

Abstract
Background K2P channels play a key role in stabilizing the resting membrane potential, thereby modulating cell excitability in the central and peripheral somatic nervous system. Whole-cell experiments revealed a riluzole-activated current (IRIL), transported by potassium, in mouse superior cervical ganglion (mSCG) neurons. The activation of this current by riluzole, linoleic acid, membrane stretch, and internal acidification, its open rectification and insensitivity to most classic potassium channel blockers, indicated that IRIL flows through channels of the TREK [two-pore domain weak inwardly rectifying K channel (TWIK)-related K channel] subfamily. Whole-ganglia and single-cell reverse transcription-PCR demonstrated the presence of TREK-1, TREK-2, and TRAAK (TWIK-related arachidonic acid-activated K+ channel) mRNA, and the expression of these three proteins was confirmed by immunocytochemistry in mSCG neurons. IRIL was enhanced by zinc, inhibited by barium and fluoxetine, but unaffected by quinine and ruthenium red, strongly suggesting that it was carried through TREK-1/2 channels. Consistently, a channel with properties identical with the heterologously expressed TREK-2 was recorded in most (75%) cell-attached patches. These results provide the first evidence for the expression of K2P channels in the mammalian autonomic nervous system, and they extend the impact of these channels to the entire nervous system.
Introduction
Mammalian two-pore domain potassium channels (K2P) are encoded by 15 known genes (KCNK), and they are distributed into six subfamilies [two-pore domain weak inwardly rectifying K+ channel (TWIK), TWIK-related K+ channel (TREK), TWIK-related acid-sensitive K+ channel (TASK), TWIK-related alkaline-activated K+ channel (TALK), TWIK-related halothane-inhibited K+ channel (THIK), and TWIK-related spinal cord K+ channel (TRESK)]. Since their discovery (Lesage et al., 1996), the biophysical, pharmacological, and putative functional properties of cloned and heterologously expressed mammalian K2P channels have been methodically investigated. Such an effort strongly contrasts with the scarce data available from native K2P channels naturally expressed in mammalian cells (for review, see Lotshaw, 2007). Among these K2P channels, the TREK subfamily [TREK-1, TREK-2, and TWIK-related arachidonic acid-activated K+ channel (TRAAK)] is considered to provide a source of background potassium channels with very weak voltage dependency and a low open probability. As such, they are involved in the maintenance of the membrane resting potential and excitability (Mathie and Veale, 2007). Their low basal activity can be strongly increased by physical and chemical stimuli such as membrane stretch (Maingret et al., 1999a; Bang et al., 2000), temperature (Maingret et al., 2000a), general anesthetics (Lesage et al., 2000b), or neuroprotective drugs (Fink et al., 1998; Duprat et al., 2000; Lesage et al., 2000b), suggesting a wide range of promising functions.
For considerable time, the ionic and molecular nature of the currents that sustain the resting membrane potential in sympathetic neurons from the superior cervical ganglion (SCG) has been pursued. A rather unpredictable and complex mechanism emerged (Lamas et al., 2002), whereby at least three voltage-dependent currents contribute to the resting current: the potassium M-current (Brown et al., 1982), the cationic h-current (Lamas, 1998), and, more recently, a persistent sodium current (Lamas et al., 2009). Because only a small percentage of all these voltage-dependent channels are open at rest, they probably have a powerful stabilizing rather than a strong settling effect on the resting membrane potential. Experiments designed to remove the contribution of the known voltage-dependent currents and the Na–K pump revealed an apparently voltage-independent “leakage” current that was transported mainly by potassium (Lamas et al., 2002). However, as in most cell types, the molecular nature of this leakage current remains to be determined.
We characterized a potassium current activated by riluzole (IRIL) that is insensitive to most classic blockers of voltage- and calcium-dependent ion currents. The electrophysiological, pharmacological, and molecular approach followed provides strong evidence that this current flows through K2P potassium channels of the TREK subfamily. Single-channel data demonstrated the strong expression of TREK-2 channels and a weaker expression of an unidentified TREK-like channel. We hypothesize that K2P channels may, at least in part, be responsible for the leak potassium current in sympathetic neurons and that they modulate their excitability.
Materials and Methods
Animal handling and all experimental procedures were approved by the Spanish Higher Research Council and the University of Vigo Scientific Committee and they conformed to Spanish and European guidelines for the protection of experimental animals (RD1201/2005; 86/609/EEC).
Cell culture.
The conditions for mouse SCG cell culture have been described previously (Martínez-Pinna et al., 2002; Romero et al., 2004; Lamas et al., 2009). Mice (20–60 d of age) were terminally anesthetized with CO2 and immediately decapitated. The SCG were removed in cold Leibowitz medium (L-15) under a binocular microscope, and after cleaning, the ganglia were chopped and incubated in collagenase (2.5 mg/ml in HBSS) for 15 min at 37°C. After washing, the ganglia were then incubated for 30 min in trypsin (1 mg/ml in Hanks). Finally, the neurons were mechanically isolated, centrifuged, and plated in 35 mm Petri dishes previously coated with laminin (10 μg/ml in EBSS). Neurons were cultured for 1–2 d at 37°C and 5% CO2 in L-15 medium containing the following: 24 mm NaHCO3, 10% fetal calf serum, 2 mm l-glutamine, 38 mm d-glucose, 100 UI/ml penicillin, 100 μg/ml streptomycin, and 50 ng/ml nerve growth factor. For electrophysiological purposes, only neurons devoid of processes were used (Romero et al., 2004).
Perforated-patch whole-cell recording.
Electrophysiological recordings were performed under continuous perfusion (∼10 ml/min) at room temperature using an Axopatch 200B amplifier (Molecular Devices). For current-clamp experiments, the “Iclamp-fast” mode (simulating bridge mode) of the same amplifier was used (Magistretti et al., 1998). Two-step fire-polished patch pipettes with a tip resistance of 2–4 MΩ were used and perforated patch obtained with freshly prepared amphotericin B (75 μg/ml) resulted in series resistance <20 MΩ (Rae et al., 1991). Junction potentials <5 mV were not corrected (Neher, 1992).
The sampling frequency was 2 kHz for voltage-clamp (filtering at 0.5 kHz) and 10 kHz for current-clamp experiments (filtering at 5 kHz); for very long-lasting recordings, data were sampled at 100 Hz (filtering at 50 Hz). Data were digitized through a Digidata 1440A using the pClamp10 software (Molecular Devices) and they were analyzed and plotted using the same software and Origin 7.5 (OriginLab Corporation). Averages represent mean ± SEM, and the statistical significance was assessed with two-sample Student's t tests.
The standard pipette solution contained the following (in mm): 90 K-acetate, 20 KCl, 3 MgCl2, 1 CaCl2, 3 EGTA, and 40 HEPES, adding ∼20 NaOH to a pH of 7.2. Standard bath solution contained the following (in mm): 140 NaCl, 3 KCl, 1 MgCl2, 2 CaCl2, 10 d-glucose, and 10 HEPES, adjusted to pH 7.2 with Tris [tris(hydroxymethyl)aminomethane]. To induce intracellular acidosis, we substituted 90 mm NaCl with 90 mm NaHCO3; changes in osmolarity were compensated for by adding sucrose. For the high-potassium bath solution, we modified the standard solution using 20 mm KCl and 120 mm NaCl. For symmetrical potassium bath solution, we used 110 mm KCl and 30 mm NaCl. To obtain a mechanical stimulus, we switched the neurons from a 290 to a 145 mOsm bath solution; both were standard solutions containing only 70 mm NaCl and their osmolarity was adjusted using mannitol. The blockers added to bath solutions included the following: 15 mm tetraethylammonium (TEA), 2 mm 4-AP, 1 mm CsCl, 100 μm CdCl, and 0.5 μm TTX to block voltage-dependent potassium, cationic, calcium, and sodium currents. All solutions used were kept between 290 and 300 mOsm saving the hypotonic solution.
Single-channel recording.
Single-channel currents were recorded using the cell-attached configuration and electrodes of ∼6 MΩ. Acquisition was set at 20 kHz and filtered at 2 kHz using the amplifier built-in filter. Threshold detection for single-channel openings was set at 50% of the maximum amplitude and patches containing more than one opening level were discarded. Single-channel mean amplitude was measured using Clampfit 10 software and openings faster than 50 μs were discarded. Pipette and bath solutions contained the following (in mm): 150 KCl, 1 MgCl2, 5 EGTA, and 10 HEPES, buffered to pH 7.2 using KOH. Open probability was calculated using the Clampfit software according to the following equation: Po = to/T, where to was the total time that the channel was found in the open state, and T is the total observation time.
Reverse transcriptase-PCR.
Total RNA was isolated from the mouse SCG and cerebral cortex samples using the RNeasy kit (QIAGEN) and the Trizol method, respectively, and the purified RNA was stored at −30°C. The quality of the total RNA was assessed through its A260/A280 absorbance ratio, and it was verified by amplification of the constitutive β-actin gene by reverse transcriptase (RT)-PCR. Reverse transcription from total RNA was performed using Moloney murine leukemia virus (M-MLV) reverse transcriptase (Invitrogen), and each reverse transcription reaction (30 μl) contained the following: 2 μg of total RNA, 3 ng/μl random primers (0.57 ng; Invitrogen), 6 μl of 2.5 mm dNTP mix, RNase-free water, 6 μl of 5× buffer (Invitrogen), 10 U of RNaseOUT recombinant ribonuclease inhibitor (Invitrogen), and 200 U of M-MLV (Invitrogen). The reactions were performed at 37°C for 60 min, followed by 42°C for 15 min and 95°C for 5 min.
First-strand cDNA from all channels was PCR amplified with the specific primers described in Table 1(Kang et al., 2004; Kang and Kim, 2006). Primers to amplify the constitutively active β-actin gene were designed using the program Primer Premier (Premier Biosoft International). Primer specificity was checked in the complete genome of Mus musculus using the basic local alignment search tool (BLAST) program (Altschul et al., 1990). The primers to amplify a 100 bp fragment of TREK-1 were designed with the PRIMER-BLAST primer designing tool. This program used Primer3 (Rozen and Skaletsky, 2000) to design the PCR primers and then it submitted them to BLAST to search against NCBI Transcript Reference Sequences of the genome of M. musculus.
Table 1.
Specific pair of primers used to amplify K2P channels and β-actin gene
| Name | Nucleotide sequence (5′–3′) | Ta (°C) | bp | Reference | |
|---|---|---|---|---|---|
| β-Actin | ActineF | F: TGCCGCATCCTCTTCCTC | 53 | 655 | This study |
| ActineR | R: CGCCTTCACCGTTCCAGT | This study | |||
| TREK-1 | TREK1F | F: CTTTGGCTTTCTACTGGCTG | 55 | 678 | This study |
| R: GACCGTTCAGATAGATGCTC | Kang and Kim, 2006 | ||||
| TREK-2 | F: GGCTAATGTCACTGCTGAGTTCC | 55 | 625 | Kang and Kim, 2006 | |
| R: ACAAGACAACACATAGTCCAATTCC | Kang et al., 2004 | ||||
| TRAAK | F: CACCACTGTAGGCTTTGGCGATTATG | 55 | 448 | Kang and Kim, 2006 | |
| R: ACTCTGCGTGTCTGAGGACTCGTCG | Kang and Kim, 2006 | ||||
| TREK-1 | TREK1scF | F: TCACTCTGACGACCATTGGA | 54 | 100 | This study |
| TREK1scR | R: ACGAGGATCCAGAACCACAC | This study |
F, Forward; R, reverse; Ta, annealing temperature.
Amplifications were performed in a Mastercycler gradient thermocycler (Eppendorf) as follows: 95°C for 5 min; and then 35 cycles at 95°C for 45 s, specific T for 1 min, 72°C for 2 min; and a final extension step at 72°C for 15 min. The PCR amplification mixture (50 μl) contained the following: 50 ng of DNA template, 10 pmol of each primer, 0.2 mm dNTPs, 1.5 mm MgCl2, 1.25 U of Taq DNA polymerase (Invitrogen), and 5 μl of 10× reaction buffer (Invitrogen). PCR products were cleaned before cloning using the Wizard SV Gel and the PCR Clean-Up System (Promega) according to the manufacturer's protocol.
Cerebral cortex tissue was used as a positive control for the PCR amplification of the channels, and β-actin expression was assessed to check the quality of the RNA. Negative controls were also performed to exclude contamination (Fig. 5A, lanes not labeled). The RT-PCR products amplified corresponding to the partial TREK-1, TREK-2, and TRAAK PCR amplicons were cloned into the vector pGEM-T (Promega) using the pGEM-T Vector System II (Promega) with JM109 competent cells and sequenced on both strands with the dideoxynucleotide chain termination method using T7 and SP6 primers.
Figure 5.

mSCG neurons express mRNAs encoding all the members of the TREK subfamily. A, Expression of mRNAs for the TREK channels in the whole mSCG and cerebral cortex as assessed by RT-PCR using specific primers for each K2P channel. DNA fragments were cloned and sequenced for confirmation. The expected band sizes were 678 bp (TREK-1, lanes 2, 3, 4, 5), 625 bp (TREK-2, lanes 7, 8, 9, 10), 448 bp (TRAAK, lanes 12, 13, 14, 15), and 655 bp (β-actin, lanes 17, 18). The first lane shows the 100 bp ladder (Takara). Negative controls are shown in lanes 6, 11, 16, and 19. B, Expression of TREK-1 (left), TREK-2 (middle), and TRAAK (right) mRNA in individual mSCG cells assessed by single-cell RT-PCR.
Single-cell PCR.
Single-cell RT-PCR was performed using QIAGEN OneStep RT-PCR kit (QIAGEN) with specific primers for TREK-1 (TREK1scF and TREK1scR), TREK-2, and TRAAK, respectively (Table 1). As a positive control, primers for β-actin were also used to detect mRNA expression. Reverse transcription was performed at 50°C for 30 min, and the PCR amplifications were then performed as follows: polymerase activation step at 95°C for 15 min; 35 cycles at 94°C for 45 s, 54°C for 1 min, 72°C for 2 min; and a final extension step at 72°C for 10 min. The RT-PCR amplification mixture (50 μl) contained 20 μl of DEPC water with the cytoplasm from a single neuron or a complete cell, 10 pmol of each primer, 0.4 mm of each dNTP, 2 μl of QIAGEN OneStep RT-PCR Enzyme Mix and 10 μl of 5× QIAGEN OneStep RT-PCR Buffer with 12.5 mm MgCl2. PCR products were visualized on 2% agarose gels with ethidium bromide.
Immunocytochemistry.
Cultured cells were fixed with paraformaldehyde (2%; for 1 h) at room temperature and then washed with PBS (Sigma-Aldrich) with gentle shaking. Permeabilization was performed by bathing the cells in PBS containing Triton X-100 (0.2%; Sigma-Aldrich) for 10 min and then washed out with PBS. Nonspecific antibody binding was blocked by incubating the cells in normal donkey serum (NDS) (10%) in PBS for 30 min at room temperature. The dishes were incubated overnight at 4°C with the primary antibodies against TREK-1, TREK-2, or TRAAK (sc-11556, sc-11560, sc-11326) diluted 1:100. After incubation, cells were rinsed in PBS (three times for 5 min each time), followed by incubation with FITC-conjugated anti-goat secondary antibody (1:200) for 1 h at room temperature; finally, they were washed with PBS (three times for 5 min each time). At the end of the procedure, the nuclei were stained with 4′,6′-diamidino-2-phenylindole (DAPI) (1:10,000 dilution for 2 min; Santa Cruz Biotechnology) and mounted (ProLong Gold; Invitrogen). Negative controls were obtained, treating cultured neurons identically without adding the primary antibody, and no neuronal labeling was observed. The same antibodies against TREK-1 and TREK-2 were carefully tested by Deng et al. (2009), and all of them have been successfully used in trigeminal ganglion cells recently (Yamamoto et al., 2009). All antibodies were purchased from Santa Cruz Biotechnology, and all antibody solutions were prepared in 2% NDS in PBS. Images were acquired with a confocal Leica SP5 microscope using LAS AF 2.0 software. The wavelengths used were 488/493–581 nm for FITC and 405/415–526 nm for DAPI.
Results
Long-lasting application of the neuroprotective agent riluzole (100 μm; 5 min) to cultured mouse SCG (mSCG) neurons clamped at −30 mV invariably induced an outward current (89.6 ± 4.8 pA; n = 6; peak current) in standard solutions (Fig. 1A, top trace). Conversely, the same application induced a hyperpolarization of −11.2 ± 2.2 mV (n = 11; peak voltage) in neurons manually fixed at −30 mV in current-clamp experiments (Fig. 1B, top trace). When the application of riluzole was sustained (≥5 min), both the outward current and the hyperpolarization reached a peak after 107.6 ± 7.6 s (n = 6) and 105 ± 19 s (n = 11), respectively, which then declined (Fig. 1A,B, top traces). A similar transient effect of riluzole has been reported for heterologously expressed TREK-1 (Duprat et al., 2000) and TREK-2 (Lesage et al., 2000b) homodimers. In these heterologously expressed channels, inhibition was observed after the initial phase of direct current activation, which was attributed to a secondary increase of cAMP by riluzole. By contrast, the activation of TRAAK, the other member of the TREK subfamily, persisted in the presence of riluzole (Duprat et al., 2000).
Figure 1.

Riluzole activates an outward current in mSCG neurons. A, Application of riluzole induced a clear transient outward current when the membrane was held at −30 mV (top trace). The amplitude of IRIL was strongly reduced at more negative membrane potentials (bottom trace). Short-duration hyperpolarizing voltage pulses were continuously applied to follow changes in conductance. B, In neurons manually current clamped at −30 mV, riluzole application induced a clear membrane hyperpolarization (top trace), an effect that was strongly reduced or absent at resting membrane potentials (bottom trace). C, The peak amplitude of the riluzole-activated current was unaffected by blockers of voltage-dependent sodium channels (a), persistent sodium channels (b), voltage-activated calcium channels (c), and hyperpolarization-activated cationic channels (d).
It is noteworthy that the effect of riluzole (100 μm) was smaller at more hyperpolarized membrane voltages, whereas at rest (−61.1 ± 1.1 mV; n = 5), a small hyperpolarization was only observed in three of five cells tested (−5.2 ± 1.3 mV; n = 3) (Fig. 1B, bottom trace). Similarly, under voltage-clamp conditions, the riluzole-activated current was strongly reduced by clamping the membrane at −50 rather than −30 mV (25.3 ± 8.1 pA; n = 4; p < 0.05) (Fig. 1A, bottom trace). To obtain a more consistent effect on the resting membrane potential, we increased the concentration of riluzole to 300 μm. In voltage clamp and at −30 mV, this concentration of riluzole evoked a current of 188.3 ± 35.7 pA (n = 8) (supplemental Fig. 1, available at www.jneurosci.org as supplemental material) and it induced a hyperpolarization of approximately −8 mV (from −62.4 ± 2.3 to −70.6 ± 2.3 mV; n = 15; p < 0.05) (data not shown) in all cells tested.
These initial results strongly suggested that IRIL was transported mainly through K2P TREK-1 and/or TREK-2 channels in SCG neurons, although the variability in the current decline over long-term exposure to riluzole prevented the involvement of TRAAK from being discarded.
Ionic nature of the riluzole-activated outward current
Riluzole inhibits voltage-activated sodium channels (Hebert et al., 1994; Reboreda et al., 2003; Wang et al., 2008), as reflected by the inhibition of transient and persistent sodium currents in cultured mSCG neurons (Lamas et al., 2009). As inhibition of a persistent sodium current would induce an apparent outward current, negative 15 mV voltage steps of 50 ms were applied at 0.4 Hz to track the variations in conductance induced by riluzole. Riluzole provoked an increase in conductance of 1.4 nS (from 2.8 ± 0.2 to 4.2 ± 0.2 nS; n = 5) (Fig. 1A, top trace), suggesting that the outward current was attributable to the opening of potassium channels rather than to the blockage of sodium channels. Long applications of riluzole (100 μm for 5 min) were used to test the effect of several blockers of nonpotassium channels, using the current induced in standard solutions at −30 mV (see above) as a control.
Persistent sodium currents in SCG neurons are sensitive to TTX and valproate (Lamas et al., 2009), yet at concentrations that strongly inhibit the sodium current (0.5 and 100 μm, respectively), neither of these compounds induced an outward current similar to that provoked by riluzole at −30 mV (Fig. 1Ca,Cb). Indeed, the amplitude of the riluzole-activated outward current recorded in the presence of TTX (97.8 ± 18.1 pA; n = 7) or valproate (76.6 ± 14.5 pA; n = 4) was not significantly different from that recorded in control conditions (p > 0.05) (see also Fig. 3E). Moreover, the hyperpolarization induced by long applications of riluzole in the presence of valproate (−16.4 ± 1.2 mV; n = 4) or TTX (−13.5 ± 2.6 mV; n = 4) in current-clamp experiments was no different from that obtained in standard solutions (p > 0.05; cells manually clamped at −30 mV) (data not shown). Riluzole also inhibits voltage-dependent calcium channels (Huang et al., 1997). However, cadmium (100 μm) did not affect the current at −30 mV in mSCG neurons, and the riluzole-activated current in the presence of cadmium was not significantly different from the control (94.8 ± 8.7 pA; n = 9) (Figs. 1Cc, 3E) (p > 0.05). The hyperpolarization-activated cation current (Ih) fulfils an important role in stabilizing the resting membrane potential in SCG cells (Lamas, 1998; Lamas et al., 2002; Romero et al., 2004). Nevertheless, the inhibition of this current with 1 mm cesium did not affect the riluzole-activated current (78.9 ± 15.7 pA; n = 4) (Figs. 1Cd, 3E) (p > 0.05).
Figure 3.

IRIL is transported through K2P channels. A–C, The riluzole-induced current was not affected by the presence of classic potassium channel blockers TEA, 4-AP, apamine, and paxilline, indicating that IRIL is not transported through voltage- or calcium-activated potassium currents. D, Barium clearly and significantly reduced the outward current evoked by riluzole. The scale bars in this figure apply to all the panels. E, Bar diagram summarizing the main results obtained using ion channel blockers. This figure represents the mean ± SEM for the peak current induced by 100 μm riluzole at a holding potential of −30 mV. **p < 0.01 versus control.
To further characterize the nature of the riluzole-activated outward current, riluzole (100 μm; 30 s) was applied to cells fixed at different membrane voltages, both in standard solutions ([K]o = 3 mm; EK = −91 mV) (Fig. 2A), as well as in a high-potassium extracellular solution ([K]o = 20 mm; EK = −43 mV) (Fig. 2B). The current–voltage curve (I–V) for IRIL in standard potassium concentrations (Fig. 2C, dots) showed a strong outward rectification that nearly vanished in 20 mm extracellular potassium (Fig. 2C, squares). A similar outwardly rectifying I–V was obtained when the riluzole-activated current was isolated using repetitive negatively progressing voltage ramps (100 mV/s, every 10 s) (Fig. 2D). The current evoked by the ramp at the peak of the riluzole effect was subtracted from that obtained before riluzole was applied to define the riluzole-activated current. The I–V strongly rectified in standard but not at symmetrical potassium concentrations (EK = 0 mV) (Fig. 2D); note that, in experiments using voltage ramps, TTX, Cd2+, and Cs+ were added to the extracellular solution. Similar outwardly rectifying I–V relationships were obtained for currents through heterologously expressed TREK-1 (Fink et al., 1996), TREK-2 (Lesage et al., 2000b), and TRAAK (Fink et al., 1998) channels at physiological potassium concentrations.
Figure 2.

IRIL is a potassium current with a reversal potential that follows the Nernst equation. A, In physiological external potassium concentrations (3 mm), short applications of riluzole evoked increasing outward currents at potentials more positive than −80 mV. In most cells tested, no inward currents could be obtained at more negative voltages. B, Inward currents were obtained at potentials below to −40 mV when the extracellular potassium concentration was elevated to 20 mm. C, Current–voltage relationships for the riluzole-induced currents shown in A and B. Note that the marked outward rectification in 3 mm potassium (dots) was strongly reduced in 20 mm potassium (squares). In both cases, the reversal potential of IRIL was close to the Nernst equilibrium potential for potassium. D, Current–voltage relationships for riluzole-induced currents obtained in response to negatively progressing voltage ramps. The current obtained in the control was subtracted from that obtained in the presence of riluzole (100 μm) (close to the peak). Note that the strong outward rectification obtained in standard solutions (EK = −91 mV) disappeared when symmetrical concentrations of potassium were used (EK = 0 mV). Recordings were acquired from two different cells.
In summary, riluzole activated an outward current with a reversal potential closely following the equilibrium potential for potassium, and the contribution of sodium, calcium, and cationic channels to IRIL can be ruled out.
Classic potassium channel blockers
K2P channels are in general insensitive to the external application of classic potassium channel blockers like TEA, 4-AP, or Cs+, but they can be blocked by barium. Once the potassium nature of the riluzole-activated current was demonstrated, we assessed the putative effect of riluzole on potassium channels other than K2P by testing the effect of several known potassium channel blockers.
In standard solutions, the current evoked by the application of riluzole (100 μm) in the presence of external TEA (15 mm; 78.9 ± 11.3 pA; n = 5) (Fig. 3A) or 4-AP (2 mm; 79 ± 13.2 pA; n = 3) (Fig. 3B) was not statistically different from the control current (p > 0.05). Although riluzole activates calcium-dependent BK (Wang et al., 2008) and SK (Grunnet et al., 2001) potassium channels, the current recorded at −30 mV was not affected by the coapplication of apamin (200 nm) and paxilline (1 μm), and the riluzole-activated current in the presence of these toxins was not significantly different from control (106.5 ± 15.7 pA; n = 6; p > 0.05) (Fig. 3C).
Among the potassium channel blockers tested, only barium (5 mm) affected the current evoked by riluzole, producing a strong reduction (37 ± 6.3 pA; 58.7%; n = 4) (Fig. 3D) (p < 0.01). It is noteworthy that barium inhibits heterologously expressed TREK-1 (Fink et al., 1996), TREK-2 (Bang et al., 2000), and TRAAK (Fink et al., 1998) channels. As shown in Figure 3, both barium and TEA (but not other blockers) strongly inhibited the steady-state outward current at −30 mV, such inhibition representing the blockage of the non-inactivating potassium M-current characteristic of SCG neurons (Hadley et al., 2003; Romero et al., 2004).
When taken together, the results obtained using channel blockers not only suggested that the riluzole-activated outward current is attributable to the modulation of K2P channels but also they rule out the contribution of other voltage-gated and calcium-dependent channels (Fig. 3E).
TREK channel modulators
There are no completely specific or selective modulators distinguishing among the different K2P channel subtypes (for review, see Lotshaw, 2007), and therefore, we tested several of the innumerable pharmacological agents that interact with them. While some enabled us to rule out the participation of a given subfamily or subtype with a degree of confidence, others strongly indicated that the TREK subfamily was the origin of the riluzole-activated outward current. To avoid the possible interactions of K2P modulators with other currents, these experiments were performed in a blockers solution (see Materials and Methods) with extended application of riluzole. In such conditions, 100 μm riluzole induced an outward current of 84.4 ± 14.2 pA (n = 5; peak current), and this value was considered as the control. Note that the outward current obtained was not statistically different from that obtained in standard solutions (p > 0.05). Moreover, a dose–response curve for riluzole in the blockers solution provided an EC50 of 139.4 μm (supplemental Fig. 1, available at www.jneurosci.org as supplemental material).
Most K2P channels are inhibited (TASK-3, TRAAK, TWIK-2, TRESK) or unaffected (THIK-1) by micromolar concentrations of zinc; only heterologously expressed TREK-1 (Czirják and Enyedi, 2006) and TREK-2 (Kim et al., 2005) are clearly activated by this ion. TASK-1 and TASK-2 were reported to be slightly inhibited (Reyes et al., 1998), unaffected (Clarke et al., 2004), or slightly enhanced (Czirják and Enyedi, 2006) by zinc. Application of zinc (100 μm) to cells bathed in the blockers solution induced a slowly developing outward current (46.3 ± 12.3 pA; n = 5; measured after 4 min application) and the riluzole-activated current was enhanced (127.5 ± 10.9 pA; n = 6; p < 0.05) (Fig. 4A). Because expressed mouse TRAAK channels are inhibited by zinc (Czirják and Enyedi, 2006), these results ruled out a major contribution of TRAAK channels to IRIL. The antidepressant fluoxetine strongly inhibits heterologously expressed TREK-1 channels at concentrations that do not affect TRAAK channels (Kennard et al., 2005; Heurteaux et al., 2006); this drug seems to also inhibit TREK-2 cannels (Kang et al., 2008). In mSCG neurons fixed at −30 mV, fluoxetine (100 μm) induced a clear inward current (−85.7 ± 11.4 pA; n = 9) and robustly reduced the riluzole-activated current in blockers solutions (30.6 ± 5.2 pA; n = 9, p < 0.01) (Fig. 4B), again pointing to a weak, if any, contribution of TRAAK channels to IRIL. The inward current induced by fluoxetine may be attributable to the inhibition of the low K2P basal activity, but also to the inhibition of delayed rectifier channels, that could be not completely blocked by our blockers solution, as previously reported (Hahn et al., 1999; Yeung et al., 1999). In general, quinine and quinidine are considered as K2P blockers (Lotshaw, 2007; Mathie and Veale, 2007), and in mSCG neurons, quinine (300 μm) induced an inward current at −30 mV (−77.5 ± 10.6 pA; n = 8) that we attributed to the reduction of remaining M-current incompletely blocked by the blockers solution. Indeed, M-current inhibition by quinine has been reported in bullfrog sympathetic neurons (Imai et al., 1999) as well as in neuroblastoma cells (Robbins et al., 1992). Although IRIL was slightly reduced in the presence of quinine (Fig. 4C), the mean current was not different from that in the control (60 ± 13.2 pA; n = 8; p > 0.05). TRAAK, but not TREK-1, homodimers are highly sensitive (IC50 of 2 μm) to the inhibition with ruthenium red (RR) (Czirják and Enyedi, 2002), but application of 10 μm RR when IRIL was totally activated did not affect the current in mSCG neurons (supplemental Fig. 2, available at www.jneurosci.org as supplemental material).
Figure 4.

TREK but not TRAAK channels provide the main contribution to IRIL. A, Application of zinc to mSCG neurons induced a slowly developing outward current and significantly increased the riluzole-activated current. B, Fluoxetine induced an inward current at −30 mV and strongly reduced IRIL. C, Quinine also provoked an inward current, but the effect on the riluzole-activated current was not statistically significant. D, Internal acidification by extracellular bicarbonate resulted in the activation of an outward current that was completely blocked by fluoxetine. E, Peak amplitude of riluzole activated currents in the presence of modulators of TREK channels. *p < 0.05 and **p < 0.01 versus control. All the experiments in this figure were carried in the blockers extracellular solution, and the membrane was held at −30 mV. The scale bars in A apply for B and C.
Strong activation of TREK-1 and TREK-2 by internal acidification (Maingret et al., 1999b; Bang et al., 2000; Duprat et al., 2000; Lesage et al., 2000b) is a characteristic that may be used to distinguish these members of the TREK subfamily from the other K2P channels (Patel and Honore, 2001). As the other member of this subfamily (TRAAK) is unaffected by internal acidification (Fink et al., 1998; Maingret et al., 1999b), this property is also useful to discriminate among members of this subfamily. We used dinitrophenol (DNP) (300 μm) as a metabolic inhibitor that decreases the intracellular ATP concentration and acidifies the cytosol. Surprisingly, DNP induced a small inward current (−30.1 ± 6.2 pA; n = 7) (data not shown) in mSCG cells clamped at −30 mV and the riluzole-evoked current in the presence of DNP was not statistically different from the control (80 ± 16.6 pA; n = 7; p > 0.05). However, internal acidification by external application of bicarbonate (Maingret et al., 1999b) induced a strong outward current (213.8 ± 22.6 pA; n = 6) that was robustly inhibited by fluoxetine (100 μm) (Fig. 4D), as expected for a current carried by TREK-1 and/or TREK-2 channels.
All members of the TREK subfamily are activated by unsaturated fatty acids (Fink et al., 1998; Patel et al., 1998; Bang et al., 2000). In mSCG neurons, application of linoleic acid (10 μm) induced a consistent and very slowly developing outward current (Danthi et al., 2003), reaching 171.5 ± 14.3 pA after 10 min application (n = 4). The current induced by linoleic acid was also strongly inhibited by fluoxetine (supplemental Fig. 3A, available at www.jneurosci.org as supplemental material). Membrane stretch is also known to activate TREK channels and TRAAK has been reported to be much less mechanosensitive than TREK-1 (Patel et al., 1998; Maingret et al., 1999a,b; Bang et al., 2000). The modification of the extracellular osmolarity from 290 to 145 mOsm induced an obvious cell swelling and evoked a fast and clear outward current (122.7 ± 22.2 pA; n = 4) in mSCG neurons fixed at −30 mV. This stretch-activated current was also very sensitive to fluoxetine in the presence of the blockers (supplemental Fig. 3B, available at www.jneurosci.org as supplemental material).
In summary, experiments using K2P modulators indicate that TRAAK channels do not contribute substantially to IRIL (Fig. 4E).
Expression of mRNA for TREK subfamily K2P channels
The mRNA for K2P channels is widely distributed through the CNS (Talley et al., 2001). TREK-1 was the first K2P channel to be confirmed in the nervous system of mammals, and it is highly expressed in the mouse and human brain (Fink et al., 1996; Lesage et al., 2000b). Also TREK-2 (Bang et al., 2000; Lesage et al., 2000b) and TRAAK (Fink et al., 1998; Lesage et al., 2000b) are strongly expressed in brain. However, the expression of mRNA for TREK channels (or even for K2P channels) in the autonomic nervous system has yet to be reported. Since the combination of electrophysiology with pharmacology did not unequivocally define whether IRIL was transported through TREK-1, TREK-2, or both, we studied the expression of their mRNA.
We first assessed the expression of mRNA for the three members of the TREK subfamily by RT-PCR and found TREK-1, TREK-2, and TRAAK mRNA in the entire ganglion (Fig. 5A). Although ganglia are relatively homogeneous structures, the possibility of contamination with non-neuronal cells could not be discarded. To circumvent this problem, single-cell RT-PCR was performed using previously identified cultured neurons. Also, in these circumstances, expression of mRNA for the three TREK members was detected (Fig. 5B). Interestingly, in these studies, the signal for TRAAK was always fainter than that of the other two.
Immunocytochemistry for TREK subfamily K2P channels in mouse SCG neurons
Having found mRNA for the three members of the TREK subfamily in mSCG neurons, we tested whether they were all translated into proteins since complete agreement between protein and mRNA localization is not always found for K2P channels (Talley et al., 2001). Nevertheless, the expression of the TREK-1 (Maingret et al., 2000b), TREK-2 (Li et al., 2005), and TRAAK (Reyes et al., 2000) proteins was reasonably consistent with the data obtained by in situ hybridization and RT-PCR.
We investigated the immunolocalization of the three members of the TREK family in cultured mSCG cells. In these cultures, neurons can be readily identified and FITC immunoreactivity (green) was observed for all the three TREK-1, TREK-2, and TRAAK channels in mSCG neurons but not in satellite cells (Fig. 6A–C, panels on the right show bright-field images of the same field to clarify cell morphology). In culture, mSCG neurons are often surrounded by one or two satellite cells having a more dense nucleus when labeled with DAPI (blue), but these were never associated with FITC fluorescence.
Figure 6.
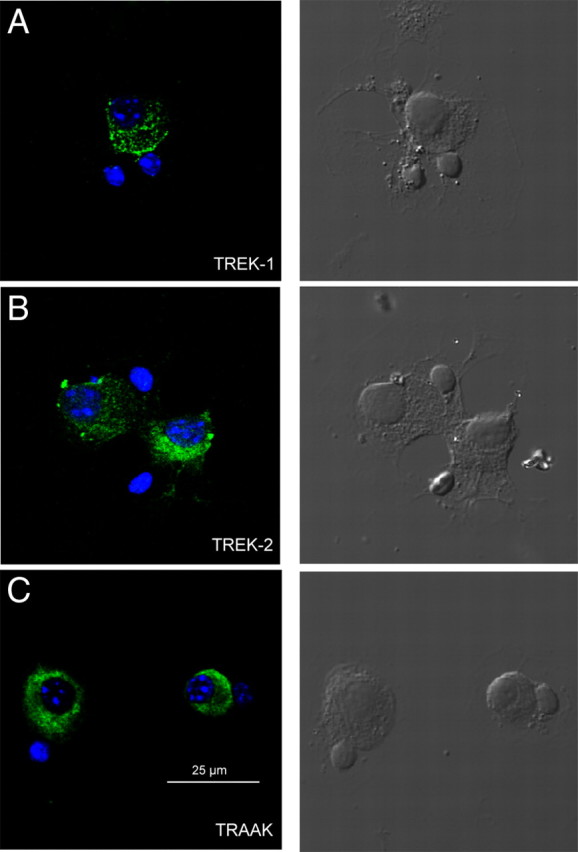
mSCG neurons express the three protein subunits of the TREK subfamily. A–C, Specific FITC-conjugated antibodies against TREK-1 (A), TREK-2 (B), and TRAAK (C) stained cultured mSCG neurons (green, left panels). DAPI-stained nuclei (blue) not surrounded by green stain belong to satellite cells that often accompanied the neurons in the culture. Satellite cells were never positive for FITC, and they can be observed under Nomarski optics in the right panels.
Single-channel recording
TREK channels have unique single-channel properties that have been used to distinguish among them (Han et al., 2003; Kang and Kim, 2006), and thus, we obtained cell-attached recordings at symmetrical concentrations of potassium (150 mm KCl) to identify which of these channels are functionally expressed in the membrane of mSCG neurons. In 16 of 32 cell-attached patches from mSCG neurons, we detected TREK-like channel activity, both at rest and well beyond the resting membrane potential (up to −100 mV). In 12 of these 16 patches (75%), a potassium channel was recorded with properties nearly identical with those reported for TREK-2 (Han et al., 2003; Kang et al., 2006). Accordingly, the single-channel conductance was 128.2 ± 6.9 pS at −60 mV and 53.8 ± 2.3 pS at +60 mV (Fig. 7Aa) (n = 12), and the mean current–voltage relationship showed a characteristic inward rectification (Fig. 7Ab) (n = 12). By contrast, in 3 of the 16 patches (18.7%), displaying activity at negative potentials, we detected a different channel (Fig. 7Ba) with milder inward rectification (Fig. 7Bb) and a single-channel conductance of 70.1 ± 2.3 and 57.1 ± 4.1 pS at −60 and +60 mV, respectively. These values do not match any heterologously expressed channel of the TREK subfamily reported previously, although they are very close to those reported for a novel TREK-like channel found in native magnocellular neurosecretory cells isolated from the supraoptic nucleus (Han et al., 2003).
Figure 7.

Large-conductance TREK-2 potassium channels were recorded in most cell-attached patches. Aa, TREK-2 cell-attached single channel currents recorded at −60, 0, and +60 mV in symmetrical 150 KCl solutions. Ab, Mean (±SEM) current–voltage relationships for 12 TREK-2 channels showing distinctive inward rectification. Ba, TREK-like single channel activity at −60, 0, and +60 mV in symmetrical solutions. Note the flickering and bursting behavior characteristic of TREK channels. Bb, Mean (±SEM) current–voltage relationships for three TREK-like channels showing weaker inward rectification. C, Closed; O, open.
In a second group of experiments, we included riluzole (300 μm) or riluzole (300 μm) plus fluoxetine (100 μm) in the recording electrode; in this group, we found TREK-2 activity in 12 of the 31 patches tested. The solution at the tip of the electrode was free of drugs to provide enough time to obtain a good cell-attached seal before the drugs reached the membrane. Once the gigaseal was obtained (time 0), the open probability was measured every 5 min by analyzing trace fragments of 2 s. The channel open probability progressively increased (from 0.02 ± 0.01 to 0.6 ± 0.05, n = 6; 5 and 25 min after patching, respectively) (supplemental Fig. 4A,C, available at www.jneurosci.org as supplemental material) when using pipettes containing riluzole. The increase in activity was almost negligible when fluoxetine was also included into the electrode (from 0.06 ± 0.05 to 0.12 ± 0.09; n = 6) (supplemental Fig. 4B,C, available at www.jneurosci.org as supplemental material). The difference between both treatments was statistically significant 15 min after the seal was obtained (p < 0.05) and was nearly maximum after 20 min (p < 0.01).
As expected from the pharmacological and single-cell PCR data, channels with properties similar to those reported for TRAAK homodimers were not found. Surprisingly, linear I–V relationships like those reported for heterologously expressed TREK-1 channels (Han et al., 2003; Kang et al., 2006) were also absent in mSCG neurons. These experiments indicated that TREK-2 and TREK-like channels are active at rest, and hence they must contribute to the resting membrane conductance in mSCG neurons.
Resting membrane potential and firing
To further investigate the importance of TREK channels in the behavior of mSCG neurons, we tested the effect of the blocker fluoxetine on the resting membrane potential. Several concentrations of fluoxetine (1, 3, 10, 30, and 100 μm) were applied to resting neurons (I = 0) and found a consistent depolarization when using 100 μm (∼12 mV; from −61.1 ± 3.9 to −48.7 ± 5.3 mV; n = 3; p = 0.015). The depolarization induced by lower concentrations of fluoxetine on the resting potential was not significant (30 μm: p = 0.06, n = 3; 10 μm: p = 0.1, n = 6; 3 μm: p = 0.24, n = 3; 1 μm: p = 0.51, n = 3).
In initial experiments, we tested the effect of fluoxetine (1, 3, 10, and 30 μm) on the amplitude and duration of the action potentials evoked by 1 s depolarizing current injections (from 50 to 350 pA in 50 pA steps). Concentrations >10 μm clearly decreased the amplitude and increased the duration of action potentials by inhibition of the voltage-activated sodium current (Hahn et al., 1999). For this reason, we tested the effect of low concentrations of fluoxetine on the number of action potentials generated by current injections. A characteristic typical of SCG neurons is their strong spike frequency adaptation when depolarized (Romero et al., 2004); however, neither 1 nor 10 μm fluoxetine significantly affected the adaptation of mSCG neurons in terms of the number of action potentials (n = 7) (supplemental Fig. 5B, available at www.jneurosci.org as supplemental material). Notwithstanding, a closer inspection of these seven neurons showed that 10 μm fluoxetine significantly reduced the latency to the first evoked action potential (from 21.6 ± 4.8 to 18.8 ± 4.6 ms; n = 7; p < 0.05; injected current, 150 pA); in the same cells, the effect of 1 μm was not significant (21.4 ± 4.8 ms; p > 0.05) (supplemental Fig. 5A, available at www.jneurosci.org as supplemental material).
Discussion
The resting membrane potential of SCG neurons lies around −60 mV and it is contributed to by several voltage-gated currents and the electrogenicity of the Na–K pump. However, as in many other cell types, a molecularly unidentified apparently voltage-independent (leak) conductance, which is mainly transported by potassium, was proposed to be the main contributor to the background current in such neurons. Here, we have studied a riluzole-activated potassium current carried by the recently discovered K2P channels that may be the molecular basis of the resting leakage current in SCG neurons. Indeed, we provide electrophysiological, pharmacological, molecular, and immunocytochemical data demonstrating that the TREK subfamily of the K2P potassium channels is the molecular correlate of the riluzole-activated current. In addition, for the first time, we demonstrate the functional expression of K2P channels in the autonomic nervous system.
A characteristic of K2P channels is their modulation by many pharmacological agents, but, despite considerable effort, selective and specific modulators are still lacking even for heterologously expressed channels. Accordingly, the few studies on putatively native K2P channels have used multichemical screening to rule out the contribution of other channels and to indirectly define the channel responsible (Talley et al., 2000; Bushell et al., 2002; Enyeart et al., 2002; Han et al., 2002). Single-channel recording has also been combined with pharmacological screening in native neurons (Han et al., 2002, 2003; Kang and Kim, 2006), and using this approach, we show here that the riluzole-activated current is mainly transported through TREK-2 channels in sympathetic neurons.
Nature of the riluzole-activated current in mSCG neurons
The reversal potential of IRIL at different potassium concentrations demonstrated that it is indeed a potassium current, and accordingly, this current was unaffected by sodium, calcium, and cationic channel modulators. Additional evidence indicated that IRIL depends on K2P but not other known potassium channels. Open rectification at physiological but not symmetrical potassium concentrations is a characteristic of several K2P channels, ruling out the participation of voltage-dependent potassium channels. Certainly, IRIL was insensitive to classic voltage-dependent and calcium-activated potassium channel blockers, although the current was reduced by millimolar concentrations of barium, a characteristic shared by several K2P potassium channels.
To ascertain which type of K2P channel is responsible for the IRIL was much more complicated. The activation by riluzole itself pointed to the TREK subfamily since, to our knowledge, this is the only K2P subfamily reported to be clearly activated by this neuroprotective agent. Even though a putative THIK-1 channel naturally expressed by mouse Purkinje neurons is inhibited (but not activated) by riluzole (Bushell et al., 2002). Open rectification and inhibition by barium and not by other classic channel blockers also support this notion. In addition, either application of linoleic acid or mechanical stimulation induced clear outward currents in blockers solutions. Finally, RT-PCR, single-cell RT-PCR, and immunocytochemical data confirmed the expression of mRNA and protein for the three members of the TREK subfamily in mSCG neurons.
Molecular counterpart of IRIL
The coexpression of several different K2P subunits in the same native neuron seems to be the norm (Han et al., 2003; Kang and Kim, 2006). Accordingly, PCR and immunocytochemistry experiments indicated that the three members of the TREK subfamily are expressed in mSCG neurons (TREK-1, TREK-2, and TRAAK). As all of them are activated by riluzole in heterologous systems, we attempted to elucidate the relative contribution of each of them to IRIL in our preparation.
We found enough pharmacological evidence to demonstrate that TRAAK channels, although expressed, are not important contributors to the current activated by riluzole in mSCG neurons. Although varying from cell to cell, the activation of the current by riluzole was transient in most cells tested, as reported for heterologously expressed TREK-1 and TREK-2 channels. This was contrary to the sustained activation reported for TRAAK channels in the same conditions.
Among the 12 functional K2P subunits known, only TREK-1 and TREK-2 channels are activated by zinc, whereas most K2P channels, including TRAAK, are inhibited (TASK-3, TASK-2, TRAAK, TWIK-2, mTRESK) or unaffected (TASK-1, THIK-1, hTRESK) by this metal ion. The strong inhibition of IRIL by fluoxetine also suggests a minor contribution of TRAAK, since it is considerably less sensitive to this drug than TREK-1 (Heurteaux et al., 2006), and TREK-2 inhibition by fluoxetine has also been reported (Kang et al., 2008). TRAAK homodimers have been reported to be strongly blocked by ruthenium red, whereas TREK-1 was practically insensitive (Czirják and Enyedi, 2002); in mSCG neurones IRIL was unaffected by 10 μm RR (supplemental Fig. 2, available at www.jneurosci.org as supplemental material). Finally, intracellular acidification induced by extracellular application of bicarbonate (Maingret et al., 1999b) provoked a dramatic outward current in mSCG neurons that was strongly blocked by fluoxetine. As both TREK-1 and TREK-2 are strongly activated by internal acidification, whereas TRAAK channels are activated by internal alkalinization (Han et al., 2003), this result indicates a poor functional expression of intracellularly alkaline-activated channels when compared with acid-activated channels in mSCG. It is generally accepted that DNP is a strong intracellular acidic stimulus, and hence it should enhance TREK-1 and TREK-2 channels. However, DNP induced an inward instead of the expected outward current and it did not affect the IRIL at all. Interestingly, a similar DNP-induced inward current (by inhibition of a putative background outward current) has been reported in rat carotid body type I cells (Buckler and Vaughan-Jones, 1998), which was associated with a very small fall in pHi (maximum decrease, 0.09; DNP, 250 μm), indicating that acidification was almost negligible and not responsible for the effects of DNP. It is tempting to speculate that, in contrast to COS cells (Duprat et al., 2000), DNP may not acidify SCG neurons.
It is far more complex to discriminate the contribution of TREK-1 and/or TREK-2 to IRIL. Homomers of these two channels have a very similar pharmacological profile, and in mSCG neurons, PCR and immunocytochemistry techniques are uninformative since they demonstrate that both subunits are expressed. The fact that IRIL was unaffected by quinine could indicate a major contribution of TREK-1 as quinine was reported to have no effect on TREK-1 (Fink et al., 1996) and TRAAK channels (Ozaita and Vega-Saenz de Miera, 2002), while strongly reduced putative TREK-2 currents (Kucheryavykh et al., 2009). However, the true effect of quinine and quinidine on TREK channels is rather uncertain as they have been reported to inhibit TREK-1 (Patel et al., 1998; Meadows et al., 2000) and TREK-2 (Lesage et al., 2000b; Kucheryavykh et al., 2009), as well as to have no effect on both these channels (Fink et al., 1996; Bang et al., 2000; Han et al., 2002).
Cell-attached patches unequivocally demonstrated the relatively profuse expression of TREK-2 channels in the membrane of mSCG neurons. In addition, a second less frequent TREK-like channel emerged with single-channel properties intermediate to those reported for TREK-1 and TREK-2. TREK-like channels have a very noisy open state and the bursting behavior characteristic of TREK channels. The conductance of this channel is lower than that of TREK-2 at negative potentials but slightly higher at positive potentials, such that they showed weaker inward rectification. These characteristics and the value of the conductance itself are nearly identical with those reported for a TREK-like channel (termed AA3) in supraoptic neurons (Han et al., 2003). The AA3 channel has been proposed to be a novel member of the TREK subfamily because cotransfection of all combinations of the three identified members of the TREK subfamily failed to produce a channel with the same characteristics. The absence of TRAAK channels in the membrane of mSCG neurons was expected from our general data, although why channels with characteristics similar to those reported for TREK-1 are not evident is unclear.
Physiological role of TREK channels in sympathetic neurons
The isolated riluzole-activated current shown here displayed a strong outward rectification at physiological potassium concentrations. This is consistent with the fact that activation of heterologously expressed TREK-1 (Duprat et al., 2000), TREK-2 (Lesage et al., 2000b), and TRAAK (Fink et al., 1998; Duprat et al., 2000) channels by riluzole mainly takes place in the outward but not the inward direction. In fact, whole-cell, outwardly rectifying I–V relationships with almost no inward current are typical of heterologously expressed TREK-1 (Fink et al., 1996; Maingret et al., 2000a; Kennard et al., 2005), TREK-2 (Bang et al., 2000; Lesage et al., 2000b), and TRAAK (Fink et al., 1998; Lesage et al., 2000a; Meadows et al., 2001) channels in physiological extracellular K+ concentrations. The small true leakage current at the resting membrane potential and the relatively positive resting membrane potential previously reported in SCG cells (Lamas et al., 2002, 2009) are also in agreement with the low open probability of TREK channels at room temperature and negative voltages (Lesage et al., 2000b). As hypothesized for other currents, K2P channels could stabilize rather than settle the resting membrane potential, thereby maintaining the negative resting potential with a very low conductance. This hypothesis is attractive from the energy saving point of view, and it would also allow neurotransmitters and other physical and chemical stimuli, in the case of TREK channels, to exert a fine modulation of this potential. This is also in agreement with the hypothesis that TREK-1 channel overexpression has a modest effect on input resistance; nevertheless, it can still modulate the resting membrane potential in cultured hippocampal neurons (Yang and Jan, 2008).
Although essentially voltage independent, the characteristic open rectification of TREK channels gives them a macroscopic I–V relationship functionally similar to that of voltage-dependent potassium M-channels. Although mechanistically different, at physiological K+ concentrations, TREK currents will exert their influence at voltages similar to those in which the voltage-dependent M-current activates (positive to approximately −60 mV). Hence, they could not only contribute to set the resting membrane potential by stabilization but also to act as a “brake” on action potential firing and to contribute to adaptation in SCG neurons, as comprehensively demonstrated for M-channels (Brown and Constanti, 1980; Lamas, 1998; Lamas et al., 2002). This could explain the strong reduction in spike frequency adaptation induced by barium (which inhibits both KCNQ and TREK channels) in mSCG neurons when compared with other more specific M-current inhibitors (Romero et al., 2004). Our results using low concentrations of fluoxetine suggest that, although adaptation is mainly under M-current control, TREK channels induce a delay in the latency of the first action potential evoked by depolarizing stimuli. To know whether TREK channels have a more important role on the adaptation than that showed here requires the discovery of a TREK-2 selective blocker. Even so, our results are congruent with TREK channels being already open and M-currents opening with a rather slow time constant on depolarization.
Finally, the modulation of the M-current by muscarinic agonists in sympathetic SCG neurons has been thoroughly studied, and it is generally accepted that the depletion of PIP2 (phosphatidylinositol 4,5-bisphosphate) accounts for the M-current inhibition (Suh and Hille, 2002; Winks et al., 2005). Recently, muscarinic inhibition of TREK-2 channels was reported in a heterologous system (Kang et al., 2006), and it is tempting to speculate that the increase in excitability provoked by muscarinic agonists in mSCG neurons may, at least in part, be attributable to this inhibition. Whether, and to what extent, the modulation of K2P channels plays a role on the synaptic performance of the sympathetic ganglia deserves to be investigated.
Footnotes
This work was supported by Spanish Ministry of Science and Innovation (MICINN) Grant BFU2008-02952/BFI (J.A.L.). S.J.R. was supported by Xunta de Galicia Grant PGIDIT-06PXIC310095PN. A.C.-M. and A.R. were supported by MICINN CONSOLIDER-INGENIO Grant CSD2008-00005. We appreciate the valuable advice of Dr. Eva Vigo and the skilful technical assistance of Vanesa Domínguez.
References
- Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- Bang H, Kim Y, Kim D. TREK-2, a new member of the mechanosensitive tandem-pore K+ channel family. J Biol Chem. 2000;275:17412–17419. doi: 10.1074/jbc.M000445200. [DOI] [PubMed] [Google Scholar]
- Brown DA, Constanti A. Intracellular observations on the effects of muscarinic agonists on rat sympathetic neurones. Br J Pharmacol. 1980;70:593–608. doi: 10.1111/j.1476-5381.1980.tb09778.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown DA, Adams PR, Constanti A. Voltage-sensitive K-currents in sympathetic neurons and their modulation by neurotransmitters. J Auton Nerv Syst. 1982;6:23–35. doi: 10.1016/0165-1838(82)90019-4. [DOI] [PubMed] [Google Scholar]
- Buckler KJ, Vaughan-Jones RD. Effects of mitochondrial uncouplers on intracellular calcium, pH and membrane potential in rat carotid body type I cells. J Physiol. 1998;513:819–833. doi: 10.1111/j.1469-7793.1998.819ba.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bushell T, Clarke C, Mathie A, Robertson B. Pharmacological characterization of a non-inactivating outward current observed in mouse cerebellar Purkinje neurones. Br J Pharmacol. 2002;135:705–712. doi: 10.1038/sj.bjp.0704518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarke CE, Veale EL, Green PJ, Meadows HJ, Mathie A. Selective block of the human 2-P domain potassium channel, TASK-3, and the native leak potassium current, IKSO, by zinc. J Physiol. 2004;560:51–62. doi: 10.1113/jphysiol.2004.070292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Czirják G, Enyedi P. Formation of functional heterodimers between the TASK-1 and TASK-3 two-pore domain potassium channel subunits. J Biol Chem. 2002;277:5426–5432. doi: 10.1074/jbc.M107138200. [DOI] [PubMed] [Google Scholar]
- Czirják G, Enyedi P. Zinc and mercuric ions distinguish TRESK from the other two-pore-domain K+ channels. Mol Pharmacol. 2006;69:1024–1032. doi: 10.1124/mol.105.018556. [DOI] [PubMed] [Google Scholar]
- Danthi S, Enyeart JA, Enyeart JJ. Modulation of native TREK-1 and Kv1.4 K+ channels by polyunsaturated fatty acids and lysophospholipids. J Membr Biol. 2003;195:147–164. doi: 10.1007/s00232-003-0616-0. [DOI] [PubMed] [Google Scholar]
- Deng PY, Xiao Z, Yang C, Rojanathammanee L, Grisanti L, Watt J, Geiger JD, Liu R, Porter JE, Lei S. GABAB receptor activation inhibits neuronal excitability and spatial learning in the entorhinal cortex by activating TREK-2 K+ channels. Neuron. 2009;63:230–243. doi: 10.1016/j.neuron.2009.06.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duprat F, Lesage F, Patel AJ, Fink M, Romey G, Lazdunski M. The neuroprotective agent riluzole activates the two P domain K+ channels TREK-1 and TRAAK. Mol Pharmacol. 2000;57:906–912. [PubMed] [Google Scholar]
- Enyeart JJ, Xu L, Danthi S, Enyeart JA. An ACTH- and ATP-regulated background K+ channel in adrenocortical cells is TREK-1. J Biol Chem. 2002;277:49186–49199. doi: 10.1074/jbc.M207233200. [DOI] [PubMed] [Google Scholar]
- Fink M, Duprat F, Lesage F, Reyes R, Romey G, Heurteaux C, Lazdunski M. Cloning, functional expression and brain localization of a novel unconventional outward rectifier K+ channel. EMBO J. 1996;15:6854–6862. [PMC free article] [PubMed] [Google Scholar]
- Fink M, Lesage F, Duprat F, Heurteaux C, Reyes R, Fosset M, Lazdunski M. A neuronal two P domain K+ channel stimulated by arachidonic acid and polyunsaturated fatty acids. EMBO J. 1998;17:3297–3308. doi: 10.1093/emboj/17.12.3297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grunnet M, Jespersen T, Angelo K, Frøkjaer-Jensen C, Klaerke DA, Olesen SP, Jensen BS. Pharmacological modulation of SK3 channels. Neuropharmacology. 2001;40:879–887. doi: 10.1016/s0028-3908(01)00028-4. [DOI] [PubMed] [Google Scholar]
- Hadley JK, Passmore GM, Tatulian L, Al-Qatari M, Ye F, Wickenden AD, Brown DA. Stoichiometry of expressed KCNQ2/KCNQ3 potassium channels and subunit composition of native ganglionic M channels deduced from block by tetraethylammonium. J Neurosci. 2003;23:5012–5019. doi: 10.1523/JNEUROSCI.23-12-05012.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hahn SJ, Choi JS, Rhie DJ, Oh CS, Jo YH, Kim MS. Inhibition by fluoxetine of voltage-activated ion channels in rat PC12 cells. Eur J Pharmacol. 1999;367:113–118. doi: 10.1016/s0014-2999(98)00955-8. [DOI] [PubMed] [Google Scholar]
- Han J, Truell J, Gnatenco C, Kim D. Characterization of four types of background potassium channels in rat cerebellar granule neurons. J Physiol. 2002;542:431–444. doi: 10.1113/jphysiol.2002.017590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han J, Gnatenco C, Sladek CD, Kim D. Background and tandem-pore potassium channels in magnocellular neurosecretory cells of the rat supraoptic nucleus. J Physiol. 2003;546:625–639. doi: 10.1113/jphysiol.2002.032094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hebert T, Drapeau P, Pradier L, Dunn RJ. Block of the rat brain IIA sodium channel alpha subunit by the neuroprotective drug riluzole. Mol Pharmacol. 1994;45:1055–1060. [PubMed] [Google Scholar]
- Heurteaux C, Lucas G, Guy N, El Yacoubi M, Thümmler S, Peng XD, Noble F, Blondeau N, Widmann C, Borsotto M, Gobbi G, Vaugeois JM, Debonnel G, Lazdunski M. Deletion of the background potassium channel TREK-1 results in a depression-resistant phenotype. Nat Neurosci. 2006;9:1134–1141. doi: 10.1038/nn1749. [DOI] [PubMed] [Google Scholar]
- Huang CS, Song JH, Nagata K, Yeh JZ, Narahashi T. Effects of the neuroprotective agent riluzole on the high voltage-activated calcium channels of rat dorsal root ganglion neurons. J Pharmacol Exp Ther. 1997;282:1280–1290. [PubMed] [Google Scholar]
- Imai S, Suzuki T, Sato K, Tokimasa T. Effects of quinine on three different types of potassium currents in bullfrog sympathetic neurons. Neurosci Lett. 1999;275:121–124. doi: 10.1016/s0304-3940(99)00775-2. [DOI] [PubMed] [Google Scholar]
- Kang D, Kim D. TREK-2 (K2P10.1) and TRESK (K2P18.1) are major background K+ channels in dorsal root ganglion neurons. Am J Physiol Cell Physiol. 2006;291:C138–C146. doi: 10.1152/ajpcell.00629.2005. [DOI] [PubMed] [Google Scholar]
- Kang D, Choe C, Kim D. Functional expression of TREK-2 in insulin-secreting MIN6 cells. Biochem Biophys Res Commun. 2004;323:323–331. doi: 10.1016/j.bbrc.2004.08.089. [DOI] [PubMed] [Google Scholar]
- Kang D, Han J, Kim D. Mechanism of inhibition of TREK-2 (K2P10.1) by the Gq-coupled M3 muscarinic receptor. Am J Physiol Cell Physiol. 2006;291:C649–C656. doi: 10.1152/ajpcell.00047.2006. [DOI] [PubMed] [Google Scholar]
- Kang D, Kim GT, Kim EJ, La JH, Lee JS, Lee ES, Park JY, Hong SG, Han J. Lamotrigine inhibits TRESK regulated by G-protein coupled receptor agonists. Biochem Biophys Res Commun. 2008;367:609–615. doi: 10.1016/j.bbrc.2008.01.008. [DOI] [PubMed] [Google Scholar]
- Kennard LE, Chumbley JR, Ranatunga KM, Armstrong SJ, Veale EL, Mathie A. Inhibition of the human two-pore domain potassium channel, TREK-1, by fluoxetine and its metabolite norfluoxetine. Br J Pharmacol. 2005;144:821–829. doi: 10.1038/sj.bjp.0706068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JS, Park JY, Kang HW, Lee EJ, Bang H, Lee JH. Zinc activates TREK-2 potassium channel activity. J Pharmacol Exp Ther. 2005;314:618–625. doi: 10.1124/jpet.105.084418. [DOI] [PubMed] [Google Scholar]
- Kucheryavykh LY, Kucheryavykh YV, Inyushin M, Shuba YM, Sanabria P, Cubano LA, Skatchkov SN, Eaton MJ. Ischemia increases TREK-2 channel expression in astrocytes: relevance to glutamate clearance. Open Neurosci J. 2009;3:40–47. doi: 10.2174/1874082000903010040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamas JA. A hyperpolarization-activated cation current (Ih) contributes to resting membrane potential in rat superior cervical sympathetic neurones. Pflugers Arch. 1998;436:429–435. doi: 10.1007/s004240050653. [DOI] [PubMed] [Google Scholar]
- Lamas JA, Reboreda A, Codesido V. Ionic basis of the resting membrane potential in cultured rat sympathetic neurons. Neuroreport. 2002;13:585–591. doi: 10.1097/00001756-200204160-00010. [DOI] [PubMed] [Google Scholar]
- Lamas JA, Romero M, Reboreda A, Sánchez E, Ribeiro SJ. A riluzole- and valproate-sensitive persistent sodium current contributes to the resting membrane potential and increases the excitability of sympathetic neurones. Pflugers Arch. 2009;458:589–599. doi: 10.1007/s00424-009-0648-0. [DOI] [PubMed] [Google Scholar]
- Lesage F, Guillemare E, Fink M, Duprat F, Lazdunski M, Romey G, Barhanin J. TWIK-1, a ubiquitous human weakly inward rectifying K+ channel with a novel structure. EMBO J. 1996;15:1004–1011. [PMC free article] [PubMed] [Google Scholar]
- Lesage F, Maingret F, Lazdunski M. Cloning and expression of human TRAAK, a polyunsaturated fatty acids-activated and mechano-sensitive K+ channel. FEBS Lett. 2000a;471:137–140. doi: 10.1016/s0014-5793(00)01388-0. [DOI] [PubMed] [Google Scholar]
- Lesage F, Terrenoire C, Romey G, Lazdunski M. Human TREK2, a 2P domain mechano-sensitive K+ channel with multiple regulations by polyunsaturated fatty acids, lysophospholipids, and Gs, Gi, and Gq protein-coupled receptors. J Biol Chem. 2000b;275:28398–28405. doi: 10.1074/jbc.M002822200. [DOI] [PubMed] [Google Scholar]
- Li ZB, Zhang HX, Li LL, Wang XL. Enhanced expressions of arachidonic acid-sensitive tandem-pore domain potassium channels in rat experimental acute cerebral ischemia. Biochem Biophys Res Commun. 2005;327:1163–1169. doi: 10.1016/j.bbrc.2004.12.124. [DOI] [PubMed] [Google Scholar]
- Lotshaw DP. Biophysical, pharmacological, and functional characteristics of cloned and native mammalian two-pore domain K+ channels. Cell Biochem Biophys. 2007;47:209–256. doi: 10.1007/s12013-007-0007-8. [DOI] [PubMed] [Google Scholar]
- Magistretti J, Mantegazza M, de Curtis M, Wanke E. Modalities of distortion of physiological voltage signals by patch-clamp amplifiers: a modeling study. Biophys J. 1998;74:831–842. doi: 10.1016/S0006-3495(98)74007-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maingret F, Fosset M, Lesage F, Lazdunski M, Honoré E. TRAAK is a mammalian neuronal mechano-gated K+ channel. J Biol Chem. 1999a;274:1381–1387. doi: 10.1074/jbc.274.3.1381. [DOI] [PubMed] [Google Scholar]
- Maingret F, Patel AJ, Lesage F, Lazdunski M, Honoré E. Mechano- or acid stimulation, two interactive modes of activation of the TREK-1 potassium channel. J Biol Chem. 1999b;274:26691–26696. doi: 10.1074/jbc.274.38.26691. [DOI] [PubMed] [Google Scholar]
- Maingret F, Patel AJ, Lesage F, Lazdunski M, Honoré E. Lysophospholipids open the two-pore domain mechano-gated K+ channels TREK-1 and TRAAK. J Biol Chem. 2000a;275:10128–10133. doi: 10.1074/jbc.275.14.10128. [DOI] [PubMed] [Google Scholar]
- Maingret F, Lauritzen I, Patel AJ, Heurteaux C, Reyes R, Lesage F, Lazdunski M, Honoré E. TREK-1 is a heat-activated background K+ channel. EMBO J. 2000b;19:2483–2491. doi: 10.1093/emboj/19.11.2483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martínez-Pinna J, Lamas JA, Gallego R. Calcium current components in intact and dissociated adult mouse sympathetic neurons. Brain Res. 2002;951:227–236. doi: 10.1016/s0006-8993(02)03165-7. [DOI] [PubMed] [Google Scholar]
- Mathie A, Veale EL. Therapeutic potential of neuronal two-pore domain potassium-channel modulators. Curr Opin Investig Drugs. 2007;8:555–562. [PubMed] [Google Scholar]
- Meadows HJ, Benham CD, Cairns W, Gloger I, Jennings C, Medhurst AD, Murdock P, Chapman CG. Cloning, localisation and functional expression of the human orthologue of the TREK-1 potassium channel. Pflugers Arch. 2000;439:714–722. doi: 10.1007/s004249900235. [DOI] [PubMed] [Google Scholar]
- Meadows HJ, Chapman CG, Duckworth DM, Kelsell RE, Murdock PR, Nasir S, Rennie G, Randall AD. The neuroprotective agent sipatrigine (BW619C89) potently inhibits the human tandem pore-domain K+ channels TREK-1 and TRAAK. Brain Res. 2001;892:94–101. doi: 10.1016/s0006-8993(00)03239-x. [DOI] [PubMed] [Google Scholar]
- Neher E. Correction for liquid junction potentials in patch clamp experiments. Methods Enzymol. 1992;207:123–131. doi: 10.1016/0076-6879(92)07008-c. [DOI] [PubMed] [Google Scholar]
- Ozaita A, Vega-Saenz de Miera E. Cloning of two transcripts, HKT4.1a and HKT4.1b, from the human two-pore K+ channel gene KCNK4: chromosomal localization, tissue distribution and functional expression. Mol Brain Res. 2002;102:18–27. doi: 10.1016/s0169-328x(02)00157-2. [DOI] [PubMed] [Google Scholar]
- Patel AJ, Honoré E. Properties and modulation of mammalian 2P domain K+ channels. Trends Neurosci. 2001;24:339–346. doi: 10.1016/s0166-2236(00)01810-5. [DOI] [PubMed] [Google Scholar]
- Patel AJ, Honoré E, Maingret F, Lesage F, Fink M, Duprat F, Lazdunski M. A mammalian two pore domain mechano-gated S-like K+ channel. EMBO J. 1998;17:4283–4290. doi: 10.1093/emboj/17.15.4283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rae J, Cooper K, Gates P, Watsky M. Low access resistance perforated patch recordings using amphotericin B. J Neurosci Methods. 1991;37:15–26. doi: 10.1016/0165-0270(91)90017-t. [DOI] [PubMed] [Google Scholar]
- Reboreda A, Sánchez E, Romero M, Lamas JA. Intrinsic spontaneous activity and subthreshold oscillations in neurones of the rat dorsal column nuclei in culture. J Physiol. 2003;551:191–205. doi: 10.1113/jphysiol.2003.039917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reyes R, Duprat F, Lesage F, Fink M, Salinas M, Farman N, Lazdunski M. Cloning and expression of a novel pH-sensitive two pore domain K+ channel from human kidney. J Biol Chem. 1998;273:30863–30869. doi: 10.1074/jbc.273.47.30863. [DOI] [PubMed] [Google Scholar]
- Reyes R, Lauritzen I, Lesage F, Ettaiche M, Fosset M, Lazdunski M. Immunolocalization of the arachidonic acid and mechanosensitive baseline TRAAK potassium channel in the nervous system. Neuroscience. 2000;95:893–901. doi: 10.1016/s0306-4522(99)00484-4. [DOI] [PubMed] [Google Scholar]
- Robbins J, Trouslard J, Marsh SJ, Brown DA. Kinetic and pharmacological properties of the M-current in rodent neuroblastoma × glioma hybrid cells. J Physiol. 1992;451:159–185. doi: 10.1113/jphysiol.1992.sp019159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romero M, Reboreda A, Sánchez E, Lamas JA. Newly developed blockers of the M-current do not reduce spike frequency adaptation in cultured mouse sympathetic neurons. Eur J Neurosci. 2004;19:2693–2702. doi: 10.1111/j.1460-9568.2004.03363.x. [DOI] [PubMed] [Google Scholar]
- Rozen S, Skaletsky HJ. Primer3 on the WWW for general users and for biologist programmers. In: Krawetz S, Misener S, editors. Bioinformatics methods and protocols: methods in molecular biology. Totowa, NJ: Humana; 2000. pp. 365–386. [DOI] [PubMed] [Google Scholar]
- Suh BC, Hille B. Recovery from muscarinic modulation of M current channels requires phosphatidylinositol 4,5-bisphosphate synthesis. Neuron. 2002;35:507–520. doi: 10.1016/s0896-6273(02)00790-0. [DOI] [PubMed] [Google Scholar]
- Talley EM, Lei Q, Sirois JE, Bayliss DA. TASK-1, a two-pore domain K+ channel, is modulated by multiple neurotransmitters in motoneurons. Neuron. 2000;25:399–410. doi: 10.1016/s0896-6273(00)80903-4. [DOI] [PubMed] [Google Scholar]
- Talley EM, Solorzano G, Lei Q, Kim D, Bayliss DA. CNS distribution of members of the two-pore-domain (KCNK) potassium channel family. J Neurosci. 2001;21:7491–7505. doi: 10.1523/JNEUROSCI.21-19-07491.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang YJ, Lin MW, Lin AA, Wu SN. Riluzole-induced block of voltage-gated Na+ current and activation of BKCa channels in cultured differentiated human skeletal muscle cells. Life Sci. 2008;82:11–20. doi: 10.1016/j.lfs.2007.10.015. [DOI] [PubMed] [Google Scholar]
- Winks JS, Hughes S, Filippov AK, Tatulian L, Abogadie FC, Brown DA, Marsh SJ. Relationship between membrane phosphatidylinositol 4,5-bisphosphate and receptor-mediated inhibition of native neuronal M channels. J Neurosci. 2005;25:3400–3413. doi: 10.1523/JNEUROSCI.3231-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto Y, Hatakeyama T, Taniguchi K. Immunohistochemical colocalization of TREK-1, TREK-2 and TRAAK with TRP channels in the trigeminal ganglion cells. Neurosci Lett. 2009;454:129–133. doi: 10.1016/j.neulet.2009.02.069. [DOI] [PubMed] [Google Scholar]
- Yang SB, Jan LY. Thrilling moment of an inhibitory channel. Neuron. 2008;58:823–824. doi: 10.1016/j.neuron.2008.06.003. [DOI] [PubMed] [Google Scholar]
- Yeung SY, Millar JA, Mathie A. Inhibition of neuronal Kv potassium currents by the antidepressant drug, fluoxetine. Br J Pharmacol. 1999;128:1609–1615. doi: 10.1038/sj.bjp.0702955. [DOI] [PMC free article] [PubMed] [Google Scholar]