

Abstract
A major goal in current neuroscience is to understand the causal links connecting protein functions, neural activity, and behavior. The cannabinoid CB1 receptor is expressed in different neuronal subpopulations, and is engaged in fine-tuning excitatory and inhibitory neurotransmission. Studies using conditional knock-out mice revealed necessary roles of CB1 receptor expressed in dorsal telencephalic glutamatergic neurons in synaptic plasticity and behavior, but whether this expression is also sufficient for brain functions is still to be determined. We applied a genetic strategy to reconstitute full wild-type CB1 receptor functions exclusively in dorsal telencephalic glutamatergic neurons and investigated endocannabinoid-dependent synaptic processes and behavior. Using this approach, we partly restored the phenotype of global CB1 receptor deletion in anxiety-like behaviors and fully restored hippocampus-dependent neuroprotection from chemically induced epileptiform seizures. These features coincided with a rescued hippocampal depolarization-induced suppression of excitation (DSE), a CB1 receptor-dependent form of synaptic plasticity at glutamatergic neurons. By comparison, the rescue of the CB1 receptor on dorsal telencephalic glutamatergic neurons prolonged the time course of DSE in the amygdala, and impaired fear extinction in auditory fear conditioning. These data reveal that CB1 receptor in dorsal telencephalic glutamatergic neurons plays a sufficient role to control neuronal functions that are in large part hippocampus-dependent, while it is insufficient for proper amygdala functions, suggesting an unexpectedly complex circuit regulation by endocannabinoid signaling in the amygdala. Our data pave the way to a better understanding of neuronal networks in the context of behavior, by fine-tuned interference with synaptic transmission processes.
Introduction
Endogenous activation of the cannabinoid CB1 receptor regulates numerous brain functions (Kano et al., 2009; Katona and Freund, 2012). The underlying cellular mechanisms have been intensively investigated, but the causal relationships between CB1 receptor signaling and neuronal circuit functions have been only partially addressed. Pharmacological and genetic approaches have demonstrated necessary roles of CB1 receptor signaling, but they do not provide evidence for its sufficient roles.
In dorsal telencephalic regions, the CB1 receptor is expressed at high levels on GABAergic interneurons, and at low levels on glutamatergic neurons (Marsicano and Lutz, 1999; Monory et al., 2006). Endogenous activation of presynaptically located CB1 receptor leads to different forms of synaptic plasticity of excitatory and inhibitory neurotransmission. This physiological role of the CB1 receptor in balancing excitation and inhibition is essential for several behaviors (Monory et al., 2006; Lafenêtre et al., 2009; Bellocchio et al., 2010; Häring et al., 2011; Dubreucq et al., 2012b; Rey et al., 2012; Ruehle et al., 2012). Thus, dissecting the cell-type-specific roles of the endocannabinoid system enables a better understanding of the excitation–inhibition balance in the brain, alterations of which may cause several neuropsychiatric diseases (Yizhar et al., 2011).
The conditional deletion of the CB1 receptor gene from dorsal telencephalic glutamatergic neurons revealed necessary roles in several physiological processes, including protection against chemically induced epileptiform seizures (Monory et al., 2006), anxiolytic properties of pharmacological cannabinoid stimulation (Rey et al., 2012), and proper extinction of cued fear (Dubreucq et al., 2012b) possibly due to dysregulated habituation to aversive stimuli (Kamprath et al., 2009). From the methodological point of view, these genetic studies were based on a “loss-of-function” approach and characterized necessary functions of the CB1 receptor gene. This approach also contains limitations (e.g., compensatory mechanisms causing misinterpretations of the phenotype observed), and does not provide information concerning the sufficient role of the cell-type-specific expression of CB1 receptor for a given function, or whether additional neuronal subtypes and circuits containing the CB1 receptor are involved in these functions. To establish causal relationships, “rescue” strategies are needed. Existing strategies can involve viral or transgenic re-expression of deleted genes (Self, 2005), but they do not exactly reproduce wild-type (WT) expression of the endogenous gene in terms of levels and cell types. To this end, a strategy using the Cre/loxP system was applied (Nagy, 2000; Balthasar et al., 2005) to restore CB1 receptor expression in a CB1 receptor-null mutant in only a subset of cells, and thereby to investigate whether such expression is sufficient for given brain functions. By rescuing the CB1 receptor gene in its endogenous genomic locus, rather than by ectopic overexpression, its function is restored at endogenous sites and levels, increasing the physiological relevance of the observed phenotypic rescue and its consequent interpretations. Thus, our present study aims at characterizing minimal requirements of CB1 receptor expression sites in selected neuronal circuits (here: dorsal telencephalic glutamatergic neurons) to guarantee WT functions in regard to synaptic plasticity and behaviors previously shown to involve CB1 receptor functions.
Materials and Methods
Animals
All experimental protocols were performed in accordance with the European Community's Council Directive of 24 November 1986 (86/609/EEC) and approved by the Ethical Committee on animal care and use of Rhineland-Palatinate, Germany, and by the Landesamt fuer Natur Umwelt und Verbraucherschutz Nordrhein-Westfalen, Germany (8.87-51.05.20.10.218). Animals were housed in a temperature-controlled and humidity-controlled room (22 ± 1°C; 50 ± 1%) with a 12 h/12 h light/dark cycle (lights on at 7:00 A.M.) and had access to food and water ad libitum. This study was performed on adult (for behavioral analyses, 3–6 months old; for electrophysiological analyses, 2–4 months old) male mice from four different mouse lines: conventional CB1-KO mice (generation and genotyping described in Marsicano et al., 2002), Stop-CB1 mice and their WT littermates, Stop-CB1 mice and their Glu-CB1 receptor rescue (Glu-CB1-RS) littermates, and mice from the complete CB1 receptor rescue (CB1-RS) line. All mouse lines used were backcrossed to C57BL/6J background for at least seven generations. Several batches of animals (either Stop-CB1 mice with WT littermates and CB1-RS mice or Glu-CB1-RS mice with Stop-CB1 littermates and CB1-RS mice) were subjected to a battery of in vivo tests. The order of different behavioral experiments was determined by the aversiveness of the test, finishing with the most aversive tests. To avoid confounding influences of social status, mice were individually housed 2 weeks before the behavioral testing started. The order of the tests was as follows: elevated-plus maze and light–dark test on separate days in the first week of testing; cued fear conditioning and extinction in the second and third week; induction of excitotoxic seizures in the fifth week. A separate batch of animals was subjected to a pain-threshold test, as this procedure interferes with fear conditioning. In our hands, the anxiety, fear, and seizure experiments did not confound each other when performed in this order (unpublished observations). Experiments were performed between 9:00 A.M. and 3:00 P.M.
Generation of the Stop-CB1 mouse line
To generate the Stop-CB1 allele, the loxP-flanked stop cassette was taken from a modified pSV-Cre plasmid (generous gift from Dr. J. K. Elmquist, Dallas) consisting of a SV40 promoter-driven neomycin-resistance (Neo) coding sequence, a HSV-TK polyadenylation sequence and two additional AATAAA sequences from the Promega pGL3-control vector (Balthasar et al., 2005). The stop cassette was inserted in the 5′ untranslated region (UTR) of the coding exon of the CB1 receptor gene, 32 nt upstream of the translational start codon, using overlap-extension PCR. Using a lambda phage DASHII genomic library constructed from E14 embryonic stem (ES) cells (Marsicano et al., 2002), the complete targeting construct was assembled by adding 7.3 kb 5′ sequences (upstream of the stop cassette) and 6.4 kb 3′ sequences (containing the CB1 receptor coding sequence) for homologous recombination in ES cells. The targeting vector (30 μg) was linearized and electroporated into v6.5 C57BL/6(F) × 129/sv(M) ES cells (Rideout et al., 2000). ES cell culture was performed as described previously (Torres and Kühn, 1997). Targeted clones were screened for homologous recombination by Southern blot analysis. To this end, radioactively labeled PCR-amplified DNA fragments derived from the 5′ and 3′ genomic regions outside of the homology arms used for the targeting construct were used as hybridization probes. Random integration of the targeting vector was excluded by using an internal probe derived from the Neo coding region. An ES cell clone with correct homologous recombination of the targeting construct was injected into C57BL/6J blastocysts to generate chimeric mice. Chimeric male founder mice were mated with C57BL/6J females, and the heterozygous offspring were backcrossed for at least seven generations to produce Stop-CB1 mice on a C57BL/6J background. Stop-CB1 animals were genotyped using the following primers: P1 5′-CAAGAAATGAGAACCGTGTC, P2 5′-TGTGTGAATCGATAGTACTAAC, P3 5′-GTTCTCCTTGAACGATGAGA (Fig. 1).
Figure 1.

Generation of a mouse line for cell-type-specific rescue of the CB1 receptor. A, Schematic representation of the WT CB1 receptor allele (top), the Stop-CB1 receptor allele after homologous recombination with the targeting vector (middle), and the rescued CB1 receptor allele after Cre recombinase-mediated excision of the stop cassette (bottom). Southern blot probes (5′ probe, 3′ probe) and the primers for genotyping (P1, P2, P3) are indicated. Black triangles, loxP sites; white box (stop), stop cassette; gray boxes, untranslated regions of the exon containing the open reading frame encoding the CB1 receptor protein; white box (CB1), open reading frame of the CB1 receptor; X, XbaI restriction site; S, SpeI restriction site. B, Southern blot analyses of neomycin-resistant (heterozygous, HET) and WT embryonic stem cell clones for left arm (top, digested with XbaI), and right arm homologous recombination (middle, digested with SpeI), and for the unique presence of the stop cassette in the CB1 receptor locus [bottom, digested with EcoRI, probe against neomycin-resistance (Neo) coding sequence in the stop cassette]. C, PCR genotyping: primers P1 and P2 produced a 543 bp band for the WT allele, and primers P2 and P3 a 462 bp band for the Stop-CB1 allele. Germ-line transmission of the rescued CB1 receptor allele (CB1-RS) resulted in a 577 bp band, as the sequence between the primers P1 and P2 contains the remaining loxP site after stop cassette excision. D, Hippocampal CB1 receptor mRNA was quantified by qPCR in WT (n = 3), Stop-CB1 mice (n = 4), and conventional CB1 receptor KO mice (CB1-KO, n = 2). CB1 receptor transcript levels in Stop-CB1 was significantly reduced compared with WT mice, but not completely lost. However, this transcript did not lead to functional CB1 receptor (Figs. 2, 3). In CB1-KO mice, no mRNA was detectable. ***p < 0.001; **p < 0.01.
Generation of mouse lines with cell-type-specific rescue of CB1 receptor
Complete CB1 receptor rescue.
By crossing Stop-CB1 mice with a mouse line expressing Cre in an early stage of preimplantation embryogenesis (EIIa-Cre; Lakso et al., 1996), the loxP-flanked stop cassette was excised during this developmental stage. Presence of the EIIa-Cre allele was determined using the primers 5′-CGGCATGGTGCAAGTTGAATA and 5′-GCGATCGCTATTTTCCATGAG, and germline transmission of the recombined allele was assessed. Mice were backcrossed with C57BL/6J mice to lose the Cre allele and were then bred to homozygosity for the recombined allele carrying one residual loxP site in the CB1 receptor 5′UTR.
Dorsal telencephalic glutamatergic CB1 receptor rescue.
To obtain selective excision of the stop cassette in dorsal telencephalic glutamatergic neurons, Stop-CB1 mice were crossed with NEX-Cre mice (Schwab et al., 2000; Goebbels et al., 2006) to obtain double mutant CB1stop/stop;NEX-Cre/+ mice. Genotyping for the NEX-Cre allele was performed as described previously (Bellocchio et al., 2010).
RNA isolation and quantitative PCR
Animals were killed by decapitation under isoflurane anesthesia. Hippocampi were quickly isolated and snap frozen on dry ice. Frozen hippocampi were transferred to tubes from a Precellys ceramic kit (1.4 mm diameter ceramic beads; 2 ml tube; Peqlab) containing homogenization buffer from the Nucleo-Spin RNAII-Kit (Macherey-Nagel; β-mercaptoethanol added; Carl Roth), and tissue was homogenized with a Precellys 24 (Peqlab) at 6000 rpm for 20 s. Total RNA was isolated using the Nucleo-Spin RNAII-Kit (Macherey-Nagel). Reverse transcription of 450 ng of DNase-treated total RNA was done using the High Capacity cDNA Reverse Transcription Kit (Applied Biosystems). In the quantitative PCR, the cDNA equivalent to 22.5 ng RNA was amplified using commercial TaqMan assays (Applied Biosystems) for mouse CB1 receptor (Cnr1; Mm00432621_s1) and glucuronidase-β (Gusb; Mm00446953_m1) with an ABI7300 real-time PCR cycler (Applied Biosystems). Reactions were performed in triplicates. Data analysis was done using the Relative Expression Software Tool (REST) (Pfaffl et al., 2002) using Gusb as reference gene.
Immunohistochemistry.
Mice were deeply anesthetized with pentobarbital, and afterward transcardially washed and perfused with PBS/heparin (5 U/ml) and 4% paraformaldehyde (PFA) solution. After isolation, the brains were postfixed for 24 h in 4% PFA solution, treated with 30% sucrose/PBS solution for 48 h, and stored at −80°C until use. For section preparation, 30-μm-thick brain slices were prepared on a cryostat Microtome HM560 (Microm), and stored at −20°C in cryoprotection solution (25% glycerin, 25% ethylene glycol, 50% PBS) until use.
All incubation steps were performed in wells of a 12-well plate (100–500 μl of solution per well) on a wave shaker (Heidolph) at room temperature. Sections were first rinsed from cryoprotection solution in PBS (10 min) and then treated twice with a 100% methanol solution containing 1.5% H2O2, each time for 20 min. After two additional 10 min washing steps in PBS, the sections were preincubated in blocking solution (4% normal goat serum, 0.3% Triton X-100 in PBS) for 1 h. After the blocking, the sections were incubated overnight with the primary antibodies (polyclonal anti-CB1 receptor from rabbit, diluted 1:1000; CB1-Rb-Af380-1, Frontier Science) and polyclonal anti-vGluT1 from guinea pig (diluted 1:500; AB5905, Millipore Bioscience Research Reagents), which were diluted in blocking solution. The next day, the sections were washed twice in PBS and treated with the endogenous biotin-blocking kit, according to the manual of Invitrogen (E21390, Invitrogen). Sections were then washed twice with PBS-T (PBS/0.1% Triton X-100), blocked for 30 min in goat-blocking reagent (Vectastain Elite ABC Kit, PK-6101, Vector Labs), and subsequently incubated for 1.5 h with the kit's biotin-labeled secondary antibody against rabbit from goat diluted (1:200) in the blocking solution. The detection was done according to the manual of Invitrogen, using streptavidin-HRP conjugate combined with Alexa Fluor 488-labeled tyramide (TSA Kit #22, Invitrogen). The treatment was followed by two 10 min washing steps in PBS and a 15 min treatment in 4% PFA solution. After another PBS step for 10 min, sections were treated twice with 100% methanol solution containing 1.5% H2O2, each for 10 min. The sections were then rehydrated twice for 10 min in PBS, followed by an incubation in blocking solution containing the HRP-conjugated anti-guinea pig-IgG from donkey (1:500; 706-035-148, Jackson Immuno Research). The incubation was followed by five 10 min washing steps in PBS. The final detection step was done for 3 min according to TSA Plus Cyanine 3 System manual (diluted 1:60; NEL744001, PerkinElmer). Sections were washed twice in PBS for 10 min and then counterstained for 10 min with DRAQ5 (BioStatus Limited) diluted 1:500 in PBS. After the counterstaining, the sections were washed twice for 10 min in PBS, and then carefully transferred into a Petri dish filled with PBS. Sections were then mounted on glass slides to dry for 1 h at 37°C. The remaining salt was washed off by dipping the slides for 2 s into distilled water. Finally, the sections were dried overnight in a dust-free environment at room temperature and covered with Mowiol 4-88 mounting medium (Roth).
Fluorescence labeling was visualized using the confocal laser-scanning microscope Zeiss LSM T-PMT 719 (Zeiss Microsystems) or Leica TCS SP5 (Leica Microsystems), equipped with appropriate excitation and emission filters for maximum separation of Cyanine 3 and Alexa Fluor 488 signals. Applying the Zeiss or Leica Confocal Software, and Adobe Photoshop (Version 7.0, Adobe), images were saved and processed. Gain, contrast, and brightness were adjusted identically for the images shown in Figure 3A–H. For the images shown in Figure 3g1–h1, the gain of the detector collecting the CB1 receptor signal was adjusted to the intensity of the fluorescence signal.
Figure 3.

Rescue of CB1 receptor protein expression in dorsal telencephalic glutamatergic neurons. A–H, Immunohistochemistry on hippocampal (A–D) and amygdalar (E–H) sections for CB1 receptor (green) and nuclear staining with DRAQ5 (blue). BA, basal amygdala; CeA, central amygdala; GC, granule cell layer; Hil, hilar region of dentate gyrus; LA, lateral amygdala; LMol, stratum lacunosum-molecularis; Mol, stratum molecularis; Pyr, CA1/CA3 pyramidal cell layer; Rad, stratum radiatum. d1, Boxed zone marked in D to show colocalization of CB1 receptor (green) with VGlut1 (red). g1, h1, Higher magnification of the BA. Scale bars: D, H, 200 μm; d1, 25 μm; h1, 10 μm. I, J, Western blot analysis of hippocampal protein extracts. I, Representative Western blot (CB1 52 kDa, tubulin (tub) 55 kDa). J, Quantification of n = 3 per genotype. #p < 0.001 versus WT and CB1-RS; +p < 0.1 versus Stop-CB1 and CB1-KO.
Western blotting.
Animals were killed by decapitation under isoflurane anesthesia. Hippocampi were isolated and homogenized in Tris-buffered saline (TBS) containing protease inhibitors (Complete, Roche Applied Science). Twenty milligrams of protein were mixed with 5× Laemmli reducing sample buffer, denatured for 5 min at 60°C, separated by 10% SDS-PAGE, and transferred onto nitrocellulose membranes (Protran, Whatman; GE Healthcare). Blocking was performed in 5% (w/v) nonfat dry milk in TBS with 0.1% Tween 20 (v/v) (TBS-T). The membrane was incubated in 1% (w/v) nonfat dry milk in TBS-T with the following antibodies: rabbit anti-CB1 primary antibody (1:500; Frontier Science) and mouse anti-tubulin (1:500,000; Sigma-Aldrich). Antibodies were detected by the appropriate HRP-conjugated secondary antibodies (Dianova) followed by ECL-detection (GE Healthcare). Chemiluminescence was visualized and quantified with the Fusion SL system (Vilber Lourmat). Mean values for each genotype were expressed relative to WT.
Autoradiography.
Animals were killed by decapitation under isoflurane anesthesia. Coronal brain sections were prepared as described in Monory et al. (2006). Autoradiography was performed as previously described (Herkenham et al., 1991). Brain sections were incubated for 2 h at 30°C in 50 mm Tris HCl, pH 7.4, containing 5% (w/v) fat-free BSA with the addition of 5 nm 3H-CP55,940 (specific activity 139.6 Ci/mmol; PerkinElmer). Nonspecific binding was determined by incubating adjacent sections in 5 nm 3H-CP55,940 in the presence of 10 μm CP55,940 (Tocris Bioscience). After the incubation, sections were washed and briefly dipped in ice-cold distilled water and dried overnight. TR Tritium Phosphor Screens (PerkinElmer) were exposed to the slides together with a tritium standard (American Radiolabeled Chemicals) for 3 d and then scanned with a Cyclone Plus Storage Phosphor System (PerkinElmer). Ligand binding to the CB1 receptor was quantified using Optiquant software (PerkinElmer). A standard curve was compiled using the tritium standard. Brain structures were identified by comparison with the mouse brain atlas (Paxinos and Franklin, 2008) and outlined manually for determination of average density. Right and left sides were pooled from four to six sections containing each structure. Unspecific binding was subtracted, and the average density was calculated for each animal. Mean values for each genotype were expressed relative to WT.
Electrophysiological recordings
As previously described (Monory et al., 2006; Lourenço et al., 2010; Kamprath et al., 2011), mice (2–4 months old) were anesthetized with isoflurane (5%) and decapitated. Their brains were rapidly removed and put into oxygenated (95% O2, 5% CO2) ice-cold artificial CSF (ACSF) containing the following (in mm): 125 NaCl, 2.5 KCl, 2 CaCl2, 10 MgCl2, 1.25 NaH2PO4, 26 NaHCO3, 16 glucose, pH 7.4. Parasagittal hippocampal or coronal amygdalar slices (300 μm thick) were cut on a vibratome (Leica Microsystems) at 4°C. The slices were allowed to equilibrate for at least 1 h at room temperature, and then transferred to a recording chamber continuously superfused with ACSF (∼1.5 ml/min).
Whole-cell voltage-clamp recordings were made at room temperature from pyramidal cells in CA1 or basolateral amygdala (BLA), neurons in the lateral section of the central amygdala (CeA), and granule cells in dentate gyrus (DG), visualized by infrared video microscopy (monochrome camera CF8/1, Kappa). Patch pipettes (3–4 MΩ) were filled with an intracellular solution containing the following (in mm): 145 CsCl, 10 HEPES, 5 EGTA, 2 MgCl2, 2 CaCl2, 2 Na2ATP, 5 phosphocreatine, 0.33 GTP, pH 7.4. Neurons were voltage-clamped at −70 mV. Cells were excluded from analysis if the access resistance changed by >20% over the course of the experiment. Recordings were made using an EPC 10.0 (HEKA Elektronik) or a Multiclamp 700B (Molecular Devices) amplifier, filtered at 0.5–1 kHz, digitized at a sampling rate of 10 kHz, and analyzed off-line using the program IGOR PRO 5.0 (Wavemetrics) or Clampfit (Molecular Devices).
After reaching a stable baseline (∼10 min after establishing the whole-cell configuration for infusion of intracellular solution), extracellular stimuli (100 μs, 50–600 μA) were delivered through a bipolar stainless-steel electrode. For CA1 and BLA experiments, the stimulation electrode was placed in the stratum radiatum for stimulation of the Schaffer collaterals, and within the local neuropil of the BLA (∼100 μm from the recorded neuron), respectively. For CeA experiments, the stimulation electrode was placed in BLA as previously described (Kamprath et al., 2011). For DG experiments, the stimulation electrode was placed in the supragranular layer (<40 μm from the cell body layer) to stimulate the mossy cell fibers (MCFs) (Chiu and Castillo, 2008). Specificity of stimulation of MCF was verified by paired-pulse depression and insensitivity to inhibition by group II metabotropic glutamate receptor agonists. Glutamatergic and GABAergic components of synaptic responses were isolated by addition of gabazine (10 μm), or picrotoxin (100 μm) and CGP55845 (50 μm), or DNQX (10 μm) and AP-5 (50 μm), respectively. Some slices were preincubated with the CB1 receptor antagonist AM251 (2 μm) at least 1 h before testing DSE.
Induction and calculation of depolarization-induced suppression of excitation and inhibition amplitude
Depolarization-induced suppression of excitation (DSE) and depolarization-induced suppression of inhibition (DSI) tests consisted of 60 evoked responses (evoked every 3 s) before the depolarization step (from −70 to 0 mV; 3 or 10 s) and 100–150 responses thereafter. At least three DSE/DSI tests were applied to each cell (Lourenço et al., 2010). Baseline was calculated from each evoked response, divided by the average of all evoked responses before the depolarization step (multiplied by 100%). DSE and DSI amplitudes were calculated from each evoked response after the depolarization step, relative to the average before the depolarization step (multiplied by 100%). Data were plotted against time. To improve readability, in Figure 6G and Figure 8, data points represent the average of 3 ePSCs. For comparative data analysis (shown in bar graphs), DSE and DSI magnitude was calculated as follows (modified from Wilson and Nicoll, 2001): Δ DSE magnitude or Δ DSI magnitude = [(x2 − x1)/x1] * 100, where x1 is mean of last five ePSC amplitudes before the depolarization, and x2 is mean of first three ePSC amplitudes immediately after the depolarization. Calculated magnitudes were statistically tested, and significant deviation from zero was considered as DSE/DSI. In view of higher variability of evoked responses in DG, we calculated x1 from the mean of last 10 ePSC amplitudes (instead of 5 ePSC amplitudes) before the depolarization. In view of prolonged time courses of DSE in BLA, we calculated x2 from the mean of three ePSC amplitudes at 160 s after the depolarization step.
Figure 6.
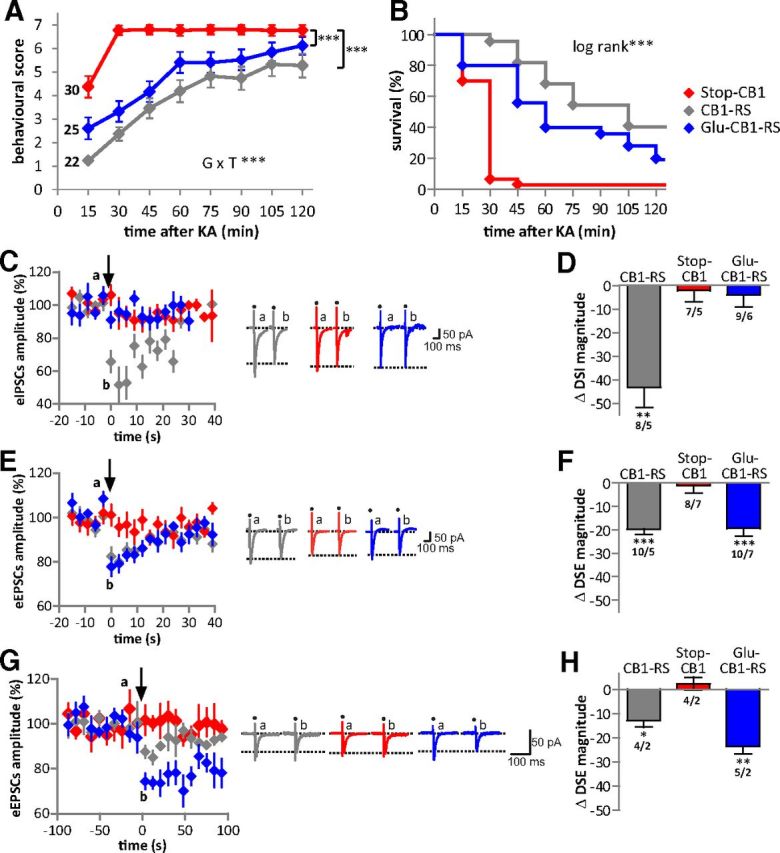
Hippocampal-dependent protection from chemically induced epileptiform seizures is rescued and hippocampal DSE is restored in Glu-CB1-RS mice. A, B, Neuroprotection against seizures induced by the excitotoxin KA was significantly rescued in Glu-CB1-RS mice. A, Behavioral scores over a period of 120 min after KA injection. GxT, interaction of genotype and time. B, Kaplan–Meier survival curves of the three genotypes after injection of KA. ***p < 0.001; animal numbers are indicated in A. C–D, DSI in hippocampal CA1 neurons. E–H, DSE in hippocampal CA1 (E–F) and dentate gyrus neurons (G–H). C, E, and G show eIPSCs and eEPSCs averaged from recordings in neurons of the three genotypes. In G, each data point represents the average of three consecutive eEPSCs. The arrows indicate application of a 3 s depolarization step. Original traces illustrate eIPSCs and eEPSCs recorded in one neuron before (a, 5 consecutive traces averaged) and after (b, 3 traces averaged) the 3 s depolarization. Dots indicate stimulation artifacts; dotted lines indicate amplitude of ePSCs after depolarization. Bar diagrams in D, F, and H show DSI and DSE magnitudes immediately after the depolarization step. Numbers indicate recorded cells/animals. Note selective rescue of DSE (vs DSI) in Glu-CB1-RS. ***p < 0.001, **p < 0.01, *p < 0.05 versus respective baseline before depolarization.
Figure 8.

Prolonged DSE in Glu-CB1-RS is BLA-specific. A–C, Time course of DSE in BLA (A), CeA (B), and hippocampal CA1 (C). Depolarization step (3 or 10 s) is indicated by an arrow. Each data point represents the average of three consecutive eEPSCs recorded under the different conditions. Numbers indicate recorded cells/animals. Note that depolarization of 10 s promotes a strong DSE and prolongation of DSE time course in BLA, but not CeA or CA1.
Elevated-plus maze
The elevated-plus maze (EPM) was a custom-made cross-shaped setup elevated 100 cm above the floor. It consisted of four arms, two opposite open arms and two opposite enclosed arms. The floor of the arms was made of white plastic, 35 cm long and 6 cm wide and the arms were interconnected by a central platform of 6 × 6 cm. Black plastic walls (20 cm high) surrounded the enclosed arms. Light intensity in the middle of the open arms was 140 lx. The animals were placed into the center of the maze, facing an enclosed arm, and were allowed to explore over 5 min (Walf and Frye, 2007). After each test, the plus-maze was cleaned with 70% ethanol. Animals were tracked using Ethovision software (Noldus) with the three body-point module (nose point, center, tail base). Time in each arm was measured when all three body points were inside the arm. Time spent in the open arms was calculated relative to time spent in all arms.
Light–dark test
The light–dark (LD) test was performed in a custom-made box (39 × 39 cm) divided in a light compartment (two-thirds of the surface area, white) and a dark compartment (one-third, black box, with a 26-cm-high lid). Light and dark compartments were directly connected by a small entrance (5 × 5 cm). Light was adjusted to 100 lx in the center of the light compartment. The animals were placed in the dark compartment and allowed to explore the apparatus for 5 min. The latency to first enter the light compartment with all four paws, percentage of time spent in the light, and number of entries to the light were assessed by trained observers blind to the genotype.
Induction of acute excitotoxic seizures
Kainic acid (KA; Ascent or Sigma-Aldrich) was dissolved in 0.9% saline and administered (30 mg/kg; i.p.) in a volume of 10 ml/kg body weight to induce epileptiform seizures. Of note, KA from Ascent induced less severe seizures than that from Sigma-Aldrich, pointing to different apparent activities regarding the induction of seizures (in Fig. 4: KA from Ascent; in Fig. 6: KA from Sigma-Aldrich). Before injection, the animals were given a light isoflurane inhalation anesthesia to reduce injection stress. A trained observer blind to the genotype of the mice monitored the severity of seizures for 2 h and scored every 15 min according to the following scale (Schauwecker and Steward, 1997; Monory et al., 2006): 0, no response; 1, immobility and staring; 2, forelimb and/or tail extension, rigid posture; 3, repetitive movements, head bobbing; 4, rearing and falling; 5, continuous rearing and falling: 6, severe clonic-tonic seizures; 7, death.
Figure 4.

CB1-RS mice show WT behavior in all tested procedures. A, Percentage of time spent in the open arms (OA) of the EPM of Stop-CB1 mice was significantly reduced compared with their WT littermates and CB1-RS animals (F(2,27) = 10.78, p < 0.001 for genotype). B, C, Neuroprotection against seizures induced by the excitotoxin KA was reduced in Stop-CB1 mice compared with WT and CB1-RS mice. B, Behavioral scores over a period of 120 min after KA injection. GxT, interaction of genotype and time (F(4.75,116.40) = 3.75, p = 0.004). C, Kaplan–Meier survival curves of the three genotypes after injection of KA [X2 (2, n = 52) = 25.29, p < 0.001 for genotype]. D–G, Conditioned freezing in response to repeated presentations of a fear-conditioned auditory stimulus at different days after training. D, Freezing response (after subtraction of baseline freezing of the same day) over the course of tone presentations per day (d1–d10, as indicated) analyzed in 20 s bins. Conditioned freezing of Stop-CB1 animals is higher compared with WT and CB1-RS. E, The total freezing response (after subtraction of baseline freezing) over the days of Stop-CB1 was stronger than that of WT or CB1-RS mice. F, Baseline freezing response before the tone started did not differ between genotypes. G, Freezing after the tone was terminated was higher in Stop-CB1 than in WT mice. Detailed statistical analysis of behavior in Table 2. ***p < 0.001, **p < 0.01, and *p < 0.05.
Cued fear conditioning
Procedures and setups were used as previously described (Kamprath and Wotjak, 2004). For conditioning, mice were placed in the conditioning chamber (Med Associates; square, 15 × 20 cm, grid floor, cleaned with 1% acetic acid) and a house light (25 lx) turned on. After 3 min, a 20 s tone (80 dB, 9 kHz sine wave, 10 ms rising and falling time) was presented to the animals. The tone coterminated with a 2 s scrambled electric foot shock of 0.6 mA. Mice were returned to their home cages 60 s later. On day (d) 1, d2, d3, and d10 after the conditioning day, conditioned mice were placed into a neutral, new environment (extinction context, custom-made Plexiglas cylinders, 15 cm diameter, with bedding, cleaned with 70% ethanol), and the house light (5 lx) was switched on. After 3 min, a 200 s continuous tone (same settings as in conditioning) was presented. Mice were returned to their home cages 60 s after the end of the tone presentation. Animals were tracked using Ethovision software. Freezing (i.e., immobility, here defined as the absence of all nonrespiratory movements) was scored with the Ethovision immobility filter set at 0.5% change of the pixels representing the mouse, with averaging over two consecutive frames (25 frames/s). Conditioned freezing, defined as freezing to the tone minus baseline freezing response of the same day, as well as baseline freezing before the tone started, and freezing after the tone presentation ended, were analyzed.
Pain threshold
Animals were placed into the conditioning chamber, and a scrambled electric foot shock of rising intensity (starting from 0 mA, 0.015 mA/s) was applied. The shock was switched off as soon as the animals jumped or vocalized. The corresponding shock intensity was defined as pain threshold.
Data analysis
The results were analyzed using SPSS 19 Statistics Software for Windows (IBM). All data are expressed as mean ± SEM. Univariate ANOVA with genotype as independent variable was used to analyze qPCR, Western blotting, autoradiography, pain threshold, and anxiety measures. Distance moved on the EPM (for EPM anxiety measures) was included in the model as covariate, but did not have significant effects and was thus excluded from the model in the final analysis. Repeated-measures ANOVAs with genotype and time (day or time bin) as independent variables were used to analyze freezing in fear extinction and seizure severity after KA injection. The Greenhouse–Geisser correction was used if the condition of sphericity was not met. Significant genotype effects were further analyzed using Bonferroni's post hoc analysis for multiple comparisons. The Kaplan–Meier method was used to evaluate survival, followed by the log rank test to identify significant differences. For electrophysiological data, DSE and DSI magnitudes (for details of calculating DSE/DSI magnitudes, see above) were analyzed with one-sample t tests using Graphpad Prism 4.0 (GraphPad Software). ePSC amplitudes were analyzed with two-way ANOVA (duration depolarization step, time course) using STATISTICA (StatSoft). Differences were considered significant at p < 0.05. Rescue was considered “fully sufficient” if the genotype of interest (CB1-RS or Glu-CB1-RS) showed a significant difference from Stop-CB1 and no significance compared with the control group (WT or CB1-RS). Values intermediate between Stop-CB1 and the control group (with significance from either both or none), were considered as “partly sufficient.” No difference from (or more extreme values than) Stop-CB1 together with a significant difference from the control group was interpreted as “no sufficiency” of the rescue.
Results
Generation of Stop-CB1 mice and global rescue of CB1 receptor function
To generate the Stop-CB1 mouse line with a silenced CB1 receptor as the default state, but with the possibility to rescue the CB1 receptor, a loxP-flanked stop cassette was inserted in the 5′ UTR upstream of the CB1 receptor translational start codon (Fig. 1A). Homologous recombination in ES cells was confirmed by Southern blot analysis using probes outside of the homology arms and a Neo probe to confirm the absence of additional random integrations in the genome (Fig. 1B). Standard genotyping of the generated Stop-CB1 mouse line was performed with PCR (Fig. 1C). For unknown reasons, the presence of the stop cassette was not sufficient to fully block transcription of CB1 receptor mRNA as revealed by quantitative PCR (Fig. 1D). However, the aim of the approach was to eliminate CB1 receptor protein. Indeed, receptor autoradiography with the radiolabeled CB1 receptor agonist 3H-CP55,940 (Fig. 2B,F,J,N,Q; Table 1), Western immunoblotting, and immunohistochemical analysis using specific CB1 receptor antibodies (Fig. 3B,F,I,J) revealed that no CB1 receptor protein was detectable in Stop-CB1 mice. Thus, Stop-CB1 mice should reveal similar phenotypes as null-mutant CB1 receptor mice, which were shown to be more susceptible to KA-induced epileptiform seizures (Marsicano et al., 2003), to display decreased exploration of open arms on an EPM (Haller et al., 2004), and to be impaired in extinction of freezing responses in cued fear conditioning tests (Marsicano et al., 2002). Consistent with the lack of CB1 receptor protein expression, Stop-CB1 mice displayed the same phenotypes (Fig. 4; Table 2), and can thus be considered as functional CB1 receptor-null mutants.
Figure 2.

Stop-CB1 allele enables rescue of the CB1 receptor ligand binding in dorsal telencephalic glutamatergic neurons. A–P, Autoradiography of CB1 receptor ligand binding using 3H-CP55,940 on brain sections of WT, Stop-CB1, CB1-RS, and Glu-CB1-RS. Black triangles, loxP sites; white box (stop), stop cassette; white box (CB1), CB1 receptor open reading frame. BLA, basolateral amygdala; Cer, cerebellum; CPu, caudate–putamen; Ctx, neocortex; EP, entopeduncular nucleus; GP, globus pallidus; Hip, hippocampus; Hy, hypothalamus; Ls, lateral septum; Ms, medial septum; Th, thalamus. Scale bar, 1 mm. Q, Quantification of bound 3H-CP55,940 in WT (n = 3–4), CB1-RS (n = 3), Glu-CB1-RS (n = 3), Stop-CB1 (n = 3); values of columns labeled with the same letter (a, b, or c) are not significantly different from each other; detailed statistical analysis in Table 1. MG, medial geniculate nucleus; VMH, ventromedial hypothalamus.
Table 1.
Statistical analysis of the quantification of 3H-CP55,940 binding in WT, CB1-RS, Stop-CB1, and Glu-CB1-RS mice
| CPu | GP | BLA | VMH | Hip | MG | |
|---|---|---|---|---|---|---|
| F | F(3,8) = 99.93 | F(3,8) = 66.00 | F(3,8) = 44.99 | F(3,8) = 188.80 | F(3,8) = 74.40 | F(3,9) = 62.10 |
| p | <0.001 | <0.001 | <0.001 | <0.001 | <0.001 | <0.001 |
| Post hoc | ||||||
| WT–CB1-RS | 0.104 | 0.679 | 1.000 | 1.000 | 1.000 | 1.000 |
| WT–Stop-CB1 | <0.001 | <0.001 | <0.001 | <0.001 | <0.001 | <0.001 |
| WT–Glu-CB1-RS | 0.001 | <0.001 | 0.002 | <0.001 | <0.001 | <0.001 |
| CB1-RS–Stop-CB1 | <0.001 | <0.001 | <0.001 | <0.001 | <0.001 | <0.001 |
| CB1-RS–Glu-CB1-RS | <0.001 | <0.001 | 0.004 | <0.001 | <0.001 | <0.001 |
| Stop-CB1–Glu-CB1-RS | 0.001 | 1.000 | 0.046 | 0.037 | 0.039 | 0.021 |
BLA, basolateral amygdala; CPu, caudate putamen; GP, globus pallidus; Hip, hippocampus; MG, medial geniculate nucleus; VMH, ventromedial hypothalamus.
Table 2.
Statistical analysis of the performance of Stop-CB1, WT, and CB1-RS mice in fear extinction
| Repeated-measures ANOVA | Factor | F | p | Post hoc G | p |
|---|---|---|---|---|---|
| For 20 s time bins | |||||
| d1 | G × T | F(9.20,124.22) = 1.40 | 0.193 | Stop-CB1 vs WT | 0.033 |
| T | F(4.60,124.22) = 28.19 | <0.001 | Stop-CB1 vs CB1-RS | 0.005 | |
| G | F(2,27) = 6.70 | 0.004 | WT vs CB1-RS | 1.000 | |
| d2 | G × T | F(11.42,154.15) = 1.76 | 0.062 | Stop-CB1 vs WT | 0.025 |
| T | F(5.71,154.15) = 16.55 | <0.001 | Stop-CB1 vs CB1-RS | 0.008 | |
| G | F(2,27) = 6.41 | 0.005 | WT vs CB1-RS | 1.000 | |
| d3 | G × T | F(9.94,134.24) = 0.88 | 0.557 | Stop-CB1 vs WT | 0.008 |
| T | F(4.97,134.24) = 16.02 | <0.001 | Stop-CB1 vs CB1-RS | 0.137 | |
| G | F(2,27) = 5.56 | 0.010 | WT vs CB1-RS | 0.723 | |
| d10 | G × T | F(8.39,113.27) = 0.76 | 0.642 | Stop-CB1 vs WT | <0.001 |
| T | F(4.20,113.27) = 7.71 | <0.001 | Stop-CB1 vs CB1-RS | 0.027 | |
| G | F(2,27) = 12.47 | <0.001 | WT vs CB1-RS | 0.120 | |
| For days | |||||
| Tone minus Baseline | G × T | F(6,81) = 1.41 | 0.220 | Stop-CB1 vs WT | <0.001 |
| T | F(3,81) = 36.02 | <0.001 | Stop-CB1 vs CB1-RS | 0.003 | |
| G | F(2,27) = 10.62 | <0.001 | WT vs CB1-RS | 1.000 | |
| Initial 20 s minus Baseline | G × T | F(6,81) = 2.17 | 0.054 | Stop-CB1 vs WT | nd |
| T | F(3,81) = 23.83 | <0.001 | Stop-CB1 vs CB1-RS | nd | |
| G | F(2,27) = 2.02 | 0.152 | WT vs CB1-RS | nd | |
| Baseline | G × T | F(6,81) = 0.39 | 0.885 | Stop-CB1 vs WT | nd |
| T | F(3,81) = 47.60 | <0.001 | Stop-CB1 vs CB1-RS | nd | |
| G | F(2,27) = 0.02 | 0.982 | WT vs CB1-RS | nd | |
| After tone | G × T | F(6,81) = 0.30 | 0.937 | Stop-CB1 vs WT | 0.004 |
| T | F(3,81) = 8.65 | <0.001 | Stop-CB1 vs CB1-RS | 0.181 | |
| G | F(2,27) = 6.52 | 0.005 | WT vs CB1-RS | 0.336 |
G, genotype; T, time; d, day; nd, not determined.
To verify the functionality of the genetic rescue approach, we crossed the Stop-CB1 line with a Cre-deleter mouse line (EIIa-Cre; Lakso et al., 1996) to obtain global rescue of CB1 receptor expression (hereafter called CB1-RS). Importantly, the mice resulting from this crossing displayed phenotypes that were indistinguishable from those of WT animals in all parameters analyzed (protection against KA-induced seizures, open arms exploration in the EPM, and extinction of conditioned fear; Fig. 4, Table 2). Moreover, brain sections from CB1-RS mice showed cannabinoid agonist binding (Fig. 2C,G,K,O) and CB1 receptor immunoreactivity (Fig. 3C,G) virtually identical to WT animals. Quantification of receptor autoradiography in several brain regions and of protein in Western blot analysis in the hippocampus confirmed that there was no difference between CB1-RS and WT (Figs. 2Q, 3I,J) in the level of bound 3H-CP55,940 and the amount of CB1 receptor protein, respectively. Therefore, Cre-mediated excision of the loxP-flanked stop cassette is a valid tool to rescue the functional expression of the CB1 receptor protein in living animals.
Rescue of CB1 receptor on dorsal telencephalic glutamatergic neurons
To obtain a selective rescue of CB1 receptor on dorsal telencephalic glutamatergic neurons, Stop-CB1 mice were crossed with a mouse line expressing Cre recombinase under the regulatory sequences of the NEX gene (Schwab et al., 2000; Monory et al., 2006), which primarily drives expression in glutamatergic neurons of the dorsal telencephalon, including neocortex and archicortex (hippocampal formation and cortical portions of the amygdala; Goebbels et al., 2006). In the resulting Glu-CB1-RS mice, binding of the synthetic CB1 receptor agonist 3H-CP55,940 revealed the rescue of the CB1 receptor in cortical areas (including neocortex, amygdala, and hippocampus), as well as subcortical regions, including thalamus, hypothalamus, and, to a large extent, the caudate–putamen (Fig. 2D,H,L,P,Q). Importantly, these areas are considered major targets of CB1 receptor-containing cortical axonal projections (Marsicano and Kuner, 2008). The very high level binding observed in WT globus pallidus was not rescued in Glu-CB1-RS mice (Fig. 2H,Q), indicating that these CB1 receptor-positive projections do not originate from the dorsal telencephalon. Quantification of CB1 receptor protein in hippocampal extracts revealed 28% of CB1 receptor in Glu-CB1-RS relative to CB1-RS animals (Fig. 3I,J). These data are in good agreement with the low levels of CB1 receptor expression in dorsal telencephalic glutamatergic neurons (Marsicano and Lutz, 1999; Kawamura et al., 2006; Marsicano and Kuner, 2008; Bellocchio et al., 2010; Steindel et al., 2013). Detailed immunohistochemical analysis of CB1 receptor expression in the hippocampus of Glu-CB1-RS mice (Fig. 3D) revealed the strongest signal in the inner third of the molecular layer of the dentate gyrus, where it colocalizes with VGluT1, a marker of glutamatergic terminals. This expression was previously described for the CB1 receptor on terminals of mossy cell glutamatergic afferent fibers, projecting to the inner third of the molecular layer (Monory et al., 2006). In the amygdala of Glu-CB1-RS mice (Fig. 3H), weak CB1 receptor immunoreactivity was detected in the basal, but not in the lateral part of the BLA. A very faint signal was detected in the central nucleus of the amygdala.
To test to what extent CB1 receptor-driven modulation of glutamatergic transmission in cortical neurons is sufficient to rescue the phenotypes observed in CB1 receptor KO animals, we subjected the Glu-CB1-RS mice to several behavioral procedures for which CB1 receptor on glutamatergic terminals was previously shown to be necessary (Monory et al., 2006; Kamprath et al., 2009; Dubreucq et al., 2012b; Rey et al., 2012).
Anxiety-like behavior
Anxiety was assessed through the EPM and the LD test. Both procedures revealed significant main effects for genotype (EPM open arm time F(2,65) = 7.43, p = 0.001; distance moved on the EPM (as a covariate) did not have a significant effect on EPM open arm time and was therefore excluded from the final analysis; LD light time F(2,67) = 5.67, p = 0.005, LD latency F(2,67) = 8.97, p < 0.001). Stop-CB1 animals spent a significantly lower percentage of time on the open arms than CB1-RS animals (p = 0.001, Fig. 5A). The percentage of time spent on the open arms of the Glu-CB1-RS animals was intermediate between that of the other genotypes. Glu-CB1-RS mice were not significantly different from CB1-RS mice (p = 0.317) and showed a trend to differ from Stop-CB1 animals (p = 0.078). In the LD test (Fig. 5B,C), Stop-CB1 animals spent less time in the light compartment (p = 0.004) and had a longer latency until the first entry (p = 0.001) than CB1-RS animals. Glu-CB1-RS animals and CB1-RS animals did not differ significantly in the time spent in the light compartment (p = 0.503) or entry latency (p = 1). The latency of Glu-CB1-RS animals to enter the light compartment was significantly shorter than that of their Stop-CB1 littermates (p = 0.003), but time spent in the light compartment was not significantly different between Glu-CB1-RS and Stop-CB1 littermates (p = 0.152). Thus, expression of the CB1 receptor on dorsal telencephalic glutamatergic neurons partially rescued the increase in anxiety observed upon CB1 receptor loss.
Figure 5.

Partial sufficiency of CB1 receptor in dorsal telencephalic glutamatergic neurons for anxiety-like behavior. A, Percentage of time spent on the open arms (OA) of the EPM of Glu-CB1-RS mice did not differ from that of CB1-RS mice and tended to be higher than that of Stop-CB1 mice, pointing to a partial rescue. B, Percentage of time spent in the light compartment of the LD box of Stop-CB1 animals is decreased compared with CB1-RS animals. Glu-CB1-RS mice did not differ significantly from the other two genotypes, indicating an intermediate anxiety level. C, Latency to enter the light compartment of the Glu-CB1-RS mice did not differ from the CB1-RS mice, but was significantly reduced compared with Stop-CB1 mice, indicating a full rescue of the anxiogenic phenotype. **p < 0.01; animal numbers are indicated in the graph.
Protection against excitotoxic epileptiform seizures
To test protection against epileptiform seizures, mice were treated with the excitotoxic agent KA, which induces activation of excitatory pathways leading to acute epileptiform seizures (Ben-Ari and Cossart, 2000). The genotypes reacted differently to KA over time (genotype–time interaction, F(5.58,206.58) = 6.72, p < 0.001; Fig. 6A). Stop-CB1 mice displayed significantly higher seizure susceptibility than CB1-RS mice (p < 0.001) and Glu-CB1-RS mice (p < 0.001), with no differences between Glu-CB1-RS and CB1-RS mice (p = 0.152). There was a significant difference in survival between Glu-CB1-RS, Stop-CB1, and CB1-RS mice (Fig. 6B; X2 (2, n = 77) = 25.20, p < 0.001). One hour after seizure induction, 40% of the Glu-CB1-RS mice survived, while only 3% of the Stop-CB1 animals were still alive. Of the CB1-RS animals, 64% survived the first hour after KA injection. Thus, CB1 receptor on dorsal telencephalic glutamatergic neurons is sufficient to provide protection against KA-induced epileptiform seizures.
DSI and DSE in the hippocampus
As the CB1 receptor in hippocampal neurons is considered as a necessary element of endogenous protection against excitotoxic epileptiform seizures in vivo (Marsicano et al., 2003; Monory et al., 2006), CB1 receptor-mediated suppression of neurotransmitter release was tested in CA1 pyramidal neurons and granule cells from the DG (Wilson and Nicoll, 2001; Ohno-Shosaku et al., 2002; Chiu and Castillo, 2008). Depolarization-induced suppression of GABAergic inhibitory (DSI; Fig. 6C,D) and glutamatergic excitatory (DSE; Fig. 6E–H) postsynaptic currents were recorded. As CB1 receptor expression in DG is limited to glutamatergic mossy cells (Monory et al., 2006, Chiu and Castillo, 2008), DSI was not tested in this region in the present study. Depolarization steps (3 s duration) of the recorded neuron produced a significant reduction of subsequently evoked GABAergic (DSI) and glutamatergic (DSE) postsynaptic currents in CB1-RS mice (DSI in CA1 neurons: t(7) = 5.05, p = 0.002; Fig. 6C,D; DSE in CA1 neurons: t(9) = 8.96, p < 0.001; Fig. 6E,F; DSE in DG neurons: t(3) = 4.75, p = 0.018; Fig. 6G,H). In Stop-CB1 mice, depolarization did not suppress the subsequent postsynaptic currents (DSI in CA1 neurons: t(6) = 0.44, p = 0.675, Fig. 6C,D; DSE in CA1 neurons: t(7) = 0.34, p = 0.742, Fig. 6E,F; DSE in DG neurons: t(3) = 0.87, p = 0.448, Fig. 6G,H), proving the absence of the CB1 receptor from the presynaptic terminals. In Glu-CB1-RS mice, DSE was present in both CA1 neurons (t(9) = 5.72, p < 0.001) and DG neurons (t(4) = 7.90, p = 0.001, Fig. 6E–H). Depolarization steps of 10 s duration resulted in DSE in CA1 neurons (t(3) = 29.33, p < 0.001) similar to that upon 3 s depolarization in these mice (see Fig. 8C). Importantly, DSI was absent (t(8) = 0.82, p = 0.435) in Glu-CB1-RS mice (Fig. 6C,D). Altogether, these data indicate that CB1 receptor expression in dorsal telencephalic glutamatergic neurons in the hippocampus is sufficient to provide endocannabinoid-dependent control of short-term synaptic plasticity of excitatory transmission.
Cued fear extinction
In a next series of experiments, the sufficient role of the CB1 receptor on dorsal telencephalic glutamatergic neurons was assessed in a cued fear-conditioning procedure. Importantly, differences in pain sensitivity were not observed between the genotypes, as tested by the first pain responses (vocalization or jump) to an electric foot shock of rising intensity in a separate batch of animals (n = 6–10 mice per genotype; WT, 0.17 ± 0.01 mA; CB1-RS, 0.17 ± 0.01 mA; Stop-CB1, 0.16 ± 0.01 mA; Glu-CB1-RS, 0.17 ± 0.01 mA; F(3,27) = 0.85, p = 0.479). CB1-RS, Stop-CB1, and Glu-CB1-RS mice were conditioned to associate a tone with a foot shock in a single tone-shock pairing. After 180 s exploration where baseline behavior was monitored, the tone was presented continuously over 200 s. Conditioned freezing in a novel context was measured on d1, d2, d3, and d10 after conditioning (Fig. 7A). The initial freezing response to the tone presentation (first 20 s bin), 24 h after conditioning, was similar for all genotypes (F(2,65) = 1.33, p = 0.272). Within-session extinction of freezing (tone-induced freezing in 20 s bins) showed a significant effect for genotype and time on all extinction days. In addition, there was a significant interaction of genotype and time on d2–d10 (p values in Table 3), indicating different reductions of the freezing response over time for the genotypes. Post hoc analysis of genotype revealed a significantly higher freezing response of Glu-CB1-RS than of Stop-CB1 and of CB1-RS mice on d1. On d2–d10, Glu-CB1-RS and Stop-CB1 animals showed a significantly higher freezing response than CB1-RS mice (Table 3). On d3, Glu-CB1-RS had a trend toward higher freezing than Stop-CB1 animals (p = 0.079). Between-session extinction (defined as the reduction in initial freezing response between the extinction days) was present in all genotypes (F(3,195) = 11.02, p < 0.001), but did not differ between the genotypes. The total freezing response to the 200 s tone presentation (main genotype effect F(2,65) = 18.70, p < 0.001) was significantly higher in Stop-CB1 and Glu-CB1-RS animals than in CB1-RS animals (Fig. 7B). Glu-CB1-RS animals displayed a nonsignificant trend (p = 0.060) toward a higher freezing response than Stop-CB1 animals. Additionally, there was a significant interaction between genotype and time for the baseline freezing response (F(6,195) = 6.57, p < 0.001), and baseline freezing significantly increased over the extinction days (Fig. 7C). On d10, Glu-CB1-RS animals displayed a significantly higher baseline freezing than both other genotypes (p < 0.001). Furthermore, Stop-CB1 and Glu-CB1-RS animals showed a significantly stronger freezing response after the tone presentation (main genotype effect F(2,65) = 14.35, p < 0.001) than CB1-RS animals (Fig. 7D; p < 0.001). In summary, CB1 receptor on dorsal telencephalic glutamatergic neurons did not rescue the KO phenotype of reduced extinction, but even led to a slightly more sustained conditioned freezing response.
Figure 7.

Insufficiency of dorsal telencephalic glutamatergic CB1 receptor for cued fear extinction and normal amygdalar DSE. A–D, Conditioned freezing in response to repeated presentations of a fear-conditioned auditory stimulus at different days after training. A, Conditioned freezing response over the course of tone presentations per day (d1–d10) analyzed in 20 s bins (after subtraction of baseline freezing on the same day). B, The total 200 s freezing response (after subtraction of baseline freezing) of Glu-CB1-RS and Stop-CB1 mice over the 10 days was higher than that of CB1-RS mice. C, Baseline freezing was similar between the genotypes, except on d10, when Glu-CB1-RS animals had higher levels than both other genotypes. D, Freezing after the tone was terminated was higher for Glu-CB1-RS and Stop-CB1 mice than for CB1-RS animals. ***p < 0.001, **p < 0.01, *p < 0.05; animal numbers are indicated in A. Detailed statistical analysis of behavior in Table 3. E, F, DSI in BLA principal neurons. E shows eIPSCs averaged from recordings in neurons of the three genotypes. The arrow indicates application of a 3 s depolarization step. Original traces illustrate eIPSCs before (a, 5 consecutive traces averaged) and after (b, 3 traces averaged) the 3 s depolarization. Dots indicate stimulation artifacts; dotted lines indicate amplitude of eIPSCs after depolarization. Bar diagrams in F show DSI magnitudes immediately after the depolarization step; numbers indicate recorded cells/animals. Note absence of DSI in Stop-CB1 and Glu-CB1-RS, and rescue of DSI in CB1-RS. G–I, DSE in BLA principal neurons. G shows averaged eEPSCs, with examples of original traces as in E; bar diagrams in H and I illustrate DSE magnitude, with numbers indicating recorded cells/animals. Note the absence of DSE in Stop-CB1 and rescue of DSE in CB1-RS (H), the rescue with extended time course of DSE in Glu-CB1-RS, and blockade of DSE by AM251 (I; imm.: immediately after depolarization; 160 s: at 160 s after depolarization). ***p < 0.001, **p < 0.01, *p < 0.05 versus respective baseline before depolarization.
Table 3.
Statistical analysis of the performance of Glu-CB1-RS, Stop-CB1, and CB1-RS mice in fear extinction
| Repeated-measures ANOVA | Factor | F | p | Post hoc G | p |
|---|---|---|---|---|---|
| For 20 s time bins | |||||
| d1 | G × T | F(10.54,342.61) = 1.40 | 0.175 | Stop-CB1 vs CB1-RS | 0.173 |
| T | F(5.27,342.61) = 26.38 | <0.001 | Stop-CB1 vs Glu-CB1-RS | 0.006 | |
| G | F(2,65) = 14.31 | <0.001 | Glu-CB1-RS vs CB1-RS | <0.001 | |
| d2 | G × T | F(13.486,438.29) = 1.92 | 0.025 | Stop-CB1 vs CB1-RS | 0.004 |
| T | F(6.74,438.29) = 23.26 | <0.001 | Stop-CB1 vs Glu-CB1-RS | 1.000 | |
| G | F(2,65) = 10.26 | <0.001 | Glu-CB1-RS vs CB1-RS | <0.001 | |
| d3 | G × T | F(11.61,377.29) = 2.42 | 0.006 | Stop-CB1 vs CB1-RS | 0.013 |
| T | F(5.80,377.29) = 29.92 | <0.001 | Stop-CB1 vs Glu-CB1-RS | 0.079 | |
| G | F(2,65) = 14.48 | <0.001 | Glu-CB1-RS vs CB1-RS | <0.001 | |
| d10 | G × T | F(11.64,378.13) = 1.96 | 0.028 | Stop-CB1 vs CB1-RS | 0.023 |
| T | F(5.82,378.13) = 19.36 | <0.001 | Stop-CB1 vs Glu-CB1-RS | 0.579 | |
| G | F(2,65) = 9.10 | <0.001 | Glu-CB1-RS vs CB1-RS | <0.001 | |
| For days | |||||
| Tone minus Baseline | G × T | F(6,195) = 1.64 | 0.137 | Stop-CB1 vs CB1-RS | 0.002 |
| T | F(3,195) = 32.29 | <0.001 | Stop-CB1 vs Glu-CB1-RS | 0.060 | |
| G | F(2,65) = 18.70 | <0.001 | Glu-CB1-RS vs CB1-RS | <0.001 | |
| Initial 20 s minus Baseline | G × T | F(6,195) = 0.99 | 0.435 | Stop-CB1 vs CB1-RS | nd |
| T | F(3,195) = 11.02 | <0.001 | Stop-CB1 vs Glu-CB1-RS | nd | |
| G | F(2,65) = 1.74 | 0.184 | Glu-CB1-RS vs CB1-RS | nd | |
| Baseline | G × T | F(6,195) = 6.57 | <0.001 | Stop-CB1 vs CB1-RS | nd |
| T | F(3,195) = 60.50 | <0.001 | Stop-CB1 vs Glu-CB1-RS | nd | |
| G | F(2,65) = 1.91 | 0.156 | Glu-CB1-RS vs CB1-RS | nd | |
| After tone | G × T | F(5.44,176.76) = 1.23 | 0.295 | Stop-CB1 vs CB1-RS | <0.001 |
| T | F(2.72,176.76) = 8.20 | <0.001 | Stop-CB1 vs Glu-CB1-RS | 1.000 | |
| G | F(2,65) = 14.35 | <0.001 | Glu-CB1-RS vs CB1-RS | <0.001 |
G, genotype; T, time; d, day; nd, not determined.
DSI and DSE in the BLA
DSE and DSI were recorded in principal neurons of the BLA (Fig. 7E–I), a crucial brain structure for conditioned fear and extinction (Quirk and Mueller, 2008; Pape and Paré, 2010). In CB1-RS mice, depolarization of the recorded neuron produced a significant reduction of the evoked GABAergic or glutamatergic postsynaptic currents (DSI: t(10) = 5.50, p < 0.001; DSE: t(13) = 5.01, p < 0.001). In Stop-CB1 mice, DSI (t(9) = 1.77, p = 0.111) and DSE (t(9) = 0.97, p = 0.357) were not detectable. In Glu-CB1-RS mice, DSI was absent (t(9) = 0.51, p = 0.622), while DSE was present (t(20) = 2.67, p = 0.015), displaying a much prolonged time course compared with that in CB1-RS (Fig. 7G,I). Averages obtained from evoked EPSCs at 160 s after the depolarization step revealed maintained DSE (Fig. 7I; t(20) = 3.64, p = 0.002). Application of the CB1 receptor antagonist AM251 abolished DSE in BLA neurons in these mice, indicating mediation by the CB1 receptor (Fig. 7I; t(11) = 0.18, p = 0.858 and t(11) = 0.40, p = 0.697, immediately after and at 160 s after the depolarization step, respectively). Increasing the duration of the depolarization step from 3 to 10 s yielded a significantly stronger DSE (t(7) = 2.69, p = 0.031; immediately after the depolarization step) and prolonged time course (t(7) = 4.58, p = 0.003; at 160 s after the depolarization step; Fig. 8A). Two-way ANOVA revealed a significant interaction of depolarization step and time course (time course–duration of depolarization step: F(112,3024) = 1.439, p = 0.002; time course: F(112,3024) = 3.157, p < 0.001, over the whole time course of DSE −40 to 300 s). The prolongation of the time course of DSE in Glu-CB1-RS mice occurred in a regionally specific manner, in that it was observed in BLA neurons, whereas DSE in CeA and CA1 (Fig. 8B,C) displayed a time course similar to that described previously in WT (Ohno-Shosaku et al., 2002; Kamprath et al., 2011).
Discussion
To gain further insights into the modulatory function of the CB1 receptor in neuronal circuits, we generated a conditional rescue mouse line, allowing selective reactivation of the CB1 receptor in specific cell populations. Similar to CB1 receptor-null mutants, the Stop-CB1 animals were shown to lack CB1 receptor protein. Absence of DSI and DSE in hippocampal and amygdalar slices demonstrated the absence of the receptor on presynaptic terminals at the synaptic network level. Behavioral tests with the Stop-CB1 mouse line reproduced several phenotypes described for CB1 receptor-null mutant animals (Marsicano et al., 2002, 2003; Haller et al., 2004). It is of note that, for unknown reasons, transcripts encoded by the CB1 receptor gene were detected by qPCR in the Stop-CB1 animals. However, cannabinoid ligand binding, protein expression, electrophysiological, and behavioral experiments did not reveal any functional CB1 receptor in Stop-CB1 mice, underlying the validity of this mouse model. Animals with rescue of the CB1 receptor throughout the whole organism (CB1-RS) did not differ from WT animals in any of the tested procedures, indicating the validity of this experimental approach. Thus, the newly generated Stop-CB1 mouse line will be a valuable tool to investigate sufficient CB1 receptor function in any given cell type to which Cre recombinase can be delivered specifically.
The present study focused on the rescue of CB1 receptor in dorsal telencephalic glutamatergic neurons by using the NEX-Cre line. As the NEX-Cre mouse line was used for cell-type-specific deletion of the CB1 receptor gene in many previous studies (Monory et al., 2006; Kamprath et al., 2009; Bellocchio et al., 2010; Häring et al., 2011; Dubreucq et al., 2012b; Metna-Laurent et al., 2012), the symmetrical use of the NEX-Cre line in these previous studies and the present study facilitates the comparison between necessary and sufficient functions of CB1 receptor in glutamatergic neurons of the dorsal telencephalon. In future studies, the use of inducible Cre mouse lines and viral Cre delivery will be advantageous to distinguish between developmental and acute effects of cell-type-specific CB1 receptor rescue. This is a relevant issue in the light of our understanding that adult behavioral dysregulation (e.g., anxiety) is strongly influenced by neurodevelopmental deficits, as exemplified for the serotonergic system (Gross et al., 2002).
In the present study, Glu-CB1-RS mice showed a partial rescue of the phenotype observed upon complete CB1 receptor loss in anxiety-like behavioral tests. Thus, CB1 receptor-dependent inhibition of dorsal telencephalic glutamate release appears to play a major role in the complex response to novel aversive environments. Although recent studies using mice with a conditional deletion of the CB1 receptor in dorsal telencephalic glutamatergic neurons (termed “cortical” glutamatergic neurons in these previous publications) reported no robust anxiety-like phenotype (Dubreucq et al., 2012b; Rey et al., 2012), CB1 receptor on these glutamatergic neurons was shown to play a necessary role for the anxiolytic effect of low doses of cannabinoids (Rey et al., 2012). These studies were performed under low aversive experimental conditions to avoid alterations of the basal state of the endocannabinoid system, whereas for the current study, more aversive experimental conditions were chosen to induce activation of the endocannabinoid system (Haller et al., 2004; Ruehle et al., 2012). Our data strongly indicate that CB1 receptor expression in dorsal telencephalic glutamatergic neurons provides substantial amelioration of the anxiogenic effects of global CB1 receptor loss.
Protection against KA-induced epileptiform seizures was reconstituted to WT levels upon rescue of the CB1 receptor on dorsal telencephalic glutamatergic neurons. Previously, loss-of-function and overexpression approaches showed the importance of the CB1 receptor for protection against excitotoxic seizures in glutamatergic terminals of the hippocampus (Monory et al., 2006; Guggenhuber et al., 2010). In human epileptic patients, the CB1 receptor is specifically downregulated on glutamatergic axon terminals in the hippocampus (Ludányi et al., 2008), underlining the importance of the preclinical data. Thus, our findings complete the picture by showing that endogenous levels of CB1 receptor-mediated reduction of excitatory signaling are sufficient for protection against excitotoxic seizures. These data are in good agreement with the findings on synaptic transmission in the underlying brain region (Ben-Ari and Cossart, 2000). In the hippocampus, DSI was completely absent, whereas DSE was fully rescued in Glu-CB1-RS mice. Thus, expression of the CB1 receptor exclusively on dorsal telencephalic glutamatergic neurons is sufficient for full restoration of hippocampal DSE, without the additional requirement of the CB1 receptor on astrocytes (Han et al., 2012) or GABAergic neurons. Altogether, these data demonstrate the sufficient role of CB1 receptor in dorsal telencephalic glutamatergic neurons to control functions that are mainly hippocampus-dependent.
In contrast, rescue of the CB1 receptor on glutamatergic neurons was not sufficient to restore extinction of conditioned fear, a behavior strongly dependent on the BLA circuitry (Pape and Paré, 2010). Previously, loss-of-function approaches showed that CB1 receptor on dorsal telencephalic glutamatergic neurons is necessary for the fear-alleviating effect of endocannabinoids (Kamprath et al., 2009; Dubreucq et al., 2012b), involving a specific balance between “passive” and “active” fear-coping strategies (Metna-Laurent et al., 2012). The unexpected insufficiency of dorsal telencephalic glutamatergic CB1 receptor to rescue extinction might indicate that CB1 receptor is required on both glutamatergic and GABAergic neurons for a coordinated control of both excitatory and inhibitory inputs within fear-regulating circuits. Moreover, in Glu-CB1-RS mice, the CB1 receptor is rescued on all dorsal telencephalic glutamatergic terminals, including those from BLA and PFC. Therefore, local GABAergic networks in neocortical and/or amygdaloid areas expressing CB1 receptor might be important for proper fear extinction. Furthermore, it is known that PFC differentially modulates fear responses during extinction (Quirk and Mueller, 2008), with the prelimbic PFC exciting amygdala output and thus increasing fear, and the infralimbic PFC inhibiting amygdala output and decreasing fear (Sierra-Mercado et al., 2011). This might be relevant, as the approach taken in the present study was unable to differentiate between glutamatergic projections from infralimbic cortex and prelimbic cortex to the amygdala, and thus, it might explain the lack of freezing reduction observed in the Glu-CB1-RS mice. Moreover, fine-tuned regulation of glutamatergic thalamic projections also controls fear extinction (Lee et al., 2012); such projections may also express CB1 receptor presynaptically, and thereby modulate glutamate release (e.g., in the amygdala). Last, mice lacking CB1 receptor on dopamine D1 receptor-expressing neurons, which are found mostly in the striatum, exhibited attenuated within-session extinction (Terzian et al., 2011), whereas mice lacking CB1 receptor on single-minded 1-expressing neurons (hypothalamus and mediobasal amygdala) showed decreased freezing, but increased active coping after fear conditioning (Dubreucq et al., 2012a). Thus, the neuronal circuits involving CB1 receptor-mediated processes are highly complex, and future experiments expanding the neuronal subtypes and brain regions where CB1 receptor function is rescued should allow the reconstitution of proper fear-extinction behavior.
The strong extinction deficit of Glu-CB1-RS mice might also be correlated with modified synaptic transmission within the BLA. In Glu-CB1-RS mice, DSI was completely absent, corroborating the cell-type specificity of the rescue. DSE was present, but displayed an extended time course compared with CB1-RS animals. Endocannabinoid levels were recently shown to determine the time course of the endocannabinoid-mediated retrograde synaptic transmission as pharmacological blockade (Pan et al., 2009), and genetic deletion (Pan et al., 2011) of monoacylglycerol lipase, the primary enzyme degrading the endocannabinoid 2-arachidonoylglycerol, resulted in changes in endocannabinoid-mediated suppression of synaptic transmission in various types of neurons in vitro (decreased magnitude and extended time course of DSE). Thus, additional mechanisms upstream or downstream of CB1 receptor signaling in the BLA might be necessary for appropriate fear extinction and synaptic network function.
The generation of this novel cell-type-specific CB1 receptor rescue mouse line will also promote the understanding of the neuroanatomy of CB1 receptor-containing projections. This is exemplified by the cannabinoid-binding experiments (Fig. 2). Here, for example, dorsal telencephalic glutamatergic rescue did not result in cannabinoid binding in globus pallidus, which indicates that there are no CB1 receptor-expressing projections onto this structure that originate from glutamatergic neurons of the dorsal telencephalon. CB1 receptor binding in the ventromedial hypothalamus, on the other hand, is reconstituted to 20% of WT levels, indicating that 80% of projections arriving in this hypothalamic nucleus do not originate from CB1 receptor-expressing dorsal telencephalic glutamatergic neurons. This fits well with the phenotype observed in mice lacking CB1 receptor only in the dorsal telencephalic glutamatergic neurons (Steiner et al., 2008), where no dysregulation of the hypothalamic–pituitary–adrenal axis was observed, whereas such dysregulation was reported for the complete CB1 receptor KO (Cota et al., 2007).
The use of the Cre/loxP system to delete or re-express genes in specific neuronal subpopulations under the endogenous regulatory elements is a very powerful tool to investigate whether the gene product is not only necessary but also sufficient to provide the full functionality in a specific circuit. Employing the rescue approach, we have shown that the CB1 receptor in dorsal telencephalic glutamatergic neurons plays a sufficient role to control neuronal functions that are in large part hippocampus-dependent, while it is insufficient for proper amygdala functions. Our experiments constitute an essential step toward the dissection of CB1 receptor function in the framework of neuronal circuits.
Footnotes
This work was supported by the Deutsche Forschungsgemeinschaft SFB-TRR 58 (subproject A04 to B.L. and H.-C.P), and by INSERM (G.M.), Region Aquitaine (G.M.), European Research Council (ENDOFOOD, ERC–2010–StG–260515 to G.M.), and a Max-Planck-Research Award (2007, to H.C.P.). We thank Stefano Gaburro for valuable discussions, and Jörg Lesting for help with statistical data analysis. We thank Andrea Conrad, Ruth Jelinek, Martin Purrio, Barbara Fackelmeier, Anisa Kosan, Claudia Schwitter (Mainz laboratory), Svetlana Kiesling, Petra Berenbock, Elke Naß, and Angelika Klinge (Muenster laboratory) for excellent technical support.
The authors declare no competing financial interests.
References
- Balthasar N, Dalgaard LT, Lee CE, Yu J, Funahashi H, Williams T, Ferreira M, Tang V, McGovern RA, Kenny CD, Christiansen LM, Edelstein E, Choi B, Boss O, Aschkenasi C, Zhang CY, Mountjoy K, Kishi T, Elmquist JK, Lowell BB. Divergence of melanocortin pathways in the control of food intake and energy expenditure. Cell. 2005;123:493–505. doi: 10.1016/j.cell.2005.08.035. [DOI] [PubMed] [Google Scholar]
- Bellocchio L, Lafenêtre P, Cannich A, Cota D, Puente N, Grandes P, Chaouloff F, Piazza PV, Marsicano G. Bimodal control of stimulated food intake by the endocannabinoid system. Nat Neurosci. 2010;13:281–283. doi: 10.1038/nn.2494. [DOI] [PubMed] [Google Scholar]
- Ben-Ari Y, Cossart R. Kainate, a double agent that generates seizures: two decades of progress. Trends Neurosci. 2000;23:580–587. doi: 10.1016/S0166-2236(00)01659-3. [DOI] [PubMed] [Google Scholar]
- Chiu CQ, Castillo PE. Input-specific plasticity at excitatory synapses mediated by endocannabinoids in the dentate gyrus. Neuropharmacology. 2008;54:68–78. doi: 10.1016/j.neuropharm.2007.06.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cota D, Steiner MA, Marsicano G, Cervino C, Herman JP, Grübler Y, Stalla J, Pasquali R, Lutz B, Stalla GK, Pagotto U. Requirement of cannabinoid receptor type 1 for the basal modulation of hypothalamic-pituitary-adrenal axis function. Endocrinology. 2007;148:1574–1581. doi: 10.1210/en.2005-1649. [DOI] [PubMed] [Google Scholar]
- Dubreucq S, Kambire S, Conforzi M, Metna-Laurent M, Cannich A, Soria-Gomez E, Richard E, Marsicano G, Chaouloff F. Cannabinoid type 1 receptors located on single-minded 1-expressing neurons control emotional behaviors. Neuroscience. 2012a;204:230–244. doi: 10.1016/j.neuroscience.2011.08.049. [DOI] [PubMed] [Google Scholar]
- Dubreucq S, Matias I, Cardinal P, Häring M, Lutz B, Marsicano G, Chaouloff F. Genetic dissection of the role of cannabinoid type-1 receptors in the emotional consequences of repeated social stress in mice. Neuropsychopharmacology. 2012b;37:1885–1900. doi: 10.1038/npp.2012.36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goebbels S, Bormuth I, Bode U, Hermanson O, Schwab MH, Nave KA. Genetic targeting of principal neurons in neocortex and hippocampus of NEX-Cre mice. Genesis. 2006;44:611–621. doi: 10.1002/dvg.20256. [DOI] [PubMed] [Google Scholar]
- Gross C, Zhuang X, Stark K, Ramboz S, Oosting R, Kirby L, Santarelli L, Beck S, Hen R. Serotonin 1A receptor acts during development to establish normal anxiety-like behaviour in the adult. Nature. 2002;416:396–400. doi: 10.1038/416396a. [DOI] [PubMed] [Google Scholar]
- Guggenhuber S, Monory K, Lutz B, Klugmann M. AAV vector-mediated overexpression of CB1 cannabinoid receptor in pyramidal neurons of the hippocampus protects against seizure-induced excitoxicity. PLoS One. 2010;5:e15707. doi: 10.1371/journal.pone.0015707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haller J, Varga B, Ledent C, Barna I, Freund TF. Context-dependent effects of CB1 cannabinoid gene disruption on anxiety-like and social behaviour in mice. Eur J Neurosci. 2004;19:1906–1912. doi: 10.1111/j.1460-9568.2004.03293.x. [DOI] [PubMed] [Google Scholar]
- Han J, Kesner P, Metna-Laurent M, Duan T, Xu L, Georges F, Koehl M, Abrous DN, Mendizabal-Zubiaga J, Grandes P, Liu Q, Bai G, Wang W, Xiong L, Ren W, Marsicano G, Zhang X. Acute cannabinoids impair working memory through astroglial CB1 receptor modulation of hippocampal LTD. Cell. 2012;148:1039–1050. doi: 10.1016/j.cell.2012.01.037. [DOI] [PubMed] [Google Scholar]
- Häring M, Kaiser N, Monory K, Lutz B. Circuit specific functions of cannabinoid CB1 receptor in the balance of investigatory drive and exploration. PLoS One. 2011;6:e26617. doi: 10.1371/journal.pone.0026617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herkenham M, Lynn AB, de Costa BR, Richfield EK. Neuronal localization of cannabinoid receptors in the basal ganglia of the rat. Brain Res. 1991;547:267–274. doi: 10.1016/0006-8993(91)90970-7. [DOI] [PubMed] [Google Scholar]
- Kamprath K, Wotjak CT. Nonassociative learning processes determine expression and extinction of conditioned fear in mice. Learn Mem. 2004;11:770–786. doi: 10.1101/lm.86104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamprath K, Plendl W, Marsicano G, Deussing JM, Wurst W, Lutz B, Wotjak CT. Endocannabinoids mediate acute fear adaptation via glutamatergic neurons independently of corticotropin-releasing hormone signaling. Genes Brain Behav. 2009;8:203–211. doi: 10.1111/j.1601-183X.2008.00463.x. [DOI] [PubMed] [Google Scholar]
- Kamprath K, Romo-Parra H, Häring M, Gaburro S, Doengi M, Lutz B, Pape HC. Short-term adaptation of conditioned fear responses through endocannabinoid signaling in the central amygdala. Neuropsychopharmacology. 2011;36:652–663. doi: 10.1038/npp.2010.196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kano M, Ohno-Shosaku T, Hashimotodani Y, Uchigashima M, Watanabe M. Endocannabinoid-mediated control of synaptic transmission. Physiol Rev. 2009;89:309–380. doi: 10.1152/physrev.00019.2008. [DOI] [PubMed] [Google Scholar]
- Katona I, Freund TF. Multiple functions of endocannabinoid signaling in the brain. Annu Rev Neurosci. 2012;35:529–558. doi: 10.1146/annurev-neuro-062111-150420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawamura Y, Fukaya M, Maejima T, Yoshida T, Miura E, Watanabe M, Ohno-Shosaku T, Kano M. The CB1 cannabinoid receptor is the major cannabinoid receptor at excitatory presynaptic sites in the hippocampus and cerebellum. J Neurosci. 2006;26:2991–3001. doi: 10.1523/JNEUROSCI.4872-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lafenêtre P, Chaouloff F, Marsicano G. Bidirectional regulation of novelty-induced behavioral inhibition by the endocannabinoid system. Neuropharmacology. 2009;57:715–721. doi: 10.1016/j.neuropharm.2009.07.014. [DOI] [PubMed] [Google Scholar]
- Lakso M, Pichel JG, Gorman JR, Sauer B, Okamoto Y, Lee E, Alt FW, Westphal H. Efficient in vivo manipulation of mouse genomic sequences at the zygote stage. Proc Natl Acad Sci U S A. 1996;93:5860–5865. doi: 10.1073/pnas.93.12.5860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S, Ahmed T, Lee S, Kim H, Choi S, Kim DS, Kim SJ, Cho J, Shin HS. Bidirectional modulation of fear extinction by mediodorsal thalamic firing in mice. Nat Neurosci. 2012;15:308–314. doi: 10.1038/nn.2999. [DOI] [PubMed] [Google Scholar]
- Lourenço J, Cannich A, Carta M, Coussen F, Mulle C, Marsicano G. Synaptic activation of kainate receptors gates presynaptic CB1 signaling at GABAergic synapses. Nat Neurosci. 2010;13:197–204. doi: 10.1038/nn.2481. [DOI] [PubMed] [Google Scholar]
- Ludányi A, Eross L, Czirják S, Vajda J, Halász P, Watanabe M, Palkovits M, Maglóczky Z, Freund TF, Katona I. Downregulation of the CB1 cannabinoid receptor and related molecular elements of the endocannabinoid system in epileptic human hippocampus. J Neurosci. 2008;28:2976–2990. doi: 10.1523/JNEUROSCI.4465-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marsicano G, Kuner R. Anatomical distribution of receptors, ligands and enzymes in the brain and in the spinal cord: circuitries and neurochemistry. In: Köfalvi A, editor. Cannabinoids and the brain. Boston: Springer US; 2008. pp. 161–201. [Google Scholar]
- Marsicano G, Lutz B. Expression of the cannabinoid receptor CB1 in distinct neuronal subpopulations in the adult mouse forebrain. Eur J Neurosci. 1999;11:4213–4225. doi: 10.1046/j.1460-9568.1999.00847.x. [DOI] [PubMed] [Google Scholar]
- Marsicano G, Wotjak CT, Azad SC, Bisogno T, Rammes G, Cascio MG, Hermann H, Tang J, Hofmann C, Zieglgänsberger W, Di Marzo V, Lutz B. The endogenous cannabinoid system controls extinction of aversive memories. Nature. 2002;418:530–534. doi: 10.1038/nature00839. [DOI] [PubMed] [Google Scholar]
- Marsicano G, Goodenough S, Monory K, Hermann H, Eder M, Cannich A, Azad SC, Cascio MG, Gutiérrez SO, van der Stelt M, López-Rodriguez ML, Casanova E, Schütz G, Zieglgänsberger W, Di Marzo V, Behl C, Lutz B. CB1 cannabinoid receptors and on-demand defense against excitotoxicity. Science. 2003;302:84–88. doi: 10.1126/science.1088208. [DOI] [PubMed] [Google Scholar]
- Metna-Laurent M, Soria-Gómez E, Verrier D, Conforzi M, Jégo P, Lafenêtre P, Marsicano G. Bimodal control of fear-coping strategies by CB1 cannabinoid receptors. J Neurosci. 2012;32:7109–7118. doi: 10.1523/JNEUROSCI.1054-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monory K, Massa F, Egertová M, Eder M, Blaudzun H, Westenbroek R, Kelsch W, Jacob W, Marsch R, Ekker M, Long J, Rubenstein JL, Goebbels S, Nave KA, During M, Klugmann M, Wölfel B, Dodt HU, Zieglgänsberger W, Wotjak CT, et al. The endocannabinoid system controls key epileptogenic circuits in the hippocampus. Neuron. 2006;51:455–466. doi: 10.1016/j.neuron.2006.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagy A. Cre recombinase: the universal reagent for genome tailoring. Genesis. 2000;26:99–109. doi: 10.1002/(SICI)1526-968X(200002)26:2<99::AID-GENE1>3.3.CO;2-2. [DOI] [PubMed] [Google Scholar]
- Ohno-Shosaku T, Tsubokawa H, Mizushima I, Yoneda N, Zimmer A, Kano M. Presynaptic cannabinoid sensitivity is a major determinant of depolarization-induced retrograde suppression at hippocampal synapses. J Neurosci. 2002;22:3864–3872. doi: 10.1523/JNEUROSCI.22-10-03864.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan B, Wang W, Long JZ, Sun D, Hillard CJ, Cravatt BF, Liu QS. Blockade of 2-arachidonoylglycerol hydrolysis by selective monoacylglycerol lipase inhibitor 4-nitrophenyl 4-(dibenzo[d][1,3]dioxol-5-yl(hydroxy)methyl)piperidine-1-carboxylate (JZL184) enhances retrograde endocannabinoid signaling. J Pharmacol Exp Ther. 2009;331:591–597. doi: 10.1124/jpet.109.158162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan B, Wang W, Zhong P, Blankman JL, Cravatt BF, Liu QS. Alterations of endocannabinoid signaling, synaptic plasticity, learning, and memory in monoacylglycerol lipase knock-out mice. J Neurosci. 2011;31:13420–13430. doi: 10.1523/JNEUROSCI.2075-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pape HC, Paré D. Plastic synaptic networks of the amygdala for the acquisition, expression, and extinction of conditioned fear. Physiol Rev. 2010;90:419–463. doi: 10.1152/physrev.00037.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paxinos G, Franklin KBJ. The mouse brain in stereotaxic coordinates, third edition: the coronal plates and diagrams. New York: Academic; 2008. [Google Scholar]
- Pfaffl MW, Horgan GW, Dempfle L. Relative expression software tool (REST©) for group-wise comparison and statistical analysis of relative expression results in real-time PCR. Nucleic Acids Res. 2002;30:e36. doi: 10.1093/nar/30.9.e36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quirk GJ, Mueller D. Neural mechanisms of extinction learning and retrieval. Neuropsychopharmacology. 2008;33:56–72. doi: 10.1038/sj.npp.1301555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rey AA, Purrio M, Viveros MP, Lutz B. Biphasic effects of cannabinoids in anxiety responses: CB1 and GABAB receptors in the balance of GABAergic and glutamatergic neurotransmission. Neuropsychopharmacology. 2012;37:2624–2634. doi: 10.1038/npp.2012.123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rideout WM, 3rd, Wakayama T, Wutz A, Eggan K, Jackson-Grusby L, Dausman J, Yanagimachi R, Jaenisch R. Generation of mice from wild-type and targeted ES cells by nuclear cloning. Nat Genet. 2000;24:109–110. doi: 10.1038/72753. [DOI] [PubMed] [Google Scholar]
- Ruehle S, Rey AA, Remmers F, Lutz B. The endocannabinoid system in anxiety, fear memory and habituation. J Psychopharmacol. 2012;26:23–39. doi: 10.1177/0269881111408958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schauwecker PE, Steward O. Genetic determinants of susceptibility to excitotoxic cell death: implications for gene targeting approaches. Proc Natl Acad Sci U S A. 1997;94:4103–4108. doi: 10.1073/pnas.94.8.4103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwab MH, Bartholomae A, Heimrich B, Feldmeyer D, Druffel-Augustin S, Goebbels S, Naya FJ, Zhao S, Frotscher M, Tsai MJ, Nave KA. Neuronal basic helix-loop-helix proteins (NEX and BETA2/Neuro D) regulate terminal granule cell differentiation in the hippocampus. J Neurosci. 2000;20:3714–3724. doi: 10.1523/JNEUROSCI.20-10-03714.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Self DW. Molecular and genetic approaches for behavioral analysis of protein function. Biol Psychiatry. 2005;57:1479–1484. doi: 10.1016/j.biopsych.2005.03.042. [DOI] [PubMed] [Google Scholar]
- Sierra-Mercado D, Padilla-Coreano N, Quirk GJ. Dissociable roles of prelimbic and infralimbic cortices, ventral hippocampus, and basolateral amygdala in the expression and extinction of conditioned fear. Neuropsychopharmacology. 2011;36:529–538. doi: 10.1038/npp.2010.184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steindel F, Lerner R, Häring M, Ruehle S, Marsicano G, Lutz B, Monory K. Neuron-type specific cannabinoid-mediated G protein signalling in mouse hippocampus. J Neurochem. 2013;124:795–807. doi: 10.1111/jnc.12137. [DOI] [PubMed] [Google Scholar]
- Steiner MA, Marsicano G, Wotjak CT, Lutz B. Conditional cannabinoid receptor type 1 mutants reveal neuron subpopulation-specific effects on behavioral and neuroendocrine stress responses. Psychoneuroendocrinology. 2008;33:1165–1170. doi: 10.1016/j.psyneuen.2008.06.004. [DOI] [PubMed] [Google Scholar]
- Terzian AL, Drago F, Wotjak CT, Micale V. The dopamine and cannabinoid interaction in the modulation of emotions and cognition: assessing the role of cannabinoid CB1 receptor in neurons expressing dopamine D1 receptors. Front Behav Neurosci. 2011;5:49. doi: 10.3389/fnbeh.2011.00049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torres R, Kühn R. Laboratory protocols for conditional gene targeting. New York: Oxford UP; 1997. [Google Scholar]
- Walf AA, Frye CA. The use of the elevated plus maze as an assay of anxiety-related behavior in rodents. Nat Protoc. 2007;2:322–328. doi: 10.1038/nprot.2007.44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson RI, Nicoll RA. Endogenous cannabinoids mediate retrograde signalling at hippocampal synapses. Nature. 2001;410:588–592. doi: 10.1038/35069076. [DOI] [PubMed] [Google Scholar]
- Yizhar O, Fenno LE, Prigge M, Schneider F, Davidson TJ, O'Shea DJ, Sohal VS, Goshen I, Finkelstein J, Paz JT, Stehfest K, Fudim R, Ramakrishnan C, Huguenard JR, Hegemann P, Deisseroth K. Neocortical excitation/inhibition balance in information processing and social dysfunction. Nature. 2011;477:171–178. doi: 10.1038/nature10360. [DOI] [PMC free article] [PubMed] [Google Scholar]