

Abstract
Anoxic depolarization of pyramidal neurons results from a large inward current that is activated, in part, by excessive glutamate release during exposure to anoxia/ischemia. Pannexin-1 (Panx1) channels can be activated both by ischemia and NMDA receptors (NMDARs), but the mechanisms of Panx1 activation are unknown. We used whole-cell recordings to show that pharmacological inhibition or conditional genetic deletion of Panx1 strongly attenuates the anoxic depolarization of CA1 pyramidal neurons in acute brain slices from rats and mice. Anoxia or exogenous NMDA activated Src family kinases (SFKs), as measured by increased phosphorylation of SFKs at Y416. The SFK inhibitor PP2 prevented Src activation and Panx1 opening during anoxia. A newly developed interfering peptide that targets the SFK consensus-like sequence of Panx1 (Y308) attenuated the anoxic depolarization (AD) without affecting SFK activation. Importantly, the NMDAR antagonists, d-APV and R-CPP, attenuated AD currents carried by Panx1, and the combined application of d-APV and 10panx (a Panx1 blocker) inhibited AD currents to the same extent as either blocker alone. We conclude that activation of NMDARs during anoxia/ischemia recruits SFKs to open Panx1, leading to sustained neuronal depolarizations.
Introduction
Loss of blood flow to the brain during stroke causes ischemia and subsequent neuronal death. Tissue in the penumbra, which surrounds the ischemic core, has a gradient of anoxia/ischemia and undergoes repetitive depolarizing events called peri-infarct depolarizations. These waves of depolarization are thought to be critical for tissue death and core expansion (Mies et al., 1993; Ohta et al., 2001). Peri-infarct depolarizations, also called anoxic depolarizations (ADs), are proposed to overwhelm the neuron's ionic homeostatic mechanisms so that loss of oxidative phosphorylation and ATP production results in neuronal death and dysfunction (Phillis et al., 1996; Lipton, 1999). The components of the AD have been investigated for decades, but little consensus has been reached on the sequelae that lead to loss of the neuronal membrane potential and cell death, likely because of the complex and multifaceted insults that result during ischemia.
In the hippocampus, enhanced glutamate release during anoxia/ischemia is likely caused by the combination of increased presynaptic vesicle exocytosis (Fleidervish et al., 2001) and reversal of the astrocytic excitatory amino acid transporter, resulting in activation of AMPA receptors and NMDA receptors (NMDARs) and consequential neuronal depolarization (Rossi et al., 2000). Our work recently identified pannexin-1 (Panx1) channels as potential contributors to ADs because Panx1 is activated directly by ischemia in an NMDAR-independent manner or by overstimulation of the NMDAR itself (Thompson et al., 2006, 2008) (but see Madry et al., 2010). Other components of the ischemic insult, such as hypoglycemia and increased extracellular K+, may also activate Panx1 channels (Kawamura et al., 2010; Santiago et al., 2011). Thus, the mechanisms of activation of Panx1 channels during the AD are likely to be diverse and have not been well characterized.
Several potential mechanisms of activation for Panx1 during anoxia and NMDAR overstimulation exist. These include truncation of the C terminal of Panx1 by caspases, as reported for apoptotic cells releasing ATP (Chekeni et al., 2010), or by posttranslational modifications such as phosphorylation or nitrosylation (Zhang et al., 2008). Panx1 activation by P2X7 receptors may be mediated by Src family kinases (SFKs) because disruption of P2X7 binding to SFKs with an interfering peptide prevented Panx1 opening by BzATP in J774 macrophages (Iglesias et al., 2008). The SFKs are also known to be physically linked to the GluN2A and GluN2B subunits of NMDARs (Yu et al., 1997) where they function to phosphorylate NMDARs and increase channel mean open time, which potentiates receptor responses during chronic pain (Salter and Pitcher, 2012). Interestingly, SFK activity increases in hippocampal neurons during ischemia (Takagi et al., 1997, 1999), raising the possibility that SFKs activate Panx1 during anoxia. Here we tested this idea in acute hippocampal slices and found that interfering with Src activation or a putative SFK phosphorylation site on Panx1 prevents anoxia-induced Panx1 opening.
Materials and Methods
Animals.
All animal care protocols were approved by the University of Calgary's Animal Care and Use Committee in accordance with the Canadian Council on Animal Care guidelines. Experiments were performed on Sprague Dawley rats (postnatal days 21–32), unless otherwise indicated, that were housed on a 12 h light/dark cycle with ad libitum access to Purina Laboratory Chow and water.
Chemicals and reagents.
All salts for making artificial CSF (aCSF; see below) were from Sigma-Aldrich. The NMDAR antagonists, d-APV (final concentration, 50 μm) and R-CPP (5 μm), as well as the pan- caspase inhibitor zVAD-FMK (100 μm, applied intracellularly) were from Tocris Bioscience. The SFK inhibitor, PP2 (10 μm); its inactive analog, PP3 (10 μm); tamoxifen; and probenecid (500 μm) were from Sigma-Aldrich. The specific mimetic peptide inhibitor of Panx1, 10panx (100 μm, sequence WRQAAFVDSY) (Pelegrin and Surprenant, 2006; Thompson et al., 2008); a scrambled control peptide to 10panx (scpanx, 100 μm; Thompson et al., 2008); the TAT-conjugated Panx1 Y308 interfering peptide, TAT-Panx1305–318 (10 μm, sequence YGRKKRRQRRRLKVYEILPTFDVLH); and a TAT control peptide (10 μm, sequence YGRKKRRQRRR) were from AnaSpec or New England Peptide. The C-terminal Panx1 antibody (α-panx1; 1:1000 final dilution, 0.25 ng/μl) was purchased from Invitrogen (catalog #488100, rabbit polyclonal). Anti-connexin-43 (α-Cx43; 1:2000 final dilution, 0.3 ng/μl) was from Abcam (catalog #ab11370, rabbit polyclonal) and was a generous gift from Dr. K. Sharkey (University of Calgary, Alberta, Canada). All drugs were made into aliquots dissolved in water or DMSO and were diluted to the final concentration in aCSF (see below; the final concentration of DMSO never exceeded 0.1%, and appropriate controls were performed as indicated).
Generation of conditional Panx1 knock-out mice.
Embryonic stem cells from agouti mice containing flox sequences flanking exon 2 of the Panx1 gene (Panx1flx/flx; accession number NM_019482.2) were purchased from the Knockout Mouse Project (KO1434, Panx1_B01) and microinjected into C57BL6/J female mice. The first generation was screened based on coat color (agouti), and expression of the Panx1flx/flx gene was confirmed by PCR using a sense primer (5′-ACCTAAGAGACGGACCTG-3′) and an antisense primer (5′-GAATCAATCCTCTGTGCC T-3′). Homozygote Panx1flx/flx were bred with C57BL6/J mice expressing cre recombinase-ERT2 under the wolfram syndrome 1 (wfs1) promoter (Jax mice; B6;C3-Tg(Wfs1-cre/ERT2)3Aibs/J; stock number 009103). Subsequent generations were screened by PCR, and homozygous Panx1flx/flx/homozygous wfs1-cre-ERT2 mice were bred and backcrossed for at least six generations to yield the conditional Panx1 knock-out mouse (FlxPanx1-Cre). The knock-out of Panx1 in weaned FlxPanx-Cre mice was initiated by two to five intraperitoneal injections of tamoxifen (300 or 100 mg/kg, respectively) at 24 h intervals. Control mice received vehicle alone (corn oil plus 5% ethanol) in parallel with male siblings that received tamoxifen. Hippocampal slices were prepared as described below from animals 48–96 h after the last intraperitoneal injection.
Acute hippocampal slice preparation.
Rats or mice were anesthetized by isoflurane inhalation in air and decapitated; the brain was extracted, blocked, and mounted on a vibrating slicer (VT1200S; Leica) while submerged in an ice-cold solution consisting of the following (in mm): 87 NaCl, 2.5 KCl, 25 NaHCO3, 0.5 CaCl2, 7 MgCl2, 1.25 NaH2PO4, 25 glucose, and 75 sucrose, saturated with 95% O2/5% CO2. Transverse hippocampal slices were cut (370 μm) and transferred into a chamber containing aCSF at 33°C for at least 1 h before experimentation. aCSF consisted of 120 mm NaCl, 26 mm NaHCO3, 3 mm KCl, 1.25 mm NaH2PO4, 1.3 mm MgCl2, 2 mm CaCl2, and 10 mm glucose and was saturated with 95% O2/5% CO2.
Electrophysiology.
Slices were transferred to a recording chamber and continually perfused with aCSF (33–35°C) at a rate of 1–2 ml per minute. Visualization of hippocampal pyramidal neurons from the CA1 region was achieved via differential interface contrast (differential interference contrast) imaging with a BX51WI microscope (Olympus). Whole-cell patch-clamp recordings were obtained using borosilicate glass microelectrodes (Sutter Instrument) with a tip resistance of 3–5 MΩ pulled using a P-1000 Flaming/Brown Micropipette Puller (Sutter Instrument) and filled with an intracellular solution containing 108 mm potassium gluconate, 2 mm MgCl2, 8 mm sodium gluconate, 8 mm KCl, 2.5 mm K2-EGTA, 4 mm K2-ATP, and 0.3 mm Na3-GTP at pH 7.25 with 10 mm HEPES. Some experiments were performed with 100 μm zVAD-FMK, with a Panx1 antibody (0.25 ng/μl, α-panx1), with the SFK inhibitor PP2 (10 μm), or with a TAT-conjugated Panx1 Y308 interfering peptide (TAT-Panx1305–318; 10 and 1 μm). For experiments where drugs were included in the pipette solution, a minimum of 10 min was allowed for intracellular equilibration. Control experiments in which the vehicle, 0.01 or 0.1% DMSO, was added to the intracellular solution were also performed when appropriate and reported as “vehicle” in the text and figures.
Anoxic conditions were achieved by switching the perfusate with aCSF equilibrated with 95% Ar/5% CO2 to displace oxygen, while maintaining pH at 7.4. These conditions typically yielded a pO2 of <5 mmHg (Thompson et al., 2006). Anoxia was applied after at least 5 min of a stable “normoxic” baseline holding current or for 10 min when blockers were applied intracellularly. Access resistance was always <25 MΩ, but if it exceeded 25 MΩ during the anoxic depolarization, the cells were discarded from further analysis.
Western blots and immunocytochemistry.
All analyses were performed on acute hippocampal slices prepared as described above. Slices were maintained in either control or anoxic aCSF, and after a 1 h exposure to drugs/anoxia, slices were quickly lysed in RIPA lysis buffer (Invitrogen) using a mini-Dounce homogenizer. Western blot analysis of hippocampal–neocortical brain slice lysates for target proteins was performed according to standard protocols. Briefly, slices were harvested with lysis buffer containing protease and phosphatase inhibitors. Total protein (25–50 μg) was loaded in each SDS-PAGE well and transferred onto PVDF membrane. Membranes were blocked using 5% skim milk powder or 1% BSA in TBS-T (10 mm Tris, 100 mm NaCl, 1% Tween 20) for 1 h at room temperature and incubated in a primary antibody for 1–2 h at room temperature (or 4°C overnight), with gentle shaking. Blots were washed three times in TBS-T and incubated in secondary antibodies for 1–2 h at room temperature, with gentle shaking. Blots were then washed two times with TBS-T, incubated in ECL-Plus (PI32132; Pierce Chemical) substrate/reagent according to the manufacturer's protocol, and imaged and quantified using a UVP Biospectrum Imaging system.
For immunocytochemical analysis of Panx1 expression in FlxPanx1-Cre mice, animals were anesthetized with an intraperitoneal injection of sodium pentobarbital (0.1 ml/100 mg body weight) and perfused by cardiac puncture with PBS, followed by 4% paraformaldehyde (PFA). The brains were harvested and postfixed in 4% PFA overnight, and antigen retrieval was performed by exposure of whole brains to 10 mm sodium citrate in 0.05% Tween, pH 6.0, at 85°C for 15–25 min. Brains were then cryoprotected with 30% sucrose for 24–48 h, cryosectioned (30 μm thickness), and floated in PBS. Slices were permeabilized with 0.5% Triton X-100 in PBS for 10 min and incubated in a blocking solution (5% donkey serum and 0.2% Triton X-100) for 1 h. Primary antibodies against Panx1 (see above) were diluted 1:40 in a blocking solution, and slices were incubated at room temperature for 24 h. After three 10 min washes in PBS, cryosections were incubated in a secondary antibody (diluted in a blocking solution), Alexa Fluor 488, for at least 4 h. Sections were washed, mounted onto chrom alum-coated slides, coverslipped with Vectashield (Vector Laboratories), and visualized with a confocal microscope (SP5; Leica).
Data analysis and statistics.
Electrophysiological data were digitized at 10 kHz and low-pass filtered at 1 kHz with a MultiClamp 700B amplifier and Digidata 1440A analog to digital convertor (Molecular Devices). Data were recorded using pCLAMP 10, Clampex 10.3, and Axoscope 10.3 (Molecular Devices) software and stored for future analysis with Clampfit 10.3, Origin 8 (OriginLab), and Excel (Microsoft). Statistical significance was determined by one-way ANOVA with the post hoc Tukey's test, two-paired t test, or two-sample t test, and significance was set at p < 0.05. All results are presented as means ± SEM.
Results
Anoxia and ischemia are potent inducers of glutamate release in the hippocampus, which consequently activates NMDAR-dependent secondary currents leading to ADs (Martin et al., 1994) and neuronal death and dysfunction (Pulsinelli et al., 1982; Hansen, 1985; Pulsinelli, 1985; Frandsen et al., 1989; Choi and Rothman, 1990; Lipton, 1999; Rossi et al., 2007). Here we exposed hippocampal slices to anoxia while recording from CA1 pyramidal neurons in the whole-cell configuration to investigate the mechanism of activation of Panx1 during the AD. We used anoxia instead of oxygen/glucose deprivation (OGD) to mimic ischemia because of substantially less swelling of the slice, allowing for longer recordings and pharmacological manipulations. Furthermore, we included 4 mm ATP in the patch pipette (postsynaptic neuron) to reduce the likelihood of direct Panx1 activation by ischemia-induced ATP depletion, allowing us to isolate NMDAR effects.
Typical responses of CA1 neurons to anoxia are shown in Figure 1A. CA1 pyramidal neurons underwent an AD 20.9 ± 2.2 min (n = 16) after removal of O2 that was manifested as a substantial inward current (initial depolarization), with a mean ± SEM peak value of −2.14 ± 0.24 nA (n = 16). The initial peak depolarization typically occurred within 10 min of onset (Fig. 1A). The anoxia-induced current then relaxed toward the baseline, leaving a residual current (mean, −1.65 ± 0.22 nA; n = 16). AD inward currents were quantified as the peak amplitude (initial depolarization, within 10 min of onset) and the residual current amplitude (average current amplitude over 10 min calculated 10 min after the peak; Fig. 1A), which allowed us to determine the severity of the anoxic insult and the magnitude of the partial recovery, respectively. Furthermore, integration of the area under the current trace provided a reliable and reproducible measure of total charge transfer across the membrane (Fig. 1D; 0.98 ± 0.16 μC at 10 min after the AD onset; n = 16 for anoxia), which we used as an indicator of the degree of ionic dysregulation induced by anoxia. A drop in neuronal input resistance of >50% was associated with the AD peak current (Thompson et al., 2006). This suggested opening of conductance pathways in the neurons that became unreliable to quantify, which is why we chose to examine charge transfer over long periods of time (i.e., 10 min).
Figure 1.

Block of pannexin-1 attenuates anoxia-induced inward currents. A, Representative whole-cell recordings from CA1 pyramidal neurons in hippocampal slices exposed to anoxia (onset indicated by arrows) with and without the presence of the Panx1 blocker 10panx (100 μm; right trace). Initial depolarization and residual currents are indicated (arrows) and were significantly attenuated by 10panx. B, Mean (red line) ± SEM (gray region) of 6–16 neurons (see C) of the AD in response to different Panx1 antagonists. Both 10panx (middle) and the intracellularly applied anti-Panx1 antibody, α-panx1(i) (0.25 ng/μl; right), reduced the AD-associated inward current, with promotion of recovery toward the baseline (dashed line). C, Quantitative analyses of the initial depolarization and residual current in the presence of Panx1 antagonists; the number of cells in each experiment are indicated in the bars. D, Cumulative net charge transfer during the anoxic inward current determined as area under the curve. Bath application of 10panx and α-panx1(i) both significantly attenuated the net charge compared with anoxia alone and their respective negative controls of bath-applied scrambled 10panx (scpanx, 100 μm) or intracellular anti-connexin 43, α-Cx43(i) (0.3 ng/μl; an α-panx1(i) control). *p < 0.05, significance compared with control; #p < 0.05, significance compared with α-Cx43(i). Error bars in B–D are SEM.
Blockers of Panx1 channels attenuate the anoxic inward current
Panx1 channels are activated in acutely isolated hippocampal neurons by both overstimulation of NMDARs and directly by ischemia (Thompson et al., 2006, 2008). To determine whether Panx1 contributes to the anoxia-induced inward current in CA1 pyramidal neurons in acute brain slices, we tested the effect of several Panx1 antagonists on the AD current. Bath application of the Panx1 inhibitor, 100 μm 10panx (Pelegrin and Surprenant, 2006; Thompson et al., 2008), significantly (p < 0.05 vs no blockers) attenuated the initial depolarization by ∼50% (Fig. 1; control AD, −2.14 ± 0.25 nA vs 10panx, −1.05 ± 0.29 nA; n = 6). The residual current (−0.24 ± 0.04 nA; n = 6) also more closely approached the preanoxic baseline when Panx1 was blocked, so that 1.41 ± 0.34 nA of inward current that normally persisted in unblocked neurons was absent. This “mean residual difference current” was calculated as the control residual current minus the 10panx residual current. Additionally, the total charge transfer when Panx1 was blocked was 0.27 ± 0.29 μC (n = 6) at 10 min after AD onset, leaving a mean difference charge transfer of 0.71 ± 0.21 μC. A scrambled version of the 10panx peptide (scpanx; n = 8) did not significantly alter the AD current amplitude (initial depolarization, −2.21 ± 0.26 nA; residual current, −1.78 ± 0.28 nA; p < 0.05) or the net charge transfer (1.07 ± 0.19 μC at 10 min; p < 0.05) compared with anoxia alone (Fig. 1C).
Other Panx1 blockers effectively attenuated the AD current, including 500 μm probenecid (Silverman et al., 2008; Ransford et al., 2009) (residual current, −0.51 ± 0.17 nA; n = 5) and an antibody against the C terminal of Panx1, α-Panx1, when included in the patch pipette (residual current, −0.30 ± 0.06 nA; n = 6; Fig. 1). Inclusion of an antibody against α-Cx43, another member of the gap junction superfamily that is not expressed in neurons (Dermietzel et al., 1989; Thompson and MacVicar, 2008), failed to affect the AD (initial depolarization, −2.30 ± 0.41 nA; residual current, −1.40 ± 0.34 nA; n = 9; p > 0.05 vs anoxia alone; Fig. 1C,D). In all cases, block of Panx1 promoted recovery of the AD current to the preanoxic baseline (Fig. 1C) and dramatically reduced total charge transfer (i.e., ionic dysregulation) across the membrane (Fig. 1D), demonstrating that Panx1 is activated during exposure of hippocampal CA1 pyramidal neurons to anoxia.
Conditional knock-out of Panx1 attenuates AD currents
Pharmacological blockers of pannexins, connexins, and innexins (the gap junction superfamily) can lack specificity and inhibit several family members (Bruzzone et al., 2005). In the case of the peptide inhibitor of Panx1, 10panx, it seems to also block connexin 46, although this protein has expression that is primarily restricted to the lens in rodents (Bruzzone et al., 2005). Nevertheless, and despite the appropriate pharmacological controls (Fig. 1C), we sought to confirm a role for Panx1 in the AD independently of pharmacological interventions. To this end, we generated conditional Panx1 knock-out mice by crossing mice with floxed exon 2 of the Panx1 gene with a tamoxifen-inducible cre-recombinase line driven by the wolfram syndrome 1 (wfs1) promoter (see Materials and Methods), which is highly expressed in the hippocampus (Luuk et al., 2008). Intraperitoneal injections of tamoxifen to 21- to 35-d-old mice induced deletion of Panx1 as indicated by loss of immunoreactivity to α-Panx1 (Fig. 2A).
Figure 2.
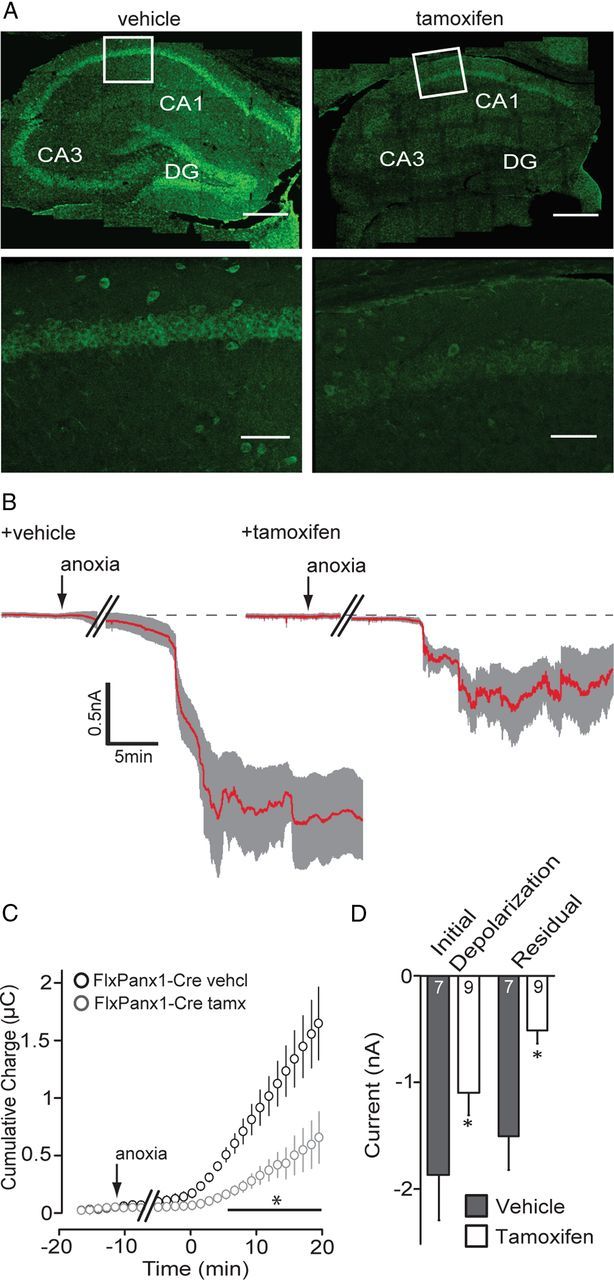
Knock-out of pannexin-1 attenuates the AD current. A, Immunoflourescence images of Panx1 expression in the hippocampus from vehicle-injected (left) and tamoxifen-injected (right) FlxPanx1-Cre mice. Top images are a 6 × 5 mosaic image taken at 20× magnification. Bottom panels are from the region indicated by the white box in the mosaic image. Scale bars: top, 300 μm; bottom, 100 μm. Note that Panx1 expression is lost in tamoxifen-treated animals, indicating knock-out. B, Mean (red line) ± SEM (gray region) whole-cell recordings of the anoxic depolarization from FlxPanx1-Cre mice treated with vehicle (left) or tamoxifen (right). C, Cumulative charge transfer during anoxia was significantly (*p < 0.05) reduced when Panx1 was knocked out. D, Quantitative analyses of the initial depolarization and residual current for Panx1 knock-out mice. Error bars are SEM.
The response of murine CA1 pyramidal neurons (from vehicle-injected FlxPanx1-Cre mice) to anoxia was similar to that measured in rat neurons (Fig. 2B–D). However, knock-out of Panx1 attenuated ADs (Fig. 2B–D); the initial depolarization was significantly (p < 0.05) smaller in the tamoxifen-injected animals compared with that in vehicle controls (initial depolarization: vehicle, −1.87 ± 0.41 nA, n = 7; tamoxifen, −1.10 ± 0.21 nA, n = 9). Furthermore, recordings from the tamoxifen-treated FlxPanx1-Cre mice had significantly reduced ionic dysregulation (charge transfer, 0.38 ± 0.08 μC 10 min after onset; n = 9) compared with vehicle-injected control animals (0.99 ± 0.16 μC; n = 7; Fig. 2C). Loss of Panx1 promoted recovery to the preanoxic baseline (i.e., residual current amplitude, −0.50 ± 0.12 nA; p < 0.05 vs vehicle control; mean residual difference current, 1.00 ± 0.31 nA; Fig. 2C). Together with the data in Figure 1, these results strongly support a role for Panx1 in inward currents and ionic dysregulation during anoxia.
Anoxia-induced opening of Panx1 is caspase cleavage independent
It was reported recently that Panx1 is activated during apoptosis by caspase cleavage of its C terminal (Chekeni et al., 2010; Sandilos et al., 2012). Anoxia can be a potent inducer of both necrosis and apoptosis, suggesting that caspase modification of the Panx1 protein could be involved in channel opening during ADs. To test the possibility of caspase-dependent activation of Panx1 in our anoxia-induced AD model, we assessed changes in total Panx1 protein after anoxia, hypothesizing that immunoblotting of hippocampal lysates with the C-terminal-directed α-Panx1 should reveal a decrease in Panx1 band density and/or a mobility shift if the Panx1 C terminus is cleaved. As shown in Figure 3A, no detectable decrease in Panx1 immunoreactivity or mobility was detectable after a 1 h exposure to anoxia.
Figure 3.

Anoxia-mediated Panx1 opening does not require caspases. A, Analysis of relative Panx1 expression after exposure of hippocampal slices to 1 h of anoxia. Note that no significant change in Panx1 levels or mobility was detected. B, Mean ± SEM traces of the anoxic depolarization for 8 neurons treated with vehicle (left; 0.1% DMSO) and 12 neurons treated with zVAD-FMK (right) included in the patch pipette. C, Quantitative summary of the initial depolarization and residual current in the presence of intracellular zVAD-FMK (dissolved in 0.1% DMSO) compared with vehicle in the pipette solution. D, Net cumulative charge across the membrane was not significantly attenuated by pan-caspase inhibition.
As an additional test for the potential role of caspases in Panx1 activation, we added the pan-caspase inhibitor zVAD-FMK (100 μm) to the patch pipette and measured AD currents in CA1 hippocampal neurons. Neurons were dialyzed for at least 10 min before exposure to anoxic aCSF, with either vehicle (0.1% DMSO) or zVAD. Intracellular zVAD, at a concentration reported to prevent Panx1 C-terminal cleavage (100 μm; Chekeni et al., 2010), did not alter the amplitude of the peak depolarization (Fig. 3B,C; initial depolarization, −1.59 ± 0.27 nA; p = 0.37; n = 12) compared with vehicle alone (initial depolarization, −1.95 ± 0.28 nA; n = 8), nor did it improve the recovery toward baseline (Fig. 3B,C; residual currents: vehicle, −1.28 ± 0.19 nA; zVAD, −1.07 ± 0.22 nA; p = 0.5). Caspase inhibition by zVAD also failed to exhibit a significant reduction in ionic dysregulation compared with vehicle alone (Fig. 3D; charge transfer, 0.75 ± 0.18 μC at 10 min from onset; p = 0.55 vs vehicle).
Src family kinases are activated during anoxia, leading to Panx1 opening
Previous work has shown that inhibition of Panx1, activated by purinergic receptors, occurs in cell lines exposed to the SFK inhibitor PP2 (Iglesias et al., 2008). This raises the possibility that SFKs regulate Panx1 opening in neurons in brain slices exposed to anoxia. We examined the Panx1 sequence and noted that the C terminus contains a putative phosphorylation site at tyrosine Y308 as part of a closely matched consensus sequence (YEEI) for SFKs (Bibbins et al., 1993; Songyang et al., 1993). Incubation of hippocampal slices in anoxia increased SFK phosphorylation at Y416 by ∼20%, and this increase was prevented by the SFK inhibitor PP2 (Fig. 4A,B) (Smart et al., 1981; Patschinsky et al., 1982). PP3, the inactive analog of PP2, was ineffective at preventing SFK phosphorylation at Y416 (Fig. 4A,B). When PP2 was included in the patch pipette, the AD current was attenuated compared with PP3 (Fig. 4C–E; residual currents: PP3, −1.29 ± 0.27 nA, n = 7; PP2, −0.55 ± 0.10 nA, n = 6). Importantly, inclusion of PP3 in the patch pipette did not produce ADs that were significantly different (p > 0.05) from the control response (i.e., compared with control samples from Fig. 1).
Figure 4.

Src family kinase inhibition during anoxia blunts the anoxic inward current. A, Src family kinase activation during anoxia was assayed by Western blot analysis of phospho-Y416 levels. Phospho-Src was increased by anoxia and blocked by the Src inhibitor PP2 (10 μm), but the inactive analog, PP3 (10 μm), was ineffective. B, Quantitative analysis of relative density of Src Y416 in A compared with GAPDH levels in the same blots. The number of individual experiments (i.e., hippocampal slices) is indicated in each bar. Note that anoxia significantly (p < 0.05) increased phospho-Y416 levels, which was blocked by PP2 but not PP3. C, Mean ± SEM traces of the anoxic depolarization with intracellular PP3 (left) and PP2 (right). D, Quantitative summary of the initial depolarization (left) and residual current (right) with Src inhibition by PP2 included in the patch pipette. PP2 significantly reduced the residual currents compared with PP3 (p < 0.05). E, Src inhibition by PP2 decreased the cumulative charge transfer compared with PP3 (p < 0.05).
We next sought to interfere with Panx1 at its SFK target sequence, Y308. This was achieved by inclusion of a small interfering peptide in the patch pipette or bath that comprised the amino acid sequence 305–318 of Panx1 conjugated to the TAT sequence for membrane permeability. The interfering peptide, TAT-Panx305–318 (see Materials and Methods), in the bath dramatically attenuated anoxia-induced ADs in a manner similar to PP2 (Fig. 5). Furthermore, to address the specificity of the Y308 site in single neurons, and to rule out possible effects of the peptide on the entire brain slice, TAT-Panx305–318 was included in the patch pipette (TAT-Panx305–318(i)), where it potently blocked the AD (Fig. 5). As an important negative control, the TAT peptide alone included in the patch pipette failed to alter the AD inward current (Fig. 5; the mean difference charge transfer was not significant vs control without TAT peptide; p = 0.86).
Figure 5.

An interfering peptide that mimics the Src consensus sequence of Panx1 attenuates the AD. A, Mean ± SEM traces of the anoxic depolarization comparing the TAT peptide alone (10 μm intracellular; left trace), bath-applied TAT-Panx305–318 (10 μm; middle trace), and TAT-Panx305–318 (1 μm; right trace) in the patch pipette. Inclusion of TAT-Panx305–318 either in the bath or the patch pipette promoted recovery of the AD current toward baseline (dashed line). B, Quantitative analyses of the initial depolarization and residual current with and without inhibition presence of TAT-Panx1305–318 compared with the TAT peptide alone. C, Net cumulative charge transfer in the presence of TAT-Panx305–318 in the bath or pipette. Error bars are SEM. *p < 0.05, significant difference versus the TAT(i) group (paired t test).
NMDARs are critical in the anoxia-induced inward current
NMDARs are important for generating ADs (Lee et al., 1999; Rossi et al., 2000) and can activate Panx1 (Thompson et al., 2008), but the mechanisms linking the two remain unknown. Our recording conditions included 4 mm ATP in the patch pipette, suggesting that activation of Panx1 during anoxia is primarily attributable to enhanced glutamate release and is occurring independently of a fall in ATP in the postsynaptic neuron. To investigate a direct role for the NMDAR in the activation of Panx1, 50 μm d-APV was included in the bath during anoxia. APV did not significantly delay the onset of the AD current (26.87 ± 5.18 min) but significantly reduced the AD peak current (Fig. 6A–C; mean difference peak current, 1.08 ± 0.29 nA; n = 11; p < 0.05 compared with control from Fig. 1), promoted recovery toward the preanoxic baseline (Fig. 6A,B; mean residual difference current, 0.35 ± 0.08 nA), and decreased charge transfer across the neuronal membrane (Fig. 6C; mean difference, 0.63 ± 0.15 μC from control). The NMDAR antagonist, 5 μm R-CPP, was similarly effective as d-APV at inhibiting the AD currents (Fig. 6B; n = 5). Coapplication of d-APV and 10panx inhibited ADs by a similar magnitude to either drug alone (Fig. 6B; mean difference peak current, 0.95 ± 0.32 nA; n = 8; p < 0.05 vs 10panx or d-APV alone), suggesting that NMDARs were required for Panx1 opening.
Figure 6.
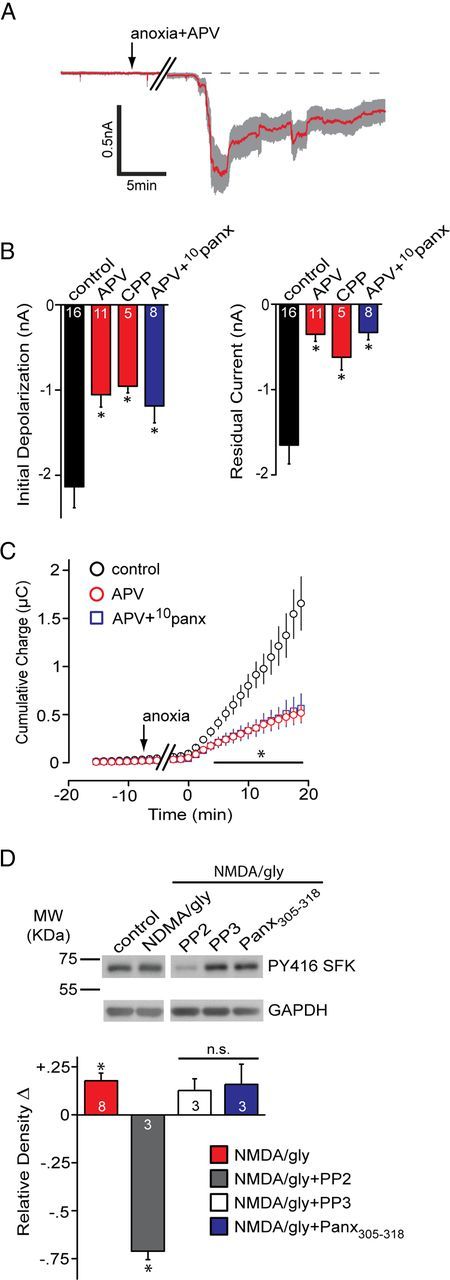
The contribution of NMDARs to Panx1 activation during the anoxic depolarization. A, Mean ± SEM trace of the anoxic depolarization during bath application of the NMDAR antagonist d-APV (50 μm). Blocking NMDARs promoted recovery toward the baseline. B, Quantitative summary of the anoxic depolarization with NMDAR block by d-APV or R-CPP (5 μm), as well as concomitant block of NMDAR and Panx1 (d-APV+10panx). Control data are the same as those presented in Figure 1. C, Net cumulative charge transfer across the membrane with NMDAR block by d-APV or R-CPP. D, Top, Src family kinase activation in the presence of bath-applied NMDA/glycine (gly) assayed by Western blot analysis of phospho-Y416 levels. Bottom, Quantitative analysis of the relative (vs control) density of phospho-Src Y416. Band densities were normalized to GAPDH and expressed relative to control, which was set at zero for each gel. Application of NMDA/glycine was significantly different than control (one-way Student's t test vs a mean of 0). PP2 was significantly different (p < 0.05) from NMDA/gly. All bands in the blot are from the same gel. Error bars are SEM.
If the NMDAR is opening Panx1 and SFKs are involved as the data above suggest, it follows that NMDAR activation should lead to SFK phosphorylation at Y416. Incubation of acute hippocampal slices in 100 μm NMDA and 10 μm glycine to activate NMDARs leads to a marked increase in SFK phospho-Y416. PP2, but not PP3 or 1 μm TAT-Panx305–318, blocked SFK activation (Fig. 6D), indicating that activation of NMDARs by exogenous NMDA (or endogenous glutamate release during anoxia; Fig. 4) leads to activation of SFKs.
Discussion
Here we have shown that anoxia, acting through NMDA receptors, activates Src kinases to open Panx1 channels and sustain the anoxic depolarization inward current. Antagonists of Panx1 channels reduced the peak amplitude of the anoxia-induced inward current and promoted recovery toward the preanoxic baseline, consequently decreasing charge transfer across the membrane. The role of Panx1 was further validated with a conditional knock-out mouse line that had confirmed lack of Panx1 expression in the hippocampus. The addition of the NMDAR antagonist d-APV together with the Panx1 blocker 10panx did not result in further inhibition of AD currents above either blocker alone. Finally, we have demonstrated that anoxia and exogenous NMDA/glycine activated Src family kinases and that SFKs were responsible for opening Panx1, possibly via phosphorylation of Y308 on the Panx1 C terminal. Together, we have identified a signaling pathway that is important for the anoxic depolarization and involves NMDARs coupling to Panx1 via SFKs.
Contribution of Panx1 to anoxia-induced neuronal depolarization and death
It was recently suggested that Panx1 does not contribute to ADs in hippocampal neurons in brain slices because the nonselective gap junction superfamily blocker carbenoxolone failed to decrease OGD/oxidative phosphorylation inhibitor-induced inward currents (Madry et al., 2010). However, Madry and colleagues were able to block dye uptake by hippocampal neurons during ADs, which is suggestive of the opening of large-pore, pannexin-like channels (Bruzzone et al., 2005; Thompson et al., 2006). Although we cannot explain why they failed to observe carbenoxolone-sensitive currents, our multipronged pharmacological and genetic approaches strongly support a role for Panx1 in the AD in hippocampal CA1 neurons in slices. One possibility is that the use of pharmacological blockers of the electron transport chain/glycolysis had direct effects on Panx1 or SFKs. Interestingly, glutamate receptor antagonists did not suppress ADs in rodent neocortex (Murphy et al., 2008) when measured directly with field electrodes or indirectly with intrinsic optical imaging, suggesting that the AD may have multiple origins that depend on brain region or developmental age.
The data presented here support our hypothesis that Panx1 functions to maintain ADs (MacVicar and Thompson, 2010). The sequence of events during ADs and subsequent neuronal death appears to be that anoxia/ischemia promotes glutamate release from presynaptic terminals (Fleidervish et al., 2001) and reversal of the astrocytic excitatory amino acid transporter (Rossi et al., 2000). Increased glutamate overstimulates NMDARs, which in turn activate SFKs and open Panx1. The depolarization of CA1 neurons during anoxia is likely initiated by ionotropic glutamate receptors, including AMPA and NMDARs (Yamamoto et al., 1997; Rossi et al., 2000). Because we maintained 4 mm ATP in the postsynaptic neuron by inclusion in the pipette, this argues against Panx1 opening induced by a fall in ATP in the postsynaptic neuron (Thompson et al., 2008). It is important to note that direct activation of Panx1 by ischemia (Thompson et al., 2006) is still possible during in vivo stroke (Zhang et al., 2008; Thompson and MacVicar, 2010). Future studies will be aimed at blocking the NMDAR during ischemia and determining the intrinsic mechanism of Panx1 activation.
In our recordings, there is a portion of the AD inward current that remains even when NMDARs, Panx1, or both are blocked. This likely results from the activation of AMPA and NMDA receptors during increased glutamate release (Yamamoto et al., 1997). Several other nonselective cation channels may govern the conductances responsible for AD currents. In addition to AMPA, NMDA receptors, and Panx1, the AD involves members of the transient receptor potential (TRP) cation channel family (Rossi et al., 2000; Aarts et al., 2003; Lipski et al., 2006; Olah et al., 2009). There are also numerous reports that Na+ influx (i.e., via Na+/Ca2+, amiloride-sensitive Na+ exchanger) and voltage-dependent Na+ channels are (Chidekel et al., 1997; Yamamoto et al., 1997; White et al., 2012) or are not (Pisani et al., 1998; Banasiak et al., 2002) involved in depolarization and death of neocortical neurons. This emphasizes the variability and complexity of the response to ischemia that likely depends on both neuronal type (cortical vs hippocampal) and model system (culture vs acute slice). Indeed, it remains a challenge of the field to identify and characterize the mechanisms and temporal contributions of these nonselective cation channels to the AD in specific neuronal populations.
Sustained anoxic depolarizations are critical for dysregulation of intracellular Ca2+ and neuronal death during excitotoxicity (Connor et al., 1988; Lee et al., 1999). If Panx1 is important in sustaining the AD, as our data suggest, it would follow that knock-out of Panx1 would be neuroprotective during stroke. Indeed, recent work confirms the hypothesis that Panx1 is important in neuronal death during stroke because an independently generated mouse with constitutive genetic knockdown of Panx1 and Panx2 resulted in significantly reduced lesion volumes in mice after middle cerebral artery occlusion (Bargiotas et al., 2011). Interestingly, death induced by Panx1 activation may be a common mechanism for many neurons, as emphasized by our recent report that Panx1 is involved in loss of enteric neurons during models of colitis and Crohn's disease (Gulbransen et al., 2012).
Mechanisms of Panx1 opening during anoxia involve Src family kinases
Here we have explored two possible mechanisms of activation of Panx1 during anoxia. The first was the possibility that Panx1 was activated by cleavage of its C terminal by caspases, as has been reported for apoptotic cells releasing ATP (via Panx1) to call immune cells and initiate phagocytosis (Chekeni et al., 2010). Whereas this interesting mechanism appears important in apoptosis, we were unable to detect either a decrease in Panx1 levels or a shift in Panx1 mobility on Western blots, suggesting that Panx1 is not being cleaved in our acute anoxic paradigm. As further support to this, no effect of the pan-caspase inhibitor zVAD-FMK, applied intracellularly at an effective concentration (Chekeni et al., 2010), on anoxia-induced inward currents was observed. We cannot, however, rule out the possibility that Panx1 cleavage occurs later under chronic anoxic exposure and contributes to localized immune responses. Interestingly, Na+ influx during ischemia is reported to activate caspase-3 and subsequent neuronal apoptosis (Banasiak et al., 2004), suggesting a link between delayed Na+ dysregulation and caspase-3-induced Panx1 opening to call phagocytotic cells (Chekeni et al., 2010), as occurs hours to days after stroke (Dirnagl et al., 1999).
The activation of Panx1 during exposure of CA1 neurons to anoxia seems to involve the Src family kinases. We demonstrate that SFKs have increased phosphorylation at Y416 during anoxia, which is a strong indicator of activation (Smart et al., 1981). The SFK inhibitor PP2 prevented this activation and significantly attenuated Panx1 activity during the AD. Interestingly, an interfering peptide that mimicked the sequence of Panx1 between 305 and 318 (and contained the only known sequence similar to the SFK consensus site on Panx1) blocked channel activation during anoxia. Together, these data support a role for SFK-mediated activation of Panx1 during anoxia via phosphorylation of Y308 at the Panx1 C terminal. Unfortunately, commercially available Panx1 antibodies, in our hands, are not able to immunoprecipitate Panx1 so that direct analysis of the phosho-tyrosine state of the channel could not be investigated.
How are SFKs being activated during anoxia? It has been described that SFKs associate with NMDARs via postsynaptic density 95 (PSD95) through interaction of the PDZ3 domain of PSD95 and the SH2 domain of SFKs (Takagi et al., 1999). This interaction seems important for augmentation of NMDAR currents in models of chronic pain (Salter and Pitcher, 2012). However, a potential reciprocal relationship where NMDARs activate SFKs is also possible, and we demonstrate here that exogenous NMDA/glycine acted similarly to anoxia, inducing phosphorylation of SFKs at Y416.
In summary, we have identified a mechanism through which overstimulation of NMDARs by anoxia leads to opening of Panx1 channels by activation of SFKs. Given the important role of NMDARs and Panx1 in neuronal death during ischemia, our work has linked these two pathways via the Src family of tyrosine kinases. Thus, targeting SFKs, Y308 on Panx1, or the Panx1 channel pore itself represents novel strategies to treat pathological neuronal depolarizations similar to those that occur during stroke.
Footnotes
The authors declare no competing financial interests.
This work was supported by grants from the Canadian Institutes for Health Research (89721), Alberta Innovates–Health Solutions, and the Canada Foundation for Innovation to R.J.T. R.J.T. holds a scholar award from Alberta Innovates–Health Solutions and a New Investigator award from the Heart and Stroke Foundation of Canada. N.L.W. holds an Alberta Innovates–Health Solutions scholarship. We thank Cheryl Sank for technical assistance and Dr. Jaideep Bains for critical reading of this manuscript.
References
- Aarts M, Iihara K, Wei WL, Xiong ZG, Arundine M, Cerwinski W, MacDonald JF, Tymianski M. A key role for TRPM7 channels in anoxic neuronal death. Cell. 2003;115:863–877. doi: 10.1016/s0092-8674(03)01017-1. [DOI] [PubMed] [Google Scholar]
- Banasiak KJ, Burenkova O, Haddad GG. Activation of voltage-sensitive sodium channels during oxygen deprivation leads to apoptotic neuronal death. Neuroscience. 2004;126:31–44. doi: 10.1016/S0306-4522(03)00425-1. [DOI] [PubMed] [Google Scholar]
- Bargiotas P, Krenz A, Hormuzdi SG, Ridder DA, Herb A, Barakat W, Penuela S, von Engelhardt J, Monyer H, Schwaninger M. Pannexins in ischemia-induced neurodegeneration. Proc Natl Acad Sci U S A. 2011;108:20772–20777. doi: 10.1073/pnas.1018262108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bibbins KB, Boeuf H, Varmus HE. Binding of the Src SH2 domain to phosphopeptides is determined by residues in both the SH2 domain and the phosphopeptides. Mol Cell Biol. 1993;13:7278–7287. doi: 10.1128/mcb.13.12.7278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruzzone R, Barbe MT, Jakob NJ, Monyer H. Pharmacological properties of homomeric and heteromeric pannexin hemichannels expressed in Xenopus oocytes. J Neurochem. 2005;92:1033–1043. doi: 10.1111/j.1471-4159.2004.02947.x. [DOI] [PubMed] [Google Scholar]
- Chekeni FB, Elliott MR, Sandilos JK, Walk SF, Kinchen JM, Lazarowski ER, Armstrong AJ, Penuela S, Laird DW, Salvesen GS, Isakson BE, Bayliss DA, Ravichandran KS. Pannexin 1 channels mediate ‘find-me’ signal release and membrane permeability during apoptosis. Nature. 2010;467:863–867. doi: 10.1038/nature09413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chidekel AS, Friedman JE, Haddad GG. Anoxia-induced neuronal injury: role of Na+ entry and Na+-dependent transport. Exp Neurol. 1997;146:403–413. doi: 10.1006/exnr.1997.6544. [DOI] [PubMed] [Google Scholar]
- Choi DW, Rothman SM. The role of glutamate neurotoxicity in hypoxic-ischemic neuronal death. Annu Rev Neurosci. 1990;13:171–182. doi: 10.1146/annurev.ne.13.030190.001131. [DOI] [PubMed] [Google Scholar]
- Connor JA, Wadman WJ, Hockberger PE, Wong RK. Sustained dendritic gradients of Ca2+ induced by excitatory amino acids in CA1 hippocampal neurons. Science. 1988;240:649–653. doi: 10.1126/science.2452481. [DOI] [PubMed] [Google Scholar]
- Dermietzel R, Traub O, Hwang TK, Beyer E, Bennett MV, Spray DC, Willecke K. Differential expression of three gap junction proteins in developing and mature brain tissues. Proc Natl Acad Sci U S A. 1989;86:10148–10152. doi: 10.1073/pnas.86.24.10148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dirnagl U, Iadecola C, Moskowitz MA. Pathobiology of ischaemic stroke: an integrated view. Trends Neurosci. 1999;22:391–397. doi: 10.1016/s0166-2236(99)01401-0. [DOI] [PubMed] [Google Scholar]
- Fleidervish IA, Gebhardt C, Astman N, Gutnick MJ, Heinemann U. Enhanced spontaneous transmitter release is the earliest consequence of neocortical hypoxia that can explain the disruption of normal circuit function. J Neurosci. 2001;21:4600–4608. doi: 10.1523/JNEUROSCI.21-13-04600.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frandsen A, Drejer J, Schousboe A. Direct evidence that excitotoxicity in cultured neurons is mediated via N-methyl-D-aspartate (NMDA) as well as non-NMDA receptors. J Neurochem. 1989;53:297–299. doi: 10.1111/j.1471-4159.1989.tb07327.x. [DOI] [PubMed] [Google Scholar]
- Gulbransen BD, Bashashati M, Hirota SA, Gui X, Roberts JA, MacDonald JA, Muruve DA, McKay DM, Beck PL, Mawe GM, Thompson RJ, Sharkey KA. Activation of neuronal P2X7 receptor-pannexin-1 mediates death of enteric neurons during colitis. Nat Med. 2012;18:600–604. doi: 10.1038/nm.2679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen AJ. Effect of anoxia on ion distribution in the brain. Physiol Rev. 1985;65:101–148. doi: 10.1152/physrev.1985.65.1.101. [DOI] [PubMed] [Google Scholar]
- Iglesias R, Locovei S, Roque A, Alberto AP, Dahl G, Spray DC, Scemes E. P2X7 receptor-Pannexin1 complex: pharmacology and signaling. Am J Physiol Cell Physiol. 2008;295:C752–C760. doi: 10.1152/ajpcell.00228.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawamura M, Jr, Ruskin DN, Masino SA. Metabolic autocrine regulation of neurons involves cooperation among pannexin hemichannels, adenosine receptors, and KATP channels. J Neurosci. 2010;17:3886–3895. doi: 10.1523/JNEUROSCI.0055-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JM, Zipfel GJ, Choi DW. The changing landscape of ischaemic brain injury mechanisms. Nature. 1999;399:A7–A14. doi: 10.1038/399a007. [DOI] [PubMed] [Google Scholar]
- Lipski J, Park TI, Li D, Lee SC, Trevarton AJ, Chung KK, Freestone PS, Bai JZ. Involvement of TRP-like channels in the acute ischemic response of hippocampal CA1 neurons in brain slices. Brain Res. 2006;1077:187–199. doi: 10.1016/j.brainres.2006.01.016. [DOI] [PubMed] [Google Scholar]
- Lipton P. Ischemic cell death in brain neurons. Physiol Rev. 1999;79:1431–1568. doi: 10.1152/physrev.1999.79.4.1431. [DOI] [PubMed] [Google Scholar]
- Luuk H, Koks S, Plaas M, Hannibal J, Rehfeld JF, Vasar E. Distribution of Wfs1 protein in the central nervous system of the mouse and its relation to clinical symptoms of the Wolfram syndrome. J Comp Neurol. 2008;509:642–660. doi: 10.1002/cne.21777. [DOI] [PubMed] [Google Scholar]
- MacVicar BA, Thompson RJ. Non-junction functions of pannexin-1 channels. Trends Neurosci. 2010;33:93–102. doi: 10.1016/j.tins.2009.11.007. [DOI] [PubMed] [Google Scholar]
- Madry C, Haglerød C, Attwell D. The role of pannexin hemichannels in the anoxic depolarization of hippocampal pyramidal cells. Brain. 2010;133:3755–3763. doi: 10.1093/brain/awq284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin RL, Lloyd HG, Cowan AI. The early events of oxygen and glucose deprivation: setting the scene for neuronal death? Trends Neurosci. 1994;17:251–257. doi: 10.1016/0166-2236(94)90008-6. [DOI] [PubMed] [Google Scholar]
- Mies G, Iijima T, Hossmann KA. Correlation between peri-infarct DC shifts and ischaemic neuronal damage in rat. Neuroreport. 1993;4:709–711. doi: 10.1097/00001756-199306000-00027. [DOI] [PubMed] [Google Scholar]
- Murphy TH, Li P, Betts K, Liu R. Two-photon imaging of stroke onset in vivo reveals that NMDA-receptor independent ischemic depolarization is the major cause of rapid reversible damage to dendrites and spines. J Neurosci. 2008;28:1756–1772. doi: 10.1523/JNEUROSCI.5128-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohta K, Graf R, Rosner G, Heiss WD. Calcium ion transients in peri-infarct depolarizations may deteriorate ion homeostasis and expand infarction in focal cerebral ischemia in cats. Stroke. 2001;32:535–543. doi: 10.1161/01.str.32.2.535. [DOI] [PubMed] [Google Scholar]
- Olah ME, Jackson MF, Li H, Perez Y, Sun HS, Kiyonaka S, Mori Y, Tymianski M, MacDonald JF. Ca2+-dependent induction of TRPM2 currents in hippocampal neurons. J Physiol. 2009;587:965–979. doi: 10.1113/jphysiol.2008.162289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patschinsky T, Hunter T, Esch FS, Cooper JA, Sefton BM. Analysis of the sequence of amino acids surrounding sites of tyrosine phosphorylation. Proc Natl Acad Sci U S A. 1982;79:973–977. doi: 10.1073/pnas.79.4.973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pelegrin P, Surprenant A. Pannexin-1 mediates large pore formation and interleukin-1beta release by the ATP-gated P2X7 receptor. EMBO J. 2006;25:5071–5082. doi: 10.1038/sj.emboj.7601378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillis JW, O'Regan MH, Estevez AY, Song D, VanderHeide SJ. Cerebral energy metabolism during severe ischemia of varying duration and following reperfusion. J Neurochem. 1996;67:1525–1531. doi: 10.1046/j.1471-4159.1996.67041525.x. [DOI] [PubMed] [Google Scholar]
- Pisani A, Calabresi P, Tozzi A, Bernardi G, Knöpfel T. Early sodium elevations induced by combined oxygen and glucose deprivation in pyramidal cortical neurons. Eur J Neurosci. 1998;10:3572–3574. doi: 10.1046/j.1460-9568.1998.00398.x. [DOI] [PubMed] [Google Scholar]
- Pulsinelli WA. Selective neuronal vulnerability: morphological and molecular characteristics. Prog Brain Res. 1985;63:29–37. doi: 10.1016/S0079-6123(08)61973-1. [DOI] [PubMed] [Google Scholar]
- Pulsinelli WA, Levy DE, Duffy TE. Regional cerebral blood flow and glucose metabolism following transient forebrain ischemia. Ann Neurol. 1982;11:499–502. doi: 10.1002/ana.410110510. [DOI] [PubMed] [Google Scholar]
- Ransford GA, Fregien N, Qiu F, Dahl G, Conner GE, Salathe M. Pannexin 1 contributes to ATP release in airway epithelia. Am J Respir Cell Mol Biol. 2009;41:525–534. doi: 10.1165/rcmb.2008-0367OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossi DJ, Oshima T, Attwell D. Glutamate release in severe brain ischaemia is mainly by reversed uptake. Nature. 2000;403:316–321. doi: 10.1038/35002090. [DOI] [PubMed] [Google Scholar]
- Rossi DJ, Brady JD, Mohr C. Astrocyte metabolism and signaling during brain ischemia. Nat Neurosci. 2007;10:1377–1386. doi: 10.1038/nn2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salter MW, Pitcher GM. Dysregulated Src upregulation of NMDA receptor activity: a common link in chronic pain and schizophrenia. FEBS J. 2012;279:2–11. doi: 10.1111/j.1742-4658.2011.08390.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandilos JK, Chiu YH, Chekeni FB, Armstrong AJ, Walk SF, Ravichandran KS, Bayliss DA. Pannexin 1, an ATP release channel, is activated by caspase cleavage of its pore-associated C-terminal autoinhibitory region. J Biol Chem. 2012;287:11303–11311. doi: 10.1074/jbc.M111.323378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santiago MF, Veliskova J, Patel NK, Lutz SE, Caille D, Charollais A, Meda P, Scemes E. Targeting pannexin1 improves seizure outcome. PLoS One. 2011;6:e25178. doi: 10.1371/journal.pone.0025178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silverman W, Locovei S, Dahl G. Probenecid, a gout remedy, inhibits pannexin 1 channels. Am J Physiol Cell Physiol. 2008;295:C761–C767. doi: 10.1152/ajpcell.00227.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smart JE, Oppermann H, Czernilofsky AP, Purchio AF, Erikson RL, Bishop JM. Characterization of sites for tyrosine phosphorylation in the transforming protein of Rous sarcoma virus (pp60v-src) and its normal cellular homologue (pp60c-src) Proc Natl Acad Sci U S A. 1981;78:6013–6017. doi: 10.1073/pnas.78.10.6013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Songyang Z, Shoelson SE, Chaudhuri M, Gish G, Pawson T, Haser WG, King F, Roberts T, Ratnofsky S, Lechleider RJ, Reel BG, Birge RB, Fajardo JE, Chou MM, Hanafusa H, Schaffhausen B, Cantle LC. SH2 domains recognize specific phosphopeptide sequences. Cell. 1993;72:767–778. doi: 10.1016/0092-8674(93)90404-e. [DOI] [PubMed] [Google Scholar]
- Takagi N, Shinno K, Teves L, Bissoon N, Wallace MC, Gurd JW. Transient ischemia differentially increases tyrosine phosphorylation of NMDA receptor subunits 2A and 2B. J Neurochem. 1997;69:1060–1065. doi: 10.1046/j.1471-4159.1997.69031060.x. [DOI] [PubMed] [Google Scholar]
- Takagi N, Cheung HH, Bissoon N, Teves L, Wallace MC, Gurd JW. The effect of transient global ischemia on the interaction of Src and Fyn with the N-methyl-D-aspartate receptor and postsynaptic densities: possible involvement of Src homology 2 domains. J Cereb Blood Flow Metab. 1999;19:880–888. doi: 10.1097/00004647-199908000-00007. [DOI] [PubMed] [Google Scholar]
- Thompson RJ, MacVicar BA. Connexin and pannexin hemichannels of neurons and astrocytes. Channels (Austin) 2008;2:81–86. doi: 10.4161/chan.2.2.6003. [DOI] [PubMed] [Google Scholar]
- Thompson RJ, Zhou N, MacVicar BA. Ischemia opens neuronal gap junction hemichannels. Science. 2006;312:924–927. doi: 10.1126/science.1126241. [DOI] [PubMed] [Google Scholar]
- Thompson RJ, Jackson MF, Olah ME, Rungta RL, Hines DJ, Beazely MA, MacDonald JF, MacVicar BA. Activation of pannexin-1 hemichannels augments aberrant bursting in the hippocampus. Science. 2008;322:1555–1559. doi: 10.1126/science.1165209. [DOI] [PubMed] [Google Scholar]
- White SH, Brisson CD, Andrew RD. Examining protection from anoxic depolarization by the drugs dibucaine and carbetapentane using whole cell recording from CA1 neurons. J Neurophsyiol. 2012;107:2083–2095. doi: 10.1152/jn.00701.2011. [DOI] [PubMed] [Google Scholar]
- Yamamoto S, Tanaka E, Shoji Y, Kudo Y, Inokuchi H, Higashi H. Factors that reverse the persistent depolarization produced by deprivation of oxygen and glucose in rat hippocampal CA1 neurons in vitro. J Neurophys. 1997;78:903–911. doi: 10.1152/jn.1997.78.2.903. [DOI] [PubMed] [Google Scholar]
- Yu XM, Askalan R, Keil GJ, 2nd, Salter MW. NMDA channel regulation by channel-associated protein tyrosine kinase Src. Science. 1997;275:674–678. doi: 10.1126/science.275.5300.674. [DOI] [PubMed] [Google Scholar]
- Zhang L, Deng T, Sun Y, Liu K, Yang Y, Zheng X. Role for nitric oxide in permeability of hippocampal neuronal hemichannels during oxygen glucose deprivation. J Neurosci Res. 2008;86:2281–2291. doi: 10.1002/jnr.21675. [DOI] [PubMed] [Google Scholar]