

Abstract
The neuronal long isoform of Fas Apoptotic Inhibitory Molecule (FAIM-L) protects from death receptor (DR)-induced apoptosis, yet its mechanism of protection remains unknown. Here, we show that FAIM-L protects rat neuronal Type II cells from Fas-induced apoptosis. XIAP has previously emerged as a molecular discriminator that is upregulated in Type II and downregulated in Type I apoptotic signaling. We demonstrate that FAIM-L requires sustained endogenous levels of XIAP to protect Type II cells as well as murine cortical neurons from Fas-induced apoptosis. FAIM-L interacts with the BIR2 domain of XIAP through an IAP-binding motif, the mutation of which impairs the antiapoptotic function of FAIM-L. Finally, we report that FAIM-L inhibits XIAP auto-ubiquitinylation and maintains its stability, thus conferring protection from apoptosis. Our results bring new understanding of the regulation of endogenous XIAP by a DR antagonist, pointing out at FAIM-L as a promising therapeutic tool for protection from apoptosis in pathological situations where XIAP levels are decreased.
Introduction
Appropriate control of death receptor (DR)-induced caspase activation is essential for neuronal survival (Choi and Benveniste, 2004). The activation of DRs induces the formation of the death-inducing signaling complex (DISC), thereby triggering apoptosis (Wilson et al., 2009). Activation of Fas DR either leads to direct caspase-3 activation in Type I cells or implicates the mitochondria through caspase-8-mediated cleavage of Bid in Type II cells (Scaffidi et al., 1998).
Antiapoptotic members of the Bcl-2 family play an important and early role in neuronal protection from apoptosis (Motoyama et al., 1995; Michaelidis et al., 1996; Berman et al., 2009). Inhibitors of apoptosis proteins (IAPs) also protect from DR activation and contain three baculovirus inhibitory repeat (BIR) domains, a UBA domain that binds to poly-ubiquitin chains, and a C-terminal Really Interesting New Gene (RING) domain that bears an E3-ligase activity (Darding and Meier, 2012). XIAP is the most potent caspase inhibitor in vitro (Eckelman and Salvesen, 2006; Srinivasula and Ashwell, 2008) and plays an important role in preventing Fas-induced apoptosis in hepatocytes (Jost et al., 2009; Ferreira et al., 2012). XIAP BIR2 domain binds to and inhibits caspase-3 and caspase-7 (Riedl et al., 2001; Silke et al., 2001; Scott et al., 2005), whereas XIAP BIR3 domain binds caspase-9 (Shiozaki et al., 2003). Also, XIAP undergoes auto-ubiquitinylation and proteasome-mediated degradation in response to apoptotic stimuli (Yang et al., 2000). XIAP deregulation has been shown extensively in peripheral nervous system-related diseases (Garrity-Moses et al., 2006).
A group of proteins designated DR antagonists have been described and are promising for the protection from neuronal cell death: c-FLIP (Irmler et al., 1997), NMP35/Lifeguard/FAIM2 (Somia et al., 1999; Fernández et al., 2007), and Fas Apoptotic Inhibitory Molecule (FAIM) (Schneider et al., 1999; Zhong et al., 2001). Nevertheless, with the exception of c-FLIP (Taoufik et al., 2007; Moubarak et al., 2010), the mechanism of action of other DR antagonists and their relevance in the nervous system remain to be clarified.
FAIM is a DR antagonist with two isoforms that display very diverse roles in the nervous system. We have reported that the short form FAIM-S is necessary for neurite outgrowth through the activation of the MAPK/ERK and NF-κB pathways (Sole et al., 2004). Whereas FAIM-S is ubiquitous and protects B lymphocytes but not neurons from Fas activation, the long form FAIM-L is exclusively expressed in neurons and protects them from DR activation (Segura et al., 2007). FAIM-L also controls Fas-induced apoptosis in midbrain dopaminergic neurons after trophic factor deprivation (Yu et al., 2008), underlining the potential interest of FAIM-L for neuroprotection in models of Parkinson disease. However, the detailed molecular mechanism through which FAIM-L protects from DR-induced apoptosis is currently unknown.
Here, we provide compelling evidence that FAIM-L interacts with XIAP through an IAP-binding motif (IBM) in the N terminus of FAIM-L, thereby impairing XIAP auto-ubiquitinylation and degradation by the proteasome. In addition, we demonstrate that, upon Fas activation, sustained XIAP protein levels are essential for FAIM-L-mediated protection from apoptosis. Our findings provide the first evidence of a neuron-specific molecule that is able to regulate XIAP auto-ubiquitinylation and degradation.
Materials and Methods
Reagents.
Recombinant human sFasL (superFasL, Enzo Life Sciences) was used at a concentration of 100 ng/ml. Fluorogenic caspase substrate Ac-DEVD-afc was purchased from Calbiochem/Merck Biosciences. Unless otherwise specified, all biochemical reagents were purchased from Sigma-Aldrich.
Cell culture.
Rat pheochromocytoma PC12 cells were grown on culture plates coated with Type I collagen (66.4 μg/ml; BD Biosciences) in DMEM supplemented with 6% heat-inactivated fetal bovine serum (FBS) and 6% heat-inactivated horse serum (Invitrogen), 10 mm HEPES, 20 U/ml penicillin, and 20 μg/ml streptomycin. HEK293T and HEK293 cells were grown in DMEM supplemented with 10% heat-inactivated FBS (Invitrogen), 20 U/ml penicillin, and 20 μg/ml streptomycin. Cultures were maintained at 37°C in a humidified atmosphere of 95% air and 5% CO2.
Primary cortical neurons culture.
Female C57BL/6 mice were killed and manipulated following the experimental protocol approved by the Vall d'Hebron Institutional Review Board. Embryonic cerebral cortices were dissected from mouse embryos at day 16 (E16). Cells were counted and resuspended in DMEM with glutamine supplemented with 5% heat-inactivated FBS and 5% heat-inactivated fetal horse serum, 20 U/ml penicillin, and 20 μg/ml streptomycin. Cells were plated in 25 mg/ml poly-l-lysine-coated plates at a density of 1.6 × 105 cells/cm2. Transduction with short hairpin lentiviruses was performed while seeding. Four hours after seeding and transduction, the medium was replaced with serum-free Neurobasal medium supplemented with 2% B27 (Invitrogen), 1% N2 (Invitrogen), 20 U/ml penicillin, 20 μg/ml streptomycin, and 0.5 mm glutamine. Culture medium was partially replaced every 3–4 d with fresh Neurobasal supplemented medium. When indicated, subsequent transductions were performed 3 d after prior transduction. Cell culture plates were kept at 37°C in a humidified incubator with 5%CO2/95%air.
Plasmids.
Fas, 3xHA-c-FLIP-L, 3xHA-Bcl-xL, 6xMyc-XIAP, 3xFLAG-FAIM-L, and 3xHA-FAIM-L cDNAs were expressed under the control of a cytomegalovirus constitutive promoter in the pcDNA3 expression vector (Invitrogen). The 6xHis-tagged ubiquitin plasmid, pMT107, was a kind gift from Dr. Dirk Bohmann (University of Rochester Medical Center) (Treier et al., 1994). The C-terminal FLAG-tagged XIAP (wild-type, deletion, and point mutants) cloned in the pEF plasmid was a kind gift from Dr. John Silke (Walter and Eliza Institute, Melbourne, Australia) and were described by Silke et al. (2002).
For RNA interference (RNAi) experiments, constructs were generated in the pSUPER.retro.puro plasmid (OligoEngine) using specific oligonucleotides against rat XIAP (sequence data available from GeneBank under accession number NM_022231) indicated by capital letters, as follows: shXIAP R1, forward, gatccccAGAATCCTATGGTGCAAGAttcaagagaTCTTGCACCATAGGATTCtttttt;
reverse, agctaaaaaAGAATCCTATGGTGCAAGAtctcttgaaTCTTGCACCATAGGATTCggg; or
shXIAP R2, forward, gatccccGTAGATAGATGGCAGTATGttcaagagaCATACTGCCATCTATCTACtttttt;
reverse, agctaaaaaGTAGATAGATGGCAGTATGtctcttgaaCATACTGCCATCTATCTACggg. A scrambled sequence was used as a control by the following oligonucleotides: Scr, forward, gatccccAGAACACGACGGAACAAGAttcaagagaTCTTGTTCCGTCGTGTTCTttttt; reverse, agctaaaaaAGAACACGACGGAACAAGAtctcttgaaTCTTGTTCCGTCGTGTTCTggg. Oligonucleotides were obtained from Sigma and were cloned between BglII/HindIII sites of pSUPER.retro.puro plasmid. Lentiviral constructs were achieved by digesting EcoR1-ClaI sites from pSUPER-sh to replace H1 promoter with H1-shRNA cassette in pLVTHM plasmid. For FAIM-L knockdown experiments, we used the lentiviral plasmid pLVTHM-shFAIM-L, which targets specifically the long form and not the short form of FAIM and has been previously validated by Segura et al. (2007).
Site-directed mutagenesis.
The XIAP E3-ubiquitin ligase mutant H467A previously described (Yang et al., 2000) has been cloned in by site-directed mutagenesis in the pcDNA3–6xMyc-XIAP plasmid using the PfuI polymerase PCR (Stratagene) and the following primers carrying the mutation and a SacI recognition site for cribling of positive colonies: forward, 5′-TATCGTTTTTGTTCCTTGTGGAGCTCTAGTCACTT-3′; reverse, 5′-AAGTGACTAGAGCTCCACAAGGAACAAAAACGATA-3′. The FAIM-L mutant (MutFAIM-L) where the first alanine was replaced by a glycine was cloned using the same protocol and the following primers carrying the mutation and a BamHI recognition site for cribling of positive colonies: forward, 5′-ACCGAATTCATGGGATCCGGAGAT-3′; reverse, 5′-ATCTCCGGATCCCATGAATTCGGT-3′. All constructs were verified by digestion and sequencing.
Lentiviral production.
Lentiviruses were produced as described previously by Segura et al. (2007).
Cell transfection and infection.
HEK293 or PC12 cells were transfected with the desired expression plasmid using Lipofectamine 2000 (Invitrogen), following the manufacturer's instructions. HEK293T were transfected using the calcium phosphate method. The total amount of transfected DNA was kept constant in all experiments by adding empty vector.
For lentiviral-based knockdown experiments, PC12 cells were seeded in 24-multiwell plates at a density of 2 × 104 cells/well. Titrated lentiviruses were added to the medium. After 4 h, the medium was changed and the transduction efficiency monitored by direct counting of GFP-positive cells. The percentage of infection reached 95%. Lentiviruses efficiently induced XIAP or FAIM-L knockdown after 3 d of transduction. Knockdown efficiency was checked by RT-PCR or Western blotting, respectively, prior cell death induction.
RT-PCR.
A total of 1 μg of total RNA was converted to first-strand cDNA using the MLV Reverse Transcriptase (Promega Biotech). The resulting cDNA was subjected to PCR analysis. PCR cycling parameters were as follows: 94°C for 2 min for one cycle, followed by 94°C for 30 s, 60°C for 30 s, and 72°C for 60 s for 30 cycles; 72°C for 5 min with xiap and the housekeeping ribosomal protein L27 primers. The PCR products were run on an agarose gel that was stained with SYBR Safe (Invitrogen). The sequences of specific primers are as follows: xiap, forward, 5′-TGAAGAAGCCAGACCGAAGA-3′; reverse, 5′-TGACTTGACTCATCCTGCGA-3′; and L27, forward, 5′-AGCTGTCATCGTGAAGAA-3′; reverse, 5′-CTTGGCGATCTTCTTCTTGCC-3′.
Cytochrome c and Smac release from the mitochondria.
Cytosolic purification was performed as previously described (Moubarak et al., 2007). Briefly, 2 × 106 PC12 cells were resuspended in a buffer containing 220 mm mannitol, 70 mm sucrose, 50 mm HEPES-KOH, pH 7.2, 10 mm KCl, 5 mm EGTA, 2 mm MgCl2, and 0.025% digitonin, and kept on ice for 5 min. Lysed cells were centrifuged (16,000 × g; 5 min; 4°C), and the supernatant was retained as the cytosolic fraction. The release of cytochrome c and Smac from the mitochondria to the cytosol was assessed by Western blot.
Western blot.
Cells were harvested and rinsed once with ice-cold PBS 1×, pH 7.2 and, unless otherwise indicated, lysed in immunoprecipitation lysis buffer (20 mm Tris, pH 7.4, 140 mm NaCl, 10% glycerol, 2 mm EDTA, 1 mm EGTA, 1% Igepal CA-630), supplemented with 1× EDTA-free Complete protease inhibitor mixture (Roche), before centrifugation at 16 000 × g, 4°C and harvesting supernatants. Protein concentration was quantified by a modified Lowry assay (DC protein assay; Bio-Rad). The cell lysates obtained were resolved by SDS-PAGE and transferred onto PVDF Immobilon-P membranes (Millipore). After blocking with TBS 1×-0.1% Tween-20 containing 5% nonfat dry milk for 1 h at room temperature, membranes were probed with the appropriate primary antibodies, before incubation for 1 h with the appropriate specific peroxidase-conjugated secondary antibody. Membranes were developed using the EZ-ECL chemiluminescence detection kit (Biological Industries) or SuperSignal Dura (Pierce/Thermo Fisher Scientific). The primary antibodies used were the following: anti-XIAP (BD Biosciences), anti-FAIM-L (in house), anti-tubulin (Sigma), anti-Bcl-xL and anti-cytochrome c (BD Biosciences), anti-HA (Roche), anti-Smac (Prosci), anti-Fas (Santa Cruz Biotechnology), anti-caspase-8, anti-rat or anti-human caspase-9, anti-caspase-3, and anti-PARP (Cell Signaling Technology). In some cases, equal loading was verified by Naphtol Blue staining of the PVDF membrane.
Immunoprecipitation.
Samples were lysed in immunoprecipitation lysis buffer as described above and lysates were quantified by a modified Lowry assay (DC protein assay; Bio-Rad). A total of 1 mg of total protein was adjusted with immunoprecipitation buffer to achieve a concentration of 1 mg/ml. A total of 40 μl of anti-FLAG M2-agarose-coupled antibody were added to each sample and incubated overnight at 4°C in an orbital shaker. Then, beads were washed five times with immunoprecipitation buffer and eluted for 30 min at 4°C with 80 μl of TBS1× containing 150 ng/ml of 3xFLAG competitor peptide. After a short spin, supernatants were carefully taken, Laemmli buffer was added, and SDS-PAGE was performed. Alternatively, for semiendogenous and endogenous immunoprecipitations, 1 mg of cleared supernatant from PC12 cells was subjected to immunoprecipitation with 10 μl of an anti-FAIM-L antibody or a control rabbit IgG, such as the anti-Myc antibody (Santa Cruz Biotechnology) overnight at 4°C. Immunocomplexes were collected with protein G-Sepharose by orbital shaking for overnight at 4°C and washed five times with immunoprecipitation lysis buffer. Beads were suspended in Laemmli buffer and boiled; then samples were resolved by SDS-PAGE. For Western blot of endogenous immunoprecipitation, the secondary anti-rabbit used was purchased from eBioscience.
Detection of Bax conformational change by immunoprecipitation.
PC12 cells were transfected with the indicated plasmids and then treated 48 h after transfection as indicated. At the end of the incubation, cells were harvested, washed once with PBS, and lysed in 1 ml of lysis buffer (10 mm HEPES, pH 7.4, 125 mm NaCl, and 1% CHAPS), supplemented with 1× EDTA-free Complete protease inhibitor mixture (Roche) and incubated for 30 min on ice. Cells were then subjected to 3 freeze-and-thaw cycles (dry ice and ethanol for 3 min, 37°C for 3 min, and vortex for 15 s) and centrifuged at 16,000 × g for 1 h at 4°C. Protein concentration of the supernatants was measured with the modified Lowry assay, and aliquots of 500 μg of each sample were brought to a volume of 500 μl with lysis buffer. Samples were incubated with 25 μl of protein G agarose beads for 1 h at 4°C to reduce the nonspecific binding to the beads and were subsequently spun down at 12,000 × g for 1 min to remove the beads. The supernatants were incubated with 2 μg of the conformation-specific anti-Bax monoclonal antibody (clone 6A7; BD Biosciences) for 2 h at 4°C, and then 25 μl of protein G agarose beads were added to the reactions and incubated at 4°C for an additional hour. Beads were recovered after short spinning, washed 6 times with lysis buffer, and boiled in 25 μl of Laemmli buffer 2×. The samples were loaded on 14% acrylamide gels and analyzed by immunoblot using a rabbit polyclonal anti-Bax antibody (Millipore) as primary antibody.
Cell viability assays.
For apoptotic nuclear morphology, cells were plated in 24-well plates and treated as indicated. Cells were then fixed with 2% paraformaldehyde and stained with 0.05 μg/ml of Hoechst 33258 for 30 min at room temperature, in a buffer composed of 2% paraformaldehyde and 1% of Igepal CA-630. Condensed or fragmented nuclei were counted as dead cells as described previously (Yuste et al., 2001). The quantification of cell death by chromatin condensation was performed in blind testing, counting at least 300 cells for each data point, and was repeated at least three times in independent experiments.
Caspase activity.
After the indicated treatments, cells were rinsed once with PBS and resuspended in lysis buffer containing 20 mm HEPES/NaOH, pH 7.2, 10% sucrose, 150 mm NaCl, 10 mm DTT, 5 mm EDTA, 1% Igepal CA-630, 0.1% CHAPS, and 1× EDTA-free Complete protease inhibitor mixture (Roche). Lysates were cleared by centrifugation at 16 000 × g for 5 min, and supernatants were quantified by the Bradford method (Bio-Rad). Assays were performed in triplicate using 25 μg of protein in the same specific lysis buffer supplemented with 25 μm of the fluorogenic substrate Ac-DEVD-afc. Plates were read in a fluorimeter using a 360 nm (40 nm bandwidth) excitation filter and a 530 nm (25 nm bandwidth) emission filter.
Detection of ubiquitinylated proteins.
Ubiquitinylated proteins were detected using the 6xHis-ubiquitin assay for mammalian cell protocol of the Tansey laboratory (www.tanseylab.com). HEK293T cells were transfected with the plasmid(s) of interest, along with the plasmid pMT107, which encodes 6xHis-tagged human ubiquitin. Cells were harvested 48 h after transfection using ice-cold PBS 1×. A small fraction of each condition was lysed in Laemmli buffer 2× and boiled at 95°C for 10 min and was later loaded by SDS-PAGE as lysates. The rest of the samples were lysed in buffer A (6 m guanidine-HCl, 0.1 m Na2HPO4/NaH2PO4, 10 mm imidazole, pH 8.0) and sonicated. Then His-tagged proteins were purified using Ni-NTA-agarose (Invitrogen), by rotation using an orbital shaker for 3 h at room temperature. Samples were spun and washed twice with buffer A, twice in buffer A/TI (1 volume of buffer A for 3 volumes of buffer TI), and once more in buffer TI (25 mm Tris-HCl, 20 mm imidazole, pH 6.8) before elution in 100 μl of Laemmli buffer 2× supplemented with 250 mm imidazole and boiling for 10 min at 95°C. Pre- (Lysates) and post- (His purification), Ni-NTA-agarose samples were analyzed by SDS-PAGE/Western blotting.
Statistical analysis.
All the experiments were repeated at least three times. Values are expressed as mean ± SD.
Results
FAIM-L protects Type II but not Type I cells from DR-induced apoptosis
To characterize FAIM-L mechanism of protection from apoptosis, we first overexpressed this protein in PC12 and HEK293 cells in parallel with two other antiapoptotic proteins. Bcl-xL is an antiapoptotic member of the Bcl-2 family that inhibits the death signal at the level of the mitochondria (Youle and Strasser, 2008) and has been used as the definitive tool to classify DR-induced apoptosis as Type I, which cannot be prevented by Bcl-xL overexpression, or Type II, which can be prevented by Bcl-xL overexpression. c-FLIP-L is a DR antagonist that competes with caspase-8 for its recruitment to the DISC after DR engagement (Lavrik and Krammer, 2012). As shown in Figure 1A, PC12 cells were sensitive to FasL-induced apoptosis, but only when de novo protein synthesis has been blocked by Actinomycin D (AD). After 24 h of treatment, the overexpression of Bcl-xL, FAIM-L, and c-FLIP-L was able to significantly inhibit sFasL plus AD-induced apoptosis (Fig. 1A). Efficiency of overexpression was checked by Western blot (data not shown). The protective effect of Bcl-xL supports that PC12 cells behave as Type II cells in response to Fas-induced apoptosis. When HEK293 cells were treated with sFasL, only c-FLIP-L, but not Bcl-xL or FAIM-L, was able to protect from sFasL or sFasL and AD treatment (Fig. 1B). Therefore, HEK293 cells respond as Type I to sFasL treatment, where activation of effector caspases by sFasL-induced activation of caspase-8 is sufficient for cell death. To explore the differences in FAIM-L protection between Type II and Type I cells, we compared the processing of caspases and their substrate poly(ADP-ribose) polymerase (PARP) in PC12 and HEK293 cells overexpressing Bcl-xL or FAIM-L. PC12 cells were treated with a combination of sFasL and AD (Fig. 1C), whereas sFasL treatment alone was sufficient for killing HEK293 cells in shorter time points (Fig. 1D). As shown in Figure 1C, the cleavage of PARP is inhibited by Bcl-xL and FAIM-L overexpression in the first 24 h of treatment, compared with pcDNA3-transfected cells where PARP is cleaved as soon as 12 h after treatment. PARP is cleaved in Bcl-xL and FAIM-L-overexpressing cells after 48 h of treatment, when apoptosis is no longer inhibited. However, cleavage of caspase-9 and caspase-3 is totally inhibited by Bcl-xL but only partially by FAIM-L overexpression, suggesting that caspases are cleaved but not active in FAIM-L-overexpressing cells. On the other hand, neither Bcl-xL nor FAIM-L overexpression inhibited PARP or caspases-8, -9, and -3 in HEK293 cells treated with sFasL between 2 and 8 h (Fig. 1D). In accordance with these results, when DEVDase activity was measured by a fluorometric assay, the activation of effector caspases was significantly inhibited by Bcl-xL and FAIM-L overexpression in Type II PC12 cells (Fig. 1E), but not Type I HEK293 cells (Fig. 1F). These data demonstrate that FAIM-L and Bcl-xL are able to protect Type II but not Type I cells from Fas-induced apoptosis.
Figure 1.
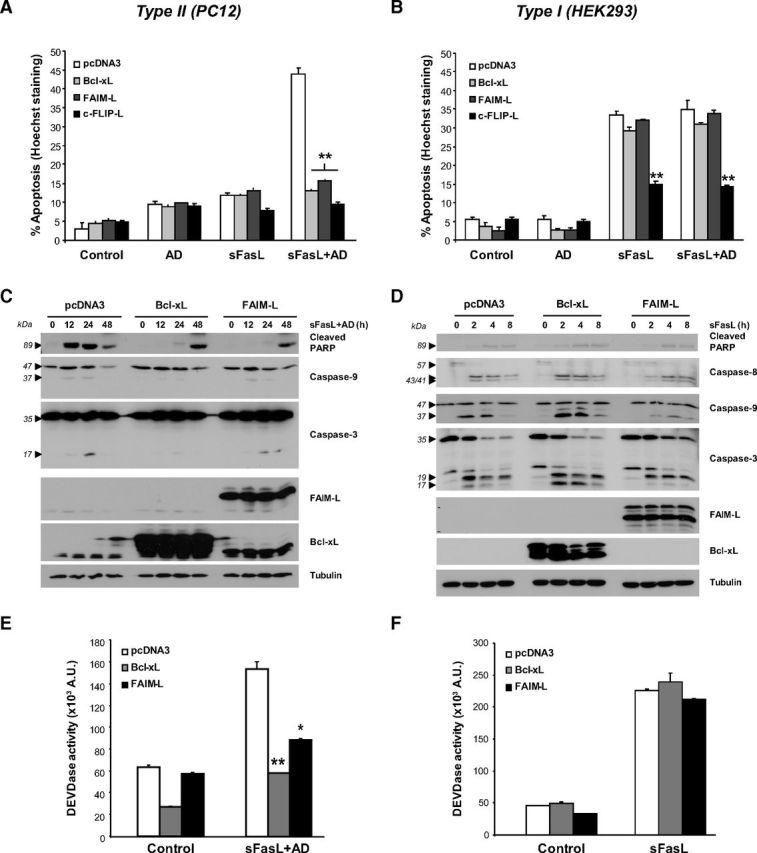
FAIM-L protects Type II but not Type I cells from Fas ligand-induced apoptosis. A, PC12 cells were transfected with pcDNA3, 3xHA-Bcl-xL, 3xFLAG-FAIM-L, or 3xHA-c-FLIP-L expression plasmids and treated with sFasL (100 ng/ml) and AD (1 nm) for 24 h. The percentage of apoptotic cells was assessed by counting nuclei with apoptotic morphology. B, HEK293 cells were transfected and treated as in A for 8 h, and the percentage of apoptotic cells was determined. C, PC12 cells were transfected with pcDNA3, 3xHA-Bcl-xL, or 3xFLAG-FAIM-L expression plasmids and treated with sFasL and AD. Cleavage of caspases and their substrate PARP was assessed by Western blot. D, HEK293 cells were transfected and treated with sFasL prior Western blot analysis as in C. E, DEVDase activity was measured in PC12 cells that were treated with sFasL and AD for 24 h. F, DEVDase activity was measured in HEK293 cells that were treated with sFasL for 8 h. Error bars indicate SD. Student test was performed, comparing transfected cells treated with sFasL or sFasL+AD with their pcDNA3-transfected counterpart. * p ≤ 0.01. **p ≤ 0.001.
Both XIAP and FAIM-L protect PC12 cells from FasL-induced apoptosis downstream Bax activation
It is unclear why FAIM-L protects Type II but not Type I cells from DR-induced apoptosis. To gain further understanding of this protection mechanism, we postulated that XIAP, a molecule that has recently emerged as a discriminator between Type I and Type II apoptosis induced by Fas (Jost et al., 2009; Varfolomeev et al., 2009), might be differentially regulated in PC12 and HEK293 cells. Indeed, even though basal levels of XIAP are somehow similar in both cells lines, XIAP protein levels were upregulated in PC12 cells as soon as 2 h after sFasL and AD treatment (Fig. 2A) and downregulated in HEK293 cells after 4 h of sFasL treatment. As reported previously in other cellular models (Deveraux et al., 1999; Conte et al., 2001), apoptosis induced by sFasL and AD treatment in PC12 cells was significantly inhibited by XIAP overexpression, at the same extent as FAIM-L overexpression (Fig. 2C). XIAP antagonizes cell death downstream of mitochondrial engagement by sequestration and inhibition of caspase-3 and processed caspase-9. To determine whether FAIM-L acts upstream or downstream mitochondrial engagement, we assessed Bax activation by immunoprecipitation of active Bax in PC12 cells transfected with Bcl-xL, XIAP, or FAIM-L and treated with sFasL and AD for 24 h. Even though all three antiapoptotic proteins efficiently inhibited FasL-induced apoptosis, only Bcl-xL overexpression prevented Bax activation. Subsequently, we assessed whether these antiapoptotic proteins are able to inhibit the release of proapoptotic factors from the mitochondrial intermembrane space. Whereas Bcl-xL overexpression impairs both cytochrome c and Smac release from the mitochondria, neither XIAP nor FAIM-L is able to do so (Fig. 2E). Therefore, since neither Bax activation nor cytochrome c/Smac release is prevented compared with pcDNA3-transfected cells (Fig. 2D,E), we conclude that both XIAP and FAIM-L exert their antiapoptotic role downstream mitochondrial engagement.
Figure 2.

XIAP and FAIM-L protect PC12 cells from FasL-induced apoptosis downstream Bax activation. A, B, Immunoblot analysis of XIAP endogenous levels in Type II PC12 cells treated with sFasL (100 ng/ml) and AD (1 nm) (A) and Type I HEK293 cells treated with sFasL (100 ng/ml) at different time points. Membranes were stained with Naphtol Blue to confirm equal loading. C, PC12 cells were transfected with pcDNA3, 3xFLAG-FAIM-L, or 6xMyc-XIAP expression plasmids and treated with sFasL (100 ng/ml) and AD (1 nm) for 24 h. Percentage of apoptosis was assessed. Error bars indicate SD. Student test was performed, comparing transfected cells treated with sFasL+AD with their pcDNA3-transfected counterpart. *p ≤ 0.01. D, PC12 cells were transfected with 3xHA-Bcl-xL, pcDNA3, 3xFLAG-FAIM-L, or 6xMyc-XIAP expression plasmids and then treated for 24 h with sFasL (100 ng/ml) and AD (1 nm). Active Bax was immunoprecipitated before immunoblotting with a total Bax antibody. *Small chain immunoglobulin position. The transfection efficiency of Bcl-xL, XIAP, and FAIM-L was assessed in the lysate fraction. E, PC12 cells were transfected as in D and then treated for 24 h with sFasL (100 ng/ml) and AD (1 nm). Cytochrome c and Smac release to the cytosol were assessed by Western blot, as well as the transfection efficiency of Bcl-xL, XIAP, and FAIM-L.
Endogenous XIAP is essential for protection by FAIM-L
Our findings that FAIM-L can only protect Type II cells where XIAP is upregulated after FasL treatment strongly suggest that FAIM-L might require sustained endogenous XIAP to exert its antiapoptotic function. To support this hypothesis, we performed knockdown experiments in PC12 cells using two different shRNA-targeting XIAPs (XIAP R1 and R2) (Fig. 3A), or FAIM-L shRNA that specifically targets FAIM-L but not FAIM-S (Segura et al., 2007) (Fig. 3B). Three days after cell transduction with lentiviral particles carrying Scrambled (Scr), XIAP shRNA R1 or R2, or FAIM-L shRNA, cells were transfected with Bcl-xL, XIAP, or FAIM-L overexpression plasmids (Fig. 3A). After 2 d of transfection, cells were treated with sFasL in combination with AD for 24 h. Right before treatment, efficiency of XIAP (Fig. 3C) and FAIM-L knockdown (Fig. 3D) was checked. As shown in Figure 3A and in accordance with results of Figures 1A and 2C, overexpression of Bcl-xL, XIAP, and FAIM-L protected Scrambled-transduced PC12 cells from sFasL and AD treatment. However, after XIAP knockdown using two different shRNA, only Bcl-xL but not FAIM-L overexpression was able to significantly protect cells from apoptosis. Interestingly, the protection exerted by Bcl-xL overexpression after XIAP silencing demonstrates that the knockdown of XIAP does not shift PC12 cells from Type II to Type I cells. These data support that, at least in our model, XIAP levels rather than mitochondrial implication is the critical factor for the protective function of FAIM-L. On the other hand, we investigated whether the endogenous levels of FAIM-L were required for the antiapoptotic role of XIAP using the same approach as in Figure 3A. As shown in Figure 3B, both XIAP and Bcl-xL overexpression is able to rescue PC12 cells that have been transduced with FAIM-L shRNA-carrying lentiviral particles and treated with sFasL and AD. Notably, the knockdown of either XIAP (Fig. 3A) or FAIM-L (Fig. 3B) sensitizes PC12 cells to the sole treatment with sFasL (compared with Scr+pcDNA3 conditions treated with sFasL). Together, we conclude that high intracellular levels of XIAP following overexpression render endogenous FAIM-L dispensable for XIAP to exert its protective role. However, endogenous XIAP is required for the antiapoptotic function of FAIM-L. These results were further reproduced in primary neuronal cultures. Ectopic expression of Bcl-xL and FAIM-L was able to protect primary cortical neurons against FasL-induced apoptosis, thereby indicating that cortical neurons behave as Type II cells. However, after XIAP knockdown, Bcl-xL but not FAIM-L overexpression was able to protect from FasL-induced apoptosis (Fig. 3E).
Figure 3.
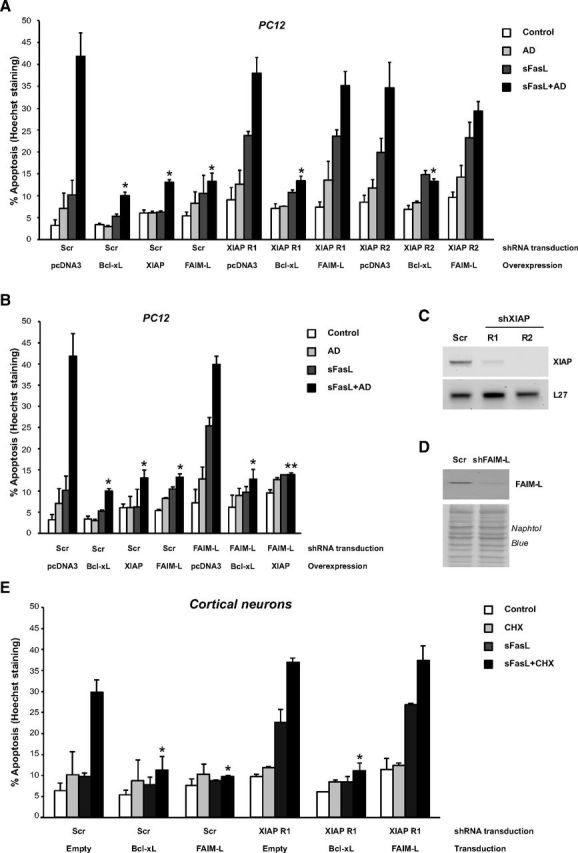
Endogenous XIAP is essential for FAIM-L to protect Type II cells. A, PC12 cells were transduced with Scrambled shRNA (Scr), shRNA XIAP R1, or shRNA XIAP R2 lentiviral particles for 3 d and then transfected with pcDNA3, 3xHA-Bcl-xL, 6xMyc-XIAP, or 3xFLAG-FAIM-L expression plasmids and treated with sFasL (100 ng/ml) and AD (1 nm) for 24 h. Percentage of apoptosis was assessed and Student test was performed, comparing Bcl-xL, XIAP, and FAIM-L-overexpressing cells treated with sFasL+AD with their control (pcDNA3)-treated counterpart. *p ≤ 0.01. B, PC12 cells were transduced as in A with Scrambled shRNA (Scr) or shRNA FAIM-L lentiviral particles and then transfected with pcDNA3, 3xHA-Bcl-xL, or 6xMyc-XIAP and treated as in A. Conditions of Scrambled-transduced cells are identical in A and B. Percentage of apoptosis was assessed and Student test was performed, comparing Bcl-xL, XIAP, and FAIM-L-overexpressing cells treated with sFasL+AD with their control (pcDNA3)-treated counterpart. **p ≤ 0.001. *p ≤ 0.01. C, RT-PCR using rat XIAP or L27 primers was performed to control XIAP knock-down efficiency. D, Immunoblot analysis followed by Naphtol Blue staining of the PVDF membrane confirmed FAIM-L knockdown efficiency. E, Cortical neurons were transduced with Scrambled shRNA (Scr) or shRNA XIAP R1 lentiviral particles for 3 d and then transduced with empty, Bcl-xL, or FAIM-L overexpression lentiviral particles for 3 additional days. Primary neurons were treated with sFasL (100 ng/ml) and cycloheximide (CHX; 1 μg/ml) for 24 h. Percentage of apoptosis was assessed and Student test was performed, comparing Bcl-xL and FAIM-L-overexpressing neurons treated with sFasL+CHX with their control (Empty)-treated counterpart. *p ≤ 0.01. All conditions represent the mean of three independent experiments.
The interaction of FAIM-L with XIAP is required for its antiapoptotic function
We sought to determine whether XIAP is a molecular partner of FAIM-L. Semi-endogenous (Fig. 4A) and endogenous immunoprecipitation experiments (Fig. 4B) were performed by immunoprecipitation of endogenous FAIM-L, using an antibody raised against the 22 amino acid sequence specific of FAIM-L (Segura et al., 2007), followed by immunoblotting of transfected 6xMyc-tagged XIAP (Fig. 4A) or endogenous XIAP (Fig. 4B). Both approaches demonstrate that endogenous FAIM-L is able to interact with XIAP. In the nervous system, FAIM-L protects from DR-induced apoptosis while its shorter isoform FAIM-S fails to protect from FasL and TNFα-induced apoptosis. Even though the physiological roles of both isoforms are different in the nervous system, their protein sequences only differ by the presence of 22 additional amino acids at the N terminus of FAIM-L (Fig. 4E). Moreover, secondary structure prediction algorithms of the FAIM-L-specific sequence do not reveal the existence of any particular domain. HEK293T cells were transfected with FLAG-tagged XIAP in combination with nontagged FAIM-L or FAIM-S. We performed XIAP immunoprecipitation followed by immunoblotting using, first, the anti-FAIM-L and then the anti-FAIM antibody, which is raised against the full-length protein, therefore detecting both isoforms. Comparison of results obtained with both antibodies clearly demonstrates that XIAP interacts with FAIM-L but not FAIM-S (Fig. 4C). This finding allows us to map the interaction of XIAP with the N-terminus of FAIM-L. Interestingly, since XIAP is a functional target of FAIM-L, the specific interaction of XIAP with FAIM-L but not FAIM-S might give a clue about the mechanism of protection of FAIM-L. IAP antagonists, such as Smac/Diablo, Omi/HtrA2, Grim, Hid, Reaper, and processed caspase-9, but also the more recently reported Nsp4 and CLPX (Verhagen et al., 2007), interact with the BIR domains of IAPs through an exposed N-terminal so-called IAP-Binding Motif (IBM) starting with an alanine. The protein sequence of mature FAIM-L starts with an alanine (Fig. 4D), which we mutated to a glycine residue to determine whether this N-terminal amino acid is required for the interaction of FAIM-L with XIAP (Fig. 4E). Consistent with our hypothesis, this mutation eliminated the interaction of FAIM-L with XIAP (Fig. 4F). HEK293T cells were transfected with FLAG-tagged XIAP together with nontagged wt FAIM-L or mut FAIM-L. After FLAG immunoprecipitation followed by immunoblotting using the anti-FAIM antibody, we show that the interaction of XIAP with FAIM-L is lost when the IBM of FAIM-L is mutated. Finally, to demonstrate that the interaction of XIAP and FAIM-L is relevant for the antiapoptotic function of FAIM-L, PC12 cells were transfected with wt FAIM-L or mut FAIM-L, then treated with sFasL and AD for 24 h. As shown in Figure 4G, the mutation of the IBM of FAIM-L abrogates its protective effect. These results indicate that FAIM-L directly interacts with XIAP through its IBM and further support the notion that XIAP endogenous levels as well as its interaction with FAIM-L are essential for the anti-apoptotic function of FAIM-L.
Figure 4.
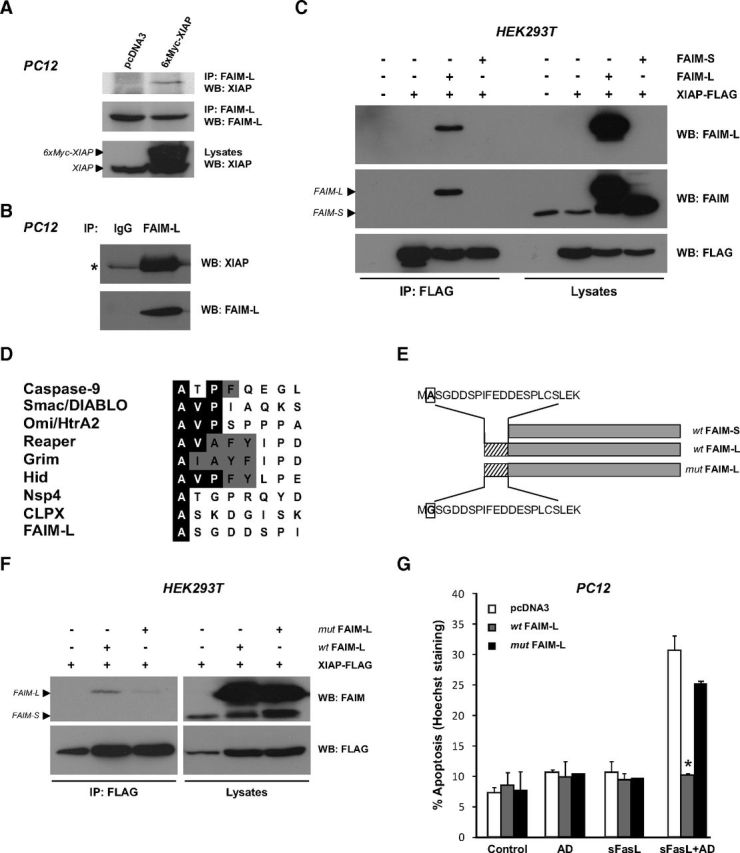
FAIM-L, but not FAIM-S, interacts with XIAP through an IBM domain. A, For semi-endogenous interaction of FAIM-L with XIAP, PC12 cells were transfected with pcDNA3 or 6xMyc-XIAP plasmids and endogenous FAIM-L was immunoprecipitated using a specific FAIM-L antibody before immunoblotting as indicated. B, Endogenous interaction of FAIM-L with XIAP was assessed in PC12 cells. FAIM-L was immunoprecipitated as in A followed by Western blotting as indicated. An anti-Myc antibody IgG was used as negative control. *Small chain Ig position. C, HEK293T cells were transfected with pcDNA3 (−), FAIM-S, FAIM-L, and XIAP-FLAG expression plasmids as indicated. FLAG-tagged XIAP was immunoprecipitated before immunoblotting with FAIM-L, FAIM, and FLAG antibodies. The FAIM-L antibody raised against the FAIM-L specific N terminus 22 amino acids only detects FAIM-L, whereas anti-FAIM (raised against full-length FAIM-S) detects both isoforms. D, Comparison of the N termini of some mammalian and Drosophila IAP-binding proteins. Identical amino acids are highlighted with a black background and similar residues with a gray background. All mature IAP-binding proteins have an N-terminal alanine residue, whereas valine in the second position and proline in the third position are common in several IAP antagonists. Alternatively, Nsp4, CLPX, reported by Verhagen et al. (2007), and FAIM-L are IAP-binding proteins whose IBM is limited only to the presence of the N-terminal alanine residue. E, Schematic representation of wild-type wt FAIM-L and wt FAIM-S isoforms, as well as the mutant FAIM-L (mut FAIM-L) where the first alanine (A, in a box) was mutated to a glycine (G). Hatched boxes represent the FAIM-L-specific 22 amino acids; gray boxes represent 100% similarity between wt FAIM-L and wt FAIM-S. F, HEK293T cells were transfected as indicated, and FLAG-tagged XIAP was immunoprecipitated as in C. G, PC12 cells were transfected with pcDNA3, wt FAIM-L, or mut FAIM-L expression plasmids and treated with sFasL (100 ng/ml) and AD (1 nm) for 24 h. Percentage of apoptotic cells was assessed. The mean of three independent experiments is represented. Error bars indicate SD. Student test was performed, comparing FAIM-L-transfected cells treated with sFasL+AD with their pcDNA3-treated counterpart. *p ≤ 0.01.
FAIM-L interacts with XIAP through its BIR2 domain
To determine the FAIM-L-binding domain of XIAP, we proceeded to coimmunoprecipitate FAIM-L with the different XIAP deletion or point mutants that are schematically represented in Figure 5A. Figure 5B shows that the interaction of FAIM-L with the BIR2 domain is very strong, and the presence of the BIR2 domain is critical for the interaction of the XIAP deletion mutants with FAIM-L. Indeed, FAIM-L was found to interact with XIAP fragments, including the BIR2 domain (lanes 2, 4, 6, 7, and 8) but does not interact with either BIR1 or BIR3 domains (lanes 3 and 5) (Fig. 5B). Because studies using BIR domains of XIAP that might disrupt the protein structure and folding have to be cautiously interpreted, we intended to verify these results using XIAP constructs with BIR domains that bear mutations in residues essential for XIAP antiapoptotic function. The XIAP D214S mutant interferes with XIAP binding to its antagonist Smac, whereas the XIAP E314S mutant interferes with both Smac and processed caspase-9 binding (Silke et al., 2002). As shown in Figure 5C, FAIM-L retains interaction with the BIR3 domain mutant of XIAP (E314S), whereas this interaction is eliminated by the mutation of the BIR2 domain (D214S) or the double mutation (D214SE314S). These results further support our findings that the BIR2 domain is necessary for the interaction of XIAP with FAIM-L. To inquire whether FAIM-L might interfere with the interaction of XIAP with its antagonist Smac, PC12 cells were transfected with full-length XIAP and XIAP point mutants, with or without the 3xHA-FAIM-L encoding plasmid (Fig. 5D). The interaction of XIAP with endogenous Smac is slightly reduced by the XIAP D214S mutant but impaired by the XIAP E314S and XIAP D214SE314S mutants (Fig. 5D, lanes 1–5), supporting previous observations of Smac interaction with the BIR3 domain of XIAP (Riedl and Shi, 2004). The overexpression of FAIM-L does not displace the interaction of XIAP with Smac (Fig. 5D, lanes 6–10), suggesting that FAIM-L acts independently of Smac interaction with XIAP and rather performs a direct action on XIAP, even though the cellular machinery is prompt to kill.
Figure 5.

FAIM-L interacts with XIAP through its BIR2 domain. A, Schematic representation of FLAG-tagged XIAP deletion and point mutants. B, HEK293T cells were transfected with FLAG-tagged XIAP and its deletion mutants as indicated together with a 3xHA-FAIM-L expression plasmid, and FLAG-tagged XIAP was immunoprecipitated before immunoblotting with HA and FLAG antibodies. C, HEK293T cells were transfected with FLAG-tagged XIAP and its point mutants as indicated together with a 3xHA-FAIM-L expression plasmid. Immunoprecipitation and Western blotting were performed as in B. D, PC12 cells were transfected with FLAG-tagged XIAP and its point mutants with or without the coexpression of the 3xHA-FAIM-L plasmid. Immunoprecipitation was performed as in B before immunoblotting with Smac, HA, and FLAG antibodies.
FAIM-L maintains XIAP stability through inhibition of auto-ubiquitinylation and degradation
Since endogenous levels of XIAP are critical for FAIM-L to protect from apoptosis, and XIAP is subject to auto-ubiquitinylation due to the E3-ligase property of its RING domain (Yang et al., 2000), we sought to determine whether FAIM-L affects XIAP auto-ubiquitinylation. First, HEK293T cells were transfected with the 6xHis-Ubiquitin-encoding plasmid together with the 6xMyc-XIAP or 6xMyc XIAP H467A mutant that abrogates its E3-ligase function. After ubiquitin pulldown, the auto-ubiquitinylation of XIAP is evident by immunoblotting with XIAP antibody (Fig. 6A). The XIAP H467A mutant lost this ubiquitinylation pattern (Fig. 6A). This result validates a ubiquitin pull-down protocol, which specifically detects XIAP auto-ubiquitinylation pattern. Subsequently, we sought to determine whether FAIM-L affects XIAP auto-ubiquitinylation (Fig. 6B). Interestingly, FAIM-L overexpression completely abrogates XIAP auto-ubiquitinylation (Fig. 6B, lane 4). To corroborate that XIAP ubiquitinylation leads to its degradation by the proteasome, cells transfected as in lanes 1–4 were treated with the proteasome inhibitor MG132 for 6 h before harvesting (lanes 5–8). As seen in Figure 6B, the strong ubiquitinylation pattern in lane 6 reveals that XIAP auto-ubiquitinylation is degradative. Lysates were immunoblotted against all transfected proteins as well as Mcl-1, to control that the lack of XIAP auto-ubiquitinylation in the presence of FAIM-L is not due to a defective proteasome. Indeed, Mcl-1 protein levels significantly increased in cells treated with MG132 compared with vehicle (Fig. 6B, lanes 5–8). We next proceeded to determine whether the inhibition of XIAP auto-ubiquitinylation and degradation by FAIM-L effectively stabilized XIAP levels. PC12 cells were transfected with 3xFLAG-FAIM-L encoding plasmid or empty vector (pcDNA3) and treated with actinomycin D for several days. Whereas XIAP protein levels drop between 2 and 4 d of AD treatment, FAIM-L overexpression maintains stable levels of endogenous XIAP (Fig. 6C). To further demonstrate that FAIM-L affects XIAP stability in primary neurons, we performed FAIM-L knockdown in cortical neurons and checked XIAP levels after several days in vitro. As seen in Figure 6D, XIAP levels drastically drop in the absence of FAIM-L. We next sought to determine whether FAIM-L knockdown, apart from resulting in XIAP ubiquitinylation and degradation, is able to induce a more rapid degradation of XIAP upon FasL treatment. Cortical neurons transduced with Scrambled or shFAIM-L lentiviral particles for 4 d were treated with sFasL for 24 h. As seen in Figure 3B for PC12 cells, the sole treatment of cortical neurons with sFasL induces apoptosis after FAIM-L knockdown (27.31% of apoptosis, data not shown). However, even though XIAP stability is slightly decreased in neurons after 4 d of shFAIM-L transduction, the treatment with sFasL for 24 h induced apoptosis without affecting any further XIAP stability (Fig. 6E). In conclusion, we report that, in Type II cells, FAIM-L overexpression reduces XIAP auto-ubiquitinylation and enhances its stability, leading to cell survival after Fas stimulation (Fig. 6F). Our results uncover the role of XIAP as a critical partner of a death receptor antagonist and shed new light on the unknown mechanism of protection of FAIM-L.
Figure 6.
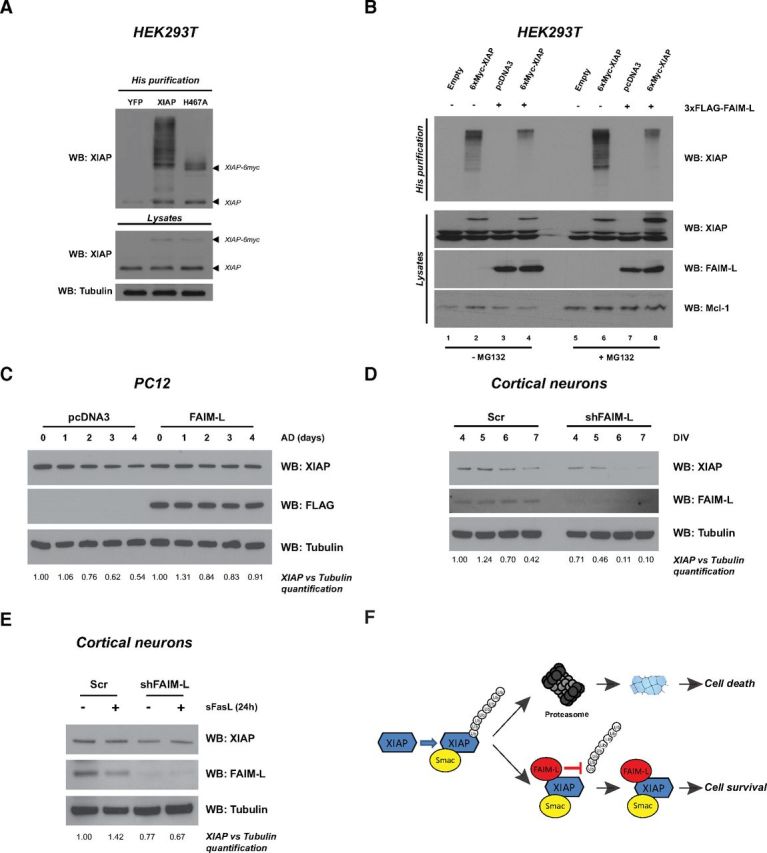
FAIM-L blocks XIAP auto-ubiquitinylation and increases XIAP stability. A, HEK293T cells were transfected with the 6xHis-ubiquitin-encoding plasmid in all conditions, together with YFP, 6xMyc-XIAP, or the 6xMyc-XIAP-H467A mutant-expressing plasmids. In vitro ubiquitinylation assay was performed, followed by XIAP blotting. B, HEK293T cells were transfected with the 6xHis-ubiquitin-encoding plasmid in all conditions, together with the 6xMyc-XIAP plasmid and the 3xFLAG-FAIM-L plasmid when indicated. Six hours before harvesting, cells were treated or not with MG132 (5 μm) and in vitro ubiquitinylation assay was performed. Lysates were loaded to confirm transfection efficiency using anti-XIAP and anti-FAIM-L antibodies. Lysates were also blotted with anti-Mcl-1 as a control of the MG132 treatment. C, PC12 cells were transfected with pcDNA3 or 3xFLAG-FAIM-L expression plasmids and treated with AD (1 nm) for several days. Cells were harvested at the time points indicated, and lysates were immunoblotted using anti-XIAP and anti-FLAG antibodies. The membrane was reblotted with anti-tubulin to ensure equal loading. D, Cortical neurons were transduced with shFAIM-L lentiviral particles upon seeding and harvested at the indicated days in vitro (DIV). Lysates were immunoblotted using an anti-XIAP antibody. The membrane was reblotted with anti-FAIM-L to assess knockdown efficiency and anti-tubulin to ensure equal loading. C, D, The experiments are each one representative of three independent experiments. E, Cortical neurons transduced with shFAIM-L lentiviral particles as in D were treated with sFasL (100 ng/ml) for 24 h, after 4 d of transduction. Lysates were immunoblotted using anti-XIAP, anti-FAIM-L, and antitubulin antibodies. C–E, The intensity of the bands relative to their respective control was quantified using the ImageJ software. F, FAIM-L mechanism of action: after an apoptotic insult, XIAP is subjected to auto-ubiquitinylation and degradation by the proteasome, leading to cell death. However, in the presence of FAIM-L, XIAP auto-ubiquitinylation is inhibited, leading to cell survival.
Discussion
Mature neurons are postmitotic cells that should survive for the lifetime of an organism, and pathological situations, such as neurodegenerative diseases, remain a major clinical challenge. The emergence of FAIM-L as an antiapoptotic protein that is specifically expressed in neurons may provide an important avenue for the conception of novel therapies. However, even though we and others have reported the function of FAIM-L in neuronal protection from DR-induced apoptosis (Segura et al., 2007; Yu et al., 2008), the understanding of its mechanism of action is still lacking. Our work puts in evidence the mechanism of action of FAIM-L by pointing out at XIAP as its critical partner for protection from FasL-induced apoptosis.
The Fas/FasL system limits inflammatory and immune response in the CNS. Although basal expression of Fas/FasL is low, its activation in various neurological disorders causes neuronal damage. The Fas/FasL pathway has roles both in maintaining the immune-suppressed status of the CNS and in inducing neuronal death in Alzheimer's disease (Morishima et al., 2001), Huntington's disease (Ferrer et al., 2000), and Parkinson's disease (Mogi et al., 1996; Ferrer et al., 2000), as well as amyotrophic lateral sclerosis (Raoul et al., 2002). In the present article, we show that FAIM-L protects neuronal Type II and not Type I cells from Fas-induced apoptosis. It is generally accepted that protection from FasL-induced apoptosis by the antiapoptotic Bcl-2 family members (e.g., Bcl-2, Bcl-xL) determines a certain cell type as “Type II.” Alternatively, protection against FasL-induced apoptosis due to loss of function of proapoptotic members of the Bcl-2 family (e.g., Bax, Bak) can also classify cells as “Type II.” In this sense, several studies performed in the nervous system have shown the relevance of antiapoptotic Bcl-2 family members in DR-induced apoptosis (Benn and Woolf, 2004).
In accordance with previous results (Jost et al., 2009), we found XIAP upregulated in Type II and downregulated in Type I cells after stimulation of the Fas receptor. Most importantly, we provide strong evidence that endogenous levels of XIAP are critical for the antiapoptotic function of FAIM-L because FAIM-L can no longer protect neuron-like cells and cortical neurons from Fas-induced apoptosis after XIAP knockdown. We have previously demonstrated that FAIM-L interacts with the Fas receptor and competes with the interaction of Fas with its adaptor FADD (Segura et al., 2007). Intriguingly, we found here that FAIM-L also acts downstream of Bax activation and release of cytochrome c and Smac from the mitochondria. These data point to a dual role of FAIM-L in the apoptotic cascade: one at the DISC level and another downstream of the mitochondria. Indeed, although FAIM-L is able to coimmunoprecipitate with both the Fas receptor and XIAP, XIAP is only able to interact with FAIM-L and not with Fas (data not shown). Also, we observe a cytosolic intracellular distribution of FAIM-L rather than an expression pattern that is restricted to the plasma membrane (Segura et al., 2007). We therefore speculate that FAIM-L interacts with the Fas receptor to impede the recruitment of FADD and then directly with XIAP at the postmitochondrial level. We report the existence of an IBM in the amino terminus of FAIM-L, which is necessary for its interaction with the BIR2 domain of XIAP and therefore for its antiapoptotic function. These findings underline the relevance of the interaction between FAIM-L and XIAP that both protect from Fas-induced apoptosis downstream Bax activation. Finally, we demonstrate that FAIM-L stabilizes XIAP through the inhibition of its auto-ubiquitinylation and degradation by the proteasome, therefore preventing cell death.
Huo et al. (2009) engineered Faim knock-out mice that were viable, even though B cells and thymocytes of mutant mice showed increased sensitivity to Fas-induced apoptosis and exacerbated liver damage in response to Fas engagement in vivo. The authors also report, in accordance with our previous results, a regulation of the physical binding of caspase-8 to the Fas receptor in thymocytes. Xiap knock-out mice are viable, and a redundancy with other members of the IAP family has been observed. Xiap-null thymocytes, splenocytes, and embryonic fibroblasts die normally in response to different apoptotic stimuli (Harlin et al., 2001). However, two types of postmitotic cells from Xiap-null mice (i.e., sympathetic neurons, Potts et al., 2003; and cardiomyocytes, Potts et al., 2005) rapidly die after microinjection of cytochrome c, whereas wild-type cells remain insensitive. Xiap-null mice have an exacerbated response to perinatal hypoxia-ischemia that leads to neuronal apoptosis and tissue loss (West et al., 2009). These observations support that, at least in mitotic cells, XIAP is a critical regulator of apoptosis. Also, apoptosis after nerve growth factor deprivation in cultured sympathetic neurons require XIAP inhibition (Potts et al., 2003). Similarly, motoneurons and neuronally differentiated PC12 pheochromocytoma cells require inhibition of XIAP to die after axotomy and growth factor withdrawal, respectively (Perrelet et al., 2004; Vyas et al., 2004). XIAP levels deregulation has been shown extensively in peripheral nervous system-related diseases and has been proposed as a therapeutic strategy (Garrity-Moses et al., 2006). For example, XIAP levels have been found decreased after motoneuron axotomy (Perrelet et al., 2002) and in mouse models of familial amyotrophic lateral sclerosis (Ishigaki et al., 2002). Moreover, XIAP levels have also been found reduced in Huntington's disease (Goffredo et al., 2005) and Alzheimer's disease (Tanaka et al., 2006). Immunohistochemical studies suggest that XIAP may be partially associated with the pathogenesis of Parkinson's disease and dementia with Lewy bodies (Kawamoto et al., 2012). Interestingly, XIAP dysfunction resulting from S-nitrosylation has also been described in Parkinson's disease (Tsang et al., 2009). Also, transgenic expression of XIAP in neurons using the neuron-specific enolase promoter (nse-XIAP) improves prognosis in a mouse model of Parkinson's disease (Crocker et al., 2003). More recently, it has been shown that XIAP is able to regulate the activation of caspase-3 that allows axonal degeneration in sensory neurons from DRG after trophic factor deprivation. Moreover, XIAP−/− mice embryos show decreased skin innervation. These data show the relevance of XIAP on the physiological regulation of caspase-3 and have important consequences for the shaping of the nervous system during development (Unsain et al., 2013). Our results suggest that FAIM-L could have potential physiological functions, apart from regulating neuronal death through death receptors. Further studies are required to demonstrate the involvement of FAIM-L in such a paradigm.
These reports are in line with our findings that XIAP regulation by FAIM-L, which inhibits XIAP auto-ubiquitinylation and degradation, is critical for the cell decision between death and survival. A growing body of evidence supports that the E3 ubiquitin ligase activity of XIAP plays a key role in its participation to cellular signaling cascades (Galbán and Duckett, 2010). Schile et al. (2008) investigated the E3 ubiquitin ligase activity of XIAP through the inactivation of the RING motif by gene targeting. Deleting the RING domain stabilized XIAP in apoptotic thymocytes, demonstrating that XIAP E3 ubiquitin ligase activity is a major determinant for its stability. These findings correlate with our results showing that the inhibition of XIAP auto-ubiquitinylation by FAIM-L is sufficient for XIAP stability. More recently, Damgaard et al. (2012) reported that the RING domain of XIAP is essential for NOD2 signaling through the XIAP-mediated ubiquitinylation of RIPK2 and the recruitment of the linear ubiquitin chain assembly complex to NOD2. Our findings, that FAIM-L is an IAP-binding protein that, unlike the antagonists Smac/DIABLO and HtrA2/Omi, serves as an XIAP “guardian,” highlight the potential use of FAIM-L as a therapeutic tool for neuronal protection in neurodegenerative diseases.
It is still unknown whether XIAP and/or FAIM-null mice are prone to neurodegenerative diseases. Many neurodegenerative diseases have a strong apoptotic component, and blocking apoptosis could have therapeutic benefits. Therefore, the inhibition of IAP degradation might constitute a powerful pharmacological target in neurodegenerative disease. Our results bring strong evidence for the role of FAIM-L in maintaining XIAP endogenous levels and protecting cells from apoptosis. In this sense, it would be interesting to determine whether a neuron-specific molecule, such as FAIM-L, which is a “guardian” of XIAP, might play a physiological role in preventing cell death upon caspase-3 activation. Our mechanistic work brings great insight into the potential use of FAIM-L as a therapeutic tool for neuronal protection from caspase activation.
Footnotes
This work was funded by the Spanish Government Ministerio de Sanidad y Consumo (Centro de Investigación Biomédica en Red sobre Enfermedades Neurodegenerativas, CB06/05/1104 to J.X.C.), Ministerio de Economía y Competitividad (SAF2010–19953 to J.X.C.; SAF2012–31485 to V.J.Y.), Instituto de Salud Carlos III (CP11/00052 to M.F.S.), and the Generalitat de Catalunya (Suport als Grups de Recerca Consolidats 2009SGR346). F.M.-F. and L.P.-F. are supported by postgraduate fellowships from the Spanish Government Ministerio de Educación y Ciencia. J.U. is supported by a postgraduate fellowship from the Generalitat de Catalunya. R.S.M. and V.J.Y. were under the Juan de la Cierva and the Ramon y Cajal programs, respectively, from the Ministerio de Educación y Ciencia (Spain), cofinanced by the European Social Fund. M.F.S. is under the Miguel Servet program from the Instituto de Salud Carlos III and cofinanced by the European Regional Development Fund. We thank Dr. Didier Trono (Geneva, Switzerland) for providing the lentiviral plasmids, Dr. John Silke for providing the XIAP mutants, and Dr. Pascal Meier for helpful discussion.
The authors declare no competing financial interests.
References
- Benn SC, Woolf CJ. Adult neuron survival strategies: slamming on the brakes. Nat Rev Neurosci. 2004;5:686–700. doi: 10.1038/nrn1477. [DOI] [PubMed] [Google Scholar]
- Berman SB, Chen YB, Qi B, McCaffery JM, Rucker EB, 3rd, Goebbels S, Nave KA, Arnold BA, Jonas EA, Pineda FJ, Hardwick JM. Bcl-x L increases mitochondrial fission, fusion, and biomass in neurons. J Cell Biol. 2009;184:707–719. doi: 10.1083/jcb.200809060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi C, Benveniste EN. Fas ligand/Fas system in the brain: regulator of immune and apoptotic responses. Brain Res Brain Res Rev. 2004;44:65–81. doi: 10.1016/j.brainresrev.2003.08.007. [DOI] [PubMed] [Google Scholar]
- Conte D, Liston P, Wong JW, Wright KE, Korneluk RG. Thymocyte-targeted overexpression of xiap transgene disrupts T lymphoid apoptosis and maturation. Proc Natl Acad Sci U S A. 2001;98:5049–5054. doi: 10.1073/pnas.081547998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crocker SJ, Liston P, Anisman H, Lee CJ, Smith PD, Earl N, Thompson CS, Park DS, Korneluk RG, Robertson GS. Attenuation of MPTP-induced neurotoxicity and behavioural impairment in NSE-XIAP transgenic mice. Neurobiol Dis. 2003;12:150–161. doi: 10.1016/S0969-9961(02)00020-7. [DOI] [PubMed] [Google Scholar]
- Damgaard RB, Nachbur U, Yabal M, Wong WW, Fiil BK, Kastirr M, Rieser E, Rickard JA, Bankovacki A, Peschel C, Ruland J, Bekker-Jensen S, Mailand N, Kaufmann T, Strasser A, Walczak H, Silke J, Jost PJ, Gyrd-Hansen M. The ubiquitin ligase XIAP recruits LUBAC for NOD2 signaling in inflammation and innate immunity. Mol Cell. 2012;46:746–758. doi: 10.1016/j.molcel.2012.04.014. [DOI] [PubMed] [Google Scholar]
- Darding M, Meier P. IAPs: guardians of RIPK1. Cell Death Differ. 2012;19:58–66. doi: 10.1038/cdd.2011.163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deveraux QL, Leo E, Stennicke HR, Welsh K, Salvesen GS, Reed JC. Cleavage of human inhibitor of apoptosis protein XIAP results in fragments with distinct specificities for caspases. EMBO J. 1999;18:5242–5251. doi: 10.1093/emboj/18.19.5242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eckelman BP, Salvesen GS. The human anti-apoptotic proteins cIAP1 and cIAP2 bind but do not inhibit caspases. J Biol Chem. 2006;281:3254–3260. doi: 10.1074/jbc.M510863200. [DOI] [PubMed] [Google Scholar]
- Fernández M, Segura MF, Solé C, Colino A, Comella JX, Ceña V. Lifeguard/neuronal membrane protein 35 regulates Fas ligand-mediated apoptosis in neurons via microdomain recruitment. J Neurochem. 2007;103:190–203. doi: 10.1111/j.1471-4159.2007.04767.x. [DOI] [PubMed] [Google Scholar]
- Ferreira KS, Kreutz C, Macnelly S, Neubert K, Haber A, Bogyo M, Timmer J, Borner C. Caspase-3 feeds back on caspase-8, Bid and XIAP in Type I Fas signaling in primary mouse hepatocytes. Apoptosis. 2012;17:503–515. doi: 10.1007/s10495-011-0691-0. [DOI] [PubMed] [Google Scholar]
- Ferrer I, Blanco R, Cutillas B, Ambrosio S. Fas and Fas-L expression in Huntington's disease and Parkinson's disease. Neuropathol Appl Neurobiol. 2000;26:424–433. doi: 10.1046/j.1365-2990.2000.00267.x. [DOI] [PubMed] [Google Scholar]
- Galbán S, Duckett CS. XIAP as a ubiquitin ligase in cellular signaling. Cell Death Differ. 2010;17:54–60. doi: 10.1038/cdd.2009.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garrity-Moses ME, Teng Q, Krudy C, Yang J, Federici T, Boulis NM. X-linked inhibitor of apoptosis protein gene-based neuroprotection for the peripheral nervous system. Neurosurgery. 2006;59:172–182. doi: 10.1227/01.NEU.0000219237.69329.B7. [DOI] [PubMed] [Google Scholar]
- Goffredo D, Rigamonti D, Zuccato C, Tartari M, Valenza M, Cattaneo E. Prevention of cytosolic IAPs degradation: a potential pharmacological target in Huntington's Disease. Pharmacol Res. 2005;52:140–150. doi: 10.1016/j.phrs.2005.01.006. [DOI] [PubMed] [Google Scholar]
- Harlin H, Reffey SB, Duckett CS, Lindsten T, Thompson CB. Characterization of XIAP-deficient mice. Mol Cell Biol. 2001;21:3604–3608. doi: 10.1128/MCB.21.10.3604-3608.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huo J, Xu S, Guo K, Zeng Q, Lam KP. Genetic deletion of faim reveals its role in modulating c-FLIP expression during CD95-mediated apoptosis of lymphocytes and hepatocytes. Cell Death Differ. 2009;16:1062–1070. doi: 10.1038/cdd.2009.26. [DOI] [PubMed] [Google Scholar]
- Irmler M, Thome M, Hahne M, Schneider P, Hofmann K, Steiner V, Bodmer JL, Schröter M, Burns K, Mattmann C, Rimoldi D, French LE, Tschopp J. Inhibition of death receptor signals by cellular FLIP. Nature. 1997;388:190–195. doi: 10.1038/40657. [DOI] [PubMed] [Google Scholar]
- Ishigaki S, Liang Y, Yamamoto M, Niwa J, Ando Y, Yoshihara T, Takeuchi H, Doyu M, Sobue G. X-linked inhibitor of apoptosis protein is involved in mutant SOD1-mediated neuronal degeneration. J Neurochem. 2002;82:576–584. doi: 10.1046/j.1471-4159.2002.00998.x. [DOI] [PubMed] [Google Scholar]
- Jost PJ, Grabow S, Gray D, McKenzie MD, Nachbur U, Huang DC, Bouillet P, Thomas HE, Borner C, Silke J, Strasser A, Kaufmann T. XIAP discriminates between Type I and Type II FAS-induced apoptosis. Nature. 2009;460:1035–1039. doi: 10.1038/nature08229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawamoto Y, Ito H, Ihara M, Takahashi R. Immunohistochemical localization of X-linked inhibitor of apoptosis protein in brainstem-type and cortical Lewy bodies. Neuroreport. 2012;23:162–167. doi: 10.1097/WNR.0b013e32834f4066. [DOI] [PubMed] [Google Scholar]
- Lavrik IN, Krammer PH. Regulation of CD95/Fas signaling at the DISC. Cell Death Differ. 2012;19:36–41. doi: 10.1038/cdd.2011.155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michaelidis TM, Sendtner M, Cooper JD, Airaksinen MS, Holtmann B, Meyer M, Thoenen H. Inactivation of bcl-2 results in progressive degeneration of motoneurons, sympathetic and sensory neurons during early postnatal development. Neuron. 1996;17:75–89. doi: 10.1016/S0896-6273(00)80282-2. [DOI] [PubMed] [Google Scholar]
- Mogi M, Harada M, Kondo T, Mizuno Y, Narabayashi H, Riederer P, Nagatsu T. The soluble form of Fas molecule is elevated in parkinsonian brain tissues. Neurosci Lett. 1996;220:195–198. doi: 10.1016/S0304-3940(96)13257-2. [DOI] [PubMed] [Google Scholar]
- Morishima Y, Gotoh Y, Zieg J, Barrett T, Takano H, Flavell R, Davis RJ, Shirasaki Y, Greenberg ME. Beta-amyloid induces neuronal apoptosis via a mechanism that involves the c-Jun N-terminal kinase pathway and the induction of Fas ligand. J Neurosci. 2001;21:7551–7560. doi: 10.1523/JNEUROSCI.21-19-07551.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Motoyama N, Wang F, Roth KA, Sawa H, Nakayama K, Nakayama K, Negishi I, Senju S, Zhang Q, Fujii S. Massive cell death of immature hematopoietic cells and neurons in Bcl-x-deficient mice. Science. 1995;267:1506–1510. doi: 10.1126/science.7878471. [DOI] [PubMed] [Google Scholar]
- Moubarak RS, Yuste VJ, Artus C, Bouharrour A, Greer PA, Menissier-de Murcia J, Susin SA. Sequential activation of poly(ADP-ribose) polymerase 1, calpains, and Bax is essential in apoptosis-inducing factor-mediated programmed necrosis. Mol Cell Biol. 2007;27:4844–4862. doi: 10.1128/MCB.02141-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moubarak RS, Solé C, Pascual M, Gutierrez H, Llovera M, Pérez-Garcia MJ, Gozzelino R, Segura MF, Iglesias-Guimarais V, Reix S, Soler RM, Davies AM, Soriano E, Yuste VJ, Comella JX. The death receptor antagonist FLIP-L interacts with Trk and is necessary for neurite outgrowth induced by neurotrophins. J Neurosci. 2010;30:6094–6105. doi: 10.1523/JNEUROSCI.0537-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perrelet D, Ferri A, Liston P, Muzzin P, Korneluk RG, Kato AC. IAPs are essential for GDNF-mediated neuroprotective effects in injured motor neurons in vivo. Nat Cell Biol. 2002;4:175–179. doi: 10.1038/ncb751. [DOI] [PubMed] [Google Scholar]
- Perrelet D, Perrin FE, Liston P, Korneluk RG, MacKenzie A, Ferrer-Alcon M, Kato AC. Motoneuron resistance to apoptotic cell death in vivo correlates with the ratio between X-linked inhibitor of apoptosis proteins (XIAPs) and its inhibitor, XIAP-associated factor 1. J Neurosci. 2004;24:3777–3785. doi: 10.1523/JNEUROSCI.0413-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Potts MB, Vaughn AE, McDonough H, Patterson C, Deshmukh M. Reduced Apaf-1 levels in cardiomyocytes engage strict regulation of apoptosis by endogenous XIAP. J Cell Biol. 2005;171:925–930. doi: 10.1083/jcb.200504082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Potts PR, Singh S, Knezek M, Thompson CB, Deshmukh M. Critical function of endogenous XIAP in regulating caspase activation during sympathetic neuronal apoptosis. J Cell Biol. 2003;163:789–799. doi: 10.1083/jcb.200307130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raoul C, Estévez AG, Nishimune H, Cleveland DW, deLapeyrière O, Henderson CE, Haase G, Pettmann B. Motoneuron death triggered by a specific pathway downstream of Fas potentiation by ALS-linked SOD1 mutations. Neuron. 2002;35:1067–1083. doi: 10.1016/S0896-6273(02)00905-4. [DOI] [PubMed] [Google Scholar]
- Riedl SJ, Shi Y. Molecular mechanisms of caspase regulation during apoptosis. Nat Rev Mol Cell Biol. 2004;5:897–907. doi: 10.1038/nrm1496. [DOI] [PubMed] [Google Scholar]
- Riedl SJ, Renatus M, Schwarzenbacher R, Zhou Q, Sun C, Fesik SW, Liddington RC, Salvesen GS. Structural basis for the inhibition of caspase-3 by XIAP. Cell. 2001;104:791–800. doi: 10.1016/S0092-8674(01)00274-4. [DOI] [PubMed] [Google Scholar]
- Scaffidi C, Fulda S, Srinivasan A, Friesen C, Li F, Tomaselli KJ, Debatin KM, Krammer PH, Peter ME. Two CD95 (APO-1/Fas) signaling pathways. EMBO J. 1998;17:1675–1687. doi: 10.1093/emboj/17.6.1675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schile AJ, García-Fernández M, Steller H. Regulation of apoptosis by XIAP ubiquitin-ligase activity. Genes Dev. 2008;22:2256–2266. doi: 10.1101/gad.1663108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider TJ, Fischer GM, Donohoe TJ, Colarusso TP, Rothstein TL. A novel gene coding for a Fas apoptosis inhibitory molecule (FAIM) isolated from inducibly Fas-resistant B lymphocytes. J Exp Med. 1999;189:949–956. doi: 10.1084/jem.189.6.949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott FL, Denault JB, Riedl SJ, Shin H, Renatus M, Salvesen GS. XIAP inhibits caspase-3 and -7 using two binding sites: evolutionarily conserved mechanism of IAPs. EMBO J. 2005;24:645–655. doi: 10.1038/sj.emboj.7600544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segura MF, Sole C, Pascual M, Moubarak RS, Perez-Garcia MJ, Gozzelino R, Iglesias V, Badiola N, Bayascas JR, Llecha N, Rodriguez-Alvarez J, Soriano E, Yuste VJ, Comella JX. The long form of Fas apoptotic inhibitory molecule is expressed specifically in neurons and protects them against death receptor-triggered apoptosis. J Neurosci. 2007;27:11228–11241. doi: 10.1523/JNEUROSCI.3462-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiozaki EN, Chai J, Rigotti DJ, Riedl SJ, Li P, Srinivasula SM, Alnemri ES, Fairman R, Shi Y. Mechanism of XIAP-mediated inhibition of caspase-9. Mol Cell. 2003;11:519–527. doi: 10.1016/S1097-2765(03)00054-6. [DOI] [PubMed] [Google Scholar]
- Silke J, Ekert PG, Day CL, Hawkins CJ, Baca M, Chew J, Pakusch M, Verhagen AM, Vaux DL. Direct inhibition of caspase 3 is dispensable for the anti-apoptotic activity of XIAP. EMBO J. 2001;20:3114–3123. doi: 10.1093/emboj/20.12.3114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silke J, Hawkins CJ, Ekert PG, Chew J, Day CL, Pakusch M, Verhagen AM, Vaux DL. The anti-apoptotic activity of XIAP is retained upon mutation of both the caspase 3- and caspase 9-interacting sites. J Cell Biol. 2002;157:115–124. doi: 10.1083/jcb.200108085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sole C, Dolcet X, Segura MF, Gutierrez H, Diaz-Meco MT, Gozzelino R, Sanchis D, Bayascas JR, Gallego C, Moscat J, Davies AM, Comella JX. The death receptor antagonist FAIM promotes neurite outgrowth by a mechanism that depends on ERK and NF-kappa B signaling. J Cell Biol. 2004;167:479–492. doi: 10.1083/jcb.200403093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Somia NV, Schmitt MJ, Vetter DE, Van Antwerp D, Heinemann SF, Verma IM. LFG: an anti-apoptotic gene that provides protection from Fas-mediated cell death. Proc Natl Acad Sci U S A. 1999;96:12667–12672. doi: 10.1073/pnas.96.22.12667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srinivasula SM, Ashwell JD. IAPs: what's in a name? Mol Cell. 2008;30:123–135. doi: 10.1016/j.molcel.2008.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka T, Isoe-Wada K, Yamamori H, Kato K, Nessa BN, Sadik GM, Takeda M. Neurobiological studies of dementia–biological markers and neuroprotective strategies for Alzheimer disease. Acta Neurol Taiwan. 2006;15:68–71. [PubMed] [Google Scholar]
- Taoufik E, Valable S, Müller GJ, Roberts ML, Divoux D, Tinel A, Voulgari-Kokota A, Tseveleki V, Altruda F, Lassmann H, Petit E, Probert L. FLIP(L) protects neurons against in vivo ischemia and in vitro glucose deprivation-induced cell death. J Neurosci. 2007;27:6633–6646. doi: 10.1523/JNEUROSCI.1091-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Treier M, Staszewski LM, Bohmann D. Ubiquitin-dependent c-Jun degradation in vivo is mediated by the δ domain. Cell. 1994;78:787–798. doi: 10.1016/S0092-8674(94)90502-9. [DOI] [PubMed] [Google Scholar]
- Tsang AH, Lee YI, Ko HS, Savitt JM, Pletnikova O, Troncoso JC, Dawson VL, Dawson TM, Chung KK. S-nitrosylation of XIAP compromises neuronal survival in Parkinson's disease. Proc Natl Acad Sci U S A. 2009;106:4900–4905. doi: 10.1073/pnas.0810595106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Unsain N, Higgins JM, Parker KN, Johnstone AD, Barker PA. XIAP regulates caspase activity in degenerating axons. Cell Rep. 2013;4:751–763. doi: 10.1016/j.celrep.2013.07.015. [DOI] [PubMed] [Google Scholar]
- Varfolomeev E, Alicke B, Elliott JM, Zobel K, West K, Wong H, Scheer JM, Ashkenazi A, Gould SE, Fairbrother WJ, et al. X chromosome-linked inhibitor of apoptosis regulates cell death induction by proapoptotic receptor agonists. 2009;284:34553–34560. doi: 10.1074/jbc.M109.040139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verhagen AM, Kratina TK, Hawkins CJ, Silke J, Ekert PG, Vaux DL. Identification of mammalian mitochondrial proteins that interact with IAPs via N-terminal IAP binding motifs. Cell Death Differ. 2007;14:348–357. doi: 10.1038/sj.cdd.4402001. [DOI] [PubMed] [Google Scholar]
- Vyas S, Juin P, Hancock D, Suzuki Y, Takahashi R, Triller A, Evan G. Differentiation-dependent sensitivity to apoptogenic factors in PC12 cells. J Biol Chem. 2004;279:30983–30993. doi: 10.1074/jbc.M400692200. [DOI] [PubMed] [Google Scholar]
- West T, Stump M, Lodygensky G, Neil JJ, Deshmukh M, Holtzman DM. Lack of X-linked inhibitor of apoptosis protein leads to increased apoptosis and tissue loss following neonatal brain injury. ASN Neuro. 2009;1:1. doi: 10.1042/AN20090005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson NS, Dixit V, Ashkenazi A. Death receptor signal transducers: nodes of coordination in immune signaling networks. Nat Immunol. 2009;10:348–355. doi: 10.1038/ni.1714. [DOI] [PubMed] [Google Scholar]
- Yang Y, Fang S, Jensen JP, Weissman AM, Ashwell JD. Ubiquitin protein ligase activity of IAPs and their degradation in proteasomes in response to apoptotic stimuli. Science. 2000;288:874–877. doi: 10.1126/science.288.5467.874. [DOI] [PubMed] [Google Scholar]
- Youle RJ, Strasser A. The BCL-2 protein family: opposing activities that mediate cell death. Nat Rev Mol Cell Biol. 2008;9:47–59. doi: 10.1038/nrm2308. [DOI] [PubMed] [Google Scholar]
- Yu LY, Saarma M, Arumäe U. Death receptors and caspases but not mitochondria are activated in the GDNF- or BDNF-deprived dopaminergic neurons. J Neurosci. 2008;28:7467–7475. doi: 10.1523/JNEUROSCI.1877-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuste VJ, Bayascas JR, Llecha N, Sánchez-López I, Boix J, Comella JX. The absence of oligonucleosomal DNA fragmentation during apoptosis of IMR-5 neuroblastoma cells: disappearance of the caspase-activated DNase. J Biol Chem. 2001;276:22323–22331. doi: 10.1074/jbc.M100072200. [DOI] [PubMed] [Google Scholar]
- Zhong X, Schneider TJ, Cabral DS, Donohoe TJ, Rothstein TL. An alternatively spliced long form of Fas apoptosis inhibitory molecule (FAIM) with tissue-specific expression in the brain. Mol Immunol. 2001;38:65–72. doi: 10.1016/S0161-5890(01)00035-9. [DOI] [PubMed] [Google Scholar]