

Abstract
Dentate granule cells, at the gate of the hippocampus, use coincidence detection of synaptic inputs to code afferent information under a sparse firing regime. In both human patients and animal models of temporal lobe epilepsy, mossy fibers sprout to form an aberrant glutamatergic network between dentate granule cells. These new synapses operate via long-lasting kainate receptor-mediated events, which are not present in the naive condition. Here, we report that in chronic epileptic rat, aberrant kainate receptors in interplay with the persistent sodium current dramatically expand the temporal window for synaptic integration. This introduces a multiplicative gain change in the input–output operation of dentate granule cells. As a result, their sparse firing is switched to an abnormal sustained and rhythmic mode. We conclude that synaptic kainate receptors dramatically alter the fundamental coding properties of dentate granule cells in temporal lobe epilepsy.
Introduction
The dentate gyrus plays a major role at the gate of the hippocampus, filtering incoming information from the entorhinal cortex (Henze et al., 2002; Acsády and Káli, 2007). A fundamental coding property of dentate granule cells (DGCs) is their sparse firing mode (Jung and McNaughton, 1993). Indeed, they behave as a coincidence detector due to the fast kinetics of excitatory synaptic events restricting integration of afferent inputs to a narrow time window (Schmidt-Hieber et al., 2007). In temporal lobe epilepsies (TLEs), the hippocampus displays important coding alterations (Lenck-Santini and Holmes, 2008) that may play a role in cognitive impairments described in patients and animal models (Hermann et al., 1997; Lenck-Santini and Holmes, 2008). However, the cellular mechanisms remain poorly understood. In animal models of TLE and human patients, neuronal tissue undergoes major reorganization; some neurons die whereas others, which are severed in their inputs or outputs, sprout and form novel aberrant connections (Coulter and Carlson, 2007; Ben-Ari et al., 2008). This phenomenon, called reactive plasticity, is well documented in the dentate gyrus where DGC axons [the so-called mossy fibers (MFs)] sprout (Tauck and Nadler, 1985; Represa et al., 1987; Sutula and Dudek, 2007) and create a powerful excitatory network between DGCs (Tauck and Nadler, 1985; Gabriel et al., 2004). We recently showed that in addition to the axonal rewiring, recurrent MFs convert the nature of glutamatergic transmission in the dentate gyrus because they operate via long-lasting kainate receptor (KAR)-mediated EPSPs (EPSPKA) not present in the naive condition (Epsztein et al., 2005). It has been suggested that AMPARs and KARs may encode different features of afferent activity (Frerking and Ohliger-Frerking, 2002; Lerma, 2006); however, the role of EPSPKA in the dynamics of synaptic integration and spike transmission between DGCs in TLE remains to be elucidated. Here, we show that in an animal model of TLE, aberrant kainate receptors in interplay with the persistent sodium current (INaP) dramatically expand the temporal window for synaptic integration in DGCs. This introduces a multiplicative gain change in the input–output operation of DGCs of chronic epileptic animals that switches their firing from a sparse to an abnormal sustained and rhythmic mode.
Materials and Methods
All experiments were approved by the Institut National de la Santé et de la Recherche Médicale Animal Care and Use Committee of May 29, 2001 (2001–464) and the European Community Council Directive of November 24, 1986 (86/609/EEC).
An animal model of temporal lobe epilepsy was obtained using adult male Wistar rats injected with pilocarpine hydrochloride, as previously described (Epsztein et al., 2005, 2010b). Experiments were performed with 34 chronic epileptic rats (5–14 months of age). Age-matched rats (27 naive rats and 3 sham rats) were used as controls (4–14 months of age).
Whole-cell recordings of DGCs from chronic epileptic and control rats were obtained using the “blind” patch-clamp technique and were performed in vitro in hippocampal slices, as previously described (Epsztein et al., 2005, 2010b). Electrodes were filled with an internal solution containing the following (in mm): 130 KMeSO4, 5 KCl, 5 NaCl, 10 HEPES-K, 2.5 MgATP, 0.3 NaGTP, 0.2 EGTA, and 0.5% biocytin, pH 7.25. Whole-cell recordings were performed in current-clamp mode (except when stated otherwise) using a Multiclamp 700B amplifier (Molecular Devices). Data were filtered at 2 kHz, digitized (20 kHz) with a Digidata 1440A (Molecular Devices) to a personal computer, and acquired using Clampex 10.1 software (PClamp, Molecular Devices). Signals were analyzed off-line using Clampfit 10.1 (Molecular Devices).
EPSPs were evoked by bulk stimulations with a bipolar NiCh electrode (NI-0.7F, Phymep) positioned in the inner one-third of the molecular layer of the dentate gyrus to stimulate either associational/commissural inputs in control rats or recurrent mossy fiber inputs in epileptic rats. EPSPs were evoked using stimuli of similar intensity in control (21.3 ± 1.4 μA, n = 56) and epileptic conditions (22.4 ± 1.1 μA, n = 88, p = 0.230) and similar duration (28.1 ± 1.4 μs, n = 56 and 28 ± 1.4 μs, n = 88, respectively; p = 0.890).
Single EPSPs were evoked (0.2 Hz) to enable their characterization by the measurement of their amplitude and half-width. In one set of experiments, EPSPs were evoked using a train of stimuli performed at 30 Hz but with a random restrained jitter of ±5, 10, and 15 ms. For this condition, we used a custom-made program to generate jittered stimulations using MATLAB (Mathworks).
As previously described (Epsztein et al., 2005, 2010b), AMPAR-mediated EPSP (EPSPAMPA) and EPSPKA were pharmacologically isolated in the presence of blockers of NMDA (40 μm d-APV or 10 μm MK801), GABAA (10 μm bicuculline or 5 μm gabazine [SR-95531]), and GABAB (5 μm CGP55845) receptors; in control rats, EPSPAMPA were recorded in the absence of a KAR antagonist. In epileptic rats, EPSPAMPA or EPSPKA were respectively pharmacologically isolated in the presence of antagonists of KA [10 μm (2S,4R)-4-methylglutamic acid (SYM 2081)] (DeVries, 2000) or AMPA [50–100 μm 1-(4-aminophenyl)-4-methyl-7,8-methylenedioxy-5H-2,3-benzodiazepine (GYKI 52466) or 10–30 μm 1-(4-aminophenyl)-3-methylcarbamyl-4-methyl-3,4-dihydro-7,8-methylenedioxy-5H-2,3-benzodiazepine hydrochloride (GYKI 53655)] (Paternain et al., 1995) receptors in addition to NMDA, GABAA, and GABAB receptor antagonists (Epsztein et al., 2005, 2010b). To further confirm the efficiency of our pharmacological conditions, additional experiments were performed in voltage-clamp mode. Control EPSPAMPA, epileptic EPSPAMPA, and EPSPKA were first recorded in current-clamp mode to adjust their amplitude (3.97 ± 0.2 mV [n = 5], 4.05 ± 0.16 mV [n = 7] and 4.02 ± 0.35 mV [n = 5], respectively; p = 0.973) (Fig. 1A). Then, recordings were switched to voltage-clamp mode [holding potential (Vh) = −70 mV] to prevent further activation of intrinsic conductances. In these conditions, EPSCs had a mean amplitude of 37.88 ± 4.11 pA (n = 5), 34.98 ± 2.9 pA (n = 7), and 32.48 ± 1.85 pA (n = 5), respectively, for control EPSCAMPA, epileptic EPSCAMPA, and epileptic EPSCKA (p = 0.605). In control cells, we confirmed that EPSCs were mediated by AMPARs, as indicated by their sensitivity to 10 μm GYKI 53655 (mean amplitude reduced by 91.85 ± 0.77%, n = 5, p = 0.001). Conversely, control EPSCs were not changed in the presence of 10 μm SYM 2081 (mean amplitude changed by −0.22 ± 1.32%, n = 5, p = 0.652) (Fig. 1A). In epileptic cells, EPSC (pharmacologically isolated in the presence of 10 μm SYM 2081) were mediated by AMPARs, as indicated by their sensitivity to 10 μm GYKI 53655 (mean amplitude reduced by 92.21 ± 0.75%, n = 7, p < 0.001) or 50 μm GYKI 52466 (mean amplitude reduced by 93.88 ± 0.48%, n = 5, p < 0.001). Conversely, epileptic EPSCs (pharmacologically isolated in the presence of 10 μm GYKI 53655) were mediated by KARs, as indicated by their sensitivity to 10 μm SYM 2081 (mean amplitude reduced by 94.51 ± 0.82%, n = 5, p < 0.001) (Fig. 1A).
Figure 1.

EPSPKA but not EPSPAMPA generate a sustained discharge in epileptic dentate granule cells. A, Average EPSPs (top) and EPSCs (bottom) recorded in control and epileptic dentate granule cells. Recordings were performed in the presence of gabazine (5 μm), CGP (5 μm), and d-APV (40 μm). Control EPSC before (black trace), after 10 μm SYM 2081 (blue trace), and after 10 μm SYM 2081 + 10 μm GYKI 53655 (purple trace). Epileptic EPSC with 10 μm SYM 2081 (green trace), and with 10 μm SYM 2081 + 10 μm GYKI 53655 (purple trace); epileptic EPSC with 10 μm GYKI 53655 (red trace), and with 10 μm GYKI 53655 + 10 μm SYM 2081 (purple trace). B, Spike discharge for control EPSPAMPA and epileptic EPSPKA at 10 or 30 Hz in DGCs. C, Input/output relationship (α = slope) for EPSPAMPA and EPSPKA (correlation coefficient r = 0.99) showing a multiplicative gain change of input–output operation for EPSPKA. D, Summation of control EPSPAMPA and EPSPKA evoked at 10 and 30 Hz. Note the EPSPKA-mediated depolarizing envelope in orange. E, Summation profile of EPSPAMPA and EPSPKA in control and epileptic conditions, evoked at 10 or 30 Hz. F, Half-width of EPSP and charge transfer of depolarizing envelope for control (c-EPSPAMPA), and epileptic EPSPs (e-EPSPAMPA and e-EPSPKA), and their correlation (r = 0.74). G, Short-term depression of EPSPs at 10 Hz (left) or 30 Hz (right). In this and following figures, spikes are truncated and electrical stimulations are represented by small dashes below the traces; *p < 0.05, **p < 0.01, ***p < 0.001.
Short-term plasticity of EPSPs was assessed by the measurement of successive EPSP amplitude throughout the train of stimulation and normalized to the amplitude of the first EPSP. Firing regularity was assessed using different analysis, as previously reported (Vervaeke et al., 2006). These included autocorrelation plots of digitally filtered raw data (low pass, 10 Hz), measurement of the interspike intervals (ISIs) and calculation of the coefficient of variation (CV = SD/mean) of the ISIs.
In one set of experiments studying the slow afterhyperpolarization current (IsAHP), recordings were performed in voltage-clamp mode (Vh = −70 mV). IsAHP was induced by a depolarizing step of current (100 ms duration, 50 pA amplitude) and was recorded in the presence of the K+ channel blocker tetraethylammonium chloride (TEA; 5 mm) in addition to AMPA, NMDA, GABAA, and GABAB receptor antagonists. IsAHP was recorded repeatedly, both before and 2 s after 30 Hz trains of EPSCKA (Ruiz et al., 2005). The effect of EPSCKA on IsAHP was tested six times per cell.
All values are given as means ± SEM. Statistical analyses were performed using SYSTAT 11 (Systat Software). For comparison between groups with normal distribution, the two-sample unpaired Student's t test was used for two groups, the one-way ANOVA with the Tukey's HSD post hoc test was used for more than two groups, and ANOVA with repeated-measures test was used for groups with dependent variables. When data were not normally distributed, the Mann–Whitney rank-sum test, the two-sample Kolmogorov–Smirnov rank-sum test, and the Friedman paired nonparametric test were used, respectively. Cumulative probability distributions were compared with the Pearson's χ2 test. The level of significance was set at p < 0.05. n refers to the number of cells.
Drugs were purchased from Sigma (TTX, pilocarpine hydrochloride, and scopolamine methyl nitrate), Tocris Bioscience [GYKI 52466, GYKI 53655, SYM 2081, bicuculline, gabazine (SR-95531), d-APV, MK 801, CGP 55845, and TEA], and Roche (diazepam).
Results
Kainate receptors increase the gain of the input–output relationship of epileptic dentate granule cells in a multiplicative manner
In epileptic DGCs, at the recurrent MF synapses, slow KAR-mediated events provide a significant part of glutamatergic synaptic transmission in addition to AMPARs (Epsztein et al., 2005, 2010b). Here, we compared the rate coded input–output relationship driven by pharmacologically isolated EPSPAMPA or EPSPKA (see Materials and Methods) (Fig. 1A) in response to frequencies of stimulation ranging from 5 to 80 Hz in control and epileptic DGCs (Fig. 1B,C). In this and the following experiments, EPSPs were evoked by electrical stimulations in the inner one-third of the molecular layer of the dentate gyrus (see Materials and Methods) at a potential close to the threshold for generating action potentials (subthreshold potential: ∼−50 mV). To get closer to the observations obtained in vivo (Epsztein et al., 2010a), small EPSPs were evoked and their amplitude rigorously controlled so that there were no significant differences of subthreshold EPSP size between control and epileptic rats (average amplitude: 3.99 ± 0.15 mV, n = 56, for control EPSPAMPA; 4 ± 0.23 mV, n = 31, for epileptic EPSPAMPA; and 3.97 ± 0.2 mV, n = 57, for epileptic EPSPKA, p = 0.114). In this condition, we observed that trains of EPSPAMPA led to a sparse firing regime (up to 5 Hz) for all frequencies tested, both in control (n = 7) and epileptic conditions (n = 10) (Fig. 1B,C). In marked contrast, EPSPKA triggered a sustained rate of discharge in epileptic DGCs (up to 13 Hz, n = 8, p < 0.001) that resulted from a change in the gain of the input–output relationship in a multiplicative manner (Silver, 2010) compared with control (3.4-fold, p < 0.001) (Fig. 1B,C). This did not result from a difference in spike threshold (−41.8 ± 1.4 mV, − 41.8 ± 0.9 mV, and −43.1 ± 2.9 mV, p = 0.684), or holding potential (−50.6 ± 1.4 mV, − 51.6 ± 1.2 mV, and −50.7 ± 2.4 mV, p = 0.910) for control EPSPAMPA (n = 14), epileptic EPSPAMPA (n = 11), and epileptic EPSPKA (n = 12).
Synaptic integration is an important determinant in the modulation of discharge regime (Magee and Johnston, 2005). We thus compared the temporal profile of summation of EPSPAMPA and EPSPKA at 10 Hz (theta) and 30 Hz (low gamma) frequencies in control and epileptic DGCs. We observed that EPSPKA (n = 16) but not EPSPAMPA (n = 32 and n = 9 from control and epileptic rats, respectively) generated a strong and sustained depolarizing envelope for both frequencies (p < 0.001) (Fig. 1D,E). This difference could not be explained by changes in short-term synaptic plasticity since EPSPAMPA and EPSPKA in control and epileptic DGCs displayed similar short-term depression (10 Hz, p = 0.152; 30 Hz, p = 0.302) (Fig. 1D,G). Alternatively, one can speculate that the high temporal integration feature of EPSPKA, compared with EPSPAMPA, results from its kinetic properties, as proposed by computational studies (Frerking and Ohliger-Frerking, 2002). Assessment of the transfer of EPSP charge, which is tightly correlated with the half-width of the synaptic event (n = 55, p < 0.001), was found to be approximately sixfold higher for EPSPKA than for EPSPAMPA (epileptic EPSPKA vs control or epileptic EPSPAMPA, p < 0.001; n = 14, n = 32, and n = 9, respectively) (Fig. 1F). Therefore, the simplest interpretation of these data is that the slower kinetics of EPSPKA allows a high temporal summation, which in turn offsets the short-term depression, contrary to fast EPSPAMPA.
Kainate but not AMPA receptors drive a rhythmic firing pattern in epileptic DGCs
Neurons translate input information via rate and temporal coding. In this set of experiments, we tested the firing regularity of DGCs driven by EPSPKA or EPSPAMPA trains. Firing regularity was assessed using different analysis (see Materials and Methods). We observed that EPSPKA, but not EPSPAMPA, triggered a robust rhythmic pattern of discharge in DGCs of epileptic rats (Fig. 2A–D). Indeed, at 30 Hz, the autocorrelation plot showed regular peaks for EPSPKA, but not for EPSPAMPA, indicating a strong spiking periodicity (Fig. 2A). In keeping with this, the plot of the distribution of the ISIs revealed a clear peak for EPSPKA (coefficient of determination R2 = 0.96, median = 150.1 ms, n = 12), but not for EPSPAMPA (n = 14 in control, and n = 12 in epileptic) (Fig. 2B); as well as a higher slope for the ISI cumulative probability curves of EPSPKA compared with EPSPAMPA (p < 0.001) (Fig. 2C). Accordingly, the CV of the ISIs was significantly lower for EPSPKA (0.38 ± 0.02, n = 12) compared with EPSPAMPA (0.57 ± 0.05, n = 6 in control, p = 0.021; 0.55 ± 0.08, n = 7 in epileptic, p = 0.036) (Fig. 2D). To test whether the rhythmic firing triggered by EPSPKA could be related to the metronomic features of the stimuli, we applied stimuli using three distinct restrained random jitters around a fixed interval (33.33 ± 5, ±10, or ±15 ms; n = 6, n = 7, and n = 7, respectively; see Materials and Methods). The use of random stimuli did not change the periodicity of firing triggered by EPSPKA as shown by both the autocorrelation plot (Fig. 2E) and the superimposition of the ISI cumulative probability curves (Δt5, Δt10, Δt15 vs Δt0; p = 0.999, p = 0.935, and p = 0.981, respectively) (Fig. 2F). Therefore, the rhythmic firing depends on the temporal features of integration of EPSPKA. Furthermore, the examination of firing pattern for different frequencies of stimulation (5–80 Hz) demonstrated that EPSPKA (n = 8), but not EPSPAMPA (n = 16), triggered a robust rhythmic activity within a wide range of frequencies (Fig. 2G). Calculation of the EPSPKA-ISI median for each frequency revealed that this value decreased following a single exponential decay fit (R2 = 0.98) versus the frequency of stimulation (Fig. 2G). This further indicates that the ISI median depends primarily on integration features of EPSPKA rather than the intervals of stimuli. All previous experiments were performed at a very restrained holding potential (−50.9 ± 1 mV, n = 36). Then, we verified that the rhythmic firing pattern could be generated by EPSPKA when the experiments were performed at a holding potential slightly more depolarized. To this end, recordings were performed at −45 ± 0.6 mV for control EPSPAMPA (n = 6), − 45 ± 0.3 mV for epileptic EPSPAMPA (n = 7), and −45 ± 0.3 mV for epileptic EPSPKA (n = 8, p = 0.998). Under these conditions, we observed that 30 Hz trains of EPSPKA triggered a sustained rate of discharge in epileptic DGCs (firing rate: 6.44 ± 0.3 Hz, n = 8) in contrast to 30 Hz trains of EPSPAMPA that led to a sparse firing regime (firing rate: 2.17 ± 0.2 and 2.47 ± 0.26 Hz, respectively, in 6 control DGCs and in 7 epileptic DGCs; p < 0.001 when compared with EPSPKA). Moreover, 30 Hz trains of EPSPKA evoked a robust rhythmic firing pattern in epileptic DGCs (ISI median: 152.6 ± 7.3 ms; CV: 0.35 ± 0.04, n = 8) contrary to 30 Hz trains of EPSPAMPA (CV: 0.62 ± 0.06 and 0.73 ± 0.04, respectively, in 6 control DGCs and in 7 epileptic DGCs; p = 0.001 when compared with EPSPKA). Therefore, a small change in the holding potential did not change the overall profile of the firing pattern generated by EPSPAMPA and EPSPKA in control and epileptic DGCs.
Figure 2.
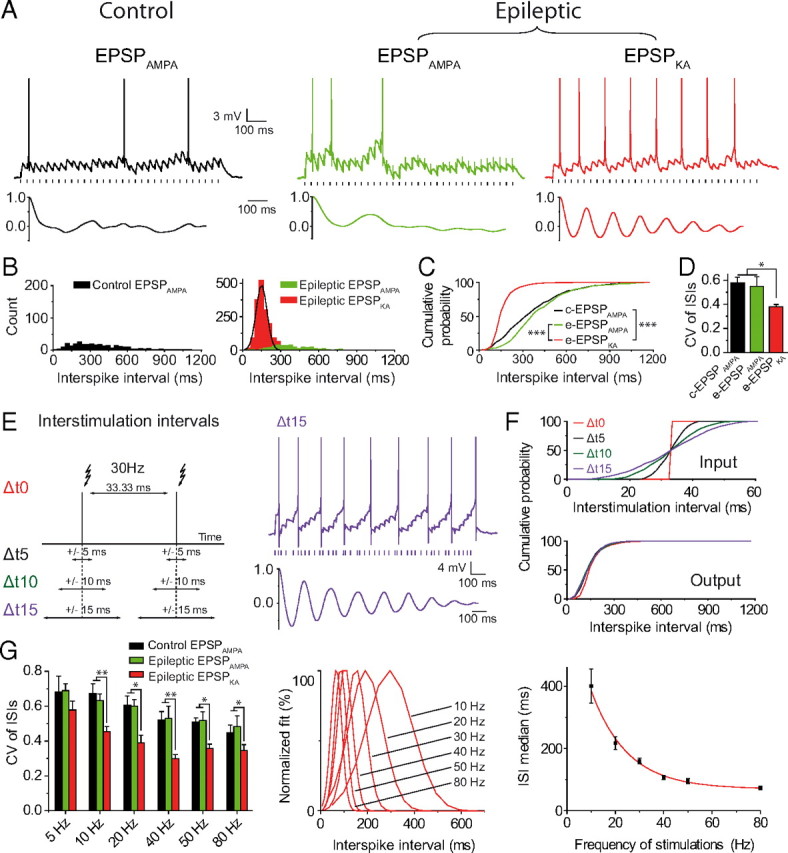
EPSPKA but not EPSPAMPA trigger a rhythmic firing pattern in epileptic dentate granule cells. A, Top, spike discharge for control EPSPAMPA, epileptic EPSPAMPA, and epileptic EPSPKA at 30 Hz in DGCs; bottom, autocorrelation plots. B, ISI distributions for control EPSPAMPA (291 ISIs), epileptic EPSPAMPA (459 ISIs), and EPSPKA (1776 ISIs) in DGCs. C, Same data as in B plotted as cumulative probabilities. D, Bar graph of CV of ISIs for EPSPAMPA and EPSPKA. E, Left, Scheme of the protocol of stimulation with restrained random jitter (Δt). Right, Spike discharge (top) in response to Δt ± 15 ms; corresponding autocorrelation plot (bottom). F, Cumulative probability plots for interstimulus intervals (top) and ISIs (bottom). G, CV of ISIs for control EPSPAMPA, epileptic EPSPAMPA, and epileptic EPSPKA at different frequencies of stimulation (left); normalized Gaussian fits of ISI distributions for EPSPKA (middle; R2 > 0.97); fit of EPSPKA-ISI median versus frequency of stimulations (right, R2 = 0.98).
The above experiments were all performed with pharmacologically isolated EPSPAMPA and EPSPKA (see Materials and Methods) (Fig. 1A). We checked that the sustained and rhythmic firing pattern observed in epileptic DGCs was not dependent on this specific pharmacological condition (Fig. 3A,B). To this end, epileptic EPSPAMPA and EPSPKA were isolated based on their kinetic features according to our previous report (in the presence of NMDA, GABAA, and GABAB receptor antagonists) (Epsztein et al., 2010b); fast EPSPs with half-width < 50 ms (37.47 ± 4.18 ms, n = 13) were categorized as EPSPAMPA, and slow EPSPs with half-width > 50 ms (66.2 ± 5.37, n = 9, p < 0.001) were categorized as EPSPKA. As expected, we observed that slow EPSPs (n = 10) generated a strong and sustained depolarizing envelope, contrary to fast EPSPs (n = 10, p < 0.001) (Fig. 3A). Moreover slow EPSPs, but not fast EPSPs, generated a high firing rate (4.8 ± 0.4 Hz for slow EPSPs, n = 6; 1.3 ± 0.2 Hz for fast EPSPs; n = 11, p < 0.001) and a rhythmic pattern of discharge in DGCs. The regularity of firing triggered by slow EPSPs was revealed by the autocorrelation plot (data not shown), a peak in the ISI distribution, a high slope in the ISI cumulative probability curves (p < 0.001) (Fig. 3B), and a low CV of ISI (0.36 ± 0.01, n = 6 for slow EPSPs; 0.66 ± 0.04, n = 7 for fast EPSPs; p < 0.001).
Figure 3.

EPSP summation and firing pattern without AMPAR, KAR, or NMDAR blockers. A, Summation (left) and summation profile (right) of slow and fast EPSPs in the absence of AMPAR or KAR antagonist at 30 Hz; insets represent single EPSP (calibration: 1 mV, 50 ms). B, ISI distribution plotted as histograms (left) and as cumulative probabilities (right), for fast EPSPs (248 ISIs) and for slow EPSPs (1011 ISIs). C, Spike discharge (top) at 30 Hz and autocorrelation plots (bottom) for EPSPs recorded without d-APV in control, and with 10 μm SYM 2081 or with 10 μm GYKI 53655 in epileptic DGCs. D, ISI distributions for no d-APV (285 ISIs), SYM 2081/no d-APV (516 ISIs), and GYKI 53655/no d-APV conditions (5903 ISIs), and CV of ISIs. E, Firing rate for the three conditions.
It is known that NMDARs driving long-lasting EPSPs can lead to burst generation in epileptic neurons (Dingledine et al., 1986; Lynch et al., 2000). Since the majority of our experiments have been performed in the presence of NMDAR antagonists (see Materials and Methods), we thus wondered whether AMPARs could promote the observed increase of excitability and the rhythmic firing pattern in DGCs when NMDARs remain functional. This hypothesis was disproved by the fact that we observed a similar low firing rate driven by EPSPAMPA at 30 Hz in the presence (1.7 ± 0.2 Hz, n = 14 in control; 1.5 ± 0.2 Hz, n = 12 in epileptic) or in the absence of blockers of NMDARs (1.5 ± 0.3 Hz, n = 9 in control, p = 0.512; 1.8 ± 0.2 Hz, n = 5 in epileptic, p = 0.411), and no rhythmic discharge in both cases (Fig. 3C–E). This confirms the critical role of KARs in this aberrant phenotype. Interestingly, we observed that KAR-mediated hyperexcitability and the rhythmic pattern of discharge were exacerbated in the presence of functional NMDARs. Indeed, we observed a stronger mean firing rate (9.1 ± 0.8 Hz, n = 8, p < 0.001) and a shorter ISI mean value (102.2 ± 6.8 ms, n = 7, p < 0.001) in the absence of NMDA receptor antagonists. Therefore, the aberrant phenotype, primarily driven by KARs, is aggravated when NMDARs is activated.
The sodium persistent current is required to trigger the kainate receptor-driven rhythmic firing pattern
Neuronal excitability is modulated by various intrinsic conductances. We have previously shown that the INaP strongly amplifies EPSPKA at a subthreshold potential in epileptic DGCs (Epsztein et al., 2010b). To test the putative role of INaP in the temporal profile of summation of synaptic inputs and the firing pattern driven by EPSPKA, a low dose of tetrodotoxin (TTX; 10–20 nm) was applied to inhibit INaP without blocking action potentials (Del Negro et al., 2005; Epsztein et al., 2010b). We observed that, at subthreshold potential, blockade of INaP strongly reduced the depolarizing envelope for EPSPKA at 10 and 30 Hz (Fig. 4A,B). Thus, bath application of a low dose of TTX reduced the charge transfer through EPSPs by 31.1 ± 6.2% (n = 6, p = 0.006) and by 29.6 ± 3.7% (n = 8, p < 0.001) for EPSPKA at 10 Hz and 30 Hz, respectively (Fig. 4A,B). Interestingly, INaP also contributed to the transient depolarization generated by EPSPAMPA for 30 Hz trains but not for 10 Hz trains (Fig. 4A,B); in the presence of TTX, the charge transfer through summed EPSPAMPA was reduced by 17.6 ± 3.8% (n = 9, p < 0.001) and by 1.8 ± 3.2% (n = 7, p = 0.724), respectively. Many studies have shown that INaP could also play a key role in the regulation of excitability and rhythmic activity (Stafstrom et al., 1985; Vervaeke et al., 2006). We observed that blockade of INaP fully prevented rhythmic spike discharge (30 Hz trains: CV = 0.55 ± 0.03, n = 11, p < 0.001; 10 Hz trains: CV = 0.6 ± 0.02, n = 7, p = 0.001) and strongly reduced the firing rate driven by EPSPKA (30 Hz trains: 1.9 ± 0.1 Hz, n = 11, p < 0.001; 10 Hz trains: 0.7 ± 0.1 Hz, n = 8, p < 0.001) (Fig. 4C–E). Therefore, INaP plays a central role in the generation of the kainate receptor-driven rhythmic firing pattern.
Figure 4.

Role of INaP in summation profiles and firing pattern for EPSPKA and EPSPAMPA in control and epileptic dentate granule cells. A, Summation (left) and summation profile (right) of control EPSPAMPA and epileptic EPSPKA at −80, − 50, or −50 mV in the presence of 10 nm TTX. Note the important voltage-dependent amplification of epileptic EPSPKA at 30 Hz. B, Charge transfer reduction after application of 10–20 nm TTX for control EPSPAMPA and epileptic EPSPKA, at 10 or 30 Hz, recorded at −50 mV. C, Spike discharge for EPSPKA at 30 Hz in the presence of 10 nm TTX (top); autocorrelation plot (bottom); ISI distributions plotted as histograms and as cumulative probabilities for EPSPKA in the absence (1776 ISIs) or in the presence (834 ISI) of 10–20 nm TTX. D, Spike discharge for EPSPKA at 10 Hz in the presence of 10 nm TTX (top); autocorrelation plot (bottom). E, Firing rate and CV of ISIs for EPSPKA in the absence or in the presence of 10–20 nm TTX at 10 or 30 Hz.
In addition to INaP, KARs can also modulate the IsAHP (Fisahn et al., 2005; Ruiz et al., 2005). We tested the putative effect of EPSCKA on IsAHP (see Materials and Methods). In our experimental conditions, a 30 Hz train of EPSCKA did not significantly modify the amplitude of IsAHP (the amplitude was changed by −5.6 ± 4.7%, n = 12, p = 0.146; data not shown), suggesting that the change of firing pattern in epileptic DGCs does not result from a direct action of KARs on IsAHP. However, we did not exclude that EPSCKA could alter IsAHP using different experimental protocols. Additional experiments should be performed to clarify this point.
In conclusion, our data show that EPSPKA, in interplay with INaP, generate a strong depolarizing envelope that conveys a sustained and rhythmic spike discharge.
Discussion
In control CA3 pyramidal cells, at the MF synapses, many studies have shown that KARs control the efficacy of spike transmission by a conjunction of presynaptic and postsynaptic actions (Contractor et al., 2001; Nicoll and Schmitz, 2005; Sachidhanandam et al., 2009). The KAR-mediated postsynaptic events display a slow kinetics compared with AMPAR-mediated ones (Castillo et al., 1997; Bureau et al., 2000; Epsztein et al., 2005). In epileptic DGCs, at the recurrent MF synapses, postsynaptic KARs provide a substantial component of glutamatergic synaptic transmission in addition to AMPARs (Epsztein et al., 2005, 2010b). In the present study, we show that abnormal postsynaptic KARs yield a multiplicative gain change in the input–output operation of epileptic DGCs. This results from the slow kinetics of EPSPKA allowing a strong integration of synaptic inputs and a long-lasting depolarizing envelope. The change of the input–output relationship leads to an alteration of the coincidence detection operation and switches the firing pattern of DGCs from a sparse to a sustained regime. Therefore, because of postsynaptic KARs, the recurrent MF synapses in epileptic DGCs appear to display some detonator features, as reported for MF synapses in control CA3 pyramidal cells (Sachidhanandam et al., 2009). Neuronal computation involves different intrinsic and synaptic conductances (Fricker and Miles, 2000; Vervaeke et al., 2006; Silver, 2010). For instance, under control conditions, slow NMDAR-mediated synaptic events can modulate the temporal precision of EPSP spike coupling (Cathala et al., 2003). Additionally, NMDAR activation can lead to burst generation in epileptic neurons (Dingledine et al., 1986; Lynch et al., 2000). In our conditions of small EPSPs, we found that NMDARs do not change the sparse firing mode driven by AMPARs in control and epileptic conditions, confirming the critical role of KARs in the aberrant firing phenotype observed in epileptic DGCs. However, NMDARs aggravate the sustained and rhythmic firing pattern driven by KARs.
Voltage-gated ion channels play an important role in shaping the integration of incoming synaptic inputs, and plasticity of intrinsic neuronal properties have been reported in various CNS disorders (Beck and Yaari, 2008). An enhanced expression of the hyperpolarization-activated channel (Ih) has been reported in DGCs in animal models of TLE and human patients (Bender et al., 2003). Because Ih tends to decrease summation of synaptic inputs (Magee and Johnston, 2005), it is unlikely that a change in this current will favor a higher synaptic integration, as observed in epileptic DGCs. Alteration of low-threshold T-type Ca2+ conductance has been also reported in DGCs from chronic epileptic rats and human patients (Beck and Yaari, 2008); however, this current is mostly inactivated at subthreshold membrane potential.
Another major candidate involved in the temporal summation of EPSPs is the INaP, since this current is activated at subthreshold potentials and prolongs the time course of EPSPs in pyramidal cells (Stafstrom et al., 1985; Fricker and Miles, 2000). Furthermore, we recently demonstrated that, in DGCs of epileptic rats, EPSPKA, but not EPSPAMPA, is specifically amplified at subthreshold potentials by INaP, leading to long-lasting plateau potentials (Epsztein et al., 2010b). We now show that in DGCs of epileptic rats, amplification of EPSPKA by INaP drastically expands the temporal window for synaptic integration, leading to a strong depolarizing envelope. Interestingly, a modest amplification by INaP was also observed for EPSPAMPA when evoked at a gamma frequency range both in DGCs of control and epileptic rats. This is likely due to the fact that, at these frequencies, summation of EPSPAMPA generates a small transient depolarizing envelope sufficient to activate INaP. This is in keeping with our previous observations showing that activation of INaP critically depends on the kinetics of depolarizing events in DGCs (Epsztein et al., 2010b). We also show that the interplay between EPSPKA and INaP is essential to trigger both a sustained and rhythmic spike discharge in DGCs of epileptic rats. Consistently, a role of INaP in rhythmic and tonic firing activities had been reported in various cell types of the CNS such as cortical neurons (Stafstrom et al., 1985; Alonso and Llinás, 1989). In DGCs of epileptic rats, no change in INaP was observed (Epsztein et al., 2010b), contrary to cortical neurons from animal models and patients with TLE (Agrawal et al., 2003; Vreugdenhil et al., 2004). Therefore, in DGCs of epileptic rats hyperexcitability is primarily determined by aberrant KAR-operated synapses, contrary to other cell types for which intrinsic neuronal bursting activities are promoted by a change of INaP (Chen et al., 2011). We have previously shown that KARs, in interplay with INaP, impose an abnormal temporal dispersion of spike discharge for low synaptic input rate in epileptic DGCs (Epsztein et al., 2010b). In striking contrast, we now show that when the input dynamic is >10 Hz, synaptic KARs, together with INaP, tune DGCs to fire with a robust rhythmic pattern of discharge. Therefore, following the modality of synaptic inputs, the firing regularity will be modulated in the opposite way by INaP in epileptic conditions. This opposite effect of INaP on the temporal precision of EPSP spike coupling is reminiscent of mechanisms described for the modulation of intrinsic firing properties in pyramidal neurons (Vervaeke et al., 2006). Many other channels, including potassium channels, can also participate in modulation of the interspike interval (Lawrence et al., 2006; Vervaeke et al., 2006). Additional experiments should be performed to further shed light on this issue in epileptic DGCs.
Dentate gyrus plays a major role as a gate at the entrance to the hippocampus (Henze et al., 2002). DGCs use a sparse and specific firing for efficient discrimination of incoming information (Leutgeb et al., 2007). This results in part from their coincidence detector function due to the fast kinetics of synaptic inputs (Schmidt-Hieber et al., 2007), and from a dynamic recruitment of perisomatic-inhibitory interneurons (Sambandan et al., 2010). In TLE, we show that the interplay between aberrant KARs and INaP, by expanding the temporal window for synaptic integration, promotes a switch from a sparse to a sustained and rhythmic firing pattern in DGCs. We propose that this aberrant phenotype may dramatically alter the fundamental operation of DGCs in TLE.
Footnotes
This work was supported by Institut National de la Santé et de la Recherche Médicale and the Agence Nationale de la Recherche (Epileptic-Code, ANR-09-BLAN-0259–01), the Ministère de l'Enseignement Supérieur et de la Recherche (to J.A. and A.P.). We thank Drs I. Bureau, P. Chavis, L. Christie, O. Manzoni, F. Molinari, and A. Represa for critical reading of the manuscript.
References
- Acsády L, Káli S. Models, structure, function: the transformation of cortical signals in the dentate gyrus. Prog Brain Res. 2007;163:577–599. doi: 10.1016/S0079-6123(07)63031-3. [DOI] [PubMed] [Google Scholar]
- Agrawal N, Alonso A, Ragsdale DS. Increased persistent sodium currents in rat entorhinal cortex layer V neurons in a post-status epilepticus model of temporal lobe epilepsy. Epilepsia. 2003;44:1601–1604. doi: 10.1111/j.0013-9580.2003.23103.x. [DOI] [PubMed] [Google Scholar]
- Alonso A, Llinás RR. Subthreshold Na+-dependent theta-like rhythmicity in stellate cells of entorhinal cortex layer II. Nature. 1989;342:175–177. doi: 10.1038/342175a0. [DOI] [PubMed] [Google Scholar]
- Beck H, Yaari Y. Plasticity of intrinsic neuronal properties in CNS disorders. Nat Rev Neurosci. 2008;9:357–369. doi: 10.1038/nrn2371. [DOI] [PubMed] [Google Scholar]
- Ben-Ari Y, Crepel V, Represa A. Seizures beget seizures in temporal lobe epilepsies: the boomerang effects of newly formed aberrant kainatergic synapses. Epilepsy Curr. 2008;8:68–72. doi: 10.1111/j.1535-7511.2008.00241.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bender RA, Soleymani SV, Brewster AL, Nguyen ST, Beck H, Mathern GW, Baram TZ. Enhanced expression of a specific hyperpolarization-activated cyclic nucleotide-gated cation channel (HCN) in surviving dentate gyrus granule cells of human and experimental epileptic hippocampus. J Neurosci. 2003;23:6826–6836. doi: 10.1523/JNEUROSCI.23-17-06826.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bureau I, Dieudonne S, Coussen F, Mulle C. Kainate receptor-mediated synaptic currents in cerebellar Golgi cells are not shaped by diffusion of glutamate. Proc Natl Acad Sci U S A. 2000;97:6838–6843. doi: 10.1073/pnas.97.12.6838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castillo PE, Malenka RC, Nicoll RA. Kainate receptors mediate a slow postsynaptic current in hippocampal CA3 neurons. Nature. 1997;388:182–186. doi: 10.1038/40645. [DOI] [PubMed] [Google Scholar]
- Cathala L, Brickley S, Cull-Candy S, Farrant M. Maturation of EPSCs and intrinsic membrane properties enhances precision at a cerebellar synapse. J Neurosci. 2003;23:6074–6085. doi: 10.1523/JNEUROSCI.23-14-06074.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S, Su H, Yue C, Remy S, Royeck M, Sochivko D, Opitz T, Beck H, Yaari Y. An increase in persistent sodium current contributes to intrinsic neuronal bursting after status epilepticus. J Neurophysiol. 2011;105:117–129. doi: 10.1152/jn.00184.2010. [DOI] [PubMed] [Google Scholar]
- Contractor A, Swanson G, Heinemann SF. Kainate receptors are involved in short- and long-term plasticity at mossy fiber synapses in the hippocampus. Neuron. 2001;29:209–216. doi: 10.1016/s0896-6273(01)00191-x. [DOI] [PubMed] [Google Scholar]
- Coulter DA, Carlson GC. Functional regulation of the dentate gyrus by GABA-mediated inhibition. Prog Brain Res. 2007;163:235–243. doi: 10.1016/S0079-6123(07)63014-3. [DOI] [PubMed] [Google Scholar]
- Del Negro CA, Morgado-Valle C, Hayes JA, Mackay DD, Pace RW, Crowder EA, Feldman JL. Sodium and calcium current-mediated pacemaker neurons and respiratory rhythm generation. J Neurosci. 2005;25:446–453. doi: 10.1523/JNEUROSCI.2237-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeVries SH. Bipolar cells use kainate and AMPA receptors to filter visual information into separate channels. Neuron. 2000;28:847–856. doi: 10.1016/s0896-6273(00)00158-6. [DOI] [PubMed] [Google Scholar]
- Dingledine R, Hynes MA, King GL. Involvement of N-Methyl-D-Aspartate Receptors in Epileptiform Bursting in the Rat Hippocampal Slice. J Physiol. 1986;380:175–189. doi: 10.1113/jphysiol.1986.sp016279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Epsztein J, Represa A, Jorquera I, Ben-Ari Y, Crépel V. Recurrent mossy fibers establish aberrant kainate receptor-operated synapses on granule cells from epileptic rats. J Neurosci. 2005;25:8229–8239. doi: 10.1523/JNEUROSCI.1469-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Epsztein J, Lee AK, Chorev E, Brecht M. Impact of spikelets on hippocampal CA1 pyramidal cell activity during spatial exploration. Science. 2010a;327:474–477. doi: 10.1126/science.1182773. [DOI] [PubMed] [Google Scholar]
- Epsztein J, Sola E, Represa A, Ben-Ari Y, Crépel V. A selective interplay between aberrant EPSPKA and INaP reduces spike timing precision in dentate granule cells of epileptic rats. Cereb Cortex. 2010b;20:898–911. doi: 10.1093/cercor/bhp156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisahn A, Heinemann SF, McBain CJ. The kainate receptor subunit GluR6 mediates metabotropic regulation of the slow and medium AHP currents in mouse hippocampal neurones. J Physiol. 2005;562:199–203. doi: 10.1113/jphysiol.2004.077412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frerking M, Ohliger-Frerking P. AMPA receptors and kainate receptors encode different features of afferent activity. J Neurosci. 2002;22:7434–7443. doi: 10.1523/JNEUROSCI.22-17-07434.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fricker D, Miles R. EPSP amplification and the precision of spike timing in hippocampal neurons. Neuron. 2000;28:559–569. doi: 10.1016/s0896-6273(00)00133-1. [DOI] [PubMed] [Google Scholar]
- Gabriel S, Njunting M, Pomper JK, Merschhemke M, Sanabria ER, Eilers A, Kivi A, Zeller M, Meencke HJ, Cavalheiro EA, Heinemann U, Lehmann TN. Stimulus and potassium-induced epileptiform activity in the human dentate gyrus from patients with and without hippocampal sclerosis. J Neurosci. 2004;24:10416–10430. doi: 10.1523/JNEUROSCI.2074-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henze DA, Wittner L, Buzsáki G. Single granule cells reliably discharge targets in the hippocampal CA3 network in vivo. Nat Neurosci. 2002;5:790–795. doi: 10.1038/nn887. [DOI] [PubMed] [Google Scholar]
- Hermann BP, Seidenberg M, Schoenfeld J, Davies K. Neuropsychological characteristics of the syndrome of mesial temporal lobe epilepsy. Arch Neurol. 1997;54:369–376. doi: 10.1001/archneur.1997.00550160019010. [DOI] [PubMed] [Google Scholar]
- Jung MW, McNaughton BL. Spatial selectivity of unit-activity in the hippocampal granular layer. Hippocampus. 1993;3:165–182. doi: 10.1002/hipo.450030209. [DOI] [PubMed] [Google Scholar]
- Lawrence JJ, Saraga F, Churchill JF, Statland JM, Travis KE, Skinner FK, McBain CJ. Somatodendritic Kv7/KCNQ/M channels control interspike interval in hippocampal interneurons. J Neurosci. 2006;26:12325–12338. doi: 10.1523/JNEUROSCI.3521-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lenck-Santini PP, Holmes GL. Altered phase precession and compression of temporal sequences by place cells in epileptic rats. J Neurosci. 2008;28:5053–5062. doi: 10.1523/JNEUROSCI.5024-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lerma J. Kainate receptor physiology. Curr Opin Pharmacol. 2006;6:89–97. doi: 10.1016/j.coph.2005.08.004. [DOI] [PubMed] [Google Scholar]
- Leutgeb JK, Leutgeb S, Moser MB, Moser EI. Pattern separation in the dentate gyrus and CA3 of the hippocampus. Science. 2007;315:961–966. doi: 10.1126/science.1135801. [DOI] [PubMed] [Google Scholar]
- Lynch M, Sayin U, Golarai G, Sutula T. NMDA receptor-dependent plasticity of granule cell spiking in the dentate gyrus of normal and epileptic rats. J Neurophysiol. 2000;84:2868–2879. doi: 10.1152/jn.2000.84.6.2868. [DOI] [PubMed] [Google Scholar]
- Magee JC, Johnston D. Plasticity of dendritic function. Curr Opin Neurobiol. 2005;15:334–342. doi: 10.1016/j.conb.2005.05.013. [DOI] [PubMed] [Google Scholar]
- Nicoll RA, Schmitz D. Synaptic plasticity at hippocampal mossy fibre synapses. Nat Rev Neurosci. 2005;6:863–876. doi: 10.1038/nrn1786. [DOI] [PubMed] [Google Scholar]
- Paternain AV, Morales M, Lerma J. Selective antagonism of AMPA receptors unmasks kainate receptor-mediated responses in hippocampal-neurons. Neuron. 1995;14:185–189. doi: 10.1016/0896-6273(95)90253-8. [DOI] [PubMed] [Google Scholar]
- Represa A, Tremblay E, Ben-Ari Y. Kainate Binding-Sites in the Hippocampal Mossy Fibers-Localization and Plasticity. Neuroscience. 1987;20:739–748. doi: 10.1016/0306-4522(87)90237-5. [DOI] [PubMed] [Google Scholar]
- Ruiz A, Sachidhanandam S, Utvik JK, Coussen F, Mulle C. Distinct subunits in heteromeric kainate receptors mediate ionotropic and metabotropic function at hippocampal mossy fiber synapses. J Neurosci. 2005;25:11710–11718. doi: 10.1523/JNEUROSCI.4041-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sachidhanandam S, Blanchet C, Jeantet Y, Cho YH, Mulle C. Kainate receptors act as conditional amplifiers of spike transmission at hippocampal mossy fiber synapses. J Neurosci. 2009;29:5000–5008. doi: 10.1523/JNEUROSCI.5807-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sambandan S, Sauer JF, Vida I, Bartos M. Associative plasticity at excitatory synapses facilitates recruitment of fast-spiking interneurons in the dentate gyrus. J Neurosci. 2010;30:11826–11837. doi: 10.1523/JNEUROSCI.2012-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt-Hieber C, Jonas P, Bischofberger J. Subthreshold dendritic signal processing and coincidence detection in dentate gyrus granule cells. J Neurosci. 2007;27:8430–8441. doi: 10.1523/JNEUROSCI.1787-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silver RA. Neuronal arithmetic. Nat Rev Neurosci. 2010;11:474–489. doi: 10.1038/nrn2864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stafstrom CE, Schwindt PC, Chubb MC, Crill WE. Properties of persistent sodium conductance and calcium conductance of layer V neurons from cat sensorimotor cortex in vitro. J Neurophysiol. 1985;53:153–170. doi: 10.1152/jn.1985.53.1.153. [DOI] [PubMed] [Google Scholar]
- Sutula TP, Dudek FE. Unmasking recurrent excitation generated by mossy fiber sprouting in the epileptic dentate gyrus: an emergent property of a complex system. Prog Brain Res. 2007;163:541–563. doi: 10.1016/S0079-6123(07)63029-5. [DOI] [PubMed] [Google Scholar]
- Tauck DL, Nadler JV. Evidence of functional mossy fiber sprouting in hippocampal-formation of kainic acid-treated rats. J Neurosci. 1985;5:1016–1022. doi: 10.1523/JNEUROSCI.05-04-01016.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vervaeke K, Hu H, Graham LJ, Storm JF. Contrasting effects of the persistent Na+ current on neuronal excitability and spike timing. Neuron. 2006;49:257–270. doi: 10.1016/j.neuron.2005.12.022. [DOI] [PubMed] [Google Scholar]
- Vreugdenhil M, Hoogland G, van Veelen CW, Wadman WJ. Persistent sodium current in subicular neurons isolated from patients with temporal lobe epilepsy. Eur J Neurosci. 2004;19:2769–2778. doi: 10.1111/j.1460-9568.2004.03400.x. [DOI] [PubMed] [Google Scholar]