

Abstract
PMS2 is one of the four susceptibility genes in Lynch syndrome (LS), the most common cancer syndrome in the world. Inherited mutations in DNA mismatch repair (MMR) genes, MLH1, MSH2, and MSH6, account for approximately 90% of LS, while a relatively small number of LS families segregate a PMS2 mutation. This and the low cancer penetrance in PMS2 families suggest that PMS2 is only a moderate or low‐risk susceptibility gene. We have previously shown that even a partial expression decrease in MLH1, MSH2, or MSH6 suggests that heterozygous LS mutation carriers have MMR malfunction in constitutive tissues. Whether and how PMS2 expression decrease affects the repair capability is not known. Here, we show that PMS2 knockdown cells retaining 19%, 33%, or 53% of PMS2 expression all have significantly reduced MMR efficiency. Surprisingly, the cells retaining expression levels comparable to PMS2 mutation carriers indicate the lowest repair efficiency.
Keywords: colorectal cancer, Lynch syndrome, mismatch repair, mRNA expression, PMS2
As a component of the MutLα (MLH1 + PMS2) heterodimer, PMS2 (postmeiotic segregation increased 2) is central in the postreplicative human DNA mismatch repair (MMR) mechanism (Kunkel & Erie, 2005). Heterozygous germline mutations in MMR genes cause Lynch syndrome (LS; MIM# 120435), the most common cancer predisposition syndrome in human (de la Chapelle, 2004; Lynch, Snyder, Shaw, Heinen, & Hitchins, 2015). Mutations in MLH1 (MIM# 120436) and MSH2 (MIM# 609309) are the most prevalent in known LS families (36.2% and 35.3%, respectively), while the number of MSH6 (MIM# 600678) and PMS2 (MIM# 600259) mutations is lower (19.4% and 9.1%, respectively; www.insight‐database.org, data accessed January 22, 2019). The absence of functional MMR protein results in DNA repair malfunction, which is the cause of early‐onset colorectal and endometrial cancers and less frequently cancer of the ovaries, stomach, pancreas, prostate, and breast in LS families (Lynch et al., 2015). However, in PMS2 families the prevalence of stomach, prostate, and breast cancers is unusually high and compared to MLH1 and MSH2 families, also the mean age of cancer onset is higher and the penetrance of colorectal and endometrial cancers lower, resembling the situation in MSH6 families (Hendriks et al., 2006; Roberts et al., 2018; Senter et al., 2008; ten Broeke et al., 2015; Truninger et al., 2005). One explanation for the lower cancer penetrance in PMS2 families might be the protein homologue MLH3, which is also able to bind and partially function with MLH1 (Cannavo et al., 2005; Korhonen, Raevaara, Lohi, & Nystrom, 2007; Senter et al., 2008; ten Broeke et al., 2015).
The diagnostic workflow to identify Lynch syndrome typically involves tumor studies and DNA analyses. The first clinical step in diagnosing LS includes tumor immunohistochemistry (IHC) and microsatellite instability (MSI) analyses followed by mutation screening dictated by the IHC and MSI results (Kansikas, Kariola, & Nystrom, 2011; Yurgelun & Hampel, 2018). While IHC is a valuable method for distinguishing PMS2 carriers, who generally lose only PMS2, from MLH1 carriers, who frequently lose both MLH1 and PMS2 protein expressions in a tumor (Niessen et al., 2009), it took a decade to differentiate sequence between PMS2 and its 15 pseudogenes for reliable sequencing and mutation detection (De Vos, Hayward, Picton, Sheridan, & Bonthron, 2004; van der Klift et al., 2010; Vaughn et al., 2010). As an outcome, the prevalence of PMS2 mutations in suspected LS families has increased according to the InSiGHT database, from approximately 2–9% during the last two decades (www.insight‐database.org). Furthermore, the population‐based PMS2 variant prevalence was recently reported to be relatively high (1:714) compared to the variant prevalence in MLH1 (1:1946) and MSH2 (1:2841; Win et al., 2017) and a strikingly high prevalence (96%) was reported from an Icelandic population using whole genome sequencing data of a colorectal cancer cohort (Haraldsdottir et al., 2017).
Nevertheless, atypical clinical phenotypes and a low cancer penetrance in PMS2 families still question its role in Lynch syndrome. Hence, this study aimed to determine the significance of PMS2 as an LS susceptibility gene by analyzing whether a partial PMS2 expression decrease, compared to the expression decrease in a Lynch syndrome mutation carrier, causes mismatch repair malfunction, as was previously shown to be the case with the susceptibility genes MLH1, MSH2, and MSH6.
Accordingly, stable short hairpin RNA (shRNA) mediated PMS2 knockdown (KD) clones were established and characterized by real‐time polymerase chain reaction (RT‐qPCR), and the clones with reduced PMS2 expression were included in functional MMR analyses (Supporting Information Materials, Materials & Methods). Each KD clone had its own specific control across all studies to avoid experimental artifacts. A prerequisite for the functional analysis was that the PMS2 messenger RNA (mRNA) expression level should be at most ~50% of the normal level, a level comparable to that of an LS mutation carrier. From a total of 16 clones, the three retaining 19%, 33%, and 53% of PMS2 mRNA expression met the criteria for further MMR analysis.
To be able to assess the repair efficiency of each clone, the nuclear proteins of the cells in chosen clones and their controls were extracted in parallel as described previously (Holmes, Clark, & Modrich, 1990; Kantelinen et al., 2010). The nuclear protein expression was analyzed by western blot analysis (WB; Supporting Information Materials, Materials & Methods; Figure 1). Despite the semiquantitative nature of the method, each KD clone clearly showed decreased PMS2 protein expression compared to the level in its respective control (NC), indicating that the amount of protein reflects the reduced mRNA expression in the cell extract. Importantly, the MLH1 protein expression was shown to be normal in the KD extracts.
Figure 1.
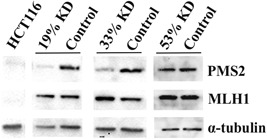
Western blot analysis of KD extracts with reduced PMS2 mRNA expression. Western blot analysis was used to evaluate the PMS2 protein expression of KD extracts and their controls. Commercial HCT116 cell line (Manassas, Virginia; ATCC® CCL‐247™) lacks both MLH1 and PMS2 protein and served as a negative control, α‐tubulin was used as a loading control. The results demonstrated decreased PMS2 protein expression in KD extracts, while the amount of MLH1 remained normal. Moreover, PMS2 protein expression increased according to mRNA expression in the clones. KD: knockdown; mRNA: messenger RNA
For a long time, PMS2 mutation prevalence was underestimated, suggesting that PMS2 is not an important LS susceptibility gene. Its significance in LS has been questioned by the atypically low cancer penetrance in PMS2 families (Senter et al., 2008; ten Broeke et al., 2015), an unclear risk for extra‐colonic cancers (Roberts et al., 2018; ten Broeke et al., 2018), as well as inconsistencies across studies due to variable cohort sizes and carrier ascertainment (compliant with Amsterdam II criteria and/or revised Bethesda Guidelines; Senter et al., 2008; ten Broeke et al., 2015; ten Broeke et al., 2018; ten Broeke, Suerink, & Nielsen, 2018). For these reasons, PMS2‐specific surveillance methods remain under debate (Espenschied et al., 2017; ten Broeke et al., 2018).
Nevertheless, over 20% of PMS2 carriers are still estimated to be missed with the conventional criteria used for LS identification (Senter et al., 2008; ten Broeke et al., 2015). The IHC‐ and MSI‐ studies help the pathogenicity assessment since up to 99% of PMS2 carriers have been shown to lack PMS2 in tumor tissue, and 98% of tumors have been characterized as MSI‐high (Goodenberger et al., 2016). Since both these tumor phenotypes signal MMR deficiency, it was anticipated that PMS2 expression decrease should also interfere with MMR capability of the cells, as was previously shown to be the case with the other LS susceptibility genes (Kansikas, Kasela, Kantelinen, & Nystrom, 2014). Indeed, the functional in vitro MMR assay (Nyström‐Lahti et al., 2002; Raevaara et al., 2003; Supporting Information Materials, Materials & Methods) demonstrated lowered MMR efficiency in cells with reduced PMS2 mRNA expression, while all controls maintained higher repair proficiencies throughout three independent assays (Figure 2a). The reduction in MMR capability in cells retaining 19%, 33%, or 53% of PMS2 mRNA expression was statistically significant (according to the Student's t test) in all, showing 21%, 15%, and 12% of average repair efficiency, while their controls showed 38%, 55%, and 48% of repair capability, respectively (P = 0.002, 0.001, and 0.0002; Figure 2a). Interestingly, the most severe repair defect was in cells retaining 53% of PMS2 expression. The relative repair efficiencies (RR%) of cells retaining 19%, 33%, and 53% of PMS2 mRNA expression were 55%, 27%, and 25%, respectively (Figure 2b), confirming the finding that the KD cells with 53% PMS2 expression had the lowest repair capability.
Figure 2.

Functional MMR efficiency of KD extracts with reduced PMS2 mRNA expression. (a) To investigate the effects of reduced PMS2 mRNA expression on the MMR capability, nuclear proteins extracted from KD cells were analyzed in the functional MMR assay. The average of absolute repair efficiencies (AR%, n = 3) of KD extracts and their controls were calculated from three repetitions and are illustrated below the gel electrophoresis picture. A significant decrease in the MMR efficiency was observed in all extracts retaining 19%, 33%, or 53% of PMS2 mRNA expression compared with their respective controls (P = 0.002; 0.001; 0.0002, respectively). (b) Relative repair efficiencies (RR%) were calculated from the AR% values of the KD extracts relative to the AR% values of their respective controls (set to 100). These results further confirm the finding that the cells with 53% PMS2 expression have the lowest repair capability. KD: knockdown; MMR: mismatch repair; mRNA: messenger RNA; SD: standard deviation
Previously, a significant reduction in MMR efficiency was detected in cells retaining 75% of MSH6, 50% of MSH2, and 25% of MLH1 mRNA expression (Kansikas et al., 2014). In this study, we show that PMS2 expression decrease also affects MMR capability but unexpectedly in a different way than what was observed with the other MMR genes. Surprisingly, the lower PMS2 expression levels (19% and 33%) had higher repair efficiencies than the carrier‐like level (53%), while in MLH1 and MSH2, 25% expression level was not enough to maintain any repair capability (25% level in MSH6 was not studied; Kansikas et al., 2014). The severity of MLH1 and MSH2 expression decrease may be due to their obligatory roles in the heterodimeric protein complexes (Acharya et al., 1996; Kondo, Horii, & Fukushige, 2001), while such a low level of PMS2 may be compensated by some homologous protein. Compensation of MutLα with MutLγ (MLH1 + MLH3) has been suggested to explain the low penetrance of PMS2 mutations (Cannavo et al., 2005; Korhonen et al., 2007). Although PMS2 is the main partner of MLH1 among MutL homologues, MutLγ has shown at least low MMR capability (Cannavo et al., 2005; Raschle, Marra, Nystrom‐Lahti, Schar, & Jiricny, 1999). The nuclear localization of MLH3 seems to be dependent on the absence of PMS2 in the cell (Korhonen et al., 2007), and while excess of MLH3 has been shown to reduce PMS2 binding to MLH1, most probably MLH3 is not a significant counterpart to MLH1 in a normal cell due to the 60‐fold higher expression of PMS2 (Cannavo et al., 2005; Kondo et al., 2001). To determine whether MLH3 could compensate MMR efficiency in 19% and 33% PMS2 KD clones and improve their repair capability compared to the 53% clone, we further studied MLH3 mRNA expression and protein expression in the clones. Although, the MLH3 mRNA expression levels were decreased, they did not differ between the KD cells (Figure S1), even though slightly lowered MLH3 protein expression in 53% KD clone compared to 19% clone was detected by WB (Figure S2). While MLH3 expression differences do not explain the lowered MMR efficiency in KD clones, it is still possible that the cells expressing only 19% or 33% of PMS2 are able to activate a “survival mechanism” that sustains MMR capability.
By using a functional approach to assess the significance of PMS2 in Lynch syndrome, we show that PMS2 expression decrease causes severe problems in mismatch repair, which is the hallmark of LS susceptibility genes.
CONFLICT OF INTERESTS
Nyström and Kansikas are inventors on patent number PCT/EP2012/062708. Nyström is a shareholder and a board member and Kansikas and Kasela are employees of LS CancerDiag Ltd.
Supporting information
Supplementary information
ACKNOWLEDGEMENT
This study was supported by contract grant sponsors Jane and Aatos Erkko Foundation.
Kasela M, Nyström M, Kansikas M. PMS2 expression decrease causes severe problems in mismatch repair. Human Mutation. 2019;40:904–907. 10.1002/humu.23756
References
REFERENCES
- Acharya, S. , Wilson, T. , Gradia, S. , Kane, M. F. , Guerrette, S. , Marsischky, G. T. , … Fishel, R. (1996). hMSH2 forms specific mispair‐binding complexes with hMSH3 and hMSH6. Proceedings of the National Academy of Sciences of the United States of America, 93(24), 13629–13634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cannavo, E. , Marra, G. , Sabates‐Bellver, J. , Menigatti, M. , Lipkin, S. M. , Fischer, F. , & Jiricny, J. (2005). Expression of the MutL homologue hMLH3 in human cells and its role in DNA mismatch repair. Cancer Research, 65(23), 10759–10766. https://doi.org/65/23/10759 [DOI] [PubMed] [Google Scholar]
- de la Chapelle, A. (2004). Genetic predisposition to colorectal cancer. Nature Reviews.Cancer, 4(10), 769–780. https://doi.org/nrc1453 [DOI] [PubMed] [Google Scholar]
- De Vos, M. , Hayward, B. E. , Picton, S. , Sheridan, E. , & Bonthron, D. T. (2004). Novel PMS2 pseudogenes can conceal recessive mutations causing a distinctive childhood cancer syndrome. American Journal of Human Genetics, 74(5), 954–964. 10.1086/420796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Espenschied, C. R. , LaDuca, H. , Li, S. , McFarland, R. , Gau, C. L. , & Hampel, H. (2017). Multigene panel testing provides a new perspective on Lynch syndrome. Journal of Clinical Oncology, 35(22), 2568–2575. 10.1200/JCO.2016.71.9260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodenberger, M. L. , Thomas, B. C. , Riegert‐Johnson, D. , Boland, C. R. , Plon, S. E. , Clendenning, M. , … Lindor, N. M. (2016). PMS2 monoallelic mutation carriers: The known unknown. Genetics in Medicine, 18(1), 13–19. 10.1038/gim.2015.27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haraldsdottir, S. , Rafnar, T. , Frankel, W. L. , Einarsdottir, S. , Sigurdsson, A. , Hampel, H. , … Stefansson, K. (2017). Comprehensive population‐wide analysis of Lynch syndrome in iceland reveals founder mutations in MSH6 and PMS2. Nature Communications, 8, 14755 10.1038/ncomms14755 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hendriks, Y. M. C. , Jagmohan–Changur, S. , van der Klift, H. M. , Morreau, H. , van Puijenbroek, M. , Tops, C. , … Wijnen, J. T. (2006). Heterozygous mutations in PMS2 cause hereditary nonpolyposis colorectal carcinoma (Lynch syndrome). Gastroenterology, 130(2), 312–322. https://doi.org/S0016‐5085(05)02232‐8 [DOI] [PubMed] [Google Scholar]
- Holmes, J., Jr , Clark, S. , & Modrich, P. (1990). Strand‐specific mismatch correction in nuclear extracts of human and Drosophila melanogaster cell lines. Proceedings of the National Academy of Sciences of the United States of America, 87(15), 5837–5841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kansikas, M. , Kariola, R. , & Nystrom, M. (2011). Verification of the three‐step model in assessing the pathogenicity of mismatch repair gene variants. Human Mutation, 32(1), 107–115. 10.1002/humu.21409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kansikas, M. , Kasela, M. , Kantelinen, J. , & Nystrom, M. (2014). Assessing how reduced expression levels of the mismatch repair genes MLH1, MSH2, and MSH6 affect repair efficiency. Human Mutation, 35(9), 1123–1127. 10.1002/humu.22605 [DOI] [PubMed] [Google Scholar]
- Kantelinen, J. , Kansikas, M. , Korhonen, M. K. , Ollila, S. , Heinimann, K. , Kariola, R. , & Nystrom, M. (2010). MutSbeta exceeds MutSalpha in dinucleotide loop repair. British Journal of Cancer, 102(6), 1068–1073. 10.1038/sj.bjc.6605531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kondo, E. , Horii, A. , & Fukushige, S. (2001). The interacting domains of three MutL heterodimers in man: HMLH1 interacts with 36 homologous amino acid residues within hMLH3, hPMS1 and hPMS2. Nucleic Acids Research, 29(8), 1695–1702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korhonen, M. K. , Raevaara, T. E. , Lohi, H. , & Nystrom, M. (2007). Conditional nuclear localization of hMLH3 suggests a minor activity in mismatch repair and supports its role as a low‐risk gene in HNPCC. Oncology Reports, 17(2), 351–354. [PubMed] [Google Scholar]
- Kunkel, T. A. , & Erie, D. A. (2005). DNA mismatch repair. Annual Review of Biochemistry, 74, 681–710. 10.1146/annurev.biochem.74.082803.133243 [DOI] [PubMed] [Google Scholar]
- Lynch, H. T. , Snyder, C. L. , Shaw, T. G. , Heinen, C. D. , & Hitchins, M. P. (2015). Milestones of Lynch syndrome: 1895–2015. Nature Reviews Cancer, 15(3), 181–194. 10.1038/nrc3878 [DOI] [PubMed] [Google Scholar]
- Niessen, R. C. , Kleibeuker, J. H. , Westers, H. , Jager, P. O. , Rozeveld, D. , Bos, K. K. , … Hofstra, R. M. (2009). PMS2 involvement in patients suspected of Lynch syndrome . Genes, Chromosomes & Cancer, 48(4), 322–329. 10.1002/gcc.20642 [DOI] [PubMed] [Google Scholar]
- Nyström‐Lahti, M. , Perrera, C. , Räschle, M. , Panyushkina‐Seiler, E. , Marra, G. , Curci, A. , … Jiricny, J. (2002). Functional analysis of MLH1 mutations linked to hereditary nonpolyposis colon cancer. Genes, Chromosomes & Cancer, 33(2), 160–167. 10.1002/gcc.1225 [DOI] [PubMed] [Google Scholar]
- Raevaara, T. E. , Vaccaro, C. , Abdel‐Rahman, W. M. , Mocetti, E. , Bala, S. , Lönnqvist, K. E. , … Nyström‐Lahti, M. (2003). Pathogenicity of the hereditary colorectal cancer mutation hMLH1 del616 linked to shortage of the functional protein. Gastroenterology, 125(2), 501–509. https://doi.org/S0016508503009053 [DOI] [PubMed] [Google Scholar]
- Raschle, M. , Marra, G. , Nystrom‐Lahti, M. , Schar, P. , & Jiricny, J. (1999). Identification of hMutLbeta, a heterodimer of hMLH1 and hPMS1. The Journal of Biological Chemistry, 274(45), 32368–32375. [DOI] [PubMed] [Google Scholar]
- Roberts, M. E. , Jackson, S. A. , Susswein, L. R. , Zeinomar, N. , Ma, X. , Marshall, M. L. , … Chung, W. K. (2018). MSH6 and PMS2 germ‐line pathogenic variants implicated in Lynch syndrome are associated with breast cancer. Genetics in Medicine, 20, 1167–1174. 10.1038/gim.2017.254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Senter, L. , Clendenning, M. , Sotamaa, K. , Hampel, H. , Green, J. , Potter, J. D. , … de la Chapelle, A. (2008). The clinical phenotype of Lynch syndrome due to germ‐line PMS2 mutations. Gastroenterology, 135(2), 419–428. 10.1053/j.gastro.2008.04.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- ten Broeke, S. W. , Brohet, R. M. , Tops, C. M. , van der Klift, H. M. , Velthuizen, M. E. , Bernstein, I. , … Wijnen, J. T. (2015). Lynch syndrome caused by germline PMS2 mutations: Delineating the cancer risk. Journal of Clinical Oncology, 33(4), 319–325. 10.1200/JCO.2014.57.8088 [DOI] [PubMed] [Google Scholar]
- ten Broeke, S. W. , Suerink, M. , & Nielsen, M. (2018). Response to roberts et al. 2018: Is breast cancer truly caused by MSH6 and PMS2 variants or is it simply due to a high prevalence of these variants in the population? Genetics in Medicine, 21, 256–257. 10.1038/s41436-018-0029-1 [DOI] [PubMed] [Google Scholar]
- ten Broeke, S. W. , van der Klift, H. M. , Tops, C. M. J. , Aretz, S. , Bernstein, I. , Buchanan, D. D. , … Win, A. K. (2018). Cancer risks for PMS2‐associated lynch syndrome. Journal of Clinical Oncology, 36(29), 2961–2968. 10.1200/JCO.2018.78.4777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Truninger, K. , Menigatti, M. , Luz, J. , Russell, A. , Haider, R. , Gebbers, J. O. , … Marra, G. (2005). Immunohistochemical analysis reveals high frequency of PMS2 defects in colorectal cancer. Gastroenterology, 128(5), 1160–1171. https://doi.org/S001650850500168X [DOI] [PubMed] [Google Scholar]
- van der Klift, H. M. , Tops, C. M. J. , Bik, E. C. , Boogaard, M. W. , Borgstein, A. M. , Hansson, K. B. M. , … Wijnen, J. T. (2010). Quantification of sequence exchange events between PMS2 and PMS2CL provides a basis for improved mutation scanning of Lynch syndrome patients. Human Mutation, 31(5), 578–587. 10.1002/humu.21229 [DOI] [PubMed] [Google Scholar]
- Vaughn, C. P. , Robles, J. , Swensen, J. J. , Miller, C. E. , Lyon, E. , Mao, R. , … Samowitz, W. S. (2010). Clinical analysis of PMS2: Mutation detection and avoidance of pseudogenes. Human Mutation, 31(5), 588–593. 10.1002/humu.21230 [DOI] [PubMed] [Google Scholar]
- Win, A. K. , Jenkins, M. A. , Dowty, J. G. , Antoniou, A. C. , Lee, A. , Giles, G. G. , & MacInnis, R. J. (2017). Prevalence and penetrance of major genes and polygenes for colorectal cancer. Cancer Epidemiology, Biomarkers & Prevention, 26(3), 404–412. 10.1158/1055-9965.EPI-16-0693 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yurgelun, M. B. , & Hampel, H. (2018). Recent advances in Lynch syndrome: Diagnosis, treatment, and cancer prevention. American Society of Clinical Oncology Educational Book. American Society of Clinical Oncology. Annual Meeting, 38, 101–109. 101‐109. 10.1200/EDBK_208341 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary information