

SUMMARY
Murine norovirus (MNoV) infects a low percentage of enteric tuft cells and can persist in these cells for months following acute infection. Both tuft cell tropism and resistance to interferon-λ (IFN-λ)-mediated clearance during persistent infection require the viral nonstructural protein 1/2 (NS1/2). We show that processing of NS1/2 yields NS1, an unconventionally secreted viral protein that is central for IFN-λ resistance. MNoV infection globally suppresses intestinal IFN-λ responses, which is attributable to secreted NS1. MNoV NS1 secretion is triggered by Caspase-3 cleavage of NS1/2, and a secreted form of human NoV NS1 is also observed. NS1 secretion is essential for intestinal infection and resistance to IFN-λ in vivo. NS1 vaccination alone protects against MNoV challenge despite the lack of induction of neutralizing anti-capsid antibodies, previously shown to confer protection. Thus, despite infecting a low number of tuft cells, NS1 secretion allows MNoV to globally suppress IFN responses and promote persistence.
Keywords: Norovirus, vaccine, secretion, NS1, IFN-λ
Graphical Abstract
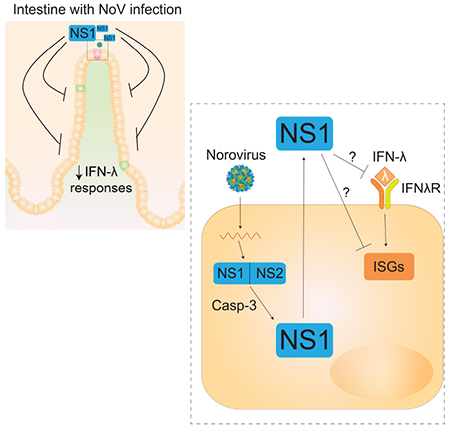
eTOC Blurb
Norovirus is an enteric virus that infects intestinal epithelial tuft cells in mice. Lee et al. discover that murine norovirus NS1 is an unconventionally secreted protein that overcomes IFN-λ-mediated norovirus control and is critical for intestinal infection in mice. Additionally, vaccination with NS1 alone confers protection, suggesting potential vaccine strategies.
INTRODUCTION
Human norovirus (HNoV) is the leading cause of viral gastroenteritis globally, causing over 70,000 deaths among patients under 5 years old annually (Lanata et al., 2013; St Clair and Patel, 2008). Currently there are no licensed vaccines or therapeutics available for this highly contagious human pathogen (Cortes-Penfield et al., 2017; Prasad et al., 2016). During the acute phase of infection HNoV causes abdominal pain, vomiting and diarrhea, which resolves within 1-2 days (Atmar et al., 2008; Rockx et al., 2002). After the acute phase, viral shedding into the stool can continue for several weeks in immunocompetent people (Gustavsson et al., 2017; Inaida et al., 2013; Kaufman et al., 2014; Teunis et al., 2015) and for years in immunocompromised individuals (Bok and Green, 2012). HNoV pathogenesis can be effectively modeled with murine norovirus (MNoV), which recapitulates several key features including viral shedding into the stool, fecal-oral transmission, intestinal epithelial cell (IEC) and myeloid cell tropism, and persistent infection for months after acute infection (Baldridge et al., 2016; Ettayebi et al., 2016; Gustavsson et al., 2017; Karandikar et al., 2016; Karst et al., 2014; Kaufman et al., 2014; Lee et al., 2017; Saito et al., 2014; Teunis et al., 2015; Wobus et al., 2004). Since MNoV readily infects mice to provide a tractable in vivo model for NoV studies, the MNoV model has facilitated identification of host immune and viral factors regulating NoV replication and pathogenesis in vivo (Baldridge et al., 2016).
Previous studies have shown that neither adaptive immunity from natural infection nor vaccination-elicited immunity are efficient at controlling NoV infection in humans or mice. NoV challenge studies in humans have shown that many individuals are equally susceptible to both primary and secondary infections, and preexisting serum antibodies to the viral capsid do not seem to be consistently associated with protective immunity (Johnson et al., 1990; Parrino et al., 1977). Vaccine trials using viral capsid proteins as immunogens have achieved some limited success in preventing HNoV infection and illness in humans. One clinical trial used HNoV-GI.1 virus-like particles (VLPs) and provided a 44% reduction in NoV infection and illness (Atmar et al., 2011). Another trial in humans using divalent GI.1 and GII.4 VLPs showed mild decreases in gastroenteritis severity and duration of viral shedding but did not reach statistical significance (Bernstein et al., 2015). Thus capsid-based vaccination may require optimization to elicit effective protection or may have inherent limitations in preventing HNoV infection. In mice, like in humans, MNoV can re-infect immunocompetent animal hosts multiple times under some conditions (Nice et al., 2015). T-cells and antibodies only exhibit limited efficacy in controlling intestinal virus despite being highly functional (Tomov et al., 2013; Tomov et al., 2017). However, it is unclear why adaptive immunity is poorly effective for controlling NoV infection.
While adaptive immunity has a limited role in controlling MNoV infection in the intestine, type III interferon (IFN-λ) is critically important for cell intrinsic innate immunity against MNoV infection. IFN-λ has specialized antiviral activities at intestinal epithelial barriers, and acts as a primary innate barrier against mucosally-transmitted viruses (Lazear et al., 2015). Endogenous IFN-λ suppresses MNoV replication in IECs several fold in wild-type mice (Baldridge et al., 2017). Treatment with exogenous IFN-λ effectively prevents and cures intestinal MNoV infection even in the absence of adaptive immunity, serving as an example of sterilizing innate immunity (Nice et al., 2015). These findings indicate a distinct role for IFN-λ in controlling intestinal MNoV replication that is independent of the antiviral activity of T-cells and antibodies.
The cellular tropism of viruses is a key factor determining viral pathogenesis and the interactions between viruses and host immune system. Recently, we discovered that a very small number of IECs are infected by MNoV and serve as the reservoir for fecal shedding and persistence (Lee et al., 2017). The rare IECs-infected by MNoV were further identified as tuft cells (Wilen et al., 2018), a specific subtype of IECs initiating type II immune responses in the intestine (Gerbe et al., 2016; Howitt et al., 2016; von Moltke et al., 2016). MNoV infects tuft cells but not other IEC types due to tuft cell-specific expression of the MNoV receptor CD300lf (Orchard et al., 2016; Wilen et al., 2018). The frequency of infected cells is extremely rare, less than 100 infected tuft cells per million IECs (<0.01% of IECs) at acute and persistent time points (Lee et al., 2017; Wilen et al., 2018). It is unclear whether the rarity of infected cells or the specific tuft cell characteristics are related to the differential contributions of innate and adaptive immunity in controlling viral infection in these cells. One hypothesis is that rare tuft cell infection by MNoV may serve as a biological niche which evades T-cells and antibody responses and acts as an immunoprivileged site (Best and Robertson, 2017).
Because intestinal NoV infection has such distinctive features, such as a tropism for rare IECs and differential contributions of innate and adaptive immune responses, a more complete understanding of how the host immune system controls NoV infection may guide development of vaccines and immunotherapeutics. In this study, we show that viral non-structural protein NS1 is unconventionally secreted via caspase-3 cleavage. This secreted NS1 is essential for intestinal pathogenesis of MNoV infection and resistance to endogenous IFN-λ. Most importantly, vaccination with NS1 provides enhanced protection compared to vaccination with the viral capsid, and thus NS1 represents a vaccine target.
RESULTS
MNoV infection globally suppresses interferon-responses in the intestine
To define host responses to MNoV infection in the intestine, we performed mRNA sequencing of intestinal tissues from uninfected wild-type mice or those infected with CR6, a persistent enteric strain of MNoV (Lee et al., 2017; Nice et al., 2013; Wilen et al., 2018). Gene expression in the ileum was assessed by gene set enrichment analysis (GSEA). Surprisingly, interferon-responsive genes including those induced by IFN-λ, IFN-α/β and IFN-γ were globally down-regulated during both acute (3 dpi) and persistent (35 dpi) infection (Figure 1A, B). The interferon-lambda response was the most negatively regulated gene-set (Figure 1C, D). Global down-regulation of interferon-stimulated genes (ISGs) was observed at 35 dpi and 3 dpi (Figure 1E and Figure S1). Given the importance of IFN-λ in control of MNoV in the intestine, we next tested whether the effect on ISGs by CR6 infection is mediated by IFN-λ acting on IECs. We employed IEC-specific Ifnlr1 conditional knock-out mice. CR6 infection down-regulated the expression of Ifitl, Oasla and Ifi35 in the ileum of control Ifnlr1f/f mice but did not have significant effect on their expression in Ifnlr1f/f-Villincre mice (Figure 1F), indicating that downregulation of IFN-λ-responsive genes in IECs contributes to global suppression of ISGs. The global repression of ISGs by CR6 infection is remarkable because we previously found that CR6 infects less than 100 tuft cells per million IECs (<0.01% of lECs) at both acute and persistent time points (Lee et al., 2017; Wilen et al., 2018). Given the extremely low frequency of infection in the intestine, the broad tissue-level effect on ISGs suggests a viral or host molecule(s) from the infected cells exerts a trans effect on broader IEC populations.
Figure 1. Global downregulation of ISG expression by CR6 infection in the intestine.
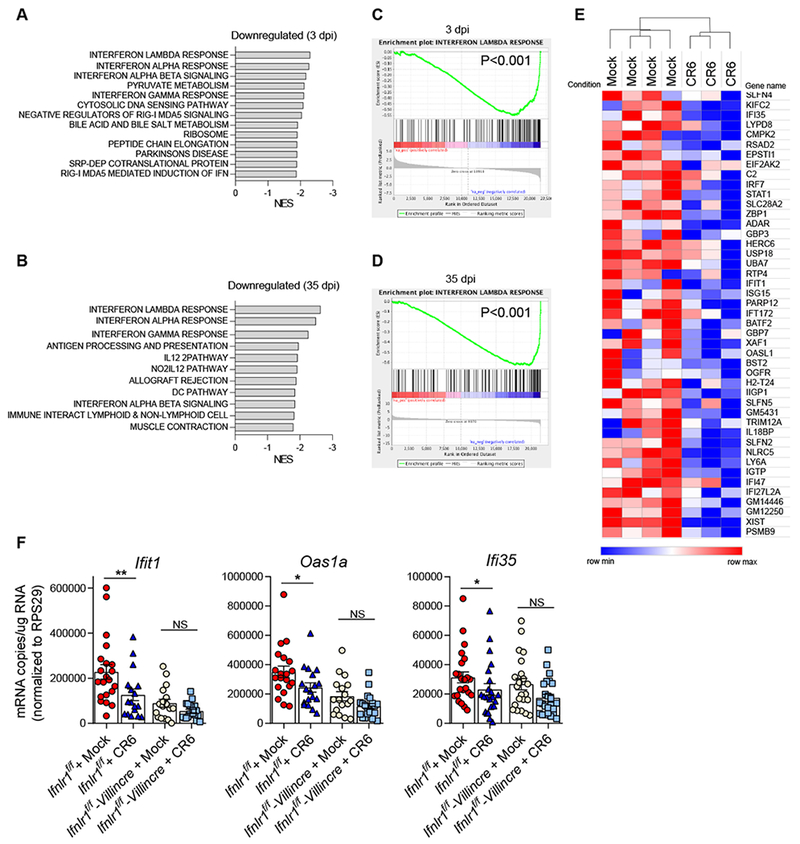
Wild-type mice were infected with 106 plaque forming units (PFU) of CR6 perorally. At 3 dpi and 35 dpi, ileum from the infected mice and the littermate control mice were collected for mRNA sequencing. (A-D) Host gene expression profile was analyzed by GSEA. Net-enrichment-scores (NES) of negatively affected gene-sets by CR6-infection at 3 dpi (A) and 35 dpi (B). Enrichment plots of Interferon lambda response genes at 3 dpi (C) and 35 dpi (D). (E) A heat-map showing downregulation of ISGs by CR6 infection at 35 dpi. (F) Ifnlr1f/f and Ifnlr1f/f-Villincre mice were infected with 106 PFU of CR6, and the expression of Ifit1, Oas1a and Ifi35 mRNA in ileum was analyzed at 35 dpi by qRT-PCR (n = 22-24 mice per group, combined from five independent experiments). Shown are means ± SEM. NS, not significant; *P < 0.05, **P < 0.01, determined by unpaired t-test. See also Figure S1.
NS1 is unconventionally secreted by Casp3-mediated cleavage during infection
Since we previously found that infection of tuft cells requires functional NS1 in addition to expression of the MNoV receptor CD300lf in tuft cells (Lee et al., 2017; Orchard et al., 2016; Wilen et al., 2018), and that CR6-infection exerts tissue-wide effects in the intestine (Figure 1), we hypothesized that NS1 is secreted to counteract the IFN-λ response. Norovirus open reading frame 1 (ORF1) translation generates a polyprotein containing NS1/2, which is initially processed by the viral protease (NS6) to release NS1/2 from the polyprotein. NS1/2 protein is further cleaved by Caspase-3 (Casp3) at two cleavage sites (Glu121 and Glu131) to produce NS1 and NS2, which were previously considered to be cytoplasmic nonstructural proteins (Figure 2A)(Sosnovtsev et al., 2006). BV2 cells, MNoV-permissive microglial cells, were infected with CR6 and the culture supernatant and total cell lysate were harvested from the cells. The cleaved form of NS1 (~17 kD) was detected in the supernatant at 12 and 16 hours post-infection (hpi) by immunoblot, but not in the lysate, while NS1/2 (~43 kD) was predominantly detected in the lysate (Figure 2B). The expression of NS1/2 peaked at 8 hpi in the lysate whereas NS1 in the supernatant was initially detected after 12 hpi. The relative distribution of NS1 and NS1/2 in the supernatant and the lysate indicated that NS1 is the major form in the supernatant, and NS1/2 is the major form in the lysate (Figure 2C). Expression of Flag-NS1/2CR6 protein in HEK293T cells was sufficient to generate secreted NS1, indicating that NS1 production does not require viral infection or expression of other viral genes (Figure 2D). To determine the mechanism of NS1 production, we generated viruses containing mutations at the predicted caspase-cleavage sites. Double mutant CR6D121/131G exhibited a profound loss of NS1 in the supernatant without affecting NS1/2 expression in the lysate. CR6D121G and CR6D131G single mutations both decreased NS1 production, indicating that both sites are functional, although the D121G mutation had a stronger effect on NS1 (Figure 2E). NS1 production was also blocked by treating cells with a pan-Caspase inhibitor, Q-VD-OPh (Figure 2F) and by genetically disrupting Casp3 in BV2 cells (Figure 2G), indicating that NS1 production is Casp3-mediated. The cleaved form of Casp3 was observed in BV2 cells during CR6 infection (Figure 2H). While most secreted proteins encode a signal peptide at the N-terminus (von Heijne, 1990), NS1 does not (Figure S2) (Nielsen, 2017). NS1-release was insensitive to treatment with Brefeldin A, which blocks the conventional secretory pathway through the Golgi apparatus (Figure 2I). These data indicate that NS1 is released through an unconventional Casp3-mediated secretion pathway. The infection-free Flag-NS1/2 expression system also enabled us to test secretion of the NS1/2 protein of HNoV GI.1, one of the two major genotypes of HNoV outbreaks (Ajami et al., 2014). Similar to NS1CR6, Flag-NS1/2GI.1 was cleaved to produce Flag-NS1GI.1 (~37 kD), and Flag-NS1GI.1 was enriched in the supernatant (Figure 2J). The expected size of NS1GI.1 without the Flag-tag is ~23 kD. This finding suggests conservation of NS1 secretion in HNoVs.
Figure 2. Caspase-3-mediated unconventional secretion of NS1 during MNoV infection.
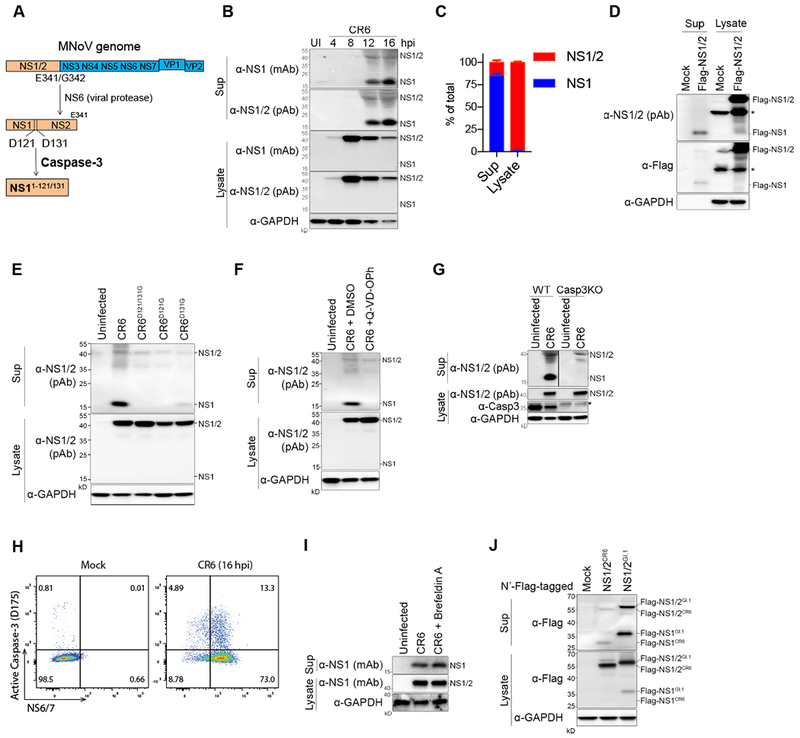
(A) Schematic depicting NS1 protein maturation. (B) Immunoblots showing NS1 secretion during CR6 infection in BV2 cells. (C) Relative expression of NS1 and NS1/2 in the supernatant (sup) and in the lysate. Band intensity of immunoblots at 12 hpi was measured from five independent experiments. Shown are means ± SD. (D) HEK293T cells were transfected with Flag-NS1/2-CR6, and Flag-NS1 secretion was examined 48 hours after transfection. (E) NS1 secretion at 12 hpi with Caspase-cleavage site mutant viruses. (F) BV2 cells were treated with Q-VD-OPh (20 nM) at 4 hpi and NS1 secretion was examined at 12 hpi. (G) Casp3KO BV2 cells were generated by CRISPR, and were infected with CR6 to access NS1 secretion. (H) Intracellular flow cytometry plots. CR6-infected BV2 cells were co-stained with cleaved-Casp3 and NS6/7 at 16 hpi. (I) Brefeldin A (5 ng/ml) treatment to block the conventional secretion pathway. The cells were incubated with Brefeldin A from 4 hpi and NS1 secretion was analyzed at 12 hpi. (J) HEK293T cells were transfected with Flag-NS1/2CR6 or Flag-NS1/2GI.I and Flag-NS1GI.1 cleavage and secretion was detected in the sup. Asterisk means non-specific bands. Representative data from at least three experiments are shown, except (I). See also Figure S2.
NS1 is secreted as a soluble monomer
To determine whether NS1 is secreted as a soluble protein or incorporated into the virion or vesicles, we performed size exclusion chromatography with the supernatant from CR6-infected BV2 cells using recombinant NS1 (rNS1) as a control (Figure 3A–B). Secreted NS1 was only detected in the low molecular weight fractions (12-15) in the same pattern as purified rNS1. VP1, the major viral capsid protein, was enriched in the intermediate (7-10) and high (3-5) molecular weight fractions. VP2, the minor capsid protein, known to be incorporated, into infectious virions, was only detected in the high molecular weight fraction (2-3). Therefore, NS1 is secreted as a soluble protein but not as a high-molecular weight vesicle or virion. Given the similar migration of rNS1 and secreted NS1 and the predicted molecular weight, secreted NS1 is likely a soluble monomer.
Figure 3. Molecular characterization of secreted NS1.
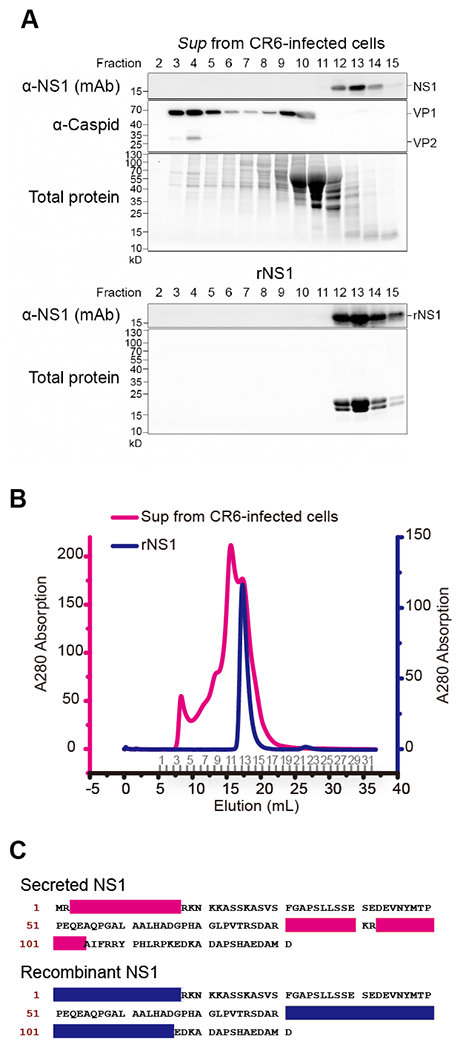
(A-B) Size exclusion chromatography with the culture supernatant from CR6-infected cells and rNS1 purified from E. coli. The samples were applied to Superdex 200 10/300 GL column for comparison. The corresponding fractions were collected in sequence as numbers indicated on the elution axis (B), then determined by either total protein staining or immunoblot with anti-NS1 and anti-capsid antibodies (A). (C) Molecular identification of secreted NS1. The sliced gel bands (Figure S3) were subjected to LC-MS/MS protein identification. The polypeptide fragments found in the MS/MS were mapped to each sample, respectively, as shown in the color used in (B). See also Figure S3.
To determine the molecular characteristics of secreted NS1, proteins from the size exclusion chromatography were subjected to LC-MS/MS (liquid chromatography with tandem mass spectrometry). Proteins at the expected size from the fractions 13 and 14 were sequenced as was rNS1 as a positive control (Figure S3). LC-MS/MS data from the supernatant mapped to amino acid sequences 3-17 and 81-104 of the NS1 protein (Figure 3C and Figure S3). The coverage of LC-MS/MS mapping using the supernatant was comparable to mapping using rNS1. In addition, secreted NS1 was detected by monoclonal antibody CM79, which recognizes the N-terminal epitope including amino acids 1-32 (Figure S3), and the N-terminally tagged NS1 was secreted with the intact tag (Figure 2D). Taken together, secreted NS1 has intact N-terminalm sequences.
NS1 secretion is essential for tuft cell infection but dispensable for non-tuft cell infection in mice
To determine the physiological role of NS1 secretion in viral replication, we assessed CR6D121/131G growth in vitro and in vivo. Mutations of caspase-cleavage sites had no effect on CR6 replication in BV2 cells (Figure 4A). In striking contrast, when CR6D121/131G was administered to mice perorally, the virus failed to replicate in the intestine or shed into the stool (Figure 4B). When CR6D121/131G was systemically delivered by intra-peritoneal injection, CR6D121/131G replicated as well as wild-type CR6 in the intestine and spleen (Figure 4C). Since tuft cells are the target cells for intestinal infection and viral shedding into stool (Wilen et al., 2018) and B-cells and myeloid cells are potential target cells for systemic infection (Grau et al., 2017; Jones et al., 2014; Wilen et al., 2018), these findings indicate that NS1 secretion is critical for enteric infection involving entry from the lumen of the intestine and tuft cell tropism, but dispensable for non-tuft cell tropism in mice. These data suggest NS1 secretion is necessary to overcome epithelial host defense barriers to establish productive infection.
Figure 4. NS1 secretion is critical for mucosal infection but dispensable for systemic infection in vivo.
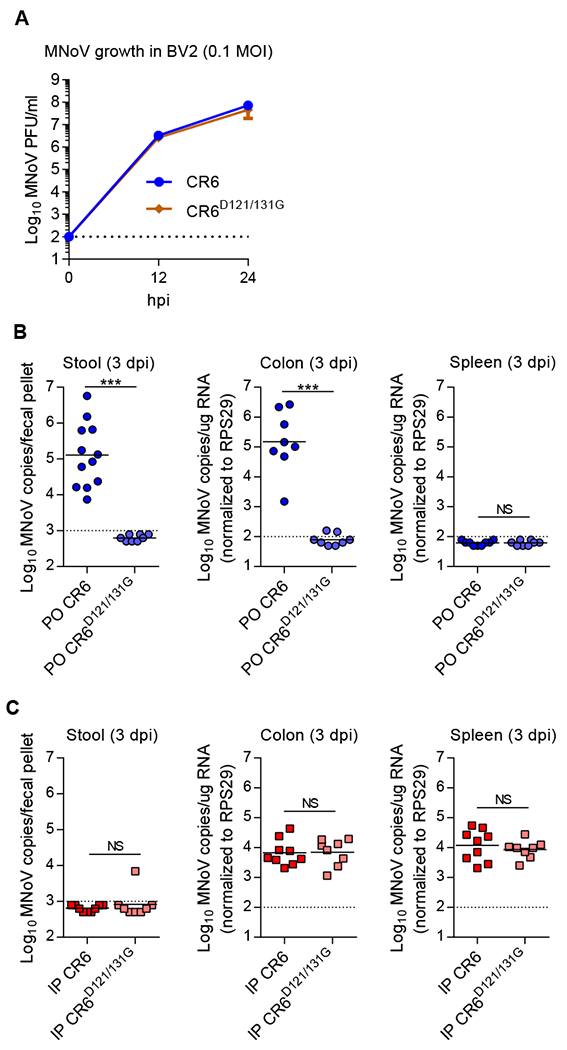
(A) BV2 cells were inoculated with CR6 or CR6D121/131G at MOI 0.1, and viral growth at 12 and 24 hpi was assessed by plaque assay. (B-C) Wild-type mice were infected with 106 PFU of the indicated viruses perorally (PO) (B) or intraperitoneally (IP) (C), and analyzed at 3 dpi. MNoV genomes in stool, colon and spleen were quantified by qRT-PCR (n = 8–11 mice per group, combined from two independent experiments). NS, not significant; ***P < 0.001, determined by Mann-Whitney test (B and C).
Secreted NS1 is critical for resistance to IFN-λ immunity in mice
Defective mutant viral replication can often be complemented by removal of the host pathway or gene regulated by a viral virulence protein (Leib et al., 2000). To determine whether removal of host genes complement the phenotype of CR6D121/131G, multiple immunodeficient mice, including Ifnlr1−/−, type III IFN receptor-deficient; Ifnar1−/−, type I IFN receptor-deficient; Ifnar1−/−, type II IFN receptor-deficient; Casp1/11−/−, inflammasome-deficient; and Rag1−/−, B- and T-cell-deficient, were infected with CR6D121/131G. Defective viral replication of CR6D121/131G was exclusively reversed in Ifnlr1−/− mice but not in the other knock-out mice tested (Figure 5A). Viral shedding of CR6D121/131G into stool was detected from acute (3 dpi) to persistent (14 dpi) time points in these mice lacking IFN-λ responsiveness (Figure 5B). CR6D121/131G infection in the intestine failed to downregulate the expression of ISGs in wild-type mice (Figure S4). CR6 Restored replication of CR6D121/131G was also observed in Ifnlr1f/f-Villincre mice by detection of shed virus (Figure 5C) and by quantification of CR6D121/131G-infected IECs (Figure 5D), indicating that deletion of IFN-λ signaling in IECs was sufficient to rescue attenuation of CR6D121/131G. We further confirmed that CR6D121/131G-infected IECs in Ifnlr1−/− mice are tuft cells (DCLK1-positive) by immunofluorescence (Figure 5E).
Figure 5. The defective viral growth of CR6D121/131G is complemented in IFN-λ signaling deficient mice.
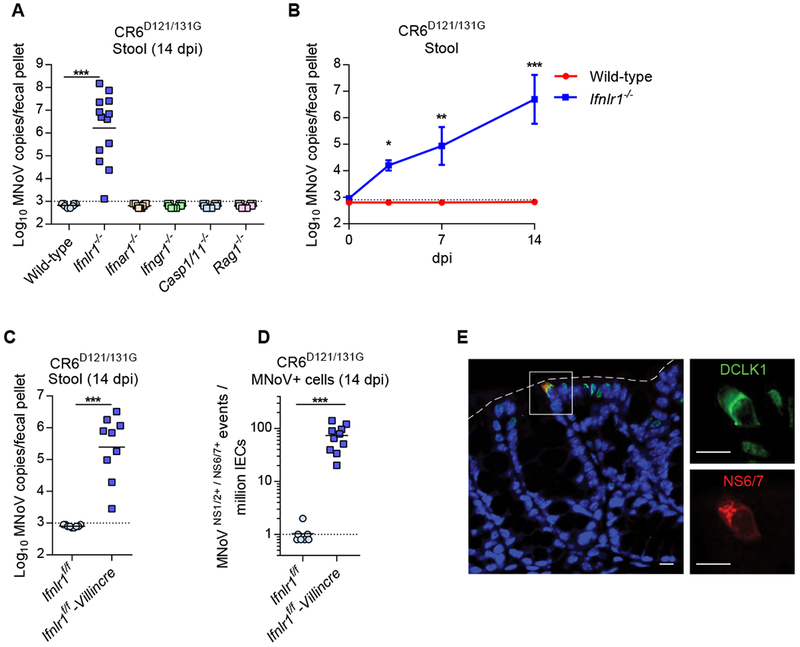
(A-E) Wild-type and the knock out mice were infected with 107 PFU of CR6D121/131G perorally. (A) Complemented viral infection of CR6D121/131G in Ifnlr1−/− mice (n = 9-13, combined from three independent experiments). (B) Viral shedding of CR6D121/131G at 3, 7 and 14 dpi in Ifnlr1−/− mice (n = 5-6, combined from two independent experiments). (C) CR6D121/131G stool shedding in Ifnlr1f/f-Villincre mice. (D) Quantification of MNoV+ cells (NS1/2+NS6/7+) in IECs (CD45−EpCAM+) by flow cytometry. (E-F) n = 9-10, combined from two independent experiments. (E) MNoV-NS6/7 co-localizes with DCLK1, a tuft cell marker, in the colon of Ifnlr1−/− mice infected with CR6D121/131G at 14 dpi. Images are representative of one of at least three independent experiments. Dashed lines represent the epithelial barrier. White boxes in the overlaid image reflect the magnified inset images. Scale bars, 10 microns. *P < 0.05; **P < 0.01; ***P < 0.001, determined by Mann-Whitney test (A, C, and D) or two-way ANOVA (B).
MNoV strain CW3 is known to induce the expression of IFN-β, IFN-λ and ISGs in the intestine (Nice et al., 2015), and we showed CR6D121/131G-infection does not suppress the ISG responses in response to CR6 infection. We therefore examined whether co-infection with CR6 or CR6D121/131G together with CW3 would inhibit CW3-induced ISG expression (Figure S4). While CR6 single-infection significantly suppressed ISG responses we did not observe an effect of CR6 infection in trans on responses to CW3 infection.
To determine whether NS1 directly inhibits IFN-λ activity or IFN-λ receptor signaling, we employed q mouse intestinal epithelial cell line encoding an ISG-reporter construct (i.e., Mx2LUC-IEC) (Figure S4). The Mx2LUC-IEC cells were pretreated with NS1 or C12R, the known viral decoy receptor for IFN-α (Smith and Alcami, 2002), then treated with IFN-α or IFN-λ. While C12R completely blocked the induction of Mx2-reporter activity specifically with IFN-α treatment, NS1 did not exhibit any inhibitory effect on IFN-α or IFN-λ. These negative data do not rule out an effect of NS1 on IFN signaling as the biology of the cell line may not reflect the biology of primary cells in the intestine
In summary, defective NS1 secretion prevents intestinal infection in mice via loss of tuft cell tropism, and infection of tuft cells was rescued in IFN-λ signaling-deficient mice. These data indicate a physiologically relevant antagonistic relationship between secreted NS1 and the IFN-λ-response, and are consistent with data indicating the global suppression of ISGs in the intestine of CR6-infected mice despite infecting only a few IECs.
Vaccination of NS1 protects mice from MNoV infection
Since NS1 is a secreted viral protein and is critical for pathogenesis in the intestine, we next tested whether NS1 is immunogenic during MNoV-infection and whether NS1 could be targeted with a vaccine. We thus assessed anti-NS1 and -VP1 serum responses in wild-type mice after both natural infection (35 dpi with perorally-administered CR6), and with immunization, 6 weeks after administration of Poly I:C adjuvant with NS1 alone, VP1-P-domain (VP1P-dom) alone, or a combination of both proteins. The P-domain of HNoV-VP1 has been considered an important immunogen for HNoV vaccine development, and neutralizing antibodies against MNoV-VP1 has been shown to limit viral infection (Chachu et al., 2008; Katpally et al., 2008; Tan et al., 2011). After three immunizations at 14 day intervals, serum was collected from the immunized mice and mice were infected with CR6 perorally (Figure 6A). Indeed, NS1 was immunogenic during natural CR6 infection. The level of α-NS1 IgG (Figure 6B) and IgA (Figure S4) in the serum was significantly increased with natural CR6 infection. Importantly, NS1 immunization significantly increased the level of α-NS1 IgG in the serum to more than 1000-fold the levels induced by natural CR6-infection (Figure 6B). The level of α-NS1 IgA in the serum was comparable between the infected group and the NS1-immunized group (Figure S4). The level of α-VP1 IgG was comparable between the infected group and the VP1P-dom-immunized group (Figure 6C). These data suggest that NS1 is not a potent immunogen during natural infection, but that α-NS1 IgG can be induced by NS1-immunization.
Figure 6. NS1-immunization prevents MNoV infection in vivo.
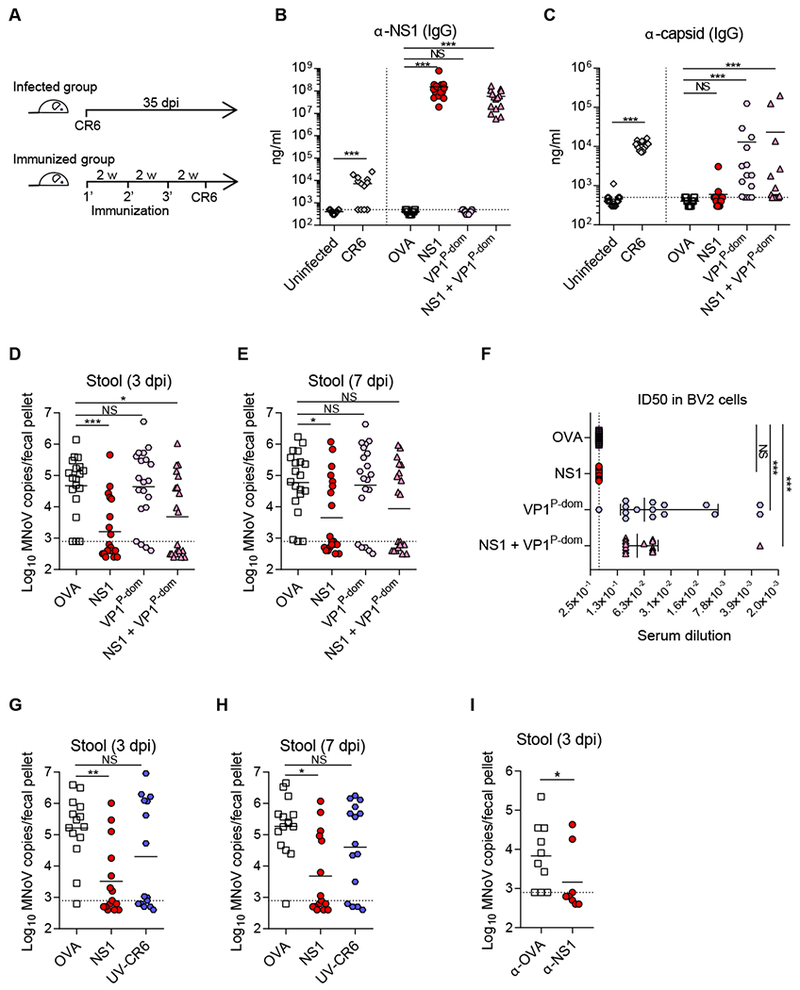
(A) Schematic outline of infection and immunization. (B-C) Concentration of α-NSI IgG (B) and α-capsid IgG (C) in the serum was measured by ELISA, and normalized by monoclonal antibodies CM79 and A6.2 respectively. (D-E) NS1-vaccination protected the mice from CR6-infection. MNoV genome from stool was quantified by qRT-PCR at 3 dpi (D) and 7 dpi (E). (F) VP1P-dom immune-sera protect CR6-infection in BV2 cells. ID50 values for protection were determined by serial dilution of the immune-sera from 1:10 to 1:100,000. (G-H) Vaccination of NS1 or UV-CR6. MNoV genome from stool was quantified by qRT-PCR at 3 dpi (G) and 7 dpi (H). (I) Passive serum transfer protected the mice from CR6 infection. Serum from the immunized mice with OVA or NS1 was transferred to naïve mice 1 week prior to CR6 infection. MNoV genome from stool was quantified by qRT-PCR at 3 dpi. (B, C, F) n = 14-15 per group, combined from three independent experiments. (D-E) n = 19-20 per group, combined from four independent experiments. (G-H) n = 10 per group, combined from two independent experiments. (I) n = 9-10 per group, combined from two independent experiments. Shown are means ± SEM. NS, not significant; *P < 0.05; **P < 0.01; ***P < 0.001, determined by Mann-Whitney test (B-C infected group, I) or Kruskal-Wallis test (B-C immunized group, D-H). See also Figure S4 and Figure S5.
We found that NS1-immunization efficiently prevented CR6 infection while VP1P-dom immunization failed to generate statistically significant protection (Figure 6D–E). There was no additive protective effect observed after combining NS1 and vP1P-dom immunization compared to immunizing with NS1 alone. The protective effect of NS1 immunization alone was also confirmed using Freund’s adjuvant (Figure S5). Even though vP1P-dom-immunized mice were not protected from CR6 infection, vP1P-dom-immune sera were still able to neutralize CR6 infection in BV2 cells (Figure 6F). In vitro, the NS1-immune sera exhibited no protection despite its activity as a vaccine antigen, indicating that NS1-immunization does not act by directly inhibiting viral replication in cells. This is consistent with the indistinguishable viral growth of wild-type and NS1-secretion-defective CR6 in BV2 cells which indicates a lack of a genetic requirement for NS1 secretion in replication per se (Figure 4A). NS1-vaccinated mice were not protected against the acute strain, CW3, infection in the intestinal and extra-intestinal organs (Figure S5), which is consistent with mucosal tissue specific protection of NS1-vaccination since CW3 does not exhibit tuft-cell tropism. It may also be that the protective epitopes are not shared between NS1 proteins of CW3 and CR6. In addition to the VP1P-dom immunogen, we used UV-irradiated CR6 (UV-CR6) as another capsid based-immunogen to mimic a VLP vaccine. Vaccination with UV-CR6 partially protected the mice but failed to reach statistical significance, whereas vaccination with NS1 significantly protected the mice from CR6 infection (Figure 6G, H). The protection of NS1 vaccination was also reconstituted by passively transferring anti-NS1 serum from the NS1-immunized mice to the unimmunized mice, indicating a-NS1 antibody response is sufficient to protect the mice against CR6 infection (Figure 6I). Taken together, natural MNoV-infection induces an NS1 antibody response, and NS1-immunization greatly amplifies the α-NS1 IgG response and can efficiently prevent mucosal infection in mice.
DISCUSSION
Here, we discovered that NS1, previously thought of as an intracellular protein, is a secreted protein that confers intestinal pathogenicity upon MNoV. NS1 secretion was specifically critical for MNoV infection of tuft cells but dispensable for non-tuft cell infection in vivo and in vitro. The NS1 secretion-defective virus completely lost infectivity with the loss of tuft cell tropism in wild-type mice, but its infectivity was restored in IFN-λ signaling-deficient mice. The combination of the tissue-wide effect of infection on IFN-responses and our genetic studies using mutant viruses and mice indicates that secreted NS1 may allow the virus to evade IFN-λ-induced responses in IECs. These data therefore support the concept that secreted NS1 is required to overcome epithelial host defense barriers mediated by IFN-λ. Most importantly, this study supports the use of NS1, a previously unrecognized target for NoV vaccine development, as an unconventional NoV vaccine strategy targeting a protein required for the critical initial steps of MNoV infection of the intestine.
The NS1 vaccine uncouples the traditional link between in vivo vaccine efficacy and in vitro antiviral activity, that has served as a reasonable basis for using NoV virus-like particles derived from the major capsid protein VP1 as a vaccine. In this study, we tested two different formats of capsid-based vaccines. One was the recombinant protein of the major capsid protein (VP1P-dom), and the other was UV-irradiated CR6. Vaccination with either partially prevented MNoV infection, but both failed to reach statistical significance under conditions in which NS1 vaccination was effective. Interestingly, the capsid vaccine successfully generated robust anti-capsid antibody levels in mice, and those antibodies neutralized MNoV in vitro despite the lack of efficacy.
The NS1 vaccine had very distinct effects compared to the capsid protein vaccines. Vaccination with NS1 alone efficiently protected mice from MNoV-infection even though the anti-NS1 sera exhibited no antiviral activity in cells. This phenotype is consistent with the differential infectivity of the NS1 secretion-defective CR6D121/131G virus in mice and cells. The NS1 secretion defect specifically dampened mucosal infection but not systemic infection in mice, and didn’t affect viral replication in cultured cells. Thus, our two distinct experimental approaches of NS1 vaccination and mutation of the NS1 cleavage site, combined to demonstrate that targeting NS1 effectively inhibits MNoV infection in the intestine without affecting viral replication in cultured cells of the myeloid lineage. It is important to note that the NS1 region is a less conserved genomic region (Baker et al., 2012; McCune et al., 2017). The NS1 region, compared to the NS2 region, exhibits amino acid variations between the MNoV strains, and more dramatic sequence diversity between NS1 of HNoVs versus MNoVs (Figure S6). The low conservation of NS1 compared to other parts of the norovirus genome suggest that this genomic region is under strong evolutionary pressure and may influence the viability of NS1 as a vaccine antigen.
It is intriguing that the capsid-based vaccines exhibited poor efficacy despite inducing neutralizing antibodies, and that MNoV efficiently evaded circulating neutralizing antibodies in mice. Recent studies from our group (Lee et al., 2017) and the Wherry group (Tomov et al., 2017) have suggested that adaptive immune responses by T- and B-cells are largely ineffective against ongoing MNoV replication in tuft cells. Immunostaining of MNoV-infected tuft cells in the intestine reveals that there is no detectable disruption of the epithelial barrier or infiltration of inflammatory cells around infected cells (Lee et al., 2017; Wilen et al., 2018), indicating minimal immune responses following MNoV-infection of tuft cells. These studies suggest that tuft cell infection may serve as a biological niche to evade adaptive immune responses, with our data here further supporting this concept in this case by showing that the initial steps in infection are not well controlled by high titer neutralizing antibodies in the circulation. This immune-ignorant phenotype might be related to the rarity of infected cells or specific aspects of tuft cell infection such as the lack of antigen presentation or limitation of viral antigen expression on the basal side of tuft cells.
MNoV NS1 secretion is unconventional that it is driven by Casp3-cleavage, and does not require a signal peptide or the conventional ER-Golgi secretion pathway. Since MNoV utilizes active Casp3 for NS1 cleavage in cultured microglial-like cells, it would be interesting to determine whether NS1 secretion is coupled with apoptosis in primary cells in vivo. Since intestinal MNoV infection was completely blocked by abolishing NS1 secretion, small molecules targeting secretion or molecular actions of NS1 may represent a potential approach to anti-NoV therapeutics. Identification of the secretion pathway for NS1 and the mechanisms underlying NS1 activity may allow design therapeutics targeting the NS1-IFN-λ axis. Targeting NS1 may represent an additional approach to the development antivirals that complements approaches targeting essential viral proteins such as the polymerase or protease.
The mRNA sequencing data in this study revealed that MNoV infection in the intestine exhibits a tissue-wide effect on ISG expression. Given the extremely low frequency of infected cells in the epithelial layer, this tissue-wide suppression of IFN-responsive genes was remarkable. It has been reported that MNoV infection is associated with other tissue-wide changes in the intestine. For example, MNoV infection is associated with triggering inflammatory bowel diseases (IBD) in genetically susceptible hosts (Basic et al., 2014; Cadwell et al., 2010), and has a tissue-wide beneficial effect on intestinal villus generation and immune remodeling in germ-free mice (Kernbauer et al., 2014). It would be interesting to investigate whether secreted NS1 plays a role in these broad NoV-associated effects on the intestine in disease-susceptible or microbiota-deficient animals.
STAR METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Herbert W. Virgin (virgin@wustl.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mice
Wild-type C57BL/6J mice were purchased from Jackson Laboratories (Bar Harbor, ME) (stock number 000664) and housed at the Washington University School of Medicine under specific-pathogen-free conditions according to university guidelines. Animal protocols were approved by the Washington University Animal Studies Committee. Knock-out mice on the C57BL/6J background were maintained in the same conditions and included the following strains: Ifnar1−/− (B6.129.Ifnar1tm1) (Muller et al., 1994), Ifngr1−/− (JAX B6.129S7-Ifngr1tm1Agt/J, stock #003288) (Huang et al., 1993), Casp 1/11−/− (JAX B6N.129S2-Casp1tm1Flv/J) (Kuida et al., 1995) and Rag1−/− (B6.129S7-Rag1tm1Mom/J) (Mombaerts et al., 1992). Ifnlr1f/f mice were generated from Ifnlr1tm1a(EUCOMM)Wtsi ES cells as described previously (Baldridge et al., 2017). Ifnlr1f/f mice were also crossed to a Deleter-Cre line (Schwenk et al., 1995) to generate an alternate Ifnlr1−/− line. Ifnlr1f/f-Villincre mice were generated by crossing Ifnlr1f/f to Villin-Cre mice for selective disruption of Ifnlr1 in intestinal epithelial cells (Madison et al., 2002). In this study, equal ratios of adult male and female mice, aged 6 to 14 weeks, were used in all experiments for all listed strains. Experimental mice were cohoused with up to 5 mice of the same sex per cage with autoclaved standard chow pellets and water provided ad libitum.
Cells
BV2 cells (derived from female mice) and HEK293T (derived from human female embryonic kidney) cells were cultured in Dulbecco’s Modified Eagle Medium (DMEM, Gibco) with 10% fetal bovine serum (FBS), 1% HEPES. For the viral growth assay and transfection, the complete media was used. For the NS1-secretion assay, VP-SFM with 1% HEPES was used. Cell lines were authenticated by microscopic morphologic evaluation and screening for mycoplasma.
Viruses
Stocks of MNoV strains CR6, CW3 and CR6D121/131G were generated from molecular clones as previously described (Strong et al., 2012). Briefly, a plasmid encoding the viral genome was transfected into HEK293T cells to generate a P0 stock, which was subsequently passaged on BV2 cells. After two passages in BV2 cells, cell cultures were frozen and thawed to liberate virions, then cultures were cleared of cellular debris and virus was concentrated by ultracentrifugation through a 30% sucrose cushion. Titers of all virus stocks were determined by plaque assay on BV2 cells.
METHOD DETAILS
MNoV infection in mice
For MNoV infections, mice were inoculated with a dose of 106 or 107 PFU of the indicated viral strain at 6 to 10 weeks of age by the oral route in a volume of 25 μl or the intraperitoneal route in a volume of 300 μl PBS. Stool and tissues were harvested into 2-ml tubes (Sarstedt, Germany) with 1-mm-diameter zirconia/silica beads (Biospec, Bartlesville, OK). Tissues were flash frozen in a bath of ethanol and dry ice and either processed on the same day or stored at −80°C.
Immunization
Wild-type mice were immunized subcutaneously with Ovalbumin (OVA), recombinant NS1 (rNS1), recombinant VPI-P-domain (rVP1Pdom) or a combination of rNS1 and rVP1Pdom. Mice were immunized three times with 2 week intervals between immunizations, then infected with 106 PFU of MNoV perorally. For all three immunization with Poly I:C as an adjuvant, 100 μg of Poly I:C (Invivogen) was pre-mixed with the immunogens in 150 μl of 1× HBSS, and the mixture was inoculated subcutaneously. For the first immunization, 30 μg of the immunogen was used, and for the second and third immunization, 15 μg of each protein was used. For the first immunization with Freund’s adjuvant, 100 μl of complete Freund’s adjuvant was pre-mixed with 30 μg of the immunogen in 100 μl 1× PBS, and the mixture was inoculated subcutaneously. For the second and third immunizations with Freund’s adjuvant, 100 μl of incomplete Freund’s adjuvant was pre-mixed with 15 μg of the immunogen in 100 μl of 1μ PBS, and the mixture was use for immunization.
For UV-irradiation of CR6, a micro-tube containing 5 × 107 PFU of CR6 in 50 ul of 1× PBS was incubated in the UV-chamber (Spectronics Corporation) at 300 mJ/cm2. The UV-irradiated CR6 was used for immunization with Poly I:C as describe before. Serum samples from mice were inactivated at 56°C fo r 30 min, then stored at −80°C until further analysis.
RNAseq data generation and analysis
Wild-type littermate C57BL/6J mice were mock-infected or infected with 106 PFU of CR6, then were sacrificed at 3 dpi or 35 dpi. Total RNA from ileum was purified, an Illumina sequencing library was generated and run on an Illumina HiSeq as previously described (Park et al., 2016). Three to four mice were included in each group. Differentially expressed genes were identified using DESeq2 (Love et al., 2014). Gene set enrichment analysis was performed as previously described (Park et al., 2016). To generate an Interferon lambda response gene set, the differential gene expression results from two independent experiments were used. Wild-type mice were intraperitoneally inoculated with either mock or IFN-λ (3 or 25 μg dose). Ileum tissues were collected one day after inoculation. The genes included in the Interferon-lambda-response gene set were all significantly differentially expressed (i.e., absolute value of log2 fold-change greater than 1 and p-value less than 0.05). The Interferon-lambda-response gene set was used to perform gene set enrichment analysis (Table S1). RNA-seq data were deposited to the European Nucleotide Archive under accession number PRJEB27021.
Cell culture and MNoV infection
For MNoV infection, BV2 cells were seeded at 1.0 × 106 cells/well of a 6-well plate. After 16 hours, the cells were infected with MNoV in 500 μl volume for 1 hour with gentle shaking at room temperature. Viral inoculum was removed and 2 ml of the media was added to the cells. For the viral growth assay, the complete media was added after virus inoculation. For the NS1-secretion assay, VP-SFM with 1% HEPES was added. At 12 and 24 hpi, the culture supernatant was harvested for the plaque assay.
For Flag-NS1/2 plasmids transfection, HEK293T cells were seeded at 2.0 × 105 cells/well of a 6-well plate. After 16 hours, cells were transfected with 2 ug of plasmid plus Lipofectamine 2000 (Invitrogen) according to the manufacturer’s protocol. Twenty four hours after transfection, the media were replaced with VP-SFM with 1% HEPES. After 16~24 hours changing the media, the supernatant and whole cell lysate were harvested for the further assays.
For NS1 treatment and Mx2-reporter assay, Mx2LUC-IEC cells were seeded at 3.0 × 104 cells/well of a 96-well white wall plate. After 16 hours, cells were pre-treated with IgG, C12R, or rNS1 at 100 ng/ml for 30 min in the CO2 incubator, then cells were treated with IFN-α or IFN-λ. After 24 hours, cells were harvested for luciferase assay (Promega).
Generating Casp3 KO BV2 cell line
Casp3 KO BV2 cell line (clone 2E10) was generated at the Genome Engineering and iPSC center at Washington University School of Medicine as previously described (Orchard et al., 2016). sgRNA (5′-ATCTCGCTCTGGTACGGATG-3′) targeting Casp3 (NCBI gene ID: 12367) was nucleofected with Cas9 into wild-type BV2 cells. Clones were screened for frameshifts by sequencing the target region with Illumina MiSeq at approximately 500× coverage.
| Amino acid | G M S S R S G T R V D A A N L |
| Wild-type | GGAATGTCATCTCGCTCTGGTACGGATGTGGACGCAGCCAACCTC |
| 2E10 allele 1 | GGAATGTCATCTCGC----GTACGGATGTGGACGCAGCCAACCTC |
| 2E10 allele 2 | GGAATGT-----------GGTACGGATGTGGACGCAGCCAACCTC |
| 2E10 allele 3 | GGAATGTCATCTCGCTCTG-TACGGATGTGGACGCAGCCAACCTC |
| 2E10 allele 4 | GGAATGTCATCTCGCTCT--TACGGATGTGGACGCAGCCAACCTC |
Detection of secreted NS1
The culture supernatant was spun down and filtered with a 0.2 μm syringe filter to remove cellular debris. The filtered supernatant was precipitated with Trichloroacetic acid (TCA, Sigma-Aldrich T9159) at final 20% concentration by volume at 4°C overnight. TCA-precipitated supernatant was centrifuged at 18,000 g for 10 min at 4°C and washed with ice-cold acetone. The protein pellets were re-suspended in 1× SDS reducing sample buffer and boiled for 5 min. Cells were lysed in RIPA buffer with the protease/phosphatase inhibitor cocktail (Invitrogen) for 10 min at 4°C and spun down at 13,000 rpm for 20 min. Protein concentration of the cell lysate was measured by Pierce BCA assay (ThermoScientific). Protein expression in the supernatant and the total cell lysate was analyzed by immunoblot. Rabbit polyclonal anti-NS1/2 (provided by Vernon Ward) and mouse monoclonal anti-NS1 (CM79) were used for immunoblot.
MNoV plaque assay
Culture supernatants from infected cells were harvested at 12 and 24 hpi and frozen at −80°C. BV2 cells were seeded at 1 × 10 6 cells/well of a six-well plate. 16 hours after seeding, 10-fold serially diluted samples were applied to each well for 1 hour with gentle rocking. Viral inoculum was aspirated and 2 ml of the methylcellulose media was added (MEM, 10% FBS, 2mM L-Gluatmine, 10 mM HEPES, and 1% methylcellulose). Plates were incubated for 60-72 hours prior to visualization with crystal violet solution (0.2% crystal violet and 20% ethanol).
Quantitative reverse transcription-PCR
As previously described (Lee et al., 2017), RNA was isolated from stool using a ZR-96 viral RNA kit (Zymo Research, Irvine, CA). RNA from tissues was isolated using TRI Reagent (Invitrogen) with a Direct-zol-96 RNA kit (Zymo Research, Irvine, CA) according to the manufacturer’s protocol. 5 μl of RNA from stool or 1 μg of total RNA from tissue was used for reverse transcription with the ImPromll reverse transcriptase enzyme (Promega, Madison, WI). MNoV TaqMan assays were performed, using a standard curve for determination of absolute viral genome copies. Primers for Ifit1 (Mm.PT.58.32674307), Oas1a (Mm.PT.58.30459792) and Ifi35 (Mm.PT.58.42591180) were purchased from IDT (Coralville, Iowa). Quantitative PCR for housekeeping gene Rps29 was performed as previously described (Baldridge et al., 2017). Standard curves for quantitative PCR assays were used to facilitate absolute quantification of transcript copy numbers.
Recombinant protein expression and purification
Construct sequence of VP1 P-domain plus hinge (219aa~541aa) was synthesized and codon optimized for expression in E. coli. This sequence was cloned into a modified pET28B vector containing a His-MBP fusion (kind gift of Dr. Neal Alto) and transformed into BL21 (DE3) E. coli. Protein was induced at an O.D. 600 of 0.6 with IPTG and incubated for 16-18 hours at 16C. Afterwards, bacterial pellets were harvested, lysed, and purified on a Nickel NTA column prior to buffer exchange to PBS and concentration.
The NS1 protein was constructed with maltose binding protein fusion in the N-terminal. The constructs were confirmed by sequencing and followed by protein expression in Escherichia coli BL21 (DE3) with IPTG induction at 18 °C overnight. Cells were pelleted by centrifugation at 10,000 g, re-suspended in lysis buffer (50 mM Tris-HCl pH=8.0, 300 mM NaCl), lysed using an EmulsiFlex-C5 homogenizer (Avestin, Canada) and centrifuged at 60,000 g to remove cell debris and precipitation. The resultant supernatant was purified using a series of affinity, ion exchange and gel filtration chromatographic columns (GE Healthcare). The MBP tag was removed after the affinity purification by TEV protease digestion. Final protein samples were subject to purity check by SDS-PAGE and concentration measurement by NanoDrop2000c spectrophotometer (Thermo Scientific).
Size exclusion chromatography
Samples from mammalian cell secretion medium were centrifuged to remove cells and debris then filtered through 0.22 μm filter unit (Merck Millipore). The clarified medium was concentrated 120× fold and buffer-exchanged to SEC buffer (25 mM Tris-HCl pH=8.0, 150 mM NaCl) via centrifugal filters (Merck Millipore). The sample was then applied to a Superdex 200 10/300 GL column (GE Healthcare) pre-equilibrated with SEC buffer. Samples from recombinant expression were also buffer-exchanged to the SEC buffer before being applied onto the column. Fractions were collected (1 mL per tube) and analyzed by SDS-PAGE with either Coomassie blue stain or immunoblot with anti-NS1 antibody.
Protein identification with mass spectrometry
Gel bands corresponding to NS1 protein were excised from the gel and digested with trypsin on the Investigator ProGest (Genomic Solutions) automated digester. The peptides were then separated by liquid chromatography (LC) and analyzed by MS/MS with Q Exactive Hybrid Quadrupole-Orbitrap Mass Spectrometer (Thermo Scientific) by fragmenting each peptide. The resulting intact and fragmentation pattern is compared to a theoretical fragmentation pattern from either MASCOT or Sequest HT to find peptides that can be used to identify the protein. The data were analyzed with the proteome discoverer version 2.0 program to identify target protein fragments.
Flow cytometry for MNoV+ intestinal epithelial cells and MNoV-infected BV2 cells
For IEC staining, the epithelial fraction of the proximal colon was isolated and stained as previously described (Lee et al., 2017). In brief, after mice were euthanized, proximal colons were collected. The tissues were washed with cold PBS twice, then chopped and transferred to new tubes. The tissues were incubated with stripping buffer (10% fetal bovine serum, 15 mM HEPES, 5 mM EDTA, 5 mM DTT in 1× HBSS) for 20 min at 37 °C and vortexed for 15 seconds at maximum setting. The dissociated cells were passed through 100 μm and 40 μm filter mesh and washed with cold PBS. The collected cells were stained with a live/dead staining kit (live/dead fixable blue dead cell stain kit, Life Technology). After live/dead staining, cells were stained with anti-EpCam-APC-Cy7 (clone G8.8, Biolegend) and anti-CD45-PacBlue (clone 30-F11, Biolegend) for 20 minutes on ice, then washed with wash buffer (2% FBS, 1 mM EDTA in PBS). For intracellular staining, cells were fixed with Cytofix/Cytoperm buffer (BD Bioscience) for 10 minutes in room temperature. After washing the fixed cells with Perm/Wash buffer (BD Bioscience) twice, and the cells were stained with anti-NS1/2 (Rabbit polyclonal antibody, provided by Vernon Ward) and anti-NS6/7 (Guinea-pig polyclonal antibody, provided by Kim Green) for 1 hour at room temperature. Cells were subsequently stained with anti-Rabbit-PE (Jackson) and anti-Guinea-pig-AF647 (Invitrogen) for one hour at room temperature. Cells were analyzed using a BD LSR II flow cytometer (BD Bioscience). Doublets were excluded by FSC-A × FSC-H and SSC-A × SSC-H analysis, and dead cells were excluded by live/dead staining. lECs were defined as EpCAM+CD45− cells, consistent with previous publications (Mahlakoiv et al., 2015).
For active Casp3 staining in BV2 cells, CR6-infected BV2 cells were trypsinized at 16 hpi, and washed once with cold 1× PBS. The cells were fixed with Cytofix/Cytoperm buffer (BD Bioscience) for 10 minutes at room temperature. After washing the fixed cells with Perm/Wash buffer (BD Bioscience) twice, the cells were stained with ant-NS6/7 (Guinea-pig polyclonal antibody, provided by Kim Green) and anti-active Casp3-AF647 (Clone C92-605, BD Bioscience). Cells were subsequently stained with anti-Guinea-pig-AF647 (Invitrogen) for one hour at room temperature. Data were processed using FACS-Diva software (BD) and FlowJo (FlowJo).
Fluorescence microscopy
Ifnlr1−/− mice infected with CR6D121/131G were sacrificed at 14 dpi. The colon was harvested, flushed with PBS followed by 10% neutral buffered formalin and then fixed overnight. Tissue was then blocked in 1% agar and embedded in paraffin for sectioning. Slides were then washed in xylene (3 washes × 5 min), isopropanol (3 washes × 5 min) and diH20 (5 min). Antigen retrieval was performed by boiling for 20 min in Dako antigen retrieval solution. Blocking was performed in TBST+10% goat serum+1%BSA. Primary antibodies were then applied in TBST overnight at 4°C (Rabbit anti-DCLK1 from Abcam 1:100; Guinea pig anti-NS6/7 gift of Kim Green 1:1000) followed by washing in TBST and incubation with secondary antibodies (1:500). Images were collected on a Zeiss LSM 880 Confocal Laser Scanning Microscope and analyzed on FIJI software. A maximum projection Z-stack was generated for each channel prior to merging.
ELISA for anti-NS1 and anti-capsid
Recombinant NS1 or CR6 viral stock were used to coat Immulon-2HB flat-bottomed ELISA plates. Plates were blocked with Assay diluent in BD OptEIA Reagent Set A (BD, #550536). Serum samples were heat-inactivated for 30 minutes at 56 °C. For anti-NS1 IgG ELISA, serum was diluted 1000-, 80,000- and 800,000-fold in the assay diluent. For anti-NS1 IgA, anti-capsid IgG and anti-capsid IgA ELISA, serum was diluted 1000-fold. Diluted serum was incubated in the coated plate for 1 hour at room temperature. Secondary antibodies, goat-anti-mouse-IgG HRP (SouthernBiotech, #1030-05) or goat-anti-mouse-IgA HRP (SouthernBiotech, #1040-05), were used at a dilution of 1:5000. For standard curve, mouse monoclonal antibodies detecting NS1 (CM79, mouse IgG1) and VP1 (A6.2, mouse IgG2b) were used.
MNoV neutralization assay
BV2 cells were seeded at 2 × 104 cells/well of a 96-well plate. One hundred PFU of CR6 were incubated with 100 μl of the diluted immune serum for 1 hour at 37 °C. The incubated mixture was inoculated to the BV2 cells. After 48 hours, cell viability was measured by Cell-Titer-Glo assay (Promega) according to the manufacturer’s protocol. ID50 value was calculated from each serum sources.
Passive transfer of immune serum
The donor mice were immunized three times with OVA or NS1 as described. The sera were heat-inactivated for 30 minutes at 56 °C, filtered through 0.2 μm pore PVDF filter, and pooled. 6-8 weeks old wild-type mice were received 500 μl of OVA or NS1-immune serum twice at 3 days interval. One week after the second serum-transfer, the mice were infected with 106 PFU of CR6 perorally. Viral genome RNA in the stool was quantified at 3 dpi.
QUANTIFICATION AND STATISTICAL ANALYSIS
Data were analyzed with Prism 7 software (GraphPad Software, San Diego, CA). In all graphs, three asterisks indicate a P value of <0.001, two asterisks indicate a P value of <0.01, one asterisk indicates a P value of <0.05, and ns indicates not significant (P > 0.05) as determined by Mann-Whitney test, one-way analysis of variance (ANOVA), one-way ANOVA with Tukey’s multiple-comparison test, or Kruskal-Wallis test, as specified in the relevant figure legends. The number of mice and/or replicates used in each experiments is also specified in the relevant figure legends.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-EpCam-APC-Cy7 (Clone G8.8) | Biolegend | Cat# 118218 |
| Anti-CD45-PacBlue (Clone 30-F11) | Biolegend | Cat# 103126 |
| Anti-NS1/2 (Rabbit polyclonal) | provided by Vernon Ward | N/A |
| Anti-NS6/7 (Guinea-pig polyclonal) | provided by Kim Green | N/A |
| Anti-NS1 (CM79, mouse monoclonal) | provided by Vernon Ward | N/A |
| Anti-capsid (A6.2, mouse monoclonal) | This study | N/A |
| Anti-cleaved Casp3 (D175)-AF647 (Clone C92-605) | BD | Cat# 560626 |
| Bacterial and Virus Strains | ||
| MNoV-CR6 | (Strong et al., 2012) | N/A |
| MNoV-CR6D121G | This study | N/A |
| MNoV-CR6D131G | This study | N/A |
| MNoV-CR6D121/131G | This study | N/A |
| MNoV-CW3 | (Strong et al., 2012) | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Recombinant IFN-λ | Bristol-Myers Squibb | N/A |
| Recombinant NS1 | This study | N/A |
| Recombinant VP1P-domain | This study | N/A |
| C12R | PBL | Cat# 12185-1 |
| Critical Commercial Assays | ||
| ZR-96 viral RNA kit | Zymo Research | Cat# R1041 |
| Direct-zol-96 RNA kit | Zymo Research | Cat# R2056 |
| ImPromll reverse transcriptase | Promega | Cat# PRA3803 |
| Luciferase Assay System | Promega | Cat# E1500 |
| PrimeTime qPCR assay (Ifi35) | IDT | Cat# Mm.PT.58.42591180 |
| PrimeTime qPCR assay (Ifit1) | IDT | Cat# Mm.PT.58.32674307 |
| PrimeTime qPCR assay (Oas1a) | IDT | Cat# Mm.PT.58.30459792 |
| Experimental Models: Cell Lines | ||
| BV2 | N/A | |
| HEK293T | N/A | |
| Mx2LUC-IEC | Inscreenex | Cat# INS-CI-1007L |
| Experimental Models: Organisms/Strains | ||
| Mouse: C57BL/6J | Jackson Laboratory | Stock # 000664 |
| Mouse: Ifnar1−/−(B6.129.Ifnar1tm1) | (Muller et al., 1994) | N/A |
| Mouse: Ifnlr1−/− (B6.Cg-Ifnlr1tm1Palu) | (Ank et al., 2008; Baldridge et al., 2017) | N/A |
| Mouse: Ifngr1−/− (JAX B6.129S7-Ifngr1tm1Agt/J, | Jackson Laboratory | Stock # 003288 |
| Mouse: Casp1/11−/− (JAX B6N.129S2-Casp1tm1Flv/J) | Jackson Laboratory | Stock # 016621 |
| Mouse: Rag1−/− (B6.129S7-Rag1tm1Mom/J) | (Mombaerts et al., 1992) | N/A |
| Mouse: Ifnlr1f/f (Ifnlr1tm1a(EUCOMM)Wtsl) | (Baldridge et al., 2017) | N/A |
| Mouse: Villin-Cre | (Madison et al., 2002) | N/A |
| Oligonucleotides | ||
| MNoV qPCR forward: 5′-CACGCCACCGATCTGTTCTG-3′ | IDT | N/A |
| MNoV qPCR reverse: 5′-GCGCTGCGCCATCACTC-3′ | IDT | N/A |
| MNoV qPCR probe: 5′-6FAM-CGCTTTGGAACAATG-MGBNFQ-3′ | Thermo Fisher Scientific | N/A |
| Rps29 qPCR forward: 5′-CAAATACGGGCTGAACATG-3′ | IDT | N/A |
| Rps29 qPCR reverse: 5′-GTCCAACTTAATGAAGCCTATGTC-3′ | IDT | N/A |
| Rps29 qPCR probe: 5′-HEX-CCTTCGCGTACTGCCGGAAGC-3′ | IDT | N/A |
| Software and Algorithms | ||
| Prism 7 software (V7.02) | GraphPad | https://www.graphpad.com/scientific-software/prism/ |
| FACSDiva software (V8.0.1) | BD | http://www.bdbiosciences.com/us/instruments/research/software/flow-cytometry-acquisition/bd-facsdiva-software/m/111112/overview |
| FlowJo (V10.1) | FlowJo | https://www.flowio.com/solutions/flowjo |
Highlights.
Murine norovirus (MNoV) infection globally suppresses IFN-λ responses in the intestine.
Non-structural protein 1 (NS1) is an unconventionally secreted MNoV protein.
NS1 is critical for overcoming IFN-λ-mediated resistance to MNoV infection in tuft cells.
Vaccination with NS1 protects mice from MNoV infection.
ACKNOWLEDGMENTS
The authors would like to thank D. Kreamalmeyer for animal care and breeding, and Anthony Orvedahl and the other members of the Virgin lab for manuscript review and discussion. We would also like to thank the Genome Engineering and iPSC center, the Flow Cytometry & Fluorescence Activated Cell Sorting Core Facility, the Molecular Microbiology Imaging Facility and the Elvie L. Taylor Histology Core Facility at Washington University School of Medicine for assistance with generating KO cell lines, cell sorting and immunohistochemistry. H.W.V. was supported by National Institutes of Health (NIH) grant U19 AI109725, and the Crohn’s and Colitis Foundation grant #326556. S.L. was supported by the Basic Science Research Program through the National Research Foundation of Korea funded by the Ministry of Education (NRF-2016R1A6A3A03012352). M.T.B. was supported by NIH grants K22 AI127846, R01 AI127552, R01 OD024917, R01 AI139314, and DDRCC grant P30 DK052574. T.J.N. was supported by NIH training grant 5T32A100716334 and postdoctoral fellowships from the Cancer Research Institute and American Cancer Society. C.B.W. was supported by NIH grant 1K08AI128043-01. R.C.O. was supported by NIH grant R00 DK116666. B.T.M. was supported by NCI-NIH award F31CA177194-01. K-W.K. was supported by NIH R37 AI049653 to G. J. Randolph. G.K.A and H.L. were supported by NIH AI120943 and AI109945. The data from this study are tabulated in the main paper and supplementary materials. All reagents are available from H.W.V under a material transfer agreement with Washington University.
DECLARATION OF INTERESTS
Dr. Virgin is an employee of and owns stock in Vir Biotechnology in San Francisco CA, and is a founder and shareholder in Casma Therapeutics in Cambridge MA. the work reported herein was not funded by either Vir biotechnology or Casma Therapeutics. Washington University School of Medicine holds patents on which Dr. Virgin is an inventor that are related to murine norovirus. Washington University School of Medicine and Dr. Virgin receive income based on licensing of these patents.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Ajami NJ, Kavanagh OV, Ramani S, Crawford SE, Atmar RL, Jiang ZD, Okhuysen PC, Estes MK, and DuPont HL (2014). Seroepidemiology of norovirus-associated travelers’ diarrhea. Journal of travel medicine 21, 6–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ank N, Iversen MB, Bartholdy C, Staeheli P, Hartmann R, Jensen UB, Dagnaes-Hansen F, Thomsen AR, Chen Z, Haugen H, et al. (2008). An important role for type III interferon (IFN-lambda/IL-28) in TLR-induced antiviral activity. Journal of immunology 180, 2474–2485. [DOI] [PubMed] [Google Scholar]
- Atmar RL, Bernstein DI, Harro CD, Al-Ibrahim MS, Chen WH, Ferreira J, Estes MK, Graham DY, Opekun AR, Richardson C, et al. (2011). Norovirus vaccine against experimental human Norwalk Virus illness. The New England journal of medicine 365, 2178–2187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atmar RL, Opekun AR, Gilger MA, Estes MK, Crawford SE, Neill FH, and Graham DY (2008). Norwalk virus shedding after experimental human infection. Emerging infectious diseases 14, 1553–1557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker ES, Luckner SR, Krause KL, Lambden PR, Clarke IN, and Ward VK (2012). Inherent structural disorder and dimerisation of murine norovirus NS1-2 protein. PloS one 7, e30534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baldridge MT, Lee S, Brown JJ, McAllister N, Urbanek K, Dermody TS, Nice TJ, and Virgin HW (2017). Expression of Ifnlr1 on Intestinal Epithelial Cells Is Critical to the Antiviral Effects of Interferon Lambda against Norovirus and Reovirus. Journal of virology 91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baldridge MT, Turula H, and Wobus CE (2016). Norovirus Regulation by Host and Microbe. Trends in molecular medicine 22, 1047–1059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basic M, Keubler LM, Buettner M, Achard M, Breves G, Schroder B, Smoczek A, Jorns A, Wedekind D, Zschemisch NH, et al. (2014). Norovirus triggered microbiota-driven mucosal inflammation in interleukin 10-deficient mice. Inflammatory bowel diseases 20, 431–443. [DOI] [PubMed] [Google Scholar]
- Bernstein DI, Atmar RL, Lyon GM, Treanor JJ, Chen WH, Jiang X, Vinje J, Gregoricus N, Frenck RW Jr., Moe CL, et al. (2015). Norovirus vaccine against experimental human GII.4 virus illness: a challenge study in healthy adults. The Journal of infectious diseases 211, 870–878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Best SM, and Robertson SJ (2017). IFN-Lambda: The Key to Norovirus’s Secret Hideaway. Cell host & microbe 22, 427–429. [DOI] [PubMed] [Google Scholar]
- Bok K, and Green KY (2012). Norovirus gastroenteritis in immunocompromised patients. The New England journal of medicine 367, 2126–2132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cadwell K, Patel KK, Maloney NS, Liu TC, Ng AC, Storer CE, Head RD, Xavier R, Stappenbeck TS, and Virgin HW (2010). Virus-plus-susceptibility gene interaction determines Crohn’s disease gene Atg16L1 phenotypes in intestine. Cell 141, 1135–1145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chachu KA, Strong DW, LoBue AD, Wobus CE, Baric RS, and Virgin H.W.t. (2008). Antibody is critical for the clearance of murine norovirus infection. Journal of virology 82, 6610–6617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cortes-Penfield NW, Ramani S, Estes MK, and Atmar RL (2017). Prospects and Challenges in the Development of a Norovirus Vaccine. Clinical therapeutics 39, 1537–1549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ettayebi K, Crawford SE, Murakami K, Broughman JR, Karandikar U, Tenge VR, Neill FH, Blutt SE, Zeng XL, Qu L, et al. (2016). Replication of human noroviruses in stem cell-derived human enteroids. Science 353, 1387–1393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerbe F, Sidot E, Smyth DJ, Ohmoto M, Matsumoto I, Dardalhon V, Cesses P, Garnier L, Pouzolles M, Brulin B, et al. (2016). Intestinal epithelial tuft cells initiate type 2 mucosal immunity to helminth parasites. Nature 529, 226–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grau KR, Roth AN, Zhu S, Hernandez A, Colliou N, DiVita BB, Philip DT, Riffe C, Giasson B, Wallet SM, et al. (2017). The major targets of acute norovirus infection are immune cells in the gut-associated lymphoid tissue. Nature microbiology 2, 1586–1591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gustavsson L, Norden R, Westin J, Lindh M, and Andersson LM (2017). Slow Clearance of Norovirus following Infection with Emerging Variants of Genotype GII.4 Strains. Journal of clinical microbiology 55, 1533–1539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howitt MR, Lavoie S, Michaud M, Blum AM, Tran SV, Weinstock JV, Gallini CA, Redding K, Margolskee RF, Osborne LC, et al. (2016). Tuft cells, taste-chemosensory cells, orchestrate parasite type 2 immunity in the gut. Science 351, 1329–1333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang S, Hendriks W, Althage A, Hemmi S, Bluethmann H, Kamijo R, Vilcek J, Zinkernagel RM, and Aguet M (1993). Immune response in mice that lack the interferon-gamma receptor. Science 259, 1742–1745. [DOI] [PubMed] [Google Scholar]
- Inaida S, Shobugawa Y, Matsuno S, Saito R, and Suzuki H (2013). The South to north variation of norovirus epidemics from 2006-07 to 2008-09 in Japan. PloS one 8, e71696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson PC, Mathewson JJ, DuPont HL, and Greenberg HB (1990). Multiple-challenge study of host susceptibility to Norwalk gastroenteritis in US adults. The Journal of infectious diseases 161, 18–21. [DOI] [PubMed] [Google Scholar]
- Jones MK, Watanabe M, Zhu S, Graves CL, Keyes LR, Grau KR, Gonzalez-Hernandez MB, Iovine NM, Wobus CE, Vinje J, et al. (2014). Enteric bacteria promote human and mouse norovirus infection of B cells. Science 346, 755–759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karandikar UC, Crawford SE, Ajami NJ, Murakami K, Kou B, Ettayebi K, Papanicolaou GA, Jongwutiwes U, Perales MA, Shia J, et al. (2016). Detection of human norovirus in intestinal biopsies from immunocompromised transplant patients. The Journal of general virology 97, 2291–2300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karst SM, Wobus CE, Goodfellow IG, Green KY, and Virgin HW (2014). Advances in norovirus biology. Cell host & microbe 15, 668–680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katpally U, Wobus CE, Dryden K, Virgin H.W.t., and Smith TJ (2008). Structure of antibody-neutralized murine norovirus and unexpected differences from viruslike particles. Journal of virology 82, 2079–2088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaufman SS, Green KY, and Korba BE (2014). Treatment of norovirus infections: moving antivirals from the bench to the bedside. Antiviral research 105, 80–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kernbauer E, Ding Y, and Cadwell K (2014). An enteric virus can replace the beneficial function of commensal bacteria. Nature 516, 94–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuida K, Lippke JA, Ku G, Harding MW, Livingston DJ, Su MS, and Flavell RA (1995). Altered cytokine export and apoptosis in mice deficient in interleukin-1 beta converting enzyme. Science 267, 2000–2003. [DOI] [PubMed] [Google Scholar]
- Lanata CF, Fischer-Walker CL, Olascoaga AC, Torres CX, Aryee MJ, Black RE, Child Health Epidemiology Reference Group of the World Health, O., and Unicef (2013). Global causes of diarrheal disease mortality in children <5 years of age: a systematic review. PloS one 8, e72788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazear HM, Nice TJ, and Diamond MS (2015). Interferon-lambda: Immune Functions at Barrier Surfaces and Beyond. Immunity 43, 15–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S, Wilen CB, Orvedahl A, McCune BT, Kim KW, Orchard RC, Peterson ST, Nice TJ, Baldridge MT, and Virgin HW (2017). Norovirus Cell Tropism Is Determined by Combinatorial Action of a Viral Non-structural Protein and Host Cytokine. Cell host & microbe 22, 449–459 e444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leib DA, Machalek MA, Williams BR, Silverman RH, and Virgin HW (2000). Specific phenotypic restoration of an attenuated virus by knockout of a host resistance gene. Proceedings of the National Academy of Sciences of the United States of America 97, 6097–6101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love MI, Huber W, and Anders S (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madison BB, Dunbar L, Qiao XT, Braunstein K, Braunstein E, and Gumucio DL (2002). Cis elements of the villin gene control expression in restricted domains of the vertical (crypt) and horizontal (duodenum, cecum) axes of the intestine. JBiolChem 277, 33275–33283. [DOI] [PubMed] [Google Scholar]
- Mahlakoiv T, Hernandez P, Gronke K, Diefenbach A, and Staeheli P (2015). Leukocyte-derived IFN-alpha/beta and epithelial IFN-lambda constitute a compartmentalized mucosal defense system that restricts enteric virus infections. PLoS pathogens 11, e1004782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCune BT, Tang W, Lu J, Eaglesham JB, Thorne L, Mayer AE, Condiff E, Nice TJ, Goodfellow I, Krezel AM, et al. (2017). Noroviruses Co-opt the Function of Host Proteins VAPA and VAPB for Replication via a Phenylalanine-Phenylalanine-Acidic-Tract-Motif Mimic in Nonstructural Viral Protein NS1/2. mBio 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mombaerts P, Iacomini J, Johnson RS, Herrup K, Tonegawa S, and Papaioannou VE (1992). RAG-1-deficient mice have no mature B and T lymphocytes. Cell 68, 869–877. [DOI] [PubMed] [Google Scholar]
- Muller U, Steinhoff U, Reis LF, Hemmi S, Pavlovic J, Zinkernagel RM, and Aguet M (1994). Functional role of type I and type II interferons in antiviral defense. Science 264, 1918–1921. [DOI] [PubMed] [Google Scholar]
- Nice TJ, Baldridge MT, McCune BT, Norman JM, Lazear HM, Artyomov M, Diamond MS, and Virgin HW (2015). Interferon-lambda cures persistent murine norovirus infection in the absence of adaptive immunity. Science 347, 269–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nice TJ, Strong DW, McCune BT, Pohl CS, and Virgin HW(2013). A singleamino-acid change in murine norovirus NS1/2 is sufficient for colonic tropism and persistence. Journal of virology 87, 327–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen H (2017). Predicting Secretory Proteins with SignalP. Methods in molecular biology 1611, 59–73. [DOI] [PubMed] [Google Scholar]
- Orchard RC, Wilen CB, Doench JG, Baldridge MT, McCune BT, Lee YC, Lee S, Pruett-Miller SM, Nelson CA, Fremont DH, et al. (2016). Discovery of a proteinaceous cellular receptor for a norovirus. Science 353, 933–936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park S, Buck MD, Desai C, Zhang X, Loginicheva E, Martinez J, Freeman ML, Saitoh T, Akira S, Guan JL, et al. (2016). Autophagy Genes Enhance Murine Gammaherpesvirus 68 Reactivation from Latency by Preventing Virus-Induced Systemic Inflammation. Cell Host Microbe 19, 91–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parrino TA, Schreiber DS, Trier JS, Kapikian AZ, and Blacklow NR (1977). Clinical immunity in acute gastroenteritis caused by Norwalk agent. The New England journal of medicine 297, 86–89. [DOI] [PubMed] [Google Scholar]
- Prasad BV, Shanker S, Muhaxhiri Z, Deng L, Choi JM, Estes MK, Song Y, Palzkill T, and Atmar RL (2016). Antiviral targets of human noroviruses. Current opinion in virology 18, 117–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rockx B, De Wit M, Vennema H, Vinje J, De Bruin E, Van Duynhoven Y, and Koopmans M (2002). Natural history of human calicivirus infection: a prospective cohort study. Clinical infectious diseases : an official publication of the Infectious Diseases Society of America 35, 246–253. [DOI] [PubMed] [Google Scholar]
- Saito M, Goel-Apaza S, Espetia S, Velasquez D, Cabrera L, Loli S, Crabtree JE, Black RE, Kosek M, Checkley W, et al. (2014). Multiple norovirus infections in a birth cohort in a Peruvian Periurban community. Clinical infectious diseases : an official publication of the Infectious Diseases Society of America 58, 483–491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwenk F, Baron U, and Rajewsky K (1995). A cre-transgenic mouse strain for the ubiquitous deletion of loxP-flanked gene segments including deletion in germ cells. Nucleic Acids Res 23, 5080–5081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith VP, and Alcami A (2002). Inhibition of interferons by ectromelia virus. Journal of virology 76, 1124–1134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sosnovtsev SV, Belliot G, Chang KO, Prikhodko VG, Thackray LB, Wobus CE, Karst SM, Virgin HW, and Green KY (2006). Cleavage map and proteolytic processing of the murine norovirus nonstructural polyprotein in infected cells. Journal of virology 80, 7816–7831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- St Clair KJ, and Patel S (2008). Intital descriptive and analytical data on an outbreak of norovirus infection at marine corps recruit depot Parris Island, South Carolina. The Journal of infectious diseases 198, 941–942; author reply 942-943. [DOI] [PubMed] [Google Scholar]
- Strong DW, Thackray LB, Smith TJ, and Virgin HW (2012). Protruding domain of capsid protein is necessary and sufficient to determine murine norovirus replication and pathogenesis in vivo. Journal of virology 86, 2950–2958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan M, Huang P, Xia M, Fang PA, Zhong W, McNeal M, Wei C, Jiang W, and Jiang X (2011). Norovirus P particle, a novel platform for vaccine development and antibody production. Journal of virology 85, 753–764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teunis PF, Sukhrie FH, Vennema H, Bogerman J, Beersma MF, and Koopmans MP (2015). Shedding of norovirus in symptomatic and asymptomatic infections. Epidemiology and infection 143, 1710–1717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomov VT, Osborne LC, Dolfi DV, Sonnenberg GF, Monticelli LA, Mansfield K, Virgin HW, Artis D, and Wherry EJ (2013). Persistent enteric murine norovirus infection is associated with functionally suboptimal virus-specific CD8 T cell responses. Journal of virology 87, 7015–7031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomov VT, Palko O, Lau CW, Pattekar A, Sun Y, Tacheva R, Bengsch B, Manne S, Cosma GL, Eisenlohr LC, et al. (2017). Differentiation and Protective Capacity of Virus-Specific CD8(+) T Cells Suggest Murine Norovirus Persistence in an Immune-Privileged Enteric Niche. Immunity 47, 723–738 e725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Heijne G (1990). The signal peptide. The Journal of membrane biology 115, 195–201. [DOI] [PubMed] [Google Scholar]
- von Moltke J, Ji M, Liang HE, and Locksley RM (2016). Tuft-cell-derived IL-25 regulates an intestinal ILC2-epithelial response circuit. Nature 529, 221–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilen CB, Lee S, Hsieh LL, Orchard RC, Desai C, Hykes BL Jr., McAllaster MR, Balce DR, Feehley T, Brestoff JR, et al. (2018). Tropism for tuft cells determines immune promotion of norovirus pathogenesis. Science 360, 204–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wobus CE, Karst SM, Thackray LB, Chang KO, Sosnovtsev SV, Belliot G, Krug A, Mackenzie JM, Green KY, and Virgin HW (2004). Replication of Norovirus in cell culture reveals a tropism for dendritic cells and macrophages. PLoS biology 2, e432. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.