

Ovary-to-testis transformation in a sex-changing fish involves transcriptomic and epigenomic reprogramming.
Abstract
Bluehead wrasses undergo dramatic, socially cued female-to-male sex change. We apply transcriptomic and methylome approaches in this wild coral reef fish to identify the primary trigger and subsequent molecular cascade of gonadal metamorphosis. Our data suggest that the environmental stimulus is exerted via the stress axis and that repression of the aromatase gene (encoding the enzyme converting androgens to estrogens) triggers a cascaded collapse of feminizing gene expression and identifies notable sex-specific gene neofunctionalization. Furthermore, sex change involves distinct epigenetic reprogramming and an intermediate state with altered epigenetic machinery expression akin to the early developmental cells of mammals. These findings reveal at a molecular level how a normally committed developmental process remains plastic and is reversed to completely alter organ structures.
INTRODUCTION
In most organisms, a fundamental dichotomy is established in early embryonic development; individuals become either female or male and maintain these fates throughout life. However, some plant and animal species exhibit remarkably diverse and plastic sexual developmental patterns (1, 2), and some even retain the ability to change sex in adulthood (3). Such functional sex change is widespread in marine fishes, appearing in 27 families (4). Among the most outstanding, and well-studied, example of sex change is the bluehead wrasse (Thalassoma bifasciatum), a small coral reef fish that undergoes rapid and complete female-to-male sex reversal in response to a social cue (5).
While the sex change process and its evolutionary advantages are well known (3, 6), there remain long-standing questions about how environmental influences initiate these dramatic changes in sexual identity and what molecular processes orchestrate this transformation (7, 8).
Across vertebrates, antagonism between core male- and female-promoting gene networks is now recognized as crucial to the establishment and maintenance of gonadal fate (9). Discoveries in mice that loss of the feminizing FOXL2 (Forkhead Box L2) transcription factor is sufficient to induce the transdifferentiation of mature ovaries into testes (10), and that testicular cells will become ovarian cells if the masculinizing transcription factor DMRT1 (Doublesex and Mab-3 Related Transcription Factor 1) is lost (11), suggest that gonadal bipotentiality is retained into adulthood and presents a mechanism through which female-male and male-female gonadal sex reversal may be controlled in fish. A key target of both factors is the gene encoding aromatase (the enzyme responsible for estrogen production), the expression of which is known to be environmentally sensitive and when inhibited, results in female-male sex reversal (12).
Epigenetic processes are suspected to be key mediators and effectors of such environmentally induced sex reversal. Temperature-sensitive sex reversal involves global methylation modification in the half-smooth tongue sole (13), with sex-specific methylation states of major sex-pathway genes inverted following sex reversal. Similarly, sex reversal in the dragon lizard (14), and temperature-dependent sex determination in red-eared slider turtles (15), appears to involve temperature-sensitive expression changes in epigenetic regulator genes of the Jumonji family, namely, kdm6b and jarid2. Therefore, a change in methylation state may facilitate reprogramming, and subsequent canalization, of sexual fate at a cellular level (9).
Sex reversal in response to social cues, as seen in the bluehead wrasse, is an especially striking example of phenotypic and sexual plasticity. Most bluehead wrasses begin their reproductive life as females but routinely reverse sex in the absence of a socially dominant male (16). Removal of the dominant terminal-phase (TP) male from its territory induces rapid and complete sex change of the largest female (5). Alternatively, bluehead wrasses may mature first as nonaggressive initial-phase (IP) males that mimic the female phenotype, and can also become TP males under the same social stimulus (16). Transitioning individuals exhibit behavioral changes within hours, and in females, complete transformation of mature ovaries to functional testes can occur in 8 to 10 days (Fig. 1). The absence of differentiated male sexual tissue in the ovaries of sex-changing wrasses implies that some form of cellular reprogramming underlies sex change in these species (17), either by a direct cellular transition (i.e., transdifferentiation) or via a dedifferentiated intermediate stage, rather than by alterations in the proportion of spermatogenic and oogenic cell populations as in the bisexual gonad or ovotestis of many other sex-changing fishes [e.g., anemone fish and bidirectional sex-changing gobies (7)].
Fig. 1. Sex change in the bluehead wrasse.
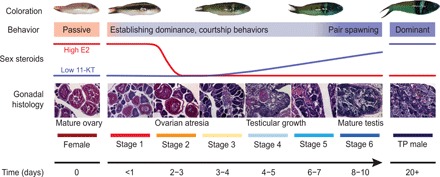
Schematic of sex change summarizing changes in external coloration, behavior, serum steroid levels, and gonadal histology across time. Within hours of removing TP males, the largest female displays aggression and male courtship behaviors, and adopts darker spawning coloration, but still has healthy ovaries (stage 1). Transitioning females establish dominance within 1 to 2 days, after which serum estrogen [17β-estradiol (E2)] levels collapse and ovarian atresia is observable (stage 2). Ovarian atresia is advanced by 3 to 4 days (stage 3). Testicular tissues are observed by days 4 to 5, and serum 11-ketotestosterone (11-KT) begins to rise (stage 4) before spermatogenesis begins by days 6 to 7 (stage 5). Within 8 to 10 days, transitioning fish are producing mature sperm and successfully reproducing with females (stage 6). Full TP male coloration develops within ~20 days. Gonadal stages are classified according to (17). Hormonal changes are predicted on the basis of patterns in the congener Thalassoma duperrey (17). For detailed descriptions of behavioral and morphological changes, see (5, 18). Photo credit: J. Godwin, North Carolina State University (fish images); H. Liu, University of Otago (histology images).
Here, we applied transcriptomic and nucleotide-level methylation approaches to identify the primary trigger and subsequent molecular cascade that orchestrates gonad remodeling in the bluehead wrasse. Our results provide a detailed molecular picture of female-to-male sex reversal in a vertebrate and underscore the role of epigenetics and pluripotency in sex determination.
RESULTS
We induced wild female bluehead wrasses to change sex in the field by removing dominant TP males from established social groups on patch reefs off the Florida coast. We collected a time series of brain and gonadal samples across the entire sex change process, assigning animals to six successive stages based on behaviors observed at the time of capture (18) and gonadal histology (Fig. 1) (17). As controls, we collected six small females that experienced the social manipulation but showed no signs of sex change (control females) and eight dominant TP males.
Transcriptome-wide gene expression patterns across sex change
We quantified transcriptome-wide gene expression across sex change using RNA sequencing (RNA-seq) and the de novo assembled transcriptome from our previous study (19) as a reference. Expression patterns across samples were visualized using principal components analysis (PCA). We observed very little variation in global gene expression in the brain (Fig. 2A and fig. S1), consistent with other studies in teleosts also showing limited sex bias in brain transcriptomic profiles (20, 21).
Fig. 2. Global gene expression changes during sex change.
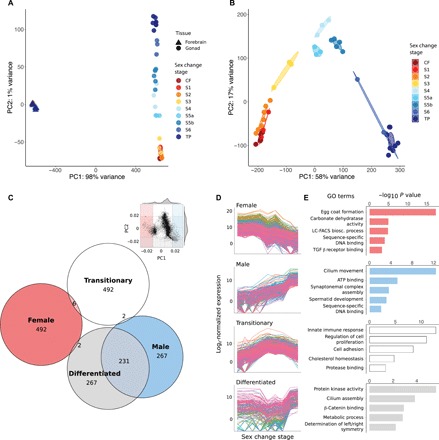
(A) PCA showing close clustering of brain samples but separation of gonad samples by sex change stage. (B) PCA (10,000 most variable transcripts) of gonad samples. The transition from ovary to testis is captured along PC1 (58% variance), whereas PC2 (17% variance) delineates fully differentiated gonads of control females (bottom left) and TP males (bottom right) from those of transitioning fish. (C) Inset: Component loadings were used to identify transcripts contributing most to PC1 and PC2. Shaded sections define 5th and 95th percentiles defining four spatial regions: “Female” (left), “Male” (right), “Differentiated” (bottom), and “Transitionary” (top). The Euler diagram shows numbers of transcripts uniquely assigned to each region. (D) Expression patterns across sex change of transcripts uniquely assigned to each of the four spatial regions, showing four distinct patterns of expression: Female (declining), Male (increasing), Transitionary (highest mid-sex change), and Differentiated (lowest mid-sex change) and (E) representative GO terms for these transcripts. LC-FACS bios. process, long-chain fatty-acyl–coenzyme A biosynthetic process; TGF β receptor binding, transforming growth factor–β receptor binding; CF, control female; S1 to S6, stages 1 to 6; TP, TP male; ATP, adenosine 5′-triphosphate.
In contrast, striking variation in expression was observed in the gonad, and samples strongly clustered by sex change stage (Fig. 2B). Samples at stage 5 (ongoing spermatogenesis) formed two distinct clusters in the PCA and were subdivided into stages 5a and 5b in further analyses. Notably, gonad samples were clearly organized, first, by sexual development from female to male (PC1) and, second, by developmental commitment (PC2) (Fig. 2B). This suggested that intermediate phases of sex change may represent unique transitional cell types rather than simply different proportions of differentiated male and female tissues. To validate this computationally, we generated “mixed” samples by combining subsampled reads from control male and female libraries in different ratios before rerunning the PCA (fig. S2A). As expected, the mixed samples are separated primarily along PC1, lying between stage 4 gonads (when male cells are first identifiable histologically) and fully differentiated TP male testes. This pattern supports the assertion that gonadal sex change involves unique transitional cell types rather than a proportional change in sexually differentiated cells.
Transitional stages are also characterized by unique expression states and functional categories. To investigate those transcripts contributing most to each extreme of PC1 (female and male) and PC2 (differentiated and transitionary), we used component loadings to select the top 500 transcripts driving each axis direction (Fig. 2C). This resulted in an extensive overlap between the “male” and “differentiated” regions of the PCA. Visualizing expression of transcripts unique to each group (Fig. 2D) confirmed that the extremes of PC1 are characterized by transcripts that are female biased and down-regulated or male biased and up-regulated across sex change, whereas PC2 is driven by transcripts with the highest expression in either transitionary stages or sexually differentiated gonads. Gene Ontology (GO) terms (Fig. 2E and data S1) associate transcripts of the “female” and “male” regions with oocyte- and sperm-specific processes, respectively, and sex-specific transcription factor activity. Unique GO terms were associated with the “transitionary” transcripts and include innate immunity, protein catabolism, cell proliferation, and adhesion processes, reflecting dynamic disassembly and rebuilding of the gonad at mid-sex change. “Transitionary” transcripts were also enriched for cholesterol homeostasis, which is a known signal of apoptotic events (22) and is affected by levels of cholesterol-derived hormones (23). Estrogen, androgen, and cortisol are all expected to undergo turnover in transitioning gonads (Fig. 1) (7, 24), consistent with the expression patterns that we observe for the relevant regulatory genes (Figs. 3 and 4). The “differentiated” region was associated with protein kinase activity, β-catenin signaling, and metabolic and developmental processes.
Fig. 3. Sex change involves transition from female- to male-specific expression and gene neofunctionalization.
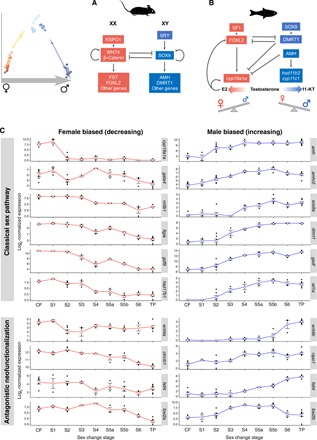
(A) Mammalian model of sex determination and development. In males, SRY (sex-determining region Y) activates SOX9 (SRY-related HMG box 9) to initiate male development while blocking expression of feminization genes that would, in turn, antagonize masculinizing expression (9). (B) Teleost model of sexual development. Diverse factors determine sex in fishes, yet conserved downstream effectors act in feminizing and masculinizing pathways to promote female or male development, respectively, while antagonizing the opposing sexual pathway. Testosterone is a prohormone and is converted to estrogen (E2) by gonadal aromatase (encoded by cyp19a1a) to promote ovarian function or to 11-KT by the products of hsd11b2 and cyp11c1 to promote testicular function. (C) Normalized expression of classical sex-pathway genes across sex change in bluehead wrasse gonads. At stage 2, cyp19a1a is sharply down-regulated, after which feminizing expression collapses as female fish transition to males. Up-regulation of masculinizing gene expression begins with amh and its receptor amhr2. Classically feminizing genes within the R-spondin/Wnt/β-catenin signaling pathway (wnt4a/b, rspo1, and two transcripts annotated as fstl4 are shown as examples), and also foxl2, are duplicated in the bluehead wrasse with orthologs showing testis-specific expression.
Specific gene expression changes
Few transcripts were differentially expressed in forebrain samples, with the greatest differential expression seen among females and males (fig. S3A and data S2). Gonadal sex change involves transition from a female-biased expression landscape to one that is male biased. This transition is not linear and is punctuated by large numbers of differentially expressed transcripts between stages 3 (advanced ovarian atresia) and 4 (early testicular development) and between stages 5 (early spermatogenesis) and 6 (spermiation) (fig. S3B). Classical ovary-specific genes [nr0b1 (dax1), figla, and gdf9] became progressively down-regulated as sex change progressed (Fig. 3).
Testis-specific genes became activated from mid-sex change. This began with amh (anti-Müllerian hormone) and its receptor amh2, which were the first male-pathway genes to show increased expression, at stage 2 and before the appearance of male tissues in the gonad, followed by other classical male-promoting genes (e.g., dmrt1, sox9, and gsdf) (Fig. 3). Stages characterized by active testicular growth and spermatogenesis (stage 4 onward) displayed increasing expression of cyp11c1 and hsd11b2, consistent with their function to convert testosterone to the more potent teleost androgen 11-ketotestosterone (11-KT) (Fig. 4).
Fig. 4. Dynamic sex-specific expression of androgenic and glucocorticoid factors.
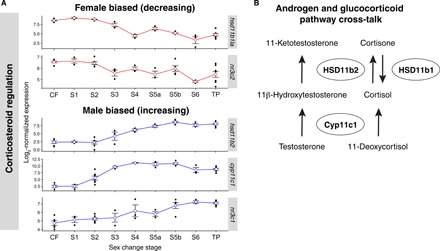
(A) Normalized gonadal expression of androgen synthesis and cortisol pathway genes across sex change. Up-regulation of cyp11c1 at stage 2 implies increased local cortisol production. Then, changes in expression of 11β-HSD enzymes at stage 3 suggest a shift from cortisone-cortisol regeneration (hsd11b1a down-regulated) to cortisol-cortisone inactivation (hsd11b2 up-regulated). Concurrent up-regulation of hsd11b2 would also promote 11-KT synthesis, the most potent teleost androgen. Genes encoding the glucocorticoid (nr3c1) and mineralocorticoid (nr3c2) receptors show opposing sex-specific expression patterns that also imply highest cortisol activity at early sex change. These expression patterns suggest a window of high cortisol activity at stage 2 that is concurrent with arrested aromatase expression and ovarian atresia, followed by the stimulation of 11-KT production by stage 3. (B) Schematic showing cross-talk between the androgen synthesis and glucocorticoid pathways. The 11β-hydroxylase enzyme (Cyp11c1, homologous to mammalian CYP11B) and two 11β-hydroxysteroid dehydrogenases (HSD11b1 and HSD11b2) are together responsible for 11-KT production and the interconversion of cortisol. Cortisol is produced via the action of Cyp11c1, whereas 11β-HSD enzymes control its interconversion with inactive cortisone, thus mediating the stress response. Cortisol itself stimulates 11β-HSD expression, resulting in the production of both 11-KT and inactive cortisone.
To identify the most upstream effectors of sex change, we focused on expression changes in the earliest phases. Notably, cyp19a1a and cyp19a1b were sharply downregulated at early sex change, in gonad (Fig. 3) and brain (fig. S3C), respectively, and remained at low levels thereafter. These genes encode the brain- and gonad-specific forms of aromatase, which converts testosterone to estradiol. The balance of estrogen [17β-estradiol (E2)] versus androgen (11-KT) production is known to control sexual fate in teleost fish (2), and aromatase inhibitors can effectively induce female-to-male sex reversal (12). Our data provide whole-transcriptome evidence that aromatase down-regulation immediately precedes a cascaded collapse in feminizing gene expression and is an early switch initiating sex change in both the brain and gonad under natural conditions.
Neofunctionalization of ovary-promoting genes and new genetic pathways are implicated in ovary-testis transformation
Many paralogous genes, arising from an ancient teleost-specific whole-genome duplication (25), show divergent sex-specific expression patterns during gonadal sex change (Fig. 3C). These include critical female-pathway genes, such as foxl2 and genes in the Rspo1/Wnt/β-catenin signaling pathway known to regulate ovarian differentiation in mammals (26). Wnt4 (wingless-type MMTV integration site family, member 4), which activates Ctnnb1 (β-catenin) and Fst (follistatin) to maintain mammalian ovarian development (Fig. 3A) (27), is duplicated in the bluehead wrasse: wnt4a was down-regulated early along with cyp19a1a, consistent with a conserved feminizing role, while its paralog wnt4b was sharply up-regulated in late sex change and is expressed only in mature testes (Fig. 3C). Furthermore, rspo1 (R-spondin–1), which stimulates Wnt4 in mammals (Fig. 3A), also showed increasing expression during testicular construction, and multiple follistatin-like genes (e.g., fstl4) showed opposing sex-specific expression patterns (Fig. 3C). Neofunctionalization of duplicated sex-pathway genes, by introducing important functional gene diversity, may underpin the notable sexual plasticity of teleost fish.
Another unexpected result from our analysis was the progressive up-regulation of genes involved in the Janus kinase–signal transducers and activators of transcription (JAK-STAT) signaling pathway, from stage 2 to 5 (table S1). JAK-STAT signaling plays an important role in determination and maintenance of male sexual fate in Drosophila melanogaster (28) but has not previously been implicated in vertebrate sexual development.
Cortisol pathways show dynamic expression across sex change
Cortisol, a glucocorticoid that controls stress-induced responses in all vertebrates, may be an important mediator of environmental sex determination (29). We therefore determined whether cortisol-pathway genes show altered expression during early sex change stages. We find that the 11β-hydroxylase gene cyp11c1, responsible for cortisol production, is up-regulated in bluehead wrasse gonads from stage 2. Furthermore, two genes encoding 11β-hydroxysteroid dehydrogenases (11β-HSD), hsd11b2 and hsd11b1-like, show differential expression across sex change but in opposite directions (Fig. 4A). The products of these genes mediate the stress response by respectively producing cortisol from inactive cortisone and vice versa, and their expression patterns imply a shift from cortisol production to inactivation from stage 3. We also observed opposite expression changes in genes encoding the glucocorticoid and mineralocorticoid receptors, nr3c1 and nr3c2 (nuclear receptor subfamily 3 group C members 1 and 2), respectively; nr3c2 matched hsd11b1la expression changes, while nr3c1 followed hsd11b2 (Fig. 4A). Together, high expression of cyp11c1, hsd11b1la, and nr3c2 at stage 2 (Fig. 4A) implies a window of high cortisol activity at the beginning of sex change, coincident with aromatase silencing and early ovarian atresia. Our data therefore suggest that dynamic cortisol production and signaling at early sex change may be a key factor triggering female-to-male transition in the bluehead wrasse.
Ovary-to-testis transformation involves extensive epigenetic reprogramming
Epigenetic marks such as DNA methylation and trimethylation of histone H3 lysine 27 (H3K27me3) are required for the specification and maintenance of cellular identity (30, 31) and are increasingly recognized as key mediators of sexual differentiation (15, 32). To explore the role of epigenetic marks in bluehead wrasse sex change, we investigated the expression profiles of genes encoding their regulatory machinery. We found that ezh2, suz12, and eed and their cofactor jarid2, components of the Polycomb repressive complex 2 (PRC2) responsible for creating H3K27me3 in vertebrates (33), were highly expressed in differentiated male and female gonads but displayed reduced expression during intermediate stages (Fig. 5A and 5D). This parallels similar findings for mammalian naïve pluripotent stem cells (34) and primordial germ cells (PGCs) (35) (the bipotential germline progenitors of egg and sperm), where H3K27me3 is also low but increases in the differentiated cells that they give rise to. The histone variant h2afz, which also features in pluripotent stem cells and PGCs, showed its highest expression at stage 3, dropping sharply at stage 4 when testicular development is first observed histologically, before recovering again (Fig. 5D). This dynamic expression pattern is interesting, given that H2A.Z colocalizes with Polycomb group (PcG) proteins at silenced developmental genes in mammalian embryonic stem cells but is dynamically redistributed during lineage commitment to potentially arbitrate developmental cell fate transitions (36). Writers and erasers of histone acetylation (acetyltransferases and deacetylases, respectively) were also expressed dynamically (fig. S4), further indicating that active chromatin modification accompanies gonadal sex change.
Fig. 5. Epigenetic factors orchestrate sex change.
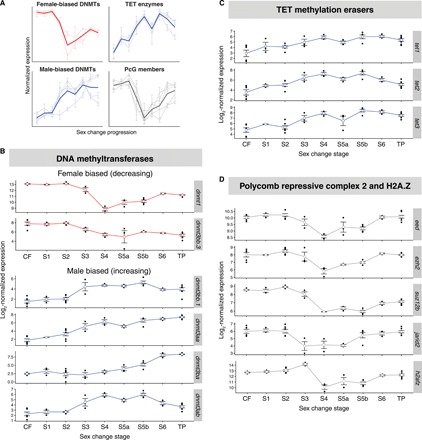
(A) Schematic summary of sex-specific expression patterns for epigenetic factors across sex change. (B) Normalized gonadal expression of DNA methyltransferase genes, showing a turnover in sex-specific expression across sex change. The maintenance methyltransferase ortholog dnmt1 is female biased and has lowest expression at mid-sex change, whereas most methyltransferase dnmt3 orthologs, responsible for de novo methylation, show increasing expression toward maleness. DNMTs, DNA methyltransferases. (C) Ten-eleven translocation (TET) methylcytosine dioxygenases, which demethylate 5-methylcytosines, show the highest expression during late sex change. (D) Genes encoding core proteins (eed, ezh2, and suz12b) and cofactors (jarid2) of the chromatin remodeling PRC2 are suppressed at mid-sex change. The variant histone H2A.Z gene (h2afz) shows a similar expression pattern.
In mammals, global reprogramming of DNA methylation is a feature of both naïve pluripotent stem cells and PGCs, a process that is driven by down-regulation of de novo DNA methyltransferase 3 (DNMT3) genes, deactivation of maintenance DNA methyltransferase activity (DNMT1), and overexpression of the ten-eleven translocation (TET) family of DNA demethylation enzymes (30). Here, we found not only reduced de novo methylation machinery at intermediate stages of sex change but also wholesale replacement of sex-specific DNMT3 orthologs during the transition from ovary to testis (Fig. 5A and 5B); dnmt3bb.3 (ortholog of mammalian Dnmt3b) showed female-specific expression that declined during sex change and was replaced by male-specific expression of dnmt3aa, dnmt3ab, dnmt3ba, and dnmt3bb.1 (orthologs of mammalian Dnmt3a and Dnmt3b). Expression of TET genes was highest at mid-late sex change (Fig. 5A and 5C). Together, this indicates that like H3K27 modification, there is an intense period of DNA methylation reprogramming in the gonad at intermediate stages of sex change. Such distinct sex-specific expression of DNA methyltransferase genes and other epigenetic modifiers was also recently observed in gonadal transcriptomes of other sex-changing fishes (37) and may be important for sexual plasticity.
Sex change involves genome-wide methylation reprogramming
To characterize the epigenetic effect of DNA methylation machinery replacement during sex change, we performed low-coverage bisulfite sequencing of DNA derived from the gonads of control females, TP males, and transitioning individuals. We found that ovary and testis showed significantly different cytosine-guanine (CG) methylation levels (70.7 and 82.1%, respectively; P < 0.001), with gonads undergoing masculinization progressively accumulating methylation (Fig. 6A). To confirm that this effect was specific to the gonad, we also tested brain tissue and found that CG methylation was relatively low (64.1%) and not sexually dimorphic.
Fig. 6. Global methylation changes and relationship between methylation and gene expression during sex change.
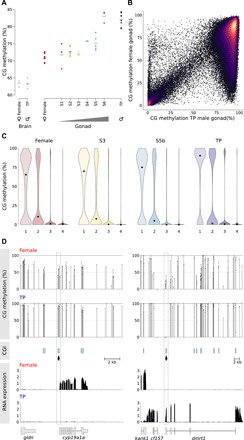
(A) Global CG methylation levels during sex change examined by low-coverage sequencing. The horizontal bar indicates the mean. Gray dots, brain samples; colored dots, gonadal samples. (B) Comparison of methylation (2-kb running windows) between female and TP male gonadal methylomes. Only probes with >100 calls were included for the analysis. (C) Violin plot showing distribution of methylation at transcription start sites (TSS) of genes classified into quartiles according to expression level (highest, 4). Each violin is scaled to the same maximum width (total area is not constant between violins) to demonstrate distributions for each quartile. Black dots denote the median. (D) Relationship between CpG methylation and RNA expression for cyp19a1a and dmrt1. CG methylation track shows methylation levels for dinucleotides with >10 calls; CGIs were predicted according to the Gardiner-Garden and Frommer criteria. Changes in TSS methylation containing CGIs (dashed box) are negatively correlated with gene expression for both genes (bottom).
To characterize where global methylation changes were occurring during sex change, we undertook deep sequencing of selected female, intermediate, and TP male libraries and compared this to a draft de novo assembly of the bluehead wrasse genome (Supplementary Materials and Methods). When 2-kb running windows were analyzed genome-wide, a notable number of low- and intermediately methylated regions in females became fully methylated in TP males (Fig. 6B). However, when partitioned in a PCA, methylomes are organized, first, by stage of sexual development from female to male (PC1) and, second, by developmental commitment (PC2) (fig. S2B), a pattern notably similar to that of the RNA-seq data (Fig. 2B). Therefore, transitionary gonads are not molecularly intermediate between differentiated ovary and testis but are distinct both in terms of global gene expression and DNA methylation.
When targeted to transcription start sites (TSS), particularly those with enriched levels of CG dinucleotides [known as CpG islands (CGIs)], DNA methylation is associated with gene silencing throughout all jawed vertebrates (38). To explore what effect the global increase in DNA methylation has on gene expression in bluehead wrasse during sex change, we binned genes into quartiles according to their expression levels and asked what levels of DNA methylation existed in their TSS (Fig. 6C). We found that DNA methylation and gene silencing were coupled in a similar fashion throughout sex change, meaning that despite the major increase in global DNA methylation during transition, DNA methylation has the capacity to enforce gene silencing at all stages of reprogramming.
The methylation patterns of key genes involved in the sex change process provides evidence for the role of DNA methylation reprogramming in gonadal transformation. A CGI linked to the aromatase (cyp19a1a) transcriptional start site (TSS) was hypermethylated as gene silencing progressed during sex change (Fig. 6D). Reciprocally, as the dmrt1 gene became up-regulated in transitioning fish, a promoter-linked CGI was progressively demethylated.
DISCUSSION
Transcriptomic and methylome analyses across sex change in the iconic bluehead wrasse have identified the triggers of socially induced sex change and enactors of gonadal metamorphosis. Our results suggest that the environmental stimulus is exerted via stress, that the subsequent steps involve repression of aromatase, and that distinctive epigenetic reprogramming is associated with reengineering ovaries into testes (Fig. 7). Importantly, this does not occur by direct transdifferentiation but involves an intermediate state with altered epigenetic machinery expression that is reminiscent of mammalian naïve pluripotent stem cells and PGCs.
Fig. 7. Mechanistic hypothesis of socially cued sex change.

Perception of a social cue (absence of a dominant male) promotes sex change in the largest female of a social group via raised cortisol. In the brain, cortisol increases isotocin expression to promote male-typical behaviors that rapidly establish social dominance. Other neuroendocrine factors may play a role [e.g., arginine vasotocin (AVT), dopamine (DA), and norepinephrine (NE)], although expression changes were not seen for their encoding genes. In the gonad, cortisol promotes transition from ovary (red) to testis (blue) via three pathways: (i) down-regulates aromatase (cyp19a1a) expression causing estrogen (E2) production to cease and feminizing expression to decline, causing ovarian atresia; (ii) up-regulates amh expression, which, as a transcription factor (TF) and a germ cell regulator, can suppress feminizing genes and promote oocyte apoptosis while promoting masculinizing expression and spermatogonial recruitment; and (iii) up-regulates androgenic genes cyp11c1 and hsd11b2 to increase 11-KT production to support testicular development. Epigenetic reprogramming, via changes in sexually dimorphic DNA methylation (represented by lollipops; open, unmethylated and filled, methylated), rewrites cellular memory of sexual fate and canalizes sex-specific expression. Photo credit: E. D’Alessandro, Oregon State University (male fish); R. Fenner, wetwebmedia.com (female fish).
We observed dynamic expression of the cortisol pathway that mediates stress. Removing the dominant TP male from a social group may represent an important social stress for large females, who must now compete for the reproductively privileged position of dominant TP male. Within minutes, a transitioning female displays aggression and courtship behaviors typical of the dominant male, along with a rapid temporary color display (a bluish coloring of the head and darkening of pectoral fin tips) that is used to establish dominance (Fig. 1). Serum cortisol levels spike during early female-male sex change in bluebanded goby (24) and male-female sex change in anemonefish (39), as well as following social rank changes in Astatotilapia (40), indicating that a stress response accompanies sexual metamorphosis and social transitions. Activation of stress pathways may also serve to meet the increased energy needs required for sex change and for maintaining social dominance (24, 40). Stress responses also vary by social rank (24), and an intriguing question is whether IP individuals exhibit a rank-dependent sensitivity to stress that may affect who undergoes sex change.
The stress response in sex-changing females is expected to elicit rapid signaling changes of brain neurotransmitters, such as arginine vasotocin, gonadotropin-releasing hormone, and norepinephrine, which have been suggested to control behavioral sex change in social wrasses (8). We observed no significant early expression changes in genes encoding these neuropeptides or their precursors in the brain, but this may reflect the limitations of transcriptomic studies performed at coarse anatomical scales (i.e., whole brain or forebrain) in detecting neural signals that may be subtle (20, 21) or highly localized (41). However, we did observe up-regulation of isotocin (it) (fig. S3C), the homolog of mammalian oxytocin, at stage 1. In fish, it is linked to territoriality and aggression (20), is up-regulated in response to cortisol (42), and so may facilitate early dominance establishment in transitioning females (Fig. 7).
Cortisol can then initiate gonadal sex change, and our data implicate pathways that promote stress-induced masculinization of genetic females in artificial settings (Fig. 7): (i) suppression of aromatase expression via glucocorticoid response elements in the cyp19a1a promoter, (ii) up-regulation of amh expression to induce germ cell apoptosis and promote maleness, and (iii) cross-talk with the androgen synthesis pathway via increased cyp11c1 and hsd11b2 expression (dual roles in 11-KT synthesis and cortisol metabolism) (29, 43). Our finding of opposing expression patterns for genes encoding glucocorticoid and mineralocorticoid receptors (nr3c1 and nr3c2) and the enzymes that control cortisol production (cyp11c1 and hsd11b1la) and inactivation (hsd11b2) implies highest cortisol production in early sex change (stage 2) (Fig. 4), consistent with observations in fishes and other vertebrates undergoing natural or temperature-induced sex reversal (14, 24, 39). Therefore, in vertebrates where the environment exerts an influence on sex, cortisol might critically link environmental stimuli with sexual fate by initiating a shift in steroidogenesis.
Following the sharp down-regulation of aromatase and estrogen production, we observed a steady collapse of the female network before a male-promoting network was progressively established. Although previous studies on protogynous wrasses (44) and protandrous anemone fish (45) are equivocal about the importance of the Foxl2 and Dmrt1 transcription factors in the sex change process, our data show that they are important later and are not triggers of sex change.
Neofunctionalization, where a gene homolog acquires a novel function following gene duplication (25), is also readily evident in our data. Important sex-pathway genes are duplicated in the bluehead wrasse and appear to have acquired new, sometimes unexpected roles. While duplicated copies of male-promoting genes (e.g., sox9) appear to have retained male-specific functions, many duplicated female-promoting genes have homologs showing a complete reversal in sex-specific expression (e.g., foxl2, wnt4, and fstl) (Fig. 3C). In particular, key components of the Rspo1/Wnt/β-catenin signaling pathway, which regulates ovarian fate in mammals, are duplicated with one homolog showing up-regulated expression during mid-to-late sex change when testicular structures are forming or have formed. This expression pattern suggests that these duplicates have acquired new roles associated with male sexual fate, notably, testicular differentiation. This flexibility in roles suggests that a less conserved female genetic network operates in bluehead wrasses and potentially other teleosts. Gene neofunctionalization following duplication allows for diversification of a standard genetic network and may be one factor contributing to the sexual plasticity that is characteristic of fishes.
The use of neofunctionalized paralogs in sex change was not restricted to hormonal and signaling pathways; we also found duplicated epigenetic machinery in bluehead wrasses that exhibited female- or male-specific expression. Global DNA methylation was remodeled, as expression of female-specific DNMTs was replaced with male-specific expression. The peak in TET expression seen at this time indicates remodeling of DNA methylation that is typical of mammalian PGCs and both naïve and classically grown pluripotent stem cells (30). The same appeared to be true for histone-modifying machinery; the PRC2 complex is associated with differentiation in mammals and showed high expression in cells belonging to committed sexual phenotypes but was deactivated in the gonads of transitional fish. Likewise, dynamic expression of histone demethylases, acetyltransferases, and variant histones further suggest active chromatin modification during gonadal sex change. Differential intron retention within Jumonji chromatin modifier genes has been associated with environmental sex reversal in a dragon lizard (14), yet we did not find consistent evidence of intron retention for Jumonji family genes in the bluehead wrasse. Our study shows comprehensive replacement of epigenetic machinery during sex change in a vertebrate and that this is analogous to epigenetic reprogramming in germline and pluripotent cells of mammals. We hypothesize that sex-specific changes in the expression of epigenetic machinery is central to plasticity of sexual phenotype seen in sex-changing fishes, where potentially a prevailing form of epigenetic memory is removed and later replaced to canalize the terminal male phenotype. Experimental inhibition of, for example, DNMTs or PRC2 activity, will be important in testing these hypotheses.
Sexual fate has long been assumed to be canalized and stable throughout life, as it generally is for mammals and birds. However, manipulation of genes in the sex determining pathway of mammalian models revealed that gonadal sex requires active maintenance via antagonistic genetic signaling to suppress pathways of the opposite sex into adulthood (10, 11). Thus, sexual fate may be inherently plastic in all vertebrates, not just in sex-changing fish.
In summary, this study reveals how environmental factors trigger gonadal sex change via a genetic cascade that reengineers ovaries into testes through an epigenetically distinct intermediate state. These findings enhance our understanding of how tissue reprogramming is controlled at the most fundamental level, and our model (Fig. 7) raises novel hypotheses about the mechanisms involved. These results also shed light on the evolution of sex determination and differentiation mechanisms in vertebrates, identifying common regulatory factors in environmentally sensitive sex determination and raising the interesting possibility that network plasticity via neofunctionalization of gene duplicates contributes to the remarkable sexual plasticity seen across teleost fishes.
MATERIALS AND METHODS
Experimental design
Female bluehead wrasses were induced to change sex by removing dominant TP males from established social groups on patch reefs off the coast of Key Largo, FL in May 2012 and May to July 2014 (5). On each reef, IP males and females larger than the 45-mm standard length were captured and sexed by examination of the sexually dimorphic genital papilla and extrusion of gametes by gentle abdominal pressure. Females were tagged and returned to home reefs. Between 0 and 15 IP males were captured from experimental reefs and relocated to distant reefs to prevent competition with females for social dominance. Within 2 days following tagging and IP male removal, TP males were removed to allow females to compete for dominance and undergo sex change. Females exhibiting TP male–typical behaviors (18) were captured at increasing time points to produce a time series of samples across the sex-change process. Sequential removal of sex changers also served to initiate sex change in the next female (5). Tagged females showing no signs of behavioral or gonadal sex change (verified by histology) were captured as controls on a range of days following TP male/sex changer removal to control for varying degrees of social upheaval. Control TP males used in reported analyses were captured from unmanipulated reefs and served as a reference. All samples were collected around the daily spawning period.
Fish were euthanized with an overdose of MS-222 (Tricaine methanesulfonate) (Sigma-Aldrich) within 2 min of capture, and the brain and gonads were dissected immediately. The brain and one gonadal lobe were preserved in RNAlater (Life Technologies Inc.) on ice, followed by storage at −20°C overnight and then −80°C until RNA extraction. The second gonadal lobe was fixed for histological analysis in 4% paraformaldehyde/1× phosphate-buffered saline (PBS) overnight at 4°C, followed by storage in 1× PBS before fixation in paraffin for histological sectioning with hematoxylin and eosin staining [Histology Laboratory, College of Veterinary Medicine, North Carolina State University (NCSU)]. Experiments were approved by the Institutional Animal Care and Use Committee at NCSU.
Gonadal sections were examined under a light microscope to determine sex change status. In total, 41 samples were partitioned into successive stages (Fig. 1) based on gonadal histology (17) and behaviors observed at the time of capture (18). Females showing no signs of behavioral or gonadal sex change (healthy ovaries with mature follicles and intact zona pellucida) served as control females. Behavioral and histological characteristics of sex change stages are summarized in Fig. 1. Sex changers at stage 5 (ongoing spermatogenesis) were further divided into stages 5a and 5b to reflect their divergent global gene expression patterns (Fig. 2B). Sample sizes for each stage were as follows: six control females (CF), three stage 1 (S1), seven stage 2 (S2), three stage 3 (S3), three stage 4 (S4), three stage 5a (S5a), five stage 5b (S5b), three stage 6 (S6), and eight TP males.
RNA-seq and whole-genome bisulfite sequencing
Brain and gonadal tissues were homogenized using TissueLyser II (Qiagen), and total RNAs were extracted with TriZol (Life Technologies), using chloroform (forebrain and midbrain) or bromochloropropane as the phase separation reagent. RNA samples were column-purified with either a NucleoSpin RNA XS kit (MACHEREY-NAGEL) after deoxyribonuclease (DNase) treatment (TURBO DNA-free Kit, Ambion) (2012 samples), or a Total RNA Purification kit (Norgen Biotek) with on-column DNase digestion (RNase-free DNase I Kit, Norgen Biotek) (2014 samples). Only the forebrain and midbrain were used for RNA extraction (removing the hindbrain containing corpus cerebelli, pons, and medulla), as these contain regions belonging to the social behavior network and the mesolimbic reward system, two neural circuits that are involved in the regulation of social decision-making (41) and likely to be key integrators and drivers of socially induced sex change.
Total RNA concentration was measured with a Qubit 2.0 Fluorometer (Life Technologies). RNA integrity was assessed with an Agilent 2100 Bioanalyzer. Sex-changing gonads consistently showed RNA profiles with a strong peak of low–molecular weight RNA, which possibly corresponds to massive 5S ribosomal RNA (rRNA) expression in atretic ovaries and masks the 18S and 28S rRNA peaks used for calculating RNA integrity numbers (RINs) in teleost ovaries and intersex gonads (46). Therefore, RIN values could not serve as useful measures of RNA integrity in sex-changing gonads of bluehead wrasses, although only samples with visibly intact rRNA peaks were used.
Library preparation and RNA-seq were performed by the Otago Genomics and Bioinformatics Facility at the University of Otago under contract to New Zealand Genomics Limited. Samples were prepared as individual Illumina TruSeq Stranded mRNA libraries. The 2012 and 2014 samples were sequenced, with multiplexing, on separate occasions. For samples collected in 2012 (three control females, three TP males, and six intersex fish), 100–base pair (bp) paired-end (PE) reads were generated over four lanes of an Illumina HiSeq 2000. For the 2014 samples (three control females, five TP males, and 21 intersex fish), 125-bp PE reads were generated over 1.5 lanes of an Illumina HiSeq 2500. Sequencing results including short read archive (SRA) accessions are summarized in data S3.
Genomic DNA for whole-genome bisulfite sequencing (WGBS) was extracted from the TRIzol-homogenized samples used for RNA-seq following the manufacturer’s instructions, with postextraction cleanup by magnetic beads (47). DNA was also extracted from the brain and gonad of additional animals collected in Belize (2013) and Florida (2016) using a dual RNA/DNA Purification kit (Norgen) or a lithium chloride protocol (48).
WGBS was undertaken using a post-bisulfite adapter tagging (PBAT) method adapted from (49). Briefly, bisulfite treatment was performed with an EZ Methylation Direct Mag Prep kit (Zymo Research) following the manufacturer’s instructions. Bisulfite treatment was performed before adapter tagging, enabling simultaneous DNA fragmentation and conversion of unmethylated cytosines. Sequencing adapters were then added by complementary strand synthesis using random heptamer priming, and last, unique molecular barcodes and sequences necessary for binding to Illumina flow cells were added to libraries by polymerase chain reaction.
Initially, we measured global DNA methylation by performing low-coverage sequencing of 6 brain (female and TP male) and 38 gonad samples (all stages) using a single-end MiSeq 100-bp protocol (Illumina) until the desired depth (at least 10,000 mapped CG calls) was attained (38). Subsequently, we performed deep sequencing on a subset of 17 gonad samples on a HiSeq 2500 instrument (Illumina) using a rapid run mode, to obtain full methylomes and nucleotide-level methylation data for replicate females, TP males, and sex changers (stage 2 to 5). Detailed sequencing and CpG quantification results are provided in data S4.
Statistical analysis
Expression quantification
Read quality was assessed in FastQC v0.11.5 (www.bioinformatics.babraham.ac.uk/projects/fastqc). Raw reads were trimmed of sequencing adaptors and low-quality bases (PHRED < 5) using Cutadapt v1.16 (50). Expression estimates for each sample were obtained using the “align_and_estimate_abundance.pl” script within the Trinity package v2.6.6 (51): For each library, trimmed reads were mapped against our published bluehead wrasse transcriptome assembly (19) using Bowtie2 v2.3.2 (52), and transcript abundances were estimated using RSEM v1.3.0 (53) with default settings. Transcript-level count matrices were generated for brain and gonad samples separately using Trinity’s “abundance_estimates_to_matrix.pl” script and the scaledTPM method.
PCA and gene set enrichment
PCA (centered and unscaled) was used to visualize transcriptome-wide expression variation within groups and among samples, following normalization by variance stabilizing transformation in DESeq2 v1.20.0 (54) in R v3.5.0 (55). The top 10,000 transcripts with greatest variance across samples were used, and confidence ellipses around barycenters were plotted using the stat_conf_ellipse() function in ggpubr v0.1.8. For gonad only, to identify the 500 transcripts contributing most to each principal component, component loadings (defined as eigenvectors scaled by the square root of the respective eigenvalues) were represented as coordinates in a Cartesian plane. Given the bimodal and skewed distribution of the values, percentiles rather than SDs were used as thresholds. Thresholds were defined as the 5th and 95th percentiles and divided the plane into four spatial regions: “Female” and “Male” represented the extremes of PC1; sexually “Differentiated” and “Transitionary” represented the extremes of PC2. Among regions, unique and shared transcripts were represented in a Euler diagram using the R package eulerr v4.1.0. To validate the results of our method, we visualized normalized expression across sex change for unique transcripts from each spatial region.
To evaluate whether intermediate stages of sex change represented a unique transitional phase, and not a simple replacement from one differentiated population to another, we replicated the above PCA analysis but included simulated mixed datasets of male and female reads. We used fastq-tools v0.8 (http://homes.cs.washington.edu/~dcjones/fastq-tools/) to randomly subsample reads from three control female and three TP male libraries, adjusting the number of initial reads to represent different proportions in the simulated datasets (male:female, 5:95, 10:90, 20:80, 40:60, 60:40, and 80:20). Transcript expression for simulated samples was quantified as above, and the original PCA results were used as a training dataset to predict principal component scores for the simulated data.
To identify the functional categories of genes uniquely associated with each spatial region, GO term enrichment analysis was performed using TopGo v2.32.0 in R and zebrafish genome annotations downloaded from Bioconductor (org.Dr.eg.db v3.6.0). Significant GO terms (P < 0.01) for “biological processes” and “molecular function” were identified using Fisher’s exact test with the “weight01” algorithm. To reduce GO term redundancy and summarize the results, GO terms and P values weighted on the basis of the scores of neighboring GO terms were used as input for REViGO (56), using SimRel as a semantic similarity measure, medium allowed similarity (0.7), and the Danio rerio GO database.
Differential expression and enrichment analyses
Differential expression analyses were performed for brain and gonad separately using a generalized linear model (GLM) framework in DESeq2. Differentially expressed transcripts were called using the Wald test in pairwise comparisons between sex change stages and control females and between neighboring stages, after fitting a single GLM to estimate size factors and dispersion across all samples per tissue. False discovery rate (FDR) was controlled at 5% to account for multiple testing, and an adjusted significance value (FDR-P) of <0.05 was used to define significant differential expression. For gonadal samples, only transcripts with a log2 fold change of >1 were considered. No fold change cutoff was applied in analyses of brain samples because of the relatively subtle expression differences observed.
Transcripts showing differential expression at each sex-change stage (compared to control females) and in comparisons between neighboring stages were searched against the Ensembl zebrafish protein database (Danio_rerio.GRCz10) (BLASTX, E-value cutoff: 10-10). Matched zebrafish protein IDs were converted to unique Ensembl zebrafish gene IDs in BioMart (57) and used for gene pathway overrepresentation analysis in DAVID v6.8 (58).
WGBS analysis and correlation with RNA-seq
Raw WGBS sequences were processed in TrimGalore! v0.4.5 (www.bioinformatics.babraham.ac.uk/projects/trim_galore/); sequencing adapters were removed, and 10 bp was trimmed from the 5′ end of reads to account for sequence biases associated with PBAT library construction, followed by removal of low-quality base calls (PHRED score < 20). Read mapping and base calling were performed in Bismark v0.19.0 (59) specifying the option --pbat. Our draft bluehead wrasse genome assembly (Supplementary Materials and Methods) was used as a reference, obtaining an average of 51.47% mapping efficiency (SD ± 3.07). BAM files were deduplicated, and reports containing CG methylation were generated. The bisulfite treatment nonconversion rate was evaluated with the frequency of non-CG methylation, with all libraries having a conversion efficiency of at least 98.94% (data S4).
CG methylation calls were analyzed in SeqMonk v1.42.0 (www.bioinformatics.babraham.ac.uk/projects/seqmonk/). Genome scaffolds were grouped into 23 pseudochromosomes, and tracks were built using in-house annotations (Supplementary Materials and Methods). Among-sample variation was examined with PCA, using 10-kb probes generated in nonoverlapping windows with a minimum of 100 CG calls. To compare among stages, the PCA was rerun with individual samples merged and using 2-kb windows with a minimum of 100 CpG calls.
To examine methylation across TSS, the longest transcript per gene was used. TSS were defined as 200 bp centered on the first nucleotide of an annotated mRNA, and a minimum of five methylation calls were applied as a threshold for inclusion. To evaluate coupling between TSS methylation and gene expression, trimmed RNA-seq reads were mapped to the reference genome using HTseq v0.9.1 (60) and imported into SeqMonk, specifying a minimum mapping quality of 60 to select only uniquely aligned reads. The SeqMonk RNA-seq quantitation pipeline was used to generate raw counts across exons of protein coding genes. Counts were corrected by transcript length and DNA contamination, and transcripts were divided into quartiles according to expression level.
To analyze methylation at individual CGs, probes of two consecutive nucleotides with a minimum of 10 methylation calls were generated and the percentage methylation measured as number of methylated calls/total calls. CGIs were identified using published methodology (61). For the regions of interest, 200-bp windows moving at 1-bp intervals were considered CGIs if the Obs/Exp value was greater than 0.6 and the GC content exceeded 60%.
Scatter plots and violin plots were drawn using ggplot2 v3.0.0 in R (62). Histograms and genome annotations were generated using Gviz v1.24.0 (63).
Supplementary Material
Acknowledgments
We thank B. Tyler, S. G. Sanchez, B. Klapheke, J. Brady, and A. Lukowsky for support in collecting samples; J. Thomas for assistance with preparing DNA; S. Fisher for aesthetic advice on Fig. 7; M. Lokman, D. Jerry, and F. Piferrer for feedback on an earlier draft; and members of the Gemmell Lab for support. Funding: This research was supported by a Marsden Fund grant (UOO1308 awarded to N.J.G.), the National Science Foundation (1257791 awarded to J.R.G. and 1257761 to B. Tyler at Indian River State College, Florida), and a University of Otago research grant awarded to N.J.G., E.V.T., and T.A.H. H.L. and O.O.-R. were supported by PhD scholarships from the Department of Anatomy at the University of Otago. E.V.T. was supported by a Royal Society of New Zealand Rutherford Postdoctoral Fellowship. Author contributions: N.J.G., J.R.G., H.L., E.V.T., T.A.H., and O.O.-R. conceived the ideas and designed the study. J.R.G., M.S.L., and H.L. collected the samples. H.L., M.S.L., O.O.-R., and O.K. performed the laboratory work. O.O.-R., E.V.T., H.L., and T.A.H. performed the bioinformatic analyses, with support from K.M.R., H.C., and M.A.B. H.C. performed the genome scaffolding and annotation. E.V.T., J.A.M.G., T.A.H., H.L., and N.J.G. wrote the manuscript, and all authors contributed editorially and approved the final version. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Sequencing data and the draft bluehead wrasse genome assembly are available from NCBI under BioProject PRJNA293777. Scripts used in the reported analyses are available on GitHub (https://github.com/hughcross/bluehead_methylome_bioinformatics/).
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/5/7/eaaw7006/DC1
Supplementary Materials and Methods
Fig. S1. Lack of sex-biased variation in global gene expression in the bluehead wrasse forebrain.
Fig. S2. Intermediate gonads are molecularly distinct from ovary and testis.
Fig. S3. Differential transcript expression in forebrain and gonad of bluehead wrasses across sex change.
Fig. S4. Writers and erasers of histone acetylation are dynamically expressed across sex change.
Table S1. Genes enriched in the JAK-STAT signaling pathway are up-regulated across sex change.
Data S1. GO enrichment detailed results.
Data S2. Differential expression statistical results.
Data S3. RNA-seq metadata for bluehead wrasse brain and gonad samples.
Data S4. WGBS metadata for bluehead wrasse gonads.
REFERENCES AND NOTES
- 1.Schärer L., The varied ways of being male and female. Mol. Reprod. Dev. 84, 94–104 (2017). [DOI] [PubMed] [Google Scholar]
- 2.Devlin R. H., Nagahama Y., Sex determination and sex differentiation in fish: An overview of genetic, physiological, and environmental influences. Aquaculture 208, 191–364 (2002). [Google Scholar]
- 3.Munday P. L., Buston P. M., Warner R. R., Diversity and flexibility of sex-change strategies in animals. Trends Ecol. Evol. 21, 89–95 (2006). [DOI] [PubMed] [Google Scholar]
- 4.Avise J. C., Mank J. E., Evolutionary perspectives on hermaphroditism in fishes. Sex. Dev. 3, 152–163 (2009). [DOI] [PubMed] [Google Scholar]
- 5.Warner R. R., Swearer S. E., Social control of sex change in the bluehead wrasse, Thalassoma bifasciatum (Pisces: Labridae). Biol. Bull. 181, 199–204 (1991). [DOI] [PubMed] [Google Scholar]
- 6.Warner R. R., The adaptive significance of sequential hermaphroditism in animals. Am. Nat. 109, 61–82 (1975). [Google Scholar]
- 7.Todd E. V., Liu H., Muncaster S., Gemmell N. J., Bending genders: The biology of natural sex change in fish. Sex. Dev. 10, 223–241 (2016). [DOI] [PubMed] [Google Scholar]
- 8.Lamm M. S., Liu H., Gemmell N. J., Godwin J. R., The need for speed: Neuroendocrine regulation of socially-controlled sex change. Integr. Comp. Biol. 55, 307–322 (2015). [DOI] [PubMed] [Google Scholar]
- 9.Capel B., Vertebrate sex determination: Evolutionary plasticity of a fundamental switch. Nat. Rev. Genet. 18, 675–689 (2017). [DOI] [PubMed] [Google Scholar]
- 10.Uhlenhaut N. H., Jakob S., Anlag K., Eisenberger T., Sekido R., Kress J., Treier A.-C., Klugmann C., Klasen C., Holter N. I., Riethmacher D., Schutz G., Cooney A. J., Lovell-Badge R., Treier M., Somatic sex reprogramming of adult ovaries to testes by FOXL2 ablation. Cell 139, 1130–1142 (2009). [DOI] [PubMed] [Google Scholar]
- 11.Matson C. K., Murphy M. W., Sarver A. L., Griswold M. D., Bardwell V. J., Zarkower D., DMRT1 prevents female reprogramming in the postnatal mammalian testis. Nature 476, 101–104 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Baroiller J.-F., D'Cotta H., The reversible sex of gonochoristic fish: Insights and consequences. Sex. Dev. 10, 242–266 (2016). [DOI] [PubMed] [Google Scholar]
- 13.Shao C., Li Q., Chen S., Zhang P., Lian J., Hu Q., Sun B., Jin L., Liu S., Wang Z., Zhao H., Jin Z., Liang Z., Li Y., Zheng Q., Zhang Y., Wang J., Zhang G., Epigenetic modification and inheritance in sexual reversal of fish. Genome Res. 24, 604–615 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Deveson I. W., Holleley C. E., Blackburn J., Marshall Graves J. A., Mattick J. S., Waters P. D., Georges A., Differential intron retention in Jumonji chromatin modifier genes is implicated in reptile temperature-dependent sex determination. Sci. Adv. 3, e1700731 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ge C., Ye J., Weber C., Sun W., Zhang H., Zhou Y., Cai C., Qian G., Capel B., The histone demethylase KDM6B regulates temperature-dependent sex determination in a turtle species. Science 360, 645–648 (2018). [DOI] [PubMed] [Google Scholar]
- 16.Godwin J., Social determination of sex in reef fishes. Semin. Cell Dev. Biol. 20, 264–270 (2009). [DOI] [PubMed] [Google Scholar]
- 17.Nakamura M., Hourigan T. F., Yamauchi K., Nagahama Y., Grau E. G., Histological and ultrastructural evidence for the role of gonadal steroid hormones in sex change in the protogynous wrasse Thalassoma duperrey. Environ. Biol. Fishes 24, 117–136 (1989). [Google Scholar]
- 18.Semsar K., Godwin J., Multiple mechanisms of phenotype development in the bluehead wrasse. Horm. Behav. 45, 345–353 (2004). [DOI] [PubMed] [Google Scholar]
- 19.Liu H., Lamm M. S., Rutherford K., Black M. A., Godwin J. R., Gemmell N. J., Large-scale transcriptome sequencing reveals novel expression patterns for key sex-related genes in a sex-changing fish. Biol. Sex Differ. 6, 26 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Todd E. V., Liu H., Lamm M. S., Thomas J. T., Rutherford K., Thompson K. C., Godwin J. R., Gemmell N. J., Female mimicry by sneaker males has a transcriptomic signature in both the brain and the gonad in a sex-changing fish. Mol. Biol. Evol. 35, 225–241 (2018). [DOI] [PubMed] [Google Scholar]
- 21.Wong R. Y., McLeod M. M., Godwin J., Limited sex-biased neural gene expression patterns across strains in Zebrafish (Danio rerio). BMC Genomics 15, 905 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Picco R., Tomasella A., Fogolari F., Brancolini C., Transcriptomic analysis unveils correlations between regulative apoptotic caspases and genes of cholesterol homeostasis in human brain. PLOS ONE 9, e110610 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kramer P. R., Wray S., 17-β-Estradiol regulates expression of genes that function in macrophage activation and cholesterol homeostasis. J. Steroid Biochem. Mol. Biol. 81, 203–216 (2002). [DOI] [PubMed] [Google Scholar]
- 24.Solomon-Lane T. K., Crespi E. J., Grober M. S., Stress and serial adult metamorphosis: Multiple roles for the stress axis in socially regulated sex change. Front. Neurosci. 7, 210 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Glasauer S. M. K., Neuhauss S. C. F., Whole-genome duplication in teleost fishes and its evolutionary consequences. Mol. Genet. Genomics 289, 1045–1060 (2014). [DOI] [PubMed] [Google Scholar]
- 26.Ungewitte E. K., Yao H. H.-C., How to make a gonad: Cellular mechanisms governing formation of the testes and ovaries. Sex Dev. 7, 7–20 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tevosian S. G., Genetic control of ovarian development. Sex Dev. 7, 33–45 (2013). [DOI] [PubMed] [Google Scholar]
- 28.Jinks T. M., Polydorides A. D., Calhoun G., Schedl P., The JAK/STAT signaling pathway is required for the initial choice of sexual identity in Drosophila melanogaster. Mol. Cell 5, 581–587 (2000). [DOI] [PubMed] [Google Scholar]
- 29.Goikoetxea A., Todd E. V., Gemmell N. J., Stress and sex: Does cortisol mediate sex change in fish? Reproduction 154, R149–R160 (2017). [DOI] [PubMed] [Google Scholar]
- 30.Lee H. J., Hore T. A., Reik W., Reprogramming the methylome: Erasing memory and creating diversity. Cell Stem Cell 14, 710–719 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Atlasi Y., Stunnenberg H. G., The interplay of epigenetic marks during stem cell differentiation and development. Nat. Rev. Genet. 18, 643–658 (2017). [DOI] [PubMed] [Google Scholar]
- 32.Piferrer F., Epigenetics of sex determination and gonadogenesis. Dev. Dyn. 242, 360–370 (2013). [DOI] [PubMed] [Google Scholar]
- 33.Margueron R., Reinberg D., The Polycomb complex PRC2 and its mark in life. Nature 469, 343–349 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Marks H., Kalkan T., Menafra R., Denissov S., Jones K., Hofemeister H., Nichols J., Kranz A., Stewart A. F., Smith A., Stunnenberg H. G., The transcriptional and epigenomic foundations of ground state pluripotency. Cell 149, 590–604 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hajkova P., Ancelin K., Waldmann T., Lacoste N., Lange U. C., Cesari F., Lee C., Almouzni G., Schneider R., Surani M. A., Chromatin dynamics during epigenetic reprogramming in the mouse germ line. Nature 452, 877–881 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Creyghton M. P., Markoulaki S., Levine S. S., Hanna J., Lodato M. A., Sha K., Young R. A., Jaenisch R., Boyer L. A., H2AZ is enriched at polycomb complex target genes in ES cells and is necessary for lineage commitment. Cell 135, 649–661 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tsakogiannis A., Manousaki T., Lagnel J., Papanikolaou N., Papandroulakis N., Mylonas C. C., Tsigenopoulos C. S., The gene toolkit implicated in functional sex in Sparidae hermaphrodites: Inferences from comparative transcriptomics. Front. Genet. 9, 749 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Peat J. R., Ortega-Recalde O., Kardailsky O., Hore T. A., The elephant shark methylome reveals conservation of epigenetic regulation across jawed vertebrates. F1000Res. 6, 526 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Godwin J. R., Thomas P., Sex change and steroid profiles in the protandrous anemonefish Amphiprion melanopus (Pomacentridae, Teleostei). Gen. Comp. Endocrinol. 91, 144–157 (1993). [DOI] [PubMed] [Google Scholar]
- 40.Maruska K. P., Social transitions cause rapid behavioral and neuroendocrine changes. Integr. Comp. Biol. 55, 294–306 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.O'Connell L. A., Hofmann H. A., Genes, hormones, and circuits: An integrative approach to study the evolution of social behavior. Front. Neuroendocrinol. 32, 320–335 (2011). [DOI] [PubMed] [Google Scholar]
- 42.Cádiz L., Román-Padilla J., Gozdowska M., Kulczykowska E., Martínez-Rodriguez G., Mancera J. M., Martos-Sitcha J. A., Cortisol modulates vasotocinergic and isotocinergic pathways in the gilthead sea bream. J. Exp. Biol. 218, 316–325 (2015). [DOI] [PubMed] [Google Scholar]
- 43.Fernandino J. I., Hattori R. S., Moreno Acosta O. D., Strüssmann C. A., Somoza G. M., Environmental stress-induced testis differentiation: Androgen as a by-product of cortisol inactivation. Gen. Comp. Endocrinol. 192, 36–44 (2013). [DOI] [PubMed] [Google Scholar]
- 44.Kobayashi Y., Horiguchi R., Nozu R., Nakamura M., Expression and localization of forkhead transcriptional factor 2 (Foxl2) in the gonads of protogynous wrasse, Halichoeres trimaculatus. Biol. Sex Differ. 1, 3 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Casas L., Saborido-Rey F., Ryu T., Michell C., Ravasi T., Irigoien X., Sex change in clownfish: Molecular insights from transcriptome analysis. Sci. Rep. 6, 35461 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Diaz de Cerio O., Rojo-Bartolomé I., Bizarro C., Ortiz-Zarragoitia M., Cancio I., 5S rRNA and accompanying proteins in gonads: Powerful markers to identify sex and reproductive endocrine disruption in fish. Environ. Sci. Technol. 46, 7763–7771 (2012). [DOI] [PubMed] [Google Scholar]
- 47.Oberacker P., Stepper P., Bond D. M., Höhn S., Focken J., Meyer V., Schelle L., Sugrue V. J., Jeunen G.-J., Moser T., Hore S. R., von Meyenn F., Hipp K., Hore T. A., Jurkowski T. P., Bio-On-Magnetic-Beads (BOMB): Open platform for high-throughput nucleic acid extraction and manipulation. PLOS Biol. 17, e3000107 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gemmell N. J., Akiyama S., An efficient method for the extraction of DNA from vertebrate tissues. Trends Genet. 12, 338–339 (1996). [DOI] [PubMed] [Google Scholar]
- 49.Peat J. R., Dean W., Clark S. J., Krueger F., Smallwood S. A., Ficz G., Kim J. K., Marioni J. C., Hore T. A., Reik W., Genome-wide bisulfite sequencing in zygotes identifies demethylation targets and maps the contribution of TET3 oxidation. Cell Rep. 9, 1990–2000 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Martin M., Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 17, 10–12 (2011). [Google Scholar]
- 51.Haas B. J., Papanicolaou A., Yassour M., Grabherr M., Blood P. D., Bowden J., Couger M. B., Eccles D., Li B., Lieber M., MacManes M. D., Ott M., Orvis J., Pochet N., Strozzi F., Weeks N., Westerman R., William T., Dewey C. N., Henschel R., Leduc R. D., Friedman N., Regev A., De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis. Nat. Protoc. 8, 1494–1512 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Langmead B., Salzberg S. L., Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Li B., Dewey C. N., RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics 12, 323 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Love M. I., Huber W., Anders S., Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.R Core Team, R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria (2018); https://www.R-project.org/.
- 56.Supek F., Bošnjak M., Škunca N., Šmuc T., REVIGO summarizes and visualizes long lists of gene ontology terms. PLOS ONE 6, e21800 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Smedley D., Haider S., Durinck S., Pandini L., Provero P., Allen J., Arnaiz O., Awedh M. H., Baldock R., Barbiera G., Bardou P., Beck T., Blake A., Bonierbale M., Brookes A. J., Bucci G., Buetti I., Burge S., Cabau C., Carlson J. W., Chelala C., Chrysostomou C., Cittaro D., Collin O., Cordova R., Cutts R. J., Dassi E., Di Genova A., Djari A., Esposito A., Estrella H., Eyras E., Fernandez-Banet J., Forbes S., Free R. C., Fujisawa T., Gadaleta E., Garcia-Manteiga J. M., Goodstein D., Gray K., Guerra-Assuncao J. A., Haggarty B., Han D. J., Han B. W., Harris T., Harshbarger J., Hastings R. K., Hayes R. D., Hoede C., Hu S., Hu Z. L., Hutchins L., Kan Z., Kawaji H., Keliet A., Kerhornou A., Kim S., Kinsella R., Klopp C., Kong L., Lawson D., Lazarevic D., Lee J. H., Letellier T., Li C. Y., Lio P., Liu C. J., Luo J., Maass A., Mariette J., Maurel T., Merella S., Mohamed A. M., Moreews F., Nabihoudine I., Ndegwa N., Noirot C., Perez-Llamas C., Primig M., Quattrone A., Quesneville H., Rambaldi D., Reecy J., Riba M., Rosanoff S., Saddiq A. A., Salas E., Sallou O., Shepherd R., Simon R., Sperling L., Spooner W., Staines D. M., Steinbach D., Stone K., Stupka E., Teague J. W., Dayem Ullah A. Z., Wang J., Ware D., Wong-Erasmus M., Youens-Clark K., Zadissa A., Zhang S. J., Kasprzyk A., The BioMart community portal: An innovative alternative to large, centralized data repositories. Nucleic Acids Res. 43, W589–W598 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Dennis G. Jr., Sherman B. T., Hosack D. A., Yang J., Gao W., Lane H. C., Lempicki R. A., DAVID: Database for annotation, visualization, and integrated discovery. Genome Biol. 4, R60 (2003). [PubMed] [Google Scholar]
- 59.Krueger F., Andrews S. R., Bismark: A flexible aligner and methylation caller for Bisulfite-Seq applications. Bioinformatics 27, 1571–1572 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Anders S., Pyl P. T., Huber W., HTSeq—A Python framework to work with high-throughput sequencing data. Bioinformatics 31, 166–169 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Gardiner-Garden M., Frommer M., CpG islands in vertebrate genomes. J. Mol. Biol. 196, 261–282 (1987). [DOI] [PubMed] [Google Scholar]
- 62.H. Wickham, ggplot2: Elegant Graphics for Data Analysis (Springer International Publishing, ed. 2, 2009). [Google Scholar]
- 63.Hahne F., Ivanek R., Visualizing genomic data using Gviz and Bioconductor. Methods Mol. Biol. 1418, 335–351 (2016). [DOI] [PubMed] [Google Scholar]
- 64.Simpson J. T., Exploring genome characteristics and sequence quality without a reference. Bioinformatics 30, 1228–1235 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Weisenfeld N. I., Yin S., Sharpe T., Lau B., Hegarty R., Holmes L., Sogoloff B., Tabbaa D., Williams L., Russ C., Nusbaum C., Lander E. S., MacCallum I., Jaffe D. B., Comprehensive variation discovery in single human genomes. Nat. Genet. 46, 1350–1355 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Bolger A. M., Lohse M., Usadel B., Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Boetzer M., Henkel C. V., Jansen H. J., Butler D., Pirovano W., Scaffolding pre-assembled contigs using SSPACE. Bioinformatics 27, 578–579 (2011). [DOI] [PubMed] [Google Scholar]
- 68.Li H., Durbin R., Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 25, 1754–1760 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Leggett R. M., Clavijo B. J., Clissold L., Clark M. D., Caccamo M., NextClip: An analysis and read preparation tool for Nextera Long Mate Pair libraries. Bioinformatics 30, 566–568 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Boetzer M., Pirovano W., Toward almost closed genomes with GapFiller. Genome Biol. 13, R56 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Bryant D. M., Johnson K., DiTommaso T., Tickle T., Couger M. B., Payzin-Dogru D., Lee T. J., Leigh N. D., Kuo T.-H., Davis F. G., Bateman J., Bryant S., Guzikowski A. R., Tsai S. L., Coyne S., Ye W. W., Freeman R. M. Jr., Peshkin L., Tabin C. J., Regev A., Haas B. J., Whited J. L., A tissue-mapped axolotl de novo transcriptome enables identification of limb regeneration factors. Cell Rep. 18, 762–776 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Haas B. J., Delcher A. L., Mount S. M., Wortman J. R., Smith R. K. Jr., Hannick L. I., Maiti R., Ronning C. M., Rusch D. B., Town C. D., Salzberg S. L., White O., Improving the Arabidopsis genome annotation using maximal transcript alignment assemblies. Nucleic Acids Res. 31, 5654–5666 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Wu T. D., Watanabe C. K., GMAP: A genomic mapping and alignment program for mRNA and EST sequences. Bioinformatics 21, 1859–1875 (2005). [DOI] [PubMed] [Google Scholar]
- 74.Kent W. J., BLAT—The BLAST-like alignment tool. Genome Res. 12, 656–664 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Camacho C., Coulouris G., Avagyan V., Ma N., Papadopoulos J., Bealer K., Madden T. L., BLAST+: Architecture and applications. BMC Bioinformatics 10, 421 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Simão F. A., Waterhouse R. M., Ioannidis P., Kriventseva E. V., Zdobnov E. M., BUSCO: Assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31, 3210–3212 (2015). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/5/7/eaaw7006/DC1
Supplementary Materials and Methods
Fig. S1. Lack of sex-biased variation in global gene expression in the bluehead wrasse forebrain.
Fig. S2. Intermediate gonads are molecularly distinct from ovary and testis.
Fig. S3. Differential transcript expression in forebrain and gonad of bluehead wrasses across sex change.
Fig. S4. Writers and erasers of histone acetylation are dynamically expressed across sex change.
Table S1. Genes enriched in the JAK-STAT signaling pathway are up-regulated across sex change.
Data S1. GO enrichment detailed results.
Data S2. Differential expression statistical results.
Data S3. RNA-seq metadata for bluehead wrasse brain and gonad samples.
Data S4. WGBS metadata for bluehead wrasse gonads.