

Abstract
Ribbon synapses continuously transmit graded membrane potential changes into changes of synaptic vesicle exocytosis and rely on intense synaptic membrane trafficking. The synaptic ribbon is considered central to this process. In the present study we asked whether tonically active ribbon synapses are associated with the generation of certain lipids, specifically the highly active signaling phospholipid phosphatidic acid (PA). Using PA-sensor proteins, we demonstrate that PA is enriched at mouse retinal ribbon synapses in close vicinity to the synaptic ribbon in situ. As shown by heterologous expression, RIBEYE, a main component of synaptic ribbons, is responsible for PA binding at synaptic ribbons. Furthermore, RIBEYE is directly involved in the synthesis of PA. Using various independent substrate binding and enzyme assays, we demonstrate that the B domain of RIBEYE possesses lysophosphatidic acid (LPA) acyltransferase (LPAAT) activity, which leads to the generation of PA from LPA. Since an LPAAT-deficient RIBEYE mutant does not recruit PA-binding proteins to artificial synaptic ribbons, whereas wild-type RIBEYE supports PA binding, we conclude that the LPAAT activity of the RIBEYE(B) domain is a physiologically relevant source of PA generation at the synaptic ribbon. We propose that PA generated at synaptic ribbons likely facilitates synaptic vesicle trafficking.
Introduction
Ribbon synapses are high-performance synapses built by certain nonspiking sensory neurons, e.g., photoreceptors, retinal bipolar cells, and inner ear hair cells. They are characterized by high rates of synaptic vesicle exocytosis, coordinated multivesicular release, and exquisite sensitivity over a wide dynamic range of signaling, including both phasic and tonic components (Wan and Heidelberger, 2011). The main site of exocytosis is characterized by unique presynaptic structures, the synaptic ribbons, which are associated with large numbers of synaptic vesicles. Ribbon-associated vesicles are primed for exocytosis and provide the active zone with a large pool of release-ready vesicles. Size, shape, and number of synaptic ribbons strongly impact synaptic signaling (Hull et al., 2006; Johnson et al., 2008; Frank et al., 2009; Jackman et al., 2009; Meyer et al., 2009; Liberman et al., 2011). Mouse models with nonanchored synaptic ribbons display defects in fast, stimulus-synchronous exocytosis and disturbances in presynaptic membrane trafficking (Khimich et al., 2005; tom Dieck et al., 2005; Buran et al., 2010; Liberman et al., 2011). Thus, synaptic ribbons are crucial for signaling at these types of synapses (Schmitz, 2009; Meyer and Moser, 2010; Wan and Heidelberger, 2011), but the detailed mechanisms how ribbons perform these functions remain elusive. RIBEYE (RE) is a protein specific to synaptic ribbons and, consequently, thought to be a major determinant of ribbon function (Schmitz et al., 2000; Magupalli et al., 2008; Schmitz, 2009). RIBEYE is composed of a unique aminoterminal A domain and a C-terminal B domain, which is largely identical to CtBP2 (Schmitz et al., 2000; Schmitz, 2009). The RIBEYE(A) domain mainly has a structural role at the synaptic ribbon (Magupalli et al., 2008), whereas the function of the RIBEYE(B) domain has been unclear. The RIBEYE(B) domain specifically binds NAD(H) and evolved from d-2-hydroxyacid dehydrogenases (Goldberg et al., 1994; Kumar et al., 2002; Nardini et al., 2003; Schmitz, 2009), indicating that the RIBEYE(B) domain could be enzymatically active. This domain shares strong sequence homology with CtBP1/BARS, a protein found at Golgi apparatus and plasma membrane/endosomes where it regulates membrane trafficking (Chinnadurai, 2002; Corda et al., 2006; Donaldson, 2009; Schmitz, 2009; Antonescu et al., 2010). CtBP1/BARS directly binds to phosphatidic acid (PA) (Yang et al., 2008) and regulates membrane trafficking at these sites, possibly by modulating, either directly or indirectly, PA synthesis (Weigert et al., 1999; Bonazzi et al., 2005; Yang et al., 2005; Corda et al., 2006; 2008; Haga et al., 2009). The role of CtBP1/BARS for PA synthesis is complex and controversial (Weigert et al., 1999; Gallop et al., 2005; Haga et al., 2009). PA, a cone-shaped lipid, favors negative membrane curvature that is needed for both membrane fusion and fission (Cazzolli et al., 2006; Oude Weernink et al., 2007). In neuroendocrine and other cells, PA is required for exocytosis (Bader and Vitale, 2009), but its role in neuronal synapses is less clear. In the present study, we asked whether PA is generated in presynaptic ribbon terminals, particularly at synaptic ribbons, where PA could mediate essential steps in synaptic vesicle trafficking.
Materials and Methods
Plasmids
Tryptophan anchoring phosphatidic acid-selective binding domain-1 PA-sensor constructs
pSFV1-EGFP was generated as described previously (Venkatesan et al., 2010). The EGFP encoding insert was amplified via PCR with forward primer (986) TTTAGATCTGCCACCATGGTGAGCAAGGGCGA and reverse primer (987) TTTGGATCCCTTGTACAGCTCGTCCAT, and subsequently cloned into the BamHI site of pSFV1 (Ashery et al., 1999; Venkatesan et al., 2010). Orientation was checked by double digestion with EcoRI/BamHI and verified by sequencing. The internal IRES of pSFV1 (Ashery et al., 1999) was deleted with BssHII/ClaI and religation after blunting with Klenow.
The tryptophan anchoring phosphatidic acid-selective binding domain-1 (TAPAS-1) encodes the 25 aminoterminal amino acids of the unique N-terminal region of the PDE4A1 cAMP-specific phosphodiesterase (PDE; 1MPLVDFFCETCSKPWLVGWWDQFKR) that specifically bind PA in a Ca2+-dependent manner (Baillie et al., 2002; Huston et al., 2006). The Ca2+-induced binding of TAPAS-1 to PA depends on the membrane insertion of a pair of tryptophan residues (WW19,20) as a central bilayer insertion unit; the Ca2+-induced membrane insertion is considered irreversible (Baillie et al., 2002, Huston et al., 2006). The subsequent TAPAS-1 constructs were cloned between the BamHI and BssHII sites of pSFV1.
TAPAS-1(WT)pSFV1-EGFP.
The TAPAS-1 wild-type (WT) insert was generated by primer annealing with forward primer (1049) TTTGGATCCATGCCTCTGGTTGACTTCTTCTGCGAGACCTGCTCCAAGCCCTGG and reverse primer (1050) TTTAAGCGCGCTTACCTTTTGAACTGGTCCCACCAGCCCACCAGCCAGGGCTTG, primer extension with Taq-polymerase, and subsequent cloning into the BamHI/BssHII sites of pSFV1-EGFP.
TAPAS-1(WW19,20AA)pSFV1-EGFP.
The TAPAS-1(WW19,20AA) insert was generated by primer annealing with forward primer (1049) TTTGGATCCATGCCTCTGGTTGACTTCTTCTGCGAGACCTGCTCCAAGCCCTGG and reverse primer (1071) TTTAAGCGCGCTTACCTTTTGAACTGGTCCGCCGCGCCCACCAGCCAGGGCTTGGAGCAGGTCTC and subsequent cloning into the BamHI/BssHII sites of pSFV1-EGFP.
TAPAS-1(WW19,20DD)pSFV1-EGFP.
The TAPAS-1(WW19,20DD) insert was generated by primer annealing with forward primer (1049) TTTGGATCCATGCCTCTGGTTGACTTCTTCTGCGAGACCTGCTCCAAGCCCTGG and reverse primer (1051) TTTAAGCGCGCTTACCTTTTGAACTGGTCGTCGTCGCCCACCAGCCAGGGCTTG and subsequent cloning into the BamHI/BssHII sites of pSFV1-EGFP.
TAPAS-1(WT)pGEX.
The TAPAS-1 wild-type insert was generated as described above with primer annealing with oligos 1049 and 1050 [see above, TAPAS-1(WT)pSFV1-EGFP] and subsequent extension of the annealed primers with Taq-polymerase. Afterward, the TAPAS-1 wild-type insert was cloned into the pJET1.2 vector (CloneJET PCR Cloning Kit; Fermentas); the orientation of the insert was checked with BamHI/NcoI and BamHI/XhoI double digests. The TAPAS-1 wild-type insert was excised via BamHI/NcoI from a respectively oriented TAPAS-1(WT)pJET clone and cloned into the BamHI/NcoI sites of pGEX-KG.
TAPAS-1(WW19,20AA)pGEX.
The TAPAS-1(WW19,20AA) insert was generated as described above with primer annealing with oligos 1049 and 1071 [see above, TAPAS-1(WW19,20AA)pSFV1-EGFP] and subsequent extension of the annealed primers with Taq-polymerase. Afterward, the TAPAS-1(WW19,20AA) insert was cloned into the pJET1.2 vector (Fermentas); the orientation of the insert was checked with BamHI/NcoI and BamHI/XhoI double digests. The TAPAS-1(WW19,20AA) insert was excised via BamHI/XhoI from a respectively oriented TAPAS-1(WW19,20AA)pJET clone and cloned into the BamHI/XhoI sites of pGEX-KG.
TAPAS-1(WW19,20DD)pGEX.
The TAPAS-1(WW19,20DD) mutant insert was generated as described above with primer annealing with oligos 1049 and 1051 [see above, TAPAS-1(WW19,20DD)pSFV1-EGFP] and subsequent extension of the annealed primers with Taq-polymerase. Afterward, the TAPAS-1(WW19,20DD) insert was cloned into the pJET1.2 vector (Fermentas); the orientation of the insert was checked with BamHI/NcoI and BamHI/XhoI double digests. The TAPAS-1(WW19,20DD) insert was excised via BamHI/NcoI from a respectively oriented TAPAS-1(WW19,20DD)pJET clone and cloned into the BamHI/NcoI sites of pGEX-KG.
Raf-1 PA-sensor constructs
Raf-1 has a distinct C-terminal bipartite PA-binding module (Gosh et al., 1996, 2003) that consists of a cluster of basic amino acids (aa398–401; RKTRH) and a subsequent cluster of hydrophobic amino acids (amino acids 404–407; ILLV). The PA-binding motif of Raf-1 works differently from the TAPAS-1 PA-binding module described above (Gosh et al., 1996, 2003) and is independent from increased intracellular Ca2+ levels. Raf-1 wild-type and PA-binding-deficient Raf-1 mutants (see below, Raf-1pmCherry-C1 and Raf-1mutpmCherry-C1) were obtained as pEGFP-C1 expression constructs using BglII and EcoRI restriction sites (Corrotte et al., 2006). The Raf WT construct encodes the 36-aa-long C-terminal PA-binding motif of human Raf-1 (Gosh et al., 1996, 2003).
Raf-1pmCherry-C1.
mCherry cDNA was amplified via PCR using pCherry-N1 as a template with forward primer (984) TTATGCTAGCTCGCCACCATGGTG and reverse primer (985) TTATAGATCTCTTGTACAGCTCGTCC, and cloned into the NheI/BglII sites of Raf1pEGFP-C1, thus replacing the EGFP tag of Raf-WT from the original vector by an mCherry tag.
Raf-1mutpmCherry-C1.
This construct has a double point mutant in the PA-binding motif of Raf-1 (L398K399T400L401 instead of wild-type R398K399T400R401) leading to strongly reduced binding of PA (Gosh et al., 1996, 2003; Corrotte at al., 2006) (data not shown). Raf-1mutpmCherry was cloned as described above for Raf-1pmCherry-C1 from Raf-1mutpmEGFP-C1.
Raf-1pGEX.
The insert was amplified by PCR using Raf-1pEGFP-C1 (Corrotte et al., 2006) as a template and the following primers: forward primer (990), TTTGGATCCATCCAGG CCTTCAGGAAT; reverse primer (991), TTTAAGCGCGCTTAGCCCTCGCACCACT). The PCR product was subcloned into the pJET1.2 vector (Fermentas); excised from a recombinant, properly oriented pJET clone with BamHI and XhoI; and cloned into the BamHI/XhoI sites of pGEX-KG.
Raf-1mutpGEX.
The insert was amplified by PCR using Raf-1mutpmEGFP-C1 as a template with forward primer (990) TTTGGATCCATCCAGGCCTTCAGGAAT and reverse primer (991) TTTAAGCGCGCTTAGCCCTCGCACCACT, and cloned into BamHI/XhoI sites of pGEX-KG as described above for Raf-1pGEX.
pGEX constructs: RE(B)-pGEX (Schmitz et al., 2000)
RE(B)G730A-pGEX was cloned with overlapping PCR using the mutant forward primer CTCATTGCCTTTGGTCGCACTGGG and mutant reverse primer ACCAAAGGCAATGAGGCCCAGTGTT, together with the respective outward primers GTTCCATGGAGATCCGCCCCCAGATCAT (forward) and GTTCTCGAGCTATTGCTCGTTGGGGT (reverse), and cloned into the NcoI and XhoI sites of pGEX-KG.
RE(B)ΔCTR-pGEX was cloned with forward primer TTTTCCATGGTTATCCGCCCCCAGATCATGA and reverse primer TTTTCTCGAGCCAA GGAGTTGAAGTAACAA and cloned in-frame into the NcoI and XhoI sites of pGEX-KG.
RE(B)F904W-pGEX, RE(B)W694F-pGEX, RE(B)W867F-pGEX, and RE(B)W911F-pGEX point mutations were generated by site-directed mutagenesis (QuikChange Site-Directed Mutagenesis Kit; Stratagene) using RE(B)-pGEX as a template (primers for F904W, forward, CAACAAAGAATGGTTTGTTACTTCAAC; reverse, GTTGAAGTAACAAACCATTCTTTGTTG; primers for W694F, forward, CGGCGGAACACATTTATGTACCAGGCCCTACG; reverse, CGTAGGGCCTGGTACAGAAATGTGTTCCGCCG; primers for W867F, forward, ACACCACACACAGCCTTTTACAGCGAACAAGC; reverse, GCTTGTTCGCTGTAAAAGGCTGTGTGTGGTGT; primers for W911F, forward, ACTTCAACTCCTTTTTCAGTCATTGACCAGC; reverse, GCTGGTCAATGACTGAAAAAGGAGTTGAAGT).
For mCtBP1-pGEX, the mCtBP1 insert was amplified with PCR using forward primer CGTGGATCCATGGGCAGCTCCCACTTGCT and reverse primer ATTAAGCTTCTACAACTGGTCACTCGTATG using a mouse muscle cDNA library as a template and cloned into the BamHI and HindIII sites of pGEX-KG.
RIBEYEpEGFP constructs: RE(B)-pEGFP (Schmitz et al., 2000)
RE(B)G730A-pEGFP was cloned by PCR using RE(B)G730A-pGEX as a template and forward primer TATTCTCGAGGTATCCGCCCCCAGATC and reverse primer ATTAGTCGACCTATTGCTCGTTGGGGTGCTC into the XhoI site of pEGFP-C1.
RE(B)-F904W-pEGFP was generated by PCR using RE(B)F904W-pGEX as a template, with forward primer TATTCTCGAGGTATCCGCCCCCAGATC and reverse primer TTTTTGGTACCCTATTGCTCGTTGGGGTGC, and cloned into the XhoI and KpnI sites of pEGFP-C1.
RE(B)ΔCTR-pEGFP was generated by PCR using RE(B)ΔCTR-pGEX as a template, with forward primer TATTCTCGAGGTATCCGCCCCCAGATC (OL380) and reverse primer TTTTTGGTACCCCAAGGAGTTGAAGTAAC (OL510), and cloned into the XhoI and KpnI sites of pEGFP-C1.
For RIBEYE(AB) with a deletion of the C-terminal region (CTR) [RE(AB)ΔCTR]-pSK, pRE(AB)ΔCTR-pSK was cloned in two subsequent steps using a viral expression vector containing RIBEYE amino acids 1–911 fused in-frame to EGFP as a starting point. First, a fragment containing amino acids 720–911 of RIBEYE fused to GFP was excised from the viral expression vector using BamHI and ClaI restriction endonucleases and cloned into the respective sites of pBluescript. In a second step, the N-terminal BamHI fragment (amino acids 1–719 of RIBEYE) of the viral expression vector was excised and inserted into the BamHI site of pBluescript containing RIBEYE amino acids 720–911 fused to EGFP, thereby creating pRE(AB)ΔCTR-pSK.
RE(AB)ΔCTR-pEGFP was cloned using a modified pEGFP-N1 vector backbone from which the EGFP-insert was cut out with AgeI and NotI and religated after filling up the sticky ends (pEGFP-N1 without EGFP). Into this vector, the EcoRI/SalI fragment from RE(AB)ΔCTR-EGFP-pSK was cloned into the respective EcoRI/SalI site. The ≈2.8 kb 5′-RIBEYE(AB) EcoRI fragment was cloned into the EcoRI site of this intermediate vector to obtain RE(AB)ΔCTR-pEGFP. The orientation of the EcoRI fragment was checked by digestion with BamHI, which releases an ∼1.3 kb fragment if the N-terminal RIBEYE fragment is in the proper orientation.
Radioactive materials
All radioactive materials were obtained from GE Healthcare except for lysophosphatidic acid (LPA), which was obtained from NEN/PerkinElmer: [14C]palmitoyl-coenzymeA (palmitoyl-CoA; CFA583; specific activity, 55 mCi/mmol; 2.04 GBq/mmol; chemical concentration, 845 μm; aqueous solution in 10 mm sodium acetate, pH 6.0). LPA (NET 1100; 1-oleoyl 2; specific activity, 47.0 Ci/mmol) dissolved in hexane-isopropanol-acetic acid (3:6:1:0.001) was lyophilized and resuspended in PBS as indicated in the respective experiments before use.
Antibodies
Anti-EGFP (T3743), polyclonal rabbit antiserum against EGFP (a kind gift from Dr. T. C. Südhof, Stanford University, Stanford, CA). RIBEYE antiserum (U2656) was described previously (Schmitz et al., 2000). Anti-GST monoclonal mouse antibody was obtained from Sigma and Invitrogen (the latter being used for the phospholipid strip assays).
Bacterial strains
For complementation assays, lysophosphatidic acid acyltransferase (LPAAT)-deficient JC201 bacteria were used (Coleman, 1990; Gallop et al., 2005). Growth of the transformed bacteria was monitored by measuring their optical density at 600 nm in a photometer (Ultrospec 2100pro; Pharmacia). For expression of bacterial fusion proteins, BL21(DE3) bacteria and, in some indicated cases, JC201 bacteria were used.
Miscellaneous materials
Palmitoyl-CoA beads were obtained from Sigma, and 1-oleoyl-2-hydroxy-sn-glycero-3-phosphate (sodium salt; LPA; 857130P) from Avanti Polar Lipids.
Procedures
Lipid dot blot
3-sn-Phosphatidic acid (Sigma; P9511) was dissolved in chloroform (stock, 10 mm). The PA was spotted in serial concentrations (100–5 nmol) on nitrocellulose membrane and allowed to dry on the membrane for 30 min. These membranes were incubated for 1 h at room temperature (RT) with PBS +1% nonfat dry milk to block nonspecific protein-binding sites. Subsequently, membranes were incubated with 1 μg/ml of TAPAS-1 wild-type or TAPAS-1 mutant (WW19,20DD or WW1920AA) fusion proteins in PBS supplemented with 1% nonfat dry milk overnight at 4°C (Baillie et al., 2002). Membranes were washed three times with 10 mm Tris and 150 mm NaCl, pH 8.0, with 0.1% Tween-20 (TBST) with gentle agitation for 10 min each following incubation with anti-GST monoclonal antibody (1:10,000 in blocking solution overnight at 4°C). After three washes with TBST (10 min each), membranes were incubated with secondary antibody (goat anti-mouse conjugated with peroxidase; 1:10,000 in blocking solution) for 1 h at RT. Membranes were again washed three times with TBST (10 min each) and further processed for chemiluminescence detection. Signals were acquired with a Bio-Rad Gel Doc apparatus.
Phospholipid strip assay
Purified TAPAS-1-GST and Raf-1-GST (1 μg/ml in 3% BSA plus 0.1% Tween-20) were incubated for 2 h at 4°C with PIP strip membranes (Invitrogen) containing various phospholipids (100 pmol for each lipid), as indicated for lipid overlay assay, that were saturated previously with 3% BSA plus 0.1% Tween-20 for 4 h at 4°C. Strips were subsequently processed for Western blot analysis with an anti-GST antibody (Invitrogen). Images were acquired with a Chemi-smart 5000 (Vilber Lourmat). Quantification of the levels of TAPAS-1-GST and Raf-1-GST binding to individual lipids was determined from the integrated densities of spots on immunoblots. Values are given as arbitrary units (mean ± SEM; n = 4 independent experiments with proteins from three distinct protein preparations).
Making of fluorescently tagged liposomes
Liposomes were essentially prepared as described by Avanti Polar Lipids (www.avantilipids.com; technical support section) with some modifications. In brief, liposomes were prepared from brain total lipid extracts (Avanti Polar Lipids; catalog #131101C) at a concentration of 2 mg/ml. In the case of phosphatidic acid containing liposomes, 10% of brain total lipid extract was replaced by phosphatidic acid (Sigma; P9511). Fluorescent labeling of liposomes was achieved by adding 1.2 μg of 2-(4,4-difluoro-5-methyl-4-bromo-3a,4a-diaza-s-indacene-3-dodecanoyl)-1-hexadecanoyl-sn-glycero-3-phosphocholine (β-Bodipy500/510 C12-HPC; Invitrogen) per milliliter of lipid mixture before lyophilization.
To prepare liposome mixtures, all components were dissolved in chloroform (7 mg/ml phosphatidic acid, 30 mg/ml brain total lipid extract, 0.6 μg/μl β-Bodipy500/510 C12-HPC) and mixed in a chloroform-cleaned glass vial. To achieve an even distribution of lipids, the final volume of lipid mixture was adjusted to at least 400 μl. Organic solvent was removed by evaporation. Once evaporated, the lipid mixture was dessicated for 15 min. The lipid cake was hydrated at a concentration of 2 mg/ml using liposome binding buffer (20 mm HEPES, 300 mm sucrose, pH 7.4) for 10 min at RT with gentle agitation. Hydration was followed by two cycles of bath sonification (5 min each) and three cycles of tip sonification (30 s each). To get unilamellar vesicles hydrated, liposomes were extruded 23 times through a 0.1 μm membrane (miniextruder; Avanti Polar Lipids) and used directly in the subsequent experiments. The overall morphology of extruded vesicles was checked on a regular basis by negative staining with uranyl acetate and subsequent electron microscopy.
Binding of PA-binding proteins to fluorescent liposomes (liposome binding assay)
Liposome binding assays were performed using equimolar amounts of the respective GST fusion proteins bound to beads (20 μg) and 200 μg of fluorescently labeled liposome mixture. Fusion proteins were incubated with liposome mixture at 4°C with constant agitation. After overnight incubation, GST beads were settled by centrifugation (1000 rpm; Eppendorf table-top centrifuge; 2 min) and the supernatant was discarded. The beads were washed three times with excess of liposome binding buffer (20 mm HEPES, 300 mm sucrose, pH 7.4). After the final wash, beads were resuspended in 500 μl of liposome binding buffer and analyzed for liposome binding using a LS55 fluorescence spectrometer (PerkinElmer). In brief, samples were excited at 500 nm, and spectra were recorded from 510 to 600 nm. For analysis, the peak region of the spectra (512–522 nm) was averaged and plotted against the relative fluorescence emitted.
Binding of palmitoyl-CoA to immobilized RIBEYE
Equimolar amounts (7 μm) of fusion protein were incubated with increasing concentrations of [1–14C]palmitoyl-CoA (1–20 μm) for 2 h at 4°C in a final volume of 50 μl of PBS. Beads were washed several times with excess volumes of PBS to remove unbound [1–14C]palmitoyl-CoA and quantified in a scintillation counter (Beckman).
Binding of RIBEYE-EGFP to palmitoyl-CoA beads
Transfected COS7 cells were collected from cell culture dishes and washed three times with PBS. Cell pellets were resuspended in PBS/2% sucrose and harvested by passage 20 times through a 22 gauge needle followed by passage 20 times through a 27 gauge needle. Care was taken that the cell extract did not heat up during this procedure. Cell extract was cleared by centrifugation (13,000 rpm; 4°C; 30 min; Biofuge Fresco tabletop centrifuge; Heraeus). For pull-down experiments, the EGFP fusion protein content of extracts obtained from transfected COS cells was monitored with a Fusion-Alpha universal microplate reader (PerkinElmer) using an excitation wavelength of 485/20 nm and a detection wavelength of 530/25 nm. Equal loading was confirmed by Western blotting. Before pull-downs, palmitoyl-CoA beads (30 μl of slurry for each pull-down) were blocked with gelatin (0.2% w/v) for 30 min at 4°C. Pull-down experiments were performed in a total volume of 300 μl. After overnight incubation (4°C) of the beads with the cell extracts, beads were washed multiple times with an excess of PBS/2% sucrose at 4°C. Quantification of pulled down EGFP fluorescence was performed with a Fusion-Alpha microplate reader as described above.
LPA-binding assay
For LPA-binding assays, 1 μl of radioactive LPA (specific activity, 47.0 Ci/mmol) was incubated with equimolar amounts of the respective eluted GST fusion proteins (all at 1 μm concentration) in a final volume of 20–30 μl. For binding analyses, samples were incubated at room temperature for 3 min, spotted on nitrocellulose, and washed several times with multiple volumes of PBS. Binding was quantified in a scintillation counter (Beckman).
LPAAT assays
Assays were performed in a final volume of 300 μl. The reaction mixture contained 20 μm 1-oleoyl LPA and 1 μm of the respective fusion proteins in PBS (volume, 290 μl). This mixture was preincubated for 3 min at room temperature. Subsequently, acyl-CoAs were added to a final concentration of 5 μm, and the reaction was performed for 3–10 min at 37°C unless indicated otherwise. The reaction was stopped by the addition of 300 μl of ice-cold stopping solution (0.8 m KCl, 0.2 m H3PO4). Lipids were extracted with 900 μl chloroform/methanol (2:1), lyophilized, redissolved in 35 μl of chloroform/methanol (2:1), and subjected to thin-layer chromatography (TLC) using a mixture of chloroform/pyridin/acetic acid (50:30:7 volume ratio). Radioactive PA generated as the result of LPAAT activity was quantified by scraping the bands from the silica plates and subsequent measurement in a scintillation counter (Beckman).
Complementation assays using LPAAT-deficient JC201 bacteria
LPAAT-deficient JC201 bacteria were electroporated with the indicated RIBEYE-pGEX constructs (Coleman, 1990; Gallop et al., 2005). LPAAT complementation in LPAAT-deficient JC201 was analyzed qualitatively by survival at 42°C and quantitatively by recording growth curves of the respective JC201 bacteria at 30 and 42°C in liquid culture (Coleman, 1990). JC201 bacteria were first cultured at 30°C (LB/ampicillin, 150 μg/ml; tetracyclin, 15 μg/ml) and then shifted to 42°C with vigorous shaking. Parallel to the shift in temperature, IPTG (isopropyl-thio-β-d-galactopyranoside) was added (final concentration, 0.1 mm). plsC, a plasmid encoding bacterial LPAAT, served as a positive control, and an empty pGEX vector served as a negative control (Coleman, 1990).
Fluorescence resonance energy transfer analyses
Fluorescence resonance energy transfer (FRET) analyses were performed with a LS55 spectrophotometer (PerkinElmer). NADH was excited through FRET by exciting tryptophan at 285 nm (excitation wavelength; excitation slit width, 2.5 nm). Emission spectra were collected from 300 to 500 nm (emission slit width, 3 nm). The maximum FRET peak was observed at 427 nm (Alpadi et al., 2008). FRET efficiencies were analyzed at 25°C in the same batch of fusion proteins in experiments performed in parallel to exclude that different qualities of fusion proteins were responsible for differences.
Coating of coverslips with poly-l-lysine and concanavalin A
Autoclaved round coverslips (25 mm diameter; Menzel-Gläser) were transferred to sterile six-well plates with 1 ml of poly-l-lysine solution (0.001% in H2O) added to each coverslip and incubated overnight. Excess of poly-l-lysine solution was removed from the coverslips by washing with PBS. To further enhance adhesion, 200 μl of concanavalin A solution (1 mg/ml H2O) was added to the poly-l-lysine coverslips and incubated for 2 h at RT followed by three washes with PBS. Treated coverslips were used immediately for cell plating.
Preparation of mouse retinal bipolar cells from adult mice
For the dissociation of retinal bipolar cells from adult mice (C57BL/6 mice of either sex), we used a dissociation procedure similar to the one described by Zhou et al. (2006) and Wan et al. (2008). Retinas were usually isolated from adult C57BL/6 mice of either sex older than 20 weeks. Isolated mouse retinas were washed once with papain digestion buffer [containing (in mm) 150 NaCl, 2.6 KCl, 1 MgSO4, 10 HEPES, pH 7.4, 10 glucose, and 0.5 CaCl2, osmolarity between 305 and 315 mOsm (LSCS)] to which 0.5 mm EDTA, 2.5 mm l-cysteine, and 10 mm HEPES, pH 7.4, were added. Each retina was gently transferred to an incubation tube containing 800 μl of digestion buffer including 40 units of cysteine-activated papain solution and incubated for 10 min at 37°C at 600 rpm. Tubes were gently inverted every 3 min. After enzymatic digestion, the papain solution was replaced with 1 ml of deactivation solution [containing 20 μl of DNase solution (1 mg/ml stock in LSCS), 100 μl 5% BSA (in LSCS), and 880 μl of LSCS] and incubated for 10 min at 37°C. Then, the deactivation solution was replaced by 1 ml of fresh LSCS. Fire-polished 1 ml pipette tips were used to triturate the retina (three to five times up and down) to completely dissociate the tissue. Cells were allowed to adhere to coated coverslips for 30 min at 37°C in a 5% CO2 incubator. The use of poly-l-lysine- and concanavalin A-coated coverslips enhanced the attachment of retinal bipolar cells with differentiated presynaptic terminals (see Figs. 1, 3, 4) (data not shown). After cell attachment, 2 ml of Neurobasal A medium (with B27 complement and 2 mm glutamine) was added to the cells and incubated at 5% CO2, 37°C until further processing. In case of virus infection, the virus was added at the indicated infectious units 30 min after the settling of the retinal cells and remained in the medium for 14–18 h. Typically, after 12 h the first fluorescent signals of the heterologous, EGFP-tagged fluorescent protein could be detected in the transduced cells under the described conditions.
Figure 1.
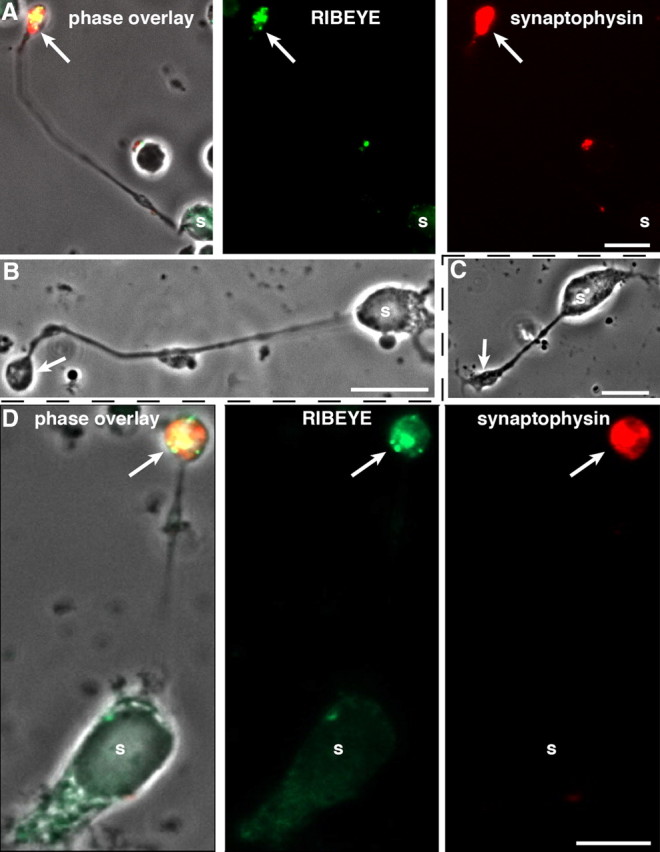
Dissociated mouse retinal bipolar cells. A–D, Characterization of isolated mouse retinal bipolar cells by immunocytochemistry (A, D) and phase contrast microscopy (B, C). Acutely dissociated (A, B) and 2 d cultured (C, D) mouse retinal bipolar cells were analyzed by immunocytochemistry (A, D) with antibodies against RIBEYE and synaptophysin as indicated. s, Soma of retinal bipolar cells. The arrows denote the presynaptic terminals of retinal bipolar cells that contain RIBEYE-labeled synaptic ribbons and synaptophysin-labeled synaptic vesicles that fill the entire presynaptic terminal. Scale bars: 10 μm.
Figure 3.
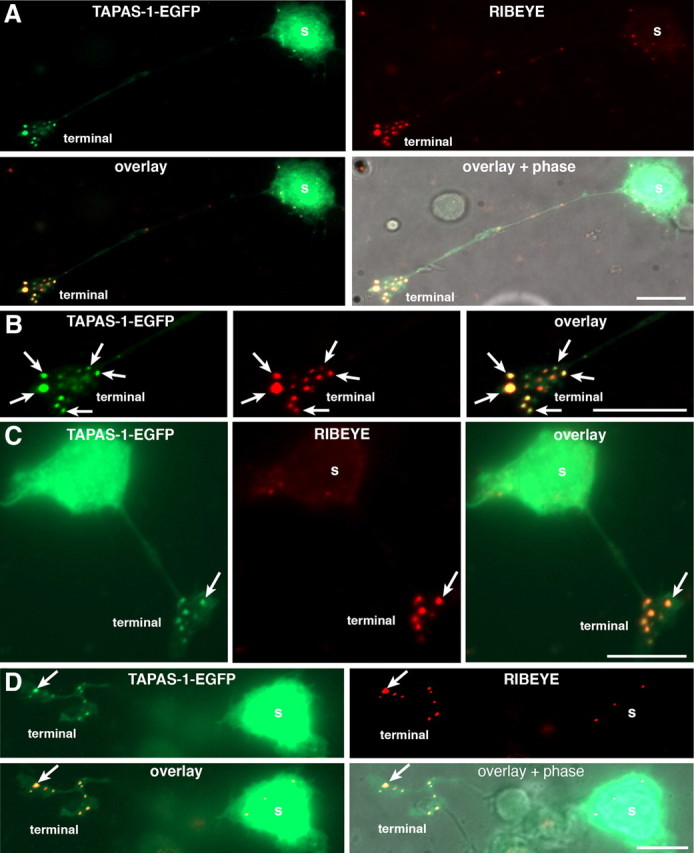
PA is enriched at synaptic ribbons. A–D, PA localization in retinal bipolar cells visualized by heterologous expression of the EGFP-tagged PA-sensor TAPAS-1 wild-type protein in isolated mouse retinal bipolar cells. EGFP-tagged TAPAS-1 wild-type protein is strongly enriched at the synaptic ribbon, which was visualized by immunolabeling with antibodies against RIBEYE (immunosignals in the red channel). Arrows point to immunolabeled synaptic ribbons, which strongly recruit the PA-sensor protein TAPAS-1. B, A high magnification of the synaptic terminal shown in A. In addition to synaptic terminals, sometimes few synaptic ribbons were also found in the cell body of dissociated bipolar cells (A, C). These somatic ribbons also showed a strong recruitment of PA-sensor proteins. As expected from an overexpression system, a diffuse TAPAS-1-EGFP signal is also present in the soma where biosynthesis of TAPAS-1-EGFP occurs. s, Soma of retinal bipolar cells. Scale bars: 10 μm (A–D).
Figure 4.
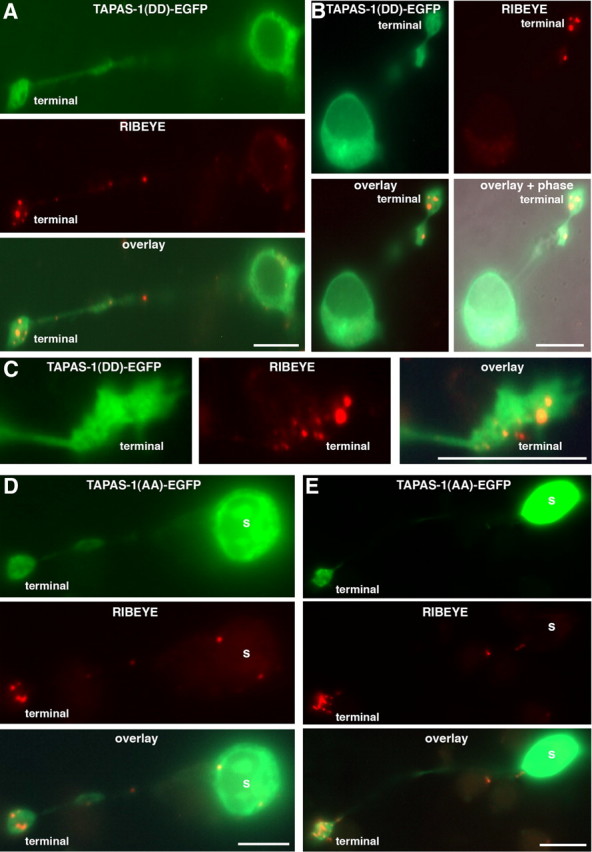
PA localization in ribbon synapses: control transfections. In contrast to wild-type PA-sensor protein (Fig. 3), PA-binding mutant sensor proteins (TAPAS-1(WW19,20DD)-EGFP; (A–C) or TAPAS-1(WW19,20AA)-EGFP; (D–E)) do not colocalize with synaptic ribbons in mouse retinal bipolar cells. This indicates that the previously observed recruitment of TAPAS-1 wild-type protein (Fig. 3) depends on enrichment of PA at the synaptic ribbons and demonstrates the specificity of the PA-sensor recruitment to synaptic ribbons (Fig. 3). TAPAS-1(DD)-EGFP, TAPAS-1(WW19,20DD)-EGFP; TAPAS-1(AA)-EGFP, TAPAS-1(WW19,20AA)-EGFP. Scale bars: A, B, D, E, 10 μm; C, 5 μm.
Generation of recombinant Semliki Forest viruses
Recombinant Semliki Forest (SLF) viruses were generated as described previously (Ashery et al., 1999; Venkatesan et al., 2010). In brief, recombinant pSFV1 plasmids were linearized with SpeI. Linearized DNA was extracted with phenol/chloroform and subsequently precipitated with ethanol. Linearized purified plasmid DNA was transcribed into RNA via in vitro transcription (mMachine SP6 kit; Ambion) according to the manufacturer's instructions. RNA was recovered by precipitation with ethanol. BHK21 cells were electroporated in Optimem medium with 10 μg recombinant RNA and 10 μg helper 2 RNA (at 360 V, 75 μF, 2 mm electroporation cuvette) to generate replication deficient recombinant SLF viruses. After 24 h in culture at 31°C (5% CO2), the virus was harvested. Virus titer and activation of virus were determined as described previously (Ashery et al., 1999; Venkatesan et al., 2010).
Fluorescence microscopy
Fluorescence microscopy was performed as described previously (Schmitz et al., 2000; Alpadi et al., 2008; Magupalli et al., 2008; Venkatesan et al., 2010) using an Axiovert200M microscope (Zeiss) or A1R confocal microscope (Nikon) as indicated.
Miscellaneous procedures
Transfection of COS cells, expression and purification of bacterial fusion proteins, and purification of synaptic ribbons were performed as described previously (Schmitz et al., 1996, 2000; Alpadi et al., 2008; Magupalli et al., 2008). Statistical analyses were performed with Student's t tests.
Results
Phosphatidic acid is highly enriched at synaptic ribbons
Synaptic ribbons are sites of intense synaptic vesicle turnover, a process that requires bending of membranes during fusion and fission with the plasma membrane. PA has been implicated in such a role; therefore, we analyzed whether PA is generated at retinal ribbon synapses. To study PA synthesis at ribbon synapses, we used a retinal bipolar cell culture from the adult mouse retina. The isolated bipolar cells contained synaptic ribbons in their presynaptic terminals as judged by immunofluorescence microscopy with antibodies against RIBEYE (Fig. 1) and by electron microscopy (data not shown). The isolated bipolar cells demonstrated a healthy morphology with a polarized distribution of synaptic proteins, e.g., synaptophysin and RIBEYE over a period of 2 d in culture (Fig. 1).
First, we tested the phospholipid specificity of two previously reported probes for PA. Recombinant TAPAS-1 of PDE4 (Baillie et al., 2002) and the PA-binding domain of Raf-1 (Corrotte et al., 2006) were incubated as GST fusion proteins on phospholipid strips, and binding to the phospholipids was quantified (Fig. 2A). While GST alone did not bind significantly to any of the lipids (data not shown), the two probes significantly bound to various negatively charged lipids with a strong preference for PA (Fig. 2A).
Figure 2.
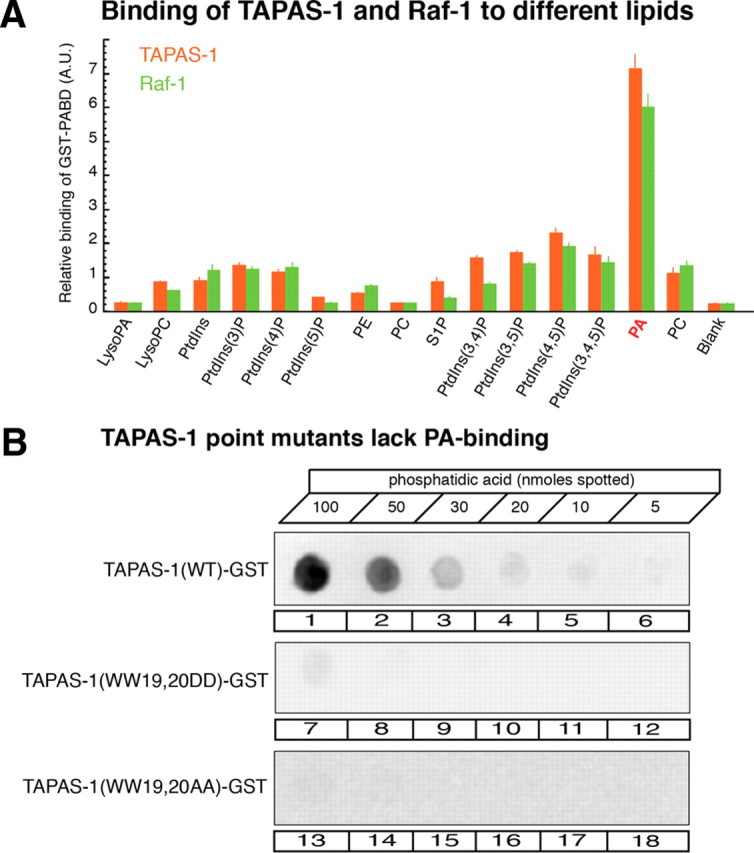
Characterization of PA-binding proteins. A, Quantitative analysis of the binding of PA-sensor proteins TAPAS-1 and Raf-1 to different phospholipids (phospholipid strip assay; 100 pmol for each spotted lipid). Quantification of the level of TAPAS-1-GST and Raf-1-GST binding to individual lipids was determined from the integrated densities of spots on immunoblots. Values are given as arbitrary units (mean ± SEM; n = 4 independent experiments with proteins from three distinct protein preparations). B, Example of lipid dot blot with the indicated amounts of PA spotted onto nitrocellulose filters (5–100 nmol). Binding of TAPAS-1 wild type (lanes 1–6), TAPAS-1(WW19,20DD) (lanes 7–12), or TAPAS-1(WW19,20AA) (lanes 13–18) was detected with anti-GST antibodies. TAPAS-1 wild-type GST significantly bound to PA but not TAPAS-1(WW19,20AA)-GST or TAPAS-1(WW19,20DD)-GST. PE, Phosphatidylethanolamine; PC, phosphatidylcholine; PtdIns, phosphatidylinositol.
Next, we expressed TAPAS-1-EGFP (using Semliki Forest virus) to monitor formation of PA in cultured retinal bipolar cells. We found that the PA probe accumulated at the presynaptic terminal where it colocalized with RIBEYE (Fig. 3). Two mutants of TAPAS-1, TAPAS-1(WW19,20AA) and TAPAS-1(WW19,20DD), that lost the ability to bind to PA in vitro (Fig. 2B) presented a diffuse distribution in bipolar cells, and the mutants failed to colocalize with RIBEYE (Fig. 4). These data demonstrate that the localization of TAPAS-1-EGFP with synaptic ribbons depends on the ability of the probe to bind to PA, and that PA is enriched at synaptic ribbons.
Since RIBEYE is a major component of synaptic ribbons, we next asked whether RIBEYE is directly involved in the binding of PA (Fig. 3). To address this question, we cotransfected COS cells, i.e., nonneuronal cells that do not contain synaptic ribbons, with full-length RIBEYE(AB)- and PA-sensor protein constructs. Heterologously expressed full-length RIBEYE(AB) forms electron-dense protein aggregates (Magupalli et al., 2008; Sheets et al., 2011) that resemble synaptic ribbons and that are denoted as artificial synaptic ribbons in the following text. For these COS cell experiments, we used mCherry-tagged Raf-1 as PA-sensor protein because Raf-1 does not require high intracellular Ca2+ levels for PA binding, a situation that may not occur in COS cells (Gosh et al., 1996, 2003; Baillie et al., 2002) (data not shown). To test these artificial synaptic ribbons for PA binding, we analyzed whether the mCherry-tagged PA-sensor protein Raf-1 is recruited to the EGFP-tagged RIBEYE aggregates (Fig. 5) and Raf-1 mutants that are unable to bind PA served as the control (Fig. 5).
Figure 5.
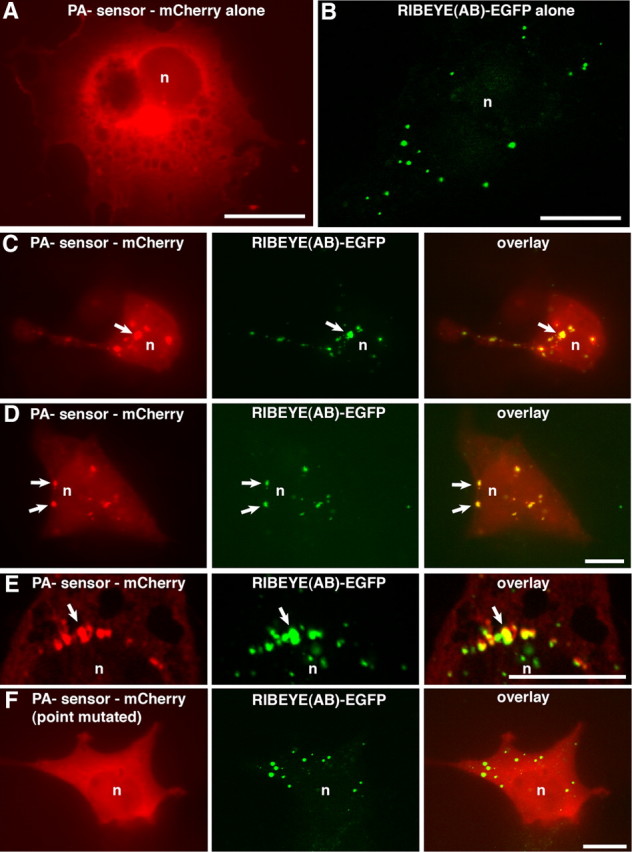
Heterologously expressed RIBEYE aggregates are sites of enrichment of PA as judged by recruitment of PA-sensor proteins. A–E, COS cells were single-transfected with either Raf-1-mCherry (A) and RIBEYE(AB)-EGFP (B) or cotransfected with RIBEYE(AB)-EGFP and PA-binding sensor protein (Raf-1-mCherry) (C–E). F, Point mutated, PA-binding-deficient protein (Raf-1mut) served as control. RIBEYE(AB) protein clusters (arrows) in transfected COS cells recruit wild-type PA-sensor protein Raf-1 (C–E), indicating enrichment of PA at the RIBEYE aggregates. PA-binding-deficient sensor protein (F; data not shown) is not recruited to RIBEYE aggregates, indicating the specificity of the recruitment. n, Nucleus; Scale bars: 10 μm.
As described previously, heterologously expressed RIBEYE generated the typical, punctate ribbon-like labeling pattern in transfected cells (Fig. 5B,C–F) (Magupalli et al., 2008; Sheets et al., 2011), whereas Raf-1 transfected alone typically shows a diffuse distribution with a slight enrichment at the perinuclear region, which is characteristic for the Golgi apparatus (Fig. 5A). In cells coexpressing RIBEYE and wild-type PA sensor, the PA probe was recruited to RIBEYE aggregates as judged by colocalization of the EGFP-tagged RIBEYE with the mCherry-tagged PA sensor Raf-1 (Fig. 5C–E). If cotransfected with RIBEYE(AB)-EGFP, Raf-1 displayed a spot-like distribution that colocalized with the RIBEYE puncta. In contrast, the PA-binding-deficient mutant Raf-1 protein was not recruited to the RIBEYE clusters and remained diffusely distributed in transfected COS cells (Fig. 5F and data not shown), demonstrating the specific recruitment of the PA sensor to RIBEYE. These experiments suggest that RIBEYE itself binds PA, which is subsequently detected by the PA sensor. Thus, RIBEYE appears to be responsible for the previously observed accumulation of PA at synaptic ribbons (see Discussion).
Does RIBEYE generate PA via LPAAT activity?
Based on these findings, we asked whether RIBEYE also has a role in the generation of PA, i.e., whether RIBEYE is directly involved in the synthesis of PA. As outlined in the introduction, RIBEYE consists of an A domain and the C-terminal B domain, which is largely identical to the corepressor CtBP2. In the present study, we tested whether the B domain of RIBEYE possesses LPAAT activity, which catalyzes the conversion of lysophosphatidic acid and specific acyl-CoAs into PA. We focused on the RIBEYE(B) domain for several reasons. First, RIBEYE(B) is highly homologous to CtBP1/BARS (Chinnadurai, 2002; Corda et al., 2006; Schmitz, 2009), and CtBP1/BARS has been suggested previously to be a LPA acyltransferase (Weigert et al., 1999; but see Gallop et al., 2005). Second, independent from these studies CtBP1/BARS has also been shown to bind to PA and to be involved in different ways in PA metabolism (Yang et al., 2008; Haga et al., 2009). Finally, the RIBEYE(B) domain is most likely exposed to the cytoplasm, where it binds NAD(H) and interacts with other components of the presynaptic terminal (Alpadi et al., 2008; Magupalli et al., 2008; Schmitz, 2009). In contrast, there are no indications that the RIBEYE(A) domain has a role in PA synthesis.
RIBEYE(B) domain specifically binds all substrates needed for LPAAT activity
First, we analyzed whether the RIBEYE(B) domain binds the substrates, i.e., acyl-CoAs and LPA, which are needed for the enzymatic LPAAT activity. We used different independent assays to determine whether RIBEYE binds palmitoyl-CoA (as acyl-CoA substrate). These assays were done with RIBEYE wild-type protein as well as with distinct point and deletion mutants of RIBEYE (Fig. 6A). (1) We tested whether purified, bead-immobilized RIBEYE(B)-GST fusion protein binds radioactive palmitoyl-CoA. Purified, bead-immobilized GST fusion protein alone served as a control in these experiments. Figure 6B shows that RIBEYE binds palmitoyl-CoA in a saturable manner, with a KD value of ∼5.5 μm. The binding of radioactive palmitoyl-CoA to RIBEYE could be blocked by the addition of an excess of “cold“ palmitoyl-CoA, but not by the addition of an equimolar excess of cold acetyl-CoA or malonyl-CoA, further indicating the specificity of binding (data not shown). (2) Vice versa, solubilized RIBEYE(B)-GST fusion protein also bound to palmitoyl-CoA immobilized to beads (Fig. 6C). Most of RIBEYE(B)-GST input bound to the palmitoyl-CoA beads, whereas only a minor fraction of the input remained unbound. In contrast, the control protein GST did not bind to bead-immobilized palmitoyl-CoA and completely remained in the supernatant (Fig. 6C). (3) Furthermore, if RIBEYE(B)-EGFP was heterologously expressed in COS cells, only RIBEYE(B)-EGFP but not EGFP alone bound to palmitoyl-CoA beads (Fig. 6D,E). The largest part of the RIBEYE(B)-EGFP input was found to be bound by the palmitoyl-CoA beads, in contrast to the control protein EGFP that did not bind at all to the palmitoyl-CoA beads (Fig. 6D). (4) We found specific NADH-dependent FRET enhancement of RIBEYE(B) by palmitoyl-CoA but not by other acyl-CoAs (Fig. 7E,F). As shown previously, RIBEYE(B) binds NAD(H) (Schmitz et al., 2000; Alpadi et al., 2008; Magupalli et al., 2008) and shows an NADH-dependent FRET effect at about 427 nm (Alpadi et al., 2008; Magupalli et al., 2008) (Fig. 7A). An NAD(H)-binding-deficient RIBEYE point mutant, RIBEYE(B)G730A, served as a negative control (Figs. 6A, 7B). Interestingly, the binding of palmitoyl-CoA to RIBEYE(B) specifically enhances the time-resolved FRET efficiency (Fig. 7E), whereas other acyl-CoAs (e.g., arachidonyl-CoA) did not produce a FRET enhancement (Fig. 7E). These findings indicate that RIBEYE shows a preferential binding for palmitoyl-CoA compared to other acyl-CoAs. Furthermore, a RIBEYE point mutant, RIBEYE(B)F904W, which has a binding deficiency for palmitoyl-CoA (Fig. 7G), did not show any FRET enhancement (Fig. 7F). This shows that the FRET enhancement was not a solvent effect of palmitoyl-CoA, but due to physical binding of palmitoyl-CoA to RIBEYE(B) domain. These data also demonstrate that binding of NADH and palmitoyl-CoA are not mutually exclusive events but can occur simultaneously. We also found strong binding of LPA to RIBEYE(B) fusion protein (Fig. 6F).
Figure 6.

A, Schematic representation of RIBEYE wild-type protein and RIBEYE point and deletion mutants analyzed in the assays. B–F, RIBEYE binds palmitoyl-CoA (B–E) and LPA (F). B, RIBEYE(B)-GST immobilized to glutathione beads binds radioactive palmitoyl-CoA in a saturable manner. GST reflects background binding. Binding of radioactive palmitoyl-CoA to RIBEYE(B)-GST fusion protein compared to GST is highly significant (p < 0.001; n = 6). C, Binding of soluble RIBEYE(B)-GST fusion protein to palmitoyl-CoA beads. RIBEYE(B)-GST fusion protein binds to palmitoyl-CoA beads, whereas GST fusion protein alone does not. Most of the input RIBEYE(B)GST (lane 2) binds to the palmitoyl-CoA beads (lane 6), whereas only a small portion of RIBEYE(B)-GST remains unbound in the supernatant (lane 4). In contrast, all of the GST input control protein (lane 1) remains unbound in the supernatant (lane 3). D, E, Palmitoyl-CoA binding to RIBEYE is enhanced by NADH. D, Palmitoyl-CoA beads bind EGFP-tagged RIBEYE(B) (lane 6, 7), while EGFP alone (control protein) does not bind (lane 4, 5). RE(B)G730A also binds palmitoyl-CoA (lane 8), but considerably less than RIBEYE wild-type protein (lane 6). Addition of NADH enhances palmitoyl-CoA-binding of RIBEYE(B)-EGFP (lane 7), but not of RIBEYE(B)G730A (lane 9) or EGFP alone (lanes 5). E, Quantification of binding of the indicated RIBEYE proteins and EGFP control protein to palmitoyl-CoA without the addition of NADH (columns 1–4) and with the addition of NADH (columns 5–8). F, Different RIBEYE-proteins were tested for their binding of LPA in the filter tests described in Materials and Methods. Error bars indicate SEM. **p < 0.01; ***p < 0.001.
Figure 7.
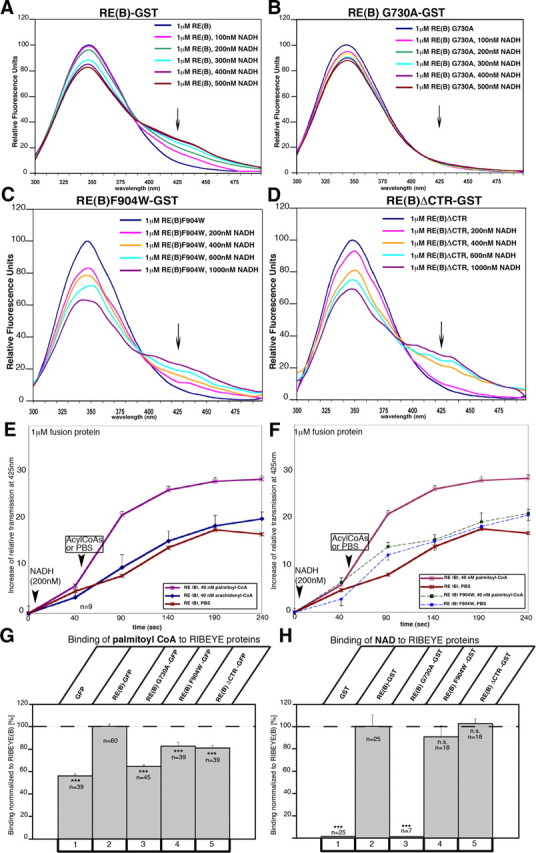
FRET analyses of different RIBEYE constructs. FRET analyses were performed as described previously (Alpadi et al., 2008; Magupalli et al., 2008) A, B, Analysis of FRET emission of RIBEYE(B)-GST (A) and of the NAD(H)-binding-deficient RIBEYE(B)G730A(B). RIBEYE(B)-GST shows a strong NADH-dependent FRET emission at 427 nm (A, arrow), whereas the NAD(H)-binding-deficient point mutant RIBEYE(B)G730A does not (B, arrow). C, D, Analysis of FRET emission of CTR mutants of RIBEYE(B)-GST: RE(B)F904W-GST (C) and RE(B)ΔCTR-GST (D). Both LPAAT-deficient mutants RE(B)F904W-GST and RE(B)ΔCTR-GST showed a robust NADH-dependent FRET (C, D, arrows) after the addition of NADH, which demonstrates proper folding of the tested proteins. CtBP1-GST served as an additional positive control and produced a strong FRET at 427 nm (data not shown); GST served as negative control protein and did not show any NADH-dependent FRET emission at 427 nm (data not shown) (see also Alpadi et al., 2008). Arrows indicate FRET emission peaks. E, F, Time-resolved analysis of FRET emission at 427 nm after addition of various reagents. Time point 0 s, Beginning of FRET emission measurement before additions of reagents; after 10 s, addition of NADH (200 nm) in all experiments; after 60 s, addition of acyl-CoA (palmitoyl-CoA, arachidonyl-CoA, acetyl-CoA; all at 40 nm) or PBS. E, The addition of palmitoyl-CoA but not of arachidonyl-CoA and acetyl-CoA (data not shown) enhanced FRET emission at 427 nm (arrowhead). F, The FRET enhancement of palmitoyl-CoA addition demonstrated in E is completely absent in the palmitoyl-CoA-binding-deficient RIBEYE(B) point mutant RIBEYE(B)F904W, indicating that the palmitoyl-CoA-induced FRET enhancement is a consequence of palmitoyl-CoA binding to RIBEYE and not result of an unspecific solvent effect. All fusion proteins were used at a concentration of 1 μm; n = 9 for RE(B); n = 5 for RE(B)F904W. Error bars indicate SEM. **p < 0.01; ***p < 0.001. G, H, Substrate-binding properties of RIBEYE proteins: RIBEYE binds palmitoyl-CoA, as shown by various fusion protein assays (Fig. 6). Different RIBEYE proteins were tested for their binding of palmitoyl-CoA (G) and NAD+ (H) as described in Materials and Methods. G, Quantification of the binding of different EGFP-tagged RIBEYE constructs to palmitoyl-CoA beads. H, Binding of NAD+ to different RIBEYE(B)-GST constructs.
RIBEYE(B) domain is a fast, NADH-stimulated LPAAT
Since all substrates necessary for the LPAAT enzymatic activity can bind to RIBEYE, we tested directly whether RIBEYE has LPAAT activity using LPA and the indicated different radioactive acyl-CoAs as substrates either in the presence or in the absence of RIBEYE(B)-GST (Fig. 8). GST alone served as control protein. RIBEYE(B) showed strong LPAAT activity in a well-characterized biochemical LPAAT assay (Weigert et al., 1999) using purified fusion protein (Fig. 8): a strong PA band was generated in TLC analyses by RIBEYE(B)-GST (Fig. 8A–G, lanes 6, 9, 11–15, 17, 22), whereas no phosphatidic acid was generated by GST alone (Fig. 8A–G, lanes 3, 5, 10, 16, 21). Remarkably, RIBEYE showed LPAAT activity only if palmitoyl-CoA was offered as acyl-CoA substrate (Fig. 8A, lane 6), but not with other acyl-CoAs, e.g., acetyl-CoA (Fig. 8A, lane 4), in support of the specificity of the reaction (Fig. 8A). Purified RIBEYE(B) fusion protein expressed in JC201 bacteria also displayed LPAAT activity in these assays, excluding the possibility that an unrelated protein copurifying with RIBEYE is responsible for the LPAAT activity (Fig. 8B, lane 9).
Figure 8.

Biochemical analysis of RIBEYE's LPAAT activity. Reaction mixes containing radioactive acyl-CoAs, LPA, and the respective RIBEYE fusion and control proteins were analyzed by TLC for the presence of PA, the end product of LPAAT activity. A, RIBEYE generates PA from LPA if palmitoyl-CoA is present as activated fatty acid donor (lane 6), but not if other acyl-CoAs are used (lane 4). GST alone does not generate PA (lanes 1, 5, 8, 16, 21). B, RE(B)-GST purified from JC201 bacteria also displayed LPAAT activity (lane 9), indicating that the LPAAT activity is not due to contaminating bacterial protein with LPAAT activity. C, The LPAAT activity of RIBEYE is fast and is continuously generating PA (lanes 11–15). D, Wild-type RIBEYE(B)-GST fusion protein as well as RIBEYE(B)G730A-GST fusion protein demonstrated LPAAT activity (lanes 17, 18), whereas the RIBEYE point and deletion mutants RIBEYE(B)F904W-GST and RIBEYE(B)ΔCTR-GST as well as GST did not (lanes 19–21). E, Addition of NADH (200 nm) enhanced the LPAAT activity of RIBEYE(B)-GST but not the LPAAT activity of the NAD(H)-binding-deficient point mutant RIBEYE(B)G730A-GST. F, Quantification of LPAAT activity. NADH addition increased LPAAT activity of RIBEYE(B) wild-type protein by 39% with no effect on RIBEYE(B)G730A. Equimolar concentrations of fusion proteins were used in all experiments (140 nm). G, Purified synaptic ribbons (Schmitz et al. 2000) showed LPAAT activity, whereas purified actin did not (lanes 22, 23). ***p < 0.001. n.s., Nonsignificant.
Time-series experiments indicated that the RIBEYE(B)-mediated LPAAT reaction is fast (Fig. 8C). Already after an incubation of 30 s, synthesis of PA could be detected by TLC. With increasing incubation times, the amount of newly synthesized PA further increased, showing ongoing LPAAT activity of RIBEYE(B) fusion protein (Fig. 8C, lanes 11–15). The RIBEYE(B)G730A point mutant also showed LPAAT activity, but the activity was considerably lower than the LPAAT activity of the wild-type protein, demonstrating that LPAAT activity is dependent on NADH binding (Fig. 8E,F). Thus, NADH binding is not required for basal LPAAT activity, but is important for upregulating LPAAT activity. In support of this view, addition of NADH enhanced LPAAT activity of RIBEYE wild-type protein by 39% (Fig. 8E,F). Synaptic ribbons purified from the bovine retina also demonstrated LPAAT activity (Fig. 8G, lane 22), emphasizing its physiological relevance to the synapse.
The specificity of RIBEYE's LPAAT activity was further confirmed by the use of defined LPAAT-deficient RIBEYE mutants. RIBEYE with mutations in the hydrophobic CTR, i.e., [RE(B)F904W and RE(B)ΔCTR, which lacks the CTR], did not show LPAAT activity in TLC analyses (Fig. 8D, lanes 19,20). Although the CTR mutants of RIBEYE did not differ from wild-type protein with respect to NAD(H) binding (Fig. 7H), they showed an altered binding profile for the LPAAT substrates, i.e., palmitoyl-CoA and LPA (Figs. 6E, 7G), and, possibly as a consequence of altered substrate binding, lack of LPAAT activity. Clearly, lack of LPAAT activity of these CTR mutants of RIBEYE is not due to overall misfolding of the mutant proteins because both CTR mutants showed a strong NADH-dependent FRET (Fig. 7C,D). These data suggest that the CTR has an important role for substrate binding and/or formation of the active center of the enzyme.
RIBEYE can rescue LPAAT activity in LPAAT-deficient JC201 bacteria
The specificity of RIBEYE's LPAAT activity was further confirmed using additional independent tests, i.e., JC201 LPAAT complementation assays (Coleman, 1990; Gallop et al., 2005). The complementation assays were performed to make sure that the LPAAT activity observed in the fusion protein assays was not due to contaminating proteins copurifying with RIBEYE fusion protein. JC201 LPAAT complementation assays (Coleman, 1990; Gallop et al., 2005) provide a readout of LPAAT activity that does not use purified fusion protein but relies on a functional rescue of an endogenous LPAAT deficiency: JC201 bacteria have a temperature-sensitive defect of the endogenous 1-acyl-sn-glycerol-3-phosphate acyltransferase and, as consequence of this LPAAT deficiency, grow only at 30°C and not at 42°C (Coleman, 1990; Gallop et al., 2005). This growth deficiency can be rescued by transformation of JC201 bacteria with recombinant plasmids that encode LPAAT activity.
In these assays, we used JC201 bacteria transformed with a plsC vector, which encodes the endogenous bacterial LPAAT enzyme, as a positive control and an empty pGEX vector as a negative control. Growth was monitored qualitatively and quantitatively at 30°C (permissive temperature) and 42°C (restrictive temperature). RIBEYE(B)-expressing JC201 bacteria survived at 42°C and showed strong growth under these conditions in liquid culture (Fig. 9C,D), demonstrating that RIBEYE(B) rescued the intrinsic LPAAT deficiency of JC201 bacteria. The growth of RIBEYE(B)-transformed JC201 bacteria was similarly strong compared to the growth of JC201 transformed with plsC, indicating efficient LPAAT complementation by RIBEYE(B) (Fig. 9B–D). In contrast, JC201 bacteria expressing GST control protein (pGEX without insert) did not grow at 42°C (Fig. 9B–D), demonstrating the specificity of the LPAAT complementation by RIBEYE. The NAD(H)-binding-deficient RIBEYE point mutant RE(B)G730A also survived at 42°C (Fig. 9B) and continued growing at 42°C, but quantitatively more weakly than wild-type RIBEYE (Fig. 9C,D). A RIBEYE point mutant, RE(B)F904W, and a RIBEYE deletion mutant, RE(B)ΔCTR, which lacks the CTR, did not complement LPAAT activity (Fig. 9B–F). Similar to the RIBEYE(B) domain, full-length RIBEYE(AB) also rescued LPAAT deficiency of JC201 bacteria, demonstrating that the RIBEYE(A) domain does not inhibit LPAAT of RIBEYE(B) (Fig. 9B,E,F). In conclusion, the finding that RIBEYE rescued LPAAT activity in LPAAT-deficient JC201 bacteria proves that RIBEYE has LPAAT activity. The LPAAT activity of RIBEYE is mediated by its B domain.
Figure 9.
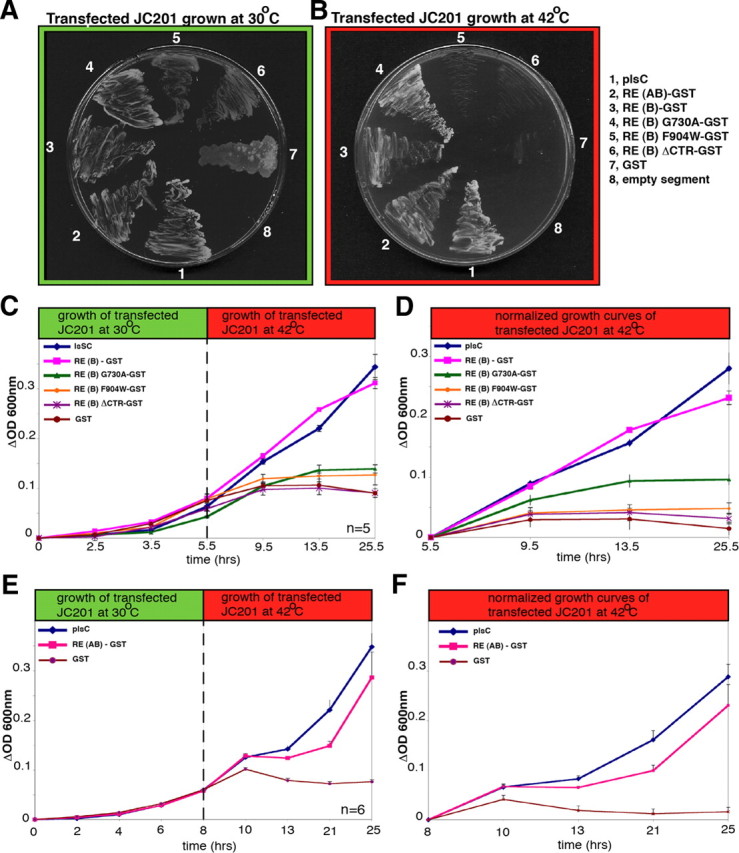
RIBEYE rescues LPAAT deficiency in JC201 bacteria. A, B, LPAAT complementation of transformed JC201 bacteria was qualitatively analyzed by growth of the respective bacteria at 42°C (on ampicillin/tetracyclin plates). Full-length RIBEYE(AB), RIBEYE(B), and RIBEYE(B)G730A complemented LPAAT deficiency, as evidenced by growth of the respective bacteria at 42°C, whereas RIBEYE(B)F904W, RIBEYE(B)ΔCTR, and GST did not. JC201 bacteria transformed with the latter RIBEYE mutants grew only at 30°C, but not at 42°C. plsC, encoding the endogenous bacterial LPAAT, served as a positive control (Coleman, 1990); an empty pGEX vector served as a negative control. C–F, LPAAT complementation of transformed JC201 bacteria was quantitatively analyzed by recording growth curves at 30°C (permissive temperature) and 42°C (restrictive temperature). plsC served as a positive control; an empty pGEX vector served as a negative control. Full-length RIBEYE(AB), RIBEYE(B), and RIBEYE(B)G730A complemented LPAAT deficiency, whereas RIBEYE(B)F904W and RIBEYE(B)ΔCTR did not complement LPAAT activity. Complementation of LPAAT deficiency by the eukaryotic RIBEYE protein was nearly as strong as LPAAT complementation by the genuine bacterial LPAAT protein encoded by plsC. n = 5 (C, D); n = 6 (E, F). Error bars indicate SEM.
An LPAAT-deficient RIBEYE mutant does not generate PA at artificial ribbons, in contrast to wild-type RIBEYE
Next, we tested whether the LPAAT-deficient RIBEYE mutant RIBEYE (AB)ΔCTR, which lacks the CTR, can generate PA in transfected COS cells. From these experiments, we expected to obtain insights as to whether the accumulation of PA at transfected RIBEYE aggregates is due to the LPAAT activity of RIBEYE or a result of unrelated events [e.g., recruitment of proteins with LPAAT or phospholipase D (PLD) activity]. Similar to full-length RIBEYE (Fig. 5) RIBEYE(AB)ΔCTR forms RIBEYE protein clusters in transfected cells (Fig. 10). While these RIBEYE clusters accumulate PA sensors (indicating PA generation) in the case of full-length RIBEYE (Fig. 10A,B), they failed to accumulate PA sensors if the LPAAT-deficient RE(AB)ΔCTR was used (Fig. 10C,D). These data suggest that the LPAAT activity of RIBEYE is probably the most significant source for PA generation at the heterologously induced RIBEYE protein clusters. As outlined in the introduction, RIBEYE is generated from the CtBP2 gene, which gives rise to two different gene products, i.e., RIBEYE, which is found only in ribbon synapses, and CtBP2, which is ubiquitously expressed (Chinnadurai, 2002; Schmitz, 2009). CtBP2 does not appear to be a major component of synaptic ribbons because Western blot analyses of purified synaptic ribbons did not show a significant CtBP2 signal, although RIBEYE was abundantly present and detectable by the anti-CtBP2 and the anti-RIBEYE(B)-domain antibodies (Fig. 11). Thus, CtBP2 probably does not contribute to the generation of PA at synaptic ribbons. The lack of PA generation at RIBEYE(AB)ΔCTR aggregates (Fig. 10A–D) supports the assumption that RIBEYE(A) domain has a structural role in building the synaptic ribbon (Magupalli et al., 2008; Schmitz, 2009) but no role in the generation and binding of PA.
Figure 10.
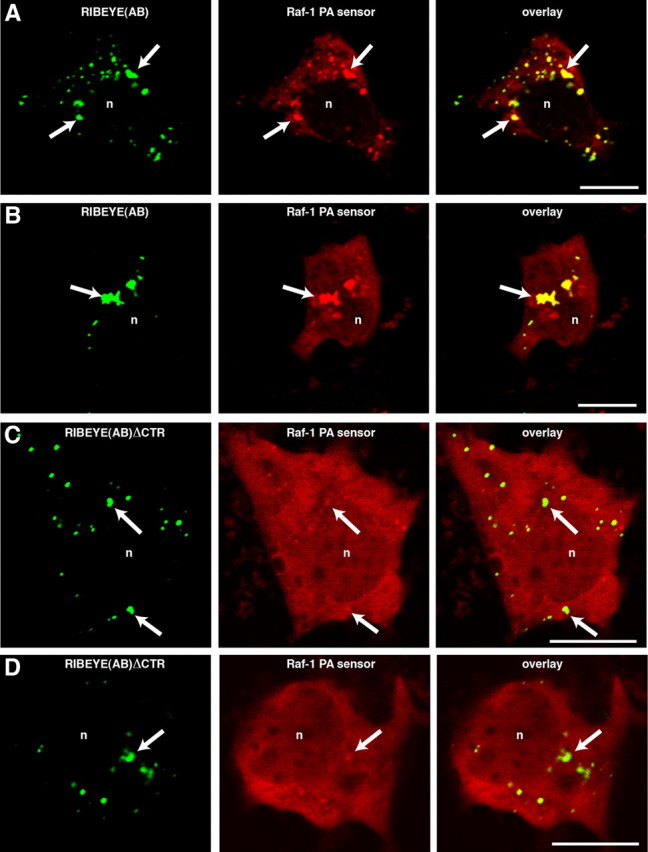
RE(AB)EGFP colocalizes with Raf-1 PA-sensor protein, whereas RE(AB)ΔCTR does not (confocal microscopy). Heterologously expressed full-length RIBEYE forms synaptic ribbon-like clusters in transfected COS cells (arrows, A–D, left) (Magupalli et al., 2008; Sheets et al., 2011). Full-length RIBEYE(AB)-EGFP generates PA as judged by the accumulation of the fluorescent PA-sensor protein Raf-1 at the ribbon-like RIBEYE clusters (A, B). The LPAAT-deficient RIBEYE(AB)ΔCTR, which lacks the CTR does not generate PA (Figs. 8D, 9) and therefore is also not able to accumulate the PA-sensor protein (C, D), although formation of artificial ribbons is not inhibited by the deletion (arrows; C, D; left). In contrast to the RIBEYE clusters formed by the wild-type protein (A, B), PA-sensor proteins were not enriched at RIBEYE clusters formed by the LPAAT-deficient RIBEYE(AB)ΔCTR (C, D). n, Nucleus. Scale bars: 10 μm.
Figure 11.

RIBEYE, but not CtBP2, is highly enriched at synaptic ribbons. Synaptic ribbons purified as described by Schmitz et al. (1996) were analyzed by Western blotting for the presence of CtBP proteins with antibodies against the B domain of RIBEYE (U2656) (Schmitz et al., 2000); CtBP1 (Becton Dickinson), and CtBP2 (Becton Dickinson). Although the antibody against CtBP2 strongly labeled RIBEYE (lane 2, arrow), no significant signal for CtBP2 was observed in purified synaptic ribbons (lane 2, arrowhead), indicating that CtBP2 is largely absent from synaptic ribbons. In contrast, CtBP1 is present in synaptic ribbons (lane 3, arrowhead). Labeling with the antibody against RIBEYE(B) domain/CtBP2 (lane 1) served as a positive control (Schmitz et al., 2000). This antibody also showed a strong RIBEYE band (arrow), but no CtBP2-band (arrowhead) in the purified ribbon fraction.
Discussion
Synaptic ribbons are hot spots of PA generation
The recruitment of PA-sensor proteins to synaptic ribbons in retinal bipolar cells demonstrated that synaptic ribbons are hot spots of PA generation. PA generation at synaptic ribbons is relevant because PA is a biologically highly active phospholipid. In addition to modulating signaling and trafficking proteins, PA affects essential membrane trafficking events due to its influence on membrane curvature (Chernomordik and Kozlov, 2005; Jenkins and Frohman, 2005; Chernomordik et al., 2006; Stace and Ktistakis, 2006; Wang et al., 2006; Zimmerberg and Kozlov, 2006; Oude Weernink et al., 2007). Depending on local conditions, PA adopts a cone-shaped geometry that favors formation of negatively curved membrane monolayers. Negative membrane curvature is required for membrane trafficking including essential aspects of exocytosis and endocytosis (McMahon and Gallop, 2005; Corda et al., 2006; Reynwar et al., 2007; Chasserot-Golaz et al., 2010). In chromaffin cells, PA is generated at sites of membrane fusion and promotes exocytosis (Zeniou-Meyer et al., 2007; Bader and Vitale, 2009; Vitale, 2010). Similarly, PA has been shown to be important for Aplysia synaptic activity (Humeau et al., 2001). Particularly, the stalk-like hemifusion intermediate is promoted by cone-shaped lipids such as PA (Chernomordik and Kozlov, 2005; Chernomordik et al., 2006). Generation of PA and negative membrane curvature are also required for endocytic membrane trafficking, for example, fission of vesicles at the Golgi apparatus or at plasma membrane/endosomes (Bonazzi et al., 2005; Yang et al., 2005; Corda et al., 2006).
We showed that RIBEYE has a central role in the binding and synthesis of PA at the synaptic ribbon. Heterologous expression of RIBEYE in COS cells suggested that RIBEYE directly binds PA. Ribbon-like structures induced in COS cells by heterologous expression of full-length RIBEYE(AB) strongly recruited PA-sensor proteins, similarly to the recruitment of PA sensors to synaptic ribbons in retinal bipolar cells. The RIBEYE(B) domain, but not the RIBEYE(A) domain, mediated PA binding because full-length RIBEYE(AB) bound PA, whereas RIBEYE(AB)ΔCTR (which contains an intact A domain) did not. We demonstrated that RIBEYE is also involved in the synthesis of PA. Principally, different pathways can generate PA: (1) hydrolysis of phospholipids via PLD (Bader and Vitale, 2009), (2) phosphorylation of diacylglycerol (DAG) by DAG kinase (Yang et al., 2011), and (3) acylation of lysophospholipids by lysophospholipid acyltransferases (Shindou et al., 2009; Shindou and Shimizu, 2009). Lysophospholipids, substrates of the latter reaction, are generated under various physiological and pathophysiological situations at various cell organelles, e.g., the endoplasmic reticulum, Golgi apparatus, endosomes, and presynaptic terminals (de Figueiredo et al., 2001; Chambers et al., 2003; Ong et al., 2005; Rigoni et al., 2005; Rossetto and Montecucco, 2008; Schmidt and Brown, 2009; Bate and Williams, 2010; Bate et al., 2010; Oliveira et al., 2010; Vitale et al., 2011).
RIBEYE is a palmitoyl-CoA-dependent LPA acyltransferase that is encoded by RIBEYE(B) domain
We demonstrated that RIBEYE is directly responsible for the synthesis of PA at the synaptic ribbon via LPAAT activity of the RIBEYE(B) domain. Using two independent approaches, we demonstrated that the RIBEYE(B) domain is an LPAAT that specifically transfers palmitoyl-CoA to lysophosphatidic acid to generate PA: (1) in vivo complementation of LPAAT deficiency in LPAAT-deficient JC201 bacteria by RIBEYE and (2) biochemical analyses using a well-established biochemical LPAAT in vitro assay system (Weigert et al., 1999) and purified RIBEYE fusion proteins.
The in vivo JC201 LPAAT complementation assay is based on the analysis of growth behavior of plasmid-transformed bacteria and is independent from experimentally purified fusion proteins that might be potentially contaminated with bacterial proteins containing LPAAT activity. These JC201 complementation assays clearly showed LPAAT activity of RIBEYE, because RIBEYE complemented the endogenous LPAAT deficiency of JC201 bacteria. LPAAT complementation assays were fully confirmed by biochemical LPAAT assays performed with purified fusion proteins isolated from LPAAT-deficient JC201 bacteria. These data exclude the possibility that an unrelated bacterial protein copurifying with RIBEYE(B) is responsible for the LPAAT activity of RIBEYE(B). The identification of RIBEYE point and deletion mutants that lack LPAAT activity further strengthens the specificity of the observed enzymatic activity. The finding that purified synaptic ribbons have LPAAT activity emphasizes the physiological relevance for PA generation in the ribbon synapse. Furthermore, we show that all substrates necessary for LPAAT activity, i.e., palmitoyl-CoA and LPA, specifically bind to RIBEYE(B) wild-type protein. Palmitoyl-CoA binding was shown by FRET-experiments, two different biochemical palmitoyl-CoA binding assays, as well as by the biochemical LPAAT assays that rely on the binding of palmitoyl-CoA. RIBEYE specifically bound long-chain palmitoyl-CoA (KD ≈ 5.5 μm), whereas short-chain acyl-CoAs and acyl-CoAs with unsaturated fatty acids were not bound to RIBEYE and not used as substrates for LPAAT activity. This further demonstrates the specificity of the enzymatic reaction.
Using point and deletion mutants, we have shown that the hydrophobic CTR of RIBEYE(B) is important for both palmitoyl-CoA binding and LPAAT activity. Mutations in the CTR also affected LPA binding, further emphasizing the central role of the CTR. Interestingly, the CTR is relatively divergent between RIBEYE(B)/CtBP2 and CtBP1 (50.6% identical residues in the CTR versus 88.5% identical residues in the crystallized portion containing the NAD(H)-binding subdomain and substrate-binding subdomain) (Kumar et al., 2002; Nardini et al., 2003). This divergence in the CTRs, which is important for LPAAT activity and substrate binding, could account for differences in LPAAT activity between RIBEYE(B)/CtBP2 and CtBP1. Weigert et al. (1999) reported LPAAT activity for CtBP1/BARS. This view was later challenged by a study by Gallop et al. (2005), who found that the observed LPAAT activity of CtBP1/BARS was due to contaminating proteins. RIBEYE and CtBP1, but not CtBP2, are present in purified synaptic ribbons (Schmitz et al., 2000; present study). Based on our data, RIBEYE appears to be the major CtBP protein with LPAAT activity at the synaptic ribbon. This suggestion is further supported by the finding that artificial synaptic ribbons induced by heterologous expression of full-length RIBEYE(AB) bind and generate PA, whereas artificial synaptic ribbons formed by LPAAT-deficient RIBEYE(AB)ΔCTR, which lack the C-terminal region of RIBEYE(B) (Fig. 6A), do not (Fig. 10). The ubiquitously expressed CtBP1 does not seem to have a major impact on PA accumulation because CtBP1 cannot overcome the LPAAT deficiency of RIBEYE(AB)ΔCTR in transfected COS cells (Fig. 10). Interestingly, the binding of NAD(H) to CtBP proteins induces structuring of the intrinsically unstructured CTR (Goldberg et al., 1994; Chinnadurai, 2002; Nardini et al., 2006; Schmitz, 2009). Thus, binding of NAD(H) and the induced structural changes of the CTR could enhance LPAAT activity of RIBEYE either by facilitating palmitoyl-CoA binding and/or by optimizing the conformation of the active center.
The synaptic ribbon as a platform for focal PA generation at the active zone: functional implications
The LPAAT activity of RIBEYE converts palmitoyl-CoA and LPA into PA, a highly potent phospholipid that strongly influences membrane trafficking. The PA-generating properties of RIBEYE suggest that PA fulfils essential roles in synaptic ribbon-dependent vesicle trafficking. Intense exocytic and endocytic membrane trafficking occurs at synaptic ribbons (Schmitz, 2009; Wan and Heidelberger, 2011). Based on its well-known role in neuroendocrine cells (Bader and Vitale, 2009), it is tempting to assume that PA directly facilitates synaptic vesicle fusion also in ribbon synapses. Synaptic vesicle fusion predominantly occurs at the base of the synaptic ribbon where vesicles are docked at the presynaptic plasma membrane. Generation of PA at this location could boost vesicle fusion, which is particularly important for high-speed ribbon synapses. It has also been proposed that vesicle–vesicle (“compound”) fusion on the synaptic ribbon contributes to signal transduction at ribbon synapses (Matthews and Sterling, 2008). PA could promote compound fusion in a similar manner as fusion of synaptic vesicles with the presynaptic plasma membrane. Mounting evidence also suggests a role of synaptic ribbons in endocytic vesicle retrieval (Lenzi et al., 2002; Logiudice et al., 2009). Ribbon-generated PA could provide negative membrane curvature needed for distinct steps of endosomal trafficking, e.g., vesicle fission, possibly similarly as proposed for CtBP1/BARS in other systems (Bonazzi et al., 2005). Which steps of exocytic and/or endocytic trafficking in the ribbon synapse depend on PA generation remains to be elucidated by future investigations.
We propose that RIBEYE is the central synaptic ribbon protein both at a structural as well as at a functional level. Previous work demonstrated that the A domain of RIBEYE has an important structural role in building the ribbon backbone (Magupalli et al., 2008; Schmitz, 2009). The present study identified RIBEYE(B) as functional effector domain of RIBEYE that works as a lipid-modifying enzyme. Through the self-aggregating properties of RIBEYE(A), a high focal concentration of RIBEYE(B)/CtBP2 is achieved at the synaptic ribbons. The high focal concentration of PA generated by LPAAT activity of RIBEYE(B) could boost fast signal transmission. Interestingly, positive feedback loops are known for the PA synthesis in other systems, e.g., activation of PLD by CtBP proteins or activation of PA-generating cascades by PA (Oude Weeernink et al., 2007; Haga et al., 2009). If applied to ribbon synapses, such an “explosive” production of PA could be relevant, e.g., for coordinated multivesicular vesicle release known to occur at ribbon synapses. Currently, few if any data are available on further PA-generating or PA-amplifying pathways in ribbon synapses.
Footnotes
This work was supported by German Research Community Research Grants SFB530 and SFB894 (F.S.) and GRK1328 (F.S., K.S.), by ANT Grant ANR-09-BLAN-0264 (N.V.), and by CNRS-Liban (N.K.). We thank Prof. Jens Rettig and Dr. Ulf Matti (Institute of Physiology, Saarland University, Homburg/Saar, Germany) for help with the generation of recombinant Semliki Forest viruses, Dr. Harvey T. McMahon (Medical Research Council, Cambridge, United Kingdom) for JC201 bacteria, Dr. Chad Grabner and Tracy Grabner (Institute for Anatomy and Cell Biology, Homburg, Germany) for critically reading this manuscript, and Sylvia Brundaler for excellent technical assistance.
The authors declare no competing financial interests.
References
- Alpadi K, Magupalli VG, Käppel S, Köblitz L, Schwarz K, Seigel GM, Sung C-H, Schmitz F. RIBEYE recruits Munc119, a mammalian ortholog of the C. elegans protein unc119, to synaptic ribbons of photoreceptor synapses. J Biol Chem. 2008;283:26461–26467. doi: 10.1074/jbc.M801625200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antonescu CN, Danuser G, Schmid SL. Phosphatidic acid plays a regulatory role in clathrin-mediated endocytosis. Mol Biol Cell. 2010;21:2944–2952. doi: 10.1091/mbc.E10-05-0421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashery U, Betz A, Xu T, Brose N, Rettig J. An efficient method for infection of adrenal chromaffin cells using the Semliki Forest virus gene expression system. Eur J Cell Biol. 1999;78:525–532. doi: 10.1016/s0171-9335(99)80017-x. [DOI] [PubMed] [Google Scholar]
- Bader MF, Vitale N. Phospholipase D in calcium-regulated exocytosis: lessons from chromaffin cells. Biochem Biophys Acta. 2009;1791:936–941. doi: 10.1016/j.bbalip.2009.02.016. [DOI] [PubMed] [Google Scholar]
- Baillie GS, Huston E, Scotland G, Hodgkin M, Gall I, Peden AH, MacKenzie C, Houslay ES, Currie R, Pettitt TR, Walmsley AR, Wakelam M.JO, Warwicker J, Houslay MD. TAPAS-1, a novel microdomain within the unique N-terminal region of he PDE4A1 cAMP-specific phosphodiesterase that allows rapid, Ca2+-triggered membrane association with selectivity for interaction with phosphatidic acid. J Biol Chem. 2002;277:28298–28309. doi: 10.1074/jbc.M108353200. [DOI] [PubMed] [Google Scholar]
- Bate C, Williams A. Amyloid-b(1–40) inhibits amyloid-b(1–42)induced activation of cytoplasmic phospholipase A2 and synapse degeneration. J Alzheimers Dis. 2010;21:985–993. doi: 10.3233/JAD-2010-100528. [DOI] [PubMed] [Google Scholar]
- Bate C, Tayebi M, Williams A. Phospholipase A2 inhibitors protect against prion and Abeta mediated synapse degeneration. Mol Neurodegner. 2010;8:5–13. doi: 10.1186/1750-1326-5-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonazzi M, Spano S, Turacchio G, Cericola C, Valente C, Colanzi A, Kweon HS, Hsu VW, Polishchuck EV, Polishchuck RS, Sallese M, Pulvirente T, Corda D, Luini A. CtBP3/BARS drives membrane fission in dynamin-independent transport pathways. Nat Cell Biol. 2005;7:570–580. doi: 10.1038/ncb1260. [DOI] [PubMed] [Google Scholar]
- Buran BN, Strenzke N, Neef A, Gundelfinger ED, Moser T, Liberman MC. Onset coding is degraded in auditory nerve fibers from mutant mice lacking synaptic ribbons. J Neurosci. 2010;30:7587–7597. doi: 10.1523/JNEUROSCI.0389-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cazzolli R, Shemon AN, Fang MQ, Hughes WE. Phospholipid signalling through phospholipase D and phosphatidic acid. IUBMB Life. 2006;58:457–461. doi: 10.1080/15216540600871142. [DOI] [PubMed] [Google Scholar]
- Chambers K, Judson B, Brown WJ. A unique lysophospholipid acyltransferase antagonist, CI-976, affects secretory and endocytic membrane trafficking pathways. J Cell Sci. 2003;118:3061–3071. doi: 10.1242/jcs.02435. [DOI] [PubMed] [Google Scholar]
- Chasserot-Golaz S, Coorssen JR, Meunier FA, Vitale N. Lipid dynamics in exocytosis. Cell Mol Neurosci. 2010;30:1335–1342. doi: 10.1007/s10571-010-9577-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chernomordik L, Kozlov MM. Membrane hemifusion: crossing a chasm in two leaps. Cell. 2005;123:375–382. doi: 10.1016/j.cell.2005.10.015. [DOI] [PubMed] [Google Scholar]
- Chernomordik LV, Zimmerberg J, Kozlov MM. Membranes of the world unite! J Cell Biol. 2006;175:201–207. doi: 10.1083/jcb.200607083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chinnadurai G. CtBP, an unconventional transcriptional corepressor in development and oncogenesis. Mol Cell. 2002;9:213–224. doi: 10.1016/s1097-2765(02)00443-4. [DOI] [PubMed] [Google Scholar]
- Coleman J. Characterization of Escherichia coli cells deficient in 1-acyl-sn-glycerol-3-phosphate acyltransferase activity. J Biol Chem. 1990;265:17215–17221. [PubMed] [Google Scholar]
- Corda D, Colanzi A, Luini A. The multiple activities of CtBP/BARS proteins: the Golgi view. Trends Cell Biol. 2006;16:167–173. doi: 10.1016/j.tcb.2006.01.007. [DOI] [PubMed] [Google Scholar]
- Corrotte M, Chasserot-Golaz S, Huang P, Du G, Ktistakis NT, Frohman MA, Vitale N, Bader MF, Grant NJ. Dynamics and function of phospholipase D and phosphatidic acid during phagocytosis. Traffic. 2006;7:365–377. doi: 10.1111/j.1600-0854.2006.00389.x. [DOI] [PubMed] [Google Scholar]
- de Figueiredo P, Doody A, Polizotto RS, Drecktrah D, Wood S, Banta M, Strang MS, Brown WJ. Inhibition of transferring recycling and endosome tubulation by phospholipase A2 antagonists. J Biol Chem. 2001;276:47361–47370. doi: 10.1074/jbc.M108508200. [DOI] [PubMed] [Google Scholar]
- Donaldson JG. Phospholipase D in endocytosis and endosomal recycling pathways. Biochim Biophys Acta. 2009;1791:845–849. doi: 10.1016/j.bbalip.2009.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank T, Khimich D, Neef A, Moser T. Mechanisms contributing to synaptic Ca2+ signals and their heterogeneity in hair cells. Proc Natl Acad Sci U S A. 2009;106:4483–4488. doi: 10.1073/pnas.0813213106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallop JL, Butler PJ, McMahon HT. Endophilin and CtBP/BARS are not acyltransferases in endocytosis or Golgi fission. Nature. 2005;438:675–678. doi: 10.1038/nature04136. [DOI] [PubMed] [Google Scholar]
- Goldberg JD, Yoshida T, Brick P. Crystal structure of a NAD-dependent d-glycerate dehydrogenase at 2.4A resolution. J Mol Biol. 1994;236:1123–1140. doi: 10.1016/0022-2836(94)90016-7. [DOI] [PubMed] [Google Scholar]
- Gosh S, Strum JC, Sciorra VA, Daniel L, Bell RM. Raf-1 kinase possess distinct binding domains for phosphatidylserine and phosphatidic acid. J Biol Chem. 1996;271:8472–8480. doi: 10.1074/jbc.271.14.8472. [DOI] [PubMed] [Google Scholar]
- Gosh S, Moore S, Bell RM, Dush M. Functional analysis of a phosphatidic acid binding domain in human Raf-1 kinase. J Biol Chem. 2003;278:45690–45696. doi: 10.1074/jbc.M302933200. [DOI] [PubMed] [Google Scholar]
- Haga Y, Miwa N, Jahanger S, Okada T, Nakamura S. CtBP1/BARS is an activator of phospholipase D1 necessary for agonist-induced macropinocytosis. EMBO J. 2009;28:1197–1207. doi: 10.1038/emboj.2009.78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hull C, Studholme K, Yazulla S, von Gersdorff H. Diurnal changes in exocytosis and the number of synaptic ribbons at active zones of an ON-bipolar cell terminal. J Neurophysiol. 2006;96:2025–2033. doi: 10.1152/jn.00364.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humeau Y, Vitale N, Chasserot-Golaz S, Dupont JL, Du G, Frohman MA, Bader MF, Poulain B. A role for phospholipase D1 in neurotransmitter release. Proc Natl Acad Sci U S A. 2001;98:15300–15305. doi: 10.1073/pnas.261358698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huston E, Gall I, Houslay TM, Houslay MD. Helix-1 of the cAMP-specific phosphodiesterase PDE4A1 regulates its phospholipase-d-dependent redistribution in response to release of Ca2+ J Cell Sci. 2006;119:3799–3810. doi: 10.1242/jcs.03106. [DOI] [PubMed] [Google Scholar]
- Jackman SL, Choi SY, Thoreson WB, Rabl K, Bartoletti TM, Kramer RH. Role of the synaptic ribbon in transmitting the cone light response. Nat Neurosci. 2009;12:303–310. doi: 10.1038/nn.2267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenkins GM, Frohman MA. Phospholipase D: a lipid centric review. Cell Mol Life Sci. 2005;62:2305–2316. doi: 10.1007/s00018-005-5195-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson SL, Forge A, Knipper M, Münkner S, Marcotti W. Tonotopic variation in the calcium dependence of neurotransmitter release and vesicle pool replenishment at mammalian auditory ribbon synapses. J Neurosci. 2008;28:7670–7678. doi: 10.1523/JNEUROSCI.0785-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khimich D, Nouvian R, Pujol R, tom Dieck S, Egner A, Gundelfinger ED, Moser T. Hair cell synaptic ribbons are essential for synchronous auditory signalling. Nature. 2005;434:889–894. doi: 10.1038/nature03418. [DOI] [PubMed] [Google Scholar]
- Kumar V, Carlson JE, Ohgi KA, Edwards TA, Rose DW, Escalante CR, Rosenfeld MG, Aggarwal AK. Transcription corepressor CtBP is an NAD+-regulated dehydrogenase. Mol Cell. 2002;10:857–869. doi: 10.1016/s1097-2765(02)00650-0. [DOI] [PubMed] [Google Scholar]
- Lenzi D, Crum J, Ellisman MH, Roberts WM. Depolarization redistributes synaptic membrane and creates a gradient of vesicles on the synaptic body at a ribbon synapse. Neuron. 2002;36:649–659. doi: 10.1016/s0896-6273(02)01025-5. [DOI] [PubMed] [Google Scholar]
- Liberman LD, Wang H, Liberman MC. Opposite gradients of ribbon size and AMPA receptor expression underlie sensitivity differences among cochlear-nerve/hair-cell synapses. J Neurosci. 2011;31:801–808. doi: 10.1523/JNEUROSCI.3389-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Logiudice L, Sterling P, Matthews G. Vesicle recycling at ribbon synapses in the finely branched axon terminals of mouse retinal bipolar neurons. Neuroscience. 2009;164:1546–1556. doi: 10.1016/j.neuroscience.2009.09.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magupalli VG, Schwarz K, Alpadi K, Natarajan S, Seigel GM, Schmitz F. Multiple RIBEYE-RIBEYE interactions create a dynamic scaffold for the formation of synaptic ribbons. J Neurosci. 2008;28:7954–7967. doi: 10.1523/JNEUROSCI.1964-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matthews G, Sterling P. Evidence that vesicles undergo compound fusion on the synaptic ribbon. J Neurosci. 2008;28:5403–5411. doi: 10.1523/JNEUROSCI.0935-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMahon HT, Gallop JL. Membrane curvature and mechanisms of dynamic cell membrane remodelling. Nature. 2005;481:590–596. doi: 10.1038/nature04396. [DOI] [PubMed] [Google Scholar]
- Meyer AC, Moser T. Structure and function of cochlear afferent innervation. Curr Opin Otolaryngol Head Neck Surg. 2010;18:441–446. doi: 10.1097/MOO.0b013e32833e0586. [DOI] [PubMed] [Google Scholar]
- Meyer AC, Frank T, Khimich D, Hoch G, Riedel D, Chapochnikov NM, Yarin NM, Harke B, Hell SW, Egner A, Moser T. Tuning of synapse number, structure and function in the cochlea. Nat Neurosci. 2009;12:444–453. doi: 10.1038/nn.2293. [DOI] [PubMed] [Google Scholar]
- Nardini M, Spano S, Cercola C, Pesce A, Massaro A, Millo E, Luini A, Corda D, Bolognesi M. CtBP/BARS: a dual-function protein involved in transcription co-repression and Golgi membrane fission. EMBO J. 2003;22:3122–3130. doi: 10.1093/emboj/cdg283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nardini M, Svergun D, Konarev PV, Spano S, Fasano M, Bracco C, Pesce A, Donadini A, Cericola C, Secundo F, Luini A, Corda D, Bolognesi M. The C-terminal domain of the transcriptional corepressor CtBP is intrinsically unstructured. Protein Sci. 2006;15:1042–1050. doi: 10.1110/ps.062115406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliveira TG, Chan RB, Tian H, Laredo M, Shui G, Staniszewski A, Zhang H, Wang L, Kim TW, Duff KE, Wenk MR, Arancio O, Di Paolo G. Phospholipase D2 ablation ameliorates Alzheimer's disease-linked synaptic dysfunction and cognitive deficits. J Neurosci. 2010;30:16419–16428. doi: 10.1523/JNEUROSCI.3317-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ong WY, Yeo JF, Faroqui AA. Distribution of calcium-independent phospholipase A2 (iPLA2) in monkey brain. J Neurocytol. 2005;34:447–458. doi: 10.1007/s11068-006-8730-4. [DOI] [PubMed] [Google Scholar]
- Oude Weernink PA, Lopez de Jesus M, Schmidt M. Phospholipase D signalling: orchestration by PIP2 and small GTPases. Naunyn Schmiedebergs Arch Pharmacol. 2007;374:399–411. doi: 10.1007/s00210-007-0131-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reynwar BJ, Illya G, Harmandaris VA, Müller MM, Kremer K, Deserno M. Aggregation and vesiculation of membrane proteins by curvature-mediated interactions. Nature. 2007;447:461–464. doi: 10.1038/nature05840. [DOI] [PubMed] [Google Scholar]
- Rigoni M, Caccin P, Gschmeissner S, Koster G, Postle AD, Rossetto O, Schiavo G, Montecucco C. Equivalent effects of snake PLA2 neurotoxins and lysophospholipid-fatty acid mixtures. Science. 2005;310:1678–1680. doi: 10.1126/science.1120640. [DOI] [PubMed] [Google Scholar]
- Rossetto O, Montecucco C. Presynaptic neurotoxins with enzymatic activities. Handb Exp Pharmacol. 2008;184:129–270. doi: 10.1007/978-3-540-74805-2_6. [DOI] [PubMed] [Google Scholar]
- Schmidt JA, Brown WJ. Lysophosphatidic acid acyltransferase 3 regulates Golgi complex structure and function. J Cell Biol. 2009;186:211–218. doi: 10.1083/jcb.200904147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitz F. The making of synaptic ribbons. Neuroscientist. 2009;15:611–624. doi: 10.1177/1073858409340253. [DOI] [PubMed] [Google Scholar]
- Schmitz F, Bechmann M, Drenckhahn D. Purification of synaptic ribbons, structural components of the active zone complex in photoreceptor synapses. J Neurosci. 1996;16:7109–7116. doi: 10.1523/JNEUROSCI.16-22-07109.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitz F, Königstorfer A, Südhof TC. RIBEYE, a component of ribbon synapses: a protein's journey through evolution provides insight into synaptic ribbon function. Neuron. 2000;28:857–872. doi: 10.1016/s0896-6273(00)00159-8. [DOI] [PubMed] [Google Scholar]
- Sheets L, Trapani JG, Mo W, Obholzer N, Nicolson T. RIBEYE is required for presynaptic CaV1.3 channel localization and afferent innervation of sensory hair cells. Development. 2011;138:1309–1319. doi: 10.1242/dev.059451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shindou H, Shimizu T. Acyl-CoA: Lysophospholipid acyltransferases. J Biol Chem. 2009;284:1–5. doi: 10.1074/jbc.R800046200. [DOI] [PubMed] [Google Scholar]
- Shindou H, Hishikawa D, Harayama T, Yuki K, Shimizu T. Recent progress on acyl CoA: lysophospholipid acyltransferase research. J Lipid Res. 2009;50:S46–S51. doi: 10.1194/jlr.R800035-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stace CL, Ktistakis NT. Phosphatidic acid- and phosphatidylserine-binding proteins. Biochim Biophys Acta. 2006;1761:913–926. doi: 10.1016/j.bbalip.2006.03.006. [DOI] [PubMed] [Google Scholar]
- tom Dieck S, Altrock WD, Kessels MM, Qualmann B, Regus H, Brauner D, Fejtova A, Bracko O, Gundelfinger ED, Brandstätter JH. Molecular dissection of the photoreceptor ribbon synapse: physical interaction of Bassoon and RIBEYE is essential for the assembly of the ribbon complex. J Cell Biol. 2005;168:825–836. doi: 10.1083/jcb.200408157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venkatesan JK, Natarajan S, Schwarz K, Mayer SI, Alpadi K, Magupalli VG, Sung CH, Schmitz F. Nicotinamide adenine dinucleotide-dependent binding of the neuronal Ca2+-sensor protein GCAP2 to photoreceptor synaptic ribbons. J Neurosci. 2010;30:6559–6576. doi: 10.1523/JNEUROSCI.3701-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vitale N. Synthesis of fusogenic lipids through activation of phospholipase D1 by GTPases and the kinase RSK2 is required for calcium-regulated exocytosis in neuroendocrine cells. Biochem Soc Trans. 2010;38:167–171. doi: 10.1042/BST0380167. [DOI] [PubMed] [Google Scholar]
- Vitale N, Thiersé D, Bader MF. Melittin promotes exocytosis in neuroendocrine cells through activation of phospholipase A2. Regul Pept. 2011;165:111–116. doi: 10.1016/j.regpep.2009.09.008. [DOI] [PubMed] [Google Scholar]
- Wan QF, Heidelberger R. Synaptic release at mammalian bipolar cell terminals. Vis Neurosci. 2011;28:109–119. doi: 10.1017/S0952523810000453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wan QF, Vila A, Zhou ZY, Heidelberger R. Synaptic vesicle dynamics in mouse rod bipolar cells. Vis Neurosci. 2008;25:523–533. doi: 10.1017/S0952523808080711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Devaiah SP, Zhang W, Welti R. Signaling functions of phosphatidic acid. Prog Lipid Res. 2006;45:250–278. doi: 10.1016/j.plipres.2006.01.005. [DOI] [PubMed] [Google Scholar]
- Weigert R, Silletta MG, Spano S, Turacchio G, Cericola C, Colanzi A, Senatore S, Mancini R, Polishchuk EV, Salmona M, Facchiano F, Burger KV, Mironov A, Luini A, Corda D. CtBP/BARS induces fission of Golgi membranes by acetylating lysophosphatidic acid. Nature. 1999;402:429–433. doi: 10.1038/46587. [DOI] [PubMed] [Google Scholar]
- Yang J, Seo J, Nair R, Han SS, Jang S, Kim K, Han K, Paik SK, Choi J, Lee S, Bae YC, Topham MK, Prescott SM, Rhee JS, Choi SY, Kim E. DGKl regulates presynaptic release during mGluR-dependent LTD. EMBO J. 2011;30:165–180. doi: 10.1038/emboj.2010.286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang JS, Lee SY, Spanò S, Gad H, Zhang L, Nie Z, Bonazzi M, Corda D, Luini A, Hsu VW. A role for BARS at the fission step of COPI vesicle formation from Golgi membrane. EMBO J. 2005;24:4133–4143. doi: 10.1038/sj.emboj.7600873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang JS, Gad H, Lee SY, Mironov A, Zhang L, Beznoussenko GV, Valente C, Turacchio G, Bonsra AN, Du G, Baldanzi G, Graziani A, Bourgoin S, Frohman MA, Luini A, Hsu VW. A role for phosphatidic acid in COPI vesicle fission yields insights into Golgi maintenance. Nat Cell Biol. 2008;10:1146–1153. doi: 10.1038/ncb1774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeniou-Meyer M, Zabari N, Ashery U, Chasserot-Golaz S, Haeberlé AM, Demais V, Bailly Y, Gottfried I, Nakanishi H, Neiman AM, Du G, Frohman MA, Bader MF, Vitale N. Phospholipase D1 production of phosphatidic acid at the plasma membrane promotes exocytosis of large dense-core granules at a late stage. J Biol Chem. 2007;282:21746–21757. doi: 10.1074/jbc.M702968200. [DOI] [PubMed] [Google Scholar]
- Zhou ZY, Wan QF, Thakur P, Heidelberger R. Capacitance measurements in the mouse rod bipolar cell identify a pool of releasable synaptic vesicles. J Neurophysiol. 2006;96:2539–2548. doi: 10.1152/jn.00688.2006. [DOI] [PubMed] [Google Scholar]
- Zimmerberg J, Kozlov MM. How proteins produce cellular membrane curvature. Nat Rev Mol Cell Biol. 2006;7:9–19. doi: 10.1038/nrm1784. [DOI] [PubMed] [Google Scholar]