

Abstract
We hypothesize that Parkinson’s disease (PD) pathogenesis can be divided into three temporal phases. During the first phase, “triggers”, such as viral infections or environmental toxins, spark the disease process in the brain and/or peripheral tissues. Triggers alone, however, may be insufficient, requiring “facilitators” like peripheral inflammation for PD pathology to develop. Once the disease manifests, “aggravators” spur further neurodegeneration and exacerbate symptoms. Aggravators are proposed to include impaired autophagy and cell-to-cell propagation of α-synuclein pathology. We believe clinical trials need to consider these three phases and target potential therapies at the appropriate stage of the disease process in order to be effective.
Keywords: Parkinson’s disease, clinical trials, inflammation, genetics, α-synuclein
A new conceptual model for understanding Parkinson’s disease
A crucial unmet need for patients with Parkinson’s disease (PD) is a disease-modifying therapy. This is particularly urgent considering that the number of people with PD is set to double over the next 20 years, with over 14 million PD cases expected worldwide by 2040 [1]. We believe that an inadequate understanding of the relative relationship and temporal sequence of factors involved in PD pathogenesis might be hampering advances of new therapies. Many past attempts to develop therapies were designed with the aim of slowing disease progression by targeting single molecular pathways in PD pathogenesis; these attempts also typically viewed PD as a uniform disease, and subsequently treated all patients with the same therapeutic regimen. The PD research field is now questioning the accuracy of considering PD as a single entity. Instead it has been suggested that PD is in fact an umbrella term given to a large a cluster of disorders encompassing a range of clinical, epidemiological, molecular, neuropathological, and genetic subtypes [2, 3]. We would argue that if research programs culminate in clinical trials that continue to target a single pathogenic mechanism in groups of patients defined by the broad clinical diagnosis of PD, they are unlikely to yield effective disease-modifying treatment. We agree with the view that PD patients are a heterogenous population and, at best, patients can be grouped into different clusters with certain shared characteristics. In this Opinion piece, we hypothesize that in a given patient, or patient cluster, the prevailing pathological mechanism will change during the different phases of PD. Based on this hypothesis, we propose a new conceptual model for PD pathogenesis. From the treatment perspective, the proposed model implies that medical interventions need to be disease-stage specific and personalized to maximize the chances of modifying the course of the disease.
Specifically, our model, first, divides the factors that contribute to neurodegeneration into three categories – triggers, facilitators, and aggravators. Second, we argue that each of these factors play distinct roles at different stages of the disease. Triggers encompass factors that enable the initiation of the disease process. These are most prominent during the period of “prodromal PD”. However, triggers alone are, in most cases, insufficient for PD to develop, as they require the presence of “facilitators” for the disease to spread to, and significantly impact, the central nervous system. We propose that this applies not only to idiopathic PD, but also to familial forms of PD, as no known genetic form has complete penetrance, and the onset of PD symptoms in the familial forms are known to occur over a wide age-span. We acknowledge that familial cases have a greater contribution of genetic factors as a facilitator event, compared to idiopathic PD. Facilitators can also be factors that sensitize cells to the toxic effects of triggers and promote a neurodegenerative cascade that may involve the formation and/or seeding of pathogenic α-synuclein. It would be necessary for these facilitators to be present during the initial stages of the disease process, when α-synuclein pathology transitions to the midbrain after initially being confined to a small number of cells in peripheral locations in the nervous system (e.g. enteric nerves) or central ones (e.g. olfactory system) . Finally, we suggest a third category of factors that contribute to PD pathogenesis. These are “aggravators”, which directly promote the neurodegenerative process by exacerbating the pathology and promoting the spread of the disease beyond the basal ganglia.
The phases where triggers, facilitators, and aggravators act do not necessarily have distinct boundaries; rather, they represent a continuum along the disease course. The proposed terminology, however, can be helpful as a conceptual framework for articulating more clearly when – across a process that typically lasts several decades – different factors act, and what are their key roles. We think our hypothesized framework will facilitate the development of selective therapies that target the right mechanism at the right time. We will use examples to illustrate how key biological mediators of PD pathophysiology can be divided into the three categories of triggers, facilitators and aggravators (Figure 1), and we will examine when is it during the disease process that they are most likely to play a critical role in PD pathogenesis.
Figure 1:
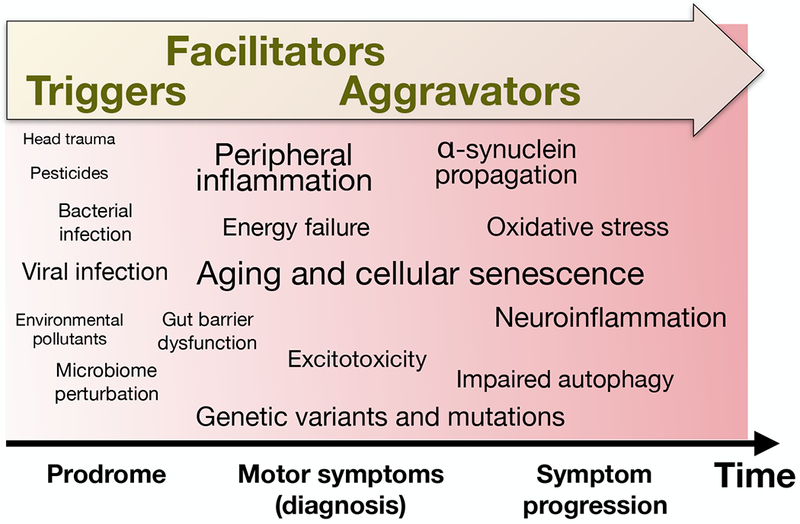
Schematic of the ‘triggers, facilitators and aggravators’ conceptual framework for redefining PD pathogenesis. We propose that triggers initiate disease pathogenesis during prodromal PD, and facilitators spread pathology to significantly impact the central nervous system. This results in the onset of PD motor symptoms, at which point aggravators continue to exacerbate cell loss and neuropathology, causing worsening symptoms and enabling the emergence of new symptoms. In the schematic, various suspected or validated factors have been positioned along the horizontal axis based approximately on the phase we consider they have the largest effect, and using font size which is roughly proportional to their estimated relative importance in PD pathogenesis.
Triggers
With the exception of relatively rare genetic forms of PD, triggers of PD are largely unknown. Yet a number of suspected triggers have been inferred from endogenous functions of candidate PD risk genes, and from findings in experimental animals. In idiopathic PD, the triggers are thought to act decades before the loss of nigrostriatal dopaminergic signaling has reached levels necessary for the onset of clinical motor symptoms [4, 5]. We propose that triggers often act transiently, with the triggering event lasting a few weeks or months and occurring relatively early in the life of individuals that will develop PD. The transient nature of such triggers makes them particularly difficult to detect, and a thorough review of patient medical histories may be necessary to identify possible triggers. Several triggers of PD have already been proposed and include pathogens, environmental toxins as well as head trauma (Table 1). Potential outcomes of triggering events may include long term gut dysfunction [6], chronic inflammation in critical tissues [7], or the direct misfolding of α-synuclein into pathogenic conformers (e.g. resulting from exposure to heavy metals) [8]. Two anatomical sites have been suggested to be of particular relevance as places of origin for PD pathology. In 2003, Braak and colleagues first proposed that the olfactory epithelium and intestines [9] might be where PD begins. This was based on the histological observation of α-synuclein aggregates in these sites in very early stage PD, and even in cases presumed to be prodromal PD [9]. Gastrointestinal dysfunction and hyposmia are core features of PD, often occurring years to decades before the emergence of motor symptoms [10]. Their involvement in the early symptoms of PD, together with the findings of α-synuclein aggregates in these tissues [9], indicate that the gastrointestinal tract and olfactory system might be trigger sites in PD pathogenesis. Below, we will use pathogens and microbiome changes, as well as pesticides and MPTP as illustrative examples of potential triggers.
Table 1:
Examples of triggers proposed to spark PD pathogenic process.
| Triggers | Examples | References |
|---|---|---|
| Gastrointestinal microbiota perturbations | Increased Lactobacillus | [21] |
| Reduced Prevotella | [21] | |
| Pathogens | Hepatitis B and C | [75] |
| Norovirus | [11] | |
| M. paratuberculosis | [53] | |
| Air pollution | Particulate matter | [76] |
| Carbon monoxide | [76] | |
| Pesticides | Rotenone | [23] |
| Paraquat | [23] | |
| Heavy metals | Iron | [77] |
| Copper | [77] | |
| Manganese | [77] | |
| Head trauma | Traumatic brain injury | [78] |
Infections and gastrointestinal microbiome
A growing number of bacterial and viral pathogens have been associated with a higher risk for developing PD. Even common gastrointestinal infections, such as norovirus, induce upregulation of α-synuclein in the gastrointestinal tract, potentially in an effort to rally an immune response [11]. Aggregated α-synuclein is present in the enteric nervous system in PD patients [12], but it has also more recently been shown to occur in healthy people, indicating that α-synuclein aggregates in the gut are not specific to PD [13, 14]. We propose that aggregation of α-synuclein in the gut is not necessarily abnormal per se, and is most often part of an appropriate, self-contained inflammatory response following infection. We speculate that what differentiates those who develop PD from those who do not might be the presence of facilitators (see below) that amplify the α-synuclein aggregates and allow for their spread along the vagus nerve, eventually causing seeding in the brainstem and Lewy pathology in the central nervous system.
Several studies have reported changes in gut microbiota in PD, including increases in Lactobacillus, Akkermansia, and Bifidobacterium, and decreases in Prevotella, Blautia and Faecalibacterium [15]. The majority of these studies suffer from the confounder that anti-parkinsonian medication might change the gut microbiome [16]. However, unmedicated patients with rapid eye movement (REM) sleep behavior disorder, who exhibit a high conversion rate to PD, display similar changes in the gut microbiota [17]. This supports the idea that microbiota alterations occur in prodromal and early PD. Moreover, α-synuclein-overexpressing mice exposed to the microbiota from PD patients display exacerbated motor dysfunction compared to those that received microbiota from healthy donors, supporting perturbation of gut microbiota as a potential PD trigger [18]. While all the aforementioned studies have focused on the gut microbiome, it is possible that analogous changes in the nasal microbiome trigger α-synuclein aggregation in the olfactory bulb. Though studies of the nasal microbiome in PD are yet to yield any convincing differences [17, 19], even if present, one should note that such suggested changes might only be present transiently during the prodrome. We speculate that a localized effect of altered microbiota causing inflammation leads to α-synuclein aggregation in enteric nerves or the olfactory system, with propagation of pathology to more central locations. Thus, gut dysbiosis and/or small intestinal bacterial overgrowth, along with increased intestinal permeability, can cause systemic inflammation, which could spread to the CNS through systemic circulation and several neural pathways [20]. The downstream effects of gastrointestinal dysbiosis could be mediated by endogenous inflammatory factors or by metabolites directly produced by the gut bacteria [21]. Metabolites secreted from gut bacteria that have been proposed to impact glial biology and neuroinflammation include short chain fatty acids and tryptophan metabolites [18]. Microbiome metabolites have been shown to induce microglia activation and motor deficits in α-synuclein transgenic mice [18]. Such metabolites may modify brain activities by regulating peripheral immune cell function and/or through activation of the vagal nerve either directly (e.g. evoke nerve responses) or indirectly (e.g. through enteroendocrine cell signaling) [22].
Pesticides
Epidemiological studies have linked pesticide exposure with increased PD risk. A recent meta-analysis reported exposure to several common pesticides is associated with ≥ 50% increased risk for developing PD [23]. Furthermore, it is plausible that exposure to more than one pesticide confers an even greater risk compared to exposure to any individual chemical [24]. In principle, pesticides could trigger PD pathogenesis via several possible mechanisms, including inhibiting mitochondrial complex I, inducing oxidative stress, stimulating inflammation, and causing α-synuclein fibrillization [25]. Human dopaminergic neurons carrying the SNCA-A53T mutation develop impaired anterograde mitochondrial transport when exposed to regulated and approved pesticides [26]. Exposure at these low levels caused impairment only in neurons carrying the PD mutation, supporting our concept that triggers (e.g. pesticides) need to be accompanied by a facilitator (e.g. PD risk mutation) to render neurons vulnerable to disease.
MPTP
Intravenous injection of the neurotoxin MPTP (e.g., through use of the synthetic analgesic drug MPPP when accidentally contaminated with MPTP) causes rapid death of nigral dopamine neurons and, consequently, PD symptoms [27]. However, the very acute nature of MPTP-induced parkinsonism limits its relevance to understanding familial or idiopathic PD. Furthermore, in the few individuals who injected MPTP intravenously and developed PD symptoms, the dose of the trigger (MPTP) was presumably so large that it overrode the need for a facilitator. Notably, not all people who injected MPTP developed parkinsonism and over thirty years ago, four cases exposed to MPTP intravenously were found to have reduced flurodopa-uptake on positron emission scans, but no clinical signs of PD [28]. This suggests that the trigger in this case (MPTP) does not always cause significant nigral neurodegeneration, possibly due to the absence of appropriate facilitators or to a lower dose being injected. While MPTP-induced parkinsonism might be illustrative of the interrelationship between a trigger and its facilitators, the relevance to aggravators in idiopathic PD is probably limited because MPTP toxicity does not induce pathological hallmarks of PD-like Lewy bodies [29].
Facilitators
We define facilitators as factors that help triggers access the nervous system or spread the pathology to more central parts of the nervous system. We propose that the facilitator role in disease pathogenesis takes place concomitantly with the triggering event, or after it, usually during the prodromal or asymptomatic phase of PD. Notably, facilitators and aggravators can either be permanent, e.g. in the form of a genetic predisposition, or transient, such as in the case of temporary gastrointestinal inflammation. PD development may depend on the simultaneous or proximal occurrence of trigger and facilitator, however, it is also conceivable that a trigger precedes the actions of a facilitator by several years. For example, pathogen-induced (trigger) inflammation in the gut resulting in localized α-synuclein accumulation and aggregation in enteric nerves [11] may not promote PD until years later, after an age-related decline in cellular energy homeostasis and lysosomal function (facilitators) has set in, which in turn could enable the propagation of α-synuclein aggregates to the brainstem.
Systemic inflammation
Inflammation in PD is present both in the periphery (systemic inflammation) and in the central nervous system [30]. Genome-wide studies further revealed several polymorphisms in inflammatory genes associated with PD [31–33]. At the molecular level, inflammatory mediators are known to promote α-synuclein misfolding and aggregation [34]. Importantly, there are also emerging, compelling links between inflammation and the spread of α-synuclein pathology, which could explain how systemic inflammation facilitates the propagation of α-synuclein pathology from peripheral tissues to central nervous system [35]. Notably, colon biopsies from PD patients exhibit increased mRNA levels for pro-inflammatory cytokines and glial markers, indicative of enteric inflammation [36]. Furthermore, patients with inflammatory bowel disease have a higher incidence of PD [37–40], which is substantially reduced by early exposure to anti-inflammatory anti-tumor necrosis factor therapy [37]. Systemic inflammation in PD is reflected by elevated serum IL-6, TNF-α and C-reactive protein, which are associated with severity or faster decline of motor symptoms [41]. In addition, there is an increased risk for PD among people who develop type 2 diabetes, especially at younger ages [42]. Since type 2 diabetes is coupled to systemic inflammation, this might suggest a facilitating role for inflammatory changes in those who happen to be exposed to disease triggers. Furthermore, general immunosuppressive and anti-inflammatory treatments reduce the risk of developing PD [43]. Taken together, systemic inflammation itself, and several chronic conditions that feature increased inflammation, might be considered facilitators of PD pathogenesis.
Mitochondrial dysfunction
Strong evidence from preclinical and clinical studies supports a role for mitochondrial dysfunction in PD pathogenesis. PD patients have reduced mitochondrial complex I activity in the substantia nigra, skeletal muscles, and platelets [44]. Furthermore, mutations in genes known to cause familial PD have been directly or indirectly implicated in causing mitochondrial dysfunction. For example, PINK1 and Parkin actively inhibit mitochondrial-derived vesicle formation and mitochondrial antigen presentation; thus mutation of these genes in autosomal recessive Parkinson’s disease may result in excessive antigen presentation that provokes an autoimmune response [45]. In addition, numerous studies demonstrate that mitochondrial dysfunction results in the chronic production of reactive oxygen species, leading to oxidative stress, and promoting both misfolding of α-synuclein and directly causing neurodegeneration [46].
Genetic facilitators
Inherited PD accounts for only 5–10% of all cases, with mutations in at least 15 genes identified as causes of relatively rare monogenic forms of the disease [47]. Leucine-rich repeat kinase 2 (LRRK2) and glucocerebrosidase (GBA) have an incomplete penetrance with 28–74% (G2019S LRRK2 mutation) [48] and 2–30% [49, 50] of patients developing PD, respectively. This suggests that these mutations, and others with incomplete penetrance might act as facilitators, requiring the added burden of a trigger for the disease to start.
LRRK2 variants are the leading heritable form of PD [47]. Mutations in LRRK2 associated with PD elevate LRRK2 kinase activity and decrease GTPase activity. Macrophages, monocytes, and neutrophils express high levels of LRRK2, implying that it contributes to defense against pathogens [51]. Therefore, people with LRRK2 variants might be more susceptible to viral or bacterial infections, including ones that have been postulated to trigger PD. A recent study, for instance, demonstrated that LRRK2 activity regulates Mycobacterium tuberculosis replication by affecting phagosome maturation in macrophages [52], and LRRK2 mutations may trigger PD pathogenesis via a dysregulation of responses to such bacteria [53]. This may be particularly relevant in the gastrointestinal tract, as there is growing evidence for a critical role of intestinal inflammation in PD pathogenesis [54]. Additionally, LRRK2 has been identified as a major susceptibility gene for Crohn’s disease, which is associated with deep transmural inflammation [55]. Collectively, LRRK2 might regulate inflammatory responses in the intestine and those perturbations in the pathway may have multiple downstream deleterious effects that facilitate the onset of PD [56]. The targets of the LRRK2 kinase, and cell model experiments, suggest that mutations can also alter vesicular trafficking, mitochondrial function, protein synthesis, cytoskeletal dynamics [57], and autophagy–functions that, when impaired, may facilitate PD pathogenesis [56].
Aggravators
We define aggravators as factors that directly contribute to the progression of existing symptoms and the onset of late-stage symptoms such as cognitive decline and falls. Aggravators tend to come into play when PD has already manifested clinically, and might affect the rate of progression, accelerating neuronal dysfunction and/or furthering the spread of α-synuclein pathology.
Impaired autophagy
Autophagy is the main cellular mechanism employed for disposal of misfolded proteins, damaged organelles, and abnormal or dysfunctional cellular components. Genome-wide studies in PD have identified several polymorphisms that are adjacent to genes involved in lysosomal function and autophagy [32, 58–62]. For example, multiple mutations in different loci of the GBA gene, that encodes a lysosomal enzyme, have been identified [63]. Failure of the lysosomal-autophagy pathway may aggravate progression of neuropathology by elevating levels of extracellular α-synuclein aggregates, and promoting cell-to-cell spread of α-synuclein pathology [64]. Consequently, improving lysosomal function and enhancing autophagy are considered viable therapeutic strategies in attempts to slow PD progression.
Neuroinflammation
Neuroinflammation has been closely linked with neurodegenerative diseases and is often characterized by the activation of glial cells and the overexpression of pro-inflammatory mediators. Glial cells are beneficial under physiological conditions as they clear cellular debris, remove toxic substances and release neurotrophic factors [65]. However, in PD, activated microglia are believed to have a deleterious effect on neurons and emit harmful levels of cytokines. This induces a vicious, self-propagating chronic neuroinflammatory state that contributes to the neurodegenerative process [66]. Activated microglia have also been shown to induce astrocytes to release neurotoxic mediators, leading to further neuronal damage [67]. The role of neuroinflammation in PD is supported by the presence of activated microglia in the substantia nigra of PD patients [68] as well as several pre-clinical studies in PD animal models [66]. Additionally, neuroinflammation is a predictive indicator in the development of non-motor symptoms and cognitive decline [69, 70], further illuminating its role as an aggravator and potential therapeutic target.
Concluding remarks: Implementing the conceptual model into PD research and therapeutic development
Our new conceptual model of triggers, facilitators, and aggravators in the development of PD pathology, builds on previously proposed gene-environment and multi-hit hypotheses for PD etiology [71, 72]. The vast array of different combinations of factors in these three categories can explain some of the clinical variability observed in patients with PD, including their symptomology, rate of progression, and response to treatments. We believe this provides further support for the emerging school of thought that considers PD not as a single uniform disease, but rather as a group of related diseases, with differing disease courses depending on – to use the terminology we offer here – the specific combination of triggers, facilitators, and aggravators present in each patient.
Perhaps the most important consequence of our proposed conceptual model is that pharmacological therapies aimed at slowing or arresting PD progression should not only be given to patients enriched for the appropriate target [73], but also be administered in the relevant phase of the disease. For example, therapies targeting foreign pathogens are unlikely to affect disease progression once PD pathogenesis is underway. Currently, there are a variety of trials targeting LRRK2 and GBA pathways; our new definition for PD pathogenesis raises the question of how effective these drugs will be once the disease is clinically evident. If LRRK2 and GBA mutations play a role as facilitators, inhibiting LRRK2 kinase activity or enhancing GBA activity might be most beneficial in prodromal or early stages of PD when facilitators are most active. This highlights the importance of continuing efforts to identify individuals at risk and/or with prodromal PD to better target factors that fall within the trigger and facilitator categories [74]. We should stress that it is also possible that certain factors act both as facilitators and aggravators. For example, increased LRRK2 kinase activity could facilitate the development of disease (e.g. by altering immune cell function after pathogen exposure), as well as aggravate progression of the disease (e.g. by disrupting neuronal autophagy). Ultimately, combination therapies addressing multiple targets, e.g. neuroinflammation and impaired autophagy, might be the best approach; however, they need to be applied during the appropriate temporal phase at which the factor is active.
A major outstanding challenge is determining the most practical way to assess the validity of the “triggers, facilitators, and aggravators” model (see Outstanding Questions). The first hurdle is developing biomarkers that accurately define sub-groups within the PD patient population so that personalized approaches for triggers, facilitators and aggravators can have successful disease-modifying outcomes. As previously described [3], changing the approach for identifying novel PD biomarkers from ‘clinical phenotype -> biomarkers’ to ‘biomarkers -> clinical phenotype validation’ has the potential to reveal biomarkers in an unbiased fashion, ideally from a large cohort of ageing individuals with and without different neurodegenerative diseases. We hope that our conceptual model, stratifying the PD pathogenic process, not only across the population but also in the temporal domain, will promote discussion among PD researchers about how to optimize the development of future disease-modifying therapies as well as identify challenges ahead that need to be addressed for this to occur. Apart from increasing the chances of successful outcomes in the development of new therapies, these discussions could lead to reconsideration of experimental therapies that previously failed – possibly because they were tested in a non-specific population of patients or during a phase when the target mechanism was not at its peak – and that may prove efficacious when applied with greater precision and specificity.
Acknowledgements
L.B. is supported by the grant 14939 from the M. J. Fox Foundation. V.L. is supported by grants from the Alzheimer’s Society of Canada (16 15), the Scottish Rite Charitable Foundation of Canada (15110), and the Department of Defense (PD170089), and a Gibby & Friends vs. Parky award. P.B. reports relevant grants from National Institutes of Health (R01DC016519–01 and 5R21NS093993–02), Department of Defense (W81XWH-17–1-0534), M. J. Fox Foundation and Cure Parkinson’s Trust. P.B. has received commercial support as a consultant from Renovo Neural, Inc., Roche, Living Cell Technologies, Teva Inc, Lundbeck A/S, AbbVie, Neuroderm, Fujifilm-Cellular Dynamics, ClearView Healthcare, FCB Health, IOS Press Partners and Capital Technologies, Inc. P.B. has received commercial support for grants/research from Roche, Renovo and Teva/Lundbeck. P.B. has ownership interests in Acousort AB and is on the steering committee of the NILO-PD trial.
References
- 1.Dorsey ER and Bloem BR (2018) The Parkinson Pandemic—A Call to Action. JAMA neurology 75 (1), 9–10. [DOI] [PubMed] [Google Scholar]
- 2.Espay AJ et al. (2017) Precision medicine for disease modification in Parkinson disease. Nature reviews neurology 13 (2), 119. [DOI] [PubMed] [Google Scholar]
- 3.Espay AJ et al. (2017) Biomarker-driven phenotyping in Parkinson’s disease: A translational missing link in disease-modifying clinical trials. Movement Disorders 32 (3), 319–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ross GW et al. (2004) Parkinsonian signs and substantia nigra neuron density in decendents elders without PD. Annals of neurology 56 (4), 532–539. [DOI] [PubMed] [Google Scholar]
- 5.Marsden CD (1990) Parkinson’s disease. Lancet 335 (8695), 948–52. [DOI] [PubMed] [Google Scholar]
- 6.Barreau F and Hugot J (2014) Intestinal barrier dysfunction triggered by invasive bacteria. Current opinion in microbiology 17, 91–98. [DOI] [PubMed] [Google Scholar]
- 7.da Fonseca DM et al. (2015) Microbiota-dependent sequelae of acute infection compromise tissue-specific immunity. Cell 163 (2), 354–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Uversky VN et al. (2001) Metal-triggered structural transformations, aggregation, and fibrillation of human α-synuclein a possible molecular link between parkinson′ s disease and heavy metal exposure. Journal of Biological Chemistry 276 (47), 44284–44296. [DOI] [PubMed] [Google Scholar]
- 9.Braak H et al. (2003) Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiology of aging 24 (2), 197–211. [DOI] [PubMed] [Google Scholar]
- 10.Schapira AH et al. (2017) Non-motor features of Parkinson disease. Nature reviews neuroscience 18 (7), 435–450. [DOI] [PubMed] [Google Scholar]
- 11.Stolzenberg E et al. (2017) A role for neuronal alpha-synuclein in gastrointestinal immunity. Journal of innate immunity 9 (5), 456–463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Barrenschee M et al. (2017) Distinct pattern of enteric phospho-alpha-synuclein aggregates and gene expression profiles in patients with Parkinson’s disease. Acta neuropathologica communications 5 (1), 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chung SJ et al. (2016) Alpha-synuclein in gastric and colonic mucosa in Parkinson’s disease: Limited role as a biomarker. Movement disorders 31 (2), 241–249. [DOI] [PubMed] [Google Scholar]
- 14.Killinger BA et al. (2018) The vermiform appendix contributes to the development of Parkinson’s disease. Science translational medicine In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Scheperjans F (2018) The prodromal microbiome. Movement disorders 33 (1), 5–7. [DOI] [PubMed] [Google Scholar]
- 16.Bedarf JR et al. (2017) Functional implications of microbial and viral gut metagenome changes in early stage L-DOPA-naïve Parkinson’s disease patients. Genome Medicine 9 (1), 39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Heintz-Buschart A et al. (2018) The nasal and gut microbiome in Parkinson’s disease and idiopathic rapid eye movement sleep behavior disorder. Movement disorders 33 (1), 88–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sampson TR et al. (2016) Gut microbiota regulate motor deficits and neuroinflammation in a model of Parkinson’s disease. Cell 167 (6), 1469–1480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pereira PA et al. (2017) Oral and nasal microbiota in Parkinson’s disease. Parkinsonism & related disorders 38, 61–67. [DOI] [PubMed] [Google Scholar]
- 20.Duerkop BA et al. (2009) Immune responses to the microbiota at the intestinal mucosal surface. Immunity 31 (3), 368–376. [DOI] [PubMed] [Google Scholar]
- 21.Sun M-F and Shen Y-Q (2018) Dysbiosis of gut microbiota and microbial metabolites in Parkinson’s Disease. Ageing research reviews 45, 53–61. [DOI] [PubMed] [Google Scholar]
- 22.Bonaz B et al. (2018) The Vagus Nerve at the Interface of the Microbiota-Gut-Brain Axis. Frontiers in neuroscience 12, 49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gunnarsson L-G and Bodin L (2017) Parkinson’s disease and occupational exposures: a systematic literature review and meta-analyses. Scandinavian journal of work, environment & health 43 (3), 197–209. [DOI] [PubMed] [Google Scholar]
- 24.Wang A et al. (2011) Parkinson’s disease risk from ambient exposure to pesticides. European journal of epidemiology 26 (7), 547–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Baltazar MT et al. (2014) Pesticides exposure as etiological factors of Parkinson’s disease and other neurodegenerative diseases—a mechanistic approach. Toxicology letters 230 (2), 85–103. [DOI] [PubMed] [Google Scholar]
- 26.Stykel MG et al. (2018) Nitration of microtubules blocks axonal mitochondrial transport in a human pluripotent stem cell model of Parkinson’s disease. The FASEB Journal 32. [DOI] [PubMed] [Google Scholar]
- 27.Langston JW et al. (1983) Chronic Parkinsonism in humans due to a product of meperidine-analog synthesis. Science 219 (4587), 979–980. [DOI] [PubMed] [Google Scholar]
- 28.Calne D et al. (1985) Positron emission tomography after MPTP: observations relating to the cause of Parkinson’s disease. Nature 317 (6034), 246. [DOI] [PubMed] [Google Scholar]
- 29.Masilamoni GJ and Smith Y (2018) Chronic MPTP administration regimen in monkeys: A model of dopaminergic and non-dopaminergic cell loss in Parkinson’s disease. Journal of Neural Transmission 125 (3), 337–363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Macchi B et al. (2015) Inflammatory and cell death pathways in brain and peripheral blood in Parkinson’s disease. CNS & neurological disorders-drug targets (formerly Current drug targets-CNS & neurological disorders) 14 (3), 313–324. [DOI] [PubMed] [Google Scholar]
- 31.Hamza TH et al. (2010) Common genetic variation in the HLA region is associated with late-onset sporadic Parkinson’s disease. Nature genetics 42 (9), 781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chang D et al. (2017) A meta-analysis of genome-wide association studies identifies 17 new Parkinson’s disease risk loci. Nature genetics 49 (10), 1511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pierce S and Coetzee GA (2017) Parkinson’s disease-associated genetic variation is linked to quantitative expression of inflammatory genes. PloS one 12 (4), e0175882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gao H-M et al. (2008) Neuroinflammation and oxidation/nitration of α-synuclein linked to dopaminergic neurodegeneration. Journal of Neuroscience 28 (30), 7687–7698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tyson T et al. (2016) Sorting out release, uptake and processing of alpha-synuclein during prion-like spread of pathology. Journal of neurochemistry 139 (S1), 275–289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Devos D et al. (2013) Colonic inflammation in Parkinson’s disease. Neurobiology of disease 50, 42–48. [DOI] [PubMed] [Google Scholar]
- 37.Peter I et al. (2018) Anti–tumor necrosis factor therapy and incidence of Parkinson disease among patients with inflammatory bowel disease. JAMA neurology, Advanced online publication- DOI: 10.1001/jamaneurol.2018.0605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Villumsen M et al. (2018) Inflammatory bowel disease increases the risk of Parkinson’s disease: a Danish nationwide cohort study 1977–2014. Gut, Advanced online publication- DOI: 10.1136/gutjnl-2018-316537 [DOI] [PubMed] [Google Scholar]
- 39.Lin J-C et al. (2016) Association between Parkinson’s disease and inflammatory bowel disease: a nationwide Taiwanese retrospective cohort study. Inflammatory bowel diseases 22 (5), 1049–1055. [DOI] [PubMed] [Google Scholar]
- 40.Weimers P et al. (2018) Inflammatory Bowel Disease and Parkinson’s Disease: A Nationwide Swedish Cohort Study. Inflammatory bowel diseases, Advanced online publication- DOI: 10.1093/ibd/izy190. [DOI] [PubMed] [Google Scholar]
- 41.Williams-Gray CH et al. (2016) Serum immune markers and disease progression in an incident Parkinson’s disease cohort (ICICLE‐PD). Movement disorders 31 (7), 995–1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.De Pablo-Fernandez E et al. (2018) Association between diabetes and subsequent Parkinson disease: A record-linkage cohort study. Neurology, Advanced online publication- DOI: 10.1212/WNL.0000000000005771 [DOI] [PubMed] [Google Scholar]
- 43.Racette BA et al. (2018) Immunosuppressants and risk of Parkinson disease. Annals of clinical and translational neurology 5 (7), 870–875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bose A and Beal MF (2016) Mitochondrial dysfunction in Parkinson’s disease. Journal of neurochemistry 139 (S1), 216–231. [DOI] [PubMed] [Google Scholar]
- 45.Matheoud D et al. (2016) Parkinson’s disease-related proteins PINK1 and Parkin repress mitochondrial antigen presentation. Cell 166 (2), 314–327. [DOI] [PubMed] [Google Scholar]
- 46.Rocha EM et al. (2018) Alpha-synuclein: pathology, mitochondrial dysfunction and neuroinflammation in Parkinson’s disease. Neurobiology of disease 109 (Part B), 249–257. [DOI] [PubMed] [Google Scholar]
- 47.Billingsley K et al. (2018) Genetic risk factors in Parkinson’s disease. Cell and tissue research 373 (1), 9–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Healy DG et al. (2008) Phenotype, genotype, and worldwide genetic penetrance of LRRK2-associated Parkinson’s disease: a case-control study. The lancet neurology 7 (7), 583–590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Anheim M et al. (2012) Penetrance of Parkinson disease in glucocerebrosidase gene mutation carriers. Neurology 78 (6), 417–420. [DOI] [PubMed] [Google Scholar]
- 50.Rana HQ et al. (2012) Age-specific Parkinson disease risk in GBA mutation carriers: information for genetic counseling. Genetics in medicine 15 (2), 146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Moehle MS et al. (2012) LRRK2 inhibition attenuates microglial inflammatory responses. Journal of Neuroscience 32 (5), 1602–1611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Härtlova A et al. (2018) LRRK2 is a negative regulator of Mycobacterium tuberculosis phagosome maturation in macrophages. The EMBO journal 37 (12), e98694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Dow CT (2014) M. paratuberculosis and Parkinson’s disease–Is this a trigger. Medical hypotheses 83 (6), 709–712. [DOI] [PubMed] [Google Scholar]
- 54.Houser MC and Tansey MG (2017) The gut-brain axis: is intestinal inflammation a silent driver of Parkinson’s disease pathogenesis? npj Parkinson’s Disease 3 (1), 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hui KY et al. (2018) Functional variants in the LRRK2 gene confer shared effects on risk for Crohn’s disease and Parkinson’s disease. Science translational medicine 10 (423), eaai7795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Alessi DR and Sammler E (2018) LRRK2 kinase in Parkinson’s disease. Science 360 (6384), 36–37. [DOI] [PubMed] [Google Scholar]
- 57.Esteves AR and Cardoso SM (2017) LRRK2 at the crossroad between autophagy and microtubule trafficking: insights into Parkinson’s disease. The Neuroscientist 23 (1), 16–26. [DOI] [PubMed] [Google Scholar]
- 58.Nalls MA et al. (2014) Large-scale meta-analysis of genome-wide association data identifies six new risk loci for Parkinson’s disease. Nature genetics 46 (9), 989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hardy J (2010) Genetic analysis of pathways to Parkinson disease. Neuron 68 (2), 201–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Coetzee GA and Pierce S (2017) The Five Dimensions of Parkinson’s Disease Genetic Risk. Journal of Parkinson’s disease 8 (1), 13–15. [DOI] [PubMed] [Google Scholar]
- 61.Simon-Sanchez J et al. (2009) Genome-wide association study reveals genetic risk underlying Parkinson’s disease. Nature genetics 41 (12), 1308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Satake W et al. (2009) Genome-wide association study identifies common variants at four loci as genetic risk factors for Parkinson’s disease. Nature genetics 41 (12), 1303. [DOI] [PubMed] [Google Scholar]
- 63.Sidransky E et al. (2009) Multicenter analysis of glucocerebrosidase mutations in Parkinson’s disease. New England journal of medicine 361 (17), 1651–1661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Lopes da Fonseca T et al. (2015) The interplay between alpha-synuclein clearance and spreading. Biomolecules 5 (2), 435–471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Gao H-M and Hong J-S (2008) Why neurodegenerative diseases are progressive: uncontrolled inflammation drives disease progression. Trends in immunology 29 (8), 357–365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Joers V et al. (2017) Microglial phenotypes in Parkinson’s disease and animal models of the disease. Progress in neurobiology 155, 57–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Liddelow SA et al. (2017) Neurotoxic reactive astrocytes are induced by activated microglia. Nature 541 (7638), 481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.McGeer P et al. (1988) Reactive microglia are positive for HLA‐DR in the substantia nigra of Parkinson’s and Alzheimer’s disease brains. Neurology 38 (8), 1285–1285. [DOI] [PubMed] [Google Scholar]
- 69.Maetzler W et al. (2007) Osteopontin is elevated in Parkinson’s disease and its absence leads to reduced neurodegeneration in the MPTP model. Neurobiology of disease 25 (3), 473–482. [DOI] [PubMed] [Google Scholar]
- 70.Menza M et al. (2010) The role of inflammatory cytokines in cognition and other non-motor symptoms of Parkinson’s disease. Psychosomatics 51 (6), 474–479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Cannon JR and Greenamyre JT (2013) Gene–environment interactions in Parkinson’s disease: specific evidence in humans and mammalian models. Neurobiology of disease 57, 38–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Sulzer D (2007) Multiple hit hypotheses for dopamine neuron loss in Parkinson’s disease. Trends in neurosciences 30 (5), 244–250. [DOI] [PubMed] [Google Scholar]
- 73.Lang AE and Espay AJ (2018) Disease modification in Parkinson’s Disease: Current approaches, challenges, and future considerations. Movement disorders 33 (5), 660–677. [DOI] [PubMed] [Google Scholar]
- 74.Postuma RB and Berg D (2016) Advances in markers of prodromal Parkinson disease. Nature reviews neurology 12 (11), 622. [DOI] [PubMed] [Google Scholar]
- 75.Kim JM et al. (2016) Association between hepatitis C virus infection and Parkinson’s disease. Movement disorders 31 (10), 1584–1585. [DOI] [PubMed] [Google Scholar]
- 76.Jayaraj RL et al. (2017) Outdoor Ambient Air Pollution and Neurodegenerative Diseases: the Neuroinflammation Hypothesis. Current environmental health reports 4 (2), 166–179. [DOI] [PubMed] [Google Scholar]
- 77.Dusek P et al. (2015) The neurotoxicity of iron, copper and manganese in Parkinson’s and Wilson’s diseases. Journal of trace elements in medicine and biology 31, 193–203. [DOI] [PubMed] [Google Scholar]
- 78.Camacho‐Soto A et al. (2017) Traumatic brain injury in the prodromal period of Parkinson’s disease: A large epidemiological study using medicare data. Annals of neurology 82 (5), 744–754. [DOI] [PMC free article] [PubMed] [Google Scholar]