

Abstract
Central neural plasticity plays a key role in pain hypersensitivity. This process is modulated by brain-derived neurotrophic factor (BDNF) and also involves the type 1a acid-sensing ion channel (ASIC1a). However, the interactions between the BDNF receptor, tropomyosin-related kinase B (TrkB), and ASIC1a are unclear. Here, we show that deletion of ASIC1 gene suppressed the sustained mechanical hyperalgesia induced by intrathecal BDNF application in mice. In both rat spinal dorsal horn neurons and heterologous cell cultures, the BDNF/TrkB pathway enhanced ASIC1a currents via phosphoinositide 3-kinase (PI3K)-protein kinase B (PKB/Akt) cascade and phosphorylation of cytoplasmic residue Ser-25 of ASIC1a, resulting in enhanced forward trafficking and increased surface expression. Moreover, in both rats and mice, this enhanced ASIC1a activity was required for BDNF-mediated hypersensitivity of spinal dorsal horn nociceptive neurons and central mechanical hyperalgesia, a process that was abolished by intrathecal application of a peptide representing the N-terminal region of ASIC1a encompassing Ser-25. Thus, our results reveal a novel mechanism underlying central sensitization and pain hypersensitivity, and reinforce the critical role of ASIC1a channels in these processes.
Introduction
Chronic pain is an expression of neuronal plasticity, which is mediated in part by activity-dependent increase of nociceptive neuron excitability and synaptic transmission in the spinal dorsal horn (SDH) (Woolf and Salter, 2000; Ji et al., 2003; Basbaum et al., 2009; Sandkühler, 2009). The molecular mechanisms underlying this nociceptive plasticity are not fully understood, but the neurotrophin, brain-derived neurotrophic factor (BDNF), and its receptor, tropomyosin-related kinase B (TrkB), have been implicated in the development of spinal central sensitization that underlies persistent pain (Pezet and McMahon, 2006; Merighi et al., 2008). Although the precise cellular processes of BDNF/TrkB signaling-mediated central sensitization remain unclear, a number of mechanisms have been proposed, including regulation of gene transcription and phosphorylation-dependent modulation of ion channels (Pezet and McMahon, 2006; Merighi et al., 2008). In particular, abundant preclinical evidence indicates that NMDA receptors (NMDARs) are crucial to central sensitization (Petrenko et al., 2003; Latremoliere and Woolf, 2009; Kuner, 2010), and BDNF sensitizes the synaptic response of NMDARs in the SDH via activation of protein kinase C (PKC) or extracellular signal-regulated kinase (Pezet et al., 2002) during persistent pain (Garraway et al., 2003; Slack et al., 2004; Zhao et al., 2006). However, clinical use of NMDAR blockers is limited by side effects resulting from suppression of the physiological functions of these receptors (Woolf, 2010).
Acid-sensing ion channels (ASICs) belong to the degenerin/epithelial Na+ channel family (Waldmann et al., 1997a). To date, at least six ASIC subunits have been identified: 1a, 1b, 2a, 2b, 3, and 4 (Krishtal, 2003). At the peripheral level, ASIC3 is important for inflammatory pain via sensing tissue acidosis (Waldmann et al., 1997b; Deval et al., 2008). ASIC3 expression and activity are enhanced by several proinflammatory mediators, including bradykinin, serotonin, nerve growth factor, and glial cell line-derived neurotrophic factor, that sensitize nociceptors (Voilley et al., 2001). At the central level, ASIC1a is the major component of ASICs and is required for acid-evoked currents in central neurons, where it contributes to neuronal excitability, synaptic plasticity, and dendritic spine development (Wemmie et al., 2002, 2004; Zha et al., 2006; Petroff et al., 2008). In the SDH, elevated ASIC1a activity has been shown to play a role in central sensitization and pain hypersensitivity (Duan et al., 2007; Mazzuca et al., 2007; Xu and Duan, 2009). However, a possible interplay between ASIC1a channels and BDNF in the development of central sensitization and pain hypersensitivity has not been investigated. In the present study, we have examined the interaction between BDNF/TrkB signaling and ASIC1a channel function, as well as its significance in central sensitization using in vitro and in vivo approaches. We found that BDNF sensitizes ASIC1a function through enhancing its surface expression and activity via the downstream phosphoinositide 3-kinase (PI3K)-protein kinase B (PKB/Akt) cascade. This study uncovers a novel mechanism underlying central sensitization and pain hypersensitivity, which involves both BDNF/TrKB pathway and ASIC1a channels, underpinning the critical role of ASIC1a channels in these processes.
Materials and Methods
Animals.
The experimental protocols were approved by the Institutional Animal Care and Use Committee of Institute of Neuroscience, Shanghai, China. All behavioral measurements were done in male, awake, unrestrained Sprague Dawley (SD) rats (∼200 g) or C57BL/6J mice (2–3 months old). Adult BDNF+/− and ASIC1−/− mice (2–3 months old) were prepared as previously described (Liebl et al., 1997; Wemmie et al., 2002). All animals were kept on a 12/12 h light/dark cycle at 22°C with food and water available ad libitum. All experimental manipulations were performed during the light-on phase of the cycle in accordance with institutional guidelines. For intrathecal injection of rats, a polyethylene 10 (PE 10) catheter was implanted at L3–L5 spinal level after the rats were anesthetized with sodium pentobarbital (50 mg/kg, i.p. injection). Intrathecal injection of mice was performed as described by Hylden and Wilcox (1980) using a 50 μl Hamilton syringe with a 22-guage needle. The needle was inserted into the intervertebral space between the L5 and L6 level of the spinal cord. A reflexive flick of the tail was considered to be a sign of the accuracy of each injection. The volume of intrathecal injection was 10 μl for rat and 5 μl for mice. A TAT-fusion peptide (TAT-Ser-25 peptide, “S25-peptide”: YGRKKRRQRRRAFASSSTLH, GL Biochem) containing the TAT (YGRKKRRQRRR) sequence, a trans-activating domain of HIV protein that can permeate the cell membrane (Schwarze et al., 1999), and its scrambled control Scr-peptide (YGRKKRRQRRRSATLFHSAS) were administered intrathecally. The mass and purity of the peptides were verified by HPLC. Peptides were dissolved in saline solution and injected at 2 mm 1 h before capsaicin treatment in mice. In capsaicin test, 0.06% capsaicin solution (10% Tween 80 in saline; 50 μl for rat, 10 μl for mouse) was injected into the subcutaneous space in the middle of the plantar of right hindpaw as shown in Figure 5C.
Figure 5.
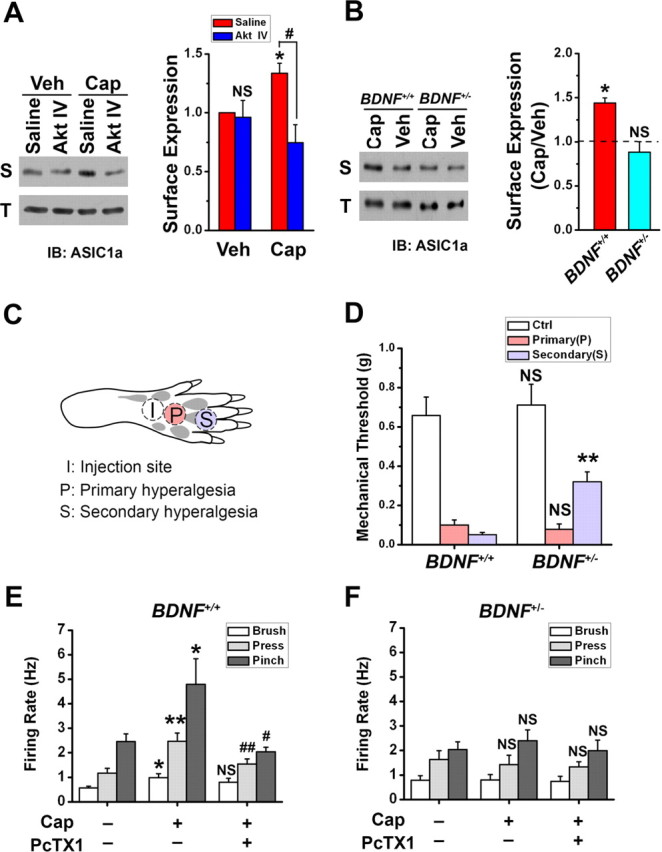
Membrane insertion of ASIC1a triggered by BDNF-Akt pathway contributes to capsaicin-induced secondary mechanical hyperalgesia. A, Akt IV abolished the increase in surface expression (S) of ASIC1a in capsaicin (Cap)-injected rats, but not in vehicle (Veh)-injected rats. The total protein level (T) of ASIC1a in SDH neurons did not change. Shown on the right is a summary (n = 3 in each group). *p < 0.05, Cap versus Veh, by Student's paired t test. NS, Akt IV versus saline in the Veh groups, and #p < 0.05, Akt IV versus saline in the Cap groups, by Student's unpaired t test. B, Representative Western blots showing that intradermal injection of capsaicin induced an increase in surface expression (S), but no change in total protein level (T), of ASIC1a in SDH of wild-type (BDNF+/+) mice. However, the change in surface expression was not detected in BDNF deletion heterozygotes (BDNF+/−). Shown on the right is a summary (dashed line indicates vehicle only; n = 3 in each group). *p < 0.05, by Student's paired t test. C, Diagram showing the sites of capsaicin injection and behavioral test in the hindpaw. Capsaicin was injected into the plantar surface of the right hindpaw (“I”). Mechanical thresholds to von Frey filament stimuli were measured at site “P” for primary hyperalgesia 15 min after capsaicin injection and at site “S” for secondary hyperalgesia 2 h after capsaicin injection. D, Capsaicin-induced mechanical hyperalgesia in the BDNF+/− (n = 9) and wild-type (WT or BDNF+/+, n = 7) littermates. Control (Ctrl) was the mechanical threshold before capsaicin injection. **p < 0.01, versus the corresponding WT, by Student's unpaired t test. E, F, Summary of in vivo single unit recording of WDR neurons in the SDH before and 2 h after capsaicin injection from the BDNF+/+ (E, n = 5) and BDNF+/− mice (F, n = 4), in response to innocuous (brushing) or noxious (pressing and pinching) mechanical stimulation. PcTX1 (10 μm, 10 μl) was directly administered to the dorsal surface of the spinal cord 15 min before recording. *p < 0.05, **p < 0.01, capsaicin (Cap) versus vehicle (no Cap, no PcTX1). #p < 0.05, ##p < 0.01, PcTX1-plus-Cap versus Cap, by Student's unpaired t test.
Behavioral tests.
Animals were assigned in treatment groups in a blinded fashion and pain response was measured in a blinded manner. Rats or mice were habituated and basal pain sensitivity was tested before drug administration or surgery. Mechanical withdrawal threshold on the plantar surface of the hindpaw was measured with a set of von Frey hairs (North Coast Medical Morgan Hill) in the range of 0.008–1.4 g (Duan et al., 2007).
In vivo single unit recordings.
Recordings were performed as previously reported (Duan et al., 2007). In brief, adult male C57BL/6J mice were initially anesthetized with sodium pentobarbital (20 mg/kg, i.p. injection). Laminectomy was performed from T13–L1 vertebrae to expose the lumbar enlargement for spinal wide dynamic range (WDR) neuron recording. Extracellular single unit recordings were made from L4–L5 with glass capillary microelectrodes (10–15 MΩ, filled with 0.5 m sodium acetate). Explorations with microelectrodes were made in the dorsal horn using an electronically controlled microstepping manipulator (Narishige) in 2 μm steps. Experiments were conducted using JL-H2003 amplifier (Jialong) and the SMUP-E software (Fudan University, Shanghai, China). Somatic stimuli were performed with innocuous brush on the skin with a cotton swab, or noxious mild pinching at the hindpaw with calibrated serrated forceps (6 N/mm2). Noxious stimulation was applied sparingly to avoid neuronal sensitization. Spinal neurons that responded with increasing firing rates to stimuli ranging from mild to noxious (brush, pressure, and pinch) were classified as WDR neurons, and were chosen for further analysis. Mechanical stimuli (brush, pressure, and pinch) were applied in ascending order with an interstimulus interval of 30 s. To evaluate the effects of PcTX1 (Peptide Institute, Osaka, Japan) on mechanical stimulation, recordings were performed at 15 min after the drug treatment.
Spinal neuron culture.
The SDH neurons from 15-d-old embryonic SD rats were isolated by a standard enzyme treatment protocol (Duan et al., 2007). Briefly, SDH neurons were dissociated in a Ca2+-free saline with sucrose (20 mm) and plated (1 × 105 cells/ml for electrophysiology and immunocytochemistry; 1 × 106 cells/ml for biochemistry) on poly-d-lysine coated cover glasses. The neurons were grown in DMEM (Invitrogen) with l-glutamine plus 10% fetal bovine serum (FBS, Invitrogen) and 10% F12 nutrient mixtures (Invitrogen). This was replaced by the neurobasal medium (Invitrogen, 2 ml) with 2% B27 serum-free supplements (Invitrogen) 1 d after cell plating. The cultures were maintained at 37°C in a 5% CO2 humidified atmosphere.
Transfection of plasmids.
Transient transfection of Chinese hamster ovary (CHO) cells or cultured SDH neurons was performed using HilyMax liposome transfection reagent (Dojindo Laboratories). When more than one plasmid was co-transfected, the amount of each plasmid was equal. Electrophysiological recordings or immunostainings were performed 16–48 h after transfection. For the GFP-ASIC1a-HA plasmid, the hemagglutinin (HA) epitope (YPYDVPDYA) of influenza virus was inserted in the extracellular loop of human ASIC1a between residues F147 and K148, and GFP was linked at the N terminus (Chen and Gründer, 2007). For ASIC1a-GFP plasmid, GFP was linked at the N terminus of human ASIC1a. The TrkB plasmid was a generous gift from Dr. Muming Poo (Institute of Neuroscience, CAS, Shanghai, China). The DN-Akt and myr-Akt plasmids were kindly provided by Dr. Yi-Zheng Wang (Institute of Neuroscience, CAS).
Electrophysiology.
Whole-cell perforated patch recording was performed using nystatin (0.4 mg/ml), which is modified from a previous study (Xu et al., 1996). Experiments were conducted using Axoclamp 700A with Digidata 1320A and the pClamp 9 software (Molecular Devices). Voltage-clamp experiments were performed. Patch pipettes (4–6 mΩ) were filled with the following (in mm): 120 KCl, 30 NaCl, 0.5 CaCl2, 1 MgCl2, 5 EGTA, 2 MgATP, and 10 HEPES, pH 7.2. The standard extracellular solution contained the following (in mm): 150 NaCl, 5 KCl, 1 MgCl2, 2 CaCl2, 10 glucose, buffered to various pH values with 10 mm HEPES.
Biotinylation assay.
Surface biotinylation was performed on cultured SDH neurons following established protocols (Mammen et al., 1997). Neurons were first stimulated with 20 ng/ml recombinant human BDNF for 5 min at 37°C. Cells were washed three times with an ice-cold PBS+/+ (PBS plus 1 mm MgCl2 and 2.5 mm CaCl2) solution, followed by the addition of 2 ml of 0.25 mg/ml NHS-biotin (Thermo Scientific) in the PBS+/+ solution to each 6 cm dish and incubated at 4°C for 30 min with gentle rocking. Cells were then washed once with the PBS+/+ solution containing 0.1 m glycine to quench the reaction. Total proteins were extracted and incubated overnight at 4°C with NeutrAvidin agarose beads (Thermo Scientific). The beads were washed three times with the PBS+/+ and bound proteins were eluted with the boiling SDS sample buffer for Western blotting.
Biotin endocytosis assays.
Cultured SDH neurons were first stimulated with 20 ng/ml recombinant human BDNF (R&D Systems) for 5 min at 37°C. To measure internalized ASIC1a channels, cultures were incubated with NHS-biotin at 37°C and allowed 3, 5, or 15 min for endocytosis. Surface biotin was stripped off with 50 mm glutathione (20 min; 4°C) and then quenched with 50 mm iodoacetamide (Waung et al., 2008). Neurons were washed in PBS+/+, lysed in RIPA buffer (10 mm Tris, pH 7.5, 300 mm sucrose, 1 mm EDTA, 1 mm sodium orthovanadate, and a protease inhibitor mixture). Ten percent volume of the lysate was removed for total (T) protein measurements, and the rest of the lysate was mixed with 30 μl of Immobilized NeutrAvidin agarose beads by rotating overnight at 4°C. Biotinylated proteins were eluted as described above.
Preparation of spinal dorsal horn membranes.
The SDH samples were homogenized in RIPA and then centrifuged at 7000 × g for 5 min to separate supernatant (S1) from the pellet (P1), which contained nuclei and cell debris and was discarded. A fraction of S1 (5%) was used to determine total protein. The rest of S1 was then centrifuged at 40,000 × g for 30 min to obtain pure cytoplasmic extract in the supernatant (S2) and crude membranes in the pellet (P2), which includes plasma and cellular organelle membranes. P2 was resuspended in PBS with a protease inhibitor mixture and sodium orthovanadate. A BCA Protein Assay kit (Bio-Rad) was used to determine protein concentrations in the total and membrane samples.
Western blotting.
Protein samples from cultured SDH neurons (DIV 10) or SDH homogenates were separated by SDS-PAGE and transferred to PVDF filters (GE Healthcare). The filters were incubated overnight at 4°C with anti-ASIC1a (1:500, Santa Cruz Biotechnology), anti-GFP (1:500, Santa Cruz Biotechnology), anti-transferrin receptor (1:1000, Invitrogen), anti-α tubulin (1:1000, Sigma), anti-Akt (1:1000, Cell Signaling Technology), or anti-Phospho-Akt (1:1000, Cell Signaling Technology) antibodies and finally visualized using an ECL solution (Thermo Scientific) followed by exposure to X-films for 1–3 min.
Immunocytochemistry.
After BDNF stimulation for 5 min, cells were immediately fixed for 20 min with 4% paraformaldehyde/PBS and blocked for 30 min with a PBS solution that contained 10% FBS. Triton X-100 (0.1%) was added to permeabilize the cells where indicated. Cells were incubated overnight with anti-HA antibody (Covance) diluted 1:1000 in 10% FBS/PBS. After rinsing with PBS, the cells were then treated with secondary antibody conjugated to Alexa 594 (1:2000, Invitrogen). To calculate the ratio of HA to GFP fluorescence, we selected the region of interest (ROI) in the GFP image that encompassed the soma of each transfected neuron. The pixel intensity of total fluorescence for each ROI was automatically measured for the GFP and Alexa 594 images and the cellular ratio of HA:GFP was calculated. This value was converted into the surface fraction by referencing to a standard curve constructed from the GFP and HA intensity values in permeabilized cells. For in vivo biotinylation staining, streptavidin-conjugated Alexa 488 (1:2000, Invitrogen) was used. NeuN antibody (1:200, Millipore) was used to label SDH neurons.
Fluorescent recovery after photobleaching.
The neurons transfected by GFP or ASIC1a-GFP (GFP was linked at the N terminus of ASIC1a) were imaged before and at 0, 120, 240, 360, and 480 s after bleaching. All GFP signals were eliminated after ∼10 s of exposure to the argon laser of the Zeiss LSM 510 Confocal microscope. Bleaching was targeted to regions near a junction of a primary and a secondary dendrite. The amount of recovery at each time point was calculated as FRAPt = Ft − F0/Fi − F0, where Ft is the fluorescence in the bleached area at time t, F0 is the fluorescence of the same field immediately after bleaching, and Fi is the initial intensity of the field before photobleaching.
Reagents and data analysis.
Recombinant human BDNF was purchased from R&D System. All drugs were purchased from Sigma unless mentioned otherwise. Results are expressed as mean ± SE (SEM). Statistical comparisons were performed using unpaired or paired Student's t tests or one-way ANOVA with Tukey post hoc tests, where p values <0.05 are considered significant.
Results
ASIC1a channel is required for sustained BDNF-induced mechanical hyperalgesia
Intrathecal injection of exogenous BDNF caused hyperalgesic responses in normal mice (Groth and Aanonsen, 2002; Yajima et al., 2005). To examine a possible role of ASIC1a channels in BDNF-mediated spinal nociceptive processing, we assessed the effect of spinally administered BDNF on mechanical nociceptive threshold of ASIC1 deletion (ASIC1−/−) and wild-type (WT or ASIC1+/+) mice. Mice were injected intrathecally with either saline only (control) or saline containing 1 μg of BDNF, and paw withdrawal threshold in response to von Frey hairs was measured at different time points after the injection. Both WT and ASIC1−/− mice developed mechanical hyperalgesia as seen by the significantly decreased withdrawal threshold at 5 and 30 min following injection in BDNF-treated mice, as compared with saline controls (Fig. 1A,B). However, the decrease was only sustained in WT mice. In ASIC1−/− mice, the enhanced mechanosensitivity started to attenuate at 30 min and was mostly diminished at 3 h after BDNF administration (Fig. 1B). Since ASIC1a is the major ASIC isoform expressed in SDH neurons (Wu et al., 2004; Duan et al., 2007), this result suggests that the ASIC1a channel plays a critical role in sustaining BDNF-induced mechanical hyperalgesia.
Figure 1.
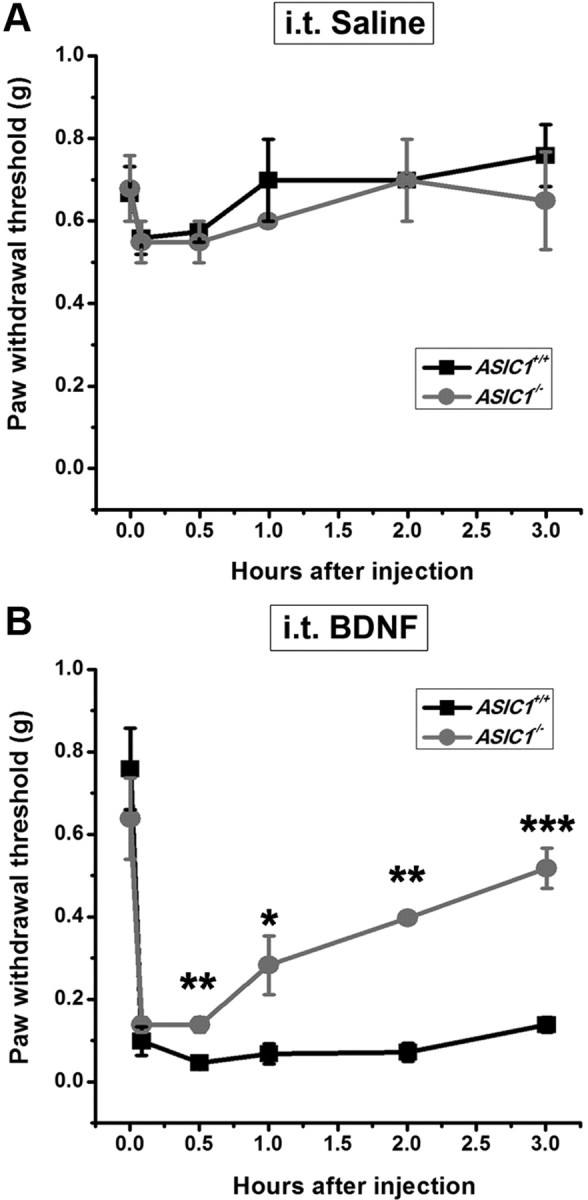
Effect of intrathecal injection of BDNF on paw withdrawal threshold to mechanical stimulus in the wild-type (ASIC1+/+) and ASIC1 deletion (ASIC1−/−) mice. A, Time course of mechanical threshold in the intrathecal saline-treated group. B, Time course of intrathecal BDNF-induced mechanical hyperalgesia. Each point represents the mean ± SEM of 5–6 mice. *p < 0.05, **p < 0.01 versus ASIC1+/+ group, by Student's unpaired t test.
PI3K/Akt pathway upregulates the activity of ASIC1a channel
To gain insights into the relationship between BDNF signaling and ASIC1a channel regulation, we examined the effect of BDNF on ASIC currents activated by lowering extracellular pH from 7.4 to 6.0 (I6.0) using perforated patches of cultured mouse SDH neurons [10–12 d in vitro (DIV)]. After a 5 min BDNF treatment (20 ng/ml), the amplitude of I6.0 increased ∼50% (146.2 ± 9.8% of the control, Fig. 2A). The current gradually returned to the original level 5–10 min after termination of the BDNF treatment (Fig. 2A). However, despite the increase in current amplitude, the pH sensitivity (pH for 50% maximal activation, pH50, was 6.42 ± 0.06 and 6.43 ± 0.08 before and after BDNF treatment, respectively; n = 7–11, p > 0.05 by paired t test) and the decay time constant (τ) of I6.0 (2.1 ± 0.4 s before vs 2.3 ± 0.4 s after BDNF treatment; n = 15, p > 0.05 by paired t test) did not change significantly, suggesting that the enhancement is unlikely due to a change in channel gating (Gao et al., 2005). By contrast, incubation of neurons with standard extracellular solution for 5 min did not affect I6.0 (Fig. 2A, ES trace). Blocking TrkB receptor activation by the pan-Trk inhibitor, K252a, and transfection of a truncated TrkB receptor, TrkB-T1, which lacks intrinsic tyrosine kinase activity (Bredt and Nicoll, 2003) and inhibits the activation of downstream signal pathways (Huang et al., 2009), both abolished the enhancing effect of BDNF on I6.0 (Fig. 2B). Moreover, blocking PI3 kinase (PI3K), the downstream effector of BDNF-TrkB signaling, with wortmannin (10 μm) or LY294002 (10 μm) also abolished the enhancement of I6.0. By contrast, the enhancing effect of BDNF was unaffected by U-73122 (2 μm), H-89 (10 μm), GF 109203X (4 μm), and KN-93 (5 μm), selective inhibitors for phospholipase C, protein kinase A, PKC, and Ca2+/calmodulin-dependent protein kinase II, respectively (Fig. 2C). Since the main target of PI3K is protein kinase B (PKB, also known as Akt), Akt inhibitor IV (10 μm) was also tested and shown to abolish BDNF-induced enhancement of I6.0 (Fig. 2D). In addition, transfection into SDH neurons of a dominant-negative mutant of Akt (DN-Akt) significantly abolished the enhancement of I6.0 (Fig. 2D). These results indicate the importance of PI3K/Akt pathway in mediating BDNF-induced enhancement of I6.0 in SDH neurons. Interestingly, inhibiting ERK activation by PD98059 (25 μm) also abolished this enhancement (Fig. 2C), agreeing with the previous reports that PI3K and ERK signaling pathways are associated in the DRG (Zhuang et al., 2004) and spinal cord (Pezet et al., 2008) during peripheral inflammation.
Figure 2.

BDNF enhances ASIC1a-mediated currents through activation of PI3K-Akt pathway. Perforated electrophysiological recordings were performed. A, The amplitude of the ASIC current (I6.0) in cultured mouse SDH neurons was enhanced by bath application of BDNF (20 ng/ml), but not a control standard extracellular solution (ES), for 5 min. Inset, Examples of ASIC current traces recorded from one neuron at different times before and after BDNF treatment. Data were normalized to the mean amplitude of I6.0 before BDNF treatment (dashed line here and in B–F). #p < 0.05, ###p < 0.001, n = 8–15, by Student's paired t test. B, Blockade of TrkB receptor with either K252a (n = 15) or by transfecting TrkB-T1 (n = 6) abolished the enhancement caused by BDNF treatment for 5 min (n = 15). ###p < 0.001, versus the amplitude of I6.0 before BDNF treatment, by Student's paired t test. **p < 0.01, ***p < 0.001, versus BDNF-treated group, by Student's unpaired t test. C, Summary of effects of various pharmacological treatments on I6.0 at 5 min after the onset of BDNF. **p < 0.01, ***p < 0.001 versus BDNF only (n = 4–15, Student's unpaired t test). NS, No significant difference. D, Blockade of Akt activity with either Akt inhibitor IV (n = 4) or by transfecting DN-Akt (n = 6) abolished the enhancement caused by BDNF. ###p < 0.001, versus before BDNF treatment, by Student's paired t test. *p < 0.05, ***p < 0.001, versus BDNF-treated group, by Student's unpaired t test. E, Sample traces and summary data showing that bath application of BDNF for 5 min (n = 7) enhanced ASIC1a current in CHO cells co-transfected with TrkB and ASIC1a, which was abolished by K252a (n = 7) and not seen if TrkB was omitted in the transfection (n = 6). ##p < 0.01, versus before BDNF treatment, by Student's paired t test. **p < 0.01, versus ASIC1a + TrkB without K252a, by Student's unpaired t test. F, Summary of effects of various pharmacological treatments on I6.0 at 5 min after the onset of BDNF in CHO cells that coexpressed ASIC1a and TrkB (same as in B). n = 5–9, **p < 0.01, versus BDNF only, by Student's unpaired t test.
Because activation of TrkB by BDNF induces elevation of intracellular Ca2+ concentration (Song et al., 1997; Seal et al., 2009), we further tested whether elevation of intracellular Ca2+ is required for BDNF-regulated enhancement of I6.0 by treating neurons with BAPTA-AM (50 μm, 30 min). However, chelating intracellular Ca2+ with BAPTA did not affect BDNF-enhanced I6.0 (Fig. 2C). Moreover, protein synthesis was not necessary for the BDNF-induced enhancement of I6.0 because blocking translation with anisomysin (40 μm) had no effect (Fig. 2C).
To firmly establish the role of PI3K/Akt cascade in regulating ASIC1a channel function, we expressed ASIC1a in CHO cells and found that BDNF (20 ng/ml, 5 min) enhanced ASIC1a currents when ASIC1a and TrkB receptor were co-transfected, an effect that was abolished by K252a treatment (Fig. 2E). No such enhancement was evident when the TrkB receptor was not co-transfected (Fig. 2E). Furthermore, consistent with the above findings from native SDH neurons, pharmacological disruption of PI3K or Akt signaling abolished the BDNF effect on I6.0 in CHO cells co-transfected with ASIC1a and TrkB receptors (Fig. 2F). Together, these results strongly support our initial hypothesis that BDNF upregulates the function of ASIC1a channels in SDH neurons. That this process involves the activation of TrkB/PI3K/Akt cascade prompted us to test whether ASIC1a trafficking is affected by BDNF, because the same cascade has previously been shown to regulate the transport of voltage-dependent calcium channels to the plasma membrane (Viard et al., 2004) and postsynaptic density protein 95 (PSD-95) to neuronal dendrites (Yoshii and Constantine-Paton, 2007).
BDNF facilitates ASIC1a trafficking to neuronal surface
To detect the surface population of ASIC1a in cultured SDH neurons (10–12 DIV), we quantified the plasma membrane levels of ASIC1a protein by performing surface biotinylation. At steady state, the fraction of the endogenous ASIC1a on cell surface was calculated to be 6.9 ± 1.6% of total ASIC1a (n = 4, data not shown), based on the amount pulled down by the NeutrAvidin beads and that in the total cell lysate. Bath application of BDNF (20 ng/ml, 5 min) significantly increased the surface bound but not the total expression level of ASIC1a by ∼50% (150.0 ± 15.6% of the untreated, Fig. 3A). Given the presence of ASIC1a in soma and dendrites (Wemmie et al., 2002), it is important to examine the subdomain distribution of ASIC1a channels on neuronal surface before and after the BDNF treatment. Since an antibody against extracellular epitope of endogenous ASIC1a is not available, we transfected cultured SDH neurons (10–12 DIV) with a recombinant ASIC1a that contains an extracellular HA tag between F147 and K148, as well as an N-terminal GFP tag (GFP-ASIC1a-HA). Despite showing a decreased proton affinity as compared with the WT ASIC1a, insertion of the HA tag did not affect the surface expression of the channel protein (Chen and Gründer, 2007). Using GFP-ASIC1a-HA, we examined the surface abundance of ASIC1a in cultured SDH neurons. Immunostaining of detergent-permeabilized cells with an anti-HA antibody showed complete colocalization between the GFP and HA signals, whereas the staining of nonpermeabilized neurons revealed GFP fluorescence throughout the cell but anti-HA staining only at the cell perimeter. To minimize the effect of neuronal heterogeneity in the SDH, we chose only transfected pyramidal neurons for analysis. After BDNF treatment, immunostaining of nonpermeabilized neurons showed increased surface expression of GFP-ASIC1a-HA in soma and dendritic shafts (Fig. 3B), a pattern that recapitulates the somatodendritic distribution of endogenous ASIC1a proteins and functional ASIC1a channels (Wemmie et al., 2002; Zha et al., 2006). The estimated ratio of mean fluorescence intensity of surface/total of the expressed GFP-ASIC1a-HA in soma and dendrite increased ∼70% (167.0 ± 17.5% of untreated, Fig. 3B) in BDNF-treated neurons. Thus, BDNF enhances ASIC1a channel membrane insertion, resulting in higher amounts of functional ASIC1a channels at the cell surface.
Figure 3.

BDNF increases ASIC1a trafficking but not internalization in vitro. A, Surface biotinylation of cultured SDH neurons treated with BDNF for 5 min. Endogenous transferrin receptor (TfR) is shown as a surface protein control and was used to normalize ASIC1a expression level in each group. Endogenous α-tubulin is shown as a cytoplasmic protein control. S, Surface; T, total. BDNF treatment for 5 min increased surface expression of ASIC1a to 150.0 ± 15.6% of the untreated control (Ctrl, n = 4, p < 0.05, vs Ctrl, by Student's paired t test). B, Surface expression of ASIC1a with extracellular HA and N-terminal GFP tags (GFP-ASIC1a-HA) transfected in cultured SDH neurons without (Ctrl) and with BDNF treatment for 5 min. Surface GFP-ASIC1a-HA was labeled using anti-HA antibody without permeabilization (surface), whereas total proteins were visualized with GFP fluorescence (total). The estimated ratio of mean fluorescence intensity of surface/total of the expressed GFP-ASIC1a-HA in BDNF-treated neurons was 167.0 ± 17.5% of Ctrl (n = 4, p < 0.05, vs Ctrl, by Student's paired t test). Shown on the right are summary data of relative surface expression normalized to the mean of the control group (n = 17 cells in each group). **p < 0.01, versus Ctrl, by Student's unpaired t test. C, Representative Western blots of internalized and total ASIC1a and Transferrin receptor (as a negative control) in high-density SDH cultures. Five minutes after treatment with control solution or BDNF (20 ng/ml), surface receptors were labeled with biotin and allowed to internalize at 37°C for 3, 5, or 15 min, after which the remaining surface biotin was stripped off. The internalized biotinylated ASIC1a was measured. In these experiments, total ASIC1a levels were not affected by the BDNF treatment (126.9 ± 10.5% of control, n = 3). Quantification of internalized/total ASIC1a and TfR levels shows endocytosis rates in control and BDNF-treated cultures (n = 3). A best-fit single exponential growth curve was used to obtain time constants (τ) for internalization of ASIC1a (6.7 ± 1.4 min for control, 5.3 ± 1.6 min for BDNF-treated).
Since decreased endocytosis could also modulate the surface abundance of ASIC1a channels, we treated cultured neurons with cytochalasin D (20 μm) to disrupt actin dynamics and thereby partially block endocytosis and recycling (Cooper, 1987). Under this condition, BDNF still enhanced I6.0 (Fig. 2C), suggesting that BDNF exerts its action independent of actin-mediated endocytosis. To directly examine the effect of BDNF on endocytosis of ASIC1a, we measured the uptake rate of surface biotinylated ASIC1a in the absence and presence of BDNF and showed that BDNF did not change the internalization rate of this channel (Fig. 3C). Therefore, it is more likely that BDNF treatment resulted in significant recruitment of ASIC1a channels to the surface of SDH neurons.
We next attempted to visualize the dynamics of BDNF-regulated ASIC1a trafficking over time via fluorescent recovery after photobleaching (FRAP). To do this, a small segment of dendrite was photobleached with an argon laser and then imaged after 120, 240, 360, 480 s with a confocal microscope to measure the recovery of GFP fluorescence intensity in the bleached area (Fig. 4A). While neurons transfected with GFP alone showed ∼100% recovery into bleached segment within 60 s (Fig. 4B), <40% recovery was achieved in the bleached segment by ASIC1a-GFP after 480 s (Fig. 4A,B), consistent with the idea that ASIC1a-GFP in dendrites does not simply diffuse in the cytoplasm. Bath application of BDNF (20 ng/ml) increased the recovery of ASIC1a-GFP in the bleached dendrite segment to an average of 80% within 480 s (Fig. 4A,B) and the increased recovery was abolished by coapplication of the Trk inhibitor K252a (Fig. 4C), revealing that BDNF facilitates ASIC1a transport. Remarkably, BDNF-regulated ASIC1a transport was eliminated by coapplication of inhibitors of PI3K (Fig. 4D) and Akt (Fig. 4E). Collectively, these data suggest that BDNF stimulates active delivery and insertion of new ASIC1a channels into the plasma membrane of SDH neurons. Importantly, the PI3K/Akt pathway seems critical for the increased surface abundance and ASIC1a activity.
Figure 4.
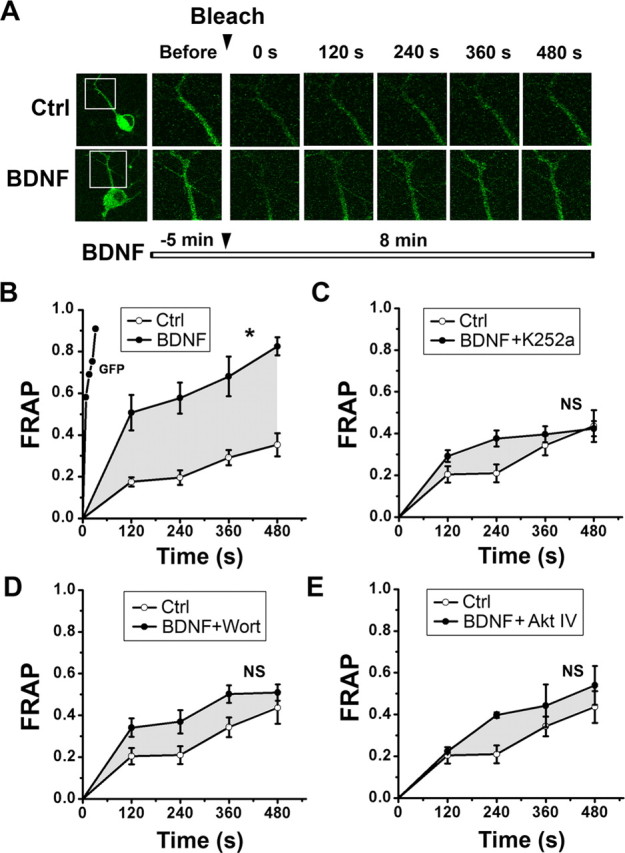
Dendritic transport of ASIC1a is facilitated by BDNF-TrkB and downstream PI3K/Akt signaling. A, Images of FRAP experiments showing that bleaching a defined dendritic segment of a neuron expressing ASIC1a-GFP resulted in a slow fluorescent recovery in 480 s (top row). Application of BDNF before bleaching resulted in faster recovery (bottom row). White rectangles label the region of the neuron that was bleached. B, FRAP was quantified by measuring the intensity of fluorescence in the frame at fixed intervals. GFP alone showed recovery (by diffusion) within 60 s (n = 5). Application of BDNF facilitated the recovery of ASIC1a-GFP (n = 8). *p < 0.05, versus Ctrl (n = 11), by one-way ANOVA with Tukey post hoc tests. C–E, Suppression of BDNF-induced ASIC1a FRAP by K252a (C, n = 6), wortmannin (Wort) (D, n = 8), and Akt IV (E, n = 6). NS, By one-way ANOVA with Tukey post hoc tests. The region between the drug-treatment and no-treatment curves is superimposed as a gray envelope on FRAP graphs for comparison.
Trafficking of ASIC1a channel is essential for BDNF-induced secondary mechanical hyperalgesia
Activity-dependent expression and release of BDNF is essential for synaptic plasticity in the CNS (Thoenen, 1995; Lu, 2003). In sensory system, activation of C-fiber by capsaicin or electrical stimulation induces BDNF release in the dorsal horn (Lever et al., 2001). To explore the functional significance of BDNF-induced and Akt-mediated ASIC1a trafficking and surface expression during pain hypersensitivity, we used capsaicin to induce pain behaviors that allows distinction between peripheral and central sensitization of mechanical pain in vivo. In particular, intradermal injection of capsaicin in the hindpaw of rats or mice induces primary mechanical hyperalgesia (several minutes after injection) at the inflamed injection region, and secondary mechanical hyperalgesia (several hours) in the neighboring surrounding healthy skin, reflecting peripheral and central sensitization, respectively (Willis, 2001). We found that capsaicin injection increased Akt phosphorylation in the SDH of WT mice but not that of BDNF+/− mice (data not shown), in which the expression level of BDNF is approximately half of the WT mice (MacQueen et al., 2001). Furthermore, surface but not total protein level of ASIC1a in the SDH was increased 2 h after capsaicin injection in rat hindpaws and the increase was attenuated by intrathecal application of Akt inhibitor IV (10 mm, 10 μl) (Fig. 5A). In vehicle-injected rats, however, Akt inhibitor IV did not affect surface expression of ASIC1a in the SDH, indicating a specific involvement of Akt in ASIC1a modulation only in the pain hypersensitized animals. Moreover, surface but not total protein level of ASIC1a was also increased in the SDH of WT mice 2 h after intradermal injection of capsaicin. This change was not observed in BDNF+/− mice (Fig. 5B), suggesting that BDNF is important for bringing about the increase in surface expression of ASIC1a following capsaicin injection.
We further tested the functional role of BDNF and its effect on ASIC1a trafficking in capsaicin-induced pain hypersensitivity. In WT mice, the mechanical threshold was 0.65 ± 0.09 g before capsaicin injection (Fig. 5D). However, 15 min after capsaicin injection, the mechanical threshold at the injection site (labeled “P” in Fig. 5C) was reduced to 0.10 ± 0.03 g (Fig. 5D, primary hyperalgesia), and, 2 h after injection, the mechanical threshold in the neighboring surrounding non-inflamed region (labeled “S” in Fig. 5C) was changed to 0.05 ± 0.01 g (Fig. 5D, secondary hyperalgesia). In BDNF+/− mice, the mechanical threshold before capsaicin injection (0.71 ± 0.11 g) was similar to that of WT littermates. However, while the primary hyperalgesia induced by capsaicin remained the same as in WT mice (0.08 ± 0.02 g, Fig. 5D), the secondary hyperalgesia was greatly attenuated (0.32 ± 0.05 g, Fig. 5D), suggesting that BDNF mainly participates in secondary (or central), but not primary (or peripheral), mechanical pain hypersensitivity.
To further address the relevance of BDNF in the development of pain hypersensitivity in the SDH, we investigated the mechanism by which BDNF contributes to nociceptive processing in SDH neurons. In vivo single unit recordings were made from WDR SDH neurons, which are responsible for nociceptive transmission and spinal flexion reflex (Jiang et al., 1995). In BDNF+/− mice, there is no difference on neuronal firing rates by mechanical stimulation before capsaicin injection, indicating that BDNF may not affect mechanical circuitry development. However, capsaicin injection strongly increased activity of WDR neurons in response to innocuous (brush, from 0.6 ± 0.1 Hz to 1.0 ± 0.2 Hz) and noxious mechanical stimuli, including press (from 1.2 ± 0.2 Hz to 2.5 ± 0.3 Hz) and pinch (from 2.5 ± 0.3 Hz to 4.5 ± 1.0 Hz) in WT mice (Fig. 5E). By contrast, capsaicin did not induce significant hyperactivity of WDR neurons in BDNF+/− mice (Fig. 5F). Because ASIC1a channel is required for hyperactivity of WDR neurons after peripheral inflammation (Duan et al., 2007), we tested its involvement in BDNF-mediated hypersensitivity of WDR neurons after capsaicin injection. Local infusion of the ASIC1a channel inhibitor, PcTX1 (10 μm, 10 μl), significantly attenuated the hyperactivity of WDR neurons in response to noxious mechanical stimuli (press: 1.5 ± 0.2 Hz; pinch: 2.0 ± 0.2 Hz; Fig. 5E) in the WT mice but did not affect the WDR neuronal firing activity in BDNF+/− mice (Fig. 5F), suggesting that ASIC1a function is important for the BDNF-mediated hypersensitivity. These data, thus, support the notion that ASIC1a is an important downstream target of BDNF in central processing of pain signals.
Phosphorylation of Ser-25 is crucial to BDNF-mediated ASIC1a trafficking
Next, we sought to identify the critical domain(s) on ASIC1a channels involved in BDNF-regulated membrane delivery and central sensitization. For this purpose, we expressed four additional ASIC isoforms, namely 1b, 2a, 1a + 2a, and 3 in CHO cells and evaluated whether BDNF regulation is unique to ASIC1a channels (Fig. 2E) or is common among various ASIC isoforms. We found that BDNF also enhanced ASIC current mediated by homomeric ASIC2a channels or heteromeric ASIC1a + ASIC2a channels (Fig. 6A), two types of ASICs in the SDH neurons in addition to the ASIC1a channel (Wu et al., 2004; Duan et al., 2007; Baron et al., 2008). By contrast, BDNF did not influence the ASIC current mediated by homomeric ASIC1b or ASIC3 channels (Fig. 6A), two types of peripheral ASICs (Waldmann et al., 1997b; Chen et al., 1998). Considering the fact that ASIC1a differs from ASIC1b only at the N terminus, we focused on the amino acid differences at this region and found that serine 25 (Ser-25) of ASIC1a and 2a is unique from other ASIC isoforms (Fig. 6B). Mutating Ser-25 of ASIC1a to alanine abolished the BDNF-mediated enhancement of ASIC1a current in CHO cells co-transfected with TrkB receptor (Fig. 6C). In addition, the surface expression level of the S25A mutant was significantly lower than that of the WT ASIC1a. By contrast, the surface expression level of the S25D mutant, which is thought to mimic phosphorylation at the mutated site, was significantly higher than that of the WT ASIC1a (Fig. 6D). These data indicate that Ser-25 is likely the major site of modulation affected by BDNF and its downstream molecules PI3K/Akt, through which they alter the phosphorylation state of ASIC1a and promote channel trafficking and consequently surface availability.
Figure 6.

Ser-25 at ASIC1a N terminus mediates BDNF-induced ASIC1a trafficking. A, Proton-activated currents in CHO cells that coexpressed TrkB with ASIC2a, ASIC1a+2a, ASIC1b, or ASIC3 before (black lines) and after (red lines) BDNF treatment (5 min). Left, Representative traces; right, summary for current amplitude after BDNF normalized to that before BDNF (dashed line; n = 5, 6, 4, and 5 for ASIC2a, ASIC1a+2a, ASIC1b, and ASIC3, respectively). BDNF increased the current mediated by ASIC2a and ASIC1a+2a but not that by ASIC1b and ASIC3. ##p < 0.01, by Student's paired t test. B, Sequence alignment of the ASIC N termini indicating that Ser-25 is present in ASIC1a and 2a but not in other ASIC subunits. C, Mutation of ASIC1a Ser-25 to Ala (S25A, n = 7) abolished BDNF-induced enhancement of ASIC1a current. Data were normalized to the mean amplitude of I6.0 before BDNF treatment (dashed line) for each cell. ##p < 0.01, by Student's paired t test. D, Representative Western blots and summary data showing that the surface expression level of S25A was significantly lower than that of WT ASIC1a (dashed line), whereas that of S25D significantly increased. Data were normalized to the surface expression level of WT ASIC1a (n = 4). #p < 0.05, ##p < 0.01, by Student's paired t test.
Blockade of ASIC1a trafficking attenuates pain sensitization
To further elucidate the role of Ser-25 in mediating ASIC1a surface expression and presumably capsaicin-induced secondary hyperalgesia, we designed a fusion peptide, TAT-Ser-25 peptide (S25-peptide), which takes advantage of the delivery potential of the TAT peptide from HIV into cells. TAT-S25 peptide or a scrambled control (Scr-peptide) was applied to cultured SDH neurons at a concentration of 50 μm for 2 h. Biotinylation treatment and subsequent Western blotting showed that incubation of S25-peptide abolished BDNF-induced increase of surface ASIC1a expression, whereas Scr-peptide had no effect (Fig. 7A,B). Further electrophysiological recording indicated that S25-peptide did not affect ASIC current density in SDH neurons under basal conditions (data not shown). However, with the treatment of S25-peptide, but not the Scr-peptide, BDNF failed to enhance ASIC current in cultured SDH neurons (Fig. 7C). Moreover, to explore the effect of S25-peptide on pain sensitization in vivo, we intrathecally injected the biotinylated S25-peptide into the lumbar SDH of rats. By monitoring biotin-streptavidin reaction, we detected the presence of the peptide in the lumbar SDH neurons (data not shown). Further experiments showed that intrathecal coinjection of BDNF with S25-peptide largely reduced BDNF-induced mechanical hyperalgesia, whereas the Scr-peptide did not exert any inhibitory effect (Fig. 8A). Moreover, intrathecal injection of S25-peptide also significantly attenuated the secondary, but not the primary, mechanical hyperalgesia after capsaicin injection in mice (Fig. 8B). These results thus support the conclusion that phosphorylation at N terminus of ASIC1a that contains Ser-25 is critical for BDNF-induced trafficking of ASIC1a channels and the central pain hypersensitivity.
Figure 7.

Effects of a TAT-fusion peptide containing ASIC1a N-terminal sequence encompassing Ser-25 on ASIC expression and activation in SDH neurons. A, B, Representative Western blots and summary showing bath application of TAT-Ser-25 peptide (S25-peptide) abolished BDNF-induced surface expression of ASIC1a. Incubation with the scrambled peptide (Scr-peptide) had no effect. S, Surface; T, total. Data were normalized to the surface expression level of ASIC1a in Ctrl group incubated with Scr-peptide. n = 3, **p < 0.01, by Student's paired t test. C, Bath incubation (2 h) of S25-peptide abolished BDNF-induced enhancement of ASIC current in cultured SDH neurons. Incubation with the Scr-peptide had no effect. Data were normalized to the mean amplitude of I6.0 before BDNF treatment (dashed line) for each neuron, n = 6 for each group. ##p < 0.01, by Student's paired t test.
Figure 8.

Effects of S25-peptide on BDNF-mediated mechanical hyperalgesia. A, Changes in paw withdrawal threshold of WT mice in response to mechanical stimuli after intrathecal coinjection of BDNF with S25-peptide (n = 5), or Scr-peptide (n = 5), or saline (n = 6). **p < 0.01, ***p < 0.001, S25-peptide versus Scr-peptide, by Student's paired t test. B, Changes in paw withdrawal threshold of mice in response to mechanical stimuli after intradermal injection of capsaicin. Intrathecal injection of S25-peptide (n = 7), but not Scr-peptide (n = 8), or saline (n = 12) 1 h before capsaicin injection significantly attenuated capsaicin-induced secondary mechanical hyperalgesia but not primary mechanical hyperalgesia. ***p < 0.001, S25-peptide versus Scr-peptide, by Student's unpaired t test.
Discussion
ASIC1a channels in SDH neurons have emerged as an attractive target in developing new analgesics (Duan et al., 2007; Mazzuca et al., 2007). The present study provides further evidence supporting a critical role of ASIC1a channels in mediating BDNF-induced central sensitization and identifies PI3K/Akt cascade as a key molecular mechanism involved in this process. We demonstrate that upregulating channel function via increasing ASIC1a membrane delivery contributes to BDNF-mediated pain plasticity in the SDH. This conclusion is based on five lines of evidence. First, spinal BDNF-induced sustained mechanical hyperalgesia is significantly attenuated in ASIC1 knock-out mice. Second, the activation of PI3K-Akt pathway by BDNF mediates the upregulation of ASIC1a function. Third, BDNF promotes forward trafficking and surface expression of ASIC1a, and this is responsible for the upregulated channel function. Fourth, upregulation of ASIC1a channel function via membrane insertion is involved in BDNF-mediated central sensitization and secondary hyperalgesia after intradermal capsaicin treatment in rodent hindpaws. Finally, we identified phosphorylation of Ser-25 at N terminus of ASIC1a as an essential step for BDNF-induced ASIC1a membrane insertion, and the Ser-25 containing channel domain plays a critical role in pain hypersensitivity (Fig. 9).
Figure 9.

Schematic diagram of the effect of BDNF/TrkB and downstream signaling pathways on central sensitization following C-fiber activity. Both BDNF and glutamate are released from presynaptic DRG neurons following C-fiber activity by peripheral capsaicin treatment. Activation of postsynaptic TrkB receptor in the spinal dorsal horn by BDNF induces intracellular PKC activation and enhances NMDAR function via phosphorylation. On the other hand, BDNF also activates intracellular PI3K/Akt pathway, and then induces ASIC1a phosphorylation and forward targeting to neuron surface. ASIC1a channels sense synaptic acidosis and cause membrane depolarization via Na+ and Ca2+ influx, which may facilitate NMDAR activation and induce central sensitization. Blockade of ASIC1a channel trafficking by fusion of S25-peptide to obstruct the phosphorylation of ASIC1a at Ser25 attenuates BDNF-mediated mechanical hyperalgesia (right). Moreover, release of BDNF from spinal microglia following peripheral nerve injury (Coull et al., 2005) may also facilitate forward trafficking of ASIC1a and then induces central sensitization.
Functional effects of BDNF on pain-signaling pathways
The neurotrophin BDNF acts as a central modulator of pain via PKC/ERK pathways (Woolf and Salter, 2000; Pezet and McMahon, 2006). However, our results indicate that PKC activity is not involved in modulating ASIC1a channel trafficking. Rather, PKC has been shown to phosphorylate NMDA receptors, which can also regulate synaptic plasticity in the SDH (Brenner et al., 2004; Kawasaki et al., 2004; Zhao et al., 2006). Accumulating evidence indicates that another downstream cascade of BDNF, PI3K-Akt pathway, which is involved in synaptic plasticity and spatial memory formation (Slack et al., 2004), also participates in pain hypersensitivity in DRG and spinal cord (Zhuang et al., 2004; Pezet et al., 2005, 2008; Sun et al., 2006; Xu et al., 2011). After capsaicin injection, phosphorylation of Akt increased robustly in the superficial dorsal horn (Sun et al., 2006), suggesting that PI3K-Akt may mediate the enhanced synaptic transmission in this region. Here, we show that activation of PI3K-Akt by BDNF facilitates ASIC1a insertion into plasma membrane of SDH neurons. Consistent with the roles of ASIC1a channel in hippocampal long-term potentiation (Wemmie et al., 2002), our data argue that upregulating ASIC1a channel function may be involved in PI3K-Akt pathway-mediated synaptic plasticity in the superficial SDH during pain hypersensitivity.
Although at 5 min after intrathecal administration, BDNF induced mechanical hyperalgesia to a similar extent in WT and ASIC1−/− mice, the enhanced mechanosensitivity slowly diminished in ASIC1−/− but not WT mice (Fig. 1B), suggesting that ASIC1a channels are essential for the maintenance, but not induction, of the mechanical hyperalgesia. Other downstream effects of BDNF signaling, such as potentiation of NMDA receptors by PKC-induced phosphorylation (Slack et al., 2004), may play a more predominant role for the induction of pain hypersensitivity. On the other hand, even though the effect of ASIC1a on BDNF-induced mechanical hyperalgesia was masked during the induction period, the 5 min BDNF treatment is sufficient to enhance surface expression and function of ASIC1a channels, which lasted for at least 5–10 min after removal of BDNF (Fig. 2A). For intrathecal injection, not only the injected BDNF was not removed, but also because BDNF is capable of stimulating its own release in vivo (Bramham and Messaoudi, 2005), sustained enhancement of ASIC1a channel insertion on the plasma membrane of SDH neurons is likely maintained for a long period of time, allowing continued hyperalgesia for hours in WT mice.
It is important to note that although BDNF plays a critical role for central sensitization and pathologic pain, the role of endogenous BDNF in acute pain still remains controversial. Noxious thermal, mechanical, and chemical (capsaicin, mustard oil) stimuli induce phosphorylation of TrkB receptors in the SDH (Pezet et al., 2002), and application of the pan-Trk inhibitor, K252a, attenuates capsaicin-induced ERK phosphorylation in the SDH (Kawasaki et al., 2004), suggesting a role of endogenous BDNF in acute pain. Indeed, the nociceptive reflex is impaired from BDNF-deficient mice (Heppenstall and Lewin, 2001; MacQueen et al., 2001). However, sequestering endogenous BDNF with a TrkB-Fc fusion protein did not affect capsaicin-induced mechanical hypersensitivity in rats (Mannion et al., 1999). Our data that the primary hyperalgesia induced by capsaicin in BDNF+/− mice remained the same as in the WT mice (Fig. 5D) are consistent with this latter observation. Considering the fact that BDNF release is triggered in dorsal horn only by electrical stimulation of C fibers with high-frequency bursts (Lever et al., 2001), which occur when the intensity of nociceptive stimuli increases over a certain threshold (Adelson et al., 1997), it is possible that the role of BDNF in modulating acute nociceptive transmission becomes significant only during intense and repetitive noxious stimulation.
Mechanisms of PI3K/Akt-induced ASIC1a trafficking
Activation of PI3K induces membrane insertion of transient receptor potential channel (Kanzaki et al., 1999) and voltage-dependent calcium channels (Viard et al., 2004), but the mechanisms involved remain unknown. A role for PI3K in the reorganization of actin cytoskeleton, which contributes to protein transport inside the cell, has also been described (Insall and Weiner, 2001). However, our results exclude this pathway because disruption of actin cytoskeleton reorganization by cytochalasin D did not affect BDNF-induced ASIC1a trafficking and functional upregulation (Fig. 2C). PI3K also activates downstream signaling, such as Akt. Several studies have reported that activation of Akt is required for ion channel trafficking (Blair et al., 1999; Lhuillier and Dryer, 2002; Wang et al., 2003). One might thus speculate that phosphorylation of ASIC1a N terminus by Akt could induce the association of the ASIC1a channel complexes with trafficking proteins to promote their translocation to the plasma membrane. Indeed, the enhancing effect of BDNF on ASIC1a cell surface expression was abolished by the mutation of Ser-25 to alanine and mimicked by substituting it with an aspartate (Fig. 6C,D), suggesting that Ser-25 is the functionally relevant phosphorylation site on ASIC1a. In addition, although there is no evidence showing that ASIC1a channel is a direct substrate of Akt, we found that ASIC1a coexists with Akt in the same protein complex using coimmunoprecipitation (data not shown). Moreover, intrathecal administration of a TAT-fusion peptide that contains the 9 aa sequence encompassing Ser-25 significantly attenuated the secondary mechanical hyperalgesia (Fig. 8B). Thus, our results support the essential role of phosphorylation at Ser-25 by Akt in BDNF-induced enhancement of ASIC1a function and secondary mechanical hyperalgesia. A direct test of this model would be a genetically modified mouse that carries a mutation in the Ser-25 phosphorylation site of ASIC1a. The prediction would be that these mice would have relatively normal ASIC1a currents in dorsal horn neurons but no modulation of this current by BDNF-mediated Akt activity.
In heterologous expression systems, we also observed that BDNF increased ASIC2a channel trafficking. Since ASIC2a facilitates ASIC1a targeting to dendritic spines in cultured hippocampal neurons (Zha et al., 2009) and BDNF-induced mechanical hyperalgesia was not completely abolished in ASIC1−/− mice (Fig. 1B), it would be interesting to further test whether ASIC2a is also regulated in a similar fashion via PI3K-Akt activation by BDNF.
Role of spinal ASIC1a in pain hypersensitivity
Since tissue acidosis during acute and chronic pain is not expected to affect pH levels in the spinal cord, it begs the question as to how ASIC1a channel participates in the nociceptive transmission. One possibility is that extracellular acidosis associated with synaptic activity may activate ASIC1a channels in the synaptic cleft (Krishtal, 2003). However, sustained exposure to acidic solution desensitizes ASIC1a channel. Desensitization begins at pH <8.0 in cultured sensory neurons, with half-maximal desensitization occurring at ∼pH 7.2 (Benson et al., 1999). When extracellular pH is held under physiological condition (∼7.4), a significant proportion of ASIC channels is tonically desensitized. Thus, BDNF-regulated ASIC1a delivery could provide a mechanism for channel reactivation during persistent nerve stimulation and prolonged tissue acidosis as occurred in the chronic pain conditions. Increased amount of functional ASIC1a channels may further increase neuronal excitability and regulate synaptic plasticity in the SDH in combination with NMDARs (Duan et al., 2007; Xu and Duan, 2009). It may also suppress endogenous analgesic opioid system (Mazzuca et al., 2007). Our present result that interfering with the interaction between ASIC1a channel and Akt produces analgesic effect underscores the importance of upregulated ASIC1a function in pain hypersensitivity. On the other hand, our results do not exclude the peripheral role of ASICs. Studies of peripheral ASICs in nociception have focused primarily on the ASIC3 subtype (Deval et al., 2010; Yu et al., 2010). However, results from a recent study using the snake toxin MitTx as a novel tool suggest that functional ASIC1a channels and ASIC1a-expressing DRG neurons also contribute to nociception (Bohlen et al., 2011). Since functional ASIC1a channels are expressed in both nociceptive and non-nociceptive somatosensory neurons (Bohlen et al., 2011), it would also be interesting to examine whether BDNF-mediated ASIC1a trafficking in DRG neurons is required for peripheral sensitization and persistent pain (Basbaum et al., 2009). Furthermore, ASIC1a is robustly expressed in lateral and basolateral nuclei of the amygdala and in the hippocampus (Wemmie et al., 2002; 2003), and overexpressing ASIC1a enhances context fear conditioning (Wemmie et al., 2004). Although speculated, it is possible that BDNF-mediated ASIC1a trafficking also plays a role in pain and pain-related discomfort and fear at the supraspinal level.
In conclusion, the present results suggest that upregulation of ASIC1a channel function by BDNF-induced ASIC1a trafficking is important to the development of central sensitization and pain hypersensitivity. Since many analgesic therapies directly targeting NMDARs have failed in clinical trials because of unacceptable CNS side effects (Woolf, 2010), similar to targeting the Src-mediated enhancement of NMDAR function (Liu et al., 2008), uncoupling Akt from ASIC1a complex, therefore, represents an new analgesic strategy because it occurs without affecting the normal BDNF/TrkB cascade and ASIC1a channel activity, and, hence, any adverse consequence directly associated with blocking the overall BDNF/TrkB pathway and ASIC1a channels is not expected to happen.
Footnotes
This study was supported by grants from the National Basic Research Program of China (2011CBA00408) and the National Natural Science Foundation of China (Nos. 30830035 and 91132303). B. Duan is a postdoctoral fellow supported by the K. C. Wong Education Foundation (Hong Kong). We thank Dr. M. J. Welsh (Howard Hughes Medical Institute, University of Iowa, Iowa City, IA) for providing ASIC1 knock-out mice, Drs. X. Zhang, L. Bao, and K.-C. Li for help of in vivo electrophysiology, Dr. Y.-Z. Wang and Ms. H. Cao for technical assistance, and Dr. L.-J. Wu for helpful comments on this manuscript.
References
- Adelson DW, Wei JY, Kruger L. Warm-sensitive afferent splanchnic C-fiber units in vitro. J Neurophysiol. 1997;77:2989–3002. doi: 10.1152/jn.1997.77.6.2989. [DOI] [PubMed] [Google Scholar]
- Baron A, Voilley N, Lazdunski M, Lingueglia E. Acid sensing ion channels in dorsal spinal cord neurons. J Neurosci. 2008;28:1498–1508. doi: 10.1523/JNEUROSCI.4975-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basbaum AI, Bautista DM, Scherrer G, Julius D. Cellular and molecular mechanisms of pain. Cell. 2009;139:267–284. doi: 10.1016/j.cell.2009.09.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benson CJ, Eckert SP, McCleskey EW. Acid-evoked currents in cardiac sensory neurons: a possible mediator of myocardial ischemic sensation. Circ Res. 1999;84:921–928. doi: 10.1161/01.res.84.8.921. [DOI] [PubMed] [Google Scholar]
- Blair LA, Bence-Hanulec KK, Mehta S, Franke T, Kaplan D, Marshall J. Akt-dependent potentiation of L channels by insulin-like growth factor-1 is required for neuronal survival. J Neurosci. 1999;19:1940–1951. doi: 10.1523/JNEUROSCI.19-06-01940.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bohlen CJ, Chesler AT, Sharif-Naeini R, Medzihradszky KF, Zhou S, King D, Sánchez EE, Burlingame AL, Basbaum AI, Julius D. A heteromeric Texas coral snake toxin targets acid-sensing ion channels to produce pain. Nature. 2011;479:410–414. doi: 10.1038/nature10607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bramham CR, Messaoudi E. BDNF function in adult synaptic plasticity: the synaptic consolidation hypothesis. Prog Neurobiol. 2005;76:99–125. doi: 10.1016/j.pneurobio.2005.06.003. [DOI] [PubMed] [Google Scholar]
- Bredt DS, Nicoll RA. AMPA receptor trafficking at excitatory synapses. Neuron. 2003;40:361–379. doi: 10.1016/s0896-6273(03)00640-8. [DOI] [PubMed] [Google Scholar]
- Brenner GJ, Ji RR, Shaffer S, Woolf CJ. Peripheral noxious stimulation induces phosphorylation of the NMDA receptor NR1 subunit at the PKC-dependent site, serine-896, in spinal cord dorsal horn neurons. Eur J Neurosci. 2004;20:375–384. doi: 10.1111/j.1460-9568.2004.03506.x. [DOI] [PubMed] [Google Scholar]
- Chen CC, England S, Akopian AN, Wood JN. A sensory neuron-specific, proton-gated ion channel. Proc Natl Acad Sci U S A. 1998;95:10240–10245. doi: 10.1073/pnas.95.17.10240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Gründer S. Permeating protons contribute to tachyphylaxis of the acid-sensing ion channel (ASIC) 1a. J Physiol. 2007;579:657–670. doi: 10.1113/jphysiol.2006.120733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper JA. Effects of cytochalasin and phalloidin on actin. J Cell Biol. 1987;105:1473–1478. doi: 10.1083/jcb.105.4.1473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coull JA, Beggs S, Boudreau D, Boivin D, Tsuda M, Inoue K, Gravel C, Salter MW, De Koninck Y. BDNF from microglia causes the shift in neuronal anion gradient underlying neuropathic pain. Nature. 2005;438:1017–1021. doi: 10.1038/nature04223. [DOI] [PubMed] [Google Scholar]
- Deval E, Noël J, Lay N, Alloui A, Diochot S, Friend V, Jodar M, Lazdunski M, Lingueglia E. ASIC3, a sensor of acidic and primary inflammatory pain. EMBO J. 2008;27:3047–3055. doi: 10.1038/emboj.2008.213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deval E, Gasull X, Noël J, Salinas M, Baron A, Diochot S, Lingueglia E. Acid-sensing ion channels (ASICs): pharmacology and implication in pain. Pharmacol Ther. 2010;128:549–558. doi: 10.1016/j.pharmthera.2010.08.006. [DOI] [PubMed] [Google Scholar]
- Duan B, Wu LJ, Yu YQ, Ding Y, Jing L, Xu L, Chen J, Xu TL. Upregulation of acid-sensing ion channel ASIC1a in spinal dorsal horn neurons contributes to inflammatory pain hypersensitivity. J Neurosci. 2007;27:11139–11148. doi: 10.1523/JNEUROSCI.3364-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao J, Duan B, Wang DG, Deng XH, Zhang GY, Xu L, Xu TL. Coupling between NMDA receptor and acid-sensing ion channel contributes to ischemic neuronal death. Neuron. 2005;48:635–646. doi: 10.1016/j.neuron.2005.10.011. [DOI] [PubMed] [Google Scholar]
- Garraway SM, Petruska JC, Mendell LM. BDNF sensitizes the response of lamina II neurons to high threshold primary afferent inputs. Eur J Neurosci. 2003;18:2467–2476. doi: 10.1046/j.1460-9568.2003.02982.x. [DOI] [PubMed] [Google Scholar]
- Groth R, Aanonsen L. Spinal brain-derived neurotrophic factor (BDNF) produces hyperalgesia in normal mice while antisense directed against either BDNF or trkB, prevent inflammation-induced hyperalgesia. Pain. 2002;100:171–181. doi: 10.1016/s0304-3959(02)00264-6. [DOI] [PubMed] [Google Scholar]
- Heppenstall PA, Lewin GR. BDNF but not NT-4 is required for normal flexion reflex plasticity and function. Proc Natl Acad Sci U S A. 2001;98:8107–8112. doi: 10.1073/pnas.141015098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang SH, Zhao L, Sun ZP, Li XZ, Geng Z, Zhang KD, Chao MV, Chen ZY. Essential role of Hrs in endocytic recycling of full-length TrkB receptor but not its isoform TrkB.T1. J Biol Chem. 2009;284:15126–15136. doi: 10.1074/jbc.M809763200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hylden JL, Wilcox GL. Intrathecal morphine in mice: a new technique. Eur J Pharmacol. 1980;67:313–316. doi: 10.1016/0014-2999(80)90515-4. [DOI] [PubMed] [Google Scholar]
- Insall RH, Weiner OD. PIP3, PIP2, and cell movement–similar messages, different meanings? Dev Cell. 2001;1:743–747. doi: 10.1016/s1534-5807(01)00086-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji RR, Kohno T, Moore KA, Woolf CJ. Central sensitization and LTP: do pain and memory share similar mechanisms? Trends Neurosci. 2003;26:696–705. doi: 10.1016/j.tins.2003.09.017. [DOI] [PubMed] [Google Scholar]
- Jiang MC, Cleland CL, Gebhart GF. Intrinsic properties of deep dorsal horn neurons in the L6–S1 spinal cord of the intact rat. J Neurophysiol. 1995;74:1819–1827. doi: 10.1152/jn.1995.74.5.1819. [DOI] [PubMed] [Google Scholar]
- Kanzaki M, Zhang YQ, Mashima H, Li L, Shibata H, Kojima I. Translocation of a calcium-permeable cation channel induced by insulin-like growth factor-I. Nat Cell Biol. 1999;1:165–170. doi: 10.1038/11086. [DOI] [PubMed] [Google Scholar]
- Kawasaki Y, Kohno T, Zhuang ZY, Brenner GJ, Wang H, Van Der Meer C, Befort K, Woolf CJ, Ji RR. Ionotropic and metabotropic receptors, protein kinase A, protein kinase C, and Src contribute to C-fiber-induced ERK activation and cAMP response element-binding protein phosphorylation in dorsal horn neurons, leading to central sensitization. J Neurosci. 2004;24:8310–8321. doi: 10.1523/JNEUROSCI.2396-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishtal O. The ASICs: signaling molecules? Modulators? Trends Neurosci. 2003;26:477–483. doi: 10.1016/S0166-2236(03)00210-8. [DOI] [PubMed] [Google Scholar]
- Kuner R. Central mechanisms of pathological pain. Nat Med. 2010;16:1258–1266. doi: 10.1038/nm.2231. [DOI] [PubMed] [Google Scholar]
- Latremoliere A, Woolf CJ. Central sensitization: a generator of pain hypersensitivity by central neural plasticity. J Pain. 2009;10:895–926. doi: 10.1016/j.jpain.2009.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lever IJ, Bradbury EJ, Cunningham JR, Adelson DW, Jones MG, McMahon SB, Marvizón JC, Malcangio M. Brain-derived neurotrophic factor is released in the dorsal horn by distinctive patterns of afferent fiber stimulation. J Neurosci. 2001;21:4469–4477. doi: 10.1523/JNEUROSCI.21-12-04469.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lhuillier L, Dryer SE. Developmental regulation of neuronal K(Ca) channels by TGFbeta1: an essential role for PI3 kinase signaling and membrane insertion. J Neurophysiol. 2002;88:954–964. doi: 10.1152/jn.2002.88.2.954. [DOI] [PubMed] [Google Scholar]
- Liebl DJ, Tessarollo L, Palko ME, Parada LF. Absence of sensory neurons before target innervation in brain-derived neurotrophic factor-, neurotrophin 3-, and TrkC-deficient embryonic mice. J Neurosci. 1997;17:9113–9121. doi: 10.1523/JNEUROSCI.17-23-09113.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu XJ, Gingrich JR, Vargas-Caballero M, Dong YN, Sengar A, Beggs S, Wang SH, Ding HK, Frankland PW, Salter MW. Treatment of inflammatory and neuropathic pain by uncoupling Src from the NMDA receptor complex. Nat Med. 2008;14:1325–1332. doi: 10.1038/nm.1883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu B. BDNF and activity-dependent synaptic modulation. Learn Mem. 2003;10:86–98. doi: 10.1101/lm.54603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacQueen GM, Ramakrishnan K, Croll SD, Siuciak JA, Yu G, Young LT, Fahnestock M. Performance of heterozygous brain-derived neurotrophic factor knockout mice on behavioral analogues of anxiety, nociception, and depression. Behav Neurosci. 2001;115:1145–1153. doi: 10.1037//0735-7044.115.5.1145. [DOI] [PubMed] [Google Scholar]
- Mammen AL, Huganir RL, O'Brien RJ. Redistribution and stabilization of cell surface glutamate receptors during synapse formation. J Neurosci. 1997;17:7351–7358. doi: 10.1523/JNEUROSCI.17-19-07351.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mannion RJ, Costigan M, Decosterd I, Amaya F, Ma QP, Holstege JC, Ji RR, Acheson A, Lindsay RM, Wilkinson GA, Woolf CJ. Neurotrophins: peripherally and centrally acting modulators of tactile stimulus-induced inflammatory pain hypersensitivity. Proc Natl Acad Sci U S A. 1999;96:9385–9390. doi: 10.1073/pnas.96.16.9385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazzuca M, Heurteaux C, Alloui A, Diochot S, Baron A, Voilley N, Blondeau N, Escoubas P, Gélot A, Cupo A, Zimmer A, Zimmer AM, Eschalier A, Lazdunski M. A tarantula peptide against pain via ASIC1a channels and opioid mechanisms. Nat Neurosci. 2007;10:943–945. doi: 10.1038/nn1940. [DOI] [PubMed] [Google Scholar]
- Merighi A, Salio C, Ghirri A, Lossi L, Ferrini F, Betelli C, Bardoni R. BDNF as a pain modulator. Prog Neurobiol. 2008;85:297–317. doi: 10.1016/j.pneurobio.2008.04.004. [DOI] [PubMed] [Google Scholar]
- Petrenko AB, Yamakura T, Baba H, Shimoji K. The role of N-methyl-d-aspartate (NMDA) receptors in pain: a review. Anesth Analg. 2003;97:1108–1116. doi: 10.1213/01.ANE.0000081061.12235.55. [DOI] [PubMed] [Google Scholar]
- Petroff EY, Price MP, Snitsarev V, Gong H, Korovkina V, Abboud FM, Welsh MJ. Acid-sensing ion channels interact with and inhibit BK K+ channels. Proc Natl Acad Sci U S A. 2008;105:3140–3144. doi: 10.1073/pnas.0712280105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pezet S, McMahon SB. Neurotrophins: mediators and modulators of pain. Annu Rev Neurosci. 2006;29:507–538. doi: 10.1146/annurev.neuro.29.051605.112929. [DOI] [PubMed] [Google Scholar]
- Pezet S, Malcangio M, Lever IJ, Perkinton MS, Thompson SW, Williams RJ, McMahon SB. Noxious stimulation induces Trk receptor and downstream ERK phosphorylation in spinal dorsal horn. Mol Cell Neurosci. 2002;21:684–695. doi: 10.1006/mcne.2002.1205. [DOI] [PubMed] [Google Scholar]
- Pezet S, Spyropoulos A, Williams RJ, McMahon SB. Activity-dependent phosphorylation of Akt/PKB in adult DRG neurons. Eur J Neurosci. 2005;21:1785–1797. doi: 10.1111/j.1460-9568.2005.04011.x. [DOI] [PubMed] [Google Scholar]
- Pezet S, Marchand F, D'Mello R, Grist J, Clark AK, Malcangio M, Dickenson AH, Williams RJ, McMahon SB. Phosphatidylinositol 3-kinase is a key mediator of central sensitization in painful inflammatory conditions. J Neurosci. 2008;28:4261–4270. doi: 10.1523/JNEUROSCI.5392-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandkühler J. Models and mechanisms of hyperalgesia and allodynia. Physiol Rev. 2009;89:707–758. doi: 10.1152/physrev.00025.2008. [DOI] [PubMed] [Google Scholar]
- Schwarze SR, Ho A, Vocero-Akbani A, Dowdy SF. In vivo protein transduction: delivery of a biologically active protein into the mouse. Science. 1999;285:1569–1572. doi: 10.1126/science.285.5433.1569. [DOI] [PubMed] [Google Scholar]
- Seal RP, Wang X, Guan Y, Raja SN, Woodbury CJ, Basbaum AI, Edwards RH. Injury-induced mechanical hypersensitivity requires C-low threshold mechanoreceptors. Nature. 2009;462:651–655. doi: 10.1038/nature08505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slack SE, Pezet S, McMahon SB, Thompson SW, Malcangio M. Brain-derived neurotrophic factor induces NMDA receptor subunit one phosphorylation via ERK and PKC in the rat spinal cord. Eur J Neurosci. 2004;20:1769–1778. doi: 10.1111/j.1460-9568.2004.03656.x. [DOI] [PubMed] [Google Scholar]
- Song HJ, Ming GL, Poo MM. cAMP-induced switching in turning direction of nerve growth cones. Nature. 1997;388:275–279. doi: 10.1038/40864. [DOI] [PubMed] [Google Scholar]
- Sun RQ, Tu YJ, Yan JY, Willis WD. Activation of protein kinase B/Akt signaling pathway contributes to mechanical hypersensitivity induced by capsaicin. Pain. 2006;120:86–96. doi: 10.1016/j.pain.2005.10.017. [DOI] [PubMed] [Google Scholar]
- Thoenen H. Neurotrophins and neuronal plasticity. Science. 1995;270:593–598. doi: 10.1126/science.270.5236.593. [DOI] [PubMed] [Google Scholar]
- Viard P, Butcher AJ, Halet G, Davies A, Nürnberg B, Heblich F, Dolphin AC. PI3K promotes voltage-dependent calcium channel trafficking to the plasma membrane. Nat Neurosci. 2004;7:939–946. doi: 10.1038/nn1300. [DOI] [PubMed] [Google Scholar]
- Voilley N, de Weille J, Mamet J, Lazdunski M. Nonsteroid anti-inflammatory drugs inhibit both the activity and the inflammation-induced expression of acid-sensing ion channels in nociceptors. J Neurosci. 2001;21:8026–8033. doi: 10.1523/JNEUROSCI.21-20-08026.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waldmann R, Champigny G, Bassilana F, Heurteaux C, Lazdunski M. A proton-gated cation channel involved in acid-sensing. Nature. 1997a;386:173–177. doi: 10.1038/386173a0. [DOI] [PubMed] [Google Scholar]
- Waldmann R, Bassilana F, de Weille J, Champigny G, Heurteaux C, Lazdunski M. Molecular cloning of a non-inactivating proton-gated Na+ channel specific for sensory neurons. J Biol Chem. 1997b;272:20975–20978. doi: 10.1074/jbc.272.34.20975. [DOI] [PubMed] [Google Scholar]
- Wang Q, Liu L, Pei L, Ju W, Ahmadian G, Lu J, Wang Y, Liu F, Wang YT. Control of synaptic strength, a novel function of Akt. Neuron. 2003;38:915–928. doi: 10.1016/s0896-6273(03)00356-8. [DOI] [PubMed] [Google Scholar]
- Waung MW, Pfeiffer BE, Nosyreva ED, Ronesi JA, Huber KM. Rapid translation of Arc/Arg3.1 selectively mediates mGluR-dependent LTD through persistent increases in AMPAR endocytosis rate. Neuron. 2008;59:84–97. doi: 10.1016/j.neuron.2008.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wemmie JA, Chen J, Askwith CC, Hruska-Hageman AM, Price MP, Nolan BC, Yoder PG, Lamani E, Hoshi T, Freeman JH, Jr, Welsh MJ. The acid-activated ion channel ASIC contributes to synaptic plasticity, learning, and memory. Neuron. 2002;34:463–477. doi: 10.1016/s0896-6273(02)00661-x. [DOI] [PubMed] [Google Scholar]
- Wemmie JA, Askwith CC, Lamani E, Cassell MD, Freeman JH, Jr, Welsh MJ. Acid-sensing ion channel 1 is localized in brain regions with high synaptic density and contributes to fear conditioning. J Neurosci. 2003;23:5496–5502. doi: 10.1523/JNEUROSCI.23-13-05496.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wemmie JA, Coryell MW, Askwith CC, Lamani E, Leonard AS, Sigmund CD, Welsh MJ. Overexpression of acid-sensing ion channel 1a in transgenic mice increases acquired fear-related behavior. Proc Natl Acad Sci U S A. 2004;101:3621–3626. doi: 10.1073/pnas.0308753101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willis WD. Role of neurotransmitters in sensitization of pain responses. Ann N Y Acad Sci. 2001;933:142–156. doi: 10.1111/j.1749-6632.2001.tb05821.x. [DOI] [PubMed] [Google Scholar]
- Woolf CJ. Overcoming obstacles to developing new analgesics. Nat Med. 2010;16:1241–1247. doi: 10.1038/nm.2230. [DOI] [PubMed] [Google Scholar]
- Woolf CJ, Salter MW. Neuronal plasticity: increasing the gain in pain. Science. 2000;288:1765–1769. doi: 10.1126/science.288.5472.1765. [DOI] [PubMed] [Google Scholar]
- Wu LJ, Duan B, Mei YD, Gao J, Chen JG, Zhuo M, Xu L, Wu M, Xu TL. Characterization of acid-sensing ion channels in dorsal horn neurons of rat spinal cord. J Biol Chem. 2004;279:43716–43724. doi: 10.1074/jbc.M403557200. [DOI] [PubMed] [Google Scholar]
- Xu Q, Fitzsimmons B, Steinauer J, Neill A, Newton AC, Hua XY, Yaksh TL. Spinal phosphinositide 3-kinase-Akt-mammalian target of rapamycin signaling cascades in inflammation-induced hyperalgesia. J Neurosci. 2011;31:2113–2124. doi: 10.1523/JNEUROSCI.2139-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu TL, Duan B. Calcium-permeable acid-sensing ion channel in nociceptive plasticity: a new target for pain control. Prog Neurobiol. 2009;87:171–180. doi: 10.1016/j.pneurobio.2009.01.004. [DOI] [PubMed] [Google Scholar]
- Xu TL, Nabekura J, Akaike N. Protein kinase C-mediated enhancement of glycine response in rat sacral dorsal commissural neurones by serotonin. J Physiol. 1996;496:491–501. doi: 10.1113/jphysiol.1996.sp021701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yajima Y, Narita M, Usui A, Kaneko C, Miyatake M, Narita M, Yamaguchi T, Tamaki H, Wachi H, Seyama Y, Suzuki T. Direct evidence for the involvement of brain-derived neurotrophic factor in the development of a neuropathic pain-like state in mice. J Neurochem. 2005;93:584–594. doi: 10.1111/j.1471-4159.2005.03045.x. [DOI] [PubMed] [Google Scholar]
- Yoshii A, Constantine-Paton M. BDNF induces transport of PSD-95 to dendrites through PI3K-AKT signaling after NMDA receptor activation. Nat Neurosci. 2007;10:702–711. doi: 10.1038/nn1903. [DOI] [PubMed] [Google Scholar]
- Yu Y, Chen Z, Li WG, Cao H, Feng EG, Yu F, Liu H, Jiang H, Xu TL. A nonproton ligand sensor in the acid-sensing ion channel. Neuron. 2010;68:61–72. doi: 10.1016/j.neuron.2010.09.001. [DOI] [PubMed] [Google Scholar]
- Zha XM, Wemmie JA, Green SH, Welsh MJ. Acid-sensing ion channel 1a is a postsynaptic proton receptor that affects the density of dendritic spines. Proc Natl Acad Sci U S A. 2006;103:16556–16561. doi: 10.1073/pnas.0608018103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zha XM, Costa V, Harding AM, Reznikov L, Benson CJ, Welsh MJ. ASIC2 subunits target acid-sensing ion channels to the synapse via an association with PSD-95. J Neurosci. 2009;29:8438–8446. doi: 10.1523/JNEUROSCI.1284-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao J, Seereeram A, Nassar MA, Levato A, Pezet S, Hathaway G, Morenilla-Palao C, Stirling C, Fitzgerald M, McMahon SB, Rios M, Wood JN. Nociceptor-derived brain-derived neurotrophic factor regulates acute and inflammatory but not neuropathic pain. Mol Cell Neurosci. 2006;31:539–548. doi: 10.1016/j.mcn.2005.11.008. [DOI] [PubMed] [Google Scholar]
- Zhuang ZY, Xu H, Clapham DE, Ji RR. Phosphatidylinositol 3-kinase activates ERK in primary sensory neurons and mediates inflammatory heat hyperalgesia through TRPV1 sensitization. J Neurosci. 2004;24:8300–8309. doi: 10.1523/JNEUROSCI.2893-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]