexpression of Doc2b, the sustained component was largely inhibited whereas the readily releasable pool was augmented. Electron microscopy revealed an increase in the total number of vesicles upon Doc2b overexpression, ruling out vesicle depletion as the cause for the reduced sustained component. Further experiments showed that, in the absence of Doc2b, the refilling of the readily releasable vesicle pools is faster, but incomplete. Faster refilling leads to an increase in the sustained component as newly primed vesicles fuse while the [Ca2+]i following stimulation is still high. We conclude that Doc2b acts to inhibit vesicle priming during prolonged calcium elevations, thus protecting unprimed vesicles from fusing prematurely, and redirecting them to refill the readily releasable pool after relaxation of the calcium signal. In sum, Doc2b favors fast, synchronized release, and limits out-of-phase secretion.
Introduction
Release of neurotransmitters depends on vesicle exocytosis driven by the formation of a ternary SNARE complex between vesicles and the plasma membrane (Jahn and Scheller, 2006). Calcium dependence is conferred to the SNAREs by proteins of the synaptotagmin (syt) family, which act together with complexin to allow the buildup of a pool of release-ready—or “primed”—vesicles, that can fuse rapidly when calcium binds to syt (Rizo et al., 2006; Martens et al., 2007; Chapman, 2008; Pang and Südhof, 2010; Walter et al., 2011).
In chromaffin cells two syt proteins participate in the exocytosis of large dense-core vesicles (LDCVs): syt-1 drives fast release (Voets et al., 2001; Nagy et al., 2006), while syt-7 augments both fast and slow release components (Schonn et al., 2008) and regulates the fusion pore (Segovia et al., 2010). Recently, proteins of the Doc2 family have come under scrutiny. Like syts, these proteins have two C2-domains, C2A and C2B, but they lack a transmembrane domain (Verhage et al., 1997). They bind to the plasma membrane in a reversible, calcium-dependent manner and thus cycle on and off of it during neuronal activity (Groffen et al., 2006). Their calcium affinity is higher than that of syt-1, with maximal binding at [Ca2+]i <1 μm (Groffen et al., 2006; Malkinson and Spira, 2006). In in vitro experiments Doc2 stimulates SNARE-dependent membrane fusion in a calcium-dependent manner (Groffen et al., 2010).
The physiological role of Doc2 proteins is enigmatic. In neurons, deletion of Doc2a/b leads to a decrease in the frequency of miniature release to 50–75%, whereas fast synchronous release remains unaffected (Groffen et al., 2010; Pang et al., 2011). Knockdown of Doc2a leads to a specific decrease in asynchronous release (Yao et al., 2011) and deletion of Doc2b reduces both the first and second phase of insulin release in β-cells (Ramalingam et al., 2012). Despite the similarity to syts in in vitro lipid mixing assays (Groffen et al., 2010), it remains unclear whether Doc2 proteins also actively trigger SNARE-dependent vesicle fusion, or whether they modulate secretion by acting on upstream steps (Pang et al., 2011; for review, see Walter et al., 2011).
In chromaffin cells, Ca2+ in the submicromolar range facilitates vesicle priming (Bittner and Holz, 1992; von Ruden and Neher, 1993; Smith et al., 1998) and augments the size of the primed vesicle pools, which support the burst phase of release. In addition, an unprimed vesicle pool (UPP) supports a later, sustained, phase of release (Ashery et al., 2000). Overexpression of Doc2b increases burst secretion leading to the suggestion that Doc2b traffics to the plasma membrane and drives calcium-dependent vesicle priming (Friedrich et al., 2008). To establish a role for Doc2b in vesicle priming and fusion, we here examined the consequences of its genetic elimination. Surprisingly, we find that Doc2b exerts dual and opposite effects on vesicle priming, such that priming is inhibited during sustained calcium elevations, but stimulated during periods of rest, resulting in overall synchronization of release.
Materials and Methods
Chromaffin cell culture.
Adrenal glands from P0–1 pups of either sex were dissected out, and chromaffin cells isolated and cultured according to previously published protocols (Sørensen et al., 2003b).
Exocytosis measurements.
Combined capacitance measurements and amperometric recordings were performed essentially as described previously (Mohrmann et al., 2010) on a Zeiss Axiovert 10 equipped with an EPC-9 amplifier (HEKA Elektronik) for patch-clamp capacitance measurements and an EPC-7 plus (HEKA Elektronik) for amperometry. The release of catecholamines was triggered either by UV flash-photolysis (JML-C2, Rapp Optoelektronik) or by sustained illumination using monochromator light (see below) of a caged-calcium compound, nitrophenyl-EGTA (Synaptic Systems), which was infused into the cell via the patch pipette, or by membrane depolarization.
Intracellular calcium concentrations were determined using a mixture of two fluorescent dyes with different Ca2+-affinities (fura-4F and furaptra, Invitrogen) (Voets, 2000; Sørensen et al., 2002), which, after calibration, allow calcium concentration measurements over a large dynamic range. For the ratiometric determination of calcium concentrations, the excitation light (Polychrome IV, TILL Photonics) was alternated between 350 and 380 nm. Emitted fluorescence was detected with a photodiode and sampled using Pulse software (HEKA Elektronik), which was also used for controlling the voltage in the pipette and performing capacitance measurements. The 350/380 fluorescence signal ratio was calibrated by infusion of cells with 8 different solutions of known calcium concentrations. The standard intracellular solution contained the following (in mm): 100 Cs-glutamate, 8 NaCl, 4 CaCl2, 32 Cs-HEPES, 2 Mg-ATP, 0.3 GTP, 5 NPE, 0.4 fura-4F, 0.4 furaptra, and 1 Vitamin C (to prevent flash-induced damage to fura dyes), pH 7.2 (osmolarity adjusted to ∼295 mOsm). To investigate the effect of basal (preflash) [Ca2+]i values on vesicle priming, in some cells we applied repeated monochromator illumination (alternating between 350 and 380 nm) to induce slow calcium uncaging and raise the basal [Ca2+]i to higher values before the uncaging flash (Voets, 2000). Calcium measurements were averaged for 20 s before the uncaging flash to determine the basal [Ca2+]i driving vesicle priming, and those values are given in the Results section, where relevant. For depolarization experiments, Ca2+ and NPE were omitted, and EGTA (0.5 mm) was added to the intracellular solution to keep a low basal [Ca2+]i. The extracellular solution was composed of the following (in mm): 145 NaCl, 2.8 KCl, 2 CaCl2, 1 MgCl2, 10 HEPES, and 11 glucose, pH 7.2 (osmolarity adjusted to ∼305 mOsm).
Amperometric recordings were performed simultaneously to membrane capacitance using carbon fibers with 5 μm in diameter (Thornel P-650/42; Cytec), insulated using the polyethylene method (Bruns, 2004). Fibers were clamped to 700 mV and currents were filtered at 2.9 kHz using an EPC-7 (HEKA) and sampled at 11.5 kHz. Information about pool sizes and fusion rates was obtained by fitting the capacitance traces with a sum of three exponential functions using custom macros written in IGOR Pro software. Data are presented as mean ± SEM and statistical comparisons were made in Graphpad Prism using unpaired, two-tailed Student's t test, unless otherwise stated.
Immunocytochemistry.
Chromaffin cells used for immunocytochemistry were cultured for 2 d on coverslips coated with poly-d-lysine (Sigma). Cells were fixated in 4% paraformaldehyde for 20 min at room temperature (RT), permeabilized in PBS with 0.1% Triton X-100 for 10 min, and blocked for 30 min in PBS containing 3% BSA. Primary antibodies (chicken anti-GFP, 1:500, Abcam; rabbit anti-Doc2, 1:100, custom made; mouse anti-VAMP2, 1:500, Synaptic Systems) were incubated for 2 h at RT followed by extensive washing in PBS before incubating the cells with secondary antibodies (goat anti-chicken Alexa Fluor 488, 1:1000; goat anti-mouse Alexa Fluor 546, 1:1000; goat anti-rabbit Alexa Fluor 647, 1:1000; all from Invitrogen) for 2 h at RT. After extensive washing the coverslips were mounted on slides using FluorSave (Calbiochem). The cells were imaged using a laser-scanning confocal microscope (Zeiss LSM 710) with a 63× oil objective.
In situ hybridization.
DIG-11-UTP-labeled RNA probes were produced using T3 (antisense probe) and T7 RNA polymerase (sense probe) from a pGEM-T-Doc2b plasmid harboring nucleotides 512–1378 of mouse Doc2b (GenBank NM_007873). Adrenal glands from E18 stage embryos were frozen on dry ice, embedded in Tissue-Tek OCT compound (Sakura Finetek) and cryosectioned at 16 μm thickness. After storage at −80°C, the tissue was fixed (10 min at RT in PBS with 4% paraformaldehyde), washed (3 × 3 min at RT in PBS), acetylated (10 min at RT in freshly prepared 100 mm triethanolamine, 66 mm HCl, and 25 mm acetic anhydride), washed again, prehybridized (2 h at RT in 50% deionized formamide, 5× SSC, 5× Denhardt's reagent, 250 μg/ml yeast tRNA, and 500 μg/ml salmon sperm DNA), and hybridized (16 h at 72°C in the same buffer supplemented with 400 ng/ml of probe). After stringent washing (2 h at 72°C in 0.2× SSC) the tissue was incubated with 5000-fold diluted anti-DIG-F(Ab) antibody conjugated to alkaline phosphatase (Roche), washed, and subsequently stained with BCIP/NBT (5-bromo-4-chloro-3-indolyl phosphate and nitro blue tetrazolium; Roche).
Electron microscopy of cultured chromaffin cells.
Chromaffin cells from WT and Doc2a/b KO mice (P0–1), both control and expressing Doc2b by means of SFV infection, were plated on coverslips (Cellocate, Eppendorf) coated with rat tail type I collagen (Sigma). Because the Doc2a/b KO line was kept homozygote for the Doc2b KO allele, for these experiments the WT animals were taken from a different mouse colony with an identical genetic background (C57BL/6). After 2 DIV, the cells were fixed for 45 min at room temperature with 2.5% glutaraldehyde in 0.1 m cacodylate buffer, pH 7.4, washed with cacodylate buffer, embedded, and analyzed as described before (Toonen et al., 2006). The distribution of chromaffin granules was analyzed in a blinded fashion for the experimental condition. Granules were defined as docked if there was no measurable distance separating them from the plasma membrane.
Results
To understand the function of endogenous Doc2b, we here studied adrenal chromaffin cells isolated from newborn (P0–1) Doc2b−/− mice (KO) obtained by crossing heterozygotes. Wild-type (Doc2b+/+) littermates (WT) were used as control, unless noted otherwise.
Deletion of Doc2b desynchronizes release
To understand the role of Doc2b in the fusion of the different vesicle pools, we first stimulated chromaffin cells with a depolarization protocol designed to distinguish between the immediately releasable pool (IRP) and the readily releasable pool (RRP) of vesicles (Fig. 1B, top). Six initial brief depolarizations (10 ms to +20 mV) cause a local increase in [Ca2+]i and release the IRP vesicles, which are positioned close to calcium channels, whereas four subsequent longer depolarizations (100 ms to +20 mV) cause global [Ca2+]i elevation and release the entire RRP (Horrigan and Bookman, 1994; Voets et al., 1999). The IRP is commonly assumed to constitute a subset of the RRP. The stimulation protocol was repeated five times at 1 min intervals (Fig. 1A) while measuring the [Ca2+]i using microfluorimetry (see Materials and Methods). Exocytosis was monitored using simultaneous membrane capacitance and amperometric measurements. Since amperometric measurements only probe a small part of the cell surface and are susceptible to differences in fiber sensitivity, we only subjected the membrane capacitance measurements to quantitative analysis. In this initial set of experiments, the pipette solution contained no added calcium, and the basal [Ca2+]i before stimulation was therefore quite low (70.0 ± 7.8 nm for WT; 63.8 ± 8.8 nm for KO).
Figure 1.
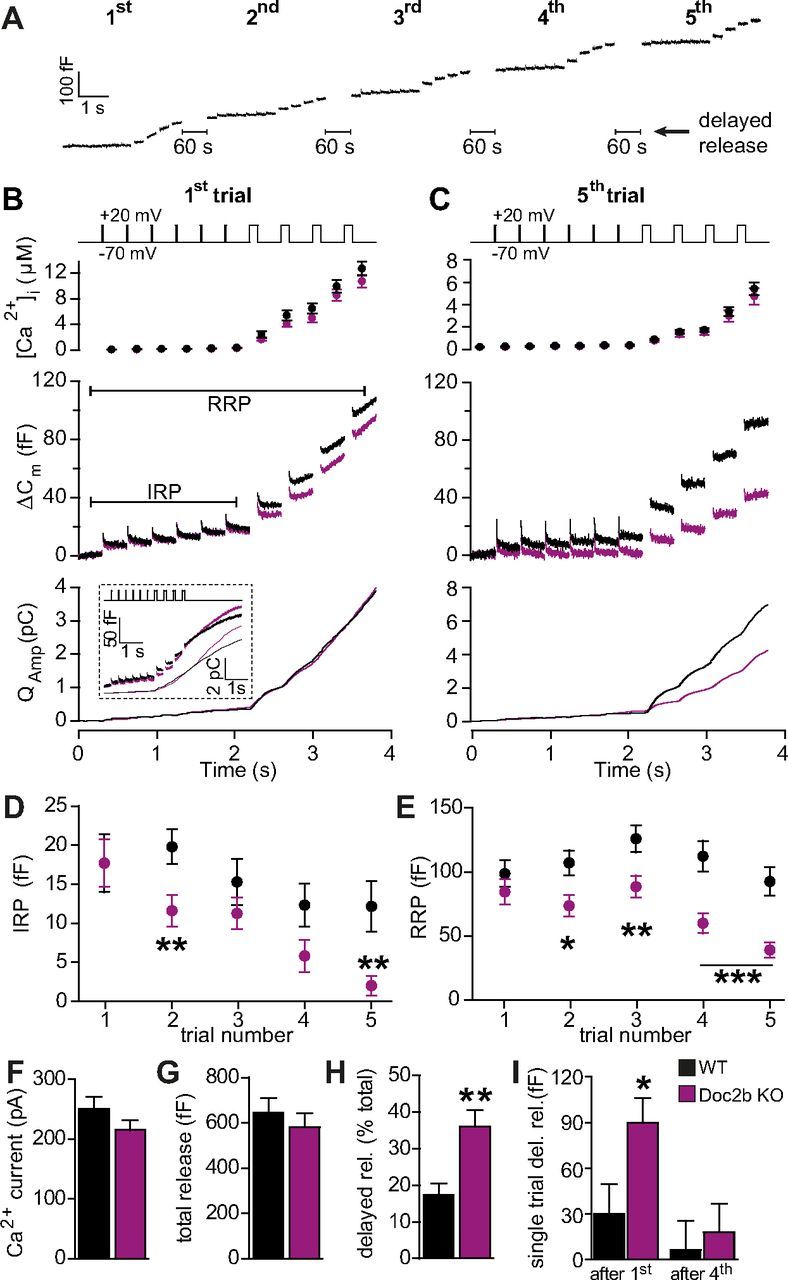
Doc2b deletion causes incomplete refilling of releasable pools. A, To probe the size of the IRP and the RRP (for more information, see Results section) a depolarization protocol, consisting of 6 brief (10 ms) and 4 long (100 ms) depolarizations to +20 mV (inserts to B and C) was repeated a total of 5 times (first to fifth trial), separated by 60 s recovery periods. Shown is an example WT cell with only a little release between trials (delayed release). B, Average results of first depolarization trial (WT: n = 28 cells, black traces; Doc2b KO: n = 27 cells, magenta traces). Top panel shows average (±SEM) [Ca2+]i measured by microfluorimetry. Middle panel shows the changes in cell membrane capacitance (mean traces for all cells). Bottom panel shows the integrated amperometric charge (mean traces). The inset shows a subset of longer recordings, directly demonstrating the stronger delayed release in Doc2b KO cells. C, As in B but displayed is the average result of the fifth trial in the same cells. D, Mean (±SEM) IRP size was reduced more over the five trials in the Doc2b KO. E, Mean (±SEM) RRP size was reduced more over the five trials in the Doc2b KO. F, The Ca2+ current amplitude (measured at the first 10 ms depolarization during the first trial) was unchanged in the Doc2b KO. G, The total release (including delayed release) was unchanged in the Doc2b KO. H, In the Doc2b KO cells, significantly more secretion happened in between trials (delayed release, see A). I, Delayed release was more prominent during initial trials. *p < 0.05; **p < 0.01; ***p < 0.001 (unpaired two-tailed Student's t test).
The first round of stimulation (6 short and 4 long depolarizations) resulted in unchanged IRP and RRP sizes and integrated amperometric charge in the Doc2b KO compared with WT cells (Fig. 1B,D,E; note that B and C display mean traces from all recorded cells). Measurements of the Ca2+ current amplitude and the [Ca2+]i increase also did not identify significant changes (Fig. 1B,F). However, when the stimulation protocol was repeated, differences between Doc2b KO and WT cells became readily apparent. In WT cells, repeating the stimulation caused a mild increase in RRP size, due to calcium-dependent priming (Voets, 2000), followed by a decrease caused by run-down (Fig. 1E). The IRP size decreased from the third stimulation (Fig. 1D). The [Ca2+]i increase was smaller during the fifth trial than during the first (Fig. 1B,C), due to Ca2+ channel inactivation in both cell types (to 103.6 ± 7.2 pA for WT and 116.3 ± 10.6 pA for KO at the fifth trial; compare withFig. 1F). This reduction in calcium influx, together with the exhaustion of upstream vesicles pools, most likely accounts for the rundown of RRP and IRP size during later trials in WT cells. In KO cells both RRP and IRP sizes displayed a stronger decrease, falling significantly below WT values already from the second stimulation (Fig. 1D,E). By the fifth stimulation, the RRP size had declined to <50% of WT values, whereas the IRP was almost empty (Fig. 1C–E). These data indicate that Doc2b KO chromaffin cells suffer from incomplete refilling of the RRP and IRP following repeated stimulation.
While performing these experiments we noted that Doc2b KO cells often secreted substantially between trials. We therefore quantified the increase in capacitance in-between measurement trials, which we dubbed “delayed release” (Fig. 1A). Adding up the delayed release to the release recorded during trials we found that the total capacitance increase across the five trials was indistinguishable in WT and KO cells (Fig. 1G), while the fraction of delayed release was significantly increased in the KO (Fig. 1H). This was primarily caused by increased delayed release following the first stimulation trials (Fig. 1I shows delayed release following the first and the fourth trial). To directly visualize this delayed release, we repeated the first stimulation in a subset of cells while extending the recording time after the last depolarization. This indeed identified an increased poststimulation secretion in Doc2b KO cells compared with WT (Fig. 1B, inset).
To gain further insight into the kinetics of fusion, we turned to calcium uncaging experiments. In the first round of stimulations we kept the initial [Ca2+]i at intermediate concentrations (299 ± 16 nm for WT; 333 ± 12 nm for KO) and then raised the [Ca2+]i abruptly to ∼20 μm. Prolonged illumination was used to both measure [Ca2+]i and further uncage calcium after the flash to sustain the calcium elevation. During the first stimulation, both WT and KO cells displayed robust secretion (Fig. 2A). Doc2b KO cells showed a nonsignificant tendency to a smaller secretory “burst” (here defined as the secretion within 0.5 s of the flash), and a larger sustained component (the rate of capacitance increase from 0.5 to 5 s after the flash; Fig. 2B), resulting in a slightly sigmoid-shaped capacitance curve. The ratio of total secretion to burst size was significantly larger in Doc2b KO cells (Fig. 2B), confirming a shift in secretion from the burst to the sustained component. When stimulating the cell again 100 s later these changes were exacerbated, leading to a significantly smaller burst and a larger sustained component (Fig. 2C,D).
Figure 2.
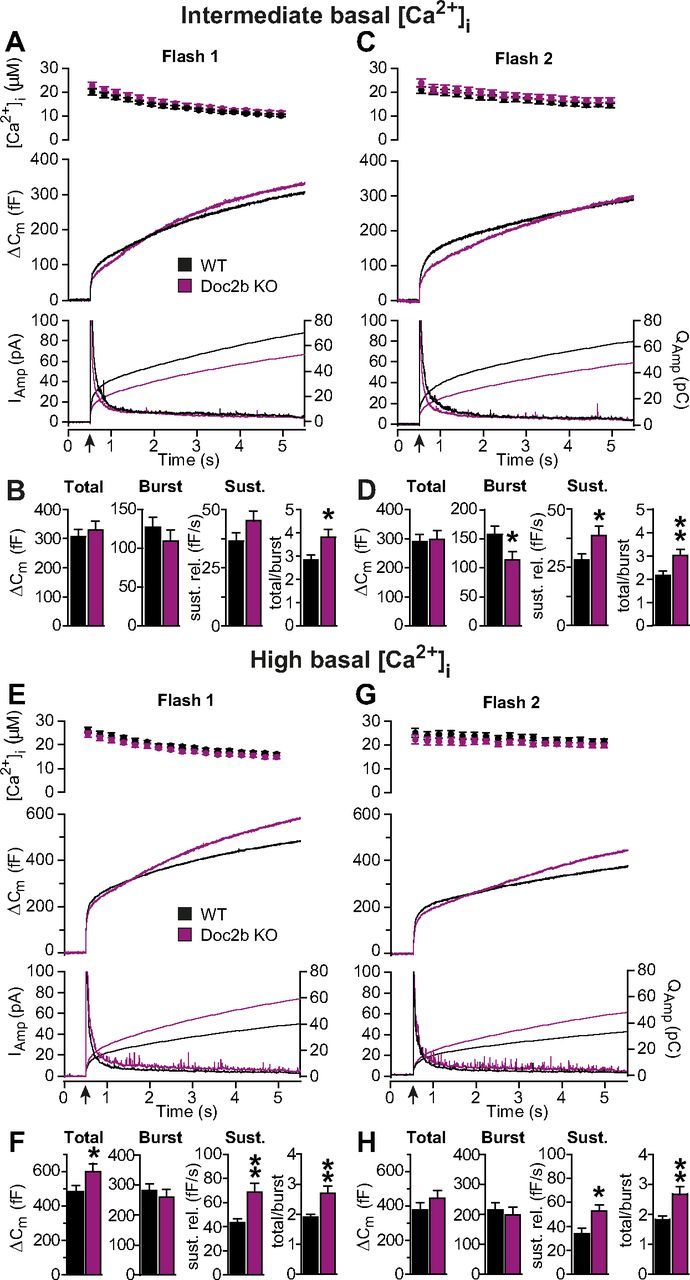
Doc2b deletion shifts secretion from the burst to the sustained component. A–D, Calcium uncaging from intermediate basal [Ca2+]i revealed a shift in secretion to a later component in the Doc2b KO. A, First calcium uncaging trial (flash at arrow; WT: n = 30 cells, black traces; Doc2b KO: n = 30 cells, magenta traces). Top panel shows average (± SEM) [Ca2+]i measured by microfluorimetry. Middle panel shows the changes in cell membrane capacitance (mean traces for all cells). Bottom panel shows the amperometric signal (mean traces); left ordinate axis: amperometric current; right ordinate axis: amperometric charge. B, Total secretion (from 0 to 5 s after the flash), burst secretion (from 0 to 0.5 s after flash) and sustained (Sust.) secretion (from 0.5–5 s after flash, linear rate) were not significantly changed. However, when the total secretion was divided by the burst, the ratio was significantly higher in the Doc2b KO. C, Second calcium uncaging trial, performed 100 s after the first in the same cells. Panels as in A. D, Burst secretion during the second trial was significantly reduced and sustained secretion, as well as the ratio between total and burst secretion, were significantly increased in the KO. E–H, Calcium uncaging from high basal [Ca2+]i revealed a significant increase in the sustained component in Doc2b KO cells. Panels as in A–D. *p < 0.05; **p < 0.01 (unpaired two-tailed Student's t test).
At higher basal [Ca2+]i, secretion starts to be prominent even before uncaging (Voets, 2000). Under these circumstances, the burst size is determined by the balance between the fusion rate, which increases with a power of three at higher [Ca2+]i and causes pool depletion, and the priming rate, which increases linearly with [Ca2+]i and causes pool refilling. To assess the influence of basal [Ca2+]i on burst size (see also below, and Fig. 5C), another round of calcium uncaging experiments was performed from a higher basal [Ca2+]i (1.02 ± 0.05 μm for WT and 0.93 ± 0.07 μm for KO). Two flash stimulations identified a significant increase in the sustained component in the Doc2b KO, and also a significant increase in total secretion and ratio of total to burst secretion (Fig. 2F,H).
Figure 5.
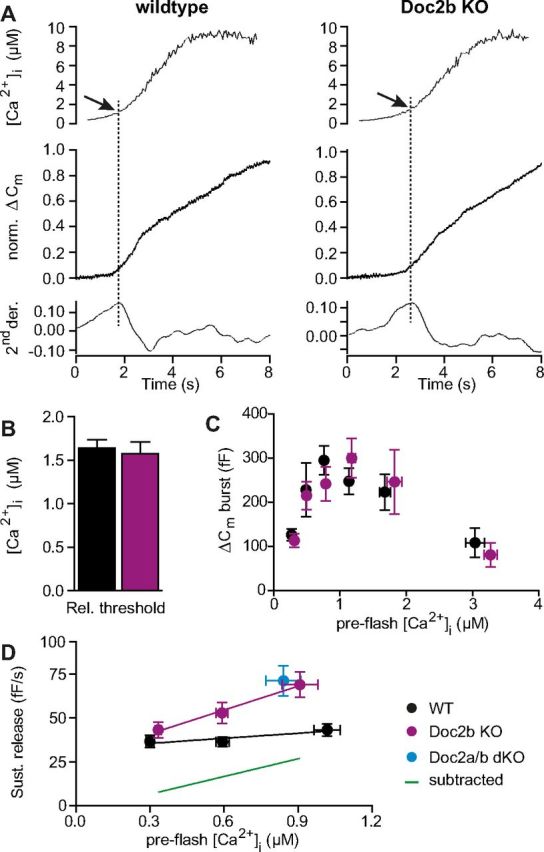
Doc2b controls the calcium dependence of the sustained component. A, Calcium ramp experiments. Slow calcium uncaging was elicited by repetitive illumination alternating between 350 and 380 nm, allowing for simultaneous calcium measurements. Top, [Ca2+]i measured by microfluorimetry (example trace). Middle, Capacitance change (example trace). Bottom, Second derivative of the capacitance with respect to time. The maximum of the second derivative was used to determine the calcium threshold for secretion (dotted line). B, The calcium threshold for secretion was unchanged in the KO (WT: n = 27 cells, black traces; Doc2b KO: n = 29 cells, magenta traces). C, Burst amplitude (0.5 s secretion after uncaging flash) plotted as a function of preflash [Ca2+]i (WT: n = 77 cells, black traces; Doc2b KO: n = 80 cells, magenta traces). No significant difference between KO and WT was identified; both cell types display the typical increase with [Ca2+]i in the submicromolar range. D, Rate of the sustained component as a function of preflash [Ca2+]i (WT: n = 94 cells, black traces; Doc2b KO: n = 94 cells, magenta traces) revealed that Doc2b exerts a calcium-dependent inhibition on sustained release. Green line: subtraction of fitted WT line from Doc2b KO fit. Blue symbol: data from Doc2a/b double KO cells (n = 15 cells, see also Fig. 4).
Calcium uncaging makes it possible to accurately assess the size and fusion kinetics of fast and slow phases of the release burst by fitting a sum of exponential components to the capacitance traces (Fig. 3A, and see Materials and Methods). These phases have been suggested to originate from the fusion of two primed vesicle pools, the RRP (see also above), and the SRP, respectively (Voets, 2000). The analysis of the cells subjected to uncaging from a high basal [Ca2+]i (Fig. 2E) did not reveal significant changes (Fig. 3B,C), although the fast burst was slightly reduced (Fig. 3B). When adding flash experiments obtained by another protocol (recovery experiment, see Fig. 6), which only allows accurate determination of the fast burst (see below), we could show that it was indeed significantly decreased, although slightly (by 17%), in the Doc2b KO cells (Fig. 3Ci), whereas the fast time constant was unchanged (Fig. 3Cii).
Figure 3.
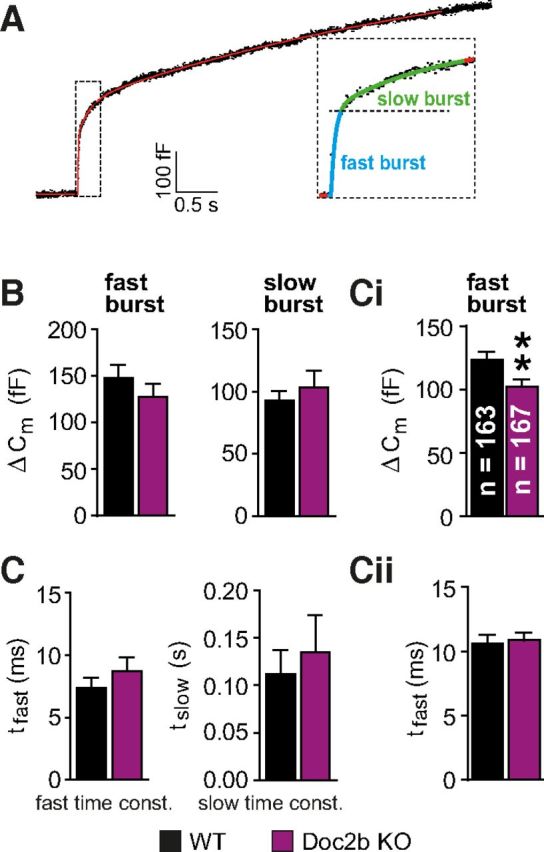
Doc2b supports a larger fast burst of release. A, Kinetic analysis was performed by fitting a sum of exponential functions to individual capacitance traces, leading to the identification of the fast and slow burst of release (inset). These are assumed to originate from two underlying vesicle pools, the RRP and the SRP. B, Releasable pool sizes and C, fusion time constants, were not significantly different in the Doc2b KO compared with WT when analyzing the experiments presented in Figure 2E. Ci, When including recordings from the “double flash protocol” (Fig. 5), the fast burst was significantly smaller in the Doc2b KO (n gives the number of cells in each group). Cii, The fast time constant was not significantly changed. **p < 0.01 (unpaired two-tailed Student's t test).
Figure 6.

Pool refilling is faster but incomplete in the absence of Doc2b. A, Double flash protocol displaying traces for WT membrane capacitance and measured [Ca2+]i. Two consecutive uncaging flashes were performed at variable interflash intervals and the fractional pool recovery assed by fitting a double exponential function to the capacitance traces. The measurement situation was modified to secure a rapid relaxation of [Ca2+]i (see Results section). B, Fast burst of release plotted as a function of interflash interval (WT: total of n = 88 cells, black traces; Doc2b KO: total n = 90 cells, magenta traces). The data were fitted with single-exponential recovery curves to all individual data points (and not to mean data). In the absence of Doc2b recovery is faster, but incomplete (statistics: see text). C, Slow component (consisting of SRP and part of the sustained component, see text) recovered much faster than the RRP in both cell types.
Uncaging experiments performed from a relatively high basal [Ca2+]i in Doc2a/b double knock-out cells (841 ± 70 nm for Doc2a/b KO, 907 ± 73 nm for WT) identified a phenotype characterized by an increase in the sustained component (Fig. 4B,C), consistent with data obtained in Doc2b single KO cells under similar circumstances (Fig. 2E,F). This finding correlates with the lack of detectable expression of Doc2a in chromaffin cells by in situ hybridization (Fig. 4A) (Friedrich et al., 2008). Thus, the increase in sustained component is not caused by compensatory upregulation of Doc2a in the absence of Doc2b.
Figure 4.
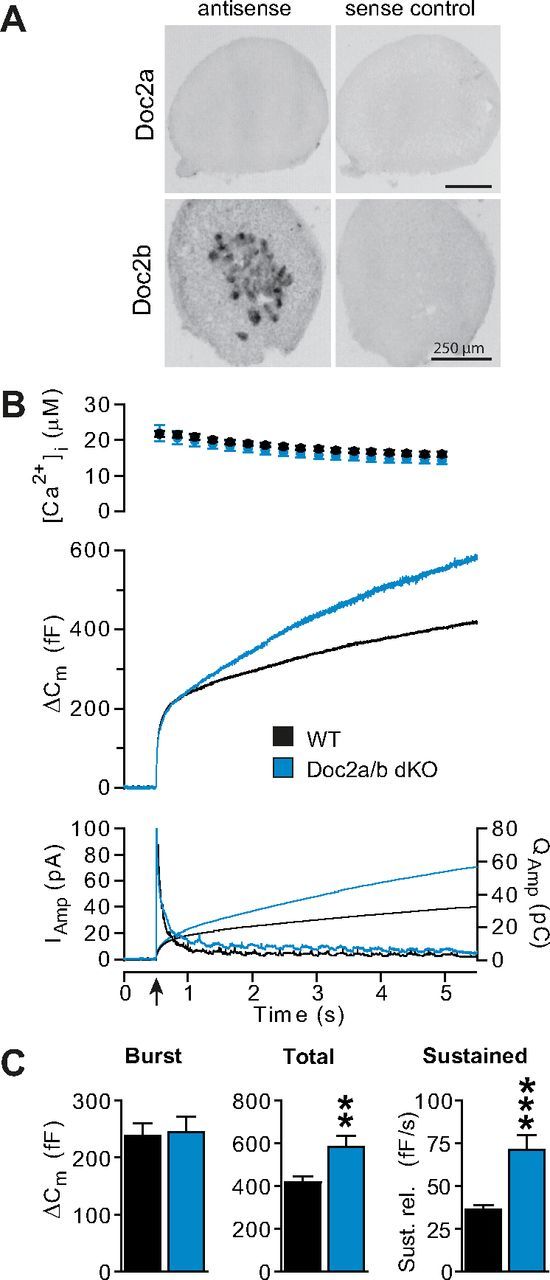
Doc2a does not compensate for the loss of Doc2b. A, In situ hybridization revealed expression of Doc2b in the mouse adrenal medulla, consisting of chromaffin cells, but no expression of Doc2a. (See also Friedrich et al., 2008). Nevertheless, we wanted to rule out that the phenotype of Doc2b KO might result from upregulation of Doc2a. B, Flash experiments performed in the Doc2a/b double KO mouse line from a high basal [Ca2+]i (841 ± 70 nm for Doc2a/b KO, 907 ± 73 nm for WT) showed a phenotype that closely resembled that of the Doc2b single KO mouse line cells (WT: n = 15 cells, black traces; Doc2a/b DKO: n = 15 cells, blue traces), characterized by a steeper sustained component (see also Fig. 5D). Panels are as in Figure 2A. C, The analysis of the exocytotic burst, total secretion, and sustained component shows that the sustained component and total release are both increased in the Doc2a/b DKO. **p < 0.01; ***p < 0.001 (unpaired two-tailed Student's t test).
Overall, our data show that Doc2b KO cells suffer from a synchronization problem: vesicles tend to fuse late during sustained calcium elevations, whereas the fast burst is slightly smaller and the refilling of the RRP is incomplete, suggesting a complex defect in the priming process.
Doc2b affects neither the calcium dependence of release nor of steady-state vesicle priming
The calcium affinity of Doc2b is markedly higher than that of syt1, and maximal lipid binding is observed at calcium concentrations <1 μm (Groffen et al., 2006; Malkinson and Spira, 2006). Since we expected that calcium binding and translocation to the membrane is a prerequisite for modulating fusion, we hypothesized that Doc2b might play a calcium-dependent role in exocytosis.
To examine the calcium dependence of fusion triggering, we illuminated the cells by rapidly alternating between 350 and 380 nm, which leads to slow calcium uncaging while allowing simultaneous ratiometric calcium measurements (Sørensen et al., 2002). The resulting calcium ramp (Fig. 5A, top) triggers a sigmoid-shaped increase in membrane capacitance (Fig. 5A, middle). The secretion threshold was determined as the [Ca2+]i at the first maximum of the second derivative of the capacitance trace (Fig. 5A, bottom), i.e., as the calcium concentration at the time of maximal secretion acceleration. This parameter is a very sensitive measurement of the calcium dependence of release at low [Ca2+]i and is affected by mutations that change the calcium affinity of synaptotagmin-1 (Sørensen et al., 2003a), or when mutating SNAP-25 to reduce its interaction with synaptotagmin-1 (Mohrmann et al., 2013). If Doc2b drives secretion at low [Ca2+]i, we would therefore expect an increase in the secretion threshold in the Doc2b KO. However, the threshold was unchanged between Doc2b KO and WT cells (Fig. 5B), which argues against a role for Doc2b in driving fusion triggering.
We next assessed the calcium dependence of steady-state vesicle priming, by plotting the burst size as a function of basal (preflash) [Ca2+]i (Fig. 5C). Increasing basal calcium causes a bell-shaped change in the size of the exocytotic burst (Voets, 2000); the increase at concentrations <1 μm is caused by a calcium-dependent priming process upstream of the primed vesicle pools (see Fig. 9A), whereas the decrease at higher [Ca2+]i is caused by the fusion of vesicles from the primed pools, causing their depletion. Our WT data (Fig. 5C) reproduce previous findings (Voets, 2000), and Doc2b KO cells display normal calcium-dependent priming at lower [Ca2+]i, ruling out an obligatory role of Doc2b in driving calcium-dependent priming upstream of the SRP (see Discussion).
Figure 9.

Doc2b inhibits vesicle priming during sustained calcium elevations and redirects vesicles to refill the RRP. A, In the presence of Doc2b, the RRP is larger; both SRP and RRP vesicles fuse at the start of stimulation. These pools are refilled from the UPP, which again is refilled from the Depot pool (data not shown; we assume that the reaction refilling the UPP is slow compared with the duration of the experiment, see Discussion). B, During sustained calcium elevations, Doc2b inhibits priming, thus limiting the number of vesicles lost from the UPP. C, During recovery, the larger UPP will allow for complete refilling of the releasable pools, assisted by the positive function of Doc2b in increasing RRP size. D, In the absence of Doc2b, at the start of stimulation the vesicles in the primed vesicle pools (RRP and SRP) will fuse. E, During sustained calcium elevations, vesicles moving unrestricted from the UPP to the SRP/RRP will fuse immediately because of the high calcium concentration, causing loss of UPP vesicles. F, Because of the partially depleted UPP, refilling of the RRP following relaxation of the calcium signal is incomplete.
When comparing the results from calcium uncaging (especially Fig. 5C) with the RRP estimated by depolarizations (Fig. 1), it appears that the latter method overestimates the RRP because, at the very low basal [Ca2+]i used in depolarization experiments, the whole burst (which is made up of SRP and RRP vesicles) should be <100 fF (Fig. 5C), while the estimate of the RRP by depolarization alone was ∼100 fF (Fig. 1E, first set of points). Indeed, with an increase in global [Ca2+]i to ∼10 μm (Fig. 1B), the SRP is expected to also fuse during this protocol (Voets, 2000). Therefore, the delayed release measured in depolarization experiments is a phase that follows fusion of both SRP and RRP vesicles, just as the sustained release phase estimated in uncaging experiments. The increases in delayed and sustained release in the Doc2b KO are therefore different reflections of one and the same phenotype.
Doc2b causes calcium-dependent inhibition of sustained release
The main effect of removing Doc2b is an increase in the rate of the sustained—or delayed—fusion component following an increase in the [Ca2+]i. When flashing from a high basal [Ca2+]i (as in Fig. 2E), Doc2b is expected to be calcium-saturated, even before uncaging, making it unlikely that the activation state of Doc2b changed following the flash. In our search for a calcium-dependent function of Doc2b we therefore asked whether the sustained component would depend on preflash [Ca2+]i. Plotting the sustained component as a function of preflash [Ca2+]i indicated almost no dependency in WT cells (Fig. 5D). Surprisingly, however, in Doc2b KO cells the sustained component displayed a clear dependence on preflash [Ca2+]i; at low preflash [Ca2+]i the rate was hardly different from WT, but at higher calcium concentrations the rate increased (Fig. 5D). Consequently, in WT cells Doc2b exerts a calcium-dependent inhibitory function on the rate of sustained release (Fig. 5D, green subtraction line). Strikingly, this inhibition appeared to almost exactly counteract the underlying calcium dependence of the sustained component (as revealed in the KO), resulting in an overall constant sustained rate at all [Ca2+]i in the WT. The sustained rate is normally assumed to report on the forward priming rate (Sørensen, 2004) indicating that, in our experiments, priming is inhibited by Doc2b.
Pool refilling is faster, but incomplete, in the absence of Doc2b
A higher rate of sustained exocytosis (as in the Doc2b KO) is commonly taken as a sign of faster vesicle priming, or pool refilling, because in the standard model for secretion, vesicle priming is rate limiting during this phase of release (Sørensen, 2004). After initial depletion of the primed vesicle pools, vesicles that prime while the [Ca2+]i is still high will immediately fuse, giving rise to sustained exocytosis. However, this interpretation is based on a specific model, and other possibilities remain. For instance, the steeper sustained component might be caused by the fusion of a separate vesicle pool and not related to primed pool recovery.
To directly probe pool recovery we designed a dual-flash protocol, where two consecutive flashes were applied with a variable interval. For short recovery times we had to ensure that the [Ca2+]i would return to baseline levels as soon as possible, to allow for a second stimulation. This was obtained by reducing illumination after the flash, and by modifying the pipette solution to contain less calcium. Before the first stimulation, monochromatic illumination was used to raise the [Ca2+]i to prime vesicles and fill the pools (772 ± 28 nm for WT; 771 ± 28 nm for KO); after the flash, the [Ca2+]i quickly relaxed with a time constant of ∼3 s (Fig. 6A, red dots). Additionally, flash intensity was adjusted between the first and second stimulations to achieve similar postflash [Ca2+]i.
These experiments showed that the fast phase of the burst recovered following an exponential process with a time constant of 49.8 ± 25.4 s in the WT cells (Fig. 6B). While the fast burst was readily detectable by its faster release kinetics, the slow burst could not be reliably separated from the sustained component. Due to the relaxing [Ca2+]i, the sustained component was not linear, but exponential with a time constant similar to the slow burst. We nevertheless estimated the slow component (consisting of the slow burst and parts of the sustained component) following the fast burst (Fig. 6C). It is evident that adrenal chromaffin cells—like central neurons (Sakaba and Neher, 2001)—have a fast releasing phase that recovers slowly (Fig. 6B) and a slowly releasing phase that recovers fast (Fig. 6C). The results show that the fast burst in Doc2b KO cells indeed recovered faster, but to a lower level than WT cells. To test whether these changes were significant, we fitted both WT and KO data with a single-exponential recovery function. We found that both the shorter time constant (9.8 ± 5.1 s for KO, p = 0.028, extra sum-of-squares F test) and the lower recovery level (60.9 ± 5.6% for KO and 105.1 ± 19.7% for WT, p = 0.0052, extra sum-of-squares F test) to be significantly different between KO and WT cells.
This finding establishes the conclusion that, in the absence of Doc2b, chromaffin cells have faster but incomplete recovery of the RRP. The incomplete recovery of this vesicle pool aligns well with the depolarization experiments (Fig. 1), and with the reduced burst size in the second stimulation when flashing from intermediate [Ca2+]i (Fig. 2A–D). Furthermore, the faster pool recovery concurs with both the increased sustained component identified in uncaging experiments and with the increased amount of delayed release between depolarization protocols (Fig. 1H).
Doc2b overexpression synchronizes release
To understand whether the changes in the Doc2b KO are the direct effect of Doc2b elimination or caused by developmental changes indirectly linked to the genetic change, we turned to rescue experiments. We used a Semliki Forest virus (SFV) construct, which expressed wild-type Doc2b followed by an internal ribosome entry site (IRES) and enhanced green fluorescent protein (EGFP) as an expression marker.
Immunostaining showed that Doc2b was overexpressed in mouse chromaffin cells after 5–6 h of infection with SFV particles (Fig. 7A,B). Electrophysiological characterization of expressing cells revealed a substantial increase in the size of the fast burst (Fig. 7C,D), and a severe decrease in the sustained component (Fig. 7C,F) compared with Doc2b KO cells. These effects go in the expected direction, i.e., in the opposite direction of Doc2b KO cells, which had a steeper rate of sustained release and a smaller fast burst (although the effect was small, Fig. 3Ci). It is interesting that Doc2b overexpression caused over-rescue: the sustained component was reduced more and the fast burst was increased more than in the WT. This is in line with the SFV being a very strong expression system (Ashery et al., 1999) and it shows that the effect of Doc2b is concentration-dependent, and not saturated by endogenous Doc2b levels. The overall effect of overexpression confirms that the knock-out phenotype is reversible and thus caused by the direct loss of Doc2b.
Figure 7.

Overexpression of Doc2b synchronizes release to the calcium signal. A, Overexpression of Doc2b using Semliki Forest virus was confirmed by immunocytochemistry. Expressing cells (GFP was an expression marker) displayed a clear signal for Doc2b. Cells were counterstained for VAMP2/synaptobrevin-2 (scale bar, 3 μm). B, Quantification of fluorescence intensity for Doc2b in KO and rescued cells. C, Uncaging experiment in Doc2b KO cells and in KO cells overexpressing Doc2b (rescue) (Doc2b KO: total of n = 20 cells, magenta traces; Doc2b KO cells expressing Doc2b: total n = 33 cells, gray traces). Top panel shows average (±SEM) [Ca2+]i measured by microfluorimetry. Bottom panel shows the changes in cell membrane capacitance (mean traces for all cells). D–H, Kinetic analysis showed that the RRP size was increased, but the sustained component markedly decreased by overexpression of Doc2b. ***p < 0.001, unpaired two-tailed Student's t test.
Vesicle number is increased upon overexpression of Doc2b
Doc2b elimination leads to an increase in the sustained component, whereas Doc2b overexpression causes a strong decrease. These changes could be caused either by a difference in the priming process itself, or by a difference in vesicular content of the cell. This is especially pertinent, as Doc2b deletion decreases the rate of spontaneous release in neurons (Groffen et al., 2010; Pang et al., 2011). Even though it is unknown whether chromaffin cells have a similar spontaneous release pathway, an effect of Doc2b on such a pathway could partly explain our data; in the knock-out, decreased spontaneous release could lead to “overfilling” of upstream pools, and thereby to an increased sustained release rate, whereas overexpression could cause upstream pool depletion and a lower sustained rate.
To investigate this point we performed electron microscopy on Doc2a/b double knock-out chromaffin cells. Both WT and KO cells displayed normal morphology, including a large pool of docked vesicles (Fig. 8A–G), and the overall number of vesicles was unchanged. However, when overexpressing Doc2b, the total number of vesicles per cell significantly increased (Fig. 8H). This was not caused by a change in the docked vesicle pool, but by a higher number of vesicles further inside the cell (Fig. 8G,I), particularly a small pool of vesicles just beneath the plasma membrane (>0–25 nm group, Fig. 8G). Vesicle diameters were not changed between the three groups (Fig. 8J). These data rule out that pool depletion or overfilling could explain the electrophysiological changes seen in the KO, or after overexpression, respectively.
Figure 8.
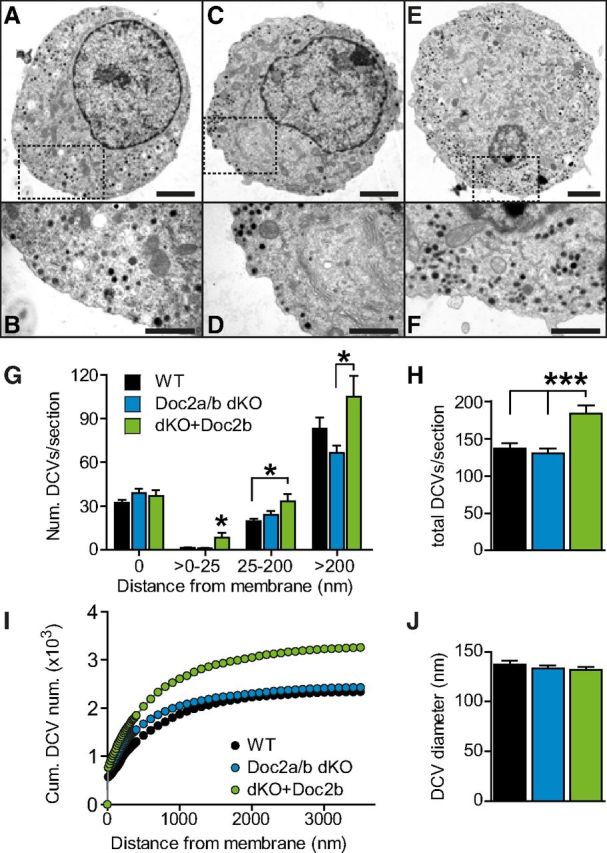
Overexpression of Doc2b accumulates vesicles beneath the plasma membrane. A–F, Electron micrographs of WT cells (A, B), Doc2a/b DKO cells (C, D), and Doc2a/b DKO cells overexpressing Doc2b (E, F). Scale bars: A,C,E, 2 μm; B,D,F, 1 μm. G, Number of dense-core vesicles according to their distance from the membrane (Doc2a/b DKO: N = 9 animals, n = 20 cells, magenta bars; WT cells N = 5 animals, n = 24 cells, black bars, Doc2a/b DKO cells expressing Doc2b: N = 9 animals, n = 18 cells, gray bars). Overexpression of Doc2b increased the number of vesicles inside the cell, especially in a layer just underneath the membrane (>0–25 nm). In contrast, docked vesicles (distance 0 nm) were unchanged in number. H, The total number of LDCVs per section was increased upon overexpression. I, Cumulative distribution of vesicles inside the cell. J, The diameter of vesicles was unchanged between conditions. *p < 0.05; ***p < 0.001 (one-way ANOVA with Tukey's post-test).
Discussion
Our data establish a function for Doc2b in shifting catecholamine release in chromaffin cells from sustained secretion to synchronized release. First, Doc2b deletion led to a slightly smaller RRP and a larger sustained component (Figs. 1–3). Second, refilling of the readily releasable pool was faster, but incomplete, in the absence of Doc2b (Figs. 1, 6). Third, in prolonged experiments, Doc2b deletion did not change the total amount of vesicle release, only its distribution between fast and delayed release components (Fig. 1). Fourth, Doc2b overexpression caused a larger fast burst and a shallower sustained component (Fig. 7). Fifth, electron microscopy identified a larger number of vesicles in Doc2b overexpressing cells (Fig. 8), showing that the effect of Doc2b on the sustained component is not secondary to vesicle depletion. The latter finding suggests that, even though Doc2b elimination does not significantly change the total amount of release over the course of an electrophysiological experiment, inhibition of release dominates when Doc2b is overexpressed, causing the retention of vesicles.
Using calcium ramps, we find no evidence for a function of Doc2b in fusing vesicles at low [Ca2+]i. Thus, despite the demonstrated ability of Doc2b to stimulate SNARE-dependent vesicle fusion in vitro (Groffen et al., 2010), our data are more consistent with an upstream role (Pang et al., 2011). Previous work using overexpression emphasized the increased burst size (Friedrich et al., 2008), but did not identify the inhibition of sustained release, which we find to be an important role for Doc2b. The accurate measurement of vesicle pool refilling during experiments encompassing multiple rounds of stimulation (Figs. 1, 5) demonstrated that endogenous Doc2b exerts dual effects on vesicle pool refilling, which ultimately favor synchronous exocytosis (see below).
Doc2b: dual functions favor synchronous release
The size of primed vesicle pools increases in parallel as a function of the [Ca2+]i at concentrations up to 0.7–1 μm, while at higher concentrations both pools decrease due to ongoing fusion (Voets, 2000). It was, therefore, concluded that both the fast and slow phases of burst release are fed by an upstream calcium-dependent process—priming—giving rise to sustained secretion after a step increase in the calcium concentration (Fig. 9A). Experiments using overexpression of Doc2b led to the suggestion that calcium-dependent Doc2b trafficking to the membrane might constitute a molecular counterpart of this process (Friedrich et al., 2008). Here, we have shown that the release burst is still calcium-dependent in the Doc2b KO mouse (Fig. 5C), ruling out the possibility that Doc2b is obligatory for the process refilling the SRP (Fig. 9A). However, the small decrease in the size of the fast burst upon Doc2b deletion, and the larger increase upon overexpression, is in line with the idea that Doc2b supports another process: the one that converts SRP vesicles to RRP vesicles (Fig. 9C).
The conclusion that Doc2b inhibits the forward priming reaction, after emptying of the primed vesicle pools, is directly supported by the observation that the sustained component is inhibited at higher basal [Ca2+]i in the presence, but not in the absence, of Doc2b (Fig. 5D). The calcium dependence of the sustained component in the Doc2b KO indicates the existence of an upstream calcium-dependent recruitment process, which is inhibited by Doc2b. Interestingly, the function of Doc2b in chromaffin cells demonstrated here appears similar to the function of rabphilin in neurons, which has been shown to delay recovery from use-dependent depression (Deak et al., 2006); in the absence of rabphilin recovery is sped up. Rabphilin is a member of the Doc2 protein family (Verhage et al., 1997), suggesting that these proteins might share the function of regulating events upstream of vesicle fusion. Alternative to the hypothesis that Doc2b inhibits priming, it might be suggested that Doc2b inhibits fusion itself at sustained high [Ca2+]i. However, fusion rates were not changed in the presence of Doc2b (Figs. 3Cii, 7G), making this suggestion unlikely. Since the downstream pools, SRP and RRP, are not reduced in the presence of Doc2b, it has to be concluded that either Doc2b only affects forward priming at high [Ca2+]i, or that Doc2b simultaneously decreases the backward priming reaction.
Doc2 binds to proteins involved in vesicle priming and fusion, including Munc13 (Orita et al., 1997; Mochida et al., 1998; Duncan et al., 1999; Higashio et al., 2008), Munc18-1 (Verhage et al., 1997), and syntaxin:SNAP-25 dimers (Sato et al., 2010; Yao et al., 2011). Recent reconstitution experiments have identified these four proteins as constituting the minimal fusion machinery on the plasma membrane, which associates with vesicular synapotagmin-1 and synaptobrevin-2/VAMP-2 to drive calcium-dependent fusion (Ma et al., 2013). It is thus conceivable that the association of Doc2b to this quaternary complex causes intermittent inhibition of vesicle priming by sequestering the fusion machinery. The interaction with SNAP-25:syntaxin dimers has been suggested to underlie the delay in vesicle priming induced by rabphilin in neurons (Deak et al., 2006). Inhibition might be released when Doc2b/rabphilin is replaced by vesicular proteins, producing a fusion competent vesicle.
In the fusion of LDCVs, CAPS might act as a functional Munc13 analog (Speese et al., 2007; Liu et al., 2008, 2010). CAPS and Munc13 probably function in a similar way by opening up closed syntaxin within the Munc18:syntaxin dimer (Betz et al., 1997; Richmond et al., 2001; Basu et al., 2005; Madison et al., 2005; Stevens et al., 2005; Khodthong et al., 2011; Ma et al., 2011). In the absence of CAPS, the sustained component of release in chromaffin cells is reduced (Liu et al., 2010). Overexpression of an open form of syntaxin bypasses the need for CAPS in setting up the RRP (Liu et al., 2010), but the sustained component remains inhibited. Thus, calcium-dependent opening of syntaxin, through Munc13/CAPS action, might underlie vesicle priming during sustained release. The similarity to the reduced sustained component and the increase in burst release upon overexpression of Doc2b (Fig. 7) suggest that Doc2b might inhibit a Munc13/CAPS-dependent step in vesicle priming, but other possibilities remain. For example, both Doc2a and Doc2b bind to tctex-1, a light chain of cytoplasmic dynein that affects the transport of intracellular vesicles (Nagano et al., 1998), although this interaction is not essential for plasma membrane association of Doc2b (Groffen et al., 2004). The increased accumulation of vesicles below the membrane in Doc2b overexpressing cells is consistent with an interaction with a motor protein.
The increase in fast burst release upon Doc2b overexpression indicates that it interacts in at least two different configurations with the fusion machinery. Whereas one interaction mode is negative, Doc2b is also able to interact constructively, by either speeding up the conversion of slow-to-fast releasing vesicles or by slowing down the opposite reaction. This interaction might involve binding of Doc2b to the already assembled priming machinery. Interestingly, the overexpression of syt-1, which competes with Doc2b for interaction with the SNARE-complex (Groffen et al., 2010), also shifts release toward the fast burst (Nagy et al., 2006). Thus, interaction with either protein might stabilize the priming machinery.
How can the complicated net effect of Doc2b during repetitive physiological stimulation, such as in Figure 1, be explained? Previous experiments demonstrated that the primed vesicle pools are refilled from an upstream vesicle pool of limited size—termed the “UnPrimed Pool”, or UPP (Ashery et al., 2000) (Fig. 9A,D). In the presence of Doc2b the refilling reaction during sustained calcium elevation is inhibited, preventing the upstream (unprimed) vesicles from priming at a time where this would lead to immediate fusion (Fig. 9B). Instead, these vesicles are saved for after the relaxation of [Ca2+]i, where the stimulatory action of Doc2b further assists the refilling of the RRP (Fig. 9C). In the absence of Doc2b (Fig. 8D--F), priming while the [Ca2+]i is still high will cause the newly primed vesicles to fuse, leading to more sustained secretion, but also to depletion of the upstream pool (UPP). This—and the lack of promotion of the SRP-to-RRP transition—in turn causes incomplete refilling when the [Ca2+]i subsides (Fig. 9F). Thus, putting a cap on the forward priming rate during sustained stimulation will spare vesicles while calcium is high, leaving more vesicles to refill the fast releasing pool during periods of rest. The UPP, in turn, is filled from a much larger depot pool, which might coincide with the process of docking (Ashery et al., 2000), even though this is unknown. Docking is a much slower process than priming (Steyer et al., 1997) which might, at least partially, account for the rundown seen during our experiments (Fig. 1).
Our study has revealed two functions for Doc2b: first, Doc2b is a priming inhibitor, which protects unprimed vesicles from fusing during sustained calcium elevations. Second, Doc2b stimulates the preferential recruitment of these vesicles into the primed vesicle pool with the highest release rate: the RRP. As illustrated in Figure 9, both mechanisms likely cooperate to favor synchronous over sustained secretion. Thus, shaping the release kinetics of a secretory cell involves an intricate interplay between promoters and inhibitors; in the case of Doc2b, both functions are found in the same protein.
Footnotes
The work was supported by the Lundbeck Foundation, the Lundbeck Foundation Center for Biomembranes in Nanomedicine, the Novo Nordisk Foundation, the Danish Medical Research Council (all to J.B.S.), the Netherlands Organization for Scientific Research, NWO (VENI 916-36-043, MEERVOUD 836-10-002 HdW, ZonMW 903-42-095 to M.V.), the European Molecular Biology Organization (EMBO; A.M.W.), and the European Union Seventh Framework Programme under grant agreements FP7-People-ITN-2008-238055 (“BrainTrain”, HdW), FP7-People-2009-IEF-254647 (“Exosyts”, to P.S.P.), HEALTH-F2-2009-242167 (“SynSys,” to J.B.S. and M.V.), HEALTH-F2-2009-241498 (“EUROSPIN”, to M.V.), and the NeuroBsik Mouse Phenomics Consortium (BSIK03053 to M.V.). Part of this research has been executed in the Systems Biology of the Synapse research program (VUmc NCA 03-03) of the Neuroscience Campus Amsterdam. We thank A.M.N. Petersen, R. Zalm, J. Wortel, and R. Dekker for excellent technical assistance.
The authors declare no competing financial interests.
References
- Ashery U, Betz A, Xu T, Brose N, Rettig J. An efficient method for infection of adrenal chromaffin cells using the Semliki Forest virus gene expression system. Eur J Cell Biol. 1999;78:525–532. doi: 10.1016/S0171-9335(99)80017-X. [DOI] [PubMed] [Google Scholar]
- Ashery U, Varoqueaux F, Voets T, Betz A, Thakur P, Koch H, Neher E, Brose N, Rettig J. Munc13-1 acts as a priming factor for large dense-core vesicles in bovine chromaffin cells. EMBO J. 2000;19:3586–3596. doi: 10.1093/emboj/19.14.3586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basu J, Shen N, Dulubova I, Lu J, Guan R, Guryev O, Grishin NV, Rosenmund C, Rizo J. A minimal domain responsible for Munc13 activity. Nat Struct Mol Biol. 2005;12:1017–1018. doi: 10.1038/nsmb1001. [DOI] [PubMed] [Google Scholar]
- Betz A, Okamoto M, Benseler F, Brose N. Direct interaction of the rat unc-13 homologue Munc13-1 with the N terminus of syntaxin. J Biol Chem. 1997;272:2520–2526. doi: 10.1074/jbc.272.4.2520. [DOI] [PubMed] [Google Scholar]
- Bittner MA, Holz RW. Kinetic analysis of secretion from permeabilized adrenal chromaffin cells reveals distinct components. J Biol Chem. 1992;267:16219–16225. [PubMed] [Google Scholar]
- Bruns D. Detection of transmitter release with carbon fiber electrodes. Methods. 2004;33:312–321. doi: 10.1016/j.ymeth.2004.01.004. [DOI] [PubMed] [Google Scholar]
- Chapman ER. How does synaptotagmin trigger neurotransmitter release? Ann Rev Biochem. 2008;77:615–641. doi: 10.1146/annurev.biochem.77.062005.101135. [DOI] [PubMed] [Google Scholar]
- Deak F, Shin OH, Tang J, Hanson P, Ubach J, Jahn R, Rizo J, Kavalali ET, Südhof TC. Rabphilin regulates SNARE-dependent re-priming of synaptic vesicles for fusion. EMBO J. 2006;25:2856–2866. doi: 10.1038/sj.emboj.7601165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duncan RR, Betz A, Shipston MJ, Brose N, Chow RH. Transient, phorbol ester-induced DOC2-Munc13 interactions in vivo. J Biol Chem. 1999;274:27347–27350. doi: 10.1074/jbc.274.39.27347. [DOI] [PubMed] [Google Scholar]
- Friedrich R, Groffen AJ, Connell E, van Weering JR, Gutman O, Henis YI, Davletov B, Ashery U. DOC2B acts as a calcium switch and enhances vesicle fusion. J Neurosci. 2008;28:6794–6806. doi: 10.1523/JNEUROSCI.0538-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groffen AJ, Brian EC, Dudok JJ, Kampmeijer J, Toonen RF, Verhage M. Ca(2+)-induced recruitment of the secretory vesicle protein DOC2B to the target membrane. J Biol Chem. 2004;279:23740–23747. doi: 10.1074/jbc.M400731200. [DOI] [PubMed] [Google Scholar]
- Groffen AJ, Friedrich R, Brian EC, Ashery U, Verhage M. DOC2A and DOC2B are sensors for neuronal activity with unique calcium-dependent and kinetic properties. J Neurochem. 2006;97:818–833. doi: 10.1111/j.1471-4159.2006.03755.x. [DOI] [PubMed] [Google Scholar]
- Groffen AJ, Martens S, Díez Arazola R, Cornelisse LN, Lozovaya N, de Jong AP, Goriounova NA, Habets RL, Takai Y, Borst JG, Brose N, McMahon HT, Verhage M. Doc2b is a high-affinity Ca2+ sensor for spontaneous neurotransmitter release. Science. 2010;327:1614–1618. doi: 10.1126/science.1183765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higashio H, Nishimura N, Ishizaki H, Miyoshi J, Orita S, Sakane A, Sasaki T. Doc2 alpha and Munc13-4 regulate Ca(2+)-dependent secretory lysosome exocytosis in mast cells. J Immunol. 2008;180:4774–4784. doi: 10.4049/jimmunol.180.7.4774. [DOI] [PubMed] [Google Scholar]
- Horrigan FT, Bookman RJ. Releasable pools and the kinetics of exocytosis in adrenal chromaffin cells. Neuron. 1994;13:1119–1129. doi: 10.1016/0896-6273(94)90050-7. [DOI] [PubMed] [Google Scholar]
- Jahn R, Scheller RH. SNAREs—engines for membrane fusion. Nat Rev Mol Cell Biol. 2006;7:631–643. doi: 10.1038/nrm2002. [DOI] [PubMed] [Google Scholar]
- Khodthong C, Kabachinski G, James DJ, Martin TF. Munc13 homology domain-1 in CAPS/UNC31 mediates SNARE binding required for priming vesicle exocytosis. Cell Metabol. 2011;14:254–263. doi: 10.1016/j.cmet.2011.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Schirra C, Stevens DR, Matti U, Speidel D, Hof D, Bruns D, Brose N, Rettig J. CAPS facilitates filling of the rapidly releasable pool of large dense-core vesicles. J Neurosci. 2008;28:5594–5601. doi: 10.1523/JNEUROSCI.5672-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Schirra C, Edelmann L, Matti U, Rhee J, Hof D, Bruns D, Brose N, Rieger H, Stevens DR, Rettig J. Two distinct secretory vesicle-priming steps in adrenal chromaffin cells. J Cell Biol. 2010;190:1067–1077. doi: 10.1083/jcb.201001164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma C, Li W, Xu Y, Rizo J. Munc13 mediates the transition from the closed syntaxin-Munc18 complex to the SNARE complex. Nat Struct Mol Biol. 2011;18:542–549. doi: 10.1038/nsmb.2047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma C, Su L, Seven AB, Xu Y, Rizo J. Reconstitution of the vital functions of Munc18 and Munc13 in neurotransmitter release. Science. 2013;339:421–425. doi: 10.1126/science.1230473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madison JM, Nurrish S, Kaplan JM. UNC-13 interaction with syntaxin is required for synaptic transmission. Curr Biol. 2005;15:2236–2242. doi: 10.1016/j.cub.2005.10.049. [DOI] [PubMed] [Google Scholar]
- Malkinson G, Spira ME. Calcium concentration threshold and translocation kinetics of EGFP-DOC2B expressed in cultured Aplysia neurons. Cell Calcium. 2006;39:85–93. doi: 10.1016/j.ceca.2005.10.007. [DOI] [PubMed] [Google Scholar]
- Martens S, Kozlov MM, McMahon HT. How synaptotagmin promotes membrane fusion. Science. 2007;316:1205–1208. doi: 10.1126/science.1142614. [DOI] [PubMed] [Google Scholar]
- Mochida S, Orita S, Sakaguchi G, Sasaki T, Takai Y. Role of the Doc2 alpha-Munc13-1 interaction in the neurotransmitter release process. Proc Natl Acad Sci U S A. 1998;95:11418–11422. doi: 10.1073/pnas.95.19.11418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohrmann R, de Wit H, Verhage M, Neher E, Sørensen JB. Fast vesicle fusion in living cells requires at least three SNARE complexes. Science. 2010;330:502–505. doi: 10.1126/science.1193134. [DOI] [PubMed] [Google Scholar]
- Mohrmann R, de Wit H, Connell E, Pinheiro PS, Leese C, Bruns D, Davletov B, Verhage M, Sørensen JB. Synaptotagmin interaction with SNAP-25 governs vesicle docking, priming, and fusion triggering. J Neurosci. 2013;33:14417–14430. doi: 10.1523/JNEUROSCI.1236-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagano F, Orita S, Sasaki T, Naito A, Sakaguchi G, Maeda M, Watanabe T, Kominami E, Uchiyama Y, Takai Y. Interaction of Doc2 with tctex-1, a light chain of cytoplasmic dynein. Implication in dynein-dependent vesicle transport. J Biol Chem. 1998;273:30065–30068. doi: 10.1074/jbc.273.46.30065. [DOI] [PubMed] [Google Scholar]
- Nagy G, Kim JH, Pang ZP, Matti U, Rettig J, Südhof TC, Sørensen JB. Different effects on fast exocytosis induced by synaptotagmin 1 and 2 isoforms and abundance but not by phosphorylation. J Neurosci. 2006;26:632–643. doi: 10.1523/JNEUROSCI.2589-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orita S, Naito A, Sakaguchi G, Maeda M, Igarashi H, Sasaki T, Takai Y. Physical and functional interactions of Doc2 and Munc13 in Ca2+-dependent exocytotic machinery. J Biol Chem. 1997;272:16081–16084. doi: 10.1074/jbc.272.26.16081. [DOI] [PubMed] [Google Scholar]
- Pang ZP, Südhof TC. Cell biology of Ca2+-triggered exocytosis. Curr Opin Cell Biol. 2010;22:496–505. doi: 10.1016/j.ceb.2010.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pang ZP, Bacaj T, Yang X, Zhou P, Xu W, Südhof TC. Doc2 supports spontaneous synaptic transmission by a Ca(2+)-independent mechanism. Neuron. 2011;70:244–251. doi: 10.1016/j.neuron.2011.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramalingam L, Oh E, Yoder SM, Brozinick JT, Kalwat MA, Groffen AJ, Verhage M, Thurmond DC. Doc2b is a key effector of insulin secretion and skeletal muscle insulin sensitivity. Diabetes. 2012;61:2424–2432. doi: 10.2337/db11-1525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richmond JE, Weimer RM, Jorgensen EM. An open form of syntaxin bypasses the requirement for UNC-13 in vesicle priming. Nature. 2001;412:338–341. doi: 10.1038/35085583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rizo J, Chen X, Araç D. Unraveling the mechanisms of synaptotagmin and SNARE function in neurotransmitter release. Trends Cell Biol. 2006;16:339–350. doi: 10.1016/j.tcb.2006.04.006. [DOI] [PubMed] [Google Scholar]
- Sakaba T, Neher E. Calmodulin mediates rapid recruitment of fast-releasing synaptic vesicles at a calyx-type synapse. Neuron. 2001;32:1119–1131. doi: 10.1016/S0896-6273(01)00543-8. [DOI] [PubMed] [Google Scholar]
- Sato M, Mori Y, Matsui T, Aoki R, Oya M, Yanagihara Y, Fukuda M, Tsuboi T. Role of the polybasic sequence in the Doc2alpha C2B domain in dense-core vesicle exocytosis in PC12 cells. J Neurochem. 2010;114:171–181. doi: 10.1111/j.1471-4159.2010.06739.x. [DOI] [PubMed] [Google Scholar]
- Schonn JS, Maximov A, Lao Y, Südhof TC, Sørensen JB. Synaptotagmin-1 and -7 are functionally overlapping Ca2+ sensors for exocytosis in adrenal chromaffin cells. Proc Natl Acad Sci U S A. 2008;105:3998–4003. doi: 10.1073/pnas.0712373105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segovia M, Alés E, Montes MA, Bonifas I, Jemal I, Lindau M, Maximov A, Südhof TC, Alvarez de Toledo G. Push-and-pull regulation of the fusion pore by synaptotagmin-7. Proc Natl Acad Sci U S A. 2010;107:19032–19037. doi: 10.1073/pnas.1014070107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith C, Moser T, Xu T, Neher E. Cytosolic Ca2+ acts by two separate pathways to modulate the supply of release-competent vesicles in chromaffin cells. Neuron. 1998;20:1243–1253. doi: 10.1016/S0896-6273(00)80504-8. [DOI] [PubMed] [Google Scholar]
- Sørensen JB. Formation, stabilisation and fusion of the readily releasable pool of secretory vesicles. Pflugers Arch. 2004;448:347–362. doi: 10.1007/s00424-004-1247-8. [DOI] [PubMed] [Google Scholar]
- Sørensen JB, Matti U, Wei SH, Nehring RB, Voets T, Ashery U, Binz T, Neher E, Rettig J. The SNARE protein SNAP-25 is linked to fast calcium triggering of exocytosis. Proc Natl Acad Sci U S A. 2002;99:1627–1632. doi: 10.1073/pnas.251673298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sørensen JB, Fernández-Chacón R, Südhof TC, Neher E. Examining synaptotagmin 1 function in dense core vesicle exocytosis under direct control of Ca2+ J Gen Physiol. 2003a;122:265–276. doi: 10.1085/jgp.200308855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sørensen JB, Nagy G, Varoqueaux F, Nehring RB, Brose N, Wilson MC, Neher E. Differential control of the releasable vesicle pools by SNAP-25 splice variants and SNAP-23. Cell. 2003b;114:75–86. doi: 10.1016/S0092-8674(03)00477-X. [DOI] [PubMed] [Google Scholar]
- Speese S, Petrie M, Schuske K, Ailion M, Ann K, Iwasaki K, Jorgensen EM, Martin TF. UNC-31 (CAPS) is required for dense-core vesicle but not synaptic vesicle exocytosis in Caenorhabditis elegans. J Neurosci. 2007;27:6150–6162. doi: 10.1523/JNEUROSCI.1466-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevens DR, Wu ZX, Matti U, Junge HJ, Schirra C, Becherer U, Wojcik SM, Brose N, Rettig J. Identification of the minimal protein domain required for priming activity of Munc13-1. Curr Biol. 2005;15:2243–2248. doi: 10.1016/j.cub.2005.10.055. [DOI] [PubMed] [Google Scholar]
- Steyer JA, Horstmann H, Almers W. Transport, docking and exocytosis of single secretory granules in live chromaffin cells. Nature. 1997;388:474–478. doi: 10.1038/41329. [DOI] [PubMed] [Google Scholar]
- Toonen RF, Kochubey O, de Wit H, Gulyas-Kovacs A, Konijnenburg B, Sørensen JB, Klingauf J, Verhage M. Dissecting docking and tethering of secretory vesicles at the target membrane. EMBO J. 2006;25:3725–3737. doi: 10.1038/sj.emboj.7601256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verhage M, de Vries KJ, Røshol H, Burbach JP, Gispen WH, Südhof TC. DOC2 proteins in rat brain: complementary distribution and proposed function as vesicular adapter proteins in early stages of secretion. Neuron. 1997;18:453–461. doi: 10.1016/S0896-6273(00)81245-3. [DOI] [PubMed] [Google Scholar]
- Voets T. Dissection of three Ca2+-dependent steps leading to secretion in chromaffin cells from mouse adrenal slices. Neuron. 2000;28:537–545. doi: 10.1016/S0896-6273(00)00131-8. [DOI] [PubMed] [Google Scholar]
- Voets T, Neher E, Moser T. Mechanisms underlying phasic and sustained secretion in chromaffin cells from mouse adrenal slices. Neuron. 1999;23:607–615. doi: 10.1016/S0896-6273(00)80812-0. [DOI] [PubMed] [Google Scholar]
- Voets T, Moser T, Lund PE, Chow RH, Geppert M, Südhof TC, Neher E. Intracellular calcium dependence of large dense-core vesicle exocytosis in the absence of synaptotagmin I. Proc Natl Acad Sci U S A. 2001;98:11680–11685. doi: 10.1073/pnas.201398798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Rüden L, Neher E. A Ca-dependent early step in the release of catecholamines from adrenal chromaffin cells. Science. 1993;262:1061–1065. doi: 10.1126/science.8235626. [DOI] [PubMed] [Google Scholar]
- Walter AM, Groffen AJ, Sørensen JB, Verhage M. Multiple Ca2+ sensors in secretion: teammates, competitors or autocrats? Trends Neurosci. 2011;34:487–497. doi: 10.1016/j.tins.2011.07.003. [DOI] [PubMed] [Google Scholar]
- Yao J, Gaffaney JD, Kwon SE, Chapman ER. Doc2 is a Ca2+ sensor required for asynchronous neurotransmitter release. Cell. 2011;147:666–677. doi: 10.1016/j.cell.2011.09.046. [DOI] [PMC free article] [PubMed] [Google Scholar]