

Abstract
Pathological, genetic, and biochemical hallmarks of Alzheimer's disease (AD) are linked to amyloid-β (Aβ) peptide aggregation. Especially misfolded Aβ42 peptide is sufficient to promote amyloid plaque formation. However, the cellular compartment facilitating the conversion of monomeric Aβ to aggregated toxic Aβ species remains unknown. In vitro models suggest lipid membranes to be the driving force of Aβ conversion. To this end, we generated two novel mouse models, expressing either membrane-anchored or nonanchored versions of the human Aβ42 peptide. Strikingly, membrane-anchored Aβ42 robustly accelerated Aβ deposition and exacerbated amyloid-associated toxicity upon crossing with Aβ precursor protein transgenic mice. These in vivo findings support the hypothesis that Aβ–membrane interactions play a pivotal role in early-onset AD as well as neuronal damage and provide evidence to study Aβ–membrane interactions as therapeutic targets.
Keywords: Alzheimer's disease, mouse model, membrane anchoring, toxicity, amyloid formation
Introduction
Alzheimer's disease (AD) is a major threat to the growing number of elderly people worldwide. Characterized clinically by memory loss and dementia, the disease is only definitely diagnosed by postmortem demonstration of specific pathological hallmarks including extracellular aggregation of amyloid-β (Aβ) peptides in senile plaques, intracellular accumulation of tau protein in neurofibrillary tangles, and loss of synapses as well as neurons.
Considerable evidence supports a critical role for Aβ peptides, namely Aβ42, in the development of AD pathology (Hardy and Selkoe, 2002). Mutations in amyloid-β protein precursor (APP) and presenilin increase the relative levels of Aβ42 and cause early-onset familial forms of AD (Haass and De Strooper, 1999). Moreover, transgenic (tg) mice exclusively expressing Aβ42 develop plaque pathology in the absence of human APP overexpression, indicating that Aβ42 is sufficient for amyloid plaque formation (McGowan et al., 2005). However, despite these studies supporting a pivotal role for Aβ42 in amyloid formation, it is currently unclear how and where monomeric Aβ initially aggregates.
Converging evidence suggests that plasma membranes may play an important role in the misfolding and aggregation of amyloidogenic proteins. First, pathological examination of AD brain material reveals early deposition on cell surface plasma membranes, forming diffuse plaques in AD subjects (Yamaguchi et al., 2000). Second, Aβ has been identified in lipid raft fractions isolated from human AD brains and APP tg mice (Lee et al., 1998; Kawarabayashi et al., 2004). Finally, the membrane can act as a catalytic site, enhancing the misfolding and aggregation of amyloidogenic proteins (Chi et al., 2008). Together, these findings suggest that Aβ association with cellular membranes may be a pivotal event in the formation and propagation of cerebral amyloidosis (Williamson and Sutherland, 2011). However, the ability for membrane-associated Aβ to enhance Aβ deposition has, thus far, not been examined in vivo.
To address this, we have generated transgenic mice that exclusively express a membrane-anchored version of the human Aβ42 peptide. To do so, the peptide was tethered to the cell membrane via a C-terminal glycosylphosphatidyl inositol (GPI) anchor. Our results demonstrate that membrane-anchored Aβ42 is targeted to sphingolipid-rich subdomains of detergent-resistant membranes (DRMs), accelerates amyloid formation, and exacerbates amyloid-associated toxicity in vivo compared with a similar but nonanchored version of Aβ42.
Materials and Methods
Cloning of pBriAβeGPI and pBriAβe expression constructs.
As a source for BriAβ42, the plasmid pAG3 BriAβ42 (Lewis et al., 2001) was used. A 491 bp fragment was amplified using the primers Aβ42 (5′-GTTGAATTTATCAGTGTGCC-3′) and XbaI Aβ42-reverse (5′-AAACTCTAGACGCTATGACAACACC-3′), introducing a unique XbaI site instead of a stop codon into the original sequence. Similarly, using the prion protein (PrP) coding plasmid pPrPHG (Fischer et al., 1996) as a template, 109 bp fragments were amplified. For pBriAβe, the primers XbaI–GPI (5′-GCGTCTAGAGACGGGAGAAGATCCTGATGAAC-3′) and BamHI-reverse (5′-TAGTGGATCCTCATCCCACGATC-3′), and for pBriAβeGPI, the primers XbaI GPI (5′-GCGTCTAGAGACGGGAGAAGATCC-3′) and BamHI-reverse were taken. After purification of the fragments, they were treated with XbaI to generate adhesive ends. Equimolar mixtures of the respective fragment combinations and pCR 2.1 Topo Vector were mixed and transfected into DH5α Escherichia coli. pCR 2.1 topo clones with correct restriction patterns were treated with BglII/BamHI to liberate 481 bp fragments either for GPI anchoring or just extended Aβ. These were ligated into the previous opened pAG3 BriAβ42 to give rise to either pAG3 BriAβe or pAG3 BriAβeGPI. After sequence confirmation, these CMV-driven plasmids were used for HEK cell transfections. To generate expression vectors for pronuclei injection, the respective pAG3 BriAβe and pAG3 BriAβeGPI were subjected to XhoI restriction digest. This liberated the coding sequence of the desired BriAβe or BriAβeGPI of 1099 bp containing start and stop codons, which were ligated into the XhoI site of the pTSC21K vector containing the Thy1 promoter (Sturchler-Pierrat et al., 1997).
Generation of transgenic mice.
Transgenic mice were generated by pronuclei injection on a C57BL/6 genetic background and kept hemizygous. Intercrossings were set up with male AβeGPI or Aβe mice and APP23 female mice all kept on C57BL/6 genetic background.
Cell lysis and protein extraction.
Cells were washed with cold PBS, scraped off the plate using a cell scraper and transferred to 1.5 ml Eppendorf tubes. Cells were subjected to centrifugation at 800 × g. Temperature during the whole procedure was maintained at 4°C. The pellets were lysed with STEN-lysis buffer (50 mm Tris pH 7.6, 150 mm NaCl, 2 mm EDTA, 0.2% Igepal CA-630, 0,1% Triton X-100 dissolved in distilled H2O) and incubated on ice for 15 min in the presence of protease inhibitor (Complete, Roche). The lysates were cleared by centrifugation at 16,000 × g for 30 min. Ten to 20 μg of protein was used for analysis via Western blots.
Crude membrane isolation and protein extraction.
Cells were washed with cold PBS and transferred to 1.5 ml Eppendorf tubes placed on ice. The cells were subjected to centrifugation at 200 × g for 5 min at 4°C. The pellet was suspended in 200 μl of cold hypotonic buffer (10 mm Tris Cl, pH 7.5, 10 mm NaCl, 0.1 mm EGTA, 25 mm β-glycerophosphate, 1 mm DTT dissolved in distilled H2O, adjusted to pH 7.4) and incubated on ice for 15 min. The cell suspension was then passed through a 1 ml syringe with a 0.6-mm-diameter needle for 15 times. Cell debris and nuclear fractions were pelleted by centrifugation at 200 × g for 15 min. at 4°C. The supernatant was transferred to new Eppendorf tubes and centrifuged at 100,000 × g for 1 h at 4°C to obtain a membrane fraction pellet. The membrane pellet samples were suspended in NuPage loading dye and analyzed by Western blot.
Western blotting.
Cell and brain extracts were analyzed on NuPage Bis-Tris mini gels (Invitrogen). Proteins were transferred to nitrocellulose membrane in a NuPage semi-dry blotting chamber and the transfer was confirmed by Ponceau-S stain. The membrane was boiled at 95°C for 5 min and subsequently blocked using 5% milk in PBS containing 0.1% Tween-20 (Carl Roth) for 1 h. Blots were incubated with 6E10 antibody [epitope amino acids 3–8 of β-amyloid; EFRHDS, Covance] solution overnight at 4°C and later incubated with secondary antibody conjugated to horseradish peroxidase (Promega) for 1 h at room temperature. Chemiluminescent peroxidase substrate (ECL Plus, GE Healthcare) was used for detection. Chemiluminescence was recorded using an ECL imager (Stella 3200, Raytest), and protein bands were quantified using the software package Aida. Statistical analysis was performed using GraphPad Prism 5. Bonferroni's post hoc test for multiple comparisons was used to determine significant differences between the protein levels of different samples analyzed on Western blot.
Cell surface biotinylation of Aβ.
Surface of intact adherent cells was treated with Biotin (Dynabeads Streptavidin Trial Kit, Invitrogen) or mock treated with PBS according to the manufacturer's instructions. The biotinylated proteins were immunoprecipitated with streptavidin beads, eluted in sample loading buffer by boiling at 95°C for 5 min. Proteins were resolved by 12% NuPage gels. Samples were analyzed using Western blot probed with anti-Aβ-specific monoclonal antibody 6E10.
Immunocytochemistry.
Cells were cultured on poly-l-lysine-coated glass coverslips to 50–60% confluence, fixed in 4% paraformaldehyde for 10 min, washed with PBS twice, and blocked with 5% BSA for 45 min. Cells were then incubated with primary antibodies (Aβ monoclonal antibody 6E10, dilution 1:1000; and N-terminally processed Aβ monoclonal antibody N/25 epitope amino acids 1–7 (DARFRHD), dilution 1:500) for 2 h in 1% BSA and kept in the dark. The cells were gently washed three times with PBS. Primary antibodies were detected by Alexa Fluor 488 (diluted 1:1000 in 1% BSA) conjugated secondary antibody (Invitrogen) and incubated for 1 h. DAPI (diluted 1:1000 in 1% BSA) (Invitrogen) was added to cells for 15 min to stain the nucleus. Coverslips were gently washed twice with distilled H2O and mounted on glass slides using Immu-Mount (Thermo Scientific). Images were acquired on confocal inverted microscope. (LSM 510 laser scanning microscope, Zeiss).
Isolation of DRM fractions.
DRMs were prepared on ice by detergent extraction using a previously described method (Naslavsky et al., 1997) with adaptations described in Baumann et al. (2007). Cell or brain extracts were suspended in 9% sucrose dissolved in PBS, homogenized with a 1 ml syringe with a 0.6-mm-diameter needle 10 times and centrifuged at 100 × g for 5 min. The protein concentration of the supernatant for the samples was determined by BCA assay, and adjusted to 1 mg/ml. Samples were extracted at 4°C with Triton X-100 (1% final) in 2 ml Eppendorf tubes and placed in a tube rotator for 2 h at 4°C. The Triton extract was mixed with 60% OptiPrep (Invitrogen) and transferred to 13.2 ml centrifuge tubes (Beckman Coulter), giving a 40% final concentration, and then was overlaid with 30% and 5% OptiPrep. Tubes were placed in a SW 41 Ti rotor (Beckman Coulter). Gradient was harvested in 400 μl aliquots after 20 h of 200,000 × g centrifugation. Fractions were suspended in sample buffer (NuPage loading dye + 10% β-mercaptoethanol), boiled for 10 min at 70°C, and analyzed on Western blot.
Phospholipase C treatment.
The cells were washed with ice-cold PBS, scraped off the plate in PBS, and transferred to 1.5 ml Eppendorf tubes placed on ice. The cells were subjected to centrifugation at 200 × g for 5 min at 4°C. The pellet was suspended in buffer (0.32 m sucrose, 20 mm Tris-HCl, and 5 mm EDTA, pH 7.5). The cell suspension was then passed through a 1 ml syringe with a 0.6-mm-diameter needle 10 times and centrifuged at 3000 × g for 10 min at 4°C. To obtain a membrane pellet, the supernatant was centrifuged at 100,000 × g for 45 min at 4°C. The membrane pellet was suspended in 30 μl of 20 mm Tris-HCl with 5 mm EDTA, pH 7.5. Fifteen microliters of membrane suspension was incubated for 2 h at 37°C with 20 U/ml recombinant Bacillus cereus phospholipase C (PLC) to release GPI-anchored membrane proteins. The remaining 15 μl were untreated and served as a control. PLC-treated and nontreated samples were centrifuged at 100,000 × g to separate the PLC-released proteins from the insoluble membrane fraction. The supernatant consisting of PLC-released proteins was subjected to 45% ammonium sulfate precipitation. The pellet and supernatant sample were suspended in buffer (NuPage loading dye + 10% β-mercaptoethanol), boiled for 10 min at 70°C, and analyzed on Western blot. For immunocytochemistry, cells were treated for 30 min with 20 U/ml recombinant B. cereus PLC at 37°C, then fixed with 4% paraformaldehyde (PFA) and stained with monoclonal anti-Aβ antibody N25 (Vandermeeren et al., 2001).
Proteinase K digestion.
One hemisphere of mouse brain was homogenized with a Precellys tissue homogenizer to render a 20% (w/v) extract in PBS. Brain extracts (30 μg) were treated with 100 μg/ml Proteinase K (PK; Roche Diagnostics) for 30 min at 37°C with agitation at 400 rpm. The protease digestion was stopped by boiling the samples at 95°C for 5 min. Samples were suspended in buffer (NuPage loading dye + 10% β-mercaptoethanol), boiled for 10 min at 70°C, and analyzed on Western blot.
In vitro thioflavin T aggregation assay.
To determine the in vitro fibrillization potential of brain extracts a Thioflavin T fibrillization kinetic was measured as described by Colby et al. (2007) with some modifications: Thioflavin T was dissolved in water generating a 200 μm stock solution. Lyophylized recombinant Aβ1–40 peptide was dissolved to a stock concentration of 5 mm in 100% DMSO and frozen at −80°C. Before use, this stock was then freshly diluted in DMSO 1:5 to 1 mm and sonified for 10 min in a water bath followed by 5 min centrifugation at 16,100 × g at room temperature. The supernatant was then further diluted to reach 250 μm Aβ1–40 in a 50% DMSO stock. Brain homogenates for this assay were prepared as follows: a crude 20% (w/v) homogenate in PBS was made, then cleared for 5 min at 800 × g and the supernatant diluted with additional PBS to yield a 10% (w/v) homogenate. BCA was performed to determine total protein content. For the kinetic measurement in 96-well format, 14 μg of brain homogenate in the presence of protease inhibitor cocktail from (Complete, Roche), 20 μm Thioflavin T, 25 μm Aβ1–40, 50 mm phosphate, and 150 mm NaCl were incubated at 37°C. Each brain homogenate was assayed in eight sealed wells of the 96-well clear-bottom plates (Greiner Bio-One). Thioflavin T fluorescence at 480 nm was measured from the plate bottom every 30 min using a BMG FLUOstar plate reader. Before each measurement, a double-orbital shaking step for 30 s with 500 rpm was performed. Increase of fluorescence over time was followed until the maximum was reached. Raw data were fitted and lag times were determined following the equations described by Nielsen et al. (2001) with GraphPad Prism 5.
Histological analysis.
Mice were perfused with PBS and excised brains post-fixed for 48 h in 4% PFA dissolved in PBS, then cryoprotected in 30% sucrose in PBS for 24 h. After freezing, 25-μm-thick coronal sections were sliced through the fixed brains using a freezing–sliding microtome. The sections were collected in 0.1 m Tris-buffered saline, pH 7.4, and stained immunohistochemically. The polyclonal antibody CN3 (Eisele et al., 2010; 1:1000) was used for immunostaining Aβ. The sections were all co-stained with Congo red and imaged under bright-field illumination. The following additional antibodies were used: rabbit polyclonal antibody against cow glial fibrillary acidic protein (GFAP; Dako; 1:1000); rabbit polyclonal antibody against ionized calcium-binding adapter molecule 1 (Iba1; Wako; 1:2000); cresyl violet; AT8-hyperphosphorylated tau (1:1000), and 5313 antibody-APP-positive dystrophic boutons (1:2000). Images were acquired on a Zeiss Axioplan 2 microscope (Carl Zeis MicroImaging).
Stereological quantification.
Stereological quantification of total parenchymal Aβ covered area was performed with the Stereologer software and a motorized x-y-z stage, which is coupled to a videomicroscopy system (Systems Planning and Analysis; Calhoun et al., 1998). Frontal, neo, and entorhinal cortex were defined using a standard mouse brain atlas (Franklin and Paxinos, 2004). Quantification was performed on the left hemisphere of the brain sections. A series of coronal-cut 25 μm free-floating sections stained for Aβ amyloid plaque deposition was analyzed. Every section starting from the frontal to the entorhinal cortex were counted to determine an overall plaque load. Thus, the analyses per transgenic animal included 10–12 sections. The amyloid load (percentage) was determined by calculating the areal fraction of parenchymal Aβ, in two-dimensional sectors at a single focal plane at 20× and 0.45 numerical aperture (NA). Statistical analysis was performed using GraphPad Prism 5. The stereological quantification scale implied a nonsymmetric distribution; therefore, the Kruskal–Wallis non-parametric ANOVA with Dunn's multiple comparisons test was performed.
Quantification of inflammatory response.
Quantification was performed on the left hemisphere of coronal brain sections. A series of 25 μm free-floating sections stained for GFAP or Iba1 were imaged using a Zeiss Axioplan 2 microscope with an Axiocam HRc color camera (Carl Zeiss MicroImaging), using a Zeiss 4× and 0.1 NA Achroplan objective and the MosaiX bundle on the Axiovision 4.7 acquisition software. Every second section starting from the frontal to the entorhinal cortex was manually delineated in ImageJ and Rényi entropy measure (Kapur et al., 1985) was used to automatically determine the threshold on the red channel of the color image. The percentage of the covered area was directly calculated by the Fiji plugin as a ratio between the selected cortex area and the thresholded structures. Statistical analysis (one-way ANOVA Bonferroni multiple-comparisons test) was done using GraphPad Prism 5.
Results
Generation of anchorless and GPI-anchored Aβ42
We modified the previously described construct BriAβ42 (Lewis et al., 2001) to assess the effect of membrane-bound Aβ on amyloidosis and associated pathologies in an AD mouse model. Expression of the BriAβ42 construct results in secretion of the encoded Aβ peptide immediately after cleavage by furin-like protease of the fusion protein in the Golgi compartment.
To anchor Aβ to the plasma membrane, the BriAβ42 construct was modified with the GPI anchor of the PrP. We exploited the fact that PrP is synthesized as a precursor protein with a classic C-terminal transmembrane domain. Under normal PrP synthesis, this domain is proteolytically processed and replaced by a GPI anchor via a transamidase reaction immediately after synthesis in the endoplasmic reticulum (ER). The cleavage signal and GPI attachment site are coded within the last 31 aa (Baumann et al., 2009; Rutishauser et al., 2009). However, only the last 24 C-terminal aa of the transmembrane domain are replaced by the GPI anchor, leaving 7 aa (SRDGRRS) added to the BriAβ42 peptide sequence (Fig. 1A). Thus, GPI-anchored Aβ42 (referred to as AβeGPI) contains this 7 aa extension. As a control, we devised an additional construct encoding a secreted C-terminally extended Aβ42 (referred to as Aβe) that will not be bound to the membrane (Fig. 1A).
Figure 1.
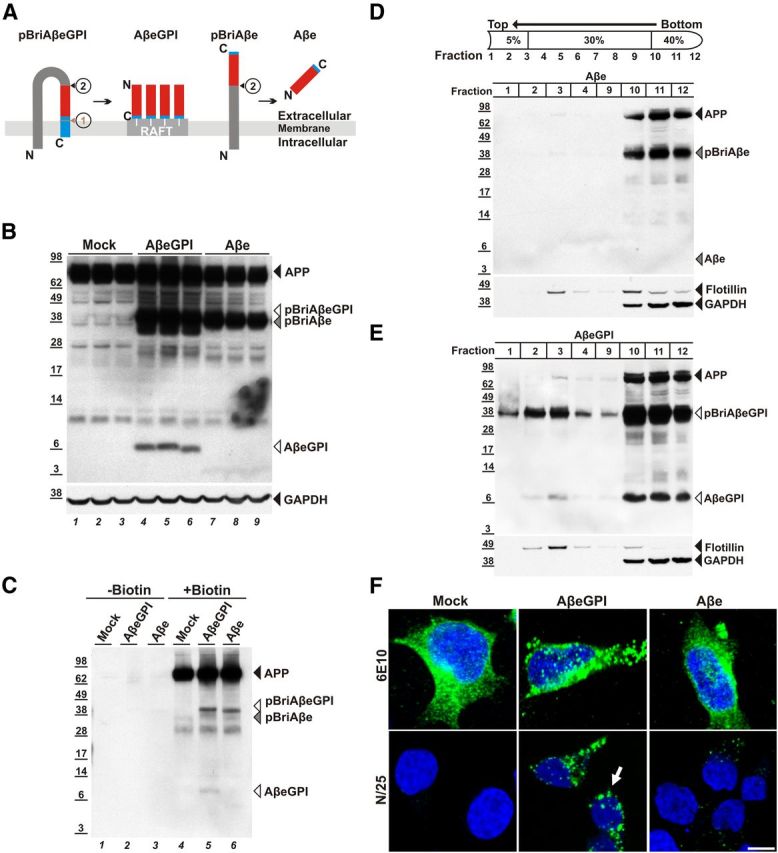
Membrane-anchored AβeGPI is stably expressed in HEK cells and localizes in rafts on cell membrane surfaces. A, Schematic representation of fusion constructs pBriAβeGPI and pBriAβe. Bri2 precursor sequence (dark gray), Aβ42 peptide (red), GPI attachment signal of PrP (blue), and raft compartment in the membrane (RAFT). ➀, Corresponding to the recognition site of the GPI-transamidase replacing the transmembrane domain of pBriAβeGPI with the GPI anchor (white lines), leaving 7 aa (SRDGRRS) as a C-terminal extension (small blue) to the Aβ sequence. ➁, Representing furin-like cleavage, releasing the N-terminus of Aβ from the pBriAβeGPI sequence. The control construct pBriAβe with the SRDGRRS extension ➁ liberating anchorless Aβe. B, Membrane extracts of HEK 293 (Mock), HEK AβeGPI, and HEK Aβe cells contain AβeGPI (lanes 4–6), and the anchored precursor proteins APP (lanes 1–9), pBriAβeGPI (lanes 4–6), and pBriAβe (lanes 7–9), but not Aβe (lanes 7–9). C, Cell surface association confirmed by surface biotinylation for AβeGPI (lane 5). Transmembrane proteins like APP (lanes 4–6), pBriAβe (lane 6), and pBriAβeGPI are also biotinylated, but soluble Aβe is not (cell lines are as denoted in B). D, Fractions of 400 μl from top to bottom of the centrifuge were collected and analyzed by Western blot and probed with Aβ antibody 6E10. HEK Aβe cells show hardly any cleaved Aβ in DRMs (fraction 3). pBriAβe, though being membrane associated, is not floating similar to APP. E, HEK AβeGPI demonstrate that the uncleaved precursor pBriAβeGPI is floating with equal buoyancy as maturely processed AβeGPI due to the GPI anchor being immediately attached after translocation in the ER (fraction 3). Flotillin-1 was used as a marker for DRMs, while the cytoplasmic protein GAPDH served as a control. F, AβeGPI is detected on the cell surface. Confocal images showing maximum projection of 0.39 μm z-stacks in nonpermeabilized HEK 293 (Mock), HEK Aβe, and HEK AβeGPI cells. The top lane is stained with 6E10 epitope (EFRHDS) detecting APP, pBriAβe, and pBriAβeGPI, while the bottom lane is stained with N/25, specifically detecting the free N terminus of processed Aβ (DAEFRHD). Only HEK AβeGPI cells show strong punctuated staining (green) on the cell surface (arrow), indicating raft association.
Expression and localization of GPI-anchored and anchorless Aβ42 in HEK293 cells
The expression and processing of the pBriAβeGPI and pBriAβe constructs function as expected in vitro after stable expression in HEK293 cells (Fig. 1B,C). Furin-like cleavage of pBriAβeGPI produced the membrane-bound AβeGPI (Fig. 1B). In contrast to Aβe, membrane-bound AβeGPI was confirmed by surface biotinylation (Fig. 1C). Other membrane-bound precursor proteins like APP, pBriAβeGPI, and pBriAβe, which were also biotinylated under these conditions, served as the internal control (Fig. 1C).
To determine whether AβeGPI was enriched in DRMs, Triton X-100 extraction from cell homogenates was performed as described previously (Lingwood and Simons, 2007). Similar to GPI-anchored PrP, AβeGPI was found in the DRM fraction, while Aβe and endogenous APP remained in the loading fraction (Fig. 1D). The pBriAβeGPI, which was not yet cleaved by furin, also associated with DRMs, while the noncleaved pBriAβe did not (Fig. 1E). These data confirm the order of events, with the transamidase attaching the GPI anchor already in the ER and targeting DRMs, while furin-like cleavage is a downstream event of the Golgi compartment. Finally, cell survival and morphology were not affected in the transfected cells, and cultures displayed similar growth rates. The monoclonal Aβ antibody N/25, recognizing the free N terminus of Aβ epitope amino acids 1–7 (DARFRHD; Vandermeeren et al., 2001; Mathews et al., 2002), allowed for detection of AβeGPI and Aβe in immunofluorescent microscopy (Fig. 1F). A small amount of Aβe was still sticking to cells, suggesting that Aβe interacts with cell membrane. In contrast to Aβe, AβeGPI displayed a strong punctuated staining on the cell membrane surface.
To test for the presence of a GPI anchor in AβeGPI-expressing cells, HEK cells stably expressing the fusion proteins were treated with PLC. PLC cleaves the phosphodiester bond in the GPI anchor and subsequently releases the GPI-anchored Aβ from the cellular membrane surface (Fig. 2A,B). Only AβeGPI was released and found in the soluble supernatant, while endogenous APP, and the peptide transmembrane domains pBriAβeGPI and pBriAβe remained unaffected by PLC treatment (Fig. 2B). The molecular mass of PLC-cleaved AβeGPI appeared similar to the molecular mass of membrane-anchored AβeGPI, due to the presence of the conjugated sugars and phosphate group on the liberated peptide. Immunofluorescence confirmed the sensitivity of AβeGPI for PLC treatment (Fig. 2C).
Figure 2.

Membrane-anchored AβeGPI is released by Phospholipase C (PLC) treatment. A, The GPI anchor with which AβeGPI (blue box representing the whole Aβe protein) is tethered to the cell membrane is depicted schematically on the left. Cleavage site of PLC is indicated at the phosphate group . EtN, ethanolamine; GlcN, glucosamine; Man, mannose; myo-Ino, myo-inositol; GalNAc, N-acetylgalactosamine. B, Cell membrane fraction and supernatants of HEK 293 cells (mock), HEK AβeGPI, and HEK Aβe, after treatment of cells with PLC (+PLC, lanes 4–6 and 10–12) or vehicle (−PLC, lanes 1–3, 7–9), were analyzed by Western blot probed with Aβ antibody 6E10. C, AβeGPI is liberated by PLC treatment. Confocal images showing maximum projection of 0.39 μm z-stacks in nonpermeabilized HEK 293 (mock), HEK Aβe, and HEK AβeGPI cells stained with N/25.
. EtN, ethanolamine; GlcN, glucosamine; Man, mannose; myo-Ino, myo-inositol; GalNAc, N-acetylgalactosamine. B, Cell membrane fraction and supernatants of HEK 293 cells (mock), HEK AβeGPI, and HEK Aβe, after treatment of cells with PLC (+PLC, lanes 4–6 and 10–12) or vehicle (−PLC, lanes 1–3, 7–9), were analyzed by Western blot probed with Aβ antibody 6E10. C, AβeGPI is liberated by PLC treatment. Confocal images showing maximum projection of 0.39 μm z-stacks in nonpermeabilized HEK 293 (mock), HEK Aβe, and HEK AβeGPI cells stained with N/25.
AβeGPI and Aβe expression in transgenic mice
The mouse Thy1 promoter (Andrä et al., 1996) was used to drive transgene expression. Mice were generated by pronuclei injections. Three tg lines for Aβe and eight tg lines for AβeGPI were established on a C57BL/6 genetic background. Two tg lines for Aβe and AβeGPI were backcrossed twice to C57BL/6 mice, and showed robust expression of their respective transgene. From each mouse strain, the higher-expressing line was chosen for further analysis (Fig. 3). In the two mouse lines, the Aβe concentration was slightly higher than the AβeGPI concentration (Fig. 3A,C). Steady-state levels of Aβe and AβeGPI did not change over time, whereas APP23 mice—a well established model for Aβ plaque deposition (Sturchler-Pierrat et al., 1997)—exhibited a dramatic increase in Aβ due to deposition of insoluble Aβ plaques beyond 9 months of age (Fig. 3B,C). Histological analysis of Aβe and AβeGPI mice displayed neither plaque formation (Fig. 3D) nor other alterations (e.g., activation of astrocytes, proliferation of microglia; data not shown) up to 24 months of age. Lack of Aβe deposition, independent of the GPI membrane anchoring, could be due to the lower levels of Aβ compared with predepositing APP23 mice (Fig. 3B) or due to the reduced aggregation propensity of this extended version of Aβ. To this end, we measured the ability of brain homogenates to induce fibril formation of monomeric Aβ. Aβe and AβeGPI extracts were compared with normal brain-derived Aβ in vitro in the presence of Thioflavin T and recombinant Aβ1–40. (Fig. 3E,F). Aβ fibrils quantitatively detected by Thioflavin T incorporation spontaneously formed after passing a lag phase during which initial fibril seeds need to form. The addition of extracts containing aggregates suitable to seed Aβ fibrillization led to a reduced lag time until maximal fluorescence was reached. This effect was clearly seen when comparing lag times from young APP23 brains before Aβ deposition to aged Aβ plaque-laden brains (curves of a representative extract are shown in Fig. 3E). Interestingly, AβeGPI transgenic extracts from young mice already showed a slightly reduced lag time, although they contain significantly less Aβ than young APP23 brains (Fig. 3C). The lag time was further decreased at old age, indicating that more AβeGPI acquires seed structure during aging of the mice. A small effect was also found for Aβe. Neither AβeGPI nor Aβe extracts showed a prolonged lag time compared with nontransgenic extracts, excluding a negative influence on Aβ fibrillization. A combination of data from different mice showed the same effects (Fig. 3F) but due to variation significance was only reached for the APP23 extract.
Figure 3.

pBriAβeGPI and pBriAβe undergo proteolytic processing, but, although tg mice do not show plaques, both promote Aβ fibrillization. A, Western blot analysis of expression levels of HEK AβeGPI (lane 1) and HEK Aβe (lane 2) cells (cell hom.), compared with brain extracts (brain hom.) from 2-month-old tg Aβe (lane 3, 4), AβeGPI (lane 5, 6), and APP23 mice (lane 7, 8) with Aβ antibody 6E10. B, Western blot analysis of brain extracts from 24-month-old tg Aβe (lane 1, 2), AβeGPI (lane 3, 4), and APP23 (lane 5, 6) mice. C, Quantification of Western blots comparing the total expression levels of Aβe and AβeGPI to Aβ (100%) as a percentage in 2- and 24-month-old tg Aβe, AβeGPI, and APP23 mice. Aβe and AβeGPI are significantly lower expressed than Aβ in 2-month-old tg APP23 mice (100%) and stay merely unchanged at 24 month in contrast to 24-month-old tg APP23 mice (∼1200%). There are significant differences between the groups, as indicated (n = 2 mice per group for all 2-month-old tg mice, and n = 3 mice per group in all 24-month-old tg mice; *p < 0.05, ***p < 0.001; ns, not significant, one-way ANOVA, Bonferroni's post hoc test for multiple comparisons). D, Histological analysis of 24-month-old tg Aβe and AβeGPI mice. Coronal sections of respective tg lines through the frontal and entorhinal cortex stained with Aβ antibody CN3 and Congo red. Amyloid deposition was observed in neither tg Aβe nor AβeGPI mice. APP23 mice at the age of 16 months served as positive control of amyloid deposition. Scale bar, 500 μm. E, Fibrillization of recombinant Aβ1–40 was monitored by incorporation of Thioflavin T. Kinetics are displayed as time versus relative fluorescence. Data points represent the mean and SE of eight technical replicates of a representative individual mouse of the indicated genotype. Brain homogenates containing fibrillization seeds reduced the lag phase until there is a steep fluorescence increase to reach plateau. F, Comparison of lag times extracted from E. At the age of 24 months, APP23 containing Aβ plaques as well as AβeGPI had reduced lag times compared with age-matched WT mice. Significant differences are only found between the groups indicated (*p < 0.05, one-way ANOVA, Bonferroni's post hoc test for multiple comparisons; 2-month-old APP23 mice, n = 4; 2-month-old WT mice, n = 3; 2-month-old AβeGPI mice, n = 4; 2-month-old Aβe mice, n = 4; 24-month-old APP23 mice, n = 2; 24-month-old WT mice, n = 2; 24-month-old AβeGPI mice, n = 4; 24-month-old Aβe mice, n = 6).
AβeGPI accelerates plaque pathology of Aβ-depositing APP23 mice
To examine whether the enhancement of fibrillization in vitro could be translated in vivo, we crossbred tg APP23 with AβeGPI mice. Aβe mice crossed with tg APP23 mice served as a control. Similar to the in vitro fibrillization experiment, there are three possible experimental outcomes. First, Aβe and AβeGPI do not form appropriate Aβ seeds, and will not interact with Aβ from APP23 mice. Therefore, the coexpressing mice would be similar to single tg APP23 mice. Second, both Aβe and AβeGPI could impede rather than promote β-amyloidosis in APP23 mice through cross-inhibition of amyloid formation (Eisenberg and Jucker, 2012). Third, Aβe and AβeGPI act as Aβ seeds in the APP23 mice and enhance plaque formation.
Double tg AβeGPI × APP23, Aβe × APP23, and single tg APP23 littermate controls were analyzed at 9 months of age (Fig. 4). Western blot analysis of brain extracts revealed a strong accumulation of APP23-derived Aβ in double tg AβeGPI × APP23 and Aβe × APP23 mice. In contrast, age-matched single tg APP23 littermates showed only very modest Aβ accumulation. Notably, double tg AβeGPI × APP23 mice accumulated more Aβ than double tg Aβe × APP23 mice, resembling the amounts of aggregated Aβ in 14-month-old single tg APP23 mice (Fig. 4A). The levels of aggregated Aβ resistant to PK treatment (Langer et al., 2011) were consistently higher in the double tg AβeGPI × APP23 mice followed by the tg Aβe × APP23 and then single tg APP23 mice (Fig. 4B).
Figure 4.
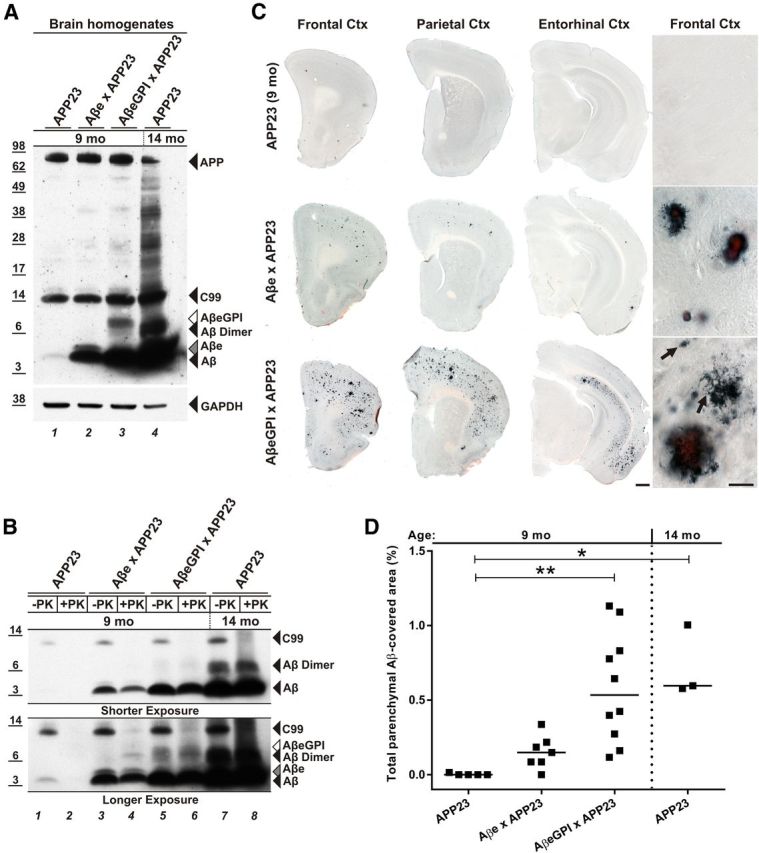
Membrane-anchored AβeGPI accelerates accumulation of Aβ in APP23 mice and leads to both congophilic and noncongophilic Aβ deposits. A, Western blot analysis of brain extracts from 9-month-old single tg APP23, double tg Aβe × APP23, AβeGPI × APP23, and 14-month-old single tg APP23 mice, with Aβ antibody 6E10. The Aβ level of 9-month-old single tg APP23 mice (lane 1) is extremely low in contrast to Aβ levels in double tg mice (lanes 2, 3), resembling the status of single tg APP23 mice at 14 months of age (lane 4). B, Brain extracts, as in A, were treated with 100 μg/ml proteinase K (+PK) and were analyzed with Western blot using 6E10. Aβ (lanes 4, 6, 8), Aβe (lane 4), and AβeGPI (lane 6) in the respective tg mice were PK resistant. Protease-sensitive APP-derived C99 fragment serves as an internal control for the efficacy of the PK treatment. C, Coronal sections through the cortex of respective tg lines at 9 months of age costained with Aβ antibody CN3 and Congo red. APP23 mice only show one to three plaques in the frontal cortex; in the double tg Aβe × APP23 mice, few plaque deposits are visible, while double tg AβeGPI × APP23 mice show robust plaque formation. Scale bar, 500 μm. Double tg mice displayed a plaque morphology, which is not seen in single tg APP23 mice at 9 months. Amorphous noncongophilic deposits were exclusively seen in double tg AβeGPI × APP23 mice (arrows). Scale bar, 50 μm. D, Stereological quantification of C, with significant differences between medians of the groups as indicated (n = 5 for APP23, n = 7 for Aβe × APP23, n = 10 for AβeGPI × APP23, n = 3 mice in the group of APP23 tg mice at 14 months of age; *p < 0.05, **p < 0.01, Kruskal–Wallis nonparametric ANOVA with Dunn's multiple-comparisons test).
Consistent with the biochemical analysis, immunohistological examination of mice brain sections demonstrated enhanced plaque formation in double tg AβeGPI × APP23 when compared with APP23 and Aβe × APP23 animals (Fig. 4C,D). Deposits were predominantly found in the frontal, parietal, and entorhinal cortices as well as the hippocampus (Fig. 4C). Notably, the amyloid deposition in 9-month-old double tg AβeGPI × APP23 mice resembled the amyloid load encountered in 14-month-old single tg APP23 mice. In contrast, the 9-month-old tg APP23 littermates were still at the initial phase of deposition with one to three plaques in the frontal cortex, quantitatively confirmed by stereological analysis (adjusted p values of Dunn's multiple-comparisons test: 9-month-old APP23 vs Aβe × APP23 mice, p = 0.9265; 9-month-old APP23 vs AβeGPI × APP23 mice, p = 0.0022; 9-month-old APP23 vs 14-month-old APP23 mice, p = 0.0132; Aβe × APP23 vs AβeGPI × APP23, p = 0.1411; Aβe × APP23 vs 14-month-old APP23, p = 0.2526; AβeGPI × APP23 vs 14-month-old APP23, p > 0.9999; Fig. 4D). Despite the lower expression level of AβeGPI compared with Aβe, double tg AβeGPI × APP23 mice developed more plaques than double tg Aβe × APP23 mice. The majority of the Aβ deposits were congophilic in the double tg mice (Fig. 4C). However, AβeGPI × APP23 mice also displayed amorphous Congo red-negative deposits (Fig. 4C).
GPI-anchored Aβ exacerbates neurotoxicity
Histological analysis of neurotoxicity indicated a displacement of neurons in the vicinity of plaques in the stratum granulosum of the dentate gyrus and CA1 stratum pyramidal of the hippocampus in 9-month-old double tg AβeGPI × APP23 mice, which was not found in the control mice (Fig. 5A). Moreover, dystrophic boutons and hyperphosphorylated tau-positive neuritic elements appeared to be increased in double tg AβeGPI × APP23 mice when compared with double tg Aβe × APP23 mice (Fig. 5A). It is important to note that even synaptic and neuritic abnormalities were found around the Congo red-negative amorphous Aβ deposits in the AβeGPI × APP23 animals (Fig. 5A). Activation of astrocytes and microglia was detected around all congophilic Aβ deposits in both double tg Aβe × APP23 and AβeGPI × APP23 mice (Fig. 5A). However, the inflammatory reaction in AβeGPI × APP23 mice was significantly higher than in all other age-matched mice and resembled aged tg APP23 mice (Sturchler-Pierrat et al., 1997). Further analysis with electron microscopy revealed no evidence for morphological changes of membrane architecture in response to membrane anchoring (data not shown).
Figure 5.
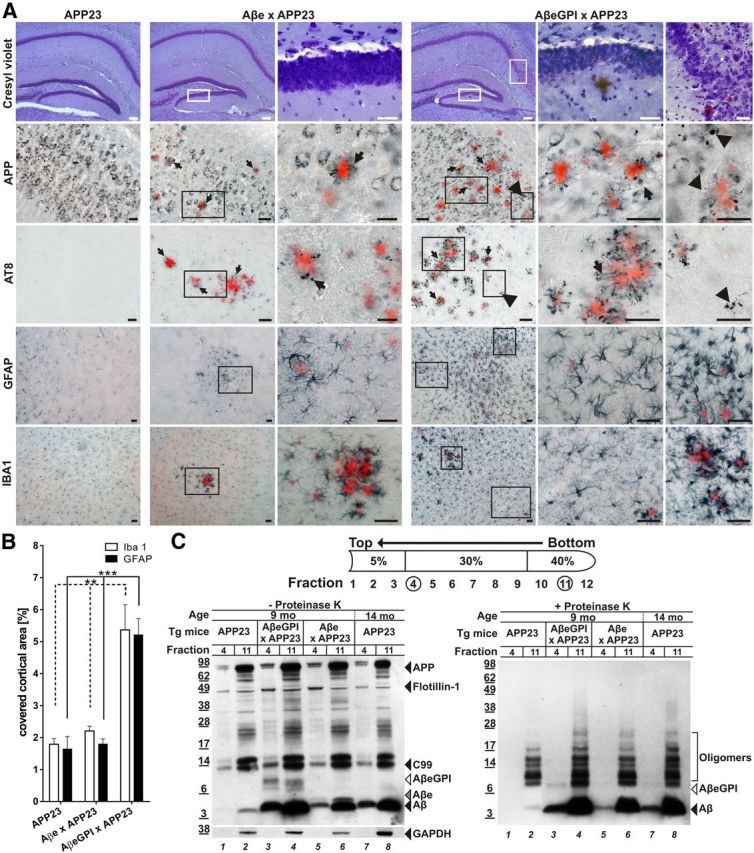
AβeGPI exacerbates amyloid-associated toxicity and promotes localization of aggregated Aβ species in DRM. A, Degenerative processes in double tg Aβe × APP23 and AβeGPI × APP23 mice. Hippocampal brain sections of 9-month-old tg mice stained with cresyl violet, anti-APP, anti-Tau (AT8), GFAP, and Iba1, respectively. All sections were additionally stained with Congo red. Higher magnifications for the double tg mice are shown in the right columns. In double tg AβeGPI × APP23 mice, displaced neurons and neuron loss are found at the sites of plaque deposition in neuronal loss at the stratum granulosum of the dentate gyrus and the CA1 stratum pyramidale of the hippocampus. Both double tg mice show dystrophic APP-positive synaptic boutons (arrows) and AT8-positive neuritic structures (arrows) in proximity to congophilic plaques. In addition, in the AβeGPI × APP23 mice APP-positive and AT8-positive dystrophic structures are found in vicinity of amorphous and Congo red-negative Aβ deposits (arrowheads). Both double tg lines display activations of astrocytes and microglia. However, in the AβeGPI × APP23 mice additional activated glia cell are found that are not restricted to the direct proximity of the congophilic deposits. Scale bar, 40 μm. B, Morphometric analysis revealed a significant increase of the area covered by Iba1 and GFAP staining in double tg AβeGPI × APP23 mice compared with Aβe × APP23 and APP23 mice (n = 5 for APP23, n = 7 for Aβe × APP23, n = 10 for AβeGPI × APP23; **p < 0.01, ***p < 0.001, one-way ANOVA Bonferroni's post hoc test for multiple comparisons). C, Left DRM extraction from brain homogenate treated with cold Triton X-100, followed by OptiPrep step density gradient. DRM proteins (fraction 4) and total protein load (fraction 11) were compared in 9-month-old tg APP23, double tg AβeGPI × APP23, Aβe × APP23, and 14-month-old tg APP23 mice by Western blot probed with 6E10. Aβ is present in all DRM fractions except for the 9-month-old APP23 predepositing mice (lanes 1 and 2). Flotillin-1 marker for DRMs; GAPDH marker for cytoplasmic protein. Right, Fractions from left treated with 100 μg/ml proteinase K (+PK) for 30 min and analyzed by Western blot probed with Aβ antibody 6E10. DRM-associated Aβ of respective tg mice (lanes 3, 5, and 7) displayed PK resistance.
PK treatment of DRM fractions prepared from the different mice revealed PK-resistant Aβ, Aβe, and AβeGPI in the DRM fractions (Fig. 5A,B). Interestingly, the highest amount of PK-resistant DRM material was found in the double tg AβeGPI × APP23 mice. This finding suggests that, in addition to increased levels of Aβ deposition, AβeGPI × APP23 mice have more membrane-associated Aβ aggregates, which have been suspected to cause neurotoxicity (Rushworth and Hooper, 2010).
Discussion
This study provides in vivo evidence for the participation of membrane-associated Aβ42 peptide in promoting Aβ aggregation and neurotoxicity. To this end, a novel tg mouse model expressing artificially AβeGPI on the membrane surface of neurons was generated. To exclude concerns that modifications at the C terminus of the Aβ42 peptide might interfere with the aggregation abilities of Aβ, we devised an extended form of secreted Aβ (Aβe) consisting of amino acid residues identical to AβeGPI, but without the GPI anchor.
The influence of biological membranes on protein aggregation has been described from in vitro studies using a variety of disease-associated, proteopathic proteins such as Aβ, α-synuclein, and PrP (Yip et al., 2001; Kazlauskaite et al., 2003; Zhu et al., 2003; Bokvist et al., 2004). More recently, a common phenomenon, known as “macromolecular crowding” has been described in which membranes locally concentrate proteins (Bokvist and Gröbner, 2007). Local protein accumulation has been shown to promote the conversion of Aβ and PrP to β-sheet-rich structures (Wang et al., 2007; Byström et al., 2008). Lipid rafts, which essentially are a membranous subcompartment, have thus received increased attention in AD and prion disease research. PrP, the fundamental component of prion infectivity, is GPI anchored and therefore a raft protein (Vey et al., 1996). APP, the source for AD-linked Aβ peptide, is proteolytically processed by β- and γ-secretase, a process reported to be raft associated (for review, see Rushworth and Hooper, 2010). Furthermore, lipid raft fractions isolated from human AD brains and transgenic mouse models showed high amounts of Aβ dimers (Kawarabayashi et al., 2004), and main components of lipid rafts have been found to be associated with amyloid fibrils, pointing to a common cellular mechanism of amyloid formation (Gellermann et al., 2005). Therefore, lipid rafts may be sites of enhanced Aβ aggregation and are important in AD pathogenesis.
In the present study, Aβ was linked to membranes via the PrP GPI anchor. This strategy was used for several reasons, as follows: (1) GPI is known to direct proteins to DRMs, resulting in higher local concentrations of the GPI-linked peptide, which might assist in the initial steps of oligomerization; (2) GPI is a very flexible domain that provides a higher degree of rotational and bending freedom for the anchored protein than a classical transmembrane domain, and this flexibility might be crucial for Aβ oligomerization; (3) GPI-anchored proteins can easily be shed, if membrane release is required for further Aβ aggregation (Borchelt et al., 1993); and (4) DRMs are found to be present in exosomes, which have already been suspected to play a role in neurodegenerative diseases like AD (Rajendran et al., 2006; Ghidoni et al., 2008; Sharples et al., 2008).
First, we used cell culture models to provide evidence that Aβ42 can be stably expressed on membrane surfaces via a GPI anchor, and that membrane-anchored Aβ42 localizes to DRMs. Cytotoxicity was not observed when AβeGPI was expressed in HEK cells. This could be due to the rapid internalization of available membrane surface AβeGPI by the cell, a process also described for PrP (Rieger et al., 1997). Moreover, it has also been reported that neuronal cell lines (e.g., N2A and PC12 cells) chronically replicate infectious prions but lack a cytotoxic phenotype (Rubenstein et al., 1984; Race et al., 1987; Bosque and Prusiner, 2000; Klöhn et al., 2003).
To investigate the consequences of Aβe and AβeGPI expression on neurons in vivo, Thy1 promoter-driven transgenic mice were generated. The levels of Aβe and AβeGPI in the mice reached only a fraction of the Aβ levels present in APP23 mice, which use the same Thy1 promoter element. Even at 24 months of age, both Aβe and AβeGPI tg mouse lines displayed no spontaneous Aβ deposition. This suggests that the critical threshold for spontaneous aggregation was not reached in the single tg Aβe and AβeGPI mice. Such a dose-dependent phenomenon has been reported for PrP. Depending on expression levels, mice overexpressing anchorless PrP spontaneously deposit PrP plaques even in the absence of an infectious seed, while mice with a lower expression level deposited PrP only after prion inoculation (Chesebro et al., 2005, 2010; Stöhr et al., 2011).
To study the amyloidogenicity of the membrane-bound Aβ peptides, we crossed single tg AβeGPI and Aβe mice with APP23 mice, and analyzed Aβ plaque deposition at the age of 9 months. Amyloid formation depends on sufficient available soluble Aβ and Aβ seeds (Harper and Lansbury, 1997). If the C-terminal extension of Aβ had interfered with the amyloid formation in the double tg mice, the onset of Aβ deposition would have been delayed (Eisenberg and Jucker, 2012). However, Aβ plaque deposition in double tg mice was found to be increased when compared with APP23 tg mice. A previous study has shown that plaque-depositing transgenic mice contain PK-resistant Aβ species (Langer et al., 2011). Consistently, PK resistance was also found in the AβeGPI × APP23 and Aβe × APP23 mice, indicating that the extended versions AβeGPI and Aβe acquired the amyloidogenic state, and were amyloid competent similar to the Aβ derived from tg APP23 mice. This observation resembles the interaction between anchorless and GPI-anchored PrP for the prion pathogenesis (Chesebro et al., 2005).
Both AβeGPI × APP23 and Aβe × APP23 tg mouse lines developed more amyloid lesions than age-matched single tg APP23 littermates, suggesting that the process of deposition starts earlier. However, despite the higher steady-state levels of Aβe compared with AβeGPI in the respective mice, AβeGPI was more efficient in promoting Aβ accumulation and plaque formation. The accelerated plaque formation in AβeGPI × APP23 mice indicates that the tethering of AβeGPI to DRMs by the GPI anchor provided a better nucleus for Aβ aggregation than soluble Aβe, which resulted in recruiting more Aβ. This is also consistent with the finding that AβeGPI showed a high propensity to induce Aβ fibrillization in vitro even at a young age.
Double tg AβeGPI × APP23 and Aβe × APP23 mice differed in amyloid morphotypes and neurotoxicity. Amorphous Congo red-negative deposits were observed only in AβeGPI × APP23 mice, not in age-matched Aβe × APP23 mice, and are likely the result of the Aβ attachment to the membrane. Congophilic, but also amorphous, deposits were surrounded by neuritic hyperphosphorylated tau-positive structures and dystrophic boutons. The inflammatory response whose controversial role in AD has been discussed before (for review, see Guillot-Sestier and Town, 2013) was significantly higher in AβeGPI × APP23 double transgenic mice. Activation of astrocytes and microglia was not restricted to direct proximity of congophilic deposits. It was completely absent in both single transgenic Aβe and AβeGPI mice. These observations are reminiscent to the distinct plaque morphology of specific prion strains with or without PrP membrane attachment. For example, the Rocky Mountain Laboratories (RML) prion strain normally deposits in wild-type mice only diffusely, with no defined plaque deposits inducing clinical signs of disease and causing lethality. When the same RML inoculation strain is replicating in a mouse with anchorless PrP, the mice develop large congophilic plaques but lack a clinical phenotype (Chesebro et al., 2005). Thus, Aβ attachment to the membrane and its localization in DRMs might be a prerequisite for its neurotoxic abilities, similar to prion disease. Up to 15 months of age, none of the AβeGPI × APP23 mice displayed a lethal phenotype. However, one cannot exclude that a lethal phenotype will occur with further aging of the mice or at higher expression levels of the AβeGPI construct.
In conclusion, the membrane-anchored Aβ mouse model described here demonstrates in vivo that lipid membranes facilitate the conversion of Aβ to aggregated, amyloid-seeding Aβ species and exacerbate amyloid-associated toxicity. Thus, Aβ–membrane interactions may play a pivotal role in the onset of AD pathogenesis and provide a new therapeutic target, which can now be experimentally tested.
Footnotes
This project was supported by the German Research Foundation DFG Grant BA2257-2 (F.B.). We thank N. Varvel, A. Marzesco, and Y. Eisele for advice; and A. Skodras, A. Bosch, U. Obermueller, I. Breuer, M. Hruscha, C. Krueger, C. Leibssle, and J. Odenthal for experimental help and support. We are grateful to M. Mercken (Johnson & Johnson Pharmaceutical Research & Development) for providing the JRF/AbN/25 Aβ antibody; to T. Golde for kindly providing the plasmid pAG3 BriAβ42; and to M. Faendrich for supplying recombinant Aβ1-40.
References
- Andrä K, Abramowski D, Duke M, Probst A, Wiederhold KH, Bürki K, Goedert M, Sommer B, Staufenbiel M. Expression of APP in transgenic mice: a comparison of neuron-specific promoters. Neurobiol Aging. 1996;17:183–190. doi: 10.1016/0197-4580(95)02066-7. [DOI] [PubMed] [Google Scholar]
- Baumann F, Tolnay M, Brabeck C, Pahnke J, Kloz U, Niemann HH, Heikenwalder M, Rülicke T, Bürkle A, Aguzzi A. Lethal recessive myelin toxicity of prion protein lacking its central domain. EMBO J. 2007;26:538–547. doi: 10.1038/sj.emboj.7601510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baumann F, Pahnke J, Radovanovic I, Rülicke T, Bremer J, Tolnay M, Aguzzi A. Functionally relevant domains of the prion protein identified in vivo. PLoS One. 2009;4:e6707. doi: 10.1371/journal.pone.0006707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bokvist M, Gröbner G. Misfolding of amyloidogenic proteins at membrane surfaces: the impact of macromolecular crowding. J Am Chem Soc. 2007;129:14848–14849. doi: 10.1021/ja076059o. [DOI] [PubMed] [Google Scholar]
- Bokvist M, Lindström F, Watts A, Gröbner G. Two types of Alzheimer's beta-amyloid (1–40) peptide membrane interactions: aggregation preventing transmembrane anchoring versus accelerated surface fibril formation. J Mol Biol. 2004;335:1039–1049. doi: 10.1016/j.jmb.2003.11.046. [DOI] [PubMed] [Google Scholar]
- Borchelt DR, Rogers M, Stahl N, Telling G, Prusiner SB. Release of the cellular prion protein from cultured cells after loss of its glycoinositol phospholipid anchor. Glycobiology. 1993;3:319–329. doi: 10.1093/glycob/3.4.319. [DOI] [PubMed] [Google Scholar]
- Bosque PJ, Prusiner SB. Cultured cell sublines highly susceptible to prion infection. J Virol. 2000;74:4377–4386. doi: 10.1128/JVI.74.9.4377-4386.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byström R, Aisenbrey C, Borowik T, Bokvist M, Lindström F, Sani MA, Olofsson A, Gröbner G. Disordered proteins: biological membranes as two-dimensional aggregation matrices. Cell Biochem Biophys. 2008;52:175–189. doi: 10.1007/s12013-008-9033-4. [DOI] [PubMed] [Google Scholar]
- Calhoun ME, Wiederhold KH, Abramowski D, Phinney AL, Probst A, Sturchler-Pierrat C, Staufenbiel M, Sommer B, Jucker M. Neuron loss in APP transgenic mice. Nature. 1998;395:755–756. doi: 10.1038/27351. [DOI] [PubMed] [Google Scholar]
- Chesebro B, Trifilo M, Race R, Meade-White K, Teng C, LaCasse R, Raymond L, Favara C, Baron G, Priola S, Caughey B, Masliah E, Oldstone M. Anchorless prion protein results in infectious amyloid disease without clinical scrapie. Science. 2005;308:1435–1439. doi: 10.1126/science.1110837. [DOI] [PubMed] [Google Scholar]
- Chesebro B, Race B, Meade-White K, Lacasse R, Race R, Klingeborn M, Striebel J, Dorward D, McGovern G, Jeffrey M. Fatal transmissible amyloid encephalopathy: a new type of prion disease associated with lack of prion protein membrane anchoring. PLoS Pathog. 2010;6:e1000800. doi: 10.1371/journal.ppat.1000800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chi EY, Ege C, Winans A, Majewski J, Wu G, Kjaer K, Lee KY. Lipid membrane templates the ordering and induces the fibrillogenesis of Alzheimer's disease amyloid-beta peptide. Proteins. 2008;72:1–24. doi: 10.1002/prot.21887. [DOI] [PubMed] [Google Scholar]
- Colby DW, Zhang Q, Wang S, Groth D, Legname G, Riesner D, Prusiner SB. Prion detection by an amyloid seeding assay. Proc Natl Acad Sci U S A. 2007;104:20914–20919. doi: 10.1073/pnas.0710152105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisele YS, Obermüller U, Heilbronner G, Baumann F, Kaeser SA, Wolburg H, Walker LC, Staufenbiel M, Heikenwalder M, Jucker M. Peripherally applied Abeta-containing inoculates induce cerebral beta-amyloidosis. Science. 2010;330:980–982. doi: 10.1126/science.1194516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisenberg D, Jucker M. The amyloid state of proteins in human diseases. Cell. 2012;148:1188–1203. doi: 10.1016/j.cell.2012.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer M, Rülicke T, Raeber A, Sailer A, Moser M, Oesch B, Brandner S, Aguzzi A, Weissmann C. Prion protein (PrP) with amino-proximal deletions restoring susceptibility of PrP knockout mice to scrapie. EMBO J. 1996;15:1255–1264. [PMC free article] [PubMed] [Google Scholar]
- Franklin KBJ, Paxinos G. The Mouse Brain in Stereotaxic Coordinates. San Diego: Academic; 2004. [Google Scholar]
- Gellermann GP, Appel TR, Tannert A, Radestock A, Hortschansky P, Schroeckh V, Leisner C, Lutkepohl T, Shtrasburg S, Röcken C, Pras M, Linke RP, Diekmann S, Fändrich M. Raft lipids as common components of human extracellular amyloid fibrils. Proc Natl Acad Sci U S A. 2005;102:6297–6302. doi: 10.1073/pnas.0407035102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghidoni R, Benussi L, Binetti G. Exosomes: the Trojan horses of neurodegeneration. Med Hypotheses. 2008;70:1226–1227. doi: 10.1016/j.mehy.2007.12.003. [DOI] [PubMed] [Google Scholar]
- Guillot-Sestier MV, Town T. Innate immunity in Alzheimer's disease: a complex affair. CNS and neurological disorders drug targets. 2013;12:593–607. doi: 10.2174/1871527311312050008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haass C, De Strooper B. The presenilins in Alzheimer's disease—proteolysis holds the key. Science. 1999;286:916–919. doi: 10.1126/science.286.5441.916. [DOI] [PubMed] [Google Scholar]
- Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer's disease: progress and problems on the road to therapeutics. Science. 2002;297:353–356. doi: 10.1126/science.1072994. [DOI] [PubMed] [Google Scholar]
- Harper JD, Lansbury PT., Jr Models of amyloid seeding in Alzheimer's disease and scrapie: mechanistic truths and physiological consequences of the time-dependent solubility of amyloid proteins. Annu Rev Biochem. 1997;66:385–407. doi: 10.1146/annurev.biochem.66.1.385. [DOI] [PubMed] [Google Scholar]
- Kapur JN, Sahoo PK, Wong ACK. A new method for gray-level picture thresholding using the entropy of the histogram. Graph Models Image Proc. 1985;29:273–285. doi: 10.1016/0734-189X(85)90125-2. [DOI] [Google Scholar]
- Kawarabayashi T, Shoji M, Younkin LH, Wen-Lang L, Dickson DW, Murakami T, Matsubara E, Abe K, Ashe KH, Younkin SG. Dimeric amyloid β protein rapidly accumulates in lipid rafts followed by apolipoprotein E and phosphorylated tau accumulation in the Tg2576 mouse model of Alzheimer's disease. J Neurosci. 2004;24:3801–3809. doi: 10.1523/JNEUROSCI.5543-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kazlauskaite J, Sanghera N, Sylvester I, Vénien-Bryan C, Pinheiro TJ. Structural changes of the prion protein in lipid membranes leading to aggregation and fibrillization. Biochemistry. 2003;42:3295–3304. doi: 10.1021/bi026872q. [DOI] [PubMed] [Google Scholar]
- Klöhn PC, Stoltze L, Flechsig E, Enari M, Weissmann C. A quantitative, highly sensitive cell-based infectivity assay for mouse scrapie prions. Proc Natl Acad Sci U S A. 2003;100:11666–11671. doi: 10.1073/pnas.1834432100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langer F, Eisele YS, Fritschi SK, Staufenbiel M, Walker LC, Jucker M. Soluble Aβ seeds are potent inducers of cerebral β-amyloid deposition. J Neurosci. 2011;31:14488–14495. doi: 10.1523/JNEUROSCI.3088-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SJ, Liyanage U, Bickel PE, Xia W, Lansbury PT, Jr, Kosik KS. A detergent-insoluble membrane compartment contains A beta in vivo. Nat Med. 1998;4:730–734. doi: 10.1038/nm0698-730. [DOI] [PubMed] [Google Scholar]
- Lewis PA, Piper S, Baker M, Onstead L, Murphy MP, Hardy J, Wang R, McGowan E, Golde TE. Expression of BRI-amyloid beta peptide fusion proteins: a novel method for specific high-level expression of amyloid beta peptides. Biochim Biophys Acta. 2001;1537:58–62. doi: 10.1016/S0925-4439(01)00054-0. [DOI] [PubMed] [Google Scholar]
- Lingwood D, Simons K. Detergent resistance as a tool in membrane research. Nat Protoc. 2007;2:2159–2165. doi: 10.1038/nprot.2007.294. [DOI] [PubMed] [Google Scholar]
- Mathews PM, Jiang Y, Schmidt SD, Grbovic OM, Mercken M, Nixon RA. Calpain activity regulates the cell surface distribution of amyloid precursor protein. Inhibition of calpains enhances endosomal generation of beta-cleaved C-terminal APP fragments. J Biol Chem. 2002;277:36415–36424. doi: 10.1074/jbc.M205208200. [DOI] [PubMed] [Google Scholar]
- McGowan E, Pickford F, Kim J, Onstead L, Eriksen J, Yu C, Skipper L, Murphy MP, Beard J, Das P, Jansen K, Delucia M, Lin WL, Dolios G, Wang R, Eckman CB, Dickson DW, Hutton M, Hardy J, Golde T. Abeta42 is essential for parenchymal and vascular amyloid deposition in mice. Neuron. 2005;47:191–199. doi: 10.1016/j.neuron.2005.06.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naslavsky N, Stein R, Yanai A, Friedlander G, Taraboulos A. Characterization of detergent-insoluble complexes containing the cellular prion protein and its scrapie isoform. J Biol Chem. 1997;272:6324–6331. doi: 10.1074/jbc.272.10.6324. [DOI] [PubMed] [Google Scholar]
- Nielsen L, Khurana R, Coats A, Frokjaer S, Brange J, Vyas S, Uversky VN, Fink AL. Effect of environmental factors on the kinetics of insulin fibril formation: elucidation of the molecular mechanism. Biochemistry. 2001;40:6036–6046. doi: 10.1021/bi002555c. [DOI] [PubMed] [Google Scholar]
- Race RE, Fadness LH, Chesebro B. Characterization of scrapie infection in mouse neuroblastoma cells. J Gen Virol. 1987;68:1391–1399. doi: 10.1099/0022-1317-68-5-1391. [DOI] [PubMed] [Google Scholar]
- Rajendran L, Honsho M, Zahn TR, Keller P, Geiger KD, Verkade P, Simons K. Alzheimer's disease beta-amyloid peptides are released in association with exosomes. Proc Natl Acad Sci U S A. 2006;103:11172–11177. doi: 10.1073/pnas.0603838103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rieger R, Edenhofer F, Lasmézas CI, Weiss S. The human 37-kDa laminin receptor precursor interacts with the prion protein in eukaryotic cells. Nat Med. 1997;3:1383–1388. doi: 10.1038/nm1297-1383. [DOI] [PubMed] [Google Scholar]
- Rubenstein R, Carp RI, Callahan SM. In vitro replication of scrapie agent in a neuronal model: infection of PC12 cells. J Gen Virol. 1984;65:2191–2198. doi: 10.1099/0022-1317-65-12-2191. [DOI] [PubMed] [Google Scholar]
- Rushworth JV, Hooper NM. Lipid rafts: linking Alzheimer's amyloid-beta production, aggregation, and toxicity at neuronal membranes. Int J Alzheimers Dis. 2010;2011:603052. doi: 10.4061/2011/603052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rutishauser D, Mertz KD, Moos R, Brunner E, Rülicke T, Calella AM, Aguzzi A. The comprehensive native interactome of a fully functional tagged prion protein. PLoS One. 2009;4:e4446. doi: 10.1371/journal.pone.0004446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharples RA, Vella LJ, Nisbet RM, Naylor R, Perez K, Barnham KJ, Masters CL, Hill AF. Inhibition of gamma-secretase causes increased secretion of amyloid precursor protein C-terminal fragments in association with exosomes. FASEB J. 2008;22:1469–1478. doi: 10.1096/fj.07-9357com. [DOI] [PubMed] [Google Scholar]
- Stöhr J, Watts JC, Legname G, Oehler A, Lemus A, Nguyen HO, Sussman J, Wille H, DeArmond SJ, Prusiner SB, Giles K. Spontaneous generation of anchorless prions in transgenic mice. Proc Natl Acad Sci U S A. 2011;108:21223–21228. doi: 10.1073/pnas.1117827108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sturchler-Pierrat C, Abramowski D, Duke M, Wiederhold KH, Mistl C, Rothacher S, Ledermann B, Bürki K, Frey P, Paganetti PA, Waridel C, Calhoun ME, Jucker M, Probst A, Staufenbiel M, Sommer B. Two amyloid precursor protein transgenic mouse models with Alzheimer disease-like pathology. Proc Natl Acad Sci U S A. 1997;94:13287–13292. doi: 10.1073/pnas.94.24.13287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vandermeeren M, Geraerts M, Pype S, Dillen L, Van Hove C, Mercken M. The functional gamma-secretase inhibitor prevents production of amyloid beta 1–34 in human and murine cell lines. Neurosci Lett. 2001;315:145–148. doi: 10.1016/S0304-3940(01)02369-2. [DOI] [PubMed] [Google Scholar]
- Vey M, Pilkuhn S, Wille H, Nixon R, DeArmond SJ, Smart EJ, Anderson RG, Taraboulos A, Prusiner SB. Subcellular colocalization of the cellular and scrapie prion proteins in caveolae-like membranous domains. Proc Natl Acad Sci U S A. 1996;93:14945–14949. doi: 10.1073/pnas.93.25.14945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang F, Yang F, Hu Y, Wang X, Jin C, Ma J. Lipid interaction converts prion protein to a PrPSc-like proteinase K-resistant conformation under physiological conditions. Biochemistry. 2007;46:7045–7053. doi: 10.1021/bi700299h. [DOI] [PubMed] [Google Scholar]
- Williamson R, Sutherland C. Neuronal membranes are key to the pathogenesis of Alzheimer's disease: the role of both raft and non-raft membrane domains. Curr Alzheimer Res. 2011;8:213–221. doi: 10.2174/156720511795256008. [DOI] [PubMed] [Google Scholar]
- Yamaguchi H, Maat-Schieman ML, van Duinen SG, Prins FA, Neeskens P, Natté R, Roos RA. Amyloid beta protein (Abeta) starts to deposit as plasma membrane-bound form in diffuse plaques of brains from hereditary cerebral hemorrhage with amyloidosis-Dutch type, Alzheimer disease and nondemented aged subjects. J Neuropathol Exp Neurol. 2000;59:723–732. doi: 10.1093/jnen/59.8.723. [DOI] [PubMed] [Google Scholar]
- Yip CM, Elton EA, Darabie AA, Morrison MR, McLaurin J. Cholesterol, a modulator of membrane-associated Abeta-fibrillogenesis and neurotoxicity. J Mol Biol. 2001;311:723–734. doi: 10.1006/jmbi.2001.4881. [DOI] [PubMed] [Google Scholar]
- Zhu M, Li J, Fink AL. The association of alpha-synuclein with membranes affects bilayer structure, stability, and fibril formation. J Biol Chem. 2003;278:40186–40197. doi: 10.1074/jbc.M305326200. [DOI] [PubMed] [Google Scholar]