

Abstract
Trans-soluble N-ethylmaleimide-sensitive factor attachment protein (SNAP) receptor (SNARE) complexes formed between the SNARE motifs of synaptobrevin II, SNAP-25, and syntaxin play an essential role in Ca2+-regulated exocytosis. Apart from the well studied interactions of the SNARE domains, little is known about the functional relevance of other evolutionarily conserved structures in the SNARE proteins. Here, we show that substitution of two highly conserved tryptophan residues within the juxtamembrane domain (JMD) of the vesicular SNARE Synaptobrevin II (SybII) profoundly impairs priming of granules in mouse chromaffin cells without altering catecholamine release from single vesicles. Using molecular dynamic simulations of membrane-embedded SybII, we show that Trp residues of the JMD influence the electrostatic surface potential by controlling the position of neighboring lysine and arginine residues at the membrane–water interface. Our observations indicate a decisive role of the tryptophan moiety of SybII in keeping the vesicles in the release-ready state and support a model wherein tryptophan-mediated protein–lipid interactions assist in bridging the apposing membranes before fusion.
Introduction
Soluble N-ethylmaleimide-sensitive factor attachment protein (SNAP) receptor (SNARE) proteins are thought to act in every intracellular trafficking pathway (Jahn and Scheller, 2006). The SNAREs, syntaxin-1, SNAP-25, and the vesicle-associated Synaptobrevin II (SybII), are central components of the exocytotic machinery in neurons and neuroendocrine cells. For precise temporal coupling between the calcium stimulus and the secretory response, a subset of vesicles is arrested in a final stage before membrane merger awaiting the calcium trigger. It is clear that the number of release-ready vesicles as well as their fusogenicity crucially depend on the presence of SNARE proteins (Schoch et al., 2001; Sørensen et al., 2003; Borisovska et al., 2005) and that the exergonic assembly of the SNARE complex brings vesicular and target membranes into close proximity (Sutton et al., 1998; Li et al., 2007). Furthermore, our previous observations have provided evidence that vesicle SNARE (v-SNARE) proteins drive Ca2+-triggered membrane fusion at a millisecond timescale and support a model wherein molecular straining by SNAREs on their transmembrane domains (TMDs) guides the vesicle from priming to exocytosis triggering and fusion pore enlargement (Kesavan et al., 2007; Guzman et al., 2010).
Yet the most probable route of SNARE-mediated membrane fusion comprises a series of events that requires not only protein–protein but also protein–lipid interactions. A common structural element of v-SNARE proteins is the presence of membranophile Trp residues within the juxtamembrane domain (JMD). Previous results suggested that the Trp residues, W89 and W90, insert into membranes (Quetglas et al., 2000) and have a critical function in preventing SybII from forming a complex with its cognate SNARE partners (Hu et al., 2002; Kweon et al., 2003). In contrast, Siddiqui et al. (2007) have shown that SybII molecules either on proteoliposomes or on synaptic vesicles are constitutively active in complex formation and deletion of both tryptophans had no effect on the efficiency of SNARE-mediated liposome fusion. Recent experiments with cortical neurons further indicated that Trp mutant proteins hardly support Ca2+-evoked transmitter release but strongly promote spontaneous vesicle exocytosis (Maximov et al., 2009). Thus, the precise role of the Trp residues in exocytosis and fusion is unclear. In general, it remains uncertain whether the JMD of SybII merely serves as a force-transmitting element between the SNARE motif and the membrane or, in addition, provides some autonomic function by means of specific protein–lipid interactions to facilitate bilayer merger.
Here, we studied the function of the Trp motif of SybII in chromaffin cell exocytosis using whole-cell membrane capacitance measurements in combination with calcium “uncaging.” Amperometry as well as lipid binding assays allowed us to gather information about kinetics of single fusion events and protein–lipid interactions, respectively. Furthermore, properties of the protein variants and their interactions with membranes were elucidated at the atomic scale using molecular dynamic (MD) simulations of the protein embedded in a vesicle membrane mimic.
We show that lipid-anchored tryptophans of SybII specifically promote priming of vesicles and control the position of the JMD of SybII as well as the electrostatics at the membrane–solvent interface by which they may facilitate the close apposition of vesicle and plasma membrane.
Materials and Methods
Mutant mice and cell culture.
Experiments were performed on mouse chromaffin cells derived from double-v-SNARE knock-out mice (dko cells; Synaptobrevin−/−/Cellubrevin−/−) (Borisovska et al., 2005) or SybII knock-out mice (SybII-ko; Synaptobrevin−/−) (Schoch et al., 2001) of either sex at E17.5–E18.5. Cells were cultured as described by Borisovska et al. (2005). Recordings were done at room temperature on days 1–3 in culture and 6–7 h after infection of cells with virus particles.
Viral constructs.
cDNAs encoding for SybII and its mutants were subcloned into the viral plasmid pSFV1 (Invitrogen) upstream of an independent open reading frame that encodes for enhanced green fluorescent protein (EGFP). EGFP labeling was used to identify infected cells. Mutant constructs carrying either amino acid insertions or replacements were generated using the overlapping primer method. All mutations were confirmed by DNA sequence analysis. Virus particles were produced as described previously (Ashery et al., 1999).
Whole-cell capacitance measurements and amperometry.
Whole-cell membrane capacitance measurements and photolysis of caged Ca2+ as well as ratiometric measurements of [Ca2+]i were performed as described previously (Borisovska et al., 2005). The extracellular Ringer's solution contained the following (in mm): 130 NaCl, 4 KCl, 2 CaCl2, 1 MgCl2, 30 glucose, 10 HEPES-NaOH, pH 7.3. The pipette solution for flash experiments contained the following (in mm): 110 Cs-glutamate, 8 NaCl, 3.5 CaCl2, 5 NP-EGTA, 0.2 fura-2, 0.3 furaptra, 2 MgATP, 0.3 Na2GTP, 40 HEPES-CsOH, pH 7.3, 320 mOsm. The flash-evoked capacitance response was approximated with the following function: f(x) = A0 + A1(1 − exp[−t/τ1]) + A2(1 − exp[−t/τ2]) + kt, where A0 represents the cell capacitance before the flash. The parameters A1, τ1, and A2, τ2, represent the amplitudes and time constants of the readily releasable pool (RRP) and the slowly releasable pool (SRP), respectively (Rettig and Neher, 2002). The exocytotic delay was defined as the time between the flash and the intersection point of the back-extrapolated fast exponential with the baseline.
Carbon fiber electrode (5 μm diameter; Amoco) manufacturing and amperometric recordings with an EPC7 amplifier (HEKA Elektronik) were done as previously described (Bruns, 2004). For Ca2+ infusion experiments, the pipette solution contained the following (in mm): 110 Cs-glutamate, 8 NaCl, 20 DPTA (diethylene triamine penta-acetic acid), 5 CaCl2, 0.2 fura-2, 0.3 furaptra, 2 MgATP, 0.3 Na2GTP, 40 HEPES-CsOH, pH 7.3, 19 μm free calcium; or 140 Cs-glutamate, 8 NaCl, 10 EGTA, 7.5 CaCl2, 0.2 fura-2, 0.3 furaptra, 2 MgATP, 0.3 Na2GTP, 20 HEPES-CsOH, pH 7.3, 350 nm free calcium. Current signals were filtered at 2 kHz and digitized gap-free at 25 kHz. Amperometric spikes with a charge ranging from 10 to 5000 fC and peak amplitude >4 pA were selected for frequency analysis, while an amplitude criterion of >7 pA was set for the analysis of single spike characteristics. To study secretion in resting chromaffin cells, a single carbon fiber was brought into direct contact and exocytosis was recorded for 180 s without stimulation in extracellular Ringer's solution. For analysis of current signals recorded in infusion experiments with 350 nm Ca2+-containing solution and in resting chromaffin cells, events exceeding the threshold of 2.5 pA (approximately five times of the SD of the baseline noise) were analyzed to consider also low-amplitude events that may occur at resting levels of [Ca]i (Fulop et al., 2005).
Immunocytochemistry.
Chromaffin cells were processed 6 h after virus infection for immunolabeling as described previously (Hannah et al., 1998). Epifluorescence pictures were acquired with an AxioCam MRm-CCD camera (Carl Zeiss) and analyzed with MetaMorph software (Universal Imaging). The affinity-purified mouse monoclonal antibody against SybII (69.1, antigen epitope amino acid position 1–14) was kindly provided by R. Jahn (MPI for Biophysical Chemistry, Göttingen, Germany). For quantification, the average intensity of the fluorescent immunolabel was determined within an area of interest comprising the outer cell perimeter.
Structured illumination microscopy and total internal reflection fluorescence microscopy.
Chromaffin cells were processed 3.5 h after virus infection to prevent strong overexpression of SybII and its variants. Cells were imaged through a 63× Plan-Apochromat (NA, 1.4) oil-immersion objective on the stage of a Zeiss Axio Observer with excitation light of 488 and 561 nm wavelengths. The ELYRA PS.1 system and ZEN software 2011 (Zeiss) were used for acquisition and processing of the images for structured illumination microscopy. Properties of SybII-fluorescent puncta in z-stacks were analyzed with the software package ImageJ, version 1.45. The weighted colocalization coefficient was analyzed after thresholding the images from the sum of intensities of SybII pixels colocalizing with cellubrevin (Ceb), compared with the overall sum of SybII pixels (Bolte and Cordelieres, 2006).
Total internal reflection fluorescence microscopic (TIRFM) pictures were taken on a stage of a Axiovert 200 (Zeiss), equipped with a total internal reflection fluorescence (TIRF) slider, an oil-immersion 100×/1.45 NA Fluar objective (Zeiss), and a solid-state laser (emission, 561 nm; 85YCA010; Melles Griot). Images were acquired with an EMCCD camera (Andor iXonEM) under control of a customized software based on LabView (National Instruments). The 1/e penetration depth (d) of the evanescent wave (wavelength, 561 nm) was calculated to be 110 nm, agreeing with experimentally determined d values for this setup as described by Pasche et al. (2012). The camera exposure time was set to 500 ms in wild-type cells and to 100 ms in overexpressing cells to obtain images of comparable signal strength. For analysis of the TIRF data, fluorescent peaks with amplitudes three times greater than the average background signal (in arbitrary units: wt, 873 ± 218; dko + SybII, 707 ± 148; dko + WW/AA, 629 ± 114) and with an area of 4–9 pixels (pixels size in object plane, 160 nm) were counted as docked granules.
Turbidity assay and spectrofluorimetry.
GST fusion constructs of SybII and its mutant proteins (amino acids 1–96) were purified from BL21 Escherichia coli cells according to standard procedures. The concentration, purity, and integrity of the recombinant fusion proteins were verified by SDS-PAGE and Coomassie staining.
For liposome preparation, chloroform solutions of brain phosphatidylcholine (PC) and phosphatidylserine (PS) (Avanti Polar Lipids) were mixed (molar ratio PC/PS, 3:1) and subsequently dried in a speed vacuum rotor for 20 min. Dried lipids were resuspended at a concentration of 2.5 mg/ml in buffer A containing the following (in mm): 100 NaCl, 20 HEPES-NaOH, pH 7.3, and 2 EDTA. Multilamellar liposomes were exposed to repeated freeze–thaw cycles and extruded by 10 passes through a Mini-Extruder (Avanti Polar Lipids) to obtain unilamellar vesicles.
Absorbance measurements were done at a wavelength of 350 nm in disposable UV-compatible plastic cuvettes. All reactions contained 20 μl of liposome solution (2.5 mg/ml) premixed with 66 μl of buffer A. After 10 s, 14 μl of protein solution was added to obtain the indicated concentration in a final reaction volume of 100 μl. Changes in absorbance were recorded with a Genesys 10UV spectrophotometer (Thermo Fisher Scientific) and digitized at a rate of 1 Hz with VisionLite software (Thermo Fisher Scientific). All measurements were performed at room temperature.
For spectrofluorimetry, fluorescence of TRP residues was elicited at 290 nm excitation (10 nm slit width) and emission spectra detected between 320 and 450 nm. SybII wild-type and mutant proteins were diluted in buffer A containing 1.4 mm liposomes. Experiments were performed in quartz cuvettes (10 mm path length), and data were acquired with a LS55 fluorescence spectrometer (PerkinElmer) at 1 nm interval and a scan rate of 10 nm/s.
Molecular dynamic simulations.
The transmembrane region of the SNARE peptide was modeled from the sequence of the synaptobrevin (NP 036795): ASQFETS 81AAKLKRKYWW 91KNLKMMIILG 101VICAIILIII 111IVYFST. As suggested by Bowen and Brunger (2006), the SNARE peptide region embedded in the membrane bilayer (in this case, residues 85–116, underlined) was modeled as an α-helix, whereas the rest was built as unstructured coil (residues 74–84). Model generation was achieved using the WHAT IF program (Vriend, 1990). Additionally, three different mutants of synaptobrevin were modeled and simulated: the double mutant W89A/W90A and the two single mutants W89A and W90A. The peptide was embedded into the membrane bilayer by the same procedure as described in our previous work (Siu and Böckmann, 2007). An asymmetric anionic membrane patch was prepared as a simple mimic of the synaptic vesicle membrane. The system was set up starting from a fully hydrated pure palmitoyl-oleoyl-PC (POPC) bilayer. Thirty-two lipids of this lipid bilayer were randomly exchanged by palmitoyl-oleoyl-PS (POPS) lipids (16 in each leaflet). The system was simulated for 175 ns at a physiological ionic concentration of 150 mm NaCl. The total charge of the system was set to 0 by adding 32 counterions. Equilibration was monitored by the area per lipid. The area per lipid decreased from 62 to 50 Å2. An asymmetric bilayer was constructed by combining one leaflet of the mixed membrane after 175 ns with one leaflet of a pure POPC lipid bilayer simulated for 100 ns. The number of lipids in the latter was reduced to cope with the total monolayer area of the mixed leaflet. In that way, different areas per lipid for the two leaflets were obtained, the mixed leaflet area per lipid being in agreement with our study of a mixed bilayer, while the area per lipid for the neutral monolayer agreed with studies on neutral bilayers at a similar ionic concentration (Böckmann et al., 2003). The asymmetric bilayer contained 51 POPC lipids in one leaflet, and 48 POPC and 16 POPS lipids in the other leaflet. This composition reflects roughly the ratio between negatively charged lipids and phosphatidylcholines reported for synaptic vesicles (Takamori et al., 2006). Before peptide insertion, the asymmetric bilayer was equilibrated for another 25 ns. Molecular dynamic simulations were performed using the GROMACS simulation package (version 4.0.2) (Hess et al., 2008) applying a combination of the GROMOS96 53a6 force field for the peptide (Oostenbrink et al., 2004) and the Berger (Berger et al., 1997) lipid force field. The SNARE membrane systems were first equilibrated with position restraints on the peptide for 200 ps. For each system, two simulations of 400 ns length each were performed, differing either in the initial tilt of the TMD region (WT simulations) or in the starting velocities. For the simulations, the Newton's equations of motion were integrated with the leapfrog algorithm using the time step of 2 fs. The neighbor list was updated every 10 steps. Long-range electrostatics was treated using the particle mesh Ewald (PME) method (Darden et al., 1993) with a direct sum cutoff at 1.2 nm, a Fourier spacing of 0.12 nm for the FFT grid, and cubic interpolation. The same cutoff distance of 0.12 nm was used for the van der Waals interactions applying long-range dispersion corrections for both energy and pressure. Periodic boundary conditions were applied in all directions. All H-bonds were constrained using the LINCS (Hess et al., 1997) or the SETTLE algorithm (Miyamoto and Kollman, 1992). The system temperature was coupled to 310 K with a coupling time constant of 0.1 ps, whereas the pressure was coupled semiisotropically to 1 bar using a time constant of 1 ps. Both couplings were performed using the Berendsen algorithm (Berendsen et al., 1984).
Interface electrostatics.
The electrostatic potential on the surface of the SNARE/membrane system was studied solving the linearized Poisson–Boltzmann equation applying DELPHI II; for the dielectric constant of water, a value of 80 was chosen, for the protein and membrane interior of 4. A 211 cubed grid with a grid resolution of 2.5 grid points per angstrom was applied for all systems. The electrostatic potential map was calculated on snapshots of the systems separated by 1 ns (in total, 3200 maps). To focus on the effect of the different positioning of the v-SNARE TMD with respect to the membrane, the N-terminal 12 residues were deleted before the calculation (i.e., residues 86–116 were taken into account). The final maps were averaged over the last 300 ns of simulation for each system.
The Gouy–Chapman equation (at physiological salt concentration) was used to estimate the effect of the basic SybII residues (close to the TMD) on the electrostatic energy between the target membrane [charge density, 0.5 e/nm2, assuming for the inner leaflet of the plasma membrane 20% PS (net charge, −1) and 2–6% phosphatidyl 4,5-bisphosphate (PIP2) (net charge, −4) (James et al., 2008; Leventis and Grinstein, 2010)] and a circular vesicle membrane patch [radius, 3.5 nm; charge density, 0.18 e/nm2 (Takamori et al., 2006)]. For calculation, 15 positive charges (representing three SybII molecules with the corresponding amino acids K85, R86, K87, K91, and K94) were set to the average distances from the vesicle membrane surface as determined from the simulations (see Fig. 10B). The SybII charges were uniformly distributed parallel to the membrane plane. No significant changes in the surface potential of areas opposing the JMD were observed (see Fig. 11A). Therefore, changes of the lipid surface potential within the contact area were not considered in this calculation.
Figure 10.

Increased insertion depth of the SybII WW/AA mutant. A, Two exemplary MD snapshots of the wild-type (SybII, top panel) and the double-mutated (W89A/W90A, bottom panel) SybII in a lipid bilayer. The protein is depicted in diagram representation. Selected amino acids are highlighted in stick representation: Lys (red), Trp and Ala89/90 in the double mutant (blue). Water molecules and lipids are drawn as balls. POPC lipids are depicted in gray (tail) and lime (head group region), and POPS molecules in pink (tail) and brown (head group). B, Distribution of basic residues with respect to the upper layer membrane surface for wild-type SybII and its mutant proteins. The graph reports the normalized (number) density of the atoms of the charged moieties of Lys85, 87, 91, 94 and of Arg86 as obtained from the MD simulations of the wild type (WT), the single mutants (WA), and the double mutant (WW/AA) Synaptobrevin II. The dashed lines indicate mean values of the corresponding frequency distribution. The densities are calculated considering the last 300 ns of each simulation, and data for the single mutants (W89, W90) were averaged. The “zero” on the x-axis represents the plane defined by the phosphorus atoms of the POPC and POPS lipids in the JMD proximal lipid leaflet. C, Residue-resolved mean distance of SybII backbone positions from the membrane-solvent interface. The inset shows the distance of simulated SybII residues as well as experimental data derived from EPR accessibility measurements (diamonds) (Kweon et al., 2003).
Figure 11.

Electrostatic potential maps of the JMD proximal membrane surface. A, The electrostatic potential averaged over 300 structures (protein plus membrane) extracted every 1 ns from the MD trajectories (neglecting the first 100 ns) at a distance of ∼1.2 nm from the membrane interface (defined by the center of mass of the lipid head groups of the JMD proximal lipid leaflet). Calculations were restricted to the residues of the JMD and TMD (residues 86–116) to minimize effects that can be obtained from insufficient sampling of the N-terminal JMD region. WT-S1 and S2 as well as WW/AA S1 and S2 are independent simulations. B, Density map of SybII backbone atoms in the membrane plane, plotted to the same scale as in A. Shown are the positions of the SybII backbone atoms (green dots), of the (simulated) N terminus (Ala74; magenta), and of the lysine residues 83 and 85 (orange; side chain positions). The TMD position is indicated by a yellow circle. The location of the negative membrane potential largely correlates with the position of the N-terminal Ala74. Additionally, lysines 83 and 85 are oriented toward the N terminus of the JMD for the WT as well as for the single mutants, but are opposed for the double mutant.
Statistical analyses.
Values are given as mean ± SEM. To determine statistically significant differences, one-way ANOVA and a Tukey–Kramer post test for comparing groups were used, if not indicated otherwise.
Results
Tryptophan residues are important for priming stability of release-ready vesicles
To test the functional importance of the membrane-proximal Trp residues, we analyzed the ability of a mutated SybII variant carrying two Trp substitutions (W89S, W90S; WW/SS) in reconstituting exocytosis of double v-SNARE-deficient cells (Borisovska et al., 2005), which are nearly devoid of secretion (Fig. 1). Whereas the SybII wild-type protein restores a robust secretory response to a step-like increase in intracellular calcium, the Trp mutant vastly fails to rescue the initial capacitance increase, referred to as exocytotic burst (EB) (Fig. 1A,B). Both components of the EB, the RRP and the SRP, are similarly affected, but no profound alteration in the subsequent sustained phase of secretion is observed (Fig. 1C). Furthermore, the kinetic rates of the exocytotic burst (RateRRP and RateSRP) remain unchanged, indicating that the Trp mutation specifically interferes with the priming of chromaffin granules. In contrast, substitution of only a single Trp residue, either W89S or W90S, does not affect secretion (Fig. 1D,E). On average, the exocytotic burst size as well as its kinetic properties are similar to dko cells expressing the wild-type SybII (Fig. 1F). Thus, a single tryptophan residue is sufficient to support exocytosis at the level of wild-type cells.
Figure 1.

Membrane-proximal tryptophan residues are important for vesicle priming. A, Schematic representation of SybII and its WW/SS mutant (W89S, W90S) on the vesicular membrane. B, Averaged flash-induced [Ca2+]i levels (top panel) and corresponding capacitance responses (bottom panel) of dko cells expressing SybII (black; n = 21) or the WW/SS variant (red; n = 20). C, The analysis of individual responses indicates a strong reduction of both pools of primed vesicles RRP and SRP, but no changes in their fusion kinetics in the absence of both tryptophan residues. D, Schematic representation of the single tryptophan mutations: W89S (left panel) and W90S (right panel). E, Averaged flash-induced [Ca2+]i levels (top panel) and corresponding capacitance responses (bottom panel) of dko cells expressing SybII (black; n = 20) or SybII variants with replaced W89S (blue; n = 21) or W90S (green; n = 21). F, The kinetic components of the secretory response remain unchanged for W89S and W90S. ***p < 0.001, one-way ANOVA. Error bars indicate SEM.
We also tested a SybII variant, in which W89 and W90 were replaced by alanines (Fig. 2), as previously studied by Maximov et al., (2009). Indeed, the phenotype of the W89A/W90A (WW/AA) mutant is remarkably similar to that of the WW/SS mutation, confirming a strong reduction of the pool of primed vesicles and no significant change in kinetics. Statistical analysis of the EB size recorded for the two mutant proteins reveals no significant difference (p = 0.701, one-way ANOVA with Tukey–Kramer post hoc testing). These results suggest that the phenotype observed for the WW/SS mutant is specifically caused by the loss of the Trp moiety rather than by the substituting amino acid. Immunofluorescence analyses of chromaffin cells using an antibody directed against SybII reveal that the SybII protein and the WW/SS mutant are efficiently expressed excluding a decreased protein level as the reason for the observed phenotype (Fig. 2D,E). In contrast, substitution of the membrane-proximal K91 and K94 by alanine residues causes missorting of the mutant protein, possibly by interference with the Ca2+-calmodulin binding to SybII and its posttranslational membrane insertion (Hassdenteufel et al., 2011).
Figure 2.

Substitution of membrane-proximal tryptophan residues with alanines also impairs vesicle priming. A, Schematic representation of synaptobrevin II W89A/W90A mutation. B, Averaged flash-induced [Ca2+]i levels (top panel) and corresponding capacitance responses (bottom panel) of dko cells expressing SybII (black; n = 23) or the WW/AA variant (orange; n = 17). The inset shows the extended scaling of the capacitance responses illustrating a similar onset of exocytosis after the stimulus (t = 0.5 s) for the mutant protein. C, The WW/AA mutant reduces the RRP and SRP pool size but leaves the kinetics of the exocytotic response unchanged. D, Exemplary images of a wild-type cell and dko cells overexpressing either sybII or mutated v-SNARE proteins. Immunosignals were detected with an affinity-purified monoclonal antibody (69.1) directed against the N terminus of sybII and are visualized after adjustment of the exposure time of the camera (wt, 1.2 s; dko + v-SNARE, 0.2 s). A mutant protein with K91A and K94A substitution (KK/AA) appears to be missorted, as judged from the homogenous distribution of the immunosignal. No exocytotic activity could be recorded with this mutant (data not shown). Scale bar, 5 μm. E, Average fluorescence intensity of wt cells and of dko cells expressing the indicated mutants or SybII. Note that expression of SybII or its mutants in dko cells leads to a 7- to 10-fold increase in protein level when compared with the wt signal. Data were normalized to the immunosignal of SybII expressed in dko cells. Numbers indicate analyzed cells. ***p < 0.001, one-way ANOVA. Error bars indicate SEM.
To determine the subcellular localization of SybII and its mutant variant in detail, we used high-resolution structured illumination microscopy (Gustafsson, 2000) and compared immunosignals of SybII and another vesicular SNARE protein, Ceb (also referred to as VAMP3) (Elferink et al., 1989; McMahon et al., 1993). SybII colocalizes well with Ceb in wild-type cells (colocalization coefficient: 0.80 ± 0.006, n = 11; Fig. 3A), which is consistent with the functional redundancy of these v-SNARE proteins in secretion from chromaffin cells (Borisovska et al., 2005). Acute viral expression of SybII or the WW/AA mutant in chromaffin cells from SybII-deficient chromaffin cells reveals a similar colocalization with Ceb (colocalization coefficient: SybII, 0.77 ± 0.016, n = 11; WW/AA, 0.76 ± 0.018, n = 10), suggesting that SybII and its mutant variant are sorted correctly to secretory granules (Fig. 3B,C). The fluorescence signals shown in the line scans have a half-width of ∼200 nm, compatible with the dimensions of single vesicular structures. By recording z-stacks (100 nm interval) through the cells, we determined the fluorescence maxima of the SybII immunosignals. The resulting fluorescence profiles reveal singular fluorescence peaks consistent with the vesicular origin of the immunosignals (Fig. 3D). To determine the mean fluorescence per granule, signals exceeding the threshold (∼6 SD of average baseline noise) were counted. For overexpression of SybII and the WW/AA mutant, signals of nearly identical amplitudes are observed, both of which being, on average, fivefold higher than in wild-type cells (Fig. 3D,E). Thus, virally expressed SybII and its mutant variant are sorted to granules with similar efficiencies.
Figure 3.
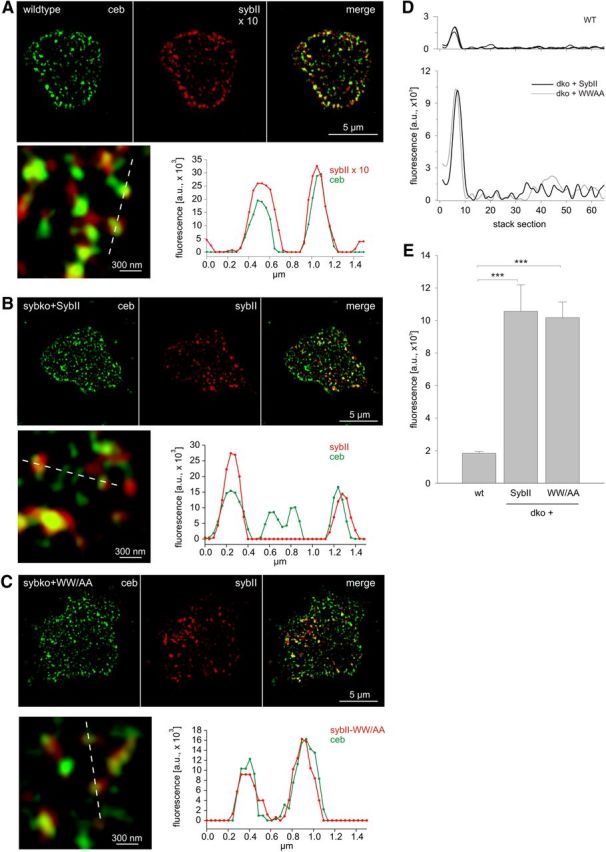
SybII and its TRP mutant are sorted to granules with similar efficiencies. Exemplary immunostainings (imaged with structured illumination microscopy) for Ceb (green) and SybII (red) in wild-type chromaffin cells (A) and Sybko cells expressing SybII (B) or the SybII-WWAA mutant protein (C). Cells were imaged within the footprint area to minimize the contribution of Golgi-derived fluorescence in virus-transfected cells. SybII fluorescence signals in wild-type cells were excited with fivefold higher laser power than in virus-transfected cells and were multiplied times 2 for display (×10). The merged images and their magnified view display a clear colocalization between Ceb and SybII (or the WWAA mutant), as also illustrated in the corresponding line scans (magnified view, dashed lines; pixel size, 40 nm). D, Exemplary fluorescence profiles of discrete SybII puncta analyzed by z-stacking through the cell. Note the different amplitudes of SybII puncta in wild-type (top panel, 3 exemplary traces) and virus-infected dko cells (bottom panel). E, The mean fluorescence intensity of single SybII puncta is similar for SybII and the WW/AA mutant and fivefold higher than in wild-type cells. For analysis, images were thresholded to values 6 SD of the background fluorescence to isolate discrete regions of interest (ROIs). On average, similar-sized ROIs in wild-type cells (0.0258 ± 0.0027 μm2; 10 cells; 1018 ROIs) and dko cells expressing SybII (0.0245 ± 0.0019 μm2; 7 cells; 250 ROIs) or the WW/AA mutant (0.0233 ± 0.0012 μm2; 12 cells; 365 ROIs) were analyzed. ***p < 0.001, one-way ANOVA. Error bars indicate SEM.
To test whether the TRP mutant interferes with the number of membrane-proximal vesicles, TIRFM was used (Fig. 4). By counting SybII-labeled puncta in the evanescent field, we find that wild-type cells exhibit on average 16 ± 1.8 granules (n = 9) within the footprint area of the cell, a result that is consistent with previous studies in mouse chromaffin cells (Speidel et al., 2005; Friedrich et al., 2008). No differences in the number of vesicles per footprint are observed for dko cells expressing SybII or the WW/AA mutant protein (Fig. 4A–C,E). Therefore, we can conclude that the TRP mutant protein exerts its phenotype downstream of vesicle docking. This result agrees well with our previous electron microscopy analyses, showing that loss of SybII and cellubrevin does not interfere with docking of secretory organelles in chromaffin cells (Borisovska et al., 2005).
Figure 4.
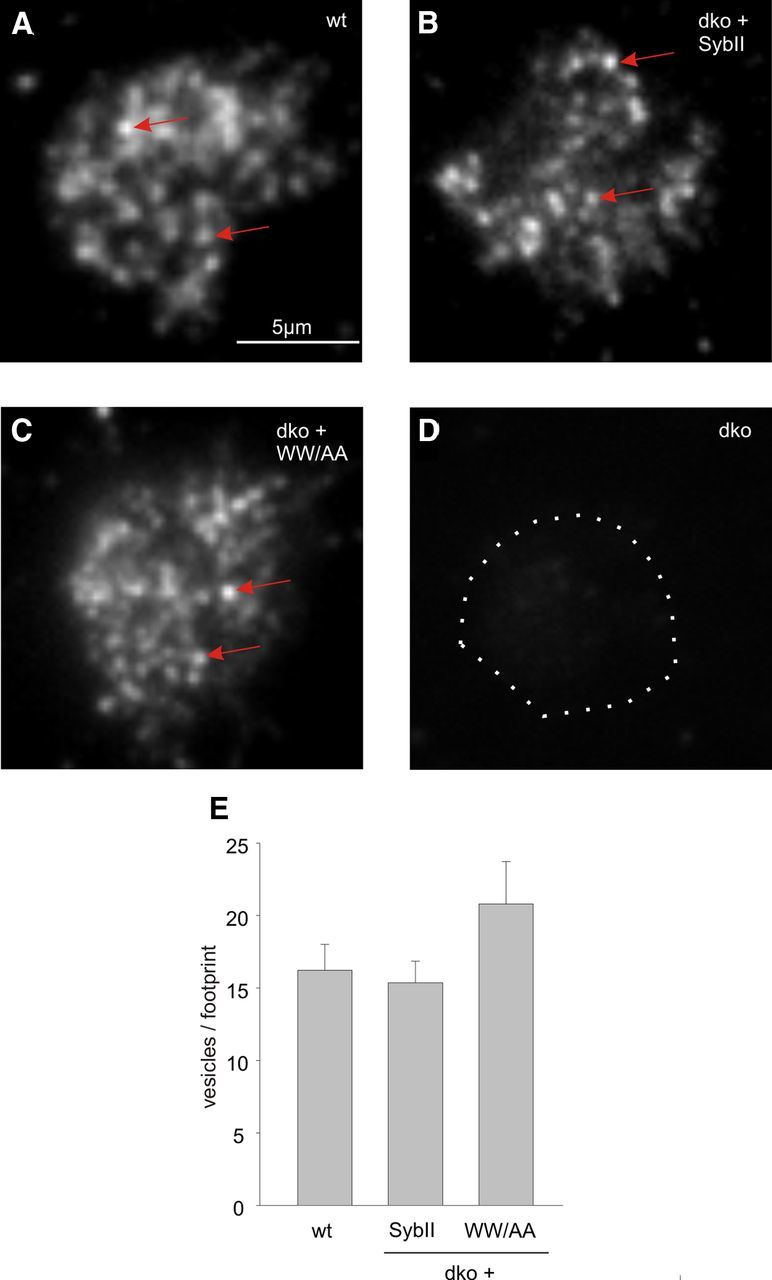
TRP mutant protein does not interfere with vesicle docking. A–C, Wild-type chromaffin cells and dko cells expressing either SybII or the WWAA mutant exhibit SybII-positive granules (exemplary granules, arrows) in the footprint of the cell visualized by evanescent illumination. D, No signals are observed in dko cells (dotted line, cell perimeter) indicating specificity of the antibody. E, The number of SybII-positive puncta is unchanged in WWAA mutant cells (n = 14) compared with SybII-expressing dko cells (n = 17) or wild-type cells (n = 9) (p = 0.314, one-way ANOVA). Error bars indicate SEM.
Recent experiments by Südhof and colleagues (Maximov et al., 2009) suggested that membrane-proximal tryptophan residues take part in the postulated “fusion clamp” because their substitution strongly increased the frequency of spontaneously occurring mEPSCs in cortical neurons. To test whether such a scenario also holds true for chromaffin cell exocytosis, we continuously recorded the membrane capacitance for up to 2 min, while dialyzing the cell with submicromolar concentrations of calcium (400–500 nm; Fig. 5B). Compared with dko cells, cells expressing SybII exhibit a steady and significant increase in membrane capacitance that is unchanged in tryptophan mutant cells (Fig. 5). To further substantiate this point, we repeated the experiment at a lower [Ca2+]i (350 nm, close to the secretion threshold of chromaffin cells) (Augustine and Neher, 1992) and with the WW/AA mutant protein for direct comparison with the neuronal data (Maximov et al., 2009). Again, no differences in the tonic capacitance response of SybII and WW/AA mutant cells are observed (Fig. 5D,E). Simultaneous amperometric recordings verify the result and exclude the possibility that potential changes in membrane capacitance are masked by concomitant changes in membrane internalization. Moreover, also in resting chromaffin cells, significant differences in the frequency of amperometric events measured in SybII and WW/AA mutant cells could not be detected (Fig. 5F). Together, our experiments reveal, compared with dko cells, a v-SNARE-dependent increase in membrane capacitance and in the frequency of amperometric signals at submicromolar [Ca]i, and provide no evidence for a role of the membrane-proximal Trp residues in the proposed fusion-clamp mechanism. Furthermore, this result counters the possibility that the observed reduction in EB size is simply the consequence of a tonically enhanced release probability leading to pool depletion.
Figure 5.
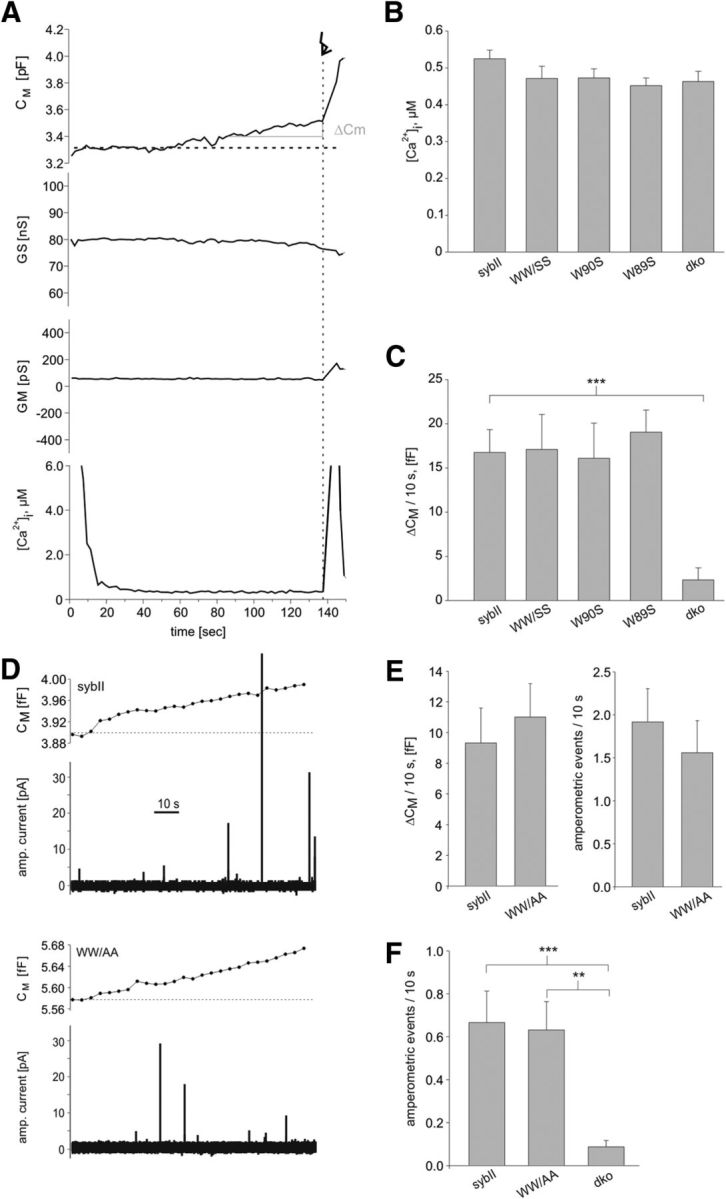
Loss of the TRP moiety in SybII does not unclamp exocytosis in chromaffin cells. A, Exemplary recording of cell membrane capacitance (Cm), access resistance (Gs), membrane resistance (Gm), and [Ca2+]i during infusion of submicromolar [Ca2+]i in a double ko cell expressing SybII. Note the shallow but steady increase in membrane capacitance (ΔCm) before the flash (arrow). The initial high [Ca] signal is due to Ca2+ indicators that leak out of the patch pipette approaching the cell. The signal is rapidly diminished due to indicator dilution after establishing cell contact and [Ca]i measurements were considered at t > 60 s. B, C, Quantification of [Ca2+]i and capacitance increase (ΔCM) over the last 10 s before flash stimulation. Note the SybII-dependent increase in ΔCM that is unchanged for the mutant proteins. Data were collected from dko cells (n = 22) and dko cells expressing SybII (n = 46), WW/SS mutant (n = 15), W89S (n = 21), W90S (n = 22). D, Measurements of membrane capacitance (top panels) and simultaneous amperometric recordings (bottom panels) from dko cells expressing sybII or the WW/AA mutant. t = 0, start of intracellular perfusion with Ca2+-containing solution (free [Ca2+]i = 350 nm) via the patch pipette. E, Both assays for secretion, membrane capacitance (left panel) and amperometry (right panel), reveal no differences between SybII (n = 22) and the WW/AA mutant (n = 22). F, Amperometric recordings in resting chromaffin cells reveals a v-SNARE-dependent increase in exocytotic activity that is similar in SybII- and WW/AA-expressing dko cells (SybII, n = 22; WW/AA, n = 25; dko, n = 8). **p < 0.005, ***p < 0.001, one-way ANOVA. Error bars indicate SEM.
Trp residues of the JMD have been implicated in membrane association of the cytoplasmic domain of SybII (Quetglas et al., 2000) and therefore may contribute to a tight molecular link between the SNARE motif and the transmembrane anchor. A similar priming defect was found for mutations that extend the juxtamembrane region of SybII by insertion of a flexible stretch of amino acids directly preceding the transmembrane domain, also referred to as “linker” mutations (Kesavan et al., 2007). Still, neither single nor double substitutions of the Trp residues cause a significant increase in the exocytotic delay (Figs. 1C,F, 2C), as it was observed for the linker extensions. This suggests that the Trp residues stabilize priming by different means than increasing the flexibility between the SNARE domain and the TMD. To further substantiate this point, we generated an alternative linker mutant that repeats the intrinsic “WWKNLK” motif instead of an extension by a flexible “KLGGSG” cassette (Fig. 6). This alteration increases the physical distance between the SNARE domain and the TMD, but preserves the original position of the Trp residues relative to the transmembrane anchor. The WWKNLK insertion diminishes the pool of primed vesicles to a comparable degree as the similarly long KLGGSG insertion (Fig. 6). These results corroborate the view that the pool size reduction caused by extension of the linker can be attributed to an increase in intramolecular flexibility between the SNARE motif and the TMD.
Figure 6.
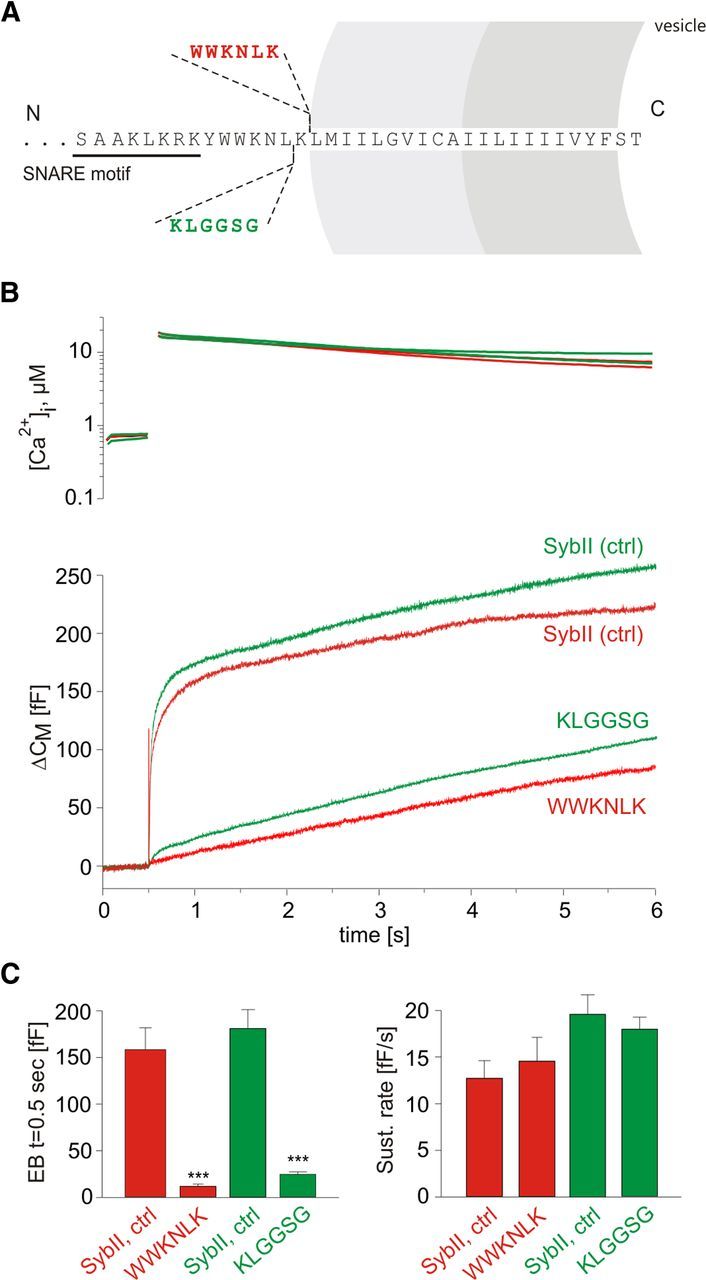
Duplication of the intrinsic WWKNLK motif reveals a similar phenotype as observed for the KLGGSG insertion. A, Schematic view of SybII domains and the amino acid insertions within the JMD of SybII. B, Averaged flash-induced [Ca2+]i levels (top panel) and corresponding capacitance responses (bottom panel) of dko cells expressing SybII (control for WWKNLK, red, n = 19; control for KLGGSG, green, n = 25) or the mutant protein carrying either an extra WWKNLK motif (red; n = 17) or a KLGGSG insertion (green; n = 41). C, Quantification of the responses reveals similarly strong reduction of the exocytotic burst (measured 0.5 s after the flash) for either type of insertion. ***p < 0.001, one-way ANOVA. Error bars indicate SEM.
Given that Trp residues form a functional landmark within the JMD of SybII, we also investigated the impact of amino acid insertions upstream of the Trp region. Insertions of 1 or 3 aa profoundly reduce the EB (Fig. 7A,B). With an insertion of 12 aa, secretion decreases to the level of dko responses, consistent with previous results by Deák et al. (2006). Even the insertion of a single amino acid (1 aa) clearly reduces RRP size without changing either the kinetics of secretion from the RRP or the SRP (Fig. 7E) nor the exocytotic delay (dko+sybII, 3.55 ± 0.5 ms; dko+1aa, 4.40 ± 0.62 ms; Fig. 7D). As shown in Figure 7C, both types of insertions decrease priming in a length-dependent fashion, consistent with a stringent length requirement of the juxtamembrane region for priming (Kesavan et al., 2007). Still, mutations upstream of the Trp residues are more effective in pool size reduction than downstream insertions (e.g., 3 aa insertion: upstream 20% of control; downstream 80% of control). An attractive explanation for this observation could be that upstream insertions either additionally interfere with the formation of a predicted α-helical secondary structures in this region (Ellena et al., 2009) or perturb interactions with auxiliary proteins that regulate fusion competence.
Figure 7.

Amino acid insertions upstream of tryptophan residues produce a strong linker length-dependent reduction of the secretory response. A, Scheme depicts the mutations introduced into SybII. Inserted amino acids are underlined. B, Averaged flash-induced [Ca2+]i levels (top panel) and corresponding capacitance responses (bottom panel) of dko cells expressing wt SybII (black; n = 24) or mutated SybII variants, 1 aa (red; n = 21), 3 aa (green; n = 15), 12 aa (blue; n = 11). The 12 aa variant fails to support any release compared with dko (gray; n = 8). C, Comparison of the linker length-dependent reduction of the exocytotic burst size (measured at t = 0.5 s after the flash) for linkers positioned upstream (red) or downstream of the TRP residues [gray; data taken from the study by Kesavan et al. (2007)] of the tryptophan residues. D, Extended scaling of the capacitance responses for control and 1 aa mutant shown in B illustrates no changes in the exocytotic delay. The gray lines represent double exponential fit of the responses. E, Quantification obtained from fitting of each individual response indicates a steep decrease in the exocytotic burst size with increasing linker length. *p < 0.05, ***p < 0.001, one-way ANOVA. Error bars indicate SEM.
Membrane-proximal Trp residues are not essential for rapid transmitter discharge from chromaffin granules
To determine the properties of single-vesicle fusion events, we assayed secretion with amperometry, a technique that resolves kinetic properties of neurotransmitter discharge from single vesicles at a millisecond timescale. For each amperometric spike, evoked by Ca2+ dialysis in SybII- and mutant-expressing cells, we determined quantal size, amplitude, and kinetic parameters. As shown in Figure 8, neither the amount of transmitter released from a granule nor the peak amplitude of the oxidative current spike changes in cells expressing mutant SybII. Furthermore, prespike signals, which precede the main amperometric spike and reflect the release of transmitter through a narrow fusion pore, are similarly resistant to a loss of the Trp moiety of SybII. Simultaneous recordings of the membrane capacitance reveal a steady increase in CM that parallels the occurrence of the amperometric signals and is unchanged in cells expressing the Trp mutants (Fig. 8A–C). Thus, exocytotic activity in response to a long-lasting calcium stimulus is unaffected by the WW substitution, which nearly abolishes the exocytotic burst. This result confirms a specific requirement of the membrane-proximal Trp residues for granule priming.
Figure 8.
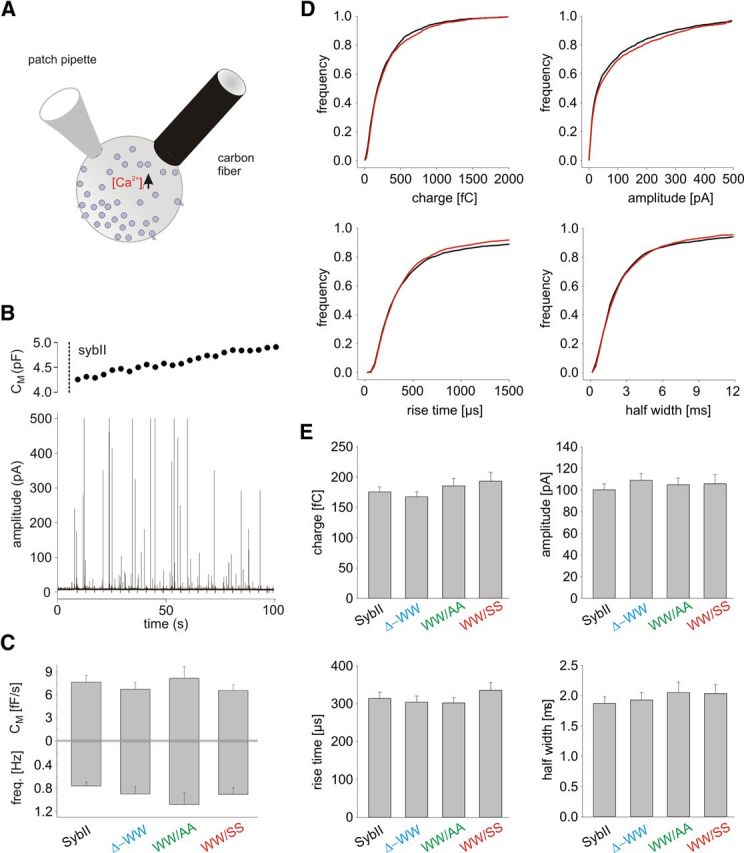
Catecholamine release from chromaffin granules is unchanged in TRP mutants. A, Schematic drawing of the experimental setup. B, Infusion of a dko cell expressing SybII (t = 5 s; cell opening; dashed line) with an intracellular solution containing 19 μm [Ca]i causes a barrage of exocytotic activity as documented by simultaneous amperometric and membrane capacitance recordings. C, Deletion (Δ−WW, n = 18) or substitution of the TRP residues (WW/AA, n = 15; WW/SS, n = 15) changes neither the amperometric event frequency nor the capacitance increase compared with SybII (n = 24). D, Comparison of amperometric spike properties recorded in SybII and WW/SS mutant protein expressing cells. Data are plotted as cumulative frequency distributions for the indicated parameters. E, Loss of the TRP moiety does not change quantal size, peak amplitude, or kinetics of single fusion events. Error bars indicate SEM.
Lipid-binding properties of the juxtamembrane region of SybII
To study whether the extent of Trp-mediated membrane association can be directly related to the degree of vesicle priming, we investigated the ability of wild-type and mutant proteins to interact with phospholipid membranes. For this, the binding properties of the corresponding GST-fusion proteins were analyzed in a liposome aggregation assay (Connell et al., 2008) that relies on the dimeric nature of GST proteins. The addition of recombinant GST-SybII (SybII: amino acids 1–96, cytoplasmic domain) to a mixture of liposomes rapidly induces liposome aggregation, measured as absorbance increase at 350 nm (Fig. 9A–C). In contrast, GST alone causes no aggregation, showing that the lipid-interacting moiety resides in the synaptobrevin molecule. By exchanging both tryptophans to serine residues, binding is strongly diminished, almost comparable with GST alone (Fig. 9A–C). The substitution of single Trp residues also decreases membrane binding, with the W90S mutation being more effective than the W89S substitution. Using the fluorescence emission of Trp residues as an alternative method to measure lipid binding, a similar Trp-dependent membrane association of SybII is observed (Fig. 9E,F). Thus, loss of both Trp residues severely impairs membrane association of the cytoplasmic domain of SybII. Still, the reduction in membrane binding observed for the single Trp mutants is not paralleled by a corresponding decrease of the exocytotic burst component, seen in our flash experiments (Fig. 1). This suggests that Trp-mediated alterations in membrane binding of the cytoplasmic domain of SybII (without TMD) do not occur with full-length SybII under in vivo conditions or are not relevant for vesicle priming.
Figure 9.
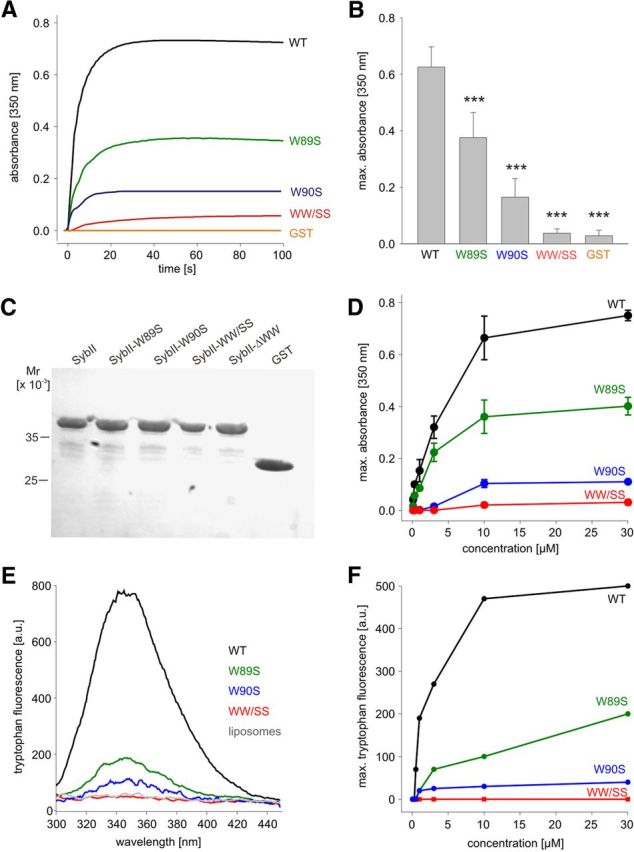
The membrane-proximal tryptophans enable phospholipid binding of the cytoplasmic domain of SybII to synthetic liposomes. A, GST-SybII (1–96 aa; 10 μm; added at t = 0 s) cross-links liposomes, measured as absorbance increase at 350 nm. Single and double TRP mutants show impaired or nearly abolished cross-linking, respectively. Glutathione S-transferase served as control. B, Maximum absorbance increase measured at a concentration of 10 μm for SybII and its mutant variants. Data were collected from nine independent experiments. C, Coomassie-stained polyacrylamide/SDS gel shows the integrity of proteins used in the turbidity assay. Ten microliters of the turbidity assay were analyzed. D, Concentration dependence of absorbance increase for SybII and its mutants. E, Fluorescence emission spectra of tryptophan residues monitored upon mixing GST-SybII 1–96 (10 μm) or its mutant variants with liposomes. Neither protein-free liposomes (gray trace) nor the WW/SS mutant (red trace) displays any significant emission. Note that fluorescence emission increased strongly for the wild-type protein (black trace) but was significantly reduced for the single mutants (green and blue trace). F, Concentration dependence of maximal tryptophan fluorescence emission for GST-SybII and the mutant proteins in the presence of liposomes. The tryptophan emission was corrected for the corresponding protein emission at the given concentration. ***p < 0.001, one-way ANOVA. Error bars indicate SEM.
A role of membrane-proximal Trp residues in controlling the position of the JMD and the electrostatic potential at the membrane–water interface
MD simulations were used to further explore the interplay between Trp residues and the phospholipid bilayer. For this purpose, a model was used wherein SybII molecules comprising the JMD together with the TMD (amino acid residues 85–116) were embedded in an asymmetric membrane bilayer (outer leaflet, PS/PC, 0.25/0.75; inner leaflet, pure PC) that roughly mimics the vesicular membrane environment (Takamori et al., 2006). As shown in Figure 10A, Trp residues reside within the hydrophilic head group region of the lipids consistent with previous electron paramagnetic resonance (EPR) and fluorescence analyses (Kweon et al., 2003; Chen et al., 2004; Bowen and Brunger, 2006), stabilized mainly by interactions between the Trp indole nitrogen and the lipid carbonyl and phosphate groups. In the case of the WW/AA mutant, however, the peptide is drawn into the membrane interior by ∼7 Å. This enables favorable contacts of the hydrophobic side chain of the lysine residues with the hydrocarbon chain of the phospholipid as well as favorable coulombic interactions of the “snorkeling” amine groups with the lipid polar head groups. Both Trp and Lys side chains prefer a head group proximal positioning inside the membrane (with free energies of −23 and −20 kJ/mol for Trp and Lys, respectively, at ∼1.3 nm from the membrane center for DOPC lipids), as shown previously (MacCallum et al., 2008). The WW/AA mutation thereby allows for an energetically more favorable positioning of the proximal lysine residues similar to what has been observed in simulations of syntaxin 1A missing a Trp motif next to the TM region (Knecht and Grubmüller, 2003). It should be noted that experimentally determined transfer free energies for partitioning of serine and alanine from water into the bilayer (interface or its interior) are very similar in contrast to that of tryptophan (e.g., water to interface in kilocalories/mole: ΔGSer, 0.13; ΔGAla, 0.17; ΔGTrp, −1.85) (Wimley and White, 1996). It is therefore likely that both types of double mutations lead to a similar positioning of the JMD of SybII with respect to the membrane–water interface, which agrees well with our functional results (Figs. 1, 2).
The repositioning of the basic residues is clearly seen in the shift of the density profile of the amine groups (e.g., K85/K87/K91/K94) plotted as a function of the distance to the membrane surface (Fig. 10B). The membrane insertion of the lysines is accompanied by an N-terminal extension of the transmembrane helix by 4–5 aa. As a consequence of the repositioning of the lysine residues, the entire JMD and the N-terminal part of the TMD moves toward the membrane center, whereas the position of the C-terminal end of the TMD remains unchanged (Fig. 10C). In contrast, the exchange of a single Trp residue results only in a comparably small shift of the amine density (Fig. 10B,C). As shown in Figure 10C, inset, diamonds, the overall position of the simulated wild-type protein within the membrane agrees well with experimental EPR results (Kweon et al., 2003) and with previous coarse-grained simulations (Durrieu et al., 2009). Notably, the Trp mutants compromise membrane association of the JMD in the biochemical experiment (Fig. 7) but not in the MD simulation. This is likely due to the stabilization of the membrane attachment of the JMD by the TMD in the MD simulation, emphasizing the necessity to study protein–lipid interaction in combination with the adjacent TMD.
For the wild-type protein, the positively charged JMD residues at the membrane–water interface establish a localized, positive electrostatic potential just above the TMD (Fig. 11) (for details of calculation, see Materials and Methods). The remaining membrane surface exhibits a net negative potential due to the content of negatively charged phospholipids (PS) in the outer membrane leaflet. The inhomogeneities of the negative surface potential are caused by electrostatic interactions between phospholipid head groups and charged SybII residues, in particular with K83 and K85 as well as with the positively charged N terminus of the modeled SybII. The positive surface potential is nearly unchanged for the single mutants (W89A and W90A) but vanished for the Trp double substitution. These results agree well with the altered position of the lysine residues relative to the membrane–water interface (Fig. 10B). The altered positioning of the lysine side chains (K83, K85) can also be seen in the density map of SybII positions plotted in the membrane plane (Fig. 11B). For the wild-type protein and the Trp single mutants, K83 and K85 (orange) are oriented toward the N terminus of the JMD (A74, magenta) but have an opposite orientation for the double mutant.
In summary, the lipid-anchored Trp residues of SybII have a profound impact on the JMD position and the electrostatic potential established at the membrane surface.
Discussion
Release of neurotransmitter molecules requires docking, priming, and fusion of secretory vesicles with the plasma membrane. While the strict requirement of SNARE proteins for exocytosis has been demonstrated beyond any reasonable doubt, the precise mechanisms by which SNARE proteins guide the vesicle throughout these stages has remained largely unclear. In particular, the interplay between proteins and lipids that should accompany the interactions between cognate SNARE partners is not understood.
In this work, we have studied how the membranophile Trp residues within the juxtamembrane region of SybII determine the secretory behavior of chromaffin cells. We found that deletion or substitution of these Trp residues reduces the pool size of primed chromaffin granules, but leaves the kinetics of stimulus–secretion coupling unchanged. The reduction in size of RRP and SRP is most likely due to an instability of highly primed vesicles, since the sustained release component that reflects forward directed priming (Voets, 2000; Rettig and Neher, 2002) is unaffected. Furthermore, we show that the mutated SybII variant is properly localized and sorted to secretory granules with similar efficiency as the SybII protein using structured illumination microscopy (Fig. 3).
Interestingly, loss of the Trp moiety does not alter the rate of v-SNARE-dependent exocytosis at submicromolar concentrations of [Ca2+]i providing no support for the notion that Trp residues in SybII act as a fusion clamp in the exocytotic process (Maximov et al., 2009). Even in resting chromaffin cell, an increase in exocytotic activity with expression of the TRP mutant protein was not detectable. Thus, TRP-mediated “clamping” of exocytosis does not seem to be a molecular concept that can be generalized for SNARE-mediated fusion processes. In close correlation, TIRF microscopy revealed that the number of membrane-proximal vesicles is unchanged in mutant cells (Fig. 4). This confirms the view that the observed reduction in EB size is not simply the consequence of tonically enhanced release leading to pool depletion at resting conditions.
Deletion or substitution of the WW residues does not change amplitude or kinetics of single fusion events, consistent with a specific requirement of the JMD of SybII for prefusional stages like priming. Moreover, we show that Trp-dependent positioning of membrane-proximal lysine residues allows for the buildup of a positive electrostatic potential at the membrane surface, which may locally reduce repulsive forces between negative surface potentials of the opposing membranes. We propose a model wherein electrostatic interactions between SybII and the membrane pave the way for priming of secretory vesicles.
Priming of secretory vesicles depends on protein–lipid interactions of SybII
It is a widely accepted notion that trans-SNARE complexes start to assemble by pairing v-SNAREs and target SNAREs from their N-terminal ends. Complex formation then progresses in a “zipper”-like fashion toward the C-terminal TMD. Since the C-terminal portion of the SNARE motif of SybII has a lower propensity to form an α-helix, folding of the trans-SNARE complex may transiently stop or slow down (Ellena et al., 2009). Such partially assembled SNARE complexes may provide the stage for priming of secretory vesicles that await the Ca2+ stimulus and then rapidly fuse in response to the triggering signal (Sørensen et al., 2006). Intriguingly, our results show that a doublet of Trp residues located at the very C-terminal end of the cytoplasmic domain of SybII facilitates exocytosis competence and priming stability of secretory vesicles, while these processes have previously been assigned to N-terminal portions of the SNARE proteins (Borisovska et al., 2005; Sørensen et al., 2006; Wadel et al., 2007; Walter et al., 2010). Thus, even early stages of the exocytotic process like priming of vesicles depend on structural properties of the membrane-proximal part of SybII. In this context, it is important to emphasize that, contrary to initial reports (Kweon et al., 2003), neither kinetics nor efficiency of SNARE complex formation is affected by Trp mutations in SybII (Siddiqui et al., 2007; Maximov et al., 2009; Hernandez et al., 2012).
Using MD simulations of SybII in a membranous environment, we find that the Trp residues determine the insertion depth of the flanking, basic residues into the membrane and by this the buildup of a local, positive electrostatic potential at the site of fusion. Upon removal of a single Trp residue, the positions of the membrane-proximal positive charges remain largely unchanged but are strongly altered when both TRP residues are substituted (Fig. 11A). This finding agrees well with our physiological observations, showing that only substitution of both Trp residues efficiently impairs the exocytotic response (Fig. 1). Thus, it seems likely that the precise arrangement of the juxtamembrane basic residues by neighboring tryptophans assists partially assembled trans-SNARE complexes in maintaining exocytosis competence, possibly by establishing a configuration that makes the unpriming reaction energetically more costly.
A local, positive membrane surface potential, as generated in the presence of the Trp residues, may reduce repulsive forces between the opposing negatively charged membranes in a “sandwich”-like fashion. This result is compatible with recent observations by Davletov and colleagues (Darios et al., 2009), showing that positively charged membrane lipids like sphingosine enhance exocytosis competence of synaptic vesicles. Furthermore, it agrees with results indicating that reduction of the net positive charge within the JMD of SybII or Syntaxin 1A inhibits secretion in β-cells and PC12 cells, respectively (Lam et al., 2008; Williams et al., 2009). Assuming a model with three SybII molecules (Domanska et al., 2010; Mohrmann et al., 2010), organized in a rosette-like structure with the TMDs pointing toward the center of the contact area (diameter, 7 nm), we estimate that the Trp-dependent positioning of positively charged residues between membranes generates an attractive potential of ∼15 kJ/mol at 1 nm intermembranous distance (considering five TMD proximal basic residues) (see Materials and Methods). The repositioning of the basic amino acids in the absence of the tryptophans diminishes this potential by ∼10 kJ/mol (=4 kBT) and may thus significantly decrease the electrostatic stabilization of the primed state.
In the context of trans-SNARE complex formation, the polybasic juxtamembrane region of Syntaxin can act synergistically in generating a local positive surface potential at the plasma membrane. Indeed, scaffolding linker regions of the SNARE proteins at the site of fusion could initiate an electrostatic tunnel that is extended by Ca2+-dependent changes in the C2 domains of Synaptotagmin (Araç et al., 2006; Bhalla et al., 2006; Zimmerberg et al., 2006; Martens and McMahon, 2008; Hui et al., 2009).
Trp residues within the JMD of SybII have further been implicated in the dimerization of SNARE complexes (Fdez et al., 2008) and the binding to the c-subunit of the V-ATPase (Di Giovanni et al., 2010). Yet, in both studies, even substitution of a single Trp residue was sufficient to impair the interaction, which renders it unlikely that these mechanisms are responsible for impaired granule priming. Recent coarse-grained modeling of SNARE-mediated fusion events also suggested that progression of SNARE zipping generates mechanical stress that can be stored in the linker region and thereby controls the self-organization of trans-SNARE complexes at a certain distance from the contact site between opposing membranes (Risselada et al., 2011). Thus, changes in the position of the JMD per se (Fig. 10) may be suitable to interfere with granule priming by changing the self-organization of the SNARE complexes relative to the fusion site.
Regardless of the exact underlying mechanism, our results suggest that the interplay between the JMD of SybII and lipids controls the JMD position and generates a positive membrane surface potential, which both may assist in priming of secretory vesicles.
In the context of the force-transmitting properties of the JMD, it stands to reason that amino acid insertions upstream of the TRP residues interfere more strongly with the secretion than similarly long downstream insertions (Fig. 7). While both types of mutations indicate a stringent length requirement of the JMD, upstream insertions might be more effective due to perturbation of an α-helical structure proposed for this region of membrane-bound SybII (amino acid position 77–88) (Ellena et al., 2009). Alternatively, one might speculate that upstream mutations interfere in addition with binding of auxiliary proteins, like complexin (Reim et al., 2001), to the SNARE complex.
In summary, the JMD of SybII establishes both a tight molecular link between the SNARE motif and the TMD as well as a local electrostatic potential at the membrane surface that are crucial for vesicle priming. Given these findings, we propose a model wherein N-terminal interactions between the SNARE proteins together with protein–lipid interactions of the cytosolic domain of SybII at its C terminus promote priming of vesicles.
Notes
Supplemental material on amperometric data presented in this paper is available at http://physiology.uni-saarland.de/Dieter_Bruns/Bruns_Publications.html. This material has not been peer reviewed.
Footnotes
This work was supported by Deutsche Forschungsgemeinschaft Grants SFB894 and GK1326 (D.B., R.A.B.) and by HOMFOR. D.B. and R.A.B. acknowledge support by the Ministry for Economy and Science of the Saarland, Germany. We are grateful to Dr. J. Rettig, Dr. D. Stevens, and S. Bruns-Engers for valuable discussions. We thank K. Klingler, R. Dufke, and M. Wirth for excellent technical assistance, and Dr. E. Krause for help with the structured illumination microscopy.
References
- Araç D, Chen X, Khant HA, Ubach J, Ludtke SJ, Kikkawa M, Johnson AE, Chiu W, Südhof TC, Rizo J. Close membrane-membrane proximity induced by Ca2+-dependent multivalent binding of synaptotagmin-1 to phospholipids. Nat Struct Mol Biol. 2006;13:209–217. doi: 10.1038/nsmb1056. [DOI] [PubMed] [Google Scholar]
- Ashery U, Betz A, Xu T, Brose N, Rettig J. An efficient method for infection of adrenal chromaffin cells using the Semliki Forest virus gene expression system. Eur J Cell Biol. 1999;78:525–532. doi: 10.1016/s0171-9335(99)80017-x. [DOI] [PubMed] [Google Scholar]
- Augustine GJ, Neher E. Calcium requirements for secretion in bovine chromaffin cells. J Physiol. 1992;450:247–271. doi: 10.1113/jphysiol.1992.sp019126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berendsen HJC, Postma JPM, Vangunsteren WF, Dinola A, Haak JR. Molecular dynamics with coupling to an external bath. J Chem Phys. 1984;81:3684–3690. [Google Scholar]
- Berger O, Edholm O, Jähnig F. Molecular dynamics simulations of a fluid bilayer of dipalmitoylphosphatidylcholine at full hydration, constant pressure, and constant temperature. Biophys J. 1997;72:2002–2013. doi: 10.1016/S0006-3495(97)78845-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhalla A, Chicka MC, Tucker WC, Chapman ER. Ca2+-synaptotagmin directly regulates t-SNARE function during reconstituted membrane fusion. Nat Struct Mol Biol. 2006;13:323–330. doi: 10.1038/nsmb1076. [DOI] [PubMed] [Google Scholar]
- Böckmann RA, Hac A, Heimburg T, Grubmüller H. Effect of sodium chloride on a lipid bilayer. Biophys J. 2003;85:1647–1655. doi: 10.1016/S0006-3495(03)74594-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolte S, Cordelières FP. A guided tour into subcellular colocalization analysis in light microscopy. J Microsc. 2006;224:213–232. doi: 10.1111/j.1365-2818.2006.01706.x. [DOI] [PubMed] [Google Scholar]
- Borisovska M, Zhao Y, Tsytsyura Y, Glyvuk N, Takamori S, Matti U, Rettig J, Südhof T, Bruns D. v-SNAREs control exocytosis of vesicles from priming to fusion. EMBO J. 2005;24:2114–2126. doi: 10.1038/sj.emboj.7600696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowen M, Brunger AT. Conformation of the synaptobrevin transmembrane domain. Proc Natl Acad Sci U S A. 2006;103:8378–8383. doi: 10.1073/pnas.0602644103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruns D. Detection of transmitter release with carbon fiber electrodes. Methods. 2004;33:312–321. doi: 10.1016/j.ymeth.2004.01.004. [DOI] [PubMed] [Google Scholar]
- Chen Y, Xu Y, Zhang F, Shin YK. Constitutive versus regulated SNARE assembly: a structural basis. EMBO J. 2004;23:681–689. doi: 10.1038/sj.emboj.7600083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connell E, Scott P, Davletov B. Real-time assay for monitoring membrane association of lipid-binding domains. Anal Biochem. 2008;377:83–88. doi: 10.1016/j.ab.2008.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darden T, York D, Pedersen L. Particle mesh Ewald—an N.log(N) method for Ewald sums in large systems. J Chem Phys. 1993;98:10089–10092. [Google Scholar]
- Darios F, Wasser C, Shakirzyanova A, Giniatullin A, Goodman K, Munoz-Bravo JL, Raingo J, Jorgacevski J, Kreft M, Zorec R, Rosa JM, Gandia L, Gutiérrez LM, Binz T, Giniatullin R, Kavalali ET, Davletov B. Sphingosine facilitates SNARE complex assembly and activates synaptic vesicle exocytosis. Neuron. 2009;62:683–694. doi: 10.1016/j.neuron.2009.04.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deák F, Shin OH, Kavalali ET, Südhof TC. Structural determinants of synaptobrevin 2 function in synaptic vesicle fusion. J Neurosci. 2006;26:6668–6676. doi: 10.1523/JNEUROSCI.5272-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Giovanni J, Boudkkazi S, Mochida S, Bialowas A, Samari N, Lévêque C, Youssouf F, Brechet A, Iborra C, Maulet Y, Moutot N, Debanne D, Seagar M, El Far O. V-ATPase membrane sector associates with synaptobrevin to modulate neurotransmitter release. Neuron. 2010;67:268–279. doi: 10.1016/j.neuron.2010.06.024. [DOI] [PubMed] [Google Scholar]
- Domanska MK, Kiessling V, Tamm LK. Docking and fast fusion of synaptobrevin vesicles depends on lipid compositions of the vesicle and the acceptor SNARE complex-containing target membrane. Biophys J [Erratum (2011) 100:269] 2010;99:2936–2946. doi: 10.1016/j.bpj.2010.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durrieu MP, Bond PJ, Sansom MS, Lavery R, Baaden M. Coarse-grain simulations of the R-SNARE fusion protein in its membrane environment detect long-lived conformational sub-states. Chemphyschem. 2009;10:1548–1552. doi: 10.1002/cphc.200900216. [DOI] [PubMed] [Google Scholar]
- Elferink LA, Trimble WS, Scheller RH. Two vesicle-associated membrane protein genes are differentially expressed in the rat central nervous system. J Biol Chem. 1989;264:11061–11064. [PubMed] [Google Scholar]
- Ellena JF, Liang B, Wiktor M, Stein A, Cafiso DS, Jahn R, Tamm LK. Dynamic structure of lipid-bound synaptobrevin suggests a nucleation-propagation mechanism for trans-SNARE complex formation. Proc Natl Acad Sci U S A. 2009;106:20306–20311. doi: 10.1073/pnas.0908317106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fdez E, Jowitt TA, Wang MC, Rajebhosale M, Foster K, Bella J, Baldock C, Woodman PG, Hilfiker S. A role for soluble N-ethylmaleimide-sensitive factor attachment protein receptor complex dimerization during neurosecretion. Mol Biol Cell. 2008;19:3379–3389. doi: 10.1091/mbc.E08-01-0010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fulop T, Radabaugh S, Smith C. Activity-dependent differential transmitter release in mouse adrenal chromaffin cells. J Neurosci. 2005;25:7324–7332. doi: 10.1523/JNEUROSCI.2042-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gustafsson MG. Surpassing the lateral resolution by a factor of two using structured illumination microscopy. J Microsc. 2000;198:82–87. doi: 10.1046/j.1365-2818.2000.00710.x. [DOI] [PubMed] [Google Scholar]
- Guzman RE, Schwarz YN, Rettig J, Bruns D. SNARE force synchronizes synaptic vesicle fusion and controls the kinetics of quantal synaptic transmission. J Neurosci. 2010;30:10272–10281. doi: 10.1523/JNEUROSCI.1551-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hannah MJ, Weiss U, Huttner WB. Differential extraction of proteins from paraformaldehyde-fixed cells: lessons from synaptophysin and other membrane proteins. Methods. 1998;16:170–181. doi: 10.1006/meth.1998.0664. [DOI] [PubMed] [Google Scholar]
- Hassdenteufel S, Schäuble N, Cassella P, Leznicki P, Müller A, High S, Jung M, Zimmermann R. Ca2+-calmodulin inhibits tail-anchored protein insertion into the mammalian endoplasmic reticulum membrane. FEBS Lett. 2011;585:3485–3490. doi: 10.1016/j.febslet.2011.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernandez JM, Stein A, Behrmann E, Riedel D, Cypionka A, Farsi Z, Walla PJ, Raunser S, Jahn R. Membrane fusion intermediates via directional and full assembly of the SNARE complex. Science. 2012;336:1581–1584. doi: 10.1126/science.1221976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hess B, Bekker H, Berendsen HJC, Fraaije JGEM. LINCS: a linear constraint solver for molecular simulations. J Comput Chem. 1997;18:1463–1472. [Google Scholar]
- Hess B, Kutzner C, van der Spoel D, Lindahl E. GROMACS 4: algorithms for highly efficient, load-balanced, and scalable molecular simulation. J Chem Theory Comput. 2008;4:435–447. doi: 10.1021/ct700301q. [DOI] [PubMed] [Google Scholar]
- Hu K, Carroll J, Fedorovich S, Rickman C, Sukhodub A, Davletov B. Vesicular restriction of synaptobrevin suggests a role for calcium in membrane fusion. Nature. 2002;415:646–650. doi: 10.1038/415646a. [DOI] [PubMed] [Google Scholar]
- Hui E, Johnson CP, Yao J, Dunning FM, Chapman ER. Synaptotagmin-mediated bending of the target membrane is a critical step in Ca2+-regulated fusion. Cell. 2009;138:709–721. doi: 10.1016/j.cell.2009.05.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jahn R, Scheller RH. SNAREs—engines for membrane fusion. Nat Rev Mol Cell Biol. 2006;7:631–643. doi: 10.1038/nrm2002. [DOI] [PubMed] [Google Scholar]
- James DJ, Khodthong C, Kowalchyk JA, Martin TF. Phosphatidylinositol 4,5-bisphosphate regulates SNARE-dependent membrane fusion. J Cell Biol. 2008;182:355–366. doi: 10.1083/jcb.200801056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kesavan J, Borisovska M, Bruns D. v-SNARE actions during Ca2+-triggered exocytosis. Cell. 2007;131:351–363. doi: 10.1016/j.cell.2007.09.025. [DOI] [PubMed] [Google Scholar]
- Knecht V, Grubmüller H. Mechanical coupling via the membrane fusion SNARE protein syntaxin 1A: a molecular dynamics study. Biophys J. 2003;84:1527–1547. doi: 10.1016/S0006-3495(03)74965-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kweon DH, Kim CS, Shin YK. Regulation of neuronal SNARE assembly by the membrane. Nat Struct Biol. 2003;10:440–447. doi: 10.1038/nsb928. [DOI] [PubMed] [Google Scholar]
- Lam AD, Tryoen-Toth P, Tsai B, Vitale N, Stuenkel EL. SNARE-catalyzed fusion events are regulated by Syntaxin1A-lipid interactions. Mol Biol Cell. 2008;19:485–497. doi: 10.1091/mbc.E07-02-0148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leventis PA, Grinstein S. The distribution and function of phosphatidylserine in cellular membranes. Annu Rev Biophys. 2010;39:407–427. doi: 10.1146/annurev.biophys.093008.131234. [DOI] [PubMed] [Google Scholar]
- Li F, Pincet F, Perez E, Eng WS, Melia TJ, Rothman JE, Tareste D. Energetics and dynamics of SNAREpin folding across lipid bilayers. Nat Struct Mol Biol. 2007;14:890–896. doi: 10.1038/nsmb1310. [DOI] [PubMed] [Google Scholar]
- MacCallum JL, Bennett WF, Tieleman DP. Distribution of amino acids in a lipid bilayer from computer simulations. Biophys J. 2008;94:3393–3404. doi: 10.1529/biophysj.107.112805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martens S, McMahon HT. Mechanisms of membrane fusion: disparate players and common principles. Nat Rev Mol Cell Biol. 2008;9:543–556. doi: 10.1038/nrm2417. [DOI] [PubMed] [Google Scholar]
- Maximov A, Tang J, Yang X, Pang ZP, Südhof TC. Complexin controls the force transfer from SNARE complexes to membranes in fusion. Science. 2009;323:516–521. doi: 10.1126/science.1166505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMahon HT, Ushkaryov YA, Edelmann L, Link E, Binz T, Niemann H, Jahn R, Südhof TC. Cellubrevin is a ubiquitous tetanus toxin substrate homologous to a putative synaptic vesicle fusion protein. Nature. 1993;364:346–349. doi: 10.1038/364346a0. [DOI] [PubMed] [Google Scholar]
- Miyamoto S, Kollman PA. Settle—an analytical version of the shake and rattle algorithm for rigid water models. J Comput Chem. 1992;13:952–962. [Google Scholar]
- Mohrmann R, de Wit H, Verhage M, Neher E, Sørensen JB. Fast vesicle fusion in living cells requires at least three SNARE complexes. Science. 2010;330:502–505. doi: 10.1126/science.1193134. [DOI] [PubMed] [Google Scholar]
- Oostenbrink C, Villa A, Mark AE, van Gunsteren WF. A biomolecular force field based on the free enthalpy of hydration and solvation: the GROMOS force-field parameter sets 53A5 and 53A6. J Comput Chem. 2004;25:1656–1676. doi: 10.1002/jcc.20090. [DOI] [PubMed] [Google Scholar]
- Pasche M, Matti U, Hof D, Rettig J, Becherer U. Docking of LDCVs is modulated by lower intracellular [Ca2+] than priming. PLoS One. 2012;7:e36416. doi: 10.1371/journal.pone.0036416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quetglas S, Leveque C, Miquelis R, Sato K, Seagar M. Ca2+-dependent regulation of synaptic SNARE complex assembly via a calmodulin- and phospholipid-binding domain of synaptobrevin. Proc Natl Acad Sci U S A. 2000;97:9695–9700. doi: 10.1073/pnas.97.17.9695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reim K, Mansour M, Varoqueaux F, McMahon HT, Südhof TC, Brose N, Rosenmund C. Complexins regulate a late step in Ca2+-dependent neurotransmitter release. Cell. 2001;104:71–81. doi: 10.1016/s0092-8674(01)00192-1. [DOI] [PubMed] [Google Scholar]
- Rettig J, Neher E. Emerging roles of presynaptic proteins in Ca++-triggered exocytosis. Science. 2002;298:781–785. doi: 10.1126/science.1075375. [DOI] [PubMed] [Google Scholar]
- Risselada HJ, Kutzner C, Grubmüller H. Caught in the act: visualization of SNARE-mediated fusion events in molecular detail. Chembiochem. 2011;12:1049–1055. doi: 10.1002/cbic.201100020. [DOI] [PubMed] [Google Scholar]
- Schoch S, Deák F, Königstorfer A, Mozhayeva M, Sara Y, Südhof TC, Kavalali ET. SNARE function analyzed in synaptobrevin/VAMP knockout mice. Science. 2001;294:1117–1122. doi: 10.1126/science.1064335. [DOI] [PubMed] [Google Scholar]
- Siddiqui TJ, Vites O, Stein A, Heintzmann R, Jahn R, Fasshauer D. Determinants of synaptobrevin regulation in membranes. Mol Biol Cell. 2007;18:2037–2046. doi: 10.1091/mbc.E07-01-0049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siu SW, Böckmann RA. Electric field effects on membranes: Gramicidin A as a test ground. J Struct Biol. 2007;157:545–556. doi: 10.1016/j.jsb.2006.10.005. [DOI] [PubMed] [Google Scholar]
- Sørensen JB, Nagy G, Varoqueaux F, Nehring RB, Brose N, Wilson MC, Neher E. Differential control of the releasable vesicle pools by SNAP-25 splice variants and SNAP-23. Cell. 2003;114:75–86. doi: 10.1016/s0092-8674(03)00477-x. [DOI] [PubMed] [Google Scholar]
- Sørensen JB, Wiederhold K, Müller EM, Milosevic I, Nagy G, de Groot BL, Grubmüller H, Fasshauer D. Sequential N- to C-terminal SNARE complex assembly drives priming and fusion of secretory vesicles. EMBO J. 2006;25:955–966. doi: 10.1038/sj.emboj.7601003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Speidel D, Bruederle CE, Enk C, Voets T, Varoqueaux F, Reim K, Becherer U, Fornai F, Ruggieri S, Holighaus Y, Weihe E, Bruns D, Brose N, Rettig J. CAPS1 regulates catecholamine loading of large dense-core vesicles. Neuron. 2005;46:75–88. doi: 10.1016/j.neuron.2005.02.019. [DOI] [PubMed] [Google Scholar]
- Sutton RB, Fasshauer D, Jahn R, Brunger AT. Crystal structure of a SNARE complex involved in synaptic exocytosis at 2.4 Å resolution. Nature. 1998;395:347–353. doi: 10.1038/26412. [DOI] [PubMed] [Google Scholar]
- Takamori S, Holt M, Stenius K, Lemke EA, Grønborg M, Riedel D, Urlaub H, Schenck S, Brügger B, Ringler P, Müller SA, Rammner B, Gräter F, Hub JS, De Groot BL, Mieskes G, Moriyama Y, Klingauf J, Grubmüller H, Heuser J, et al. Molecular anatomy of a trafficking organelle. Cell. 2006;127:831–846. doi: 10.1016/j.cell.2006.10.030. [DOI] [PubMed] [Google Scholar]
- Voets T. Dissection of three Ca2+-dependent steps leading to secretion in chromaffin cells from mouse adrenal slices. Neuron. 2000;28:537–545. doi: 10.1016/s0896-6273(00)00131-8. [DOI] [PubMed] [Google Scholar]
- Vriend G. WHAT IF—a molecular modeling and drug design program. J Mol Graphics. 1990;8:52–56. doi: 10.1016/0263-7855(90)80070-v. [DOI] [PubMed] [Google Scholar]
- Wadel K, Neher E, Sakaba T. The coupling between synaptic vesicles and Ca2+ channels determines fast neurotransmitter release. Neuron. 2007;53:563–575. doi: 10.1016/j.neuron.2007.01.021. [DOI] [PubMed] [Google Scholar]
- Walter AM, Wiederhold K, Bruns D, Fasshauer D, Sørensen JB. Synaptobrevin N-terminally bound to syntaxin-SNAP-25 defines the primed vesicle state in regulated exocytosis. J Cell Biol. 2010;188:401–413. doi: 10.1083/jcb.200907018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams D, Vicôgne J, Zaitseva I, McLaughlin S, Pessin JE. Evidence that electrostatic interactions between vesicle-associated membrane protein 2 and acidic phospholipids may modulate the fusion of transport vesicles with the plasma membrane. Mol Biol Cell. 2009;20:4910–4919. doi: 10.1091/mbc.E09-04-0284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wimley WC, White SH. Experimentally determined hydrophobicity scale for proteins at membrane interfaces. Nat Struct Biol. 1996;3:842–848. doi: 10.1038/nsb1096-842. [DOI] [PubMed] [Google Scholar]
- Zimmerberg J, Akimov SA, Frolov V. Synaptotagmin: fusogenic role for calcium sensor? Nat Struct Mol Biol. 2006;13:301–303. doi: 10.1038/nsmb0406-301. [DOI] [PubMed] [Google Scholar]