

Abstract
Cerebellar motor coordination and cerebellar Purkinje cell synaptic function require metabotropic glutamate receptor 1 (mGluR1, Grm1). We used an unbiased proteomic approach to identify protein partners for mGluR1 in cerebellum and discovered glutamate receptor δ2 (GluRδ2, Grid2, GluΔ2) and protein kinase Cγ (PKCγ) as major interactors. We also found canonical transient receptor potential 3 (TRPC3), which is also needed for mGluR1-dependent slow EPSCs and motor coordination and associates with mGluR1, GluRδ2, and PKCγ. Mutation of GluRδ2 changes subcellular fractionation of mGluR1 and TRPC3 to increase their surface expression. Fitting with this, mGluR1-evoked inward currents are increased in GluRδ2 mutant mice. Moreover, loss of GluRδ2 disrupts the time course of mGluR1-dependent synaptic transmission at parallel fiber–Purkinje cells synapses. Thus, GluRδ2 is part of the mGluR1 signaling complex needed for cerebellar synaptic function and motor coordination, explaining the shared cerebellar motor phenotype that manifests in mutants of the mGluR1 and GluRδ2 signaling pathways.
Introduction
Cerebellar Purkinje cells are crucial for motor control. In the molecular layer, Purkinje cells receive a major glutamatergic input from granule cell parallel fibers, and this synapse has many critical postsynaptic receptors, including metabotropic glutamate receptor 1 (mGluR1) and GluRδ2. mGluR1 and its signaling cascade play essential roles (Levenes et al., 1998; Knöpfel and Grandes, 2002; Hartmann and Konnerth, 2009). Mutations in mGluR1 impair motor coordination, cerebellar learning, proper wiring of climbing fibers to Purkinje cells, and long-term depression (LTD) (Aiba et al., 1994; Conquet et al., 1994; Kano et al., 1997). Glutamate binding to mGluR1 activates phospholipase C (PLC) via Gαq to produce inositol 1,4,5-trisphosphate (IP3) and diacylglycerol (DAG) (Hartmann et al., 2004). IP3 then mobilizes intracellularly stored Ca2+, and conventional protein kinase Cs (PKCs) are key effectors for DAG and Ca2+ (Nishizuka, 1992). Another major effector of mGluR1 in Purkinje cells is canonical transient receptor potential 3 (TRPC3), which is activated by DAG or phospholipase D, and inactivated by PKC phosphorylation (Trebak et al., 2003; Venkatachalam et al., 2003; Glitsch, 2010).
Activation of mGluR1 by parallel fiber stimulation or application of a group I mGluR agonist, (RS)-3,5-dihydroxyphenylglycine (DHPG), induces slow-onset EPSCs (sEPSCs) (Batchelor and Garthwaite, 1993; Batchelor et al., 1994). Gene knock-out (KO) of TRPC3 enriched in cerebellar Purkinje cells completely abolishes sEPSCs, indicating that TRPC3 mediates this mGluR1-induced signaling (Hartmann et al., 2008). Importantly, loss of any component in the mGluR1 cascade, Gαq (Offermanns et al., 1997; Hartmann et al., 2004), PLCβ4 (Hashimoto et al., 2001a; Miyata et al., 2001), IP3 receptor (Matsumoto et al., 1996; Inoue et al., 1998), or PKCγ (Chen et al., 1995; Kano et al., 1995) or TRPC3 (Hartmann et al., 2008), results in cerebellar ataxia often associated with defective LTD and multiple climbing fiber innervation of Purkinje cells.
GluRδ2 protein has homology to ionotropic glutamate receptors; however, GluRδ2 neither binds glutamate nor conducts current (Schmid and Hollmann, 2008; MacLean, 2009; Mandolesi et al., 2009; Armstrong et al., 2011; Hirano, 2012; Yuzaki, 2011). GluRδ2 is highly expressed in cerebellar Purkinje cells and is localized specifically to parallel fiber–Purkinje cell (PF–PC) synapses (Araki et al., 1993; Lomeli et al., 1993). Genetic studies have identified critical roles for GluRδ2 in cerebellar function. Loss-of-function mutations in GluRδ2 result in ataxia, multiple innervation of Purkinje cells by climbing fibers, and loss of LTD (Kashiwabuchi et al., 1995; Hashimoto et al., 2001b). Morphological studies reveal that the loss of GluRδ2 also causes misalignment of parallel fiber presynaptic active zones with Purkinje cell postsynaptic densities (PSDs) (Kashiwabuchi et al., 1995; Kurihara et al., 1997; Takeuchi et al., 2005). This latter observation is explained by a synaptogenic activity of GluRδ2 (Uemura and Mishina, 2008; Kuroyanagi et al., 2009); the extracellular N-terminal domain of GluRδ2 links to neurexin through the secreted synapse organizer cerebellin-1 (Matsuda et al., 2010; Uemura et al., 2010; Joo et al., 2011; Matsuda and Yuzaki, 2011).
Defects in mGluR1 signaling and GluRδ2 mutations cause overlapping phenotypes; however, the functional relationship of these two pathways is unclear. Here, we report physical and functional interactions between GluRδ2 and mGluR1 pathways.
Materials and Methods
Antibodies.
We used following antibodies: rabbit anti-mGluR1a (07-617; Millipore), mouse anti-mGluR1a (clone G209-488; BD Biosciences), rabbit anti-mGluR1a/b (AGC-006; Alamone Labs), anti-GluRδ2 (AB-1514; Millipore; GluRδ2-Rb-Af500-1; Frontier Institute), anti-PKCγ (sc-211; Santa Cruz Biotechnology), anti-PKCα (sc-208; Santa Cruz Biotechnology), anti-PKCβII (sc-210; Santa Cruz Biotechnology), anti-mGluR2/3 (AB1553; Millipore), anti-GluA2 (MAB-397; Millipore), anti-GluN1 (clone 54.1; BD Biosciences Pharmingen), and anti-TRPC3 (ACC-016; Alamone Labs).
Mutant mice.
Animal experiments were conducted under guidelines of the Institutional Animal Care and Use Committee at Eli Lilly & Co. Hotfoot-4J (ho-4J) mice were obtained from The Jackson Laboratory and maintained at Taconic Farms and Eli Lilly & Co. Heterozygous male and female mice were mated to obtain homozygous ho-4J mice. The mGluR1 KO mouse line B6(129)-T801〈tm1〉 was obtained from Deltagen. The animals were backcrossed to CD-1 for >20 generations.
Immunoprecipitation.
To identify coimmunoprecipitated proteins by mass spectroscopy, we used a time-controlled transcardiac perfusion crosslinking method (Schmitt-Ulms et al., 2004). Briefly, deeply anesthetized rats were perfused with 3% formaldehyde. The cerebella were rapidly removed and frozen by immersion in liquid nitrogen. Cerebellar tissues were homogenized in buffer: 50 mm NH4Cl and 40 mm Tris-Cl, pH 8.0, with Complete protease inhibitors (Roche) by Polytron. The homogenate was spun at 43,000 × g, and the pellet was resuspended in buffer: 50 mm Tris-Cl, pH 8.0, and 30 mm NaCl. The homogenate was solubilized with 1% SDS at 55°C for 5 min and then spun at 100,000 × g for 1 h at 4°C. SDS in the lysate was neutralized with 5 vol of 1% Triton X-100 in 50 mm Tris-Cl, pH 8.0, 30 mm NaCl. After centrifugation at 100,000 × g for 1 h at 4°C, antibodies for immunoprecipitation (IP) were incubated overnight at 4°C. Protein A Sepharose (GE Healthcare) was added for 2 h. The resin was washed with RIPA buffer (20 mm NaCl, 0.6% deoxycholate, 0.6% NP-40, and 20 mm, Tris pH8.0), followed by PBS. The beads were eluted with Laemli's buffer with 10 mg/ml DTT at 55°C for 30 min, followed by 95°C for 10 min.
For analytical IP, the postnuclear fraction was solubilized in buffer containing 0.2% Triton X-100, 0.32 m sucrose and 3 mm HEPES, pH 7.4, and then the soluble fraction was precleared with protein A Sepharose. After centrifugation at 100,000 × g for 1 h, 1–5 μg of antibody was added for 2–3 h at 4°C. Protein A Sepharose was added and mixed for 1 h, and the resin was washed with 0.2% Triton X-100. Beads were eluted with Laemli's buffer containing 10 mg/ml DTT at 55°C for 30 min, followed by 95°C for 10 min.
Mass spectroscopy.
Mass spectroscopy was done as described previously (Kato et al., 2007). Briefly, polyacrylamide gels were stained with silver, and protein bands were excised and destained with 1% H2O2 (Sumner et al., 2002). Gel pieces were reduced, alkylated (Hale et al., 2004), and digested with trypsin (20 μg/ml) overnight at 37°C. The extracted peptides were purified with a Ziptip (C-18) (Millipore) and loaded onto an HPLC (HP 1100 Nanopump) with a reverse-phase C-18 column connected to a mass spectrometer (LTQ-FT; ThermoFinnigan).
Quantification of protein expression levels.
Mouse cerebella were homogenized in buffer (HB) (0.32 m sucrose and 3 mm HEPES, pH 7.4) and centrifuged at 20,000 × g for 20 min at 4°C. The resultant pellets were suspended in HB. Five micrograms of protein were separated by SDS-PAGE in quadruplicate and then immunoblotted after protein transfer to PVDF membranes. Bands were visualized by HRP-conjugated secondary antibody and ECL-plus reagent (GE Healthcare) and were quantified using NIH Image-J.
Subcellular fractionation.
Subcellular fractions were prepared by differential centrifugation as described previously (Jo et al., 1999). Mouse cerebella were homogenized in buffer I (0.32 m sucrose, 3 mm HEPES-Na, pH 7.4, and 0.1 mg/ml PMSF). The homogenate was centrifuged at 1000 × g to produce a pellet (P1) and a supernatant (S1). P1 pellet was resuspended in buffer I and centrifuged at 1000 × g to obtain crude nuclear fraction (P1′) and a supernatant (S1′). The combined supernatant (S1 + S1′) was centrifuged at 12,000 × g for 15 min to produce a pellet (P2). The P2 fraction was resuspended in homogenization buffer, overlaid on the discontinuous sucrose density gradient (0.8, 1.0, and 1.2 m), and spun at 58,000 × g for 2 h. The synaptosomal fraction was collected from the interface between 1.0 and 1.2 m. Synaptosomal fraction was treated once or twice with ice-cold 0.5% Triton X-100 in 6 mm Tris-Cl, pH 7.5, and then centrifuged to obtain the PSD I and PSD II pellets.
Triton X-100 solubility assay.
Mouse cerebella were homogenized in HB, centrifuged at 100,000 × g, and then resuspended in 10 vol of the original tissue weight. Triton X-100 was added to 2.5% final and mixed for 1 h at 4°C. Solubilized samples were centrifuged at 100,000 × g, and the supernatant was removed. The pellet was suspended in the 10× original tissue volume of HB. Proteins in both samples were separated by SDS-PAGE, and proteins of interest were quantified by immunoblotting as described above.
Quantification of surface receptors.
Slice biotinylation was done as described previously (Kato et al., 2010). Briefly, 300–400 μm mouse cerebellar slices were incubated in slicing buffer (in mm: 124 NaCl, 26 NaHCO3, 3 KCl, 10 glucose, 0.5 CaCl2, and 4 MgCl2) for 30 min and then recovered in biotinylation solution (slicing solution except [CaCl2] and [MgCl2] were raised to 2.3 and 1.3 mm, respectively) for 30 min at room temperature. Slices were then preincubated in ice-cold biotinylation solution for ∼1 min. Surface proteins were labeled with sulfo-NHS-SS-biotin (1.5 mg/ml; Pierce) for 30 min on ice, and the reaction was quenched with glycine (50 mm). Hippocampi and cerebella were homogenized with Tris buffer (TB) (50 mm Tris, pH 7.4, and 2 mm EGTA) and then sonicated. To isolate the membrane fraction, homogenates were centrifuged at 100,000 g for 20 min, and the pellets were resuspended in TB containing NaCl [TN (TB plus 100 mm NaCl)]. Membrane were solubilized with 0.4% SDS in TN for 5 min at 4°C and then neutralized with 10-fold excess of Triton X-100. The lysate was cleared by centrifuging at 100,000 × g for 20 min. A total of 20 μl of ULTRA link Neutravidin resin (Roche) was added and incubated at 4°C for 2 h. Nonbound internal protein solution was removed. Beads were washed with RIPA buffer, and biotinylated surface proteins were eluted by boiling for 5 min in Laemli's buffer containing DTT (7.7 mg/ml). Eluted proteins and internal proteins were separated by SDS-PAGE and detected by immunoblotting.
Immunohistochemistry.
Deeply anesthetized mice were perfused with PBS and then fixed with ice-cold 4% paraformaldehyde. Cerebella were removed, postfixed in 4% PBS for 48 h at room temperature, and then were soaked in 70% ethanol at 4°C overnight. After tissue blocks were dehydrated by sequential changes treatment with organic solvents (95% ethanol/5% methanol 100% ethanol and then xylene), paraffin was infused at 58°C for 2 h. Cerebellar sections were cut (2 μm) and adhered onto plus-charged slide glasses. After drying, the sections were incubated at 56°C for 10 min and then deparaffinized in xylene and rehydrated with decreasing concentrations of ethanol in water. To retrieve the antigens, sections were autoclaved at 100°C for 30 min in 10 mm sodium citrate plus 0.05% Tween 20. The sections were dried and treated with 0.2N HCl for 20 min. For GluRδ2 staining, sections were then treated with 4 m guanidine thiocyanate in 10 mm Tris-Cl, pH 7.5. The sections were rinsed with PBS and blocked with 10% normal goat serum. Primary antibodies were added overnight. Immunosignals were visualized by secondary antibodies labeled with Alexa Fluor 488 or Alexa Fluor 555.
Electrophysiology.
Whole-cell patch-clamp recordings were made from Purkinje cells in acute cerebellar slices (250 μm thick) as described previously (Takayasu et al., 2004). Sagittal cerebellar slices (250 μm) were obtained from mice using a vibrating microslicer (World Precision Instruments). Whole-cell patch-clamp recordings were made from visualized Purkinje cells using infrared illumination and differential interference contrast optics. The recording pipette (3–5 MΩ resistance) was filled with the internal solution (150 mm Cs-gluconate, 8 mm NaCl, 2 mm Mg-ATP, 10 mm HEPES, 0.1 mm spermine, and 5 mm QX-314, pH 7.2, with osmolarity at 295 mOsm), and recordings were made in voltage-clamp mode at a holding potential of −80 mV. Data with access resistances >50 MΩ were discarded. Concentric bipolar tungsten stimulating electrodes were placed in the molecular layer to activate parallel fiber inputs to Purkinje cells. For recording of fast EPSCs (fEPSCs) in Purkinje cells, single square pulses (100 μs) were delivered in the presence of bicuculline (20 μm). The stimulation intensity was determined to evoke 500 ± 150 pA fEPSC without spikes. After 5 min perfusion with CNQX (20 μm) plus bicuculline (20 μm), to completely block fEPSC, five square pulses (100 μs) at 100 Hz were delivered to evoke sEPSCs. To measure mGluR1-dependent components, CPCCOEt [(−)-ethyl (7E)-7-hydroxyimino-1,7a-dihydrocyclopropa[b]chromene-1a-carboxylate] (200 μm) was added for 5 min, and then stimulations were repeated as for sEPSCs. fEPSC decays were fitted with two exponentials, and the weighted tau (τw) was calculated according to following: τw = (τ1 × a1) + (τ2 × a2), where a1 and a2 are the relative amplitudes of the two exponential components. The full-width at half-maximum (FWHM) of the sEPSC is the duration between the two time points on the CPCCOEt-sensitive sEPSC trace at which the sEPSC reaches half of the peak amplitude. To measure extrasynaptic mGluR1/TRPC3 signaling in cerebellar slices, we made whole-cell patch-clamp recordings from Purkinje cells and measured currents evoked by GABA (1 mm) alone and then by DHPG (20 and 50 μm) in the presence of an inhibitor mixture (300 nm tetrodotoxin, 20 μm bicuculline, and 40 μm CNQX) with cells voltage clamped at −80 mV. To obtain reproducible DHPG responses, we continuously perfused the inhibitor mixture and applied DHPG at >5 min intervals.
Results
Identification of interactions among mGluR1, GluRδ2, and PKCγ
To identify cerebellar proteins that interact with mGluR1, we immunoprecipitated mGluR1a, which is the major mGluR1 splice variant (Pin et al., 1992; Soloviev et al., 1999). The immunopurified proteins were separated by SDS-PAGE, stained with silver, and analyzed by mass spectrometry (Fig. 1A). We detected eight specific proteins interacting with mGluR1. Notably, we found GluRδ2 and PKCγ in the mGluR1 immunoprecipitate. We therefore performed precipitations of the GluRδ2 complex and identified 11 proteins, which included mGluR1 and PKCγ (Fig. 1B). Follow-up analytical experiments confirmed that mutual mGluR1–GluRδ2–PKCγ interactions are readily identified by any of the three immunoprecipitating antibodies (Fig. 1C–E,G). To evaluate specificity, we tested whether other mGluRs or ionotropic glutamate receptors are in the immunoprecipitates. Figure 1F shows that mGluR2/3, GluA2, and GluN1 were not detected in immunoprecipitates using anti-mGluR1, anti-GluRδ2, or anti-PKCγ. We also found that two other conventional PKCs in cerebellum, PKCα, and PKCβII do not interact with mGluR1 or GluRδ2 (Fig. 1H). Furthermore, we found that the PKCγ is not coprecipitated by mGluR1 or GluRδ2 antibodies in cortex, in which these glutamate receptors are much more weakly expressed (Fig. 1H).
Figure 1.
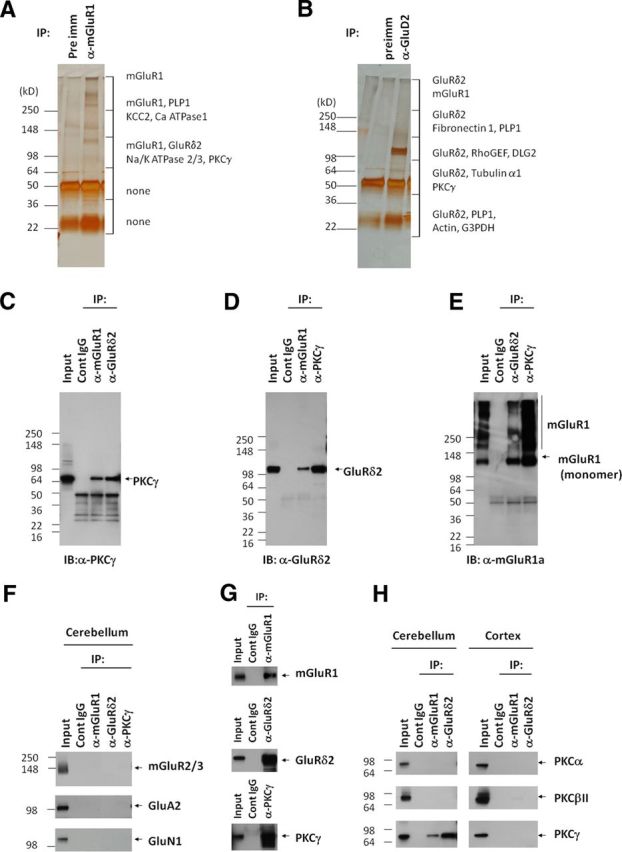
mGluR1, GluRδ2, and PKCγ form protein complexes in cerebellum. A, B, Silver staining of proteins coimmunoprecipitated with mGluR1a or anti-GluRδ2. Each protein lane was excised, divided into six pieces, and analyzed by mass spectroscopy. Proteins identified by their tryptic fragments are indicated. Contaminating IgG, keratin, and trypsin were omitted. C, PKCγ coimmunoprecipitated with mGluR1 or GluRδ2. D, GluRδ2 coimmunoprecipitated with mGluR1 or PKCγ. E, mGluR1a coimmunoprecipitated with GluRδ2 or PKCγ. F, Other metabotropic and ionotropic glutamate receptors did not coimmunoprecipitate with mGluR1, GluRδ2, or PKCγ, confirming the specificity of the IP. G, mGluR1 and GluRδ2 did not associate with other PKC subtypes; H, PKCγ did not associate with mGluR1 or GluRδ2 in cerebral cortex.
The mGluR1–GluRδ2–PKCγ protein complex also contains TRPC3
Because TRPC3 is the downstream effector of mGluR1 in generating Purkinje cell sEPSCs (Hartmann et al., 2008), we examined whether TRPC3 associates with mGluR1–GluRδ2–PKCγ. Indeed, we identified mGluR1 (Fig. 2A1), GluRδ2 (Fig. 2B1), and PKCγ (Fig. 2C1) in cerebellar samples immunoprecipitated with anti-TRPC3. To test whether GluRδ2 mediates formation of the overall protein complex, we used the hypomorphic GluRδ2 mutant mouse line ho-4J (Lalouette et al., 2001; Matsuda and Yuzaki, 2002), which has virtually no detectable GluRδ2 (Fig. 2B2). Levels of mGluR1 in TRPC3 and PKCγ immunoprecipitates from wild-type and ho-4J cerebella are similar, indicating that these interactions do not require wild-type expression levels of GluRδ2 (Fig. 2A2). The GluRδ2 mutation also does not affect the TRPC3–PKC interaction (Fig. 2C2). The lack of PKCγ signal in GluRδ2 immunoprecipitate from ho-4J cerebellar lysate confirms assay specificity (Fig. 2C2).
Figure 2.

TRPC3 associates with mGluR–GluRδ2–PKCγ in cerebellum. Triton X-100-solubilized postnuclear membrane fractions from either wild-type or mutant mice were immunoprecipitated and blotted as indicated. A, mGluR1 coimmunoprecipitated with TRPC3 from either wild-type (A1) or GluRδ2 (A2) mutant cerebella. B1, GluRδ2 coimmunoprecipitated with TRPC3. B2, In ho-4J mice, a truncated GluRδ2 is expressed at very low levels. C, PKCγ associates with mGluR1 or TRPC3 in the presence (C1) or absence (C2) of GluRδ2. D, TRPC3 and GluRδ2 interact in the presence (D1) or absence (D2) of mGluR1. E, PKCγ associates with GluRδ2 and TRPC3, which is not affected by the presence or absence of mGluR1. F, GluK2/3 did not interact with mGluR1, GluRδ2, or TRPC3.
To assess the requirement of mGluR1 in protein complex formation, we used mGluR1-KO cerebella. GluRδ2 was detected in the TRPC3 immunoprecipitate in both wild-type and mGluR1-KO at similar levels (Fig. 2D). Similarly, PKCγ was in GluRδ2- and TRPC3-containing protein complexes in both wild-type and mGluR1-KO mice (Fig. 2E). Lack of PKCγ in mGluR1 immunoprecipitates from mGluR1-KO (Fig. 2E2) and failure of kainate receptor GluK2/3 to interact with mGluR1, GluRd2, or TRPC3 (Fig. 2F) further confirm assay specificity. Together, mGluR1, GluRδ2, PKCγ, and TRPC3 associate in cerebellum, and loss of GluRδ2 or mGluR1 does not affect interactions between the remaining proteins.
Levels of TRPC3 or PKCγ are not affected in mGluR1-KO or GluRδ2ho-4J/ ho-4J mice
We next compared the levels of mGluR1 interacting proteins and representative synaptic proteins in mGluR1 or GluRδ2 hypomorphic (ho-4J) mice. There was no change in the levels of TRPC3, mGluR1, synaptophysin, or PSD-95 in ho-4J mouse cerebellum. An increase in the level of the major AMPA receptor subunit GluA2 was observed in ho-4J (Fig. 3A), which is consistent with a recent report (Yamasaki et al., 2011). Levels of PKCγ, TRPC3, GluRδ2, GluA2, PSD-95, and synaptophysin were not changed in the mGluR1-KO (Fig. 3B). Therefore, loss of either GluRδ2 or mGluR1 does not affect levels of their associated proteins.
Figure 3.

Neither GluRδ2 nor mGluR1 mutation affects cerebellar levels of interacting proteins. A, GluRδ2 mutation results in increased levels of GluA2 but does not affect levels of mGluR1, TRPC3, or other synaptic proteins. B, Mutation of GluR1 does not affect levels of any of the proteins analyzed. Error bars indicate SEM. Statistical significance with respect to GluRδ2+/+ or mGluR1+/+ (t test). n = 4 for each sample. *p < 0.05.
Neither GluRδ2 nor mGluR1 loss grossly alters protein complex cellular localization
We performed immunofluorescent localization of mGluR1, GluRδ2, PKCγ, and TRPC3 in the cerebella of wild-type, ho-4J, and mGluR1-KO mice. mGluR1, GluRδ2, PKCγ, and TRPC3 are all enriched in the molecular layer with punctate staining (Fig. 4). In the magnified images, GluRδ2 shows discrete staining in puncta, which partially colocalize with mGluR1 (Fig. 4A). mGluR1 is distributed more widely than GluRδ2. PKCγ is enriched in the cell bodies and dendrites of Purkinje cells. mGluR1 partially colocalizes with PKCγ (Fig. 4B). TRPC3 also partially colocalizes with mGluR1. We did not detect changes in any of these distributions in mGluR1-KO or ho-4J mice (Fig. 4A–C). The staining specificities of GluRδ2 and mGluR1 antibodies were confirmed by their elimination in the corresponding mutant mice.
Figure 4.

GluRδ2 or mGluR1 mutation does not affect immunofluorescent distribution of the interacting proteins. A–C, Immunofluorescent double labeling of sagittal cerebellar sections; the bottom panels are magnified images of the molecular layer. Specificity of staining is confirmed by elimination of immunosignal in corresponding mutant mice. A, Punctate GluRδ2 staining partially colocalizes with mGluR1 in molecular layer of wild type. The molecular layer staining patterns for GluRδ2 and mGluR1 were not dramatically altered in the mGluR1-KO or GluRδ2ho-4J/ho-4J, respectively. B, PKCγ shows strong labeling of Purkinje cell bodies, dendrites, and neuropil. The colabeling of mGluR1 and PKCγ in the molecular layer was not dramatically altered in the GluRδ2ho-4J/ho-4J, and the PKCγ distribution was not changed in the mGlu1-KO. C, mGluR1 and TRPC3 partially colocalize in wild-type mouse. No obvious difference in the staining patterns was detected in either GluRδ2ho-4J/ho-4J or mGluR1-KO. ML, Molecular layer; PC, Purkinje cell.
The subcellular localization of TRPC3 is affected in GluRδ2ho-4J/ho-4J
We used biochemical fractionation to detect more subtle effects of loss of GluRδ2 or mGluR1 on localization of the interacting proteins. In wild-type mice, we found that mGluR1, GluRδ2, PKCγ, and TRPC3 are all present in synaptosomal and PSD fractions (Fig. 5A). Of the four interacting proteins, TRPC3 shows the least PSD enrichment. In mGluR1-KO, we found no change in the synaptic fractionation profile of the other protein components. In the ho-4J mice, we noted a selective reduction of TRPC3 in the most insoluble PSD II fraction (Fig. 5A). To assess this more quantitatively, we measured the ratio of the insoluble to the soluble proteins in the presence of Triton X-100. We found that TRPC3 in the Triton X-100-soluble fraction is increased approximately threefold in ho-4J mice (Fig. 5B,C). We did not detect a significant difference in the fractionation of either mGluR1 or PKCγ in ho-4J mice (Fig. 5B,C).
Figure 5.

In GluRδ2ho-4J/ho-4J, TRPC3 partially redistributes to the Triton X-100-soluble fraction. A, Synaptosomal (Syp) and PSD fractions of mouse cerebella from wild-type, GluRδ2ho-4J/ho-4J, and mGluR1-KO were prepared and immunoblotted. mGluR1, GluRδ2, PKCγ, and TRPC3 are detected in the synaptosomal and PSD fractions. The blotting profiles of PSD-95 and synaptophysin validate the subcellular fractionations. B, Cerebellar homogenates were treated with 2.5% Triton X-100, followed by ultracentrifugation to yield supernatant (Sup) and pellet (Ppt). C, A greater percentage of TRPC3 was detected in Triton X-100-soluble fraction in GluRδ2ho-4J/ho-4J than in wild type (GluRδ2+/+). Error bars indicate SEM. Statistical significance with respect to GluRδ2+/+ (t test). n = 4 for each sample. *p < 0.05.
Loss of GluRδ2 increases surface expression of mGluR1 and TRPC3
We next asked whether GluRδ2 regulates the cell-surface expression of mGluR1 and TRPC3. Brain slices were treated with a cell-impermeable biotinylation reagent, and surface proteins were purified on Neutravidin agarose. Because the surface expression of the minor mGluR1b splice variant is regulated differently from mGluR1a (which we have denoted mGluR1) (Ciruela et al., 1999; Kumpost et al., 2008), we tested both variants. Interestingly, surface expression of mGluR1b and TRPC3 were enhanced in GluRδ2ho-4J/ho-4J (Fig. 6). The C-terminal tail of mGluR1a, but not that of mGluR1b, has an intracellular retention signal (Ciruela et al., 1999). Fitting with this, the surface fraction of mGluR1b is larger than that of mGluR1a (Fig. 6). The lack of this retention signal may allow greater access of mGluR1b to the cell surface in the GluRδ2 mutant. Note that the surface level of AMPA receptor subunit GluA2 and that of the kainate receptor subunit GluK2/3 are not changed in ho-4J mice (Fig. 6).
Figure 6.

Effects of mGluR1 and GluRδ2 mutation on surface expression of interacting proteins. Mouse cerebellar slices were treated with a membrane-impermeable biotinylation reagent to mark cell-surface proteins. Total, internal, and surface proteins were resolved by SDS-PAGE and subjected to immunoblotting. GluRδ2 mutation increased surface expression of TRPC3 and mGluR1, whereas other components were unchanged. mGluR1b is the shorter splice variant of mGluR1. β3-Tubulin and/or synaptophysin served as internal protein controls. Error bars indicate SEM. Statistical significance with respect to GluRδ2+/+ (t test). n = 4 for mGluR1, mGluR1b, TRPC3, and GluK2/3. n = 3 for GluA2, synaptophysin, and β3-tubulin. *p < 0.05.
The onset of Purkinje cells sEPSC is delayed in ho-4J mice
mGluR1 activity in cerebellar Purkinje cells is translated into a sEPSC, mediated by TRPC channels (Kim et al., 2003; Hartmann et al., 2008). Importantly, loss of TRPC3 abolishes sEPSCs and results in ataxic gait (Hartmann et al., 2008). Because the subcellular localization of TRPC3 and mGluR1 are perturbed with loss of GluRδ2, we wondered how this might influence the mGluR1-dependent sEPSC. We compared PF–PC responses using whole-cell patch-clamp recordings from Purkinje cells in cerebellar slices from ho-4J or wild-type mice. To evaluate the specific effects on the sEPSC, we first recorded AMPA-receptor-mediated fEPSCs evoked by single-shock stimulation in the presence of bicuculline (20 μm) and then in the same cells monitored the mGluR1-dependent sEPSC evoked by five shocks at 100 Hz in the presence of bicuculline plus CNQX (20 μm) using the same stimulus intensity. To confirm the mGluR1-dependent component to the response, we bath applied the mGluR1 antagonist CPCCOEt (200 μm) together with bicuculline plus CNQX at the end of each experiment (Fig. 7A1,A2).
Figure 7.
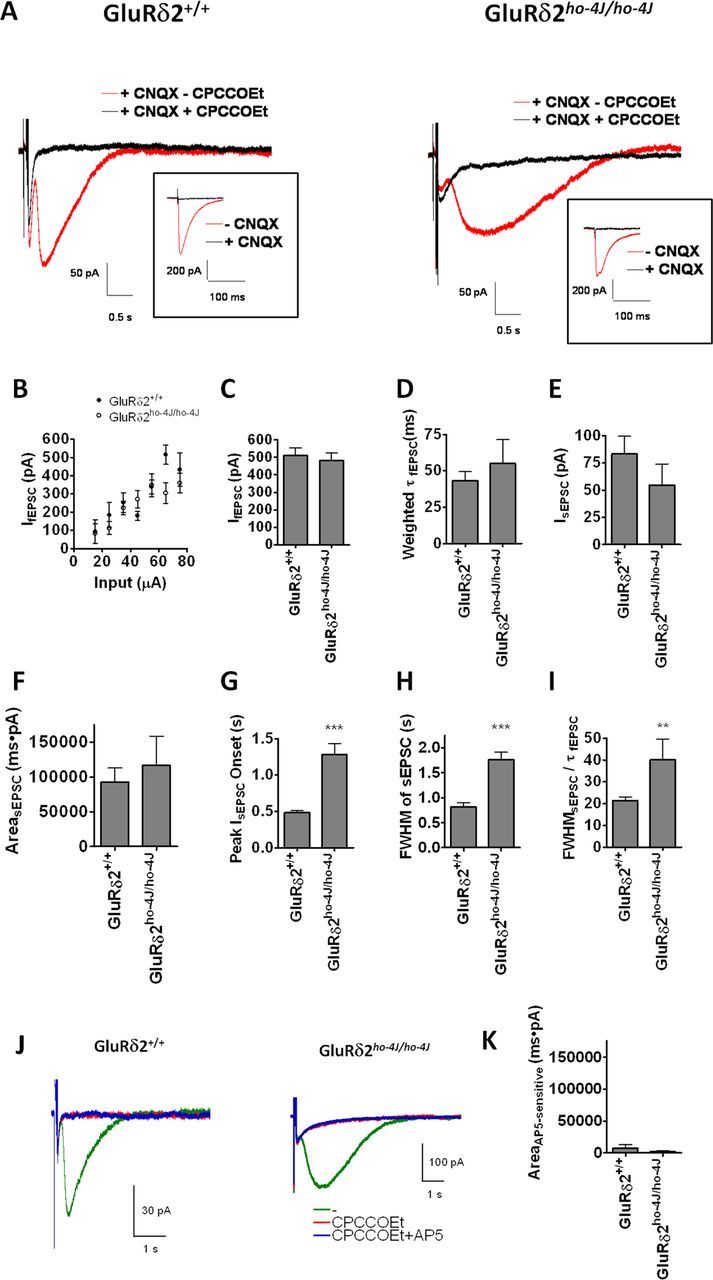
GluRδ2ho-4J/ho-4J alters the time course of the mGluR1-dependent sEPSC at PF–PC synapses. A, Typical traces of sEPSCs recorded from wild-type or GluRδ2ho-4J/ho-4J mice. Application of the mGluR1 antagonist CPCCOEt blocks sEPSCs. Inset shows corresponding fEPSCs, which are blocked by CNQX. B, GluRδ2ho-4J/ho-4J shows a similar fEPSC input–output relationship as wild type. C, Stimulation intensity for fEPSCs and sEPSCs was set to evoke similar fEPSC amplitudes (∼500 pA). D, The decay constant of fEPSCs was not affected by GluRδ2 mutation. E, The amplitude of sEPSCs was not changed significantly by GluRδ2 mutation. F, The integrated area of the CPCCOEt-sensitive sEPSC was not affected by GluRδ2 mutation. G, The onset of the sEPSC was significantly slowed in GluRδ2ho-4J/ho-4J. The average duration between the stimulus and the peak of the sEPSC was calculated. H, The average FWHM for the CPCCOEt-sensitive sEPSC was calculated. I, FWHM of sEPSC was normalized by the decay constant of fEPSC. The kinetics of sEPSCs was specifically slowed in GluRδ2ho-4J/ho-4J mice. Error bars indicate SEM. C–F, GluRδ2+/+ (n = 18) and GluRδ2ho-4J/ho-4J (n = 11). G–I, GluRδ2+/+ (n = 18) and GluRδ2ho-4J/ho-4J (n = 7). The waveforms of sEPSC in 4 of 11 samples recorded from GluRδ2ho-4J/ho-4J Purkinje cells were too small to evaluate, so we omitted these samples from G–I. J, K, NMDA receptors are not involved in the slower synaptic responses at PF–PC synapses in GluRδ2ho-4J/ho-4J. J, Typical sEPSC traces from wild-type and GluRδ2ho-4J/ho-4J in the presence or absence of 100 μm AP-5. All responses were evoked by five pulses at 100 Hz in the presence of 20 μm bicuculline and 40 μm CNQX. K, Quantified AP-5-sensitive charge transfer. Statistical significance with respect to GluRδ2+/+ (t test). **p < 0.01, ***p < 0.001.
First, we tested the input–output relationship of fEPSCs evoked by stimulation at 20–80 μA, which shows a linear relationship from 100 to 500 pA in both wild-type and ho-4J mice (Fig. 7B). One previous study reported no significant difference in the input–output curve of fEPSC in wild-type and GluRδ2-KO (Kashiwabuchi et al., 1995), whereas another reported a reduction of fEPSCs in GluRδ2-KO (Kurihara et al., 1997). Despite the increase in AMPA receptor levels (Fig. 3), which is consistent with a recent report (Yamasaki et al., 2011), we find no change in the input–output for fEPSCs in the GluRδ2 hypomorph. This observation may indicate homeostatic compensation of fEPSCs in GluRδ2-deficient/GluRδ2-hypomorphic mice, in which PF–PC synapse numbers are reduced (Kurihara et al., 1997) and AMPA receptors are increased (Yamasaki et al., 2011). To record reliable sEPSCs, we applied a relatively strong stimulation, which evokes ∼500 pA fEPSCs without spikes, for both wild-type and ho-4J slices (Fig. 7C). The weighted tau of the fEPSC did not show a significant difference between the two genotypes (Fig. 7D). The amplitude of the sEPSC trended to decrease in the mutant, although this was not statistically significant (Fig. 7E). The area of the sEPSC, which represents charge transfer, was also not affected by loss of GluRδ2 (Fig. 7F). Interestingly, the onset of the sEPSC, which is the mGluR1-dependent component, was significantly slowed in GluRδ2ho-4J/ho-4J mice. We found significant differences in all relevant measurements, including onset of sEPSC peak (Fig. 7G), FWHM of sEPSCs (Fig. 7H), and FWHM of sEPSCs normalized by the fEPSC decay constant (Fig. 7I). These results indicate that GluRδ2 is not involved in the fEPSC and is critical for generating the appropriate time course of the mGluR1-dependent sEPSC. After the application of CPCCOEt, we observed some residual small responses, which were slower in ho-4J mice (Fig. 7A). We tested for potential involvement of NMDA receptors in this response, but AP-5 did not block the responses (Fig. 7J,K). The residual responses after CPCCOEt application likely reflect increased extracellular K+ after parallel fiber stimulation (Batchelor and Garthwaite, 1993).
Extrasynaptic mGluR1-mediated responses are increased in the Purkinje cells of GluRδ2ho-4J/ho-4J mice
To quantify extrasynaptic mGluR1/TRPC3 function (Tempia et al., 2001; Hartmann et al., 2008), we measured currents in voltage-clamped Purkinje cells after bath application (40 s) of DHPG, a group I mGluR1 agonist (Fig. 8A). The DHPG-evoked responses were stable when the drug was applied at >5 min intervals (data not shown). At 20 and 50 μm, DHPG evoked concentration-dependent responses that were blocked by CPCCOEt (Fig. 8A). As a control, we also measured GABA-evoked currents from the same neurons (Fig. 8A). The peak current amplitude evoked by 20 or 50 μm DHPG were significantly increased in GluRδ2ho-4J/ho-4J mice, whereas GABA-evoked currents were not different (Fig. 8B1–B3). The peak amplitude of the DHPG-evoked responses (normalized to that by GABA-evoked responses) was significantly increased (Fig. 8B4,B5). We also quantified the area under the curve, which corresponds to total charge transfer, and observed a significant increase in the DHPG-evoked, but not GABA-evoked, responses recorded from GluRδ2ho-4J/ho-4J Purkinje cells (Fig. 8C1–C5).
Figure 8.
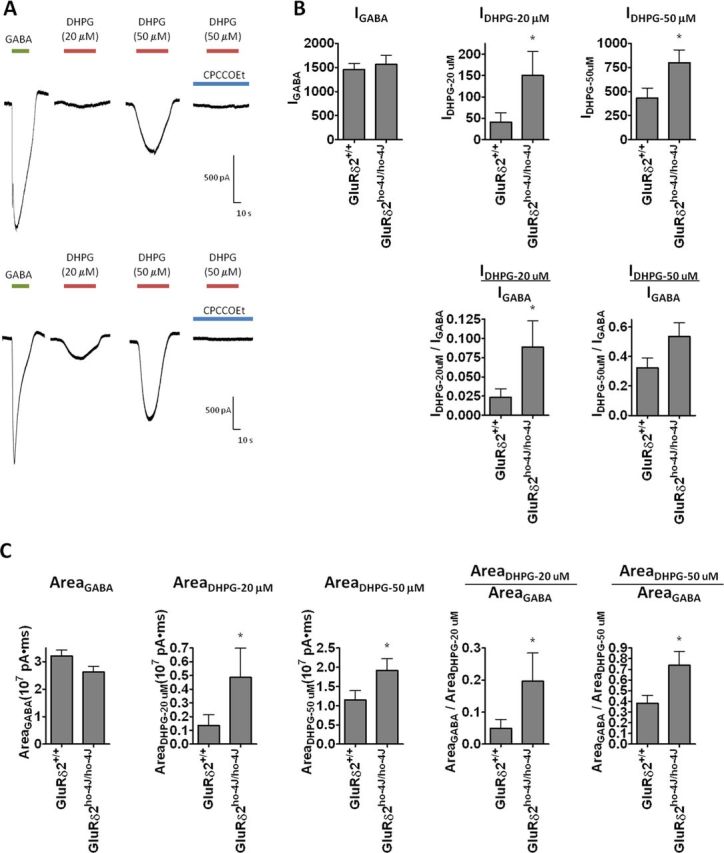
DHPG-evoked currents are increased in Purkinje cells from GluRδ2ho-4J/ho-4J mice. A, Typical traces recorded from wild-type or GluRδ2ho-4J/ho-4J Purkinje cells evoked by GABA (1 mm), DHPG (20 and 50 μm), or CPCCOEt (200 μm) plus DHPG (50 μm). B, Quantified peak amplitude of the GABA- and DHPG-evoked responses. C, Quantified area under the curve of GABA- and DHPG-evoked responses. Error bars indicate SEM. Statistical significance with respect to GluRδ2+/+ (t test). *p < 0.05.
Discussion
Here we demonstrate that mGluR1 and its effector molecules PKCγ and TRPC3 interact with GluRδ2 in cerebellum. Importantly, GluRδ2 is critical for generating the appropriate time course of synaptic mGluR1 signaling. This observation fits with the common phenotypes of GluRδ2 and mGluR1 mutant mice. Altered biochemical properties of cerebellar TRPC3 and mGluR1, Triton X-100 solubility, and surface expression in ho-4J mice indicate that GluRδ2 plays roles in the proper localization of mGluR1 and TRPC3, and this likely explains the abnormal sEPSC timing in GluRδ2 mutant mice. In contrast, neither GluRδ2 nor mGluR1 deficiency affects the other interactions or the levels of the other components, suggesting that neither GluRδ2 nor mGluR1 functions as a formal scaffolding protein or auxiliary subunit.
Convergence of the mechanisms in the manifestation of cerebellar phenotypes in mGluR1 and GluRδ2 signaling pathways
Studies of several KO mice (mGluR1, Gαq, PLCβ4, PKCγ, and TRPC3) demonstrate that mGluR1 signaling cascade is crucial for cerebellar function by regulating synaptic physiology and plasticity of Purkinje neurons (Hartmann and Konnerth, 2009). Interestingly, loss of function in GluRδ2 or its newly found ligand Clbn1 yield phenotypes resembling those in the mGluR1-signaling pathway (Matsuda et al., 2010; Uemura et al., 2010). Our unbiased proteomic approach demonstrated that key binding partners of mGluR1 are GluRδ2 and PKCγ. Notably, the IP experiments worked in all directions: IP with anti-mGluR1 contained GluRδ2 and PKCγ, IP with anti-GluRδ2 contained mGluR1 and PKCγ, and IP with anti-PKCγ contained mGluR1 and GluRδ2 (Fig. 1). We also confirmed the binding specificity using mGluR1 and GluRδ2-KO mice. These interactions are selective, because we did not identify other ionotropic/metabotropic GluRs in the immunoprecipitates. mGluR1–PKCγ–TRPC3 interactions may facilitate functional coupling of this signaling cascade. PKCs are classical effectors of Gαq-coupled GPCRs (Knöpfel and Grandes, 2002); TRPC3 is responsible for mGluR1-mediated synaptic cation influx into Purkinje neurons at PF–PC synapses (Hartmann et al., 2008), and PKC phosphorylation negatively regulates TRPC3 channel (Venkatachalam et al., 2003).
Formation and localization of mGluR1–GluRδ2–PKCγ–TRPC3 proteins
mGluR1–TRPC3–PKCγ interactions do not require wild-type levels of GluRδ2ho-4J/ho-4J, and mutation of GluRδ2 does not abolish postsynaptic localization of TRPC3/mGluR1. These observations suggest the involvement of additional scaffolding proteins, which could include TRPC3 or PKCγ. Because mGluR1 and GluRδ2 do not interact in cerebral cortex, the relevant scaffolding proteins are likely enriched in cerebellum. Furthermore, the C-terminal region of GluRδ2 contains at least three domains for the interactions with PDZ proteins (Roche et al., 1999; Uemura et al., 2004; Yawata et al., 2006). The PDZ-binding domain at the C terminus, designated as the T site, interacts with several PSD proteins, including PSD-93, PTPMEG, and delphilin (Roche et al., 1999; Hironaka et al., 2000; Miyagi et al., 2002). The second PDZ-binding domain in the middle of the C-terminal region, designated as the S segment, interacts with Shank scaffold proteins (Uemura et al., 2004). The membrane-proximal domain of the C-terminal region binds to PICK1 (Yawata et al., 2006). Lack of the Shank-binding S segment results in a moderate decrease of GluRδ2 at the PSD (Yasumura et al., 2008). Clearly, multiple scaffold proteins localize GluRδ2 at postsynaptic sites of PF–PC synapses. Interacting proteins for mGluR1 have also been identified. The N termini of postsynaptically localized Homer-1, Homer-2, and Homer-3 bind to the cytoplasmic tail of mGluR1a (Xiao et al., 2000; Ehrengruber et al., 2004). Although these scaffolding proteins interact with GluRδ2 and mGluR1, we did not detect them in our IP–mass spectroscopy studies. This discrepancy may be explained by our use of formaldehyde fixation followed by SDS solubilization. Formaldehyde fixation may not efficiently crosslink PDZ proteins/ligand complexes.
Physiological relevance of GluRδ2–mGluR1–PKCγ–TRPC3 interactions
Mutation of GluRδ2 increases levels of Triton X-100-soluble, surface-exposed TRPC3/mGluR1 and augments DHPG-evoked responses. In contrast, the amplitude of synaptic mGluR1/TRPC3-mediated sEPSCs in GluRδ2ho-4J/ho-4J is not increased but their onset is slowed. These results indicate that loss of GluRδ2 increases extrasynaptic mGluR1/TRPC3. Potential mechanisms are as follows. In the GluRδ2-KO mouse, cerebellar architecture is grossly normal, and Purkinje cells elaborate dendritic arbors and spines (Kashiwabuchi et al., 1995; Hashimoto et al., 2001b); however, the KO mouse has misaligned PF–PC synapses, and numerous free spines emerge (Kurihara et al., 1997). This morphological abnormality is explained by the lack of interaction between the PF–PC synapse organizers neurexin–Clbn1–GluRδ2 (Matsuda et al., 2010; Uemura et al., 2010; Matsuda and Yuzaki, 2011; Lee et al., 2012). The generation of free spines was also reported in GluRδ2ho-4J/ho-4J (Lalouette et al., 2001). Reduction of functional PF–PC synapses may be homeostatically compensated by an increase in surface mGluR1/TRPC3. Indeed, synaptic responses mediated by AMPA receptors at PF–PC synapses are not changed (Kashiwabuchi et al., 1995) (Fig. 7) or may even be decreased (Kurihara et al., 1997), whereas AMPA receptor expression levels and the localization of AMPA receptors at PF–PC synapses are increased (Yamasaki et al., 2011) (Fig. 6). This observation implies that abnormal PF–PC synapse formation results in homeostatic compensation of AMPA-receptor-mediated responses as well.
In GluRδ2ho-4J/ho-4J mice, fEPSCs are normal, and the onset of sEPSCs is delayed. The precise mechanism for this selective change is not clear. sEPSCs are elicited by strong synaptic stimulation, which causes “spillover” of released glutamate. Accordingly, sEPSC are significantly increased by inhibitors of glutamate transporters (Reichelt and Knöpfel, 2002). Our data indicate that the time course, but not the amplitude or the total charge transfer of sEPSCs, is impaired in the GluRδ2ho-4J/ho-4J. These results suggest that the altered localization of TRPC3/mGluR1 and/or the altered synapse formation in GluRδ2 mutants result in delayed onset and increased duration of sEPSCs. This latter notion fits with the reported ultrastructural defects in synapse formation with loss of GluRδ2 (Kashiwabuchi et al., 1995; Kurihara et al., 1997; Takeuchi et al., 2005).
The protein interactions among mGluR1, GluRδ2, PKCγ, and TRPC3 may also be relevant to human movement disorders, especially spinocerebellar ataxia (SCA), which can involve abnormality of Purkinje cell function. More than 30 genetic type of SCA have been identified and dominantly inherited mutations in PKCγ cause SCA14. Most disease-causing mutations in PKCγ occur in the C1 domain, which binds DAG and is necessary for translocation and regulation of PKCγ. Although the C1 domain mutants display kinase activity, they are unable to phosphorylate and thereby inactivate TRPC channels in vivo (Adachi et al., 2008). This failure to phosphorylate TRPC3 channels alters Ca2+ homeostasis, which likely contributes to neurodegeneration (Matilla-Dueñas et al., 2010). This fits with the progressive Purkinje cell loss in moonwalker mice whose TRPC3 channel is mutated such that cannot be phosphorylated and inactivated by PKCγ (Becker et al., 2009). Furthermore, auto-antibodies to GluRδ2 are found in patients with acute cerebellar ataxia, acute cerebellitis, and steroid-responsive chronic cerebellitis (Shiihara et al., 2007; Shimokaze et al., 2007; Kubota and Takahashi, 2008; Fukuoka et al., 2012; Kinno et al., 2012; Matsumoto et al., 2012). Also, auto-antibodies to mGluR1 are associated with neoplastic cerebellar ataxia (Marignier et al., 2010; Sillevis Smitt et al., 2000; Coesmans et al., 2003). Pharmacological regulation of the GluRδ2/mGluR1/PKCγ/TRPC network identified here might therefore be therapeutic.
Footnotes
A.S.K., M.D.K., E.S.N., and J.T.R.I. are full-time employees of Eli Lilly & Co. D.S.B. and E.R.S. were full-time employees of Eli Lilly & Co. when this research was conducted.
References
- Adachi N, Kobayashi T, Takahashi H, Kawasaki T, Shirai Y, Ueyama T, Matsuda T, Seki T, Sakai N, Saito N. Enzymological analysis of mutant protein kinase Cgamma causing spinocerebellar ataxia type 14 and dysfunction in Ca2+ homeostasis. J Biol Chem. 2008;283:19854–19863. doi: 10.1074/jbc.M801492200. [DOI] [PubMed] [Google Scholar]
- Aiba A, Kano M, Chen C, Stanton ME, Fox GD, Herrup K, Zwingman TA, Tonegawa S. Deficient cerebellar long-term depression and impaired motor learning in mGluR1 mutant mice. Cell. 1994;79:377–388. [PubMed] [Google Scholar]
- Araki K, Meguro H, Kushiya E, Takayama C, Inoue Y, Mishina M. Selective expression of the glutamate receptor channel delta 2 subunit in cerebellar Purkinje cells. Biochem Biophys Res Commun. 1993;197:1267–1276. doi: 10.1006/bbrc.1993.2614. [DOI] [PubMed] [Google Scholar]
- Armstrong CL, Duffin CA, McFarland R, Vogel MW. Mechanisms of compartmental Purkinje cell death and survival in the lurcher mutant mouse. Cerebellum. 2011;10:504–514. doi: 10.1007/s12311-010-0231-4. [DOI] [PubMed] [Google Scholar]
- Batchelor AM, Garthwaite J. Novel synaptic potentials in cerebellar Purkinje cells: probable mediation by metabotropic glutamate receptors. Neuropharmacology. 1993;32:11–20. doi: 10.1016/0028-3908(93)90124-l. [DOI] [PubMed] [Google Scholar]
- Batchelor AM, Madge DJ, Garthwaite J. Synaptic activation of metabotropic glutamate receptors in the parallel fibre–Purkinje cell pathway in rat cerebellar slices. Neuroscience. 1994;63:911–915. doi: 10.1016/0306-4522(94)90558-4. [DOI] [PubMed] [Google Scholar]
- Becker EB, Oliver PL, Glitsch MD, Banks GT, Achilli F, Hardy A, Nolan PM, Fisher EM, Davies KE. A point mutation in TRPC3 causes abnormal Purkinje cell development and cerebellar ataxia in moonwalker mice. Proc Natl Acad Sci U S A. 2009;106:6706–6711. doi: 10.1073/pnas.0810599106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C, Kano M, Abeliovich A, Chen L, Bao S, Kim JJ, Hashimoto K, Thompson RF, Tonegawa S. Impaired motor coordination correlates with persistent multiple climbing fiber innervation in PKC gamma mutant mice. Cell. 1995;83:1233–1242. doi: 10.1016/0092-8674(95)90148-5. [DOI] [PubMed] [Google Scholar]
- Ciruela F, Soloviev MM, McIlhinney RA. Cell surface expression of the metabotropic glutamate receptor type 1alpha is regulated by the C-terminal tail. FEBS Lett. 1999;448:91–94. doi: 10.1016/s0014-5793(99)00341-5. [DOI] [PubMed] [Google Scholar]
- Coesmans M, Smitt PA, Linden DJ, Shigemoto R, Hirano T, Yamakawa Y, van Alphen AM, Luo C, van der Geest JN, Kros JM, Gaillard CA, Frens MA, de Zeeuw CI. Mechanisms underlying cerebellar motor deficits due to mGluR1-autoantibodies. Ann Neurol. 2003;53:325–336. doi: 10.1002/ana.10451. [DOI] [PubMed] [Google Scholar]
- Conquet F, Bashir ZI, Davies CH, Daniel H, Ferraguti F, Bordi F, Franz-Bacon K, Reggiani A, Matarese V, Cond é F, Collingridge GL, Crépel F. Motor deficit and impairment of synaptic plasticity in mice lacking mGluR1. Nature. 1994;372:237–243. doi: 10.1038/372237a0. [DOI] [PubMed] [Google Scholar]
- Ehrengruber MU, Kato A, Inokuchi K, Hennou S. Homer/Vesl proteins and their roles in CNS neurons. Mol Neurobiol. 2004;29:213–227. doi: 10.1385/MN:29:3:213. [DOI] [PubMed] [Google Scholar]
- Fukuoka T, Takeda H, Ohe Y, Deguchi I, Takahashi Y, Tanahashi N. Anti-glutamate receptor delta2 antibody-positive migrating focal encephalitis. Clin Neurol Neurosurg. 2012 doi: 10.1016/j.clineuro.2012.03.026. Advance online publication. Retrieved September 17, 2012. [DOI] [PubMed] [Google Scholar]
- Glitsch MD. Activation of native TRPC3 cation channels by phospholipase D. FASEB J. 2010;24:318–325. doi: 10.1096/fj.09-134973. [DOI] [PubMed] [Google Scholar]
- Hale JE, Butler JP, Gelfanova V, You JS, Knierman MD. A simplified procedure for the reduction and alkylation of cysteine residues in proteins prior to proteolytic digestion and mass spectral analysis. Anal Biochem. 2004;333:174–181. doi: 10.1016/j.ab.2004.04.013. [DOI] [PubMed] [Google Scholar]
- Hartmann J, Konnerth A. Mechanisms of metabotropic glutamate receptor-mediated synaptic signaling in cerebellar Purkinje cells. Acta Physiol (Oxf) 2009;195:79–90. [Google Scholar]
- Hartmann J, Blum R, Kovalchuk Y, Adelsberger H, Kuner R, Durand GM, Miyata M, Kano M, Offermanns S, Konnerth A. Distinct roles of Gαq and Gα11 for Purkinje cell signaling and motor behavior. J Neurosci. 2004;24:5119–5130. doi: 10.1523/JNEUROSCI.4193-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartmann J, Dragicevic E, Adelsberger H, Henning HA, Sumser M, Abramowitz J, Blum R, Dietrich A, Freichel M, Flockerzi V, Birnbaumer L, Konnerth A. TRPC3 channels are required for synaptic transmission and motor coordination. Neuron. 2008;59:392–398. doi: 10.1016/j.neuron.2008.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashimoto K, Miyata M, Watanabe M, Kano M. Roles of phospholipase Cbeta4 in synapse elimination and plasticity in developing and mature cerebellum. Mol Neurobiol. 2001a;23:69–82. doi: 10.1385/MN:23:1:69. [DOI] [PubMed] [Google Scholar]
- Hashimoto K, Ichikawa R, Takechi H, Inoue Y, Aiba A, Sakimura K, Mishina M, Hashikawa T, Konnerth A, Watanabe M, Kano M. Roles of glutamate receptor δ2 subunit (GluRδ2) and metabotropic glutamate receptor subtype 1 (mGluR1) in climbing fiber synapse elimination during postnatal cerebellar development. J Neurosci. 2001b;21:9701–9712. doi: 10.1523/JNEUROSCI.21-24-09701.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirano T. Glutamate-receptor-like molecule GluRdelta2 involved in synapse formation at parallel fiber–Purkinje neuron synapses. Cerebellum. 2012;11:71–77. doi: 10.1007/s12311-010-0170-0. [DOI] [PubMed] [Google Scholar]
- Hironaka K, Umemori H, Tezuka T, Mishina M, Yamamoto T. The protein-tyrosine phosphatase PTPMEG interacts with glutamate receptor delta 2 and epsilon subunits. J Biol Chem. 2000;275:16167–16173. doi: 10.1074/jbc.M909302199. [DOI] [PubMed] [Google Scholar]
- Inoue T, Kato K, Kohda K, Mikoshiba K. Type 1 inositol 1,4,5-trisphosphate receptor is required for induction of long-term depression in cerebellar Purkinje neurons. J Neurosci. 1998;18:5366–5373. doi: 10.1523/JNEUROSCI.18-14-05366.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jo K, Derin R, Li M, Bredt DS. Characterization of MALS/Velis-1, -2, and -3: a family of mammalian LIN-7 homologs enriched at brain synapses in association with the postsynaptic density-95/NMDA receptor postsynaptic complex. J Neurosci. 1999;19:4189–4199. doi: 10.1523/JNEUROSCI.19-11-04189.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joo JY, Lee SJ, Uemura T, Yoshida T, Yasumura M, Watanabe M, Mishina M. Differential interactions of cerebellin precursor protein (Cbln) subtypes and neurexin variants for synapse formation of cortical neurons. Biochem Biophys Res Commun. 2011;406:627–632. doi: 10.1016/j.bbrc.2011.02.108. [DOI] [PubMed] [Google Scholar]
- Kano M, Hashimoto K, Chen C, Abeliovich A, Aiba A, Kurihara H, Watanabe M, Inoue Y, Tonegawa S. Impaired synapse elimination during cerebellar development in PKC gamma mutant mice. Cell. 1995;83:1223–1231. doi: 10.1016/0092-8674(95)90147-7. [DOI] [PubMed] [Google Scholar]
- Kano M, Hashimoto K, Kurihara H, Watanabe M, Inoue Y, Aiba A, Tonegawa S. Persistent multiple climbing fiber innervation of cerebellar Purkinje cells in mice lacking mGluR1. Neuron. 1997;18:71–79. doi: 10.1016/s0896-6273(01)80047-7. [DOI] [PubMed] [Google Scholar]
- Kashiwabuchi N, Ikeda K, Araki K, Hirano T, Shibuki K, Takayama C, Inoue Y, Kutsuwada T, Yagi T, Kang Y, Aizawa S, Mishina M. Impairment of motor coordination, Purkinje cell synapse formation, and cerebellar long-term depression in GluR delta 2 mutant mice. Cell. 1995;81:245–252. doi: 10.1016/0092-8674(95)90334-8. [DOI] [PubMed] [Google Scholar]
- Kato AS, Zhou W, Milstein AD, Knierman MD, Siuda ER, Dotzlaf JE, Yu H, Hale JE, Nisenbaum ES, Nicoll RA, Bredt DS. New transmembrane AMPA receptor regulatory protein isoform, γ-7, differentially regulates AMPA receptors. J Neurosci. 2007;27:4969–4977. doi: 10.1523/JNEUROSCI.5561-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato AS, Gill MB, Ho MT, Yu H, Tu Y, Siuda ER, Wang H, Qian YW, Nisenbaum ES, Tomita S, Bredt DS. Hippocampal AMPA receptor gating controlled by both TARP and cornichon proteins. Neuron. 2010;68:1082–1096. doi: 10.1016/j.neuron.2010.11.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SJ, Kim YS, Yuan JP, Petralia RS, Worley PF, Linden DJ. Activation of the TRPC1 cation channel by metabotropic glutamate receptor mGluR1. Nature. 2003;426:285–291. doi: 10.1038/nature02162. [DOI] [PubMed] [Google Scholar]
- Kinno R, Yamazaki T, Yamamoto M, Takahashi Y, Fukui T, Kinugasa E. Cerebellar symptoms in a case of acute limbic encephalitis associated with autoantibodies to glutamate receptors delta2 and epsilon2. Clin Neurol Neurosurg. 2012 doi: 10.1016/j.clineuro.2012.06.011. Advance online publication. Retrieved September 17, 2012. [DOI] [PubMed] [Google Scholar]
- Knöpfel T, Grandes P. Metabotropic glutamate receptors in the cerebellum with a focus on their function in Purkinje cells. Cerebellum. 2002;1:19–26. doi: 10.1007/BF02941886. [DOI] [PubMed] [Google Scholar]
- Kubota M, Takahashi Y. Steroid-responsive chronic cerebellitis with positive glutamate receptor delta 2 antibody. J Child Neurol. 2008;23:228–230. doi: 10.1177/0883073807307973. [DOI] [PubMed] [Google Scholar]
- Kumpost J, Syrova Z, Kulihova L, Frankova D, Bologna JC, Hlavackova V, Prezeau L, Kralikova M, Hruskova B, Pin JP, Blahos J. Surface expression of metabotropic glutamate receptor variants mGluR1a and mGluR1b in transfected HEK293 cells. Neuropharmacology. 2008;55:409–418. doi: 10.1016/j.neuropharm.2008.06.073. [DOI] [PubMed] [Google Scholar]
- Kurihara H, Hashimoto K, Kano M, Takayama C, Sakimura K, Mishina M, Inoue Y, Watanabe M. Impaired parallel fiber→Purkinje cell synapse stabilization during cerebellar development of mutant mice lacking the glutamate receptor δ2 subunit. J Neurosci. 1997;17:9613–9623. doi: 10.1523/JNEUROSCI.17-24-09613.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuroyanagi T, Yokoyama M, Hirano T. Postsynaptic glutamate receptor delta family contributes to presynaptic terminal differentiation and establishment of synaptic transmission. Proc Natl Acad Sci U S A. 2009;106:4912–4916. doi: 10.1073/pnas.0900892106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lalouette A, Lohof A, Sotelo C, Guénet J, Mariani J. Neurobiological effects of a null mutation depend on genetic context: comparison between two hotfoot alleles of the delta-2 ionotropic glutamate receptor. Neuroscience. 2001;105:443–455. doi: 10.1016/s0306-4522(01)00193-2. [DOI] [PubMed] [Google Scholar]
- Lee SJ, Uemura T, Yoshida T, Mishina M. GluRδ2 assembles four neurexins into trans-synaptic triad to trigger synapse formation. J Neurosci. 2012;32:4688–4701. doi: 10.1523/JNEUROSCI.5584-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levenes C, Daniel H, Crépel F. Long-term depression of synaptic transmission in the cerebellum: cellular and molecular mechanisms revisited. Prog Neurobiol. 1998;55:79–91. doi: 10.1016/s0301-0082(97)00096-8. [DOI] [PubMed] [Google Scholar]
- Lomeli H, Sprengel R, Laurie DJ, Köhr G, Herb A, Seeburg PH, Wisden W. The rat delta-1 and delta-2 subunits extend the excitatory amino acid receptor family. FEBS Lett. 1993;315:318–322. doi: 10.1016/0014-5793(93)81186-4. [DOI] [PubMed] [Google Scholar]
- MacLean DM. The δ2 glutamate-like receptor undergoes similar conformational changes as other ionotropic glutamate receptors. J Neurosci. 2009;29:6767–6768. doi: 10.1523/JNEUROSCI.1456-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandolesi G, Cesa R, Autuori E, Strata P. An orphan ionotropic glutamate receptor: the delta2 subunit. Neuroscience. 2009;158:67–77. doi: 10.1016/j.neuroscience.2008.02.050. [DOI] [PubMed] [Google Scholar]
- Marignier R, Chenevier F, Rogemond V, Sillevis Smitt P, Renoux C, Cavillon G, Androdias G, Vukusic S, Graus F, Honnorat J, Confavreux C. Metabotropic glutamate receptor type 1 autoantibody-associated cerebellitis: a primary autoimmune disease? Arch Neurol. 2010;67:627–630. doi: 10.1001/archneurol.2010.51. [DOI] [PubMed] [Google Scholar]
- Matilla-Dueñas A, Sánchez I, Corral-Juan M, Dávalos A, Alvarez R, Latorre P. Cellular and molecular pathways triggering neurodegeneration in the spinocerebellar ataxias. Cerebellum. 2010;9:148–166. doi: 10.1007/s12311-009-0144-2. [DOI] [PubMed] [Google Scholar]
- Matsuda K, Yuzaki M. Cbln family proteins promote synapse formation by regulating distinct neurexin signaling pathways in various brain regions. Eur J Neurosci. 2011;33:1447–1461. doi: 10.1111/j.1460-9568.2011.07638.x. [DOI] [PubMed] [Google Scholar]
- Matsuda K, Miura E, Miyazaki T, Kakegawa W, Emi K, Narumi S, Fukazawa Y, Ito-Ishida A, Kondo T, Shigemoto R, Watanabe M, Yuzaki M. Cbln1 is a ligand for an orphan glutamate receptor delta2, a bidirectional synapse organizer. Science. 2010;328:363–368. doi: 10.1126/science.1185152. [DOI] [PubMed] [Google Scholar]
- Matsuda S, Yuzaki M. Mutation in hotfoot-4J mice results in retention of delta2 glutamate receptors in ER. Eur J Neurosci. 2002;16:1507–1516. doi: 10.1046/j.1460-9568.2002.02219.x. [DOI] [PubMed] [Google Scholar]
- Matsumoto H, Okabe S, Hirakawa-Yamada M, Takahashi Y, Satoh N, Igeta Y, Hashida H. Steroid-responsive focal epilepsy with focal dystonia accompanied by glutamate receptor delta2 antibody. J Neuroimmunol. 2012;249:101–104. doi: 10.1016/j.jneuroim.2012.04.009. [DOI] [PubMed] [Google Scholar]
- Matsumoto M, Nakagawa T, Inoue T, Nagata E, Tanaka K, Takano H, Minowa O, Kuno J, Sakakibara S, Yamada M, Yoneshima H, Miyawaki A, Fukuuchi Y, Furuichi T, Okano H, Mikoshiba K, Noda T. Ataxia and epileptic seizures in mice lacking type 1 inositol 1,4,5-trisphosphate receptor. Nature. 1996;379:168–171. doi: 10.1038/379168a0. [DOI] [PubMed] [Google Scholar]
- Miyagi Y, Yamashita T, Fukaya M, Sonoda T, Okuno T, Yamada K, Watanabe M, Nagashima Y, Aoki I, Okuda K, Mishina M, Kawamoto S. Delphilin: a novel PDZ and formin homology domain-containing protein that synaptically colocalizes and interacts with glutamate receptor δ2 subunit. J Neurosci. 2002;22:803–814. doi: 10.1523/JNEUROSCI.22-03-00803.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyata M, Kim HT, Hashimoto K, Lee TK, Cho SY, Jiang H, Wu Y, Jun K, Wu D, Kano M, Shin HS. Deficient long-term synaptic depression in the rostral cerebellum correlated with impaired motor learning in phospholipase C beta4 mutant mice. Eur J Neurosci. 2001;13:1945–1954. doi: 10.1046/j.0953-816x.2001.01570.x. [DOI] [PubMed] [Google Scholar]
- Nishizuka Y. Intracellular signaling by hydrolysis of phospholipids and activation of protein kinase C. Science. 1992;258:607–614. doi: 10.1126/science.1411571. [DOI] [PubMed] [Google Scholar]
- Offermanns S, Hashimoto K, Watanabe M, Sun W, Kurihara H, Thompson RF, Inoue Y, Kano M, Simon MI. Impaired motor coordination and persistent multiple climbing fiber innervation of cerebellar Purkinje cells in mice lacking Galphaq. Proc Natl Acad Sci U S A. 1997;94:14089–14094. doi: 10.1073/pnas.94.25.14089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pin JP, Waeber C, Prezeau L, Bockaert J, Heinemann SF. Alternative splicing generates metabotropic glutamate receptors inducing different patterns of calcium release in Xenopus oocytes. Proc Natl Acad Sci U S A. 1992;89:10331–10335. doi: 10.1073/pnas.89.21.10331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reichelt W, Knöpfel T. Glutamate uptake controls expression of a slow postsynaptic current mediated by mGluRs in cerebellar Purkinje cells. J Neurophysiol. 2002;87:1974–1980. doi: 10.1152/jn.00704.2001. [DOI] [PubMed] [Google Scholar]
- Roche KW, Ly CD, Petralia RS, Wang YX, McGee AW, Bredt DS, Wenthold RJ. Postsynaptic density-93 interacts with the δ2 glutamate receptor subunit at parallel fiber synapses. J Neurosci. 1999;19:3926–3934. doi: 10.1523/JNEUROSCI.19-10-03926.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmid SM, Hollmann M. To gate or not to gate: are the delta subunits in the glutamate receptor family functional ion channels? Mol Neurobiol. 2008;37:126–141. doi: 10.1007/s12035-008-8025-0. [DOI] [PubMed] [Google Scholar]
- Schmitt-Ulms G, Hansen K, Liu J, Cowdrey C, Yang J, DeArmond SJ, Cohen FE, Prusiner SB, Baldwin MA. Time-controlled transcardiac perfusion cross-linking for the study of protein interactions in complex tissues. Nat Biotechnol. 2004;22:724–731. doi: 10.1038/nbt969. [DOI] [PubMed] [Google Scholar]
- Shiihara T, Kato M, Konno A, Takahashi Y, Hayasaka K. Acute cerebellar ataxia and consecutive cerebellitis produced by glutamate receptor delta2 autoantibody. Brain Dev. 2007;29:254–256. doi: 10.1016/j.braindev.2006.09.004. [DOI] [PubMed] [Google Scholar]
- Shimokaze T, Kato M, Yoshimura Y, Takahashi Y, Hayasaka K. A case of acute cerebellitis accompanied by autoantibodies against glutamate receptor delta2. Brain Dev. 2007;29:224–226. doi: 10.1016/j.braindev.2006.08.011. [DOI] [PubMed] [Google Scholar]
- Sillevis Smitt P, Kinoshita A, De Leeuw B, Moll W, Coesmans M, Jaarsma D, Henzen-Logmans S, Vecht C, De Zeeuw C, Sekiyama N, Nakanishi S, Shigemoto R. Paraneoplastic cerebellar ataxia due to autoantibodies against a glutamate receptor. N Engl J Med. 2000;342:21–27. doi: 10.1056/NEJM200001063420104. [DOI] [PubMed] [Google Scholar]
- Soloviev MM, Ciruela F, Chan WY, McIlhinney RA. Identification, cloning and analysis of expression of a new alternatively spliced form of the metabotropic glutamate receptor mGluR1 mRNA1. Biochim Biophys Acta. 1999;1446:161–166. doi: 10.1016/s0167-4781(99)00083-4. [DOI] [PubMed] [Google Scholar]
- Sumner LW, Wolf-Sumner B, White SP, Asirvatham VS. Silver stain removal using H2O2 for enhanced peptide mass mapping by matrix-assisted laser desorption/ionization time-of-flight mass spectrometry. Rapid Commun Mass Spectrom. 2002;16:160–168. doi: 10.1002/rcm.555. [DOI] [PubMed] [Google Scholar]
- Takayasu Y, Iino M, Ozawa S. Roles of glutamate transporters in shaping excitatory synaptic currents in cerebellar Purkinje cells. Eur J Neurosci. 2004;19:1285–1295. doi: 10.1111/j.1460-9568.2004.03224.x. [DOI] [PubMed] [Google Scholar]
- Takeuchi T, Miyazaki T, Watanabe M, Mori H, Sakimura K, Mishina M. Control of synaptic connection by glutamate receptor δ2 in the adult cerebellum. J Neurosci. 2005;25:2146–2156. doi: 10.1523/JNEUROSCI.4740-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tempia F, Alojado ME, Strata P, Knöpfel T. Characterization of the mGluR(1)-mediated electrical and calcium signaling in Purkinje cells of mouse cerebellar slices. J Neurophysiol. 2001;86:1389–1397. doi: 10.1152/jn.2001.86.3.1389. [DOI] [PubMed] [Google Scholar]
- Trebak M, Vazquez G, Bird GS, Putney JW., Jr The TRPC3/6/7 subfamily of cation channels. Cell Calcium. 2003;33:451–461. doi: 10.1016/s0143-4160(03)00056-3. [DOI] [PubMed] [Google Scholar]
- Uemura T, Mishina M. The amino-terminal domain of glutamate receptor delta2 triggers presynaptic differentiation. Biochem Biophys Res Commun. 2008;377:1315–1319. doi: 10.1016/j.bbrc.2008.10.170. [DOI] [PubMed] [Google Scholar]
- Uemura T, Mori H, Mishina M. Direct interaction of GluRdelta2 with Shank scaffold proteins in cerebellar Purkinje cells. Mol Cell Neurosci. 2004;26:330–341. doi: 10.1016/j.mcn.2004.02.007. [DOI] [PubMed] [Google Scholar]
- Uemura T, Lee SJ, Yasumura M, Takeuchi T, Yoshida T, Ra M, Taguchi R, Sakimura K, Mishina M. Trans-synaptic interaction of GluRdelta2 and Neurexin through Cbln1 mediates synapse formation in the cerebellum. Cell. 2010;141:1068–1079. doi: 10.1016/j.cell.2010.04.035. [DOI] [PubMed] [Google Scholar]
- Venkatachalam K, Zheng F, Gill DL. Regulation of canonical transient receptor potential (TRPC) channel function by diacylglycerol and protein kinase C. J Biol Chem. 2003;278:29031–29040. doi: 10.1074/jbc.M302751200. [DOI] [PubMed] [Google Scholar]
- Xiao B, Tu JC, Worley PF. Homer: a link between neural activity and glutamate receptor function. Curr Opin Neurobiol. 2000;10:370–374. doi: 10.1016/s0959-4388(00)00087-8. [DOI] [PubMed] [Google Scholar]
- Yamasaki M, Miyazaki T, Azechi H, Abe M, Natsume R, Hagiwara T, Aiba A, Mishina M, Sakimura K, Watanabe M. Glutamate receptor δ2 is essential for input pathway-dependent regulation of synaptic AMPAR contents in cerebellar Purkinje cells. J Neurosci. 2011;31:3362–3374. doi: 10.1523/JNEUROSCI.5601-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yasumura M, Uemura T, Yamasaki M, Sakimura K, Watanabe M, Mishina M. Role of the internal Shank-binding segment of glutamate receptor delta2 in synaptic localization and cerebellar functions. Neurosci Lett. 2008;433:146–151. doi: 10.1016/j.neulet.2008.01.009. [DOI] [PubMed] [Google Scholar]
- Yawata S, Tsuchida H, Kengaku M, Hirano T. Membrane-proximal region of glutamate receptor δ2 subunit is critical for long-term depression and interaction with protein interacting with C kinase 1 in a cerebellar Purkinje neuron. J Neurosci. 2006;26:3626–3633. doi: 10.1523/JNEUROSCI.4183-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuzaki M. Cbln1 and its family proteins in synapse formation and maintenance. Curr Opin Neurobiol. 2011;21:215–220. doi: 10.1016/j.conb.2011.01.010. [DOI] [PubMed] [Google Scholar]