

Abstract
Protein tyrosine phosphatase 1B (PTP1B) counteracts leptin signaling and is a therapeutic target for obesity and diabetes. Here we found that LIM domain only 4 (LMO4) inhibits PTP1B activity by increasing the oxidized inactive form of PTP1B. Mice with neuronal ablation of LMO4 have elevated PTP1B activity and impaired hypothalamic leptin signaling, and a PTP1B inhibitor normalized PTP1B activity and restored leptin control of circulating insulin levels. LMO4 is palmitoylated at its C-terminal cysteine, and deletion of this residue prevented palmitoylation and retention of LMO4 at the endoplasmic reticulum and abolished its inhibitory effect on PTP1B. Importantly, LMO4 palmitoylation is sensitive to metabolic stress; mice challenged with a brief high-fat diet or acute intracerebroventricular infusion of saturated fatty acid had less palmitoylated LMO4, less oxidized PTP1B, and increased PTP1B activity in the hypothalamus. Thus, unleashed PTP1B activity attributable to loss of LMO4 palmitoylation may account for rapid loss of central leptin signaling after acute exposure to saturated fat.
Introduction
Leptin is a peptide hormone produced by adipocytes in proportion to the extent of adiposity (Zhang et al., 1994; Frederich et al., 1995). Leptin receptors are present on many cells, but the main homeostatic effect of leptin occurs via neurons of the mediobasal hypothalamus (Lee et al., 1996; Woods and Stock, 1996). Central leptin signaling regulates metabolism by suppressing feeding and increasing energy expenditure, promoting lipolysis and thermogenesis (Ahima and Flier, 2000). Central leptin also regulates glucose homeostasis by enhancing hypothalamic insulin signaling important to control hepatic glucose production (Morton et al., 2005; Koch et al., 2010) and increasing sympathetic outflow to peripheral tissues, including the adrenal gland and pancreas, to suppress insulin secretion (Frohman and Bernardis, 1971; Bloom and Edwards, 1975).
In diet-induced obesity, circulating leptin levels are elevated (hyperleptinemia) but the central response to leptin is defective, a phenomenon known as leptin resistance (Frederich et al., 1995). Leptin signals through receptors that couple to the tyrosine kinase Janus kinase 2 (JAK2) that phosphorylates and activates the transcription factor signal transducer and activator of transcription (STAT3). Suppressor of cytokine signaling 3 (SOCS3) and protein tyrosine phosphatase 1B (PTP1B) are two endogenous inhibitors of leptin signaling. SOCS3 binds and inhibits the JAK2 kinase (Bjørbaek et al., 1999), whereas PTP1B dephosphorylates and inactivates JAK2 (Zabolotny et al., 2002). Both SOCS3 and PTP1B levels are increased in mice with long-term high-fat diet (HFD)-induced obesity (Münzberg et al., 2004; White et al., 2009) and contribute to the loss of leptin signaling. PTP1B is also dynamically regulated by oxidation of its catalytic domain that suppresses its phosphatase activity (Mahadev et al., 2001; Meng et al., 2002). Whether this mechanism also contributes to loss of leptin signaling in diet-induced obesity resulting in insulin resistance has not been explored.
The LIM domain only 4 (LMO4) protein contains two LIM domains, each consisting of two zinc finger motifs. LMO4 is expressed in neurons but not in glia (Chen et al., 2007b). LMO4 works as a transcription cofactor in the nucleus (Visvader et al., 2001; Duquette et al., 2010), as well as a signaling modulator at the plasma membrane (Novotny-Diermayr et al., 2005; Chen et al., 2007b; Schock et al., 2008; Gomez-Smith et al., 2010; Zhou et al., 2012). Previously, we reported that mice with neuron-specific ablation of LMO4, CaMK2αCre/LMO4flox (hereafter called LMO4KO) mice, develop early-onset adiposity attributable to reduced sympathetic outflow to adipose tissue, progressing to hyperphagia and obesity (Zhou et al., 2012). These mice have impaired leptin-dependent STAT3 activation in the hypothalamus, although the mechanism for this deficit was not known. Here, we report that LMO4KO mice are also insulin resistant, have impaired glucose homeostasis, and develop hyperglycemia attributable to impaired central leptin signaling that fails to lower circulating insulin levels. The mechanism whereby LMO4 regulates leptin signaling appears to work through a direct interaction and inhibition of PTP1B. Furthermore, we found that this interaction and inhibition is regulated by palmitoylation of LMO4, a process that is inhibited by elevated saturated fatty acid. These findings suggest that LMO4 is a novel endogenous inhibitor of PTP1B and a metabolic sensor that controls central leptin signaling and glucose homeostasis.
Materials and Methods
Animal care.
All procedures were approved by the animal care and use committee of the University of Ottawa (Ottawa, Ontario, Canada). LMO4KO mice (CaMK2αCre/LMO4flox mice) were bred in a CD1 background as described previously (Schock et al., 2008; Qin et al., 2012; Zhou et al., 2012). HFD consisted of 60% kilocalories from fat (TD.06414 Adjusted Calories Diet; Harlan Laboratories) fed to the mice starting at 2 months of age for 7 d. In all studies, male mice were used. For paired feeding, mice were housed individually from the age of 9 to 17 weeks. Each day, LMO4 mutant and littermate control mice were provided the average amount of food consumed by age-matched control mice. Mice were fed twice daily: one-third of food was given at 9:00 A.M., and two-thirds were given at 5 P.M. to ensure that mice never underwent long periods of fasting.
Intracerebroventricular infusion.
Anesthetized mice were placed in a stereotactic apparatus. The sagittal sinus was displaced laterally, and a 21 gauge stainless-steel guide cannula (Plastics One) was lowered directly on the midline, 1.7 mm posterior to the bregma and 5.6 mm ventral to the dura, and fixed to the skull with anchor screws and dental acrylic. Leptin (10 μg) was infused over 7 d to the third ventricle by an osmotic minipump (Alza Corporation). Some mice received a single-dose (30 μg/kg weight in 1 μl) intracerebroventricular injection of trodusquemine (Ahima et al., 2002; Lantz et al., 2010) or vehicle (saline) immediately before leptin pump installation. Palmitic acid (1 μl of 500 nm; Sigma) prepared as described previously (Listenberger et al., 2001) or vehicle was stereotactically injected to the third ventricle.
Glucose tolerance test.
Mice fasted overnight (∼16 h) with access to water ad libitum were tested at 10:00 A.M. Basal blood glucose sampled from the saphenous vein was measured using a standard glucometer before and after mice receiving a glucose bolus (2 g/kg body weight of 20% d-glucose) by intraperitoneal injection.
Insulin tolerance test.
Mice were fasted for 4 h before the insulin tolerance test (ITT), performed between 2:00 P.M. and 5:00 P.M. Human recombinant insulin (catalog #91077C; Sigma), diluted in sterile saline was administered by intraperitoneal injection at 0.75 U/kg. Blood glucose levels were monitored as described above.
Blood pressure measurement.
Mice underwent 1 week of habituation to the tail-cuff apparatus (P-2000 Blood Pressure Analysis System; Visitech Systems), after which basal arterial pressure and pulse rate were sampled 10 times per day for a total of 3 d and averaged for the final results.
Norepinephrine turnover assay.
The norepinephrine turnover (NETO) assay was performed as described previously (Zhou et al., 2012).
Acute hypothalamic slice wedges and leptin response.
Three 400 μm coronal vibratome sections from bregma −1.1 to −2.3 mm were obtained per mouse, and square hypothalamic wedges were removed, incubated in aCSF at room temperature for 20 min and at 37°C for 1 h, followed by leptin (62.5 nm) treatment for various times, as indicated. For each time point, six hypothalamic wedges were pooled from two mice (containing all three hypothalamic wedges per mouse to avoid rostrocaudal variation).
Cell culture and transfections.
F11 neuronal cells were maintained and transfected using Lipofectamine2000 (Invitrogen) with LMO4 wild-type (WT) or ΔC165 mutant cDNA or control scrambled shRNA or LMO4-specific shRNA expression vectors as described previously (Chen et al., 2007a,b; Duquette et al., 2010; Gomez-Smith et al., 2010). The Flag–LMO4 WT and ΔC165 were generated by PCR amplification from the pcDNA3–mLMO4 with primers (forward, 5′-AAGCGGCCGCAATGGTGAATCCGGGCAGCA-3′; WT reverse, 5′-CTTATCTAGAAGCTTTCATCAG-3′; ΔC165 reverse, 5′-CTTATCTAGAAGCTTTCAGCAG-3′), and the amplified cDNA fragments were digested with restriction enzymes NotI and XbaI and subcloned into the pFLAG–CMV2 vector. The yellow fluorescent protein (YFP)-tagged PTP1B D/A mutant was a gift from Dr. Tarik Issad (Institute Cochin, Paris, France).
Palmitoylation assay.
This assay was performed according to a biotin exchange protocol (Drisdel et al., 2006), kindly provided by Dr. William Green's laboratory (University of Chicago, Chicago, IL).
Modified cysteinyl-labeling assay to detect reversible oxidation of PTP1B.
The assay was performed as described by Boivin and colleagues (Boivin et al., 2008; Boivin and Tonks, 2010).
Endoplasmic reticulum and nuclear and cytosolic fraction extraction.
Endoplasmic reticulum (ER) fractions were obtained from stimulated (as indicated) F11 cell lysates using Endoplasmic Reticulum Enrichment Kit (catalog #10088K; Imgenex) according to the protocol of the manufacturer. Nuclear and cytoplasmic cell fractions were obtained as described previously (Chen et al., 2007a) and used for immunoblot analysis, as indicated.
PTP1B phosphatase activity assay.
PTP1B phosphatase activity was measured with the PhosphoSeek PTP1B Assay Kit (BioVision) in extracts from hypothalamic wedges or F11 cells according to the instructions of the manufacturer with PTP1B enzyme and phosphatase inhibitor as positive and negative controls, respectively.
GST pull-down, immunoprecipitation, and immunoblot analyses.
F11 cells transfected with Flag–LMO4 together with GST–PTP1B WT or a substrate-trapping mutant in which the acidic residue aspartate 181 is replaced by an alanine (D181A, D/A) [gifts from Dr. Michel Tremblay, McGill University, Montreal, Quebec, Canada (Stuible et al., 2008)] were harvested in lysis buffer and pulled down with glutathione beads according to the protocol of the manufacturer (Invitrogen). To detect oxidized PTP1B (oxi-PTP1B), PTP1B was immunoprecipitated with rabbit anti-PTP1B antibody (Cell Signaling Technology) conjugated to protein G-Sepharose and immunoblotted against a mouse antibody specific to oxi-PTP active site (R&D Systems). Antibodies specific to phosphorylated (Y705) STAT3, phosphorylated (Y1007/1008) JAK2, STAT3, and JAK2 were obtained from Cell Signaling Technology. Mouse monoclonal anti-emerin antibody (AG45; Abcam) labeled the nuclear fraction, and calnexin antibody (AF18; Santa Cruz Biotechnology) labeled the ER fraction. Immunoblots were performed as described previously (Gomez-Smith et al., 2010), and an average of three blots were quantitated using NIH ImageJ software.
Statistical analyses.
All results are expressed as mean ± SEM and analyzed using SPSS Software (IBM). For between-group comparisons, a two-tailed unpaired Student's t test was used. For multiple comparisons, ANOVA with post hoc least significant difference test was used for between-group comparisons. p < 0.05 was considered significant.
Results
Impaired glucose homeostasis in LMO4KO mice
LMO4KO mice have a deficit in central leptin signaling associated with early-onset adiposity by 2 months of age. These mice become hyperleptinemic, hyperphagic, and obese by 4 months of age when fed regular chow (Zhou et al., 2012). Because adiposity and loss of central leptin signaling are associated with impaired glucose homeostasis (Morton and Schwartz, 2011), we asked whether LMO4 mutant mice are insulin resistant and diabetic. The response to a glucose tolerance test (GTT) was normal at 2 months but was impaired by 4 months. By 6 months, LMO4KO mice were overtly hyperglycemic (Fig. 1A). Although at 2 months the GTT appeared normal, these mice already showed reduced sensitivity to insulin, as determined by an ITT (Fig. 1B). Moreover, insulin levels were twofold higher in serum of LMO4KO mice compared with littermate WT controls at 2 months of age and were further elevated at 4 months (Fig. 1C).
Figure 1.

LMO4KO mice exhibit impaired glucose homeostasis and insulin resistance in association with reduced sympathetic outflow. A, GTT. B, ITT. C, Serum insulin levels. (A–C, n = 9–12 per group). D, NETO assay. E, Blood pressures (D, E, n = 8 per group, aged 2 months). *p < 0.05.
Reduced sympathetic outflow to the pancreas and adrenal gland in LMO4KO mice
Insulin secretion from the pancreas is inhibited by stimulating sympathetic innervation to the pancreas (Bloom and Edwards, 1975) and by elevated circulating catecholamines released from the adrenal gland (Frohman and Bernardis, 1971). Sympathetic outflow to the periphery is activated by hypothalamic neurons in response to leptin signaling (Enriori et al., 2011). Thus, elevated circulating insulin levels in LMO4KO mice may be a consequence of impaired central leptin signaling and reduced sympathetic tone to the pancreas and the adrenal gland. Indeed, sympathetic outflow to the pancreas and to the adrenal glands measured using the NETO assay was reduced at 2 months of age in LMO4KO mice (Fig. 1D). Consistent with reduced sympathetic drive, resting blood pressure was significantly lower in LMO4KO mice (Fig. 1E), although heart rate was not different (control, 645 ± 65 bpm; LMO4KO, 651 ± 33 bpm).
Caloric restriction does not rescue insulin resistance in LMO4KO mice
To determine to what extent impaired glucose homeostasis and insulin sensitivity are a consequence of increased adiposity and feeding or are directly related to impaired central leptin signaling, we undertook a paired-feeding regimen to restrict caloric intake in LMO4KO mice. LMO4KO mice were pair-fed with littermate controls starting at the age of 2 months, because at this age food intake and total body weight are not different between WT and mutant mice (Zhou et al., 2012). Paired feeding for 2 months not only prevented excess weight gain (Fig. 2A); it further normalized fat content (Fig. 2B). Although paired feeding improved glucose handling at 4 months compared with ad libitum-fed mutant mice (compare Fig. 2C with 1A), insulin sensitivity remained impaired (Fig. 2D) and serum insulin levels remained elevated in LMO4KO mice (Fig. 2E). These results indicate that LMO4 function in the brain is required to maintain peripheral insulin sensitivity.
Figure 2.

Paired feeding prevents adiposity and obesity but not insulin resistance in LMO4KO mice. A, Body weights during caloric restriction by paired feeding (n = 12 per group). B, Lean and adipose tissue masses after paired feeding. WAT, Retroperitoneal white adipose tissue; isWAT, intrascapular white adipose tissue; inWAT, inguinal white adipose tissue; G muscle, gastrocnemius muscle. GTT (C), ITT (D), and (E) serum insulin levels were measured after paired feeding (C–E, n = 9 per group). *p < 0.05.
Impaired central leptin signaling in LMO4KO mice is associated with elevated PTP1B activity in the hypothalamus
Previously, we reported that far fewer phospho-STAT3-positive ventromedial hypothalamic neurons could be detected in LMO4KO mice than in littermate controls after intracerebroventricular leptin infusion (Zhou et al., 2012). To elucidate the mechanism whereby loss of LMO4 impairs leptin signaling, we compared the leptin response of hypothalamic wedges by immunoblot analysis. Leptin caused a progressive activation and phosphorylation of JAK2 and STAT3 in WT mice, but, to our surprise, the opposite effect was observed in LMO4KO mice (Fig. 3A). The levels of SOCS3 and PTP1B were not elevated in LMO4KO hypothalamic wedges (Fig. 3A) and could not account for the paradoxical reduction in JAK2 and STAT3 phosphorylation in response to leptin signaling. This suggests that leptin signaling normally inactivates an endogenous PTP and that this ligand-dependent mechanism is lost with LMO4 ablation.
Figure 3.

Impaired central leptin signaling in LMO4KO mice is associated with elevated PTP1B activity. A, Immunoblot analysis of leptin signaling in hypothalamic wedges. To measure oxi-PTP1B with a pan-oxi-PTP antibody, PTP1B was immunoprecipitated (IP) with rabbit anti-PTP1B antibody and then the immunoblot (IB) probed with mouse anti-oxi-PTP antibody. B, PTP1B activity in hypothalamic wedges. n = 6 per time point. *p < 0.05 compared with baseline; **p < 0.05 compared with the previous value.
PTP1B activity is reversibly inhibited by oxidation (Mahadev et al., 2001; Meng et al., 2002). Insulin signaling is known to transiently oxidize and inactivate PTP1B to enhance the signaling cascade (Mahadev et al., 2001). We asked whether leptin would have a similar effect. Leptin treatment caused a transient increase in oxi-PTP1B (Fig. 3A) that was accompanied by a suppression of PTP1B phosphatase activity in WT hypothalamic wedges (Fig. 3B). In contrast, leptin did not alter oxi-PTP1B levels (Fig. 3A) or suppress PTP1B phosphatase activity (Fig. 3B) in LMO4KO hypothalamic wedges. PTP1B phosphatase activity remained fourfold higher in LMO4KO hypothalamic wedges compared with WT controls (Fig. 3 B). Thus, these results indicate that LMO4 is required for leptin-dependent inactivation of PTP1B.
LMO4 interacts with PTP1B and promotes oxi-PTP1B
Immunoprecipitation analysis showed that LMO4 interacts with PTP1B, and this interaction was stronger using a substrate-trapping mutant PTP1B (D/A) than for WT PTP1B (Fig. 4A). The substrate-trapping mutant has markedly diminished catalytic activity but retains substrate-binding ability, allowing the identification of PTP1B-targeted substrates (Flint et al., 1997). This result suggested that LMO4 might be tyrosine phosphorylated and a substrate of PTP1B phosphatase. Immunoprecipitation of Flag-tagged LMO4 followed by immunoblot analysis using an antibody to phosphotyrosine showed, to our knowledge for the first time, that LMO4 is tyrosine phosphorylated (Fig. 4B). Pretreating F11 cells with pervanadate, a pan-inhibitor of tyrosine phosphatases, further increased the level of pTyr–LMO4 (Fig. 4B, lane 3).
Figure 4.
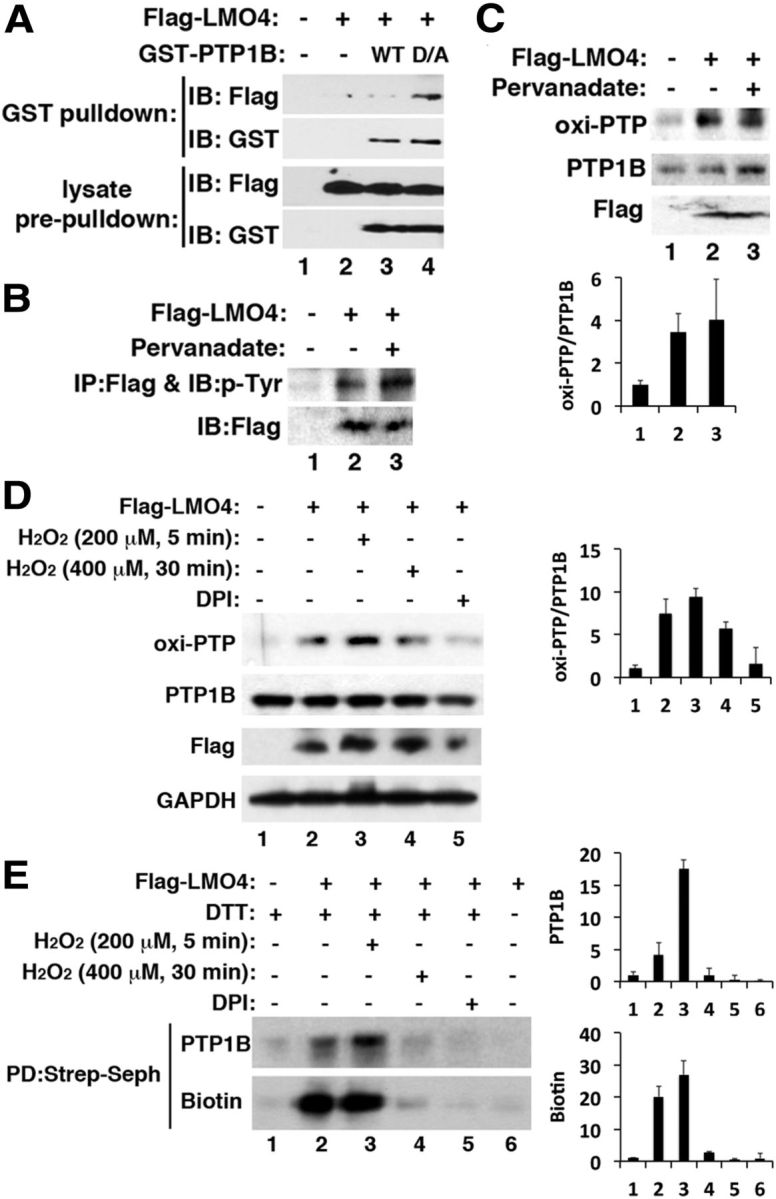
LMO4 interacts with PTP1B and promotes oxidation of PTP1B. A, GST pull-down showed that GST–PTP1B interacts with Flag–LMO4. B, Immunoprecipitation (IP) with anti-Flag antibody followed by immunoblot (IB) probed with anti-phosphorylated tyrosine antibody showed that LMO4 is tyrosine phosphorylated. C, Overexpression of Flag–LMO4 elevated the levels of oxi-PTP1B (compare lanes 2, 3 with lane 1). D, Brief, but not prolonged, exposure to H2O2 increases oxi-PTP1B levels. Treatment with the NADPH oxidase inhibitor DPI (10 μm for 30 min) prevented increased oxi-PTP1B. E, Cysteinyl-labeling assay (Boivin et al., 2008) using biotinylated iodoacetylpolyethyleneoxide to detect the reversibly oxidized cysteine residue after streptavidin Sepharose (Strep-Seph) pull-down (PD) and immunoblot with PTP1B antibody or biotin revealed similar changes in PTP1B oxidation as in D.
Because we found that loss of LMO4 prevents leptin-induced oxidation and transient inhibition of PTP1B (Fig. 3), we next overexpressed LMO4 and observed increased PTP1B oxidation (Fig. 4C). Acute exposure to H2O2 further increased PTP1B oxidation (Fig. 4D), but this increase was blunted with prolonged exposure to higher levels of H2O2 (Fig. 4D). Similar changes were observed using the cysteinyl-labeling assay (Fig. 4E), an alternative method to detect PTP1B oxidation (Boivin et al., 2008). Of note, the NADPH oxidase inhibitor diphenylene iodonium (DPI) prevented PTP1B oxidation (Fig. 4D,E). Together, these results suggest that LMO4 stabilizes oxi-PTP1B to inhibit its activity.
LMO4-dependent inhibition of PTP1B is regulated by LMO4 palmitoylation
The inhibitory effect of LMO4 on PTP1B activity appears to be unique to LMO4. Overexpression of the close homolog LMO2 had little effect on PTP1B activity (data not shown). The C-terminal cysteine (C165) is unique to LMO4 (Chen et al., 2002). Cysteine residues are targets of posttranslational modifications, such as palmitoylation, a process that affects protein trafficking and interaction with the plasma membrane and ER (Linder and Deschenes, 2007). Sequence analysis (http://csspalm.biocuckoo.org/) indicated a high probability of C165 being palmitoylated. Using a biotin exchange protocol (Drisdel et al., 2006), we confirmed that LMO4 is palmitoylated (Fig. 5A). Furthermore, deletion of C165 (ΔC165) blocked LMO4 palmitoylation (Fig. 5A, compare lanes 1, 2), indicating that this is the principal residue modified by palmitoylation.
Figure 5.
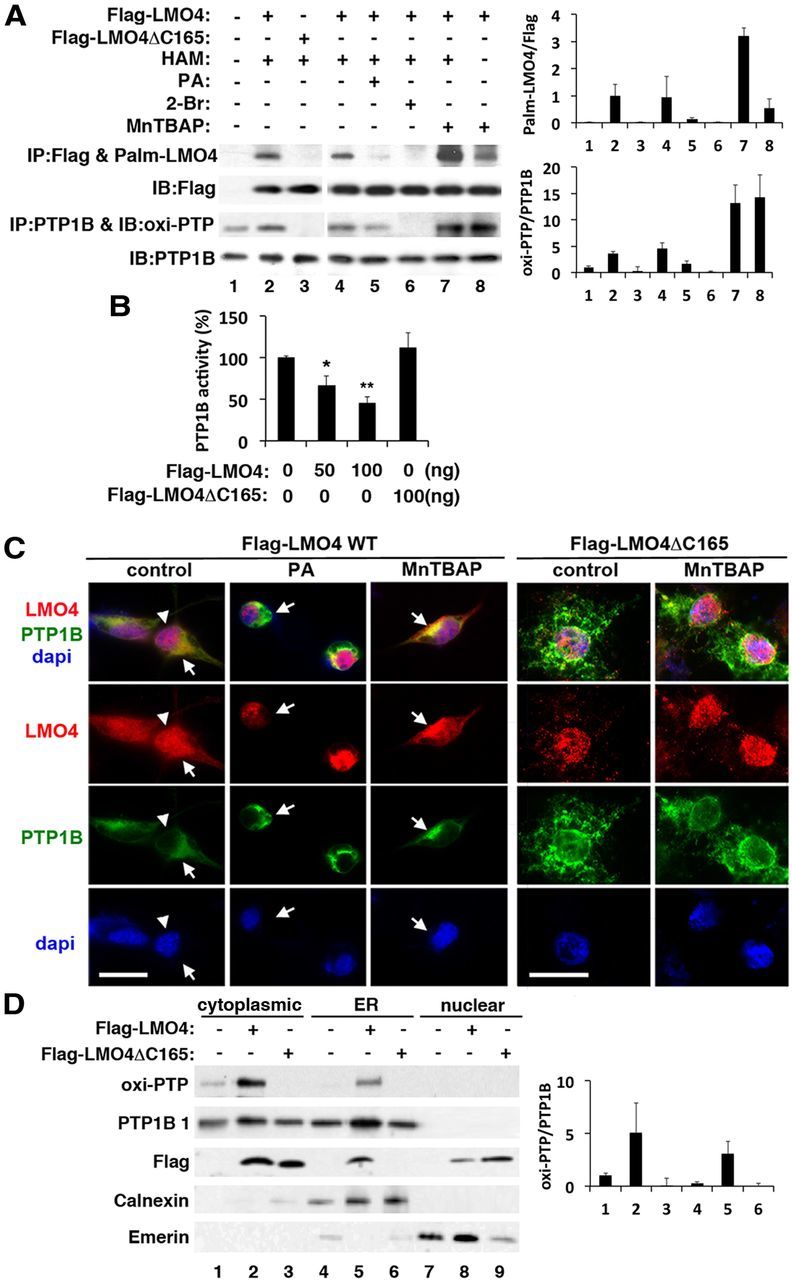
Palmitoylation of LMO4 regulates its subcellular localization and inhibition of PTP1B activity. A, Immunoblot (IB) analysis reveals that LMO4 is palmitoylated (Palm) and correlates with oxi-PTP1B levels. Lane 7 is a negative control that leaves out hydroxylamine (HAM) from the assay. Palmitic acid (PA; 400 μm), 2-bromo-hexadecanoic acid (2-Br; 50 μm), and MnTBAP (10 μm) were applied to cells for 6 h. B, PTP1B activity is suppressed by overexpression of WT but not mutant (ΔC165) LMO4. *p < 0.05 compared with baseline; **p < 0.05 compared with the previous value. n = 6 per group. C, Palmitoylation controls cytoplasmic retention of LMO4. YFP-tagged PTP1B was present in the cytoplasm at the ER. The palmitoylation-defective mutant LMO4ΔC165 was detected in the nucleus even in the presence of MnTBAP. Colocalization of LMO4 (red) with PTP1B (green) appears yellow. Nuclei are revealed by DAPI staining (blue). Scale bars, 15 μm. D, Immunoblot analysis confirmed the presence of Flag–LMO4 and PTP1B in the ER. Calnexin is an ER marker, and emerin is a nucleus-enriched marker. Flag–LMO4ΔC165 was not retained in the ER fraction but was present in the nuclear fraction. IP, Immunoprecipitation.
Protein palmitoylation is sensitive to oxidants resulting from metabolic stress; saturated fatty acid, such as palmitic acid, was reported to inhibit palmitoylation, whereas removal of oxidative stress promoted palmitoylation (Burgoyne et al., 2012). We also found that LMO4 palmitoylation was blocked by culturing neuronal cells in palmitic acid or the inhibitor of palmitoylation 2-bromo-palmitate (Fig. 5A, lanes 4, 5, respectively) and increased by treating cells with manganese (III) tetrakis (4-benzoic acid) porphyrin chloride (MnTBAP) (Fig. 5A, lane 6), a reactive oxygen species (ROS) scavenger that accelerates the conversion of superoxide to H2O2 (Kohli et al., 2007).
Of note, LMO4 palmitoylation was correlated with the level of oxi-PTP1B (Fig. 5A). Overexpression of the palmitoylation-deficient mutant LMO4ΔC165 failed to increase oxi-PTP1B levels, unlike WT LMO4 (Fig. 5A, compare lanes 1, 2). Consistent with this observation, overexpression of WT LMO4 dose dependently inhibited PTP1B activity, whereas overexpression of LMO4ΔC165 had no effect (Fig. 5B). Of note, LMO4 overexpression had no effect on the activity of T-cell protein tyrosine phosphatase (TC-PTP), the nearest homolog of PTP1B (data not shown).
Immunofluorescence analysis confirmed that LMO4 retention in the cytoplasm is sensitive to the redox state of the cell (Fig. 5C). Palmitic acid treatment reduced the cytoplasmic localization and caused nuclear accumulation of LMO4, whereas the ROS scavenger MnTBAP increased the cytoplasmic retention of LMO4. Importantly, the palmitoylation-deficient mutant LMO4ΔC165 was not retained in the cytoplasm under basal conditions or in the presence of MnTBAP. That the LMO4ΔC165 mutant protein did not colocalize with PTP1B, unlike WT LMO4, is consistent with its inability to inhibit PTP1B activity.
To confirm that LMO4 is present together with PTP1B and oxi-PTP1B in the ER, immunoblot analysis was performed using a purified ER fraction (Fig. 5D). Flag-tagged WT LMO4 was detected in the ER fraction together with oxi-PTP1B (Fig. 5D, lane 2), as well as the nuclear fraction (lane 8). In contrast, the LMO4ΔC165 mutant was not detected in the ER fraction (lane 3) but was present in the nuclear fraction (lane 9).
Together, these results indicate that LMO4 palmitoylation is sensitive to metabolic stress and is required for LMO4 to inhibit PTP1B activity by maintaining PTP1B in an oxidized inactivated state.
Metabolic stress of HFD reduces LMO4 palmitoylation and increases PTP1B activity
HFD impairs leptin and insulin signaling and leads to obesity and diabetes, and blocking hypothalamic PTP1B activity restores leptin and insulin signaling (Picardi et al., 2008). We explored whether loss of LMO4-dependent inhibition of PTP1B activity might underlie diet-induced leptin and insulin resistance. Indeed, we found that hypothalamic PTP1B activity was increased after 4 h of intracerebroventricular infusion with palmitic acid (Fig. 6A) or after 1 week of HFD (Fig. 6C). Elevated PTP1B activity was associated with lower levels of oxi-PTP1B and a reduction in LMO4 palmitoylation (Fig. 6B,D). Hypothalamic PTP1B levels were not different after these treatments. Acute palmitic acid infusion and short-term HFD had no effect on TC-PTP activity (data not shown).
Figure 6.

LMO4 palmitoylation (Palm) and inhibition of PTP1B activity are sensitive to saturated fatty acid. Hypothalamic PTP1B activity was increased 4 h after intracerebroventricular infusion of palmitic acid (A) (PA; 1 μl of 500 nm) or 7 d after an HFD (C). Immunoblot analyses showed that LMO4 palmitoylation and oxi-PTP1B levels were reduced by PA (B) or HFD (D). *p < 0.05; n = 5–6 mice per group, aged 6–8 weeks.
The PTP1B inhibitor trodusquemine restores leptin-dependent signaling and regulation of circulating insulin levels in LMO4KO mice
To test whether leptin signaling in LMO4-deficient cells could be restored by inhibiting PTP1B activity, F11 cells transfected with LMO4-specific or control scrambled shRNA were treated with the PTP1B inhibitor trodusquemine (Lantz et al., 2010; Krishnan et al., 2011) for 30 min before leptin treatment. Leptin caused an increase in oxi-PTP1B levels in control shRNA-transfected F11 cells (Fig. 7A, compare lanes 1, 2) but not in LMO4 knockdown cells (Fig. 7A, compare lanes 5, 6). Basal oxi-PTP1B levels were lower in LMO4 knockdown cells (Fig. 7A, compare lanes 1, 5). Trodusquemine rescued the effect of leptin to increase oxi-PTP1B in LMO4 knockdown cells (Fig. 7A, compare lanes 6, 8) and restored leptin-induced JAK2 phosphorylation (Fig. 7A, compare lanes 6, 8). Trodusquemine had no effect on TC-PTP activity (data not shown), as reported previously (Lantz et al., 2010).
Figure 7.

The PTP1B inhibitor trodusquemine (Trodu) restores leptin-dependent signaling and regulation of circulating insulin levels in LMO4KO mice. A, Immunoblot analysis of the response to leptin (30 min) with or without pretreatment with trodusquemine (10 μm, 30 min). Ctl, Control. B, Hypothalamic PTP1B activity in response to trodusquemine with and without leptin. C, Serum insulin in response to leptin with and without trodusquemine. D, The effect of intracerebroventricular administration of trodusquemine and/or leptin on body weight. B–D, n = 5–8 mice per group, aged 2 months. *p < 0.05 compared with WT saline; #p < 0.05 compared with LMO4KO saline; $p < 0.05, not significant (NS).
One week after trodusquemine treatment, PTP1B activity in LMO4KO hypothalamus was lowered to levels observed in saline-treated WT mice (Fig. 7B). We next tested whether this treatment could also restore the central response to leptin. Leptin infusion significantly lowered serum insulin levels in WT mice, but this leptin response was absent in LMO4KO mice. However, intracerebroventricular infusion of trodusquemine was able to restore the leptin-dependent suppression of serum insulin levels in LMO4KO mice (Fig. 7C). Of note, a single dose of trodusquemine (30 μg/kg) administered intracerebroventricularly did not cause a significant weight loss after 7 d in either the WT or LMO4KO mice (Fig. 7D), and blood glucose levels were also not different (data not shown). These results indicate that PTP1B inhibition by trodusquemine is sufficient to restore leptin signaling and its control of circulating insulin levels.
Discussion
We have shown, for the first time to our knowledge, that LMO4 is an endogenous inhibitor of the protein tyrosine phosphatase PTP1B in the hypothalamus. Importantly, this inhibition is sensitive to metabolic stress and is blunted by acute exposure to saturated fat or a short-term HFD. Because PTP1B inhibits leptin signaling, loss of LMO4-dependent suppression of PTP1B activity may contribute to diet-induced loss of central leptin signaling, resulting in impaired insulin sensitivity and glucose handling.
Neuron-specific ablation of the insulin receptor has a limited effect on circulating insulin levels and peripheral insulin sensitivity (Brüning et al., 2000), whereas impaired glucose homeostasis and markedly elevated peripheral insulin levels were observed in mice with neuron-specific ablation of leptin receptor (McMinn et al., 2005). Because circulating insulin levels were significantly elevated in LMO4KO mice at 2 months of age, before overt weight gain and elevated plasma levels of leptin (Zhou et al., 2012), we surmised that a central defect in leptin signaling might account for elevated circulating insulin levels. Hypothalamic leptin signaling activates sympathetic outflow (Enriori et al., 2011), and increased sympathetic outflow suppresses insulin secretion from the pancreas (Frohman and Bernardis, 1971; Bloom and Edwards, 1975). Sympathetic outflow to the pancreas and adrenal gland, as revealed by the NETO assay, is impaired in LMO4KO mice, suggesting an intrinsic deficit in central leptin-dependent control of insulin secretion. Indeed, leptin infusion to the third ventricle over 7 d caused a marked reduction in circulating insulin levels in WT mice, but this response was absent in LMO4KO mice (Fig. 7). Importantly, the central leptin-induced suppression of circulating insulin levels was fully restored by a single intracerebroventricular dose of trodusquemine, a small-molecule noncompetitive inhibitor of PTP1B (Lantz et al., 2010; Krishnan et al., 2011).
Trodusquemine is a spermine metabolite of cholesterol isolated from the dogfish shark liver that was identified in the search for naturally occurring antimicrobial compounds. Unexpectedly, trodusquemine suppressed feeding and caused fat-specific weight loss in mice with diet-induced obesity (Ahima et al., 2002; Lantz et al., 2010), leading to its identification as a noncompetitive inhibitor of PTP1B (but not TC-PTP). Trodusquemine has undergone phase I clinical trials for its anti-obesity effect (Nguyen et al., 2013). It is important to note that the low dose of trodusquemine administered by intracerebroventricular injection in our study is the dose used in rats that was reported to have a limited transient effect on feeding and no net effect on body weight 7 d after intracerebroventricular administration (Ahima et al., 2002). The effect of trodusquemine on restoring leptin-dependent control of circulating insulin levels in LMO4KO mice may result from a central effect on sympathetic outflow to suppress insulin secretion and/or from improved peripheral insulin sensitivity associated with leptin-mediated weight loss. It will be important to elucidate how trodusquemine inhibits PTP1B activity and determine whether other related compounds have similar or better effects (Salmi et al., 2008). Inhibition of PTP1B activity holds promise for the treatment of obesity-associated diabetes.
Resistance to leptin action in the CNS is a hallmark of HFD-induced obesity and insulin resistance (Schwartz and Porte, 2005). HFD increases hypothalamic PTP1B and induces leptin resistance through leptin-dependent and -independent mechanisms (White et al., 2009). A leptin-independent mechanism involves HFD-induced inflammation and activation of nuclear factor-κB signaling (Cai et al., 2005; Zabolotny et al., 2008), which then upregulates PTP1B (Yu et al., 2013). Elevated hypothalamic expression of SOCS3 and PTP1B, molecules that terminate leptin signaling, account for impaired central leptin signaling after a chronic HFD (Münzberg et al., 2004; White et al., 2009). Mice with neuron-specific ablation of PTP1B have improved peripheral insulin sensitivity, are resistant to diet-induced obesity, and are hypersensitive to leptin (Bence et al., 2006). However, even acute (30 min) exposure of the hypothalamus to a saturated fatty acid, such as palmitic acid, whose circulating and hypothalamic levels rise rapidly after a single high-fat meal (Posey et al., 2009), attenuates leptin-induced STAT3 phosphorylation and suppression of feeding (Kleinridders et al., 2009). This rapid effect of palmitic acid is unlikely to occur through altered expression of SOCS3 or PTP1B.
PTP1B activity is dynamically regulated (inhibited) through oxidation of a cysteine residue in the catalytic domain, a reversible process controlled by several signals (Lee et al., 1998; Gross et al., 1999; Mahadev et al., 2001). For example, insulin signaling causes a rapid increase in hydrogen peroxide that transiently oxidizes and inhibits PTP1B to sustain insulin signaling (Mahadev et al., 2001). Similarly, our study showed that leptin signaling also suppresses PTP1B activity by increasing the inactive form of PTP1B, oxi-PTP1B (Fig. 3). Importantly, this acute effect of leptin was absent in LMO4KO hypothalamic wedges. Neuron-specific ablation of LMO4 was associated with a fourfold elevated PTP1B activity with no change in PTP1B protein levels (Fig. 3), whereas LMO4 overexpression dose dependently inhibited PTP1B activity (Fig. 5). Moreover, we found that LMO4 is tyrosine phosphorylated and is a likely substrate of PTP1B because LMO4 was pulled down together with a GST-tagged substrate-trapping mutant of PTP1B. It remains to be determined which tyrosine residue in LMO4 is phosphorylated and whether tyrosine phosphorylation of LMO4 is required for its interaction and inhibition of PTP1B.
How LMO4 inhibits PTP1B activity remains to be elucidated. One possibility is that LMO4 stabilizes oxi-PTP1B. Indeed, lower oxi-PTP1B levels were observed in neuronal cells after knockdown of LMO4, whereas increased oxi-PTP1B levels were observed with overexpression of LMO4 (Figs. 4–6). LMO4 carries four cysteine-rich zinc fingers in its two LIM domains (Chen et al., 2002), and zinc finger proteins are known to be highly susceptible to oxidation at cysteine residues (Webster et al., 2001). Whether transient ROS production during leptin or insulin signaling could oxidize cysteines of the zinc fingers and cause stabilization of the PTP1B/LMO4 complex awaits additional studies.
LMO4 was initially identified as a nuclear cofactor interacting with many transcriptional factors to regulate gene expression (Sugihara et al., 1998; Visvader et al., 2001; Aizawa et al., 2004; Song et al., 2009; Duquette et al., 2010). Later studies reported that LMO4 is also present in the cytoplasm and interacts with membrane-bound receptors and kinases to regulate cellular functions in response to extracellular signals (Novotny-Diermayr et al., 2005; Bong et al., 2007; Chen et al., 2007b; Schaffar et al., 2008; Schock et al., 2008; Gomez-Smith et al., 2010; Zhou et al., 2012). Our previous studies showed that subcellular localization and cytoplasmic/nuclear shuttling of LMO4 is dynamically regulated by stresses and extracellular signals (Chen et al., 2007b). Here, we identified that palmitoylation targets LMO4 to cell membranes (e.g., membranes of the ER and plasma membrane), providing a mechanism whereby LMO4 participates in signaling at the plasma membrane (Novotny-Diermayr et al., 2005; Chen et al., 2007b; Gomez-Smith et al., 2010). Moreover, palmitoylation of LMO4 is also dynamically regulated, and this process is required to actively keep LMO4 at the ER. Deletion of the C-terminal cysteine of LMO4 (LMO4Δ165) prevents its palmitoylation and retention in the ER (Fig. 5). PTP1B is predominantly localized at the ER (Frangioni et al., 1992), and preventing LMO4 retention at the ER would also prevent LMO4 from interacting with and stabilizing oxi-PTP1B. We found that reduced LMO4 palmitoylation was accompanied with lowered oxi-PTP1B levels and elevated PTP1B activity after acute exposure of the hypothalamus to the saturated fatty acid palmitic acid or to a short-term HFD (Fig. 6). These findings could account for the previously reported rapid reduction of leptin signaling in the hypothalamus observed 30 min after palmitic acid treatment (Kleinridders et al., 2009).
To our knowledge, ours is the first study to link reduced oxi-PTP1B levels in the hypothalamus with acute exposure to palmitic acid or to a short-term exposure to HFD. These treatments also reduced palmitoylation of LMO4. Protein palmitoylation is inhibited by excessive ROS production caused by palmitic acid treatment (Burgoyne et al., 2012). Conversely, the ROS scavenger MnTBAP promoted LMO4 palmitoylation and cytoplasmic retention and increased oxi-PTP1B levels (Fig. 5). Unlike DPI that blocks superoxide production by NADPH oxidase and prevents PTP1B oxidation, MnTBAP promotes H2O2 production from superoxide, and this likely contributes to increased oxi-PTP1B. We reported that LMO4 has a half-life of 45 min (Chen et al., 2007b). Thus, given its short half-life, LMO4 could rapidly and dynamically regulate PTP1B activity in response to numerous stresses.
Previously, we observed that purinergic receptor activation by elevated extracellular ATP caused increased nuclear translocation of LMO4 (Chen et al., 2007a). Because purinergic signaling increases ROS production (Kawamura et al., 2012; Wang and Sluyter, 2013), this is likely to reduce LMO4 palmitoylation and ER retention. Of note, glial cells called tanycytes that line the third ventricle and secrete ATP in response to elevated extracellular glucose (Frayling et al., 2011). Thus, under conditions of chronic hyperglycemia, activation of purinergic receptors on nearby hypothalamic neurons may contribute to leptin resistance by increasing PTP1B activity through loss of LMO4 ER retention and LMO4-dependent inhibition. Future studies would be required to examine whether this does take place in vivo.
Footnotes
This work was supported by Canadian Diabetes Association (CDA) Grant OG-3-11-3520-HC, the Ontario Research Fund, the Canada Foundation for Innovation, Heart and Stroke Foundation of Canada Grant-in-Aid NA6301 and Grant G-13-0002596, Canadian Institutes of Health Research (CIHR) Grant MOP-179197, and Heart and Stroke Foundation of Ontario (HSFO) Centre for Stroke Recovery Catalyst grants (H.H.C.). H.C.C. is supported by the Henry J. M. Barnett Research Scholarship and the New Investigator Award from the Heart and Stroke Foundation of Canada and by an Early Researcher Award from the Ontario Ministry of Research and Innovation. A.F.R.S. is supported by a Discovery Grant from the Natural Sciences and Engineering Research Council of Canada (NSERC), CIHR Grant MOP-259068, and CDA. Z.Q. is supported by an NSERC graduate scholarship. K.K. is supported by an Alexander Graham Bell Canada Graduate NSERC Scholarship.
The authors declare no competing financial interests.
References
- Ahima RS, Flier JS. Leptin. Annu Rev Physiol. 2000;62:413–437. doi: 10.1146/annurev.physiol.62.1.413. [DOI] [PubMed] [Google Scholar]
- Ahima RS, Patel HR, Takahashi N, Qi Y, Hileman SM, Zasloff MA. Appetite suppression and weight reduction by a centrally active aminosterol. Diabetes. 2002;51:2099–2104. doi: 10.2337/diabetes.51.7.2099. [DOI] [PubMed] [Google Scholar]
- Aizawa H, Hu SC, Bobb K, Balakrishnan K, Ince G, Gurevich I, Cowan M, Ghosh A. Dendrite development regulated by CREST, a calcium-regulated transcriptional activator. Science. 2004;303:197–202. doi: 10.1126/science.1089845. [DOI] [PubMed] [Google Scholar]
- Bence KK, Delibegovic M, Xue B, Gorgun CZ, Hotamisligil GS, Neel BG, Kahn BB. Neuronal PTP1B regulates body weight, adiposity and leptin action. Nat Med. 2006;12:917–924. doi: 10.1038/nm1435. [DOI] [PubMed] [Google Scholar]
- Bjørbaek C, El-Haschimi K, Frantz JD, Flier JS. The role of SOCS-3 in leptin signaling and leptin resistance. J Biol Chem. 1999;274:30059–30065. doi: 10.1074/jbc.274.42.30059. [DOI] [PubMed] [Google Scholar]
- Bloom SR, Edwards AV. The release of pancreatic glucagon and inhibition of insulin in response to stimulation of the sympathetic innervation. J Physiol. 1975;253:157–173. doi: 10.1113/jphysiol.1975.sp011185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boivin B, Tonks NK. Analysis of the redox regulation of protein tyrosine phosphatase superfamily members utilizing a cysteinyl-labeling assay. Methods Enzymol. 2010;474:35–50. doi: 10.1016/S0076-6879(10)74003-9. [DOI] [PubMed] [Google Scholar]
- Boivin B, Zhang S, Arbiser JL, Zhang ZY, Tonks NK. A modified cysteinyl-labeling assay reveals reversible oxidation of protein tyrosine phosphatases in angiomyolipoma cells. Proc Natl Acad Sci U S A. 2008;105:9959–9964. doi: 10.1073/pnas.0804336105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bong YS, Lee HS, Carim-Todd L, Mood K, Nishanian TG, Tessarollo L, Daar IO. ephrinB1 signals from the cell surface to the nucleus by recruitment of STAT3. Proc Natl Acad Sci U S A. 2007;104:17305–17310. doi: 10.1073/pnas.0702337104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brüning JC, Gautam D, Burks DJ, Gillette J, Schubert M, Orban PC, Klein R, Krone W, Müller-Wieland D, Kahn CR. Role of brain insulin receptor in control of body weight and reproduction. Science. 2000;289:2122–2125. doi: 10.1126/science.289.5487.2122. [DOI] [PubMed] [Google Scholar]
- Burgoyne JR, Haeussler DJ, Kumar V, Ji Y, Pimental DR, Zee RS, Costello CE, Lin C, McComb ME, Cohen RA, Bachschmid MM. Oxidation of HRas cysteine thiols by metabolic stress prevents palmitoylation in vivo and contributes to endothelial cell apoptosis. FASEB J. 2012;26:832–841. doi: 10.1096/fj.11-189415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai D, Yuan M, Frantz DF, Melendez PA, Hansen L, Lee J, Shoelson SE. Local and systemic insulin resistance resulting from hepatic activation of IKK-beta and NF-kappaB. Nat Med. 2005;11:183–190. doi: 10.1038/nm1166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen HH, Yip JW, Stewart AF, Frank E. Differential expression of a transcription regulatory factor, the LIM domain only 4 protein Lmo4, in muscle sensory neurons. Development. 2002;129:4879–4889. doi: 10.1242/dev.129.21.4879. [DOI] [PubMed] [Google Scholar]
- Chen HH, Xu J, Safarpour F, Stewart AF. LMO4 mRNA stability is regulated by extracellular ATP in F11 cells. Biochem Biophys Res Commun. 2007a;357:56–61. doi: 10.1016/j.bbrc.2007.03.113. [DOI] [PubMed] [Google Scholar]
- Chen HH, Schock SC, Xu J, Safarpour F, Thompson CS, Stewart AF. Extracellular ATP-dependent upregulation of the transcription cofactor LMO4 promotes neuron survival from hypoxia. Exp Cell Res. 2007b;313:3106–3116. doi: 10.1016/j.yexcr.2007.04.026. [DOI] [PubMed] [Google Scholar]
- Drisdel RC, Alexander JK, Sayeed A, Green WN. Assays of protein palmitoylation. Methods. 2006;40:127–134. doi: 10.1016/j.ymeth.2006.04.015. [DOI] [PubMed] [Google Scholar]
- Duquette PM, Zhou X, Yap NL, MacLaren EJ, Lu JJ, Wallace VA, Chen HH. Loss of LMO4 in the retina leads to reduction of GABAergic amacrine cells and functional deficits. PLoS One. 2010;5:e13232. doi: 10.1371/journal.pone.0013232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enriori PJ, Sinnayah P, Simonds SE, Garcia Rudaz C, Cowley MA. Leptin action in the dorsomedial hypothalamus increases sympathetic tone to brown adipose tissue in spite of systemic leptin resistance. J Neurosci. 2011;31:12189–12197. doi: 10.1523/JNEUROSCI.2336-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flint AJ, Tiganis T, Barford D, Tonks NK. Development of “substrate-trapping” mutants to identify physiological substrates of protein tyrosine phosphatases. Proc Natl Acad Sci U S A. 1997;94:1680–1685. doi: 10.1073/pnas.94.5.1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frangioni JV, Beahm PH, Shifrin V, Jost CA, Neel BG. The nontransmembrane tyrosine phosphatase PTP-1B localizes to the endoplasmic reticulum via its 35 amino acid C-terminal sequence. Cell. 1992;68:545–560. doi: 10.1016/0092-8674(92)90190-N. [DOI] [PubMed] [Google Scholar]
- Frayling C, Britton R, Dale N. ATP-mediated glucosensing by hypothalamic tanycytes. J Physiol. 2011;589:2275–2286. doi: 10.1113/jphysiol.2010.202051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frederich RC, Hamann A, Anderson S, Löllmann B, Lowell BB, Flier JS. Leptin levels reflect body lipid content in mice: evidence for diet-induced resistance to leptin action. Nat Med. 1995;1:1311–1314. doi: 10.1038/nm1295-1311. [DOI] [PubMed] [Google Scholar]
- Frohman LA, Bernardis LL. Effect of hypothalamic stimulation on plasma glucose, insulin, and glucagon levels. Am J Physiol. 1971;221:1596–1603. doi: 10.1152/ajplegacy.1971.221.6.1596. [DOI] [PubMed] [Google Scholar]
- Gomez-Smith M, Qin Z, Zhou X, Schock SC, Chen HH. LIM domain only 4 protein promotes granulocyte colony-stimulating factor-induced signaling in neurons. Cell Mol Life Sci. 2010;67:949–957. doi: 10.1007/s00018-009-0223-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gross S, Knebel A, Tenev T, Neininger A, Gaestel M, Herrlich P, Böhmer FD. Inactivation of protein-tyrosine phosphatases as mechanism of UV-induced signal transduction. J Biol Chem. 1999;274:26378–26386. doi: 10.1074/jbc.274.37.26378. [DOI] [PubMed] [Google Scholar]
- Kawamura H, Kawamura T, Kanda Y, Kobayashi T, Abo T. Extracellular ATP-stimulated macrophages produce macrophage inflammatory protein-2 which is important for neutrophil migration. Immunology. 2012;136:448–458. doi: 10.1111/j.1365-2567.2012.03601.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleinridders A, Schenten D, Könner AC, Belgardt BF, Mauer J, Okamura T, Wunderlich FT, Medzhitov R, Brüning JC. MyD88 signaling in the CNS is required for development of fatty acid-induced leptin resistance and diet-induced obesity. Cell Metab. 2009;10:249–259. doi: 10.1016/j.cmet.2009.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koch C, Augustine RA, Steger J, Ganjam GK, Benzler J, Pracht C, Lowe C, Schwartz MW, Shepherd PR, Anderson GM, Grattan DR, Tups A. Leptin rapidly improves glucose homeostasis in obese mice by increasing hypothalamic insulin sensitivity. J Neurosci. 2010;30:16180–16187. doi: 10.1523/JNEUROSCI.3202-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohli R, Pan X, Malladi P, Wainwright MS, Whitington PF. Mitochondrial reactive oxygen species signal hepatocyte steatosis by regulating the phosphatidylinositol 3-kinase cell survival pathway. J Biol Chem. 2007;282:21327–21336. doi: 10.1074/jbc.M701759200. [DOI] [PubMed] [Google Scholar]
- Krishnan N, Fu C, Pappin DJ, Tonks NK. H2S-Induced sulfhydration of the phosphatase PTP1B and its role in the endoplasmic reticulum stress response. Sci Signal. 2011;4:ra86. doi: 10.1126/scisignal.2002329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lantz KA, Hart SG, Planey SL, Roitman MF, Ruiz-White IA, Wolfe HR, McLane MP. Inhibition of PTP1B by trodusquemine (MSI-1436) causes fat-specific weight loss in diet-induced obese mice. Obesity (Silver Spring) 2010;18:1516–1523. doi: 10.1038/oby.2009.444. [DOI] [PubMed] [Google Scholar]
- Lee GH, Proenca R, Montez JM, Carroll KM, Darvishzadeh JG, Lee JI, Friedman JM. Abnormal splicing of the leptin receptor in diabetic mice. Nature. 1996;379:632–635. doi: 10.1038/379632a0. [DOI] [PubMed] [Google Scholar]
- Lee SR, Kwon KS, Kim SR, Rhee SG. Reversible inactivation of protein-tyrosine phosphatase 1B in A431 cells stimulated with epidermal growth factor. J Biol Chem. 1998;273:15366–15372. doi: 10.1074/jbc.273.25.15366. [DOI] [PubMed] [Google Scholar]
- Linder ME, Deschenes RJ. Palmitoylation: policing protein stability and traffic. Nat Rev Mol Cell Biol. 2007;8:74–84. doi: 10.1038/nrm2084. [DOI] [PubMed] [Google Scholar]
- Listenberger LL, Ory DS, Schaffer JE. Palmitate-induced apoptosis can occur through a ceramide-independent pathway. J Biol Chem. 2001;276:14890–14895. doi: 10.1074/jbc.M010286200. [DOI] [PubMed] [Google Scholar]
- Mahadev K, Zilbering A, Zhu L, Goldstein BJ. Insulin-stimulated hydrogen peroxide reversibly inhibits protein-tyrosine phosphatase 1b in vivo and enhances the early insulin action cascade. J Biol Chem. 2001;276:21938–21942. doi: 10.1074/jbc.C100109200. [DOI] [PubMed] [Google Scholar]
- McMinn JE, Liu SM, Liu H, Dragatsis I, Dietrich P, Ludwig T, Boozer CN, Chua SC., Jr Neuronal deletion of Lepr elicits diabesity in mice without affecting cold tolerance or fertility. Am J Physiol Endocrinol Metab. 2005;289:E403–E411. doi: 10.1152/ajpendo.00535.2004. [DOI] [PubMed] [Google Scholar]
- Meng TC, Fukada T, Tonks NK. Reversible oxidation and inactivation of protein tyrosine phosphatases in vivo. Mol Cell. 2002;9:387–399. doi: 10.1016/S1097-2765(02)00445-8. [DOI] [PubMed] [Google Scholar]
- Morton GJ, Schwartz MW. Leptin and the central nervous system control of glucose metabolism. Physiol Rev. 2011;91:389–411. doi: 10.1152/physrev.00007.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morton GJ, Gelling RW, Niswender KD, Morrison CD, Rhodes CJ, Schwartz MW. Leptin regulates insulin sensitivity via phosphatidylinositol-3-OH kinase signaling in mediobasal hypothalamic neurons. Cell Metab. 2005;2:411–420. doi: 10.1016/j.cmet.2005.10.009. [DOI] [PubMed] [Google Scholar]
- Münzberg H, Flier JS, Bjørbaek C. Region-specific leptin resistance within the hypothalamus of diet-induced obese mice. Endocrinology. 2004;145:4880–4889. doi: 10.1210/en.2004-0726. [DOI] [PubMed] [Google Scholar]
- Nguyen LK, Matallanas D, Croucher DR, von Kriegsheim A, Kholodenko BN. Signalling by protein phosphatases and drug development: a systems-centred view. FEBS J. 2013;280:751–765. doi: 10.1111/j.1742-4658.2012.08522.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novotny-Diermayr V, Lin B, Gu L, Cao X. Modulation of the interleukin-6 receptor subunit glycoprotein 130 complex and its signaling by LMO4 interaction. J Biol Chem. 2005;280:12747–12757. doi: 10.1074/jbc.M500175200. [DOI] [PubMed] [Google Scholar]
- Picardi PK, Calegari VC, Prada PO, Moraes JC, Araújo E, Marcondes MC, Ueno M, Carvalheira JB, Velloso LA, Saad MJ. Reduction of hypothalamic protein tyrosine phosphatase improves insulin and leptin resistance in diet-induced obese rats. Endocrinology. 2008;149:3870–3880. doi: 10.1210/en.2007-1506. [Erratum (2013) 154:1667 (Prada, Patrícia de Oliveira corrected to Prada, Patricia Oliveira)] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Posey KA, Clegg DJ, Printz RL, Byun J, Morton GJ, Vivekanandan-Giri A, Pennathur S, Baskin DG, Heinecke JW, Woods SC, Schwartz MW, Niswender KD. Hypothalamic proinflammatory lipid accumulation, inflammation, and insulin resistance in rats fed a high-fat diet. Am J Physiol Endocrinol Metab. 2009;296:E1003–E1012. doi: 10.1152/ajpendo.90377.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin Z, Zhou X, Gomez-Smith M, Pandey NR, Lee KF, Lagace DC, Béïque JC, Chen HH. LIM domain only 4 (LMO4) regulates calcium-induced calcium release and synaptic plasticity in the hippocampus. J Neurosci. 2012;32:4271–4283. doi: 10.1523/JNEUROSCI.6271-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salmi C, Loncle C, Vidal N, Laget M, Letourneux Y, Brunel JM. Antimicrobial activities of 3-amino- and polyaminosterol analogues of squalamine and trodusquemine. J Enzyme Inhib Med Chem. 2008;23:860–865. doi: 10.1080/14756360701809910. [DOI] [PubMed] [Google Scholar]
- Schaffar G, Taniguchi J, Brodbeck T, Meyer AH, Schmidt M, Yamashita T, Mueller BK. LIM-only protein 4 interacts directly with the repulsive guidance molecule A receptor Neogenin. J Neurochem. 2008;107:418–431. doi: 10.1111/j.1471-4159.2008.05621.x. [DOI] [PubMed] [Google Scholar]
- Schock SC, Xu J, Duquette PM, Qin Z, Lewandowski AJ, Rai PS, Thompson CS, Seifert EL, Harper ME, Chen HH. Rescue of neurons from ischemic injury by PPARgamma requires a novel essential cofactor LMO4. J Neurosci. 2008;28:12433–12444. doi: 10.1523/JNEUROSCI.2897-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz MW, Porte D., Jr Diabetes, obesity, and the brain. Science. 2005;307:375–379. doi: 10.1126/science.1104344. [DOI] [PubMed] [Google Scholar]
- Song MR, Sun Y, Bryson A, Gill GN, Evans SM, Pfaff SL. Islet-to-LMO stoichiometries control the function of transcription complexes that specify motor neuron and V2a interneuron identity. Development. 2009;136:2923–2932. doi: 10.1242/dev.037986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuible M, Dubé N, Tremblay ML. PTP1B regulates cortactin tyrosine phosphorylation by targeting Tyr446. J Biol Chem. 2008;283:15740–15746. doi: 10.1074/jbc.M710534200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugihara TM, Bach I, Kioussi C, Rosenfeld MG, Andersen B. Mouse deformed epidermal autoregulatory factor 1 recruits a LIM domain factor, LMO-4, and CLIM coregulators. Proc Natl Acad Sci U S A. 1998;95:15418–15423. doi: 10.1073/pnas.95.26.15418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Visvader JE, Venter D, Hahm K, Santamaria M, Sum EY, O'Reilly L, White D, Williams R, Armes J, Lindeman GJ. The LIM domain gene LMO4 inhibits differentiation of mammary epithelial cells in vitro and is overexpressed in breast cancer. Proc Natl Acad Sci U S A. 2001;98:14452–14457. doi: 10.1073/pnas.251547698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang B, Sluyter R. P2X7 receptor activation induces reactive oxygen species formation in erythroid cells. Purinergic Signal. 2013;9:101–112. doi: 10.1007/s11302-012-9335-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webster KA, Prentice H, Bishopric NH. Oxidation of zinc finger transcription factors: physiological consequences. Antioxid Redox Signal. 2001;3:535–548. doi: 10.1089/15230860152542916. [DOI] [PubMed] [Google Scholar]
- White CL, Whittington A, Barnes MJ, Wang Z, Bray GA, Morrison CD. HF diets increase hypothalamic PTP1B and induce leptin resistance through both leptin-dependent and -independent mechanisms. Am J Physiol Endocrinol Metab. 2009;296:E291–E299. doi: 10.1152/ajpendo.90513.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woods AJ, Stock MJ. Leptin activation in hypothalamus. Nature. 1996;381:745. doi: 10.1038/381745a0. [DOI] [PubMed] [Google Scholar]
- Yu IC, Lin HY, Liu NC, Sparks JD, Yeh S, Fang LY, Chen L, Chang C. Neuronal androgen receptor regulates insulin sensitivity via suppression of hypothalamic NF-kappaB-mediated PTP1B expression. Diabetes. 2013;62:411–423. doi: 10.2337/db12-0135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zabolotny JM, Bence-Hanulec KK, Stricker-Krongrad A, Haj F, Wang Y, Minokoshi Y, Kim YB, Elmquist JK, Tartaglia LA, Kahn BB, Neel BG. PTP1B regulates leptin signal transduction in vivo. Dev Cell. 2002;2:489–495. doi: 10.1016/S1534-5807(02)00148-X. [DOI] [PubMed] [Google Scholar]
- Zabolotny JM, Kim YB, Welsh LA, Kershaw EE, Neel BG, Kahn BB. Protein-tyrosine phosphatase 1B expression is induced by inflammation in vivo. J Biol Chem. 2008;283:14230–14241. doi: 10.1074/jbc.M800061200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Proenca R, Maffei M, Barone M, Leopold L, Friedman JM. Positional cloning of the mouse obese gene and its human homologue. Nature. 1994;372:425–432. doi: 10.1038/372425a0. [DOI] [PubMed] [Google Scholar]
- Zhou X, Gomez-Smith M, Qin Z, Duquette PM, Cardenas-Blanco A, Rai PS, Harper ME, Tsai EC, Anisman H, Chen HH. Ablation of LMO4 in glutamatergic neurons impairs leptin control of fat metabolism. Cell Mol Life Sci. 2012;69:819–828. doi: 10.1007/s00018-011-0794-3. [DOI] [PMC free article] [PubMed] [Google Scholar]