

Abstract
The yeast Saccharomyces cerevisiae is widely used in industrial biotechnology for the production of fuels, chemicals, food ingredients, food and beverages, and pharmaceuticals. To obtain high‐performing strains for such bioprocesses, it is often necessary to test tens or even hundreds of metabolic engineering targets, preferably in combinations, to account for synergistic and antagonistic effects. Here, we present a method that allows simultaneous perturbation of multiple selected genetic targets by combining the advantage of CRISPR/Cas9, in vivo recombination, USER assembly and RNA interference. CRISPR/Cas9 introduces a double‐strand break in a specific genomic region, where multiexpression constructs combined with the knockdown constructs are simultaneously integrated by homologous recombination.
We show the applicability of the method by improving cis,cis‐muconic acid production in S. cerevisiae through simultaneous manipulation of several metabolic engineering targets.
The method can accelerate metabolic engineering efforts for the construction of future cell factories.
Keywords: cis,cis‐muconic acid; CRISPR/Cas9; genome editing; metabolic engineering; RNA interference; USER (uracil‐specific excision reagent); Saccharomyces cerevisiae
1. INTRODUCTION
Industrial biotechnology uses cell factories to produce therapeutical proteins, antibiotics, enzymes, fuels, and chemicals. To achieve favorable process economics, one needs to optimize the cell factories, where performance metrics as titer, rate, and yield are improved. Strain development programs for the products that are not native to the host are very costly and take a long time. The required investment in biotechnology companies that develop novel strains and processes is typically above $50 Mio. During the strain development, hundreds to thousands of strain variants are engineered in iterative design‐build‐test cycles. High‐throughput strain construction and screening in the range of 105–106variants are possible when a biosensor indicating the product presence is available (Zhang, Jensen, & Keasling, 2015); however, this is seldom the case. Hence, the main course of action remains laborious manual strain construction via polymerase chain reaction (PCR), cloning, and transformations. The cloning and strain construction is typically performed at 10–50‐μl scale, where the high cost of specialized reagents also contributes to the high price of the strain development.
Metabolic engineering research requires tools for multiplex genome editing that would allow simultaneous upregulation and downregulation of multiple genes in a combinatorial way. Clustered regularly interspaced short palindromic repeats system with associated nuclease Cas9 (CRISPR/Cas9) system has dramatically simplified genome editing in yeasts, particularly for performing gene overexpression, mutations, and deletions (Lian, HamediRad, & Zhao, 2018; Stovicek, Holkenbrink, & Borodina, 2017). Convenient CRIPSR/Cas‐based genetic tools have been developed for Saccharomyces cerevisiae that enable integration of several gene expression cassettes into multiple locior simultaneous deletion of multiple genes in a single transformation (Bao et al., 2014; Generoso, Gottardi, Oreb, & Boles, 2016; Horwitz et al., 2015; Jakočiūnas et al., 2015; Ryan et al., 2014; Verwaal, Buiting‐Wiessenhaan, Dalhuijsen, & Roubos, 2018). The CRISPR/Cas systems are efficient in editing not only haploid laboratory strains but also diploid and polyploid strains of S. cerevisiae important for brewing and bioethanol applications (Denby et al., 2018; Lian, Bao, Hu, & Zhao, 2018; Stovicek, Borodina, & Forster, 2015). It has also been illustrated in multiple studies how overexpressions, deletions, and mutations can be performed in a single transformation (Jakočiu̅nas et al., 2015; Lian, HamediRad, Hu, & Zhao, 2017; Mans et al., 2015).
Controlled downregulation of gene expression, however, remains a challenge. Gene downregulation is often a more desirable metabolic engineering strategy than complete gene inactivation, and, in case of essential genes, the only option. Catalytically inactivated dCas9, also in a variant coupled to a transcriptional repressor, has been applied for downregulation, but typically multiple gRNA binding sites need to be tested to obtain the desired repression level (Deaner & Alper, 2017; Jensen et al., 2017; Zalatan et al., 2015). Alternatively, RNA interference (RNAi) has been demonstrated to allow more precise control of gene downregulation (Crook, Schmitz, & Alper, 2013; Drinnenberg et al., 2009; Si, Luo, Bao, & Zhao, 2014; Suk et al., 2011).
In this study, we aimed to develop a method that would allow multiplex upregulation and downregulation of several genes by combining the advantages of the CRISPR/Cas9 system and RNAi. The level of upregulation and downregulation can be tuned by selecting promoters of different strengths. To illustrate the applicability of the method, we optimized the cell for production of a prospective chemical molecule cis,cis‐muconic acid (CCM).
2. MATERIALS AND METHODS
2.1. Strains, media, and chemicals
S. cerevisiae CEN.PK strains used in this study are listed in Table S1. The strain of Naumovozyma castellii CLIB290 was received from Centre International de Ressources Microbiennes, Institut National de la Recherche Agronomique (INRA), France. Yeast strains were grown in synthetic complete (SC) medium, synthetic drop‐out (SD) medium, defined mineral medium or synthetic fed‐batch medium Sc.syn‐1000 (M2P labs GmbH, Germany) at 30°C. SC and SD media and agar plates were prepared using premixed drop‐out powders from Sigma‐Aldrich. The defined mineral medium was prepared as described previously (Jensen et al., 2014). Escherichia coli strain DH5α was used as a host for plasmid propagation. E. coli cells were grown at 37°C in Luria–Bertani medium containing 100 μg ml−1 ampicillin. The chemicals were obtained, if not indicated otherwise, from Sigma‐Aldrich. Nourseothricin was obtained from WERNER BioAgents GmbH (Germany). Phusion U Hot Start DNA polymerase and PhusionHot Start II DNA polymerase were purchased from Thermo Fisher Scientific.
2.2. Biobricks amplification and plasmids construction
The oligonucleotides, biobricks, and plasmids used in this study are listed in Tables S2, S3, S4, and S5, respectively. Oligonucleotides were synthesized by Integrated DNA Technologies, Inc (Leuven, Belgium).
A plasmid containing Cas9 and gRNA plasmid for targeting CAN1.Y locus was obtained from Addgene (DiCarlo et al., 2013).
The genes AGO1 and DCR1 that encode correspondingly for the Argonaute and Dicer proteins were amplified from genomic DNA of Naumovozyma castellii. The genes, encoding Klebsiella pneumoniae KpAroY.B (AAY57854.1), KpAroY.D (AAY57856.1), KpAroY.Ciso ([BAH20873.1], Candida albicans CaCatA (XP_722784.1), and Podospora anserina PaAroZ (XP_001905369) were synthesized by GeneArt (Life Technologies) in versions codon‐optimized for S. cerevisiae. KpAroY.B and KpAroY.D encode B and D subunits of the protocatechuic acid decarboxylase (PCA‐DC), whereas KpAroY.Ciso encodes an isoform of subunit C of PCA‐DC. CaCatA encodes the catechol 1,2‐dioxygenase (CDO), and PaAroZ encodes the dehydroshikimate dehydratase (3‐DHDS). Plasmids expressing CaCatA, PaAroZ, KpAroY. B, KpAroY.D, and KpAroY. Ciso were previously constructed and described in Skjoedt et al. (2016). TKL1 encodes the enzyme transketolase from S. cerevisiae. ZWF1 and ARO1 ∆aroE genes were from S. cerevisiae. The S. cerevisiae aro4 K229L encoded a feedback‐resistant 3‐deoxy‐D‐arabino‐heptulosonate‐7‐phosphate (DAHP) synthase with an amino acid change Aro4pK229L. The gene was as described in Rodriguez, Kildegaard, Li, Borodina, and Nielsen (2015).
All DNA fragments (Table S4) were amplified by PCR using Phusion U Hot Start DNA Polymerase (Thermo Fisher Scientific) with primers containing suitable overhangs for USER‐cloning and templates as described in Tables S2 and S3. The amplified products were cloned along with strong constitutive promoters into EasyClone integrative plasmids by USER cloning (Jensen et al., 2014). DNA manipulations in E. coli were carried out according to standard procedures. The clones with correct inserts were identified by colony PCR, and the plasmids were isolated from overnight E. coli cultures and confirmed by sequencing. The list of the constructed vectors can be found in Table S5.
For the construction of overexpression cassettes for in vivo assembly, there are five part types in our assembly standard (promoters, genes, terminators, upstream homology arm, and downstream homology arm). The specific overhangs flanking individual parts were designed and introduced at 5′ end of the forward and reverse primers as described in Table S3. All DNA parts were PCR amplified using Phusion U DNA polymerase according to the manufacturer's instructions. DNA fragments were gel purified and were assembled by consecutive procedures of USER reaction, T4 ligation, and PCR amplification of the assembled expression cassettes as follows: 17 μl of gel‐purified DNA fragments containing similar molar ratio of all parts was mixed with 2 μl of CutSmartTM buffer and 1 μl of USER enzyme (New England BioLabs). The mixes were incubated for 25 min at 37°C followed by 10 min at 25°C. After USER reaction was complete, 1 μl of T4 ligase, 3 μl of ligase buffer, and 6 μl of water were added. The mix was incubated for 5 min at room temperature. Two to three microliter of this ligation mix were used as a template for the final PCR reaction in order to amplify the whole expression cassette. The fragments were purified from the gel and used for yeast transformation (0.7 pmoles per transformation). For fragments smaller than 500 bp, ca. 2 pmoles of the fragment were used per transformation.
2.3. Construction of shRNAs
The small hairpin RNA (shRNA) constructs were composed of two DNA fragments. The first fragment contained approximately 250 bp sense sequence of the target gene under the control of the constitutive promoter and an 81‐bp sequence spanning intron 1 from Schizosaccharomyces pombe rad9. The second fragment contained the antisense sequence of the target gene together with terminator and an 81‐bp sequence of intron 1 from S. pombe rad9. Sense, anti‐sense, promoter, and terminator fragments were amplified by PCR. The corresponding fragments for generating sense and antisense cassettes were assembled via USER‐ligation‐PCR as described above. The intron sequence was implemented in the primer overhang.
Sense and antisense DNA fragments were introduced together with UP‐ and DW‐fragments for CAN‐1 and were assembled into the genome of S. cerevisiae at CAN‐1 locus via homologous recombination.
2.4. Construction of dsRNA
To generate double‐stranded RNA (dsRNA) constructs, the target gene was PCR amplified and assembled with PGK1p and TEF1p promoters, and ADH1t and RPM9t terminators in convergent direction via USER‐ligation‐PCR as above.
2.5. Yeast strains construction
All strains used in this study are listed in Table S1. The integrative plasmids were NotI‐linearized and transformed into S. cerevisiae cells using the lithium acetate protocol (Gietz & Woods, 2002). The cells were selected on SD medium selecting for URA, HIS, LEU and TRP markers. For the selection of strains carrying KanMXsyn and CloNatMXsyn, the ammonium sulfate in the SD medium was replaced with 1 g L−1 monosodium glutamate. The medium was supplemented with 200 μg ml−1 G418 sulfate and 100 μg ml−1 nourseothricin. The correct transformants were confirmed by PCR using primers described in Supplementary Table S2.
2.6. Single cell measurements of fluorescence
Colonies of S. cerevisiae strains to be tested were inoculated into 24 deep‐well plates (EnzyScreen, NL) containing 2‐ml SC medium at 30°C with 300 rpm. After approximately 24 hr, the cells were harvested and washed twice with water. The cell pellet was resuspended in 1 ml of phosphate‐buffered saline buffer. Cells were analyzed on BD FACSAria equipped with three solid‐state diode lasers: air‐cooled Coherent™ Sapphire™ solid‐state diode laser (488 nm, 100 mW), air‐cooled Coherent™ Yellow Green laser (561 nm, 100 mW), and an air‐cooled Coherent™ Deep Blue laser (445 nm, 50 mW). The following filters were used: FITC‐A, PE‐Cy5‐A, and mCFP‐A for the analysis of emission from yellow fluorescent proteins (YFP), red fluorescent proteins (RFP), and cyan fluorescent proteins (CFP), respectively. Compensation was performed according to the manufacturer's protocol (BD FACSAria II User's Guide).
Flow cytometry data were analyzed and interpreted using FlowJo software.
2.7. Muconic acid production in S. cerevisiae
At least 12 single colonies of each transformant were cultivated in 24‐well plate with air‐penetrable lids (EnzyScreen, NL) to test for the production of CCM. The colonies were inoculated in 1‐ml SD medium without uracil, histidine, and leucine and grown at 30°C with 250 rpm agitation at 5‐cm orbit cast for 24 hr; 300 μl of the overnight culture was used to inoculate 3 ml of defined mineral medium (pH 6.0) in 24‐deep well plate and incubated for 72 hr at the same conditions as above. Experiments were done in triplicates. At the end of the cultivation, OD600 was measured in microplate reader BioTek Synergy MX (BioTek). The culture broth was spun down at 3,500 g, and the supernatant was analyzed for CCM concentration using High‐performance liquid chromatography (HPLC).
2.8. Quantification of CCM and its intermediates by HPLC
The samples were diluted five times with water and then analyzed for 45 min using Aminex HPX‐87H ion exclusion column with eluent 1‐mM H2SO4 flow of 0.6 ml min−1. The temperature of the column was 60°C. The UV detector (Dionex) was used for detection of CCM (250 nm), PCA (220 nm), and catechol (220 nm). CCM, PCA, and catechol concentrations were quantified by comparison with the standard calibration curve.
2.9. qRT‐PCR analysis
The expression level of ZWF1 in recombinant yeast strains was determined by quantitative real‐time PCR (qRT‐PCR). Samples for RNA isolation were taken from the cells grown in the mineral medium for 24 hr in triplicates. Sampling procedure and total RNA extraction were performed as previously described (Kildegaard et al., 2014). The first strand cDNA synthesis was performed using Oligo (dt)12–18 Primer and SuperScript™ II Reverse Transcriptase from Invitrogen following the manufacturer's manual. qRT‐PCR analysis of cDNA was carried out in triplicate using Brilliant III Ultra‐Fast SYBR® Green QPCR Master Mix (Agilent Technologies) on a Stratagene Mx3005P (Agilent Technologies). The reactions were performed in 20‐μl final volume with 10 μl of 2x SYBR Green QPCR master mix, 0.5 μl of each upstream and downstream primers, 0.3 μl of reference dye, 2 μl of cDNA template (10 ng), and 6.7 μl of nuclease‐free PCR‐grade water. The thermal cycling conditions were 95°C, 10 min followed by 40 cycles of 95°C for 20 sec, and 60°C for 22 sec, then 1 cycle of 95°C for 1 min, 55°C for 30 min, and 95°C for 30 sec. The gene copy numbers were measured relative to that of a housekeeping gene (ALG9). Oligos used for qRT‐PCR are listed in Table S2.The fold change in gene expression of ZWF1 was determined by relative quantification, and the calculations were made using double delta method (ΔΔCt), where ΔΔCt = (ΔCtE − ΔCtC).
2.10. Growth test in 96‐well microtiter plates
Precultures were prepared by inoculating a single colony in 0.5 ml defined mineral medium (pH 6.0) in 96‐deep well plate (Enzyscreen). The plate was incubated at 30°C with 250 rpm agitation at 5‐cm orbit cast overnight. Five microliter of the overnight cultures were inoculated into 150 μl of fresh medium in a new 96‐well flat bottom plate (Greiner). The plate was sealed with Breathe‐Easy® sealing membrane (Sigma‐Aldrich) and incubated at 30°C with shaking in the BioTek ELx808 microplate reader (BioTek), and the absorbance was measured at 630 nm wavelength every 10 min for 42 hr. Experiments were done in five biological replicates, and the maximum specific growth rates were calculated in the exponential growth phase.
3. RESULTS AND DISCUSSION
3.1. Validation of the method for simultaneous expression of multiple genes at different levels
We aimed to develop a method that would allow simultaneous perturbation of multiple genetic targets. For this, we decided to combine the advantages of CRISPR/Cas9, in vivo recombination, USER assembly, and RNAi. CRISPR/Cas9 system was used to introduce a double‐strand break into a specific genome region, then overexpression and RNAi knock‐down constructs were assembled and integrated into this genome region by homologous recombination. To enable the assembly, we designed 60 bp synthetic homologous recombination (SHR) sequences like following. We have used the UPTAG and DNTAG sequences from yeast knockout libraries to design the SHR sequences. We recombined 20 bp‐UPTAG and DNTAG sequences from yeast knock‐out library (Giaever and Nislow 2014) to obtain final sequences of 60 bp. These sequences were BLASTed against S. cerevisiae genome to select the sequences with low homology that were used as overhang sequences for assembly.
The gene BioBricks included standard 6–8 bp USER overhangs for easy assembly with promoters and terminators (Figure 1a). The promoter biobricks included standard 18 bp overhang (L1) at the 5′‐end and 6–8 bp USER overhang at 3′‐end. Similarly, the terminator biobricks also included standard 6–8 bp USER overhang and 18 bp overhang (L2) at 5′‐ and 3′‐end, respectively. The standard overhangs L1 and L2 were combined with the SHR sequences and used as primers for amplification of the assembled expression cassettes. This design allows reusing a standard set of primers for amplification of different genes, so the genes can be combined with different promoter/terminator pair. There is also a standard set of primers for amplification of expression cassettes that can be combined in the desired order. We used a range of promoters of different strengths (Table S2) and terminators. In order to validate the method for expressing multiple genes, we introduced three fluorescent protein‐coding genes (CFP, YFP, and RFP) under control of promoters of varying strength. A S. cerevisiae strain CEN.PK2‐1C (Mata ura3 his3 leu2 trp1) expressing Cas9p (TRP1 selection) was transformed with gRNA (LEU2 selection) targeting CAN1 site and with three overexpression cassettes, marker cassette (KlURA3), and up‐ and down‐fragments of CAN1. The CAN1 site was chosen because it allows easy validation of correct integration on selective plates, but as such, any site can be used. For example, intergenic sites reported as EasyClone sites can be used (Jessop‐Fabre et al., 2016). The selection marker can be omitted as well if desired; this will, however, lead to a slightly higher number of nonedited clones. Transformants were selected on drop‐out plates without tryptophan, leucine, and uracil. The correct integration into the CAN1 site was investigated by replicating the colonies on SC‐arg + canavanine plates, where only strains with disrupted CAN1 gene can survive. More than 95% of the colonies could grow on SC‐arg + can.
Figure 1.
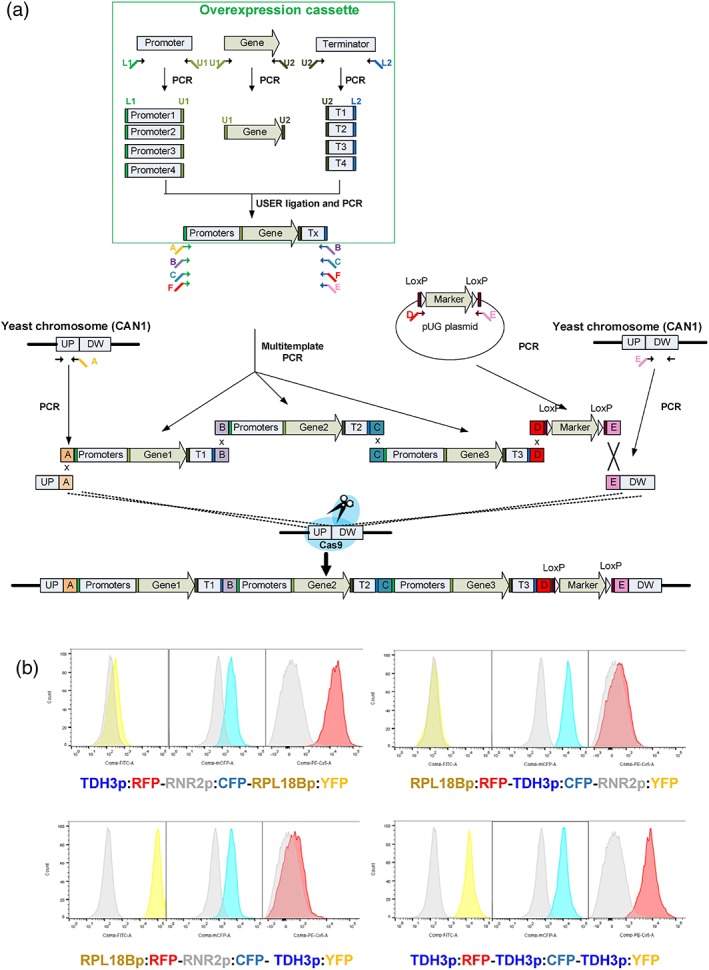
Method for expression of multiple genes. (a) Overview of the CRISPR/Cas9‐RNA interference workflow for expressing multiple genes. First, expression constructs are assembled using USER cloning‐ligation‐PCR. The promoter and terminator are chosen to obtain the desired gene expression level. In the next step, the expression constructs are transformed into Cas9‐expressing yeast strain, along with upstream and downstream repair fragments and a selective marker. (b) Fluorescent cytometry analysis of four Saccharomyces cerevisiae strains, where genes encoding for red (RFP), cyan (CFP), and yellow (YFP) fluorescent proteins were expressed under control of promoters with different strengths [Colour figure can be viewed at wileyonlinelibrary.com]
Furthermore, multiplex PCR was performed to verify the correct assembly, at least 70% of the tested strains were correct according to PCR. The fluorescence levels were evaluated by fluorescent cytometry. The four designed strains expressed all three RFP, CFP, and YFP proteins at the levels that corresponded to promoter strength (TDH3p > RPL18Bp > RNR2p; Figure 1b).
In the past few years, several CRISPR/Cas9 mediated multiplex genome engineering approaches were demonstrated. Mans et al. (2015) explored the potential of CRISPR/Cas9 to combine gene deletion with the simultaneous in vivo assembly and chromosomal integration of multiple DNA fragments. A strain carrying a double ACS1 and ACS2 deletion combined with six gene cassettes expressing the Enterococcus faecalis pyruvate dehydrogenase (PDH) complex (aceF, lplA2, lplA, pdhB, lpd, and pdhA) was constructed in a single transformation with 100% efficiency. In another study, Jakočiu̅nas et al. (2015) developed CasEMBLR, a tool for highly efficient and marker‐free assembly and integration of multiple DNA components into genomic loci. One step assembly and integration of the carotenoid pathway (CrtYB, CrtI, and CrtE) from 15 DNA parts (upstream homology arm, promoter, CDS, terminator, and downstream homology arm) into three targeted loci (ADE2, HIS3, and URA3) was demonstrated with the 31% efficiency. Furthermore, CasEMBLR was also used to assemble and integrate the five‐part assembly of the ARO4* and ARO7* expression cassettes into genomic PDC5 and ARO10 loci with an average efficiency of 58%. Our method is not essentially different from the previous studies but provides an advantage of standardized design of primer overhangs and consequently facilitates combinatorial assembly of genes and promoters/terminators.
3.2. Validation of the method for downregulation of gene expression using RNAi
RNAi machinery is present in multiple eukaryotes, including some yeast species, such as Naumovozyma castellii (Crook et al., 2013; Drinnenberg et al., 2009; Suk et al., 2011). Although S. cerevisiae does not harbor an active RNAi pathway, this pathway can be restored by introducing Argonaute (AGO1) and Dicer (DCR1) genes from Naumovozyma castellii into the genome of S. cerevisiae. In this study, we sought to reconstitute the RNAi machinery in S. cerevisiae to allow controlled downregulation of multiple target genes. We first implemented AGO1 and DCR1 from Naumovozyma castellii into S. cerevisiae through genomic integration and further expressed Cas9 in the engineered strain from a CEN/ARS plasmid (Figure 2a). For the proof‐of‐concept, we chose to use fluorescent proteins as a reporter system. Three fluorescent protein‐encoding genes under control of strong constitutive promoters were integrated into the genome of the yeast strain with AGO1/DCR1/Cas9 to obtain strain ST3135 for testing RNAi.
Figure 2.
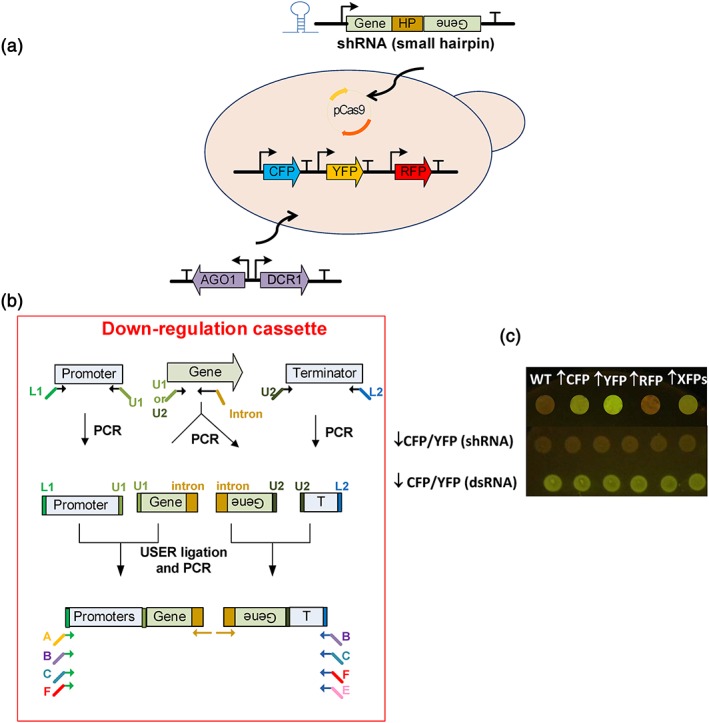
Method for downregulation of target genes. (a) Two heterologous genes AGO1 and DCR1 from Naumovozyma castellii were overexpressed in a yeast strain already expressing Cas9 and CFP‐YFP‐RFP genes. (b) Schematic illustration of USER assembly of the downregulation cassette. (c) Fluorescence images of yeast colonies expressing either individual fluorescent proteins, three fluorescent proteins (XFP), or expressing XFPs, and a downregulation construct for CFP/YFP [Colour figure can be viewed at wileyonlinelibrary.com]
To test the capability of RNA silencing in S. cerevisiae, we evaluated two different approaches, shRNAs and dsRNAs, to silence CFP and YFP. Due to the nucleotide sequence homology between CFP and YFP, we designed shRNA and dsRNA constructs to target both genes simultaneously. The shRNA constructs contained inverted repeats of 250‐bp parts of the target gene with a hairpin in between (Figure 2b). The dsRNA construct contained the target gene flanked by convergent promoters to generate a dsRNA transcript. Both silencing constructs were under the control of strong constitutive promoters. A significant knockdown of CFP/YFP expression was observed with shRNA construct of CFP/YFP, but not with dsRNA construct (Figure 2c). These results confirmed that the RNAi mechanism is functional in S. cerevisiae, and the highest level of RNA silencing was obtained from hairpin constructs, which was in line with the previous reports. Drinnenberg et al. (2009) restored the functional RNAi system in S. cerevisiae by heterologous expression of AGO1 and DCR1. The two constructs shRNA and dsDNA were designed to silence a green fluorescent protein (GFP) reporter, and shRNA has been reported to be the stronger silencing construct compared with dsRNA, both at RNA and fluorescence levels. Furthermore, Crook et al. (2013) studied several design principles for the construction of hairpin RNA expression cassettes and reported that the RNAi efficiency was improved with increasing hairpin length and demonstrated the effectiveness of RNAi by testing several genetic targets for improvement of itaconic acid production in three strains of S. cerevisiae.
In our study, the hairpin length of approximately 250 bp was used. It should also be noted that in vivo assembly of sense and antisense fragments provides a more straightforward approach to introduce shRNA compared with the cloning of inverted repeats via restriction‐ligation cloning in E. coli as in Yoshimatsu and Nagawa (1989).
3.3. Engineering CCM production through multiplex engineering
In the previous study, we have constructed a S. cerevisiae CCM producing strain ST3058 (Skjoedt et al., 2016). ST3058 expresses a three‐step heterologous pathway consisting of a gene encoding dehydroshikimate dehydratase (3‐DHS) from Podosporaanserine (PaAroZ), the genes encoding three different subunits of PCA‐DC from Klebsiella pneumonia (KpAroY.B, KpAroY.Ciso, KpAroY.D), and the gene encoding catechol 1,2‐dioxygenase (CDO) from Candida albicans (CaCatA; Figure 3a). It has been reported that PCA‐DC was a rate‐limiting step for the CCM flux (Curran, Leavitt, Karim, & Alper, 2013; Weber et al., 2012). For this reason, we integrated KpAroY.B and KpAroY.Ciso genes in multiple copies into long 113 terminal repeats (LTRs) of retrotransposon of the TY4 family (Maury et al., 2016). As the transformants were expected to have different copy numbers of the expression vector, we screened 12 randomly selected clones to test for CCM production. The best isolate of ST3058 produced 400 mg L−1 CCM in defined mineral medium and was chosen for evaluating the CRISPR/Cas9‐RNAi method. We implemented Cas9, AGO1, and DCR1 into the best isolate of ST3058, resulting in strain ST3639 that was suitable for testing our method.
Figure 3.
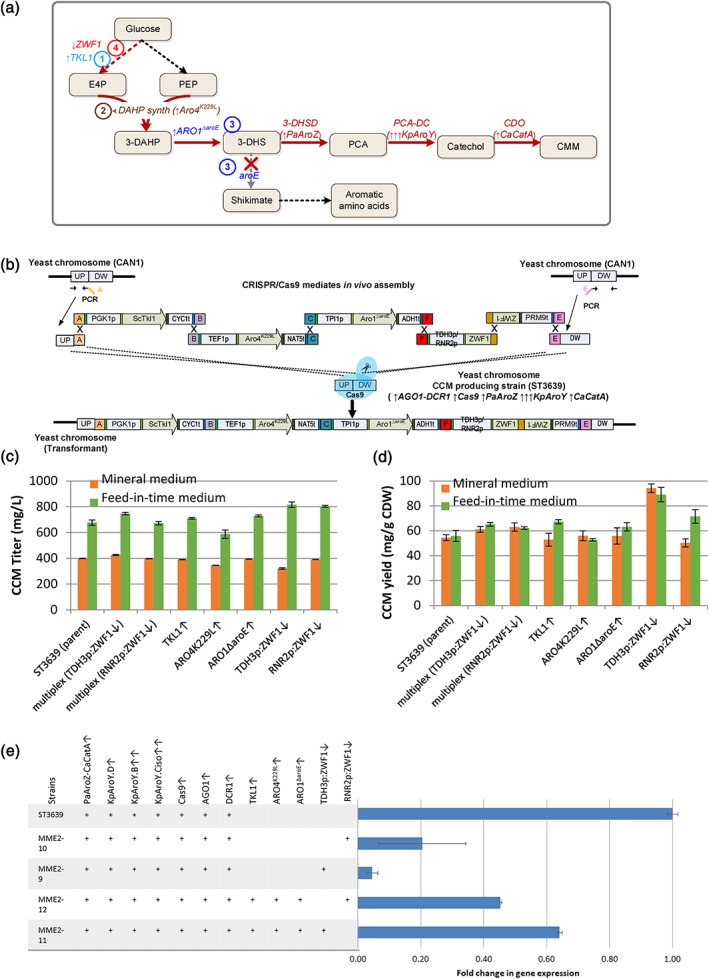
Application of CRISPR/Cas9‐RNA interference method for engineering cis,cis‐muconic acid production in Saccharomyces cerevisiae. (a) Muconic biosynthesis pathway in yeast. (b) Schematic illustration of the seven‐part assembly of the three overexpression cassettes for TKL1, ARO4 K229L, ARO1 ΔaroE, one downregulation cassette of ZWF1, and homologous recombination with chromosomal target site CAN1. (c, d) Average cis,cis‐muconic acid titers and yields, respectively, in the parent strain ST3639 and engineered strains with either expression of TKL1, ARO4 K229L, ARO1 ΔaroE, downregulation of ZWF1 or multiplex expression of all combinations. Cultivations were performed in biological triplicates, and error bars represent the standard deviation of the average (n = 3). (e) qRT‐PCR analyses. Fold change in gene expression of engineered strains compared with the parent strain ST3639. ↑ indicates that a gene was expressed in a copy, ↑↑ indicates that a gene was expressed in several copies, ↓ indicates downregulation of ZWF1 under control of either TDH3p or RNR2p promoters. Error bars represent the standard deviation of duplicates [Colour figure can be viewed at wileyonlinelibrary.com]
For the test, we designed to vary the expression of four native genes that could influence the CCM flux: TKL1 encoding transketolase, ARO4 K229L encoding tyrosine‐feedback‐resistant allele of phospho‐2‐dehydro‐3‐deoxyheptonate aldolase, ARO1 ∆aroE encoding a pentafunctional AROM protein ARO1 without the dehydrogenase domain AROE, and ZWF1 encoding glucose‐6‐phosphate dehydrogenase (Figure 3b). We generated seven strain variants that carried overexpressions of either TKL1, ARO4 K229L, ARO1 ΔaroE or downregulations of ZWF1, or a combination of overexpressions and downregulation. For verification of correct assembly and integration, multiplex PCR of a minimum of 12 colonies per transformation was used. On the basis of genotyping, we obtained engineering efficiencies of at least 85% for in vivo assembly and integration of three DNA fragments (upstream homology arm, single expression cassette, and downstream homology arm), whereas 55% efficiency was obtained for combinatorial multiplex genome integration of seven DNA fragments. Several strain variants, that is, strains with downregulation of ZWF1, had higher CCM titer and specific yield than the parental strain ST3639. The improvement in CCM production in the engineered strains was more pronounced on feed‐in‐time medium simulating carbon‐limited fed‐batch conditions than in a standard batch medium. Overexpression of either TKL1 or ARO1 ΔaroE and downregulation of ZWF1 with either strong or weak promoter (TDH3p and RNR2p) improved the titer by 5–21%, and the specific yield by 11–60% when the strains were grown on feed‐in‐time medium (Figure 3c,d). Contrary, overexpression of the ARO4 K229L gene had no positive effect on CCM titer and yield. We also measured the μmax of the four strains with ZWF1 downregulation. No significant difference was observed in the ZWF1 downregulation strains in comparison with the reference strain (Figure S1). However, ZWF1 downregulation did result in a reduction of the biomass yield in comparison with the reference strain. This observation might explain the significant improvement in specific CCM yield in strains with downregulation of ZWF1 (TDH3p). The downregulation of ZWF1 gene was investigated by qRT‐PCR (Figure 3e). In the strain, where the only implemented modification was ZWF1 downregulation, the expression level decreased by 80% or 95% when weak and strong promoters were driving shRNA expression, respectively. In the strain, where additional three genes were overexpressed, the downregulation of ZWF1 was at 35% or 55%, again depending on the promoter for shRNA. The positive effects of TKL1 overexpression and ZWF1 downregulation on CCM production are in agreement with a previous report, where ZWF1 was though deleted rather than downregulated (Curran et al., 2013; Weber et al., 2012). Both genes are involved in the pentose phosphate pathway, and the modification of their expression possibly improved the supply of the aromatic amino acids precursor—erythrose 4‐phosphate. The positive effects of these modifications need to be further confirmed in fed‐batch fermentations in controlled bioreactors.
In the past few years, there has been a growing interest in applying CRISPR methods for combinatorial metabolic engineering. Vanegas, Lehka, and Mortensen (2017) developed a Cas9/dCas9 based system, SWITCH, which allows S. cerevisiae strains to alternate between a genetic engineering state and a pathway control state. The Cas9 system was first used in the genetic engineering state to implement the five genes necessary for naringenin production into the chromosome. Next, the cells were switched to pathway control state by replacing the Cas9 expression cassette with dCas9 expression cassette. At this state, the naringenin production was further optimized by dCas9‐mediated downregulation of an essential gene TSC13 to prevent for formation of a by‐product. However, the SWITCH approach only allows the cells to be in either a genetic engineering or a pathway control state at a time.
In another study, Lian et al. (2017) developed a trifunctional CRISPR system that combines one nuclease‐deficient CRISPR protein fused with an activation domain for transcriptional activation (CRISPRa), a second nuclease‐deficient CRISPR protein fused with a repression domain for transcriptional interference (CRISPRi), and a third catalytically active CRISPR protein for gene deletion (CRISPRd) in the same cells. Lian et al. characterized several CRISPR orthologs in S. cerevisiae and further optimized for transcriptional regulation by engineering the corresponding effector domains. The optimal design of the trifunctional CRISPR system was using nuclease‐deficient Cpf1 from Lachnospiraceae bacterium (dLbCpf1‐VP) for CRISPRa, nuclease‐deficient Cas9 from Streptococcus pyogenes (dSpCas9‐RD1153) for CRISPRi, and Cas9 from Staphylococcus aureus (SaCas9) for CRISPRd. As a proof‐of‐ concept, the trifunctional CRISPR system was used to increase β‐carotene production via simultaneous upregulation of HMG1, downregulation of ERG9, and deletion of ROX1. Furthermore, 2.5‐fold improvement in the display of an endoglucanase on the yeast surface was obtained by combinatorial optimization of several metabolic targets. At this point, the selection of efficient gRNA for CRISPRi remains a challenge and multiple variants need to be tested. This increases the number of strains that need to be constructed for testing downregulation targets or combinations of downregulation targets with overexpression targets.
During this work, a study was published by Si et al. (2017) that reported a combination of RNAi and CRISPR/Cas9 for constructing S. cerevisiae strains with overexpressions and downregulations. The authors used δ‐regions for integration of the constructs, and hence the obtained strains are not defined as in our method but have varying numbers of different expression/downregulation cassettes integrated. Si et al. applied dsDNA constructs for RNAi, whereas in our study, shDNA were shown to be more effective for downregulating gene expression.
Our method combines the advantages of RNAi for precise downregulation, of CRISPR/Cas9 for efficient genomic integration and of yeast homologous recombination for the multiple fragment assembly. The method is convenient for testing defined combinations of multiple upregulation and downregulation targets for metabolic engineering. The method can facilitate the strain development efforts by increasing the throughput and decreasing the cost of strain construction. In the future, it can be further applied for generating combinatorial libraries of strain variants by using mixes of BioBricks rather than specific BioBricks. The library approach is particularly attractive if a high‐throughput method for screening the strain libraries is available, as is the case with muconic acid, where a biosensor has been reported (Skjoedt et al., 2016).
CONFLICT OF INTEREST
The authors declare no conflict of interests.
Supporting information
Figure S1. The maximum specific growth rate (μmax) of the recombinant strains grown in defined mineral medium in 96‐well plate. Data bars show the mean and error bars show the standard deviations calculated from five biological replicates.
Table S1. List of strains
Table S2. List of primers
USER‐specific overhangs are marked in bold, standardized overhangs L1 and L2 are marked in blue and green texts, respectively. SHR overhang are underlined.
Table S3. Summary of overhangs used for amplification of biobrick parts for in vivo assembly. USER‐specific overhangs are marked in bold, standardized overhangs L1 and L2 are marked in blue and green texts, respectively. Intron sequences are in orange texts. AAAACA – Kozak sequence, ATG– start codon, TCA – stop codon, (N)n – gene (promoter)‐specific sequence.
Table S4. List of BioBricks
Table S5. List of plasmids
ACKNOWLEDGEMENTS
This work was supported by the Novo Nordisk Foundation (grant agreements NNF15OC0016592 and NNF10CC1016517). The authors would like to thank Dushica Arsovska and Laura Dato for help with FACS analysis and Tim Snoek for help with FlowJo software.
Kildegaard KR, Tramontin LRR, Chekina K, et al. CRISPR/Cas9‐RNA interference system for combinatorial metabolic engineering of Saccharomyces cerevisiae . Yeast. 2019;36:237–247. 10.1002/yea.3390
REFERENCES
- Bao, Z. , Xiao, H. , Liang, J. , Zhang, L. , Xiong, X. , Sun, N. , … Zhao, H. (2014). Homology‐integrated CRISPR–Cas (HI‐CRISPR) system for one‐step multigene disruption in Saccharomyces cerevisiae . ACS Synthetic Biology, 4, 585–594. 10.1021/sb500255k [DOI] [PubMed] [Google Scholar]
- Crook, N. C. , Schmitz, A. C. , & Alper, H. S. (2013). Optimization of a yeast RNA interference system for controlling gene expression and enabling rapid metabolic engineering. ACS Synthetic Biology, 3, 307–313. [DOI] [PubMed] [Google Scholar]
- Curran, K. A. , Leavitt, J. M. , Karim, A. S. , & Alper, H. S. (2013). Metabolic engineering of muconic acid production in Saccharomyces cerevisiae . Metabolic Engineering, 15, 55–66. 10.1016/j.ymben.2012.10.003 [DOI] [PubMed] [Google Scholar]
- Deaner, M. , & Alper, H. S. (2017). Systematic testing of enzyme perturbation sensitivities via graded dCas9 modulation in Saccharomyces cerevisiae . Metabolic Engineering, 40, 14–22. 10.1016/j.ymben.2017.01.012 [DOI] [PubMed] [Google Scholar]
- Denby, C. M. , Li, R. A. , Vu, V. T. , Costello, Z. , Lin, W. , Chan, L. J. G. , … Keasling, J. D. (2018). Industrial brewing yeast engineered for the production of primary flavor determinants in hopped beer. Nature Communications, 9, 965 10.1038/s41467-018-03293-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiCarlo, J. E. , Norville, J. E. , Mali, P. , Rios, X. , Aach, J. , & Church, G. M. (2013). Genome engineering in Saccharomyces cerevisiae using CRISPR‐Cas systems. Nucleic Acids Research, 41, 4336–4343. 10.1093/nar/gkt135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drinnenberg, I. A. , Weinberg, D. E. , Xie, K. T. , Mower, J. P. , Wolfe, K. H. , Fink, G. R. , & Bartel, D. P. (2009). RNAi in budding yeast. Science, 326, 544–550. 10.1126/science.1176945 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Generoso, W. C. , Gottardi, M. , Oreb, M. , & Boles, E. (2016). Simplified CRISPR‐Cas genome editing for Saccharomyces cerevisiae . Journal of Microbiological Methods, 127, 203–205. 10.1016/j.mimet.2016.06.020 [DOI] [PubMed] [Google Scholar]
- Giaever, G. , & Nislow, C. (2014). The Yeast Deletion Collection: A Decade of Functional Genomics. Genetics, 197, 451–465. 10.1534/genetics.114.161620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gietz, R. D. , & Woods, R. A. (2002). Transformation of yeast by lithium acetate/single‐stranded carrier DNA/polyethylene glycol method. Methods Enzymology, 350, 87–96. 10.1016/S0076-6879(02)50957-5 [DOI] [PubMed] [Google Scholar]
- Horwitz, A. A. , Walter, J. M. , Schubert, M. G. , Kung, S. H. , Hawkins, K. , Platt, D. M. , … Newman, J. D. (2015). Efficient multiplexed integration of synergistic alleles and metabolic pathways in yeasts via CRISPR‐Cas. Cell Systems, 1, 88–96. 10.1016/j.cels.2015.02.001 [DOI] [PubMed] [Google Scholar]
- Jakočiūnas, T. , Bonde, I. , Herrgård, M. , Harrison, S. J. , Kristensen, M. , Pedersen, L. E. , … Keasling, J. D. (2015). Multiplex metabolic pathway engineering using CRISPR/Cas9 in Saccharomyces cerevisiae . Metabolic Engineering, 28, 213–222. 10.1016/j.ymben.2015.01.008 [DOI] [PubMed] [Google Scholar]
- Jakočiu̅nas, T. , Rajkumar, A. S. , Zhang, J. , Arsovska, D. , Rodriguez, A. , Jendresen, C. B. , … Keasling, J. D. (2015). CasEMBLR: Cas9‐facilitated multiloci genomic integration of in vivo assembled DNA parts in Saccharomyces cerevisiae . ACS Synthetic Biology, 4, 1226–1234. 10.1021/acssynbio.5b00007 [DOI] [PubMed] [Google Scholar]
- Jensen, E. D. , Ferreira, R. , Jakočiūnas, T. , Arsovska, D. , Zhang, J. , Ding, L. , … Keasling, J. D. (2017). Transcriptional reprogramming in yeast using dCas9 and combinatorial gRNA strategies. Microbial Cell Factories, 16, 46 10.1186/s12934-017-0664-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen, N. B. , Strucko, T. , Kildegaard, K. R. , David, F. , Maury, J. , Mortensen, U. H. , … Borodina, I. (2014). EasyClone: method for iterative chromosomal integration of multiple genes in Saccharomyces cerevisiae . FEMS Yeast Research, 14, 238–248. 10.1111/1567-1364.12118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jessop‐Fabre, M. M. , Jakočiūnas, T. , Stovicek, V. , Dai, Z. , Jensen, M. K. , Keasling, J. D. , & Borodina, I. (2016). EasyClone‐MarkerFree: A vector toolkit for marker‐less integration of genes into Saccharomyces cerevisiae via CRISPR‐Cas9. Biotechnology Journal, 11, 1110–1117. 10.1002/biot.201600147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kildegaard, K. R. , Hallström, B. M. , Blicher, T. H. , Sonnenschein, N. , Jensen, N. B. , Sherstyk, S. , … Forster, J. (2014). Evolution reveals a glutathione‐dependent mechanism of 3‐hydroxypropionic acid tolerance. Metabolic Engineering, 26C, 57–66. [DOI] [PubMed] [Google Scholar]
- Lian, J. , Bao, Z. , Hu, S. , & Zhao, H. (2018). Engineered CRISPR/Cas9 system for multiplex genome engineering of polyploid industrial yeast strains. Biotechnology and Bioengineering, 115, 1630–1635. 10.1002/bit.26569 [DOI] [PubMed] [Google Scholar]
- Lian, J. , HamediRad, M. , Hu, S. , & Zhao, H. (2017). Combinatorial metabolic engineering using an orthogonal tri‐functional CRISPR system. Nature Communications, 8, 1688 10.1038/s41467-017-01695-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lian, J. , HamediRad, M. , & Zhao, H. (2018). Advancing metabolic engineering of Saccharomyces cerevisiae using the CRISPR/Cas System. Biotechnology Journal, 13, 1700601 10.1002/biot.201700601 [DOI] [PubMed] [Google Scholar]
- Mans, R. , van Rossum, H. M. , Wijsman, M. , Backx, A. , Kuijpers, N. G. , van den Broek, M. , … Daran, J. M. (2015). CRISPR/Cas9: A molecular Swiss army knife for simultaneous introduction of multiple genetic modifications in Saccharomyces cerevisiae . FEMS Yeast Research, 15(2), fov004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maury, J. , Germann, S. M. , Baallal Jacobsen, S. A. , Jensen, N. B. , Kildegaard, K. R. , Herrgård, M. J. , … Borodina, I. (2016). EasyCloneMulti: A set of vectors for simultaneous and multiple genomic integrations in Saccharomyces cerevisiae . PLoS ONE, 11, e0150394 10.1371/journal.pone.0150394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez, A. , Kildegaard, K. R. , Li, M. , Borodina, I. , & Nielsen, J. (2015). Establishment of a yeast platform strain for production of p‐coumaric acid through metabolic engineering of aromatic amino acid biosynthesis. Metabolic Engineering, 31, 181–188. 10.1016/j.ymben.2015.08.003 [DOI] [PubMed] [Google Scholar]
- Ryan, O. W. , Skerker, J. M. , Maurer, M. J. , Li, X. , Tsai, J. C. , Poddar, S. , … Cate, J. H. D. (2014). Selection of chromosomal DNA libraries using a multiplex CRISPR system. eLife, 3, e03703 10.7554/eLife.03703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Si, T. , Chao, R. , Min, Y. , Wu, Y. , Ren, W. , & Zhao, H. (2017). Automated multiplex genome‐scale engineering in yeast. Nature Communications, 8, 15187 10.1038/ncomms15187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Si, T. , Luo, Y. , Bao, Z. , & Zhao, H. (2014). RNAi‐assisted genome evolution in Saccharomyces cerevisiae for complex phenotype engineering. ACS Synthetic Biology, 4, 283–291. 10.1021/sb500074a [DOI] [PubMed] [Google Scholar]
- Skjoedt, M. L. , Snoek, T. , Kildegaard, K. R. , Arsovska, D. , Eichenberger, M. , Goedecke, T. J. , … Keasling, J. D. (2016). Engineering prokaryotic transcriptional activators as metabolite biosensors in yeast. Nature Chemical Biology, 12(11), 951–958. 10.1038/nchembio.2177 [DOI] [PubMed] [Google Scholar]
- Stovicek, V. , Borodina, I. , & Forster, J. (2015). CRISPR–Cas system enables fast and simple genome editing of industrial Saccharomyces cerevisiae strains. Metabolic Engineering Communications, 2, 13–22. 10.1016/j.meteno.2015.03.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stovicek, V. , Holkenbrink, C. , & Borodina, I. (2017). CRISPR/Cas system for yeast genome engineering: Advances and applications. FEMS Yeast Research, 17(5). 10.1093/femsyr/fox030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suk, K. , Choi, J. , Suzuki, Y. , Ozturk, S. B. , Mellor, J. C. , Wong, K. H. , … Roth, F. P. (2011). Reconstitution of human RNA interference in budding yeast. Nucleic Acids Research, 39, e43 10.1093/nar/gkq1321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanegas, K. G. , Lehka, B. J. , & Mortensen, U. H. (2017). SWITCH: A dynamic CRISPR tool for genome engineering and metabolic pathway control for cell factory construction in Saccharomyces cerevisiae . Microbial Cell Factories, 16, 25 10.1186/s12934-017-0632-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verwaal, R. , Buiting‐Wiessenhaan, N. , Dalhuijsen, S. , & Roubos, J. A. (2018). CRISPR/Cpf1 enables fast and simple genome editing of Saccharomyces cerevisiae . Yeast, 35, 201–211. 10.1002/yea.3278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber, C. , Brückner, C. , Weinreb, S. , Lehr, C. , Essl, C. , & Boles, E. (2012). Biosynthesis of cis,cis‐muconic acid and its aromatic precursors, catechol and protocatechuic acid, from renewable feedstocks by Saccharomyces cerevisiae . Applied and Environmental Microbiology, 78, 8421–8430. 10.1128/AEM.01983-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshimatsu, T. , & Nagawa, F. (1989). Control of gene expression by artificial introns in Saccharomyces cerevisiae . Science, 244, 1346–1348. 10.1126/science.2544026 [DOI] [PubMed] [Google Scholar]
- Zalatan, J. G. , Lee, M. E. , Almeida, R. , Gilbert, L. A. , Whitehead, E. H. , la Russa, M. , … Lim, W. A. (2015). Engineering complex synthetic transcriptional programs with CRISPR RNA scaffolds. Cell, 160, 339–350. 10.1016/j.cell.2014.11.052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, J. , Jensen, M. K. , & Keasling, J. D. (2015). Development of biosensors and their application in metabolic engineering. Current Opinion in Chemical Biology, 28, 1–8. 10.1016/j.cbpa.2015.05.013 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. The maximum specific growth rate (μmax) of the recombinant strains grown in defined mineral medium in 96‐well plate. Data bars show the mean and error bars show the standard deviations calculated from five biological replicates.
Table S1. List of strains
Table S2. List of primers
USER‐specific overhangs are marked in bold, standardized overhangs L1 and L2 are marked in blue and green texts, respectively. SHR overhang are underlined.
Table S3. Summary of overhangs used for amplification of biobrick parts for in vivo assembly. USER‐specific overhangs are marked in bold, standardized overhangs L1 and L2 are marked in blue and green texts, respectively. Intron sequences are in orange texts. AAAACA – Kozak sequence, ATG– start codon, TCA – stop codon, (N)n – gene (promoter)‐specific sequence.
Table S4. List of BioBricks
Table S5. List of plasmids