

Abstract
The PAX3 gene encodes a member of the PAX family of transcription factors that is characterized by a highly conserved paired box motif. The PAX3 protein is a transcription factor consisting of an N-terminal DNA binding domain (containing a paired box and homeodomain) and a C-terminal transcriptional activation domain. This protein is expressed during development of skeletal muscle, central nervous system and neural crest derivatives, and regulates expression of target genes that impact on proliferation, survival, differentiation and motility in these lineages. Germline mutations of the murine Pax3 and human PAX3 genes cause deficiencies in these developmental lineages and result in the Splotch phenotype and Waardenburg syndrome, respectively. Somatic genetic rearrangements that juxtapose the PAX3 DNA binding domain to the transcriptional activation domain of other transcription factors deregulate PAX3 function and contribute to the pathogenesis of the soft tissue cancers alveolar rhabdomyosarcoma and biphenotypic sinonasal sarcoma. The wild-type PAX3 protein is also expressed in other cancers related to developmental lineages that normally express this protein and exerts phenotypic effects related to its normal developmental role.
Keywords: Transcription factor, Paired box, Homeodomain, Alternative splicing, Target gene, Skeletal muscle development, Central nervous system development, Neural crest, Melanocyte, Waardenburg syndrome, Splotch, Fusion oncoprotein, Rhabdomyosarcoma, Soft tissue sarcoma, Melanoma
1. Introduction
The PAX3 gene (paired box gene 3; also known as WS1, WS3, CDHS and HUP2) encodes a member of the paired box or PAX family of transcription factors. This family is characterized by a highly conserved paired DNA binding domain first identified in Drosophila segmentation genes (Tremblay and Gruss, 1994). There are nine human members (PAX1-PAX9) and nine homologous murine members (Pax1-Pax9) that are grouped into four subfamilies. PAX3 and PAX7 (along with Pax3 and Pax7) comprise a subfamily termed group III, based on a high degree of sequence homology and similar genomic and functional organization (Stuart et al., 1994). The nine human PAX genes are located at non-contiguous loci throughout the human genome, and most are located on separate chromosomes.
1.1. PAX3 gene location and structure
The human PAX3 gene is present in the q36.1 region on the long arm of chromosome 2 (Table 1). The PAX3 gene consists of 10 exons (Barber et al., 1999) within a 100 kb region, spanning from location 222,199,888 bp to 222,298,996 bp (Ensembl assembly release GRCh38.p10) (Fig. 1). Exons 2, 3 and 4 encode the conserved paired domain, whereas exons 5 and 6 encode the conserved octapeptide and homeodomain, and exons 7 and 8 encode the transactivation domain (Baldwin et al., 1995).
Table 1.
Human PAX3 and mouse Pax3 - chromosome location, transcripts and proteins
| Species | Ensembl Gene | Chrom | Transcript |
Protein |
||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Name | Ensembl ID | Pre-spliced (nt) | Exons | Spliced (nt) | UniProt | Length (aa) | Mass (Da) | |||
| Human | ENSG00000135903 | 2q36.1 | P23760 | |||||||
| PAX3-201 (PAX3b) | ENST00000258387.5 | 5,230 | 5 | 958 | 206 | 22,743 | ||||
| PAX3-202 (PAX3h) | ENST00000336840.10 | 98,209 | 9 | 2038 | 407 | 45,217 | ||||
| PAX3-203 (PAX3g) | ENST00000344493.8 | 99,109 | 8 | 3109 | 403 | 44,822 | ||||
| PAX3-204 (PAX3c) | ENST00000350526.8 | 98,862 | 8 | 3610 | 479 | 52,968 | ||||
| PAX3-205 (PAX3e) | ENST00000392069.6 | 99,094 | 10 | 3170 | 505 | 55,975 | ||||
| PAX3-206 (PAX3d) | ENST00000392070.6 | 98,011 | 9 | 2258 | 484 | 53,464 | ||||
| PAX3-207 (PAX3i) | ENST00000409551.7 | 97,538 | 9 | 1782 | 483 | 53,336 | ||||
| PAX3-208 (PAX3a) | ENST00000409828.7 | 5,117 | 4 | 1254 | 215 | 25,136 | ||||
| Mouse | ENSMUSG00000004872 | 1 | P24610 & | Q8BRF1 | ||||||
| Pax3-201 (PAX3b) | ENSMUST00000004994.15 | 95,572 | 9 | 3078 | 484 | 53,445 | ||||
| Pax3-202 (PAX3a) | ENSMUST00000087086.6 | 95,865 | 8 | 3863 | 479 | 52,948 | ||||
Figure 1. Schematic representation of PAX3 gene, mRNA and protein organization.
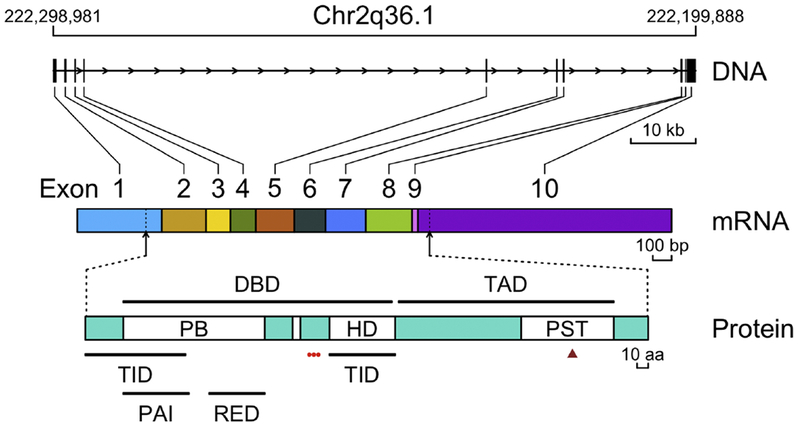
The PAX3 gene structure is depicted based on transcript PAX3-205. The exons of PAX3 are named using Arabic numerals, and the horizontal arrows indicate the direction of transcription. The locations of start and stop codons are indicated by the dashed lines and vertical arrows. Representative sizes are shown by the thin bracketed horizontal line segments beneath the gene, mRNA and protein diagrams. Conserved domains are shown as open boxes within the protein diagram, and functional domains are shown as thick horizontal lines above and below the protein diagram. The red dots and brown triangle beneath the protein diagram corresponds to phosphorylation and monoubiquitination sites, respectively. Abbreviations: PB, paired box domain; HD, homeodomain; PST, proline-, serine- and threonine-rich region; PAI, N-terminal subdomain; RED, C-terminal subdomain; DBD, DNA binding domain; TAD, transcription activation domain; TID, transcription inhibition domain.
1.2. PAX3 mRNA structure and splicing
As a result of alternative processing and splicing, at least ten distinct PAX3 isoforms have been detected at the mRNA level (Table 1, Fig. 2). The longest isoform is PAX3e, which comprises 10 exons (Barber et al., 1999) and encodes for a 505 amino acid protein. The consensus GT donor at the 5’ end of intron 9 is not found in species other than human, and thus the longest isoforms produced by other species, such as mouse, correspond to PAX3c and PAX3d (Barber et al., 1999; Parker et al., 2004). These latter two isoforms consist of 8 or 9 exons, respectively, and are formed by events that suppress the exon 8 to exon 9 splice or the exon 9 to exon 10 splice and thereby result in extension of the transcripts into intron 8 or intron 9, respectively. The three long isoforms (PAX3c, PAX3d, and PAX3e) contain all known characterized functional domains and differ at the extreme C-terminal end. PAX3c and PAX3d appear to be the predominant isoforms expressed in melanoma (Matsuzaki et al., 2005), and PAX3d is expressed at higher levels than PAX3c in a series of cancer cell lines (Parker et al., 2004). In functional studies, PAX3c and PAX3d similarly stimulated endpoints such as cell growth in melanocytes whereas PAX3e inhibited these activities (Wang et al., 2006). Furthermore, a comparison of PAX3c and PAX3d in assays of transcriptional function revealed that both isoforms have stimulatory activity, with PAX3d having greater activity than PAX3c (Barber et al., 1999; Barr et al., 1999).
Figure 2. Exons organization in PAX3 mRNA isoforms.
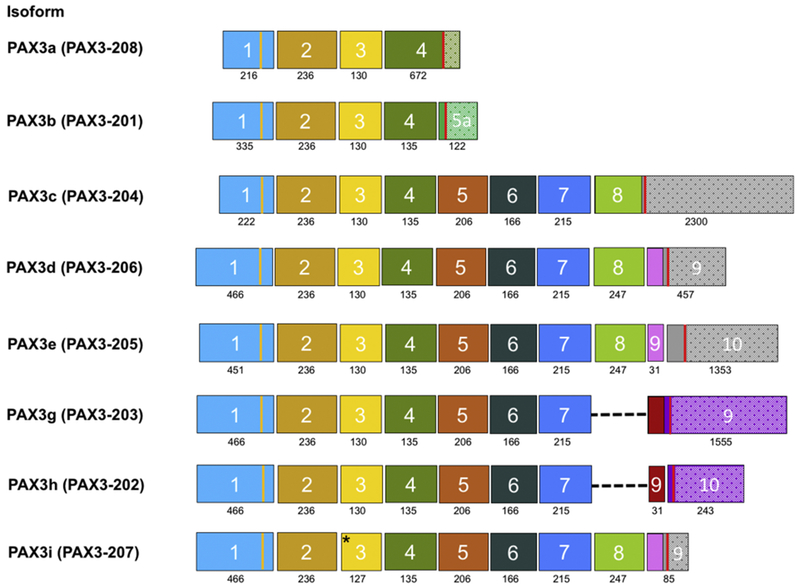
The exons are represented with colored boxes. For each transcript, the exon number is indicated inside the box, and the exon size (in base pairs) is showed below the box. The vertical yellow line in exon 1 indicates the start codon, whereas the vertical red line in the last exon indicates the stop codon. The dashed line indicates regions where exon 8 is spliced out, and the out of frame exons that follow are shown in a different color. For each variant, the 3’ UTR in the last exon is represented by stippling. The asterisk in exon 3 of the PAX3i (PAX3-207) isoform indicates the alternative splice that removes three base pairs.
Several shorter PAX3 isoforms have also been detected (Fig. 2). The PAX3g and PAX3h isoforms include exon 9 or exons 9 and 10, respectively, but differ from the PAX3e or PAX3d isoforms due to a splicing event that joins exon 7 to exon 9 and thereby skips exon 8. In addition to removing exon 8, which encodes a portion of the transactivation domain, the translational reading frame is altered in the transition from exon 7 to exon 9, and thus novel protein sequence is introduced at the C-terminal end of these isoforms. A dominant negative function for Pax3g is suggested by the absence of transcriptional activity and the ability to inhibit activity of the full-length protein (Pritchard et al., 2003). Though PAX3g may inhibit several biological endpoints, such as growth and migration of melanocytes, PAX3h appears to stimulate these endpoints (Wang et al., 2006). The two shortest isoforms, PAX3a and PAX3b, consist of 4 or 5 exons, respectively, and are formed by alternative splicing events within intron 4. These splicing events result in extension of the mRNA into intron 4 or splicing to a novel cryptic exon within intron 4. These two alternative PAX3 transcripts lack the 3’ exons encoding the homeodomain and the transactivation domain, and thus form truncated proteins, which functionally demonstrate either no activity or only inhibition of biological endpoints such as melanocyte growth and migration (Wang et al., 2006).
Studies of human PAX3 and murine Pax3 transcripts identified a frequent alternative splicing event that either includes or excludes the codon CAG as the 3’ splice acceptor at the 5’ end of exon 3 (Vogan et al., 1996). This alternative splicing event results in the presence or absence of a glutamine residue within the paired box motif. The two isoforms (Q+ and Q-) resulting from this alternative splicing are co-expressed at all embryonic stages at which Pax3 is expressed. This alternative splice is also found in several tumors in which PAX3 is expressed, including in the PAX3-FOXO1 fusion transcripts expressed in alveolar rhabdomyosarcoma. The level of the Q+ isoform is approximately two-fold higher than that of the Q− isoform, and there is no significant variation in this ratio in various developmental time points and tumor types. In assays of transcriptional function with several PAX3 DNA binding sites, the Q− isoform has similar or greater DNA binding and transactivation function than the corresponding Q+ isoform (Du et al., 2005). Though most studies of this splicing event did not establish which PAX3 isoforms contain this alternative splice, limited sequencing studies of full-length human cDNAs identified this splicing event as a variant of the PAX3d isoform, and this spliced form has been separately termed the PAX3i isoform. However, more comprehensive full-length sequencing studies are needed to determine whether this alternative splicing event occurs in other PAX3 isoforms.
2. PAX3 Protein
2.1. PAX3 functional domains
The PAX3 protein is a transcription factor characterized by an N-terminal DNA binding domain and a C-terminal transactivation domain (Fig. 1). The DNA binding domain consists of a paired box (PB), octapeptide and homeodomain (HD), and the transactivation domain contains a proline-, serine- and threonine (PST)-rich region.
The PB consists of a stretch of 128 amino acids that is well-conserved in evolution (Czerny et al., 1993; Bopp et al., 1989). The PB is formed by N-terminal (PAI) and C terminal (Kim et al.) subdomains (PAI+RED=PAIRED) (Fig. 1), each consisting of a three-helix fold that includes a helix-turn-helix (HTH) motif, with the C-terminal helix of each HTH making base-specific contacts in the major groove of DNA. The PB recognizes highly related DNA sequences (TCACGC/G, with minor variability seen for each PAX orthologue) (Apuzzo and Gros, 2007).
The HD is typically 60 amino acids long and contains three alpha-helices. PAX3 and three other PAX members (PAX4, PAX6, and PAX7) possess this DNA binding domain, while PAX2, PAX5 and PAX8 contain a rudimentary domain consisting of the first alpha-helix (Tremblay and Gruss, 1994). Most HDs recognize sequences containing a TAAT core motif. HDs found in PAX proteins cooperatively dimerize on palindromic sites of the type 5’-TAAT (N)2-3 ATTA-3’, either as a homodimer or as a heterodimer with another homeodomain (Wilson et al., 1993; Buckingham and Relaix, 2007). Cooperative interactions are present between the HD and PB domains, and are responsible for the functional interdependence of these two domains in DNA binding (Underhill and Gros, 1997; Apuzzo and Gros, 2007).
The C-terminus of PAX3 contains a 78 amino acid long PST-rich region reminiscent of transcriptional activation domains of other transcription factors, such as OCT-2 and CTF-1 (Tanaka and Herr, 1990; Chalepakis et al., 1994b). This C-terminal PAX3 region confers transcriptional activation in reporter assays. Though the C-terminal PAX7 region also has transactivating activity, there are subtle functional differences between these PAX3 and PAX7 regions (Bennicelli et al., 1999). These C-terminal transactivation domains appear to be potently inhibited by transcriptional repression domains located in both the HD and the N-terminus (including the first half of the PB) of PAX3 and PAX7.
2.2. DNA binding properties of PAX3
Early studies demonstrated that PAX3 binds in vitro to the e5 sequence present upstream of the Drosophila even–skipped gene. This sequence contains PB and HD binding sites that include GTTCC and ATTA motifs, respectively (Chalepakis et al., 1994a). Efficient binding of PAX3 to the e5 sequence requires the presence of both PB and HD binding sites. Furthermore, an intervening region of 2-10 bp between the PB and HD binding sites is essential for efficient PAX3 binding (Chalepakis et al., 1994c).
Cyclic amplification and selection of targets (CASTing) methodology revealed that optimal consensus DNA sequences recognized by the PAX3 PB domain include TCGTCAC(G/A)C(T/C/A)(T/C)(C/A/T)A and CGTTCACG(G/C)TT (Chalepakis and Gruss, 1995; Epstein et al., 1996). These sequences are similar to consensus DNA-binding sequences for the PAX2 and PAX6 PB (Epstein et al., 1994a), and the closely related Drosophila paired PB. A search for optimal binding sequences for the combined PB and HD domains of PAX3 identified ATTA and GTNNN motifs in the majority of isolated binding sites, and the sequence ATTA-(N)n-GTTAT in 20% of the binding sites (Phelan and Loeken, 1998). Comparison of PAX3 and PAX7 binding sites suggested that these proteins bind identical DNA motifs. However, while PAX7 can activate target gene expression from combined PB/HD or HD alone, PAX3 is not effective in inducing transcription from genes with only HD motifs (Soleimani et al., 2012).
2.3. PAX3 interacting proteins
The role of PAX3 as a transcriptional regulator is clarified by the identification of other proteins that interact with PAX3 and modulate its transcriptional activity. First, PAX3 is capable of forming homodimers (PAX3/PAX3) and heterodimers (PAX3/PAX7) that can influence DNA binding to a palindromic HD binding site (P2-TAATCAATTA) (Schafer et al., 1994). Furthermore, PAX3 can bind to other transcription factors such as SOX10, ETS1, GLI2, and ZIC2, thereby resulting in synergistic activation of downstream genes (Lang and Epstein, 2003; Himeda et al., 2013; Kubic et al., 2015a). In addition, the Hippo pathway effectors TAZ and YAP65 bind and function as coactivators of PAX3 to regulate expression of PAX3 target genes, such as MITF (Murakami et al., 2006; Manderfield et al., 2014). To extend these findings to chromatin structure, PAX3 also interacts with PAX3/7BP, an essential adaptor linking PAX3 and PAX7 with the H3K4 methyltransferase complex, which activates target gene expression and proliferation in muscle precursor cells (Diao et al., 2012).
PAX3 can also function as a transcriptional inhibitory factor by binding co-repressors. Putative co-repressors interacting with PAX3 and inhibiting PAX3-dependent transcriptional activation include DAXX, RB1, HIRA, Calmyrin (EF-hand calcium-binding protein), KAP1 and Grg4 (Groucho co-repressor) (Magnaghi et al., 1998; Wiggan et al., 1998; Hollenbach et al., 1999; Hollenbach et al., 2002; Lang et al., 2005; Hsieh et al., 2006). DAXX, HIRA and RB1 interact with the PAX3 HD, while Calmyrin and KAP1 interact with the PAX3 PB. DAXX and Calmyrin interactions with PAX3 appear to also involve the octapeptide motif. These co-repressors may alter chromatin configuration at a subset of PAX3 target genes (e.g., HIRA), inhibit the ability of PAX3 to bind to its DNA recognition sequences (e.g., Calmyrin), compete with heterochromatin protein 1 for binding with PAX3 on target promoters (e.g., KAP1) and modulate PAX3 transcription activity through direct interaction (e.g., DAXX).
Pax3 may also exert other functions through protein-protein interactions independent of its role as a transcription factor. In particular, Pax3 associates with the p53 and Mdm2 proteins, and stimulates Mdm2-mediated ubiquitination and degradation of the p53 protein (Wang et al., 2011). These effects can be generated by the isolated Pax3 PB or HD, and do not require the Pax3 transactivation domain.
2.4. PAX3 target genes
PAX3 binding sites are located in a variety of genomic locations. Some are present in or near target genes, such as the 5’ proximal promoter (e.g., MET and MITF), the first intron (e.g., CXCR4 and NRCAM), and the 3’ untranslated region (UTR) (e.g., NF1) (Epstein et al., 1994b; Watanabe et al., 1998; Galibert et al., 1999; Lagha et al., 2008; Kubic et al., 2015b). PAX3 binding sites were also identified in further upstream and downstream regions. DMRT2, MYOD1, and MYF5 are regulated by PAX3 via binding sites located 18 kb, 20 kb, and 57.5 kb, respectively, upstream of the corresponding transcription start sites (Kucharczuk et al., 1999; Bajard et al., 2006; Cao et al., 2010).
In contrast, PAX3 regulates FGFR4 expression by binding to a site located 19.2 kb downstream from the FGFR4 translation start site and 6.5 kb downstream of the last exon (Lagha et al., 2008). Though PAX3 functions as a transcriptional activator for most target genes, PAX3 may act as a transcriptional repressor of some targets, such as MBP. In addition, some genes with PAX3 binding sites (e.g., NF1) do not show altered expression in PAX3-deficient cells, suggesting that not all PAX3 binding sites are functional or other changes compensate for lack of PAX3 binding (Lakkis et al., 1999).
In addition to the above-described candidate gene approaches, genome-wide screening of genomic DNA with a CASTing strategy identified PAX3 binding sites in genes including ITM2A, FATH, FLT1, TGFA, BVES, and EN2 (Barber et al., 2002). Subsequent analysis of selected genes revealed that these binding sites confer PAX3-dependent regulation. In addition, array-based expression profiling of PAX3- and control-transfected medulloblastoma cells identified additional putative PAX3 downstream targets such as STX, TIMP3, MARCKS, RELA, BMP5, PTPRJ, GOS1, SLAP, IL6R, and ID4 (Mayanil et al., 2001). Similar microarray studies in melanocytes revealed that different PAX3 isoforms (PAX3c, PAX3e, and PAX3g) regulate distinct but overlapping sets of genes associated with differentiation, proliferation, migration, adhesion, apoptosis or angiogenesis (Wang et al., 2007).
PAX3 target genes can be divided into various groups based on their biological function (Table 2). One group of genes is associated with muscle development (e.g., MET, MYF5, MYOD1, WNT1, DIRT2 and FGFR4). A second group of genes is associated with neural and melanocyte development (e.g., MITF, TYRP1, RET, TBX2, NGN2, HES1, NRCAM, and TGFB2). In addition, PAX3 also targets alpha-satellite repeats of centromeres and mediates the formation of heterochromatin (Bulut-Karslioglu et al., 2012), extending the function of PAX3 beyond regulation of protein coding genes to the regulation of chromatin structure (Wu et al., 2015).
Table 2.
Representative PAX3 target genes.
| Symbol | Biochemical Category | Representative Functions | PAX3 binding location |
|---|---|---|---|
| CXCR4 | Chemokine receptor | Motility | 1st intron |
| DMRT2 | Transcription Factor | Differentiation | 5’ enhancer |
| FGFR4 | Receptor tyrosine kinase | Proliferation, differentiation, migration, metabolism | 3’ enhancer |
| HES1 | Transcription Factor | Proliferation, differentiation | 5’ promoter |
| MET | Receptor Tyrosine Kinase | Proliferation, invasion, survival | 5’ promoter |
| MITF | Transcription Factor | Differentiation, proliferation, survival, melanogenesis | 5’ promoter |
| MYF5 | Transcription Factor | Muscle differentiation | 5’ enhancer |
| MYOD1 | Transcription Factor | Muscle differentiation | 5’ enhancer |
| NF1 | GTPase-activating protein | Differentiation, control of cell growth, melanogenesis | 3’ UTR |
| NGN2 | Transcription Factor | Differentiation | 5’ promoter |
| NRCAM | Cell adhesion molecule | Intercellular adhesion | 1st Intron |
| RET | Receptor Tyrosine Kinase | Proliferation, migration, differentiation | 5’ promoter |
| TGFB2 | Ligand | Growth suppression | 5’ promoter |
| TYRP1 | Enzyme | Cell growth, melanogenesis | 5’ promoter |
| WNT1 | Ligand | Proliferation, differentiation | 5’ promoter |
2.5. Phosphorylation, ubiquitination and acetylation of PAX3 protein
PAX3 contains three major sites of phosphorylation: serines 201, 205 and 209 (Dietz et al., 2009; Dietz et al., 2011) (Fig. 2). The kinases that phosphorylate PAX3 includes GSK3β for serine 201 and CK2 for serines 205 and 209 (Dietz et al., 2011; Iyengar et al., 2012). Variation of phosphorylation states appears to occur in an ordered fashion during myogenesis. In melanoma cells, phosphorylation by GSK3β increases the stability of the PAX3 protein (Kubic et al., 2012). Furthermore, pharmacologic studies revealed that phosphorylation of the N-terminal PAX3 region regulates the transcriptional function of the PAX3-FOXO1 fusion protein in rhabdomyosarcoma (see Section 4.2) (Amstutz et al., 2008). The phosphorylation status of these serine residues affects the oncogenic function of the wild-type PAX3 protein in melanoma and the PAX3-FOXO1 fusion protein in rhabdomyosarcoma, and pharmacologic or genetic manipulation of the phosphorylation status results in changes in phenotypes such as proliferation, transformation and invasion (Iyengar et al., 2014; Loupe et al., 2015).
In addition to phosphorylation, the PAX3 protein is also modified by ubiquitination and acetylation. Lysines 437 and 475 in the C-terminal region of PAX3 are important for monoubiquitination and subsequent degradation of the PAX3 protein (Boutet et al., 2007). Both TAF1 (a major subunit of the TFIID transcriptional initiation complex) and anaphase-promoting complex/cyclosome E3 ligase participate in this degradation process (Boutet et al., 2010; Cao et al., 2015). These same two lysines (437 and 475) in the PAX3 protein can be acetylated. Acetylated PAX3 is a substrate of SIRT1 deacetylase and may contribute to decreased stem cell proliferation and increased neurogenic activity (Ichi et al., 2011).
3. Expression and function of PAX3 during development
3.1. Pax3 expression in myogenic precursors
Pax3 is a major regulator of embryonic myogenesis (Buckingham and Relaix, 2007). Pax3 expression is first seen at embryonic day 8.5 in the pre-somitic paraxial mesoderm before segmentation occurs to give rise to the somites (Bober et al., 1994; Goulding et al., 1994; Williams and Ordahl, 1994). As somites form, Pax3 is initially expressed throughout the somites, and later is confined to the dorsal epithelial portion, which is the dermomyotome (Goulding et al., 1991). Pax3 expression becomes concentrated in the epaxial and hypaxial regions of the dermomyotome (Tajbakhsh and Buckingham, 2000; Buckingham and Relaix, 2007). When muscle precursor cells detach from the dermomyotome to form the underlying differentiated muscle of the myotome, Pax3 expression is downregulated and myogenic transcription factors are activated (Myf5 and Myod1) (Tajbakhsh and Buckingham, 2000). Another population of Pax3-expressing cells detach from the dermomyotome and migrate to distant myogenic sites, such as the limbs, diaphragm, and hypoglossal cord (Franz et al., 1993; Bober et al., 1994; Goulding et al., 1994; Tajbakhsh et al., 1997; Tremblay et al., 1998). Finally, cells expressing Pax3 and Pax7 derived from the central dermomyotome provide a pool of myogenic progenitors for muscle growth during fetal development.
During the later stages of fetal development, myogenic progenitors expressing Pax3 and Pax7 form the satellite cells embedded under the basal lamina of muscle fibers. These cells contribute to muscle growth during the postnatal period and muscle regeneration thereafter. Though Pax7 is expressed by most adult satellite cells, which remain in a quiescent state until activated by injury, a subset expresses Pax3, often with coexpression of Pax7. The proportion of satellite cells that expresses Pax3 varies in different muscles; for example, Pax3-expressing satellite cells are frequent in the forelimb and diaphragm but not in the hind limb (Buckingham et al., 2003; Relaix et al., 2006).
3.2. Pax3 expression in developing nervous system
Pax3 expression is first observed during formation of the neural tube at embryonic day 8.5 In particular, Pax3 is expressed in the dorsal region of the neural groove and, as the neural groove deepens and closes, Pax3 continues to be expressed in the dorsal region of the neural tube (Goulding et al., 1991). This expression extends along the neural tube from the forebrain to the posterior neurospore. The localization of Pax3 expression to the dorsal half of the neural tube gives rise to a sharp ventral border between the basal and alar plates visible up through embryonic day 13 (Goulding et al., 1991). As the neural tube thickens, Pax3 expression is confined to proliferative cells in the inner ventricular zone and is downregulated as these cells migrate superficially. In addition, Pax3 expression is turned off in a rostral to caudal direction as neural tube development proceeds. Similarly, though Pax3 is initially expressed in neuroepithelium of the dorsolateral walls of the forebrain, midbrain and hindbrain, this expression becomes progressively restricted to more caudal regions during embryonic days 12-13 (Goulding et al., 1991).
3.3. Pax3 expression in neural crest and derivatives
The neural crest is a multipotent population of cells that arises at the border between neural and non-neural ectoderm and gives rise to variety of cells and tissues in the developing vertebrate (Knecht and Bronner-Fraser, 2002; Milet and Monsoro-Burq, 2012). During neural induction, Pax3 is expressed at the lateral and posterior edges of the neural plate (and then the closed neural tube), the region from which the neural crest cells emerge and migrate (Monsoro-Burq et al., 2005). Pax3 is subsequently found to be expressed by a variety of cell types derived from the neural crest, such as melanoblasts, dorsal root ganglia, and Schwann cell precursors. For melanoblasts, Pax3 expression is present during migration to the skin and persists in the developing hair follicles (Hornyak et al., 2001; Blake and Ziman, 2005). In addition, Pax3-expressing cells related to the melanoblast lineage also contribute to the development of multiple inner ear components (Freyer et al., 2011; Kim et al., 2014). In Schwann cell precursors, Pax3 is restricted to proliferating cells and functions to regulate differentiation and associated myelination (Kioussi et al., 1995). Finally, Pax3 is also expressed in a subpopulation of cranial neural crest cells and in structures derived from these cells, such as mesenchyme of the mandible and maxilla.
3.4. Pax3 mutation during development
The significance of Pax3 in development is further evidenced by analyses of germline mutations involving the Pax3 gene. The splotch phenotype is found in several spontaneous mutant mouse strains as well as in chemical- or radiation-induced strains, and is characterized by white patches in the belly, tail and feet in the heterozygous state and more significant phenotypic changes in the homozygous state (Russell, 1947; Moase and Trasler, 1992). In addition, as described below, there is a related human disease termed Waardenburg syndrome. Point mutations and deletions affecting functional domains of the mouse Pax3 gene were identified in the splotch mouse. Functional studies showed that these mutations alter or abolish Pax3 transcriptional activity, and result in a loss of biological function. Different Splotch alleles (such as Splotch4H, Splotch2H, Splotch-delayed) are caused by different Pax3 mutations, which combine with differences in genetic background and environmental factors to result in varying phenotypic severity (Epstein et al., 1991; Vogan et al., 1996).
In heterozygous splotch mice, the characteristic white spots are attributed to defects in neural crest cells resulting in localized deficiencies in pigment-forming melanocytes. Mice homozygous for these Pax3 mutations demonstrate embryonic lethality around embryonic day 15. There are prominent neural tube closure defects, such as spina bifida and exencephaly. Multiple neural crest-derived structures are absent or abnormal, including dorsal root ganglia, enteric ganglia, and melanocytes. Though Pax3 may not be specifically required for neural crest migration, loss of function contributes to defects in neural crest specification, proliferation, survival and/or differentiation. The severity of neural crest defects increases in posterior regions of the body, resulting in a progressive loss of cardiac, trunk, and caudal neural crest (Relaix et al., 2004). Loss of the cardiac neural crest cells results in heart malformations, due to the contributions of these cells to heart innervation and structures of the cardiac outflow tract. Some of these malformations, such as neural tube defects, can be rescued by crosses with p53-deficient mice, thus revealing that p53 inactivation and corresponding inhibition of p53-mediated apoptosis is a major function of Pax3 during early neural development (Pani et al., 2002; Wang et al., 2011). Finally, there are significant defects in muscle development in homozygous Splotch mice. The limb musculature fails to develop whereas the axial musculature shows varying degrees of abnormality. This hypoplasia in the limb musculature is associated with increased cell death in myogenic precursors in the lateral dermomyotome (Borycki et al., 1999) and diminished migration of any surviving precursors into limb buds (Bober et al., 1994; Goulding et al., 1994; Daston et al., 1996). This muscle defect is attributed to reduced expression of Met, a Pax3 target gene (Bladt et al., 1995; Epstein et al., 1995; Daston et al., 1996; Yang et al., 1996).
3.5. Regulation of Pax3 expression
Deletion studies in transgenic mice revealed that the region located 1.6 kb upstream of the Pax3 transcription start site contains regulatory sequences sufficient for induction of Pax3 expression and restriction to the hindbrain and trunk (Natoli et al., 1997; Degenhardt et al., 2010). In this sequence, two evolutionarily conserved regions named NCE1 and NCE2 were found to be important for Pax3 expression in the neural tube and neural crest cells (Fig. 3) (Degenhardt et al., 2010). Anterior and posterior expression of Pax3 are supported by NCE1 and NCE2, respectively (Sanchez-Ferras et al., 2012). NCE1 contains binding sites for Pbx factors and is required for hindbrain expression (Phelan et al., 1995; Naini et al., 2008). NCE2 is recognized by Cdx factors (Sanchez-Ferras et al., 2012) and is thus indirectly activated by the Wnt signaling pathway (Sanchez-Ferras et al., 2012). In addition, NCE2 contains response elements for the sonic Hedgehog pathway, and is responsible for restriction of Pax3 expression to the lateral neural plate and dorsal neural tube and repression in the ventral neural tube (Goulding et al., 1993; Sanchez-Ferras et al., 2012). Both NCE1 and NCE2 contain binding sites for Tead2 and the POU class III factors Brn1 and Brn2 (Pruitt et al., 2004), which are proposed to maintain rather than induce Pax3 expression.
Figure 3. Elements regulating PAX3 expression.
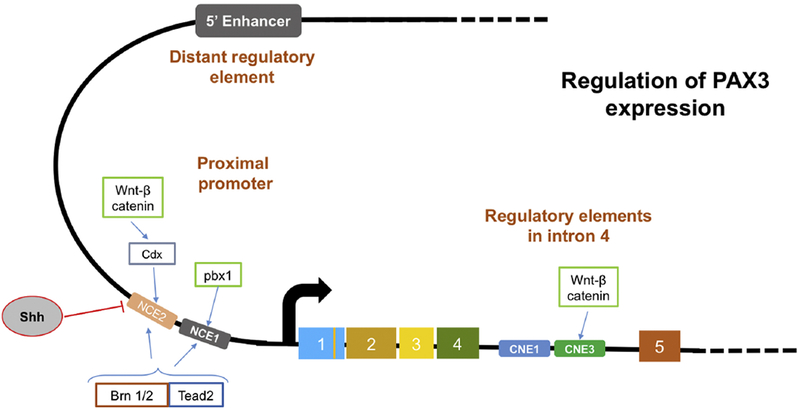
The cis-acting elements, their position relative to the PAX3 gene and relevant transcription factors involved in the regulation of PAX3 expression are shown (see section 3.5 for details).
The finding that deletion of NCE1 and NCE2 does not significantly alter Pax3 expression in mice indicates that additional regulatory sequences support Pax3 expression. An additional region regulating neural tube and neural crest expression is present in the fourth intron of the Pax3 gene (Degenhardt et al., 2010; Moore et al., 2013) and contains two conserved non-coding elements, CNE1 and CNE3 (or ECR2), which serve as enhancers of Pax3 expression in the developing central nervous system. This latter element contains TCF/LEF binding sites, supporting an additional direct regulation of Pax3 by the Wnt signaling pathway (Degenhardt et al., 2010). CNE3 also binds to Nkx6.1, which serves as a repressor and restricts Pax3 expression to the dorsal region of the neural tube (Moore et al., 2013). In addition to this intronic element, an enhancer located 6 kb upstream of the Pax3 transcription start site mediates Pax3 expression in the hypaxial somite (Fig. 3) (Brown et al., 2005). Taken together, the regulatory elements adjacent to the Pax3 promoter, in the fourth intron and in the far upstream region provide evidence for the complexity and a time- and site-specific fine tuning of Pax3 expression during development.
In addition to these elements regulating Pax3 expression at the transcriptional level, there are several microRNAs that regulate Pax3 expression at the post-transcriptional level. There are binding sites for miR-1, mir-27b, miR-128a, and miR-206 within the 3’ UTR of the Pax3 mRNA (Crist et al., 2009; Goljanek-Whysall et al., 2011; Motohashi et al., 2012). Expression of one or more of these microRNA’s will decrease Pax3 mRNA stability and/or translation, resulting in decreased Pax3 expression (Boutet et al., 2012). The muscle-specific microRNAs are induced by Myodl in the somites (Hirai et al., 2010), and miR-27b is inhibited by Pitx2c in early myogenic precursors and then later expressed in differentiating embryonic myotome and activated satellite cells (Lozano-Velasco et al., 2011). Whereas Pax3 is expressed in the absence of these microRNAs to promote proliferation and migration and to inhibit differentiation, upregulation of one or more of these microRNAs leads to Pax3 downregulation, thereby inducing differentiation and further inhibiting proliferation and migration. In subsequent myogenic stem cell populations, mir-206 and miR-128a help maintain the quiescent state by inhibiting expression of Pax3 and other developmental genes (Goljanek-Whysall et al., 2011; Motohashi et al., 2012). In a subpopulation of quiescent stem cells, Pax3 is expressed despite the presence of mir-206 by generating “resistant” transcripts with alternative polyadenylation sites, which remove the miR-206 binding sites from the Pax3 3’ UTR (Boutet et al., 2012).
4. PAX3 in human disease
4.1. PAX3 germline mutations in Waardenburg syndrome
The importance of PAX3 in neural tube, neural crest and muscle development is also confirmed by the molecular genetic findings in Waardenburg syndrome. This syndrome is a group of four autosomal dominant genetic disorders (WS1, WS2, WS3 and WS4), which are characterized by variable features in affected individuals including hearing loss, eye abnormalities and pigmentation disorders (Waardenburg, 1951). WS1 is also frequently characterized by a midfacial alteration called dystopia canthorum (Farrer et al., 1994), and WS3 (Klein-Waardenburg syndrome) is comparable to WS1 with additional musculoskeletal abnormalities affecting the upper limbs. WS1 and WS3 are frequently caused by mutations in PAX3, whereas WS2 is caused by mutations in MITF and WS4 is caused by mutations in EDN3, EDNRB, or SOX10. Though these mutations were initially detected by directed Sanger sequencing of polymerase chain reaction (PCR)-amplified exons from selected genes, more recent next generation sequencing approaches can screen larger panels of genes related to Waardenburg syndrome and other genetic causes of hearing loss (Xiao et al., 2016).
Heterozygous mutations in the PAX3 gene are observed in nearly 80% of WS1 cases (Fig. 4), whereas partial or total deletion of PAX3 and contiguous genes are often observed in WS3 cases. Smaller mutations can also occur in WS3 and could be either heterozygous or homozygous (Hoth et al., 1993; Tassabehji et al., 1995). Approximately 100 sequence changes in the PAX3 gene have been reported in these disorders, and very few of these changes are recurrent. These reported sequence changes include missense mutations (38%), small deletions (20%), nonsense mutations (15%), gross deletion (11%), splicing mutations (8%), and small insertions (8%) (Jalilian et al., 2015). The majority of these PAX3 mutations are localized to exons 2 through 6 with the highest frequency in exon 2 (Pingault et al., 2010). These frequently mutated regions alter the structure of the paired domain or homeodomain, and thus affect DNA binding function. Mutations were not found in exons 9 and 10, and only a few mutations were reported in exons 1, 7, and 8, including mutations that affect the transactivation domain (Baldwin et al., 1995; Carey et al., 1998; Jalilian et al., 2015). There is no correlation between the mutation type, location and severity of the phenotype (Baldwin et al., 1995; Tassabehji et al., 1995). In a subset of WS1 cases, such as those recently reported in the Chinese and Korean populations, the proband’s PAX3 mutation was not detected in either parent, suggesting the occurrence of a de novo mutational event (Wang et al., 2010; Jang et al., 2015). However, the report of two WS1-affected siblings in which the shared PAX3 mutation was not present in either parent also points to the possibility of germinal mosaicism in rare cases (Chen et al., 2018).
Figure 4. Distribution of PAX3 mutations in Waardenburg syndrome.
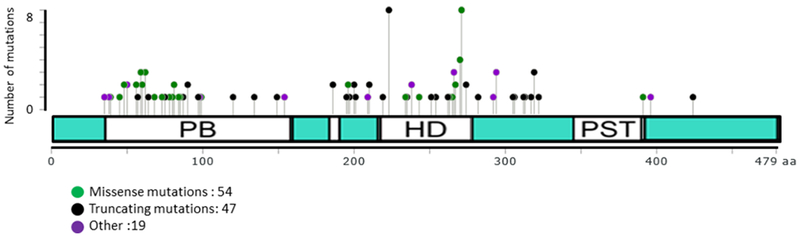
The displayed genetic changes include missense mutations, truncating mutations (nonsense and frameshift deletion) and others (frameshift duplications, frameshift mutation, insertions, and deletions). The diagram was generated using Mutation Mapper software (Vohra and Biggin, 2013) and data were partially collected from (Pingault et al., 2010).
PAX3 mutations have been also been identified in rare disorders related to Waardenburg syndrome. Craniofacial-deafness-hand syndrome (CDHS) has been described in one family with a mutation in PAX3 exon 2 (Asher et al., 1996). This autosomal dominant syndrome is considered to be an allelic variant of Waardenburg syndrome and is characterized by craniofacial anomalies, profound sensorineural deafness, and ulnar deviation of the hand with contractures of the fingers (Sommer et al., 1983). A rare case has also been described in which a child had heterozygous mutations for both PAX3 and MITF (Glass, 1990). One parent had WS1 caused by a mutation in the splicing site of PAX3 intron 4 and the other parent had WS2 caused by a pathogenic variant in MITF. The child showed significantly more pigmentary defects than his parents, suggesting an additive effect of these two gene alterations.
4.2. Gene fusions in alveolar rhabdomyosarcoma
Alveolar rhabdomyosarcoma (ARMS) is an aggressive pediatric soft tissue sarcoma that occurs mainly in the extremities and trunk (Wexler LH, 1997). In most ARMS cases, there is a recurrent t(2;13)(q35;q14) chromosomal translocation that breaks PAX3 and joins it to FOXO1 (Fig. 5A), which encodes a member of the forkhead (or FOX) transcription factor family (Seidal et al., 1982; Turc-Carel et al., 1986; Douglass et al., 1987; Wang-Wuu et al., 1988). The t(2; 13) breakpoint always disrupts the seventh intron of PAX3 and the first intron of FOXO1 (Davis et al., 1995; Barr et al., 1998). By swapping portions of the two genes, the 2;13 translocation thereby generates a PAX3-FOXO1 fusion gene that is transcribed into a PAX3-FOXO1 fusion transcript (Barr et al., 1993; Galili et al., 1993; Shapiro et al., 1993). In this fusion transcript, the 5’ end of the PAX3 coding sequence fuses in frame with the 3’ end of the FOXO1 coding sequence to yield a PAX3-FOXO1 fusion protein, which is 836 amino acids in length. The t(2; 13) breakpoint is positioned such that the N-terminal PAX3 DNA binding domain is preserved, and separated from most of the PAX3 transactivation domain. In addition, the translocation breakpoint disrupts the forkhead DNA binding domain, but the FOXO1 transcriptional activation domain remains intact. This arrangement results in fusion of regions encoding the N-terminal PAX3 DNA-binding domain and the C-terminal FOXO1 transactivation domain to create a novel fusion transcription factor.
Figure 5. Comparison of wild-type and fusion products in ARMS and SNS.
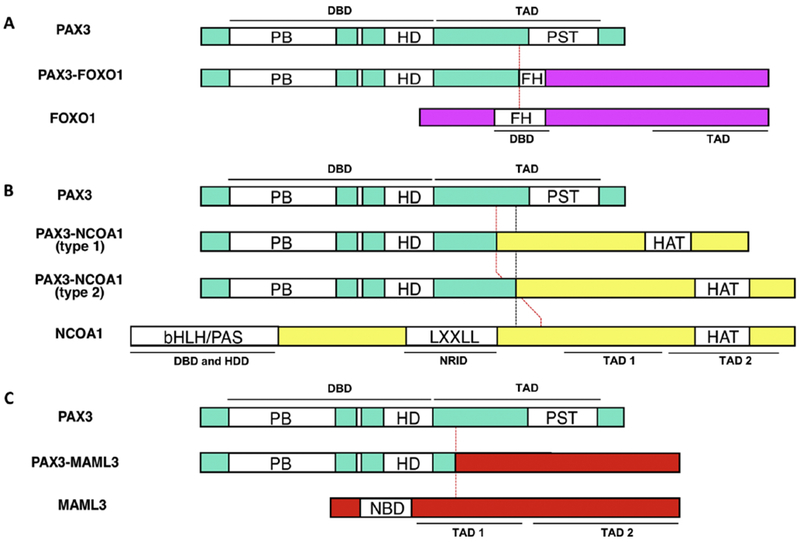
A. PAX3-FOXO1 fusion generated by 2;13 translocation in ARMS (and rarely in SNS). B. PAX3-NCOA1 and PAX3-NCOA2 fusions generated by 2;2 and 2;8 translocations in ARMS (and rarely in SNS). C. PAX3-MAML3 fusion generated by 2;4 translocation in SNS. Conserved domains are shown as opened boxes, while functional domains are indicated as horizontal lines. The vertical dashed lines indicate translocation fusion points. Abbreviations: DBD, DNA binding domain; TAD, transactivation domain; HDD, heterodimerization domain; NRID, nuclear receptor interaction domain; PB, paired box; HD, homeodomain; FH, forkhead domain; bHLH/PAS, basic helix-loop-helix/Per/ARNT/Sim homologous domain; LXXLL, typical LXXLL α-helix motifs; HAT, histone acetyltransferase; NBD, Notch-binding domain.
The PAX3-FOXO1 fusion results in increased activity at both the functional and expression levels. Transcriptional studies revealed that, compared to the wild-type PAX3 protein, the PAX3-FOXO1 fusion protein is a stronger activator of genes with PAX3 binding sites (Bennicelli et al., 1995; Fredericks et al., 1995; Bennicelli et al., 1996). This enhanced transcriptional activity is related to decreased sensitivity of the FOXO1 activation domain to inhibitory effects of N-terminal PAX3 domains when compared to the effects of these N-terminal domains on the C-terminal PAX3 activation domain. In addition to generating a potent transcription factor, the translocation also results in high-level expression of this fusion protein, which is associated with higher steady-state transcript levels of PAX3-FOXO1 mRNA relative to wild-type PAX3 transcript levels (Davis and Barr, 1997). This overexpression is usually not attributable to changes in PAX3-FOXO1 copy number. Nuclear runoff analysis confirmed that PAX3-FOXO1 is more actively transcribed than wild type PAX3, and thus PAX3-FOXO1 overexpression is due to increased copy number-independent transcription.
PAX3-FOXO1 and related gene fusions can be detected in tumors by various methodologies, including reverse transcription-PCR, fluorescence in situ hybridization or next generation sequencing (Duan et al., 2012; Shern et al., 2014). Whereas 60% of ARMS tumors have a PAX3-FOXO1 fusion, an additional 20% have a 1;13 translocation that fuses the closely related PAX7 gene to FOXO1 and the remaining 20% do not express either of these two fusions (Barr et al., 2002). The far majority of this latter ARMS group is truly fusion-negative, but a small subset has variant fusions of PAX3 to other proteins. In one rare variant, PAX3 is fused to the activation domain of FOXO4, another forkhead transcription factor that is similar in structure and function to FOXO1. In two other rare variants, PAX3 fuses with activation domains from NCOA1 or NCOA2, two members of the nuclear receptor transcriptional coactivator family (Sumegi et al., 2010) (Fig. 5B). Finally, whole genome sequencing studies identified a single ARMS case in which the PAX3 DNA binding domain is fused with the C-terminal region of INO80D, which is a subunit of the ATP dependent chromatin remodeling complex (Shern et al., 2014).
The PAX3-FOXO1 fusion protein contributes to tumorigenesis by binding to PAX3 binding sites in target genes, and causing an aberrant change in expression of these downstream targets. In ARMS cells, PAX3-FOXO1 generally functions as a transcriptional activator and increases expression of target genes. Many of these target genes were identified by microarray-based expression profiling following introduction of a PAX3-FOXO1 expression construct into non-ARMS cells or introduction of 5’ PAX3-directed siRNA into ARMS cells (Tomescu et al., 2004; Nabarro et al., 2005; Davicioni et al., 2006; Ebauer et al., 2007). More recently, ChIP-Seq approaches were used to identify genomic regions that bind to the PAX3-FOXO1 protein in ARMS cells (Cao et al., 2010; Gryder et al., 2017). This latter approach revealed that PAX3 and E-box DNA binding motifs were highly enriched in genomic regions bound by PAX3-FOXO1. Furthermore, the vast majority of these bound regions were located >2.5 kb distal to the nearest transcriptional start site. Of note, numerous PAX3-FOXO1-associated regions previously defined by standard promoter-based analyses were not confirmed by these ChIP studies. Integration of PAX3-FOXO1 binding studies with additional ChIP studies showed that PAX3-FOXO1 often binds along with MYOD1, MYOG and MYCN to set up super enhancers in the vicinity of target genes in ARMS cells. In addition, PAX3-FOXO1 interacts with proteins, such as CHD4 and BRD4, involved in defining chromatin structure; these interactions are critical to PAX3-FOXO1-mediated downstream effects as indicated by the high sensitivity of PAX3-FOXO1-positive ARMS cells to small molecules or siRNAs directed against these proteins.
Activation of target genes by PAX3-FOXO1 alters multiple biological signaling pathways, including proliferation, cell death, myogenic differentiation, and migration. Introduction of PAX3-FOXO1 expression constructs into a number of cell lines results in transforming behavior in culture and tumorigenesis in vivo (Lam et al., 1999; Anderson et al., 2001). This deregulated proliferative activity is associated with increased expression of multiple target genes such as MYCN and MET, and generally requires additional collaborating events to ensure optimal high-level MYCN expression and to inhibit the TP53 and RB1 pathways (Naini et al., 2008; Xia et al., 2009; Cao et al., 2010; Pandey et al., 2017). In addition to these proliferative effects, experiments with siRNA or shRNA expression constructs that decrease PAX3-FOXO1 expression indicate that PAX3-FOXO1 also inhibits cell death and myogenic differentiation (Ebauer et al., 2007; Kikuchi et al., 2008; Xia et al., 2009). The apoptotic function is mediated by the downstream targets including TFAP2B and FGFR4. PAX3-FOXO1 induces expression of genes such as MYOD1 that stimulate myogenesis as well as target genes such as JARID2 that inhibit differentiation, resulting in a complicated scenario in which PAX3-FOXO1 facilitates entry into myogenesis and variably blocks the final steps (Graf Finckenstein et al., 2008; Cao et al., 2010; Calhabeu et al., 2013; Walters et al., 2014). Finally, a role in local and distal dissemination is indicated by the stimulation of basement membrane invasion in vitro and lung colonization in vivo when PAX3-FOXO1 is introduced into murine myoblasts. These invasive and metastatic effects require expression of the target gene CNR1 (Marshall et al., 2011) and are associated with a larger program of pro-invasive mRNA expression changes (Loupe et al., 2016a). An additional PAX3-FOXO1 target that contributes to metastasis is CXCR4, which encodes a chemokine receptor that modulates homing to the bone marrow (Libura et al., 2002). Finally, the fusion protein promotes aneuploidy by inducing gene expression changes that override regulatory cell cycle checkpoints and dysregulate maintenance of chromosome number and structure (Loupe et al., 2016b). In addition to protein-encoding target genes, PAX3-FOXO1 also regulates expression of a series of microRNAs that impact on the above-described cellular functions, such as differentiation (Loupe et al., 2016a; Loupe et al., 2016b; Loupe et al., 2017; Hanna et al., 2018).
A mouse model that develops ARMS tumors was developed using a conditional knock-in approach that generates a Pax3-Foxo1 fusion in specific myogenic lineages (Keller et al., 2004). Of the lineages studied, the fetal myoblast, a prenatal myogenic progenitor defined by Myf6 regulatory elements, showed the highest susceptibility to ARMS formation (Abraham et al., 2014). Though ARMS tumorigenicity in the original mice was low, tumor frequency is substantially increased in Pax3-Foxo1 mice homozygous for Cdkn2a or Trp53 deletion, confirming the need for additional collaborating events (Keller et al., 2004). Homozygosity of Pax3-Foxo1 was also necessary to achieve enhanced tumor formation suggesting the need for increased Pax3-Foxo1 and/or decreased wild-type Pax3 dosage in tumorigenesis.
4.3. Gene fusions in biphenotypic sinonasal sarcoma
Biphenotypic sinonasal sarcoma (BSNS) is a low-grade malignancy occurring in adults that is defined by both myogenic and neural differentiation (Lewis et al., 2012). This cancer is associated with a recurrent t(2;4)(q35;q31.1) chromosomal translocation that fuses PAX3 to the MAML3 gene (Fig. 5C), which encodes a member of the mastermind-like family of transcriptional coactivators for the Notch signaling pathway (Wang et al., 2014). In a smaller subset of BSNS cases, PAX3 is rearranged without MAML3 involvement, and some of these variant cases contain a PAX3-NCOA1 or PAX3-FOXO1 fusion (Fritchie et al., 2016; Huang et al., 2016; Wong et al., 2016). In the characteristic PAX3-MAML3 fusion, the N-terminal PAX3 DNA binding domain is fused with the C-terminal MAML3 transactivation domain to generate a powerful activator of target genes with PAX3 binding sites. The transactivation potential of PAX3-MAML3 is similar to that of PAX3-FOXO1, but multiple characteristic PAX3-FOXO1 targets (such as FGFR4 and MYCN) are not comparably expressed in BSNS. Though overlapping fusions are found in both ARMS and BSNS, the biological differences suggest that the target cell environment significantly modulates the transcriptional output.
4.4. Wild-type PAX3 in other cancers
Wild-type PAX3 is highly expressed in multiple tumor types, which are often related to developmental lineages in which wild-type PAX3 is normally expressed. In particular, PAX3 is expressed in tumors related to neural tube-derived lineages, such as medulloblastoma and glioblastoma, and tumors related to neural crest-derived lineages, such as melanoma, malignant nerve sheath tumor, neurofibroma and Ewing’s sarcoma (Schulte et al., 1997; Scholl et al., 2001; Gershon et al., 2005; He et al., 2010). PAX3 is also expressed in several tumors associated with myogenic differentiation, including embryonal rhabdomyosarcoma and Wilms tumor with a myogenic component (Frascella et al., 1998; Barr et al., 1999; Hueber et al., 2009). Finally, PAX3 expression has also been noted in several other cancer types, such as breast carcinoma, gastric carcinoma and osteosarcoma, without a clear connection to PAX3-expressing developmental lineages (Tan et al., 2014; Zhang et al., 2015; Liu et al., 2017). Clinical correlative studies have further suggested that PAX3 expression may be associated with a worse prognosis in a subset of these cancers, such as glioma and gastric carcinoma (Chen et al., 2012; Zhang et al., 2015).
The oncogenic role of wild-type PAX3 has been studied in several cancer model systems, including glioma and melanoma cell lines. In these studies, PAX3 expression was downregulated by RNA interference or antisense approaches or upregulated by cDNA complementation (Mayanil et al., 2001; He et al., 2005; Xia et al., 2013). In cell culture experiments, these manipulations of PAX3 expression reveal that PAX3 contributes to the control of proliferation, apoptosis, differentiation and motility. Complementary in vivo xenograft studies demonstrate that PAX3 contributes to tumorigenicity and metastasis. Downstream transcriptional targets of PAX3 involved in these phenotypic effects include MET, which encodes a cell surface receptor involved in multiple oncogenic signaling pathways (Mascarenhas et al., 2010). These findings thus suggest that PAX3 has a major regulatory role in the development, maintenance and progression of these tumors, which may be related to its role in normal development.
5. Acknowledgments
This review and the corresponding Gene Wiki article are written as part of the Gene Wiki Review series--a series resulting from a collaboration between the journal GENE and the Gene Wiki Initiative. The Gene Wiki Initiative is supported by National Institutes of Health (GM089820). Additional support for Gene Wiki Reviews is provided by Elsevier, the publisher of GENE. The authors were supported by the Intramural Research Program of the National Cancer Institute and the Joanna McAfee Childhood Cancer Foundation. SB is a recipient of a postdoctoral fellowship from the Fonds de recherche du Québec – Santé (FRQS).
Abbreviations:
- ARMS
alveolar rhabdomyosarcoma
- BSNS
biphenotypic sinonasal sarcoma
- CASTing
cyclic amplification and selection of targets
- ChIP-Seq
chromatin immunoprecipitation sequencing
- HD
homeodomain
- HTH
helix-turn-helix
- PAI
N-terminal paired box subdomain
- PB
paired box
- PCR
polymerase chain reaction
- PST
proline, serine and threonine
- Q+
glutamine-including
- Q−
glutamine-excluding
- RED
C-terminal paired box subdomain
- UTR
untranslated region
- WS
Waardenburg syndrome
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declarations of interest: none
The corresponding Gene Wiki entry for this review can be found here: https://en.wikipedia.org/wiki/PAX3
References
- Abraham J, Nunez-Alvarez Y, Hettmer S, Carrio E, Chen HI, Nishijo K, Huang ET, Prajapati SI, Walker RL, Davis S, Rebeles J, Wiebush H, McCleish AT, Hampton ST, Bjornson CR, Brack AS, Wagers AJ, Rando TA, Capecchi MR, Marini FC, Ehler BR, Zarzabal LA, Goros MW, Michalek JE, Meltzer PS, Langenau DM, LeGallo RD, Mansoor A, Chen Y, Suelves M, Rubin BP and Keller C, 2014. Lineage of origin in rhabdomyosarcoma informs pharmacological response. Genes Dev 28, 1578–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amstutz R, Wachtel M, Troxler H, Kleinert P, Ebauer M, Haneke T, Oehler-Janne C, Fabbro D, Niggli FK and Schafer BW, 2008. Phosphorylation regulates transcriptional activity of PAX3/FKHR and reveals novel therapeutic possibilities. Cancer Res 68, 3767–76. [DOI] [PubMed] [Google Scholar]
- Anderson J, Ramsay A, Gould S and Pritchard-Jones K, 2001. PAX3-FKHR induces morphological change and enhances cellular proliferation and invasion in rhabdomyosarcoma. Am J Pathol 159, 1089–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Apuzzo S and Gros P, 2007. Cooperative interactions between the two DNA binding domains of Pax3: helix 2 of the paired domain is in the proximity of the amino terminus of the homeodomain. Biochemistry 46, 2984–93. [DOI] [PubMed] [Google Scholar]
- Asher JH Jr., Sommer A, Morell R and Friedman TB, 1996. Missense mutation in the paired domain of PAX3 causes craniofacial-deafness-hand syndrome. Hum Mutat 7, 30–5. [DOI] [PubMed] [Google Scholar]
- Bajard L, Relaix F, Lagha M, Rocancourt D, Daubas P and Buckingham ME, 2006. A novel genetic hierarchy functions during hypaxial myogenesis: Pax3 directly activates Myf5 in muscle progenitor cells in the limb. Genes Dev 20, 2450–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baldwin CT, Hoth CF, Macina RA and Milunsky A, 1995. Mutations in PAX3 that cause Waardenburg syndrome type I: ten new mutations and review of the literature. Am J Med Genet 58, 115–22. [DOI] [PubMed] [Google Scholar]
- Barber TD, Barber MC, Cloutier TE and Friedman TB, 1999. PAX3 gene structure, alternative splicing and evolution. Gene 237, 311–9. [DOI] [PubMed] [Google Scholar]
- Barber TD, Barber MC, Tomescu O, Barr FG, Ruben S and Friedman TB, 2002. Identification of target genes regulated by PAX3 and PAX3-FKHR in embryogenesis and alveolar rhabdomyosarcoma. Genomics 79, 278–84. [DOI] [PubMed] [Google Scholar]
- Barr FG, Fitzgerald JC, Ginsberg JP, Vanella ML, Davis RJ and Bennicelli JL, 1999. Predominant expression of alternative PAX3 and PAX7 forms in myogenic and neural tumor cell lines. Cancer Res 59, 5443–8. [PubMed] [Google Scholar]
- Barr FG, Galili N, Holick J, Biegel JA, Rovera G and Emanuel BS, 1993. Rearrangement of the PAX3 paired box gene in the paediatric solid tumour alveolar rhabdomyosarcoma. Nat Genet 3, 113–7. [DOI] [PubMed] [Google Scholar]
- Barr FG, Nauta LE and Hollows JC, 1998. Structural analysis of PAX3 genomic rearrangements in alveolar rhabdomyosarcoma. Cancer Genet Cytogenet 102, 32–9. [DOI] [PubMed] [Google Scholar]
- Barr FG, Qualman SJ, Macris MH, Melnyk N, Lawlor ER, Strzelecki DM, Triche TJ, Bridge JA and Sorensen PH, 2002. Genetic heterogeneity in the alveolar rhabdomyosarcoma subset without typical gene fusions. Cancer Res 62, 4704–10. [PubMed] [Google Scholar]
- Bennicelli JL, Advani S, Schafer BW and Barr FG, 1999. PAX3 and PAX7 exhibit conserved cis-acting transcription repression domains and utilize a common gain of function mechanism in alveolar rhabdomyosarcoma. Oncogene 18, 4348–56. [DOI] [PubMed] [Google Scholar]
- Bennicelli JL, Edwards RH and Barr FG, 1996. Mechanism for transcriptional gain of function resulting from chromosomal translocation in alveolar rhabdomyosarcoma. Proc Natl Acad Sci U S A 93, 5455–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennicelli JL, Fredericks WJ, Wilson RB, Rauscher FJ 3rd and Barr FG, 1995. Wild type PAX3 protein and the PAX3-FKHR fusion protein of alveolar rhabdomyosarcoma contain potent, structurally distinct transcriptional activation domains. Oncogene 11, 119–30. [PubMed] [Google Scholar]
- Bladt F, Riethmacher D, Isenmann S, Aguzzi A and Birchmeier C, 1995. Essential Role for the C-Met Receptor in the Migration of Myogenic Precursor Cells into the Limb Bud. Nature 376, 768–771. [DOI] [PubMed] [Google Scholar]
- Blake JA and Ziman MR, 2005. Pax3 transcripts in melanoblast development. Development Growth & Differentiation 47, 627–635. [DOI] [PubMed] [Google Scholar]
- Bober E, Brandsaberi B, Ebensperger C, Wilting J, Balling R, Paterson BM, Arnold HH and Christ B, 1994. Initial Steps of Myogenesis in Somites Are Independent of Influence from Axial Structures. Development 120, 3073–3082. [DOI] [PubMed] [Google Scholar]
- Bopp D, Jamet E, Baumgartner S, Burri M and Noll M, 1989. Isolation of two tissue-specific Drosophila paired box genes, Pox meso and Pox neuro. EMBO J 8, 3447–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borycki AG, Li J, Jin FZ, Emerson CP and Epstein JA, 1999. Pax3 functions in cell survival and in pax7 regulation. Development 126, 1665–1674. [DOI] [PubMed] [Google Scholar]
- Boutet SC, Biressi S, Iori K, Natu V and Rando TA, 2010. Taf1 regulates Pax3 protein by monoubiquitination in skeletal muscle progenitors. Mol Cell 40, 749–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boutet SC, Cheung TH, Quach NL, Liu L, Prescott SL, Edalati A, Iori K and Rando TA, 2012. Alternative polyadenylation mediates microRNA regulation of muscle stem cell function. Cell Stem Cell 10, 327–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boutet SC, Disatnik MH, Chan LS, Iori K and Rando TA, 2007. Regulation of Pax3 by proteasomal degradation of monoubiquitinated protein in skeletal muscle progenitors. Cell 130, 349–62. [DOI] [PubMed] [Google Scholar]
- Brown CB, Engleka KA, Wenning J, Min Lu M and Epstein JA, 2005. Identification of a hypaxial somite enhancer element regulating Pax3 expression in migrating myoblasts and characterization of hypaxial muscle Cre transgenic mice. Genesis 41, 202–9. [DOI] [PubMed] [Google Scholar]
- Buckingham M, Bajard L, Chang T, Daubas P, Hadchouel J, Meilhac S, Montarras D, Rocancourt D and Relaix F, 2003. The formation of skeletal muscle: from somite to limb. J Anat 202, 59–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buckingham M and Relaix F, 2007. The role of Pax genes in the development of tissues and organs: Pax3 and Pax7 regulate muscle progenitor cell functions. Annual Review of Cell and Developmental Biology 23, 645–673. [DOI] [PubMed] [Google Scholar]
- Bulut-Karslioglu A, Perrera V, Scaranaro M, de la Rosa-Velazquez IA, van de Nobelen S, Shukeir N, Popow J, Gerle B, Opravil S, Pagani M, Meidhof S, Brabletz T, Manke T, Lachner M and Jenuwein T, 2012. A transcription factor-based mechanism for mouse heterochromatin formation. Nat Struct Mol Biol 19, 1023–30. [DOI] [PubMed] [Google Scholar]
- Calhabeu F, Hayashi S, Morgan JE, Relaix F and Zammit PS, 2013. Alveolar rhabdomyosarcoma-associated proteins PAX3/FOXO1A and PAX7/FOXO1A suppress the transcriptional activity of MyoD-target genes in muscle stem cells. Oncogene 32, 651–62. [DOI] [PubMed] [Google Scholar]
- Cao J, Dai X, Wan L, Wang H, Zhang J, Goff PS, Sviderskaya EV, Xuan Z, Xu Z, Xu X, Hinds P, Flaherty KT, Faller DV, Goding CR, Wang Y, Wei W and Cui R, 2015. The E3 ligase APC/C(Cdh1) promotes ubiquitylation-mediated proteolysis of PAX3 to suppress melanocyte proliferation and melanoma growth. Sci Signal 8, ra87. [DOI] [PubMed] [Google Scholar]
- Cao L, Yu Y, Bilke S, Walker RL, Mayeenuddin LH, Azorsa DO, Yang F, Pineda M, Helman LJ and Meltzer PS, 2010. Genome-wide identification of PAX3-FKHR binding sites in rhabdomyosarcoma reveals candidate target genes important for development and cancer. Cancer Res 70, 6497–508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carey ML, Friedman TB, Asher JH Jr. and Innis JW, 1998. Septo-optic dysplasia and WS1 in the proband of a WS1 family segregating for a novel mutation in PAX3 exon 7. J Med Genet 35, 248–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chalepakis G, Goulding M, Read A, Strachan T and Gruss P, 1994a. Molecular basis of splotch and Waardenburg Pax-3 mutations. Proc Natl Acad Sci U S A 91, 3685–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chalepakis G and Gruss P, 1995. Identification of DNA recognition sequences for the Pax3 paired domain. Gene 162, 267–70. [DOI] [PubMed] [Google Scholar]
- Chalepakis G, Jones FS, Edelman GM and Gruss P, 1994b. Pax-3 contains domains for transcription activation and transcription inhibition. Proc Natl Acad Sci U S A 91, 12745–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chalepakis G, Wijnholds J and Gruss P, 1994c. Pax-3-DNA interaction: flexibility in the DNA binding and induction of DNA conformational changes by paired domains. Nucleic Acids Res 22, 3131–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, Xia L, Wu X, Xu L, Nie D, Shi J, Xu X, Ni L, Ju S, Wu X, Zhu H and Shi W, 2012. Clinical significance and prognostic value of PAX3 expression in human glioma. J Mol Neurosci 47, 52–8. [DOI] [PubMed] [Google Scholar]
- Chen K, Zhan Y, Wu X, Zong L and Jiang H, 2018. Germinal mosaicism of PAX3 mutation caused Waardenburg syndrome type I. Int J Pediatr Otorhinolaryngol 104, 200–204. [DOI] [PubMed] [Google Scholar]
- Crist CG, Montarras D, Pallafacchina G, Rocancourt D, Cumano A, Conway SJ and Buckingham M, 2009. Muscle stem cell behavior is modified by microRNA-27 regulation of Pax3 expression. Proc Natl Acad Sci U S A 106, 13383–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Czerny T, Schaffner G and Busslinger M, 1993. DNA sequence recognition by Pax proteins: bipartite structure of the paired domain and its binding site. Genes Dev 7, 2048–61. [DOI] [PubMed] [Google Scholar]
- Daston G, Lamar E, Olivier M and Goulding M, 1996. Pax-3 is necessary for migration but not differentiation of limb muscle precursors in the mouse. Development 122, 1017–1027. [DOI] [PubMed] [Google Scholar]
- Davicioni E, Finckenstein FG, Shahbazian V, Buckley JD, Triche TJ and Anderson MJ, 2006. Identification of a PAX-FKHR gene expression signature that defines molecular classes and determines the prognosis of alveolar rhabdomyosarcomas. Cancer Res 66, 6936–46. [DOI] [PubMed] [Google Scholar]
- Davis RJ and Barr FG, 1997. Fusion genes resulting from alternative chromosomal translocations are overexpressed by gene-specific mechanisms in alveolar rhabdomyosarcoma. Proc Natl Acad Sci U S A 94, 8047–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis RJ, Bennicelli JL, Macina RA, Nycum LM, Biegel JA and Barr FG, 1995. Structural characterization of the FKHR gene and its rearrangement in alveolar rhabdomyosarcoma. Hum Mol Genet 4, 2355–62. [DOI] [PubMed] [Google Scholar]
- Degenhardt KR, Milewski RC, Padmanabhan A, Miller M, Singh MK, Lang D, Engleka KA, Wu M, Li J, Zhou D, Antonucci N, Li L and Epstein JA, 2010. Distinct enhancers at the Pax3 locus can function redundantly to regulate neural tube and neural crest expressions. Dev Biol 339, 519–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diao Y, Guo X, Li Y, Sun K, Lu L, Jiang L, Fu X, Zhu H, Sun H, Wang H and Wu Z, 2012. Pax3/7BP is a Pax7- and Pax3-binding protein that regulates the proliferation of muscle precursor cells by an epigenetic mechanism. Cell Stem Cell 11, 231–41. [DOI] [PubMed] [Google Scholar]
- Dietz KN, Miller PJ and Hollenbach AD, 2009. Phosphorylation of serine 205 by the protein kinase CK2 persists on Pax3-FOXO1, but not Pax3, throughout early myogenic differentiation. Biochemistry 48, 11786–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dietz KN, Miller PJ, Iyengar AS, Loupe JM and Hollenbach AD, 2011. Identification of serines 201 and 209 as sites of Pax3 phosphorylation and the altered phosphorylation status of Pax3-FOXO1 during early myogenic differentiation. Int J Biochem Cell Biol 43, 936–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Douglass EC, Valentine M, Etcubanas E, Parham D, Webber BL, Houghton PJ, Houghton JA and Green AA, 1987. A specific chromosomal abnormality in rhabdomyosarcoma. Cytogenet Cell Genet 45, 148–55. [DOI] [PubMed] [Google Scholar]
- Du S, Lawrence EJ, Strzelecki D, Rajput P, Xia SJ, Gottesman DM and Barr FG, 2005. Co-expression of alternatively spliced forms of PAX3, PAX7, PAX3-FKHR and PAX7-FKHR with distinct DNA binding and transactivation properties in rhabdomyosarcoma. Int J Cancer 115, 85–92. [DOI] [PubMed] [Google Scholar]
- Duan F, Smith LM, Gustafson DM, Zhang C, Dunlevy MJ, Gastier-Foster JM and Barr FG, 2012. Genomic and clinical analysis of fusion gene amplification in rhabdomyosarcoma: a report from the Children’s Oncology Group. Genes Chromosomes Cancer 51, 662–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebauer M, Wachtel M, Niggli FK and Schafer BW, 2007. Comparative expression profiling identifies an in vivo target gene signature with TFAP2B as a mediator of the survival function of PAX3/FKHR. Oncogene 26, 7267–81. [DOI] [PubMed] [Google Scholar]
- Epstein DJ, Vekemans M and Gros P, 1991. Splotch (Sp2h), a Mutation Affecting Development of the Mouse Neural-Tube, Shows a Deletion within the Paired Homeodomain of Pax-3. Cell 67, 767–774. [DOI] [PubMed] [Google Scholar]
- Epstein J, Cai J, Glaser T, Jepeal L and Maas R, 1994a. Identification of a Pax paired domain recognition sequence and evidence for DNA-dependent conformational changes. J Biol Chem 269, 8355–61. [PubMed] [Google Scholar]
- Epstein JA, Cai JX and Maas RM, 1994b. Pax3 Recognizes a Sequence within the 3’utr of the Murine Neurofibromatosis Gene Nf1. Circulation 90, 635–635. [Google Scholar]
- Epstein JA, Lam P, Jepeal L, Maas RL and Shapiro DN, 1995. Pax3 Inhibits Myogenic Differentiation of Cultured Myoblast Cells. Journal of Biological Chemistry 270, 11719–11722. [DOI] [PubMed] [Google Scholar]
- Epstein JA, Shapiro DN, Cheng J, Lam PY and Maas RL, 1996. Pax3 modulates expression of the c-Met receptor during limb muscle development. Proc Natl Acad Sci U S A 93, 4213–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farrer LA, Arnos KS, Asher JH Jr., Baldwin CT, Diehl SR, Friedman TB, Greenberg J, Grundfast KM, Hoth C, Lalwani AK and et al. , 1994. Locus heterogeneity for Waardenburg syndrome is predictive of clinical subtypes. Am J Hum Genet 55, 728–37. [PMC free article] [PubMed] [Google Scholar]
- Franz T, Kothary R, Surani MAH, Halata Z and Grim M, 1993. The Splotch Mutation Interferes with Muscle Development in the Limbs. Anatomy and Embryology 187, 153–160. [DOI] [PubMed] [Google Scholar]
- Frascella E, Toffolatti L and Rosolen A, 1998. Normal and rearranged PAX3 expression in human rhabdomyosarcoma. Cancer Genet Cytogenet 102, 104–9. [DOI] [PubMed] [Google Scholar]
- Fredericks WJ, Galili N, Mukhopadhyay S, Rovera G, Bennicelli J, Barr FG and Rauscher FJ 3rd, 1995. The PAX3-FKHR fusion protein created by the t(2;13) translocation in alveolar rhabdomyosarcomas is a more potent transcriptional activator than PAX3. Mol Cell Biol 15, 1522–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freyer L, Aggarwal V and Morrow BE, 2011. Dual embryonic origin of the mammalian otic vesicle forming the inner ear. Development 138, 5403–5414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fritchie KJ, Jin L, Wang X, Graham RP, Torbenson MS, Lewis JE, Rivera M, Garcia JJ, Schembri-Wismayer DJ, Westendorf JJ, Chou MM, Dong J and Oliveira AM, 2016. Fusion gene profile of biphenotypic sinonasal sarcoma: an analysis of 44 cases. Histopathology 69, 930–936. [DOI] [PubMed] [Google Scholar]
- Galibert MD, Yavuzer U, Dexter TJ and Goding CR, 1999. Pax3 and regulation of the melanocyte-specific tyrosinase-related protein-1 promoter. J Biol Chem 274, 26894–900. [DOI] [PubMed] [Google Scholar]
- Galili N, Davis RJ, Fredericks WJ, Mukhopadhyay S, Rauscher FJ 3rd, Emanuel BS, Rovera G and Barr FG, 1993. Fusion of a fork head domain gene to PAX3 in the solid tumour alveolar rhabdomyosarcoma. Nat Genet 5, 230–5. [DOI] [PubMed] [Google Scholar]
- Gershon TR, Oppenheimer O, Chin SS and Gerald WL, 2005. Temporally regulated neural crest transcription factors distinguish neuroectodermal tumors of varying malignancy and differentiation. Neoplasia 7, 575–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glass RM, 1990. The progress and prospects of psychiatry. JAMA 264, 2549–50. [PubMed] [Google Scholar]
- Goljanek-Whysall K, Sweetman D, Abu-Elmagd M, Chapnik E, Dalmay T, Hornstein E and Munsterberg A, 2011. MicroRNA regulation of the paired-box transcription factor Pax3 confers robustness to developmental timing of myogenesis. Proc Natl Acad Sci U S A 108, 11936–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goulding M, Lumsden A and Paquette AJ, 1994. Regulation of Pax-3 Expression in the Dermomyotome and Its Role in Muscle Development. Development 120, 957–971. [DOI] [PubMed] [Google Scholar]
- Goulding MD, Chalepakis G, Deutsch U, Erselius JR and Gruss P, 1991. Pax-3, a Novel Murine DNA-Binding Protein Expressed during Early Neurogenesis. Embo Journal 10, 1135–1147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goulding MD, Lumsden A and Gruss P, 1993. Signals from the notochord and floor plate regulate the region-specific expression of two Pax genes in the developing spinal cord. Development 117, 1001–16. [DOI] [PubMed] [Google Scholar]
- Graf Finckenstein F, Shahbazian V, Davicioni E, Ren YX and Anderson MJ, 2008. PAX-FKHR function as pangenes by simultaneously inducing and inhibiting myogenesis. Oncogene 27, 2004–14. [DOI] [PubMed] [Google Scholar]
- Gryder BE, Yohe ME, Chou HC, Zhang X, Marques J, Wachtel M, Schaefer B, Sen N, Song Y, Gualtieri A, Pomella S, Rota R, Cleveland A, Wen X, Sindiri S, Wei JS, Barr FG, Das S, Andresson T, Guha R, Lal-Nag M, Ferrer M, Shern JF, Zhao K, Thomas CJ and Khan J, 2017. PAX3-FOXO1 Establishes Myogenic Super Enhancers and Confers BET Bromodomain Vulnerability. Cancer Discov 7, 884–899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanna JA, Garcia MR, Lardennois A, Leavey PJ, Maglic D, Fagnan A, Go JC, Roach J, Wang YD, Finkelstein D and Hatley ME, 2018. PAX3-FOXO1 drives miR-486-5p and represses miR-221 contributing to pathogenesis of alveolar rhabdomyosarcoma. Oncogene 37, 1991–2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He S, Yoon HS, Suh BJ and Eccles MR, 2010. PAX3 Is extensively expressed in benign and malignant tissues of the melanocytic lineage in humans. J Invest Dermatol 130, 1465–8. [DOI] [PubMed] [Google Scholar]
- He SJ, Stevens G, Braithwaite AW and Eccles MR, 2005. Transfection of melanoma cells with antisense PAX3 oligonucleotides additively complements cisplatin-induced cytotoxicity. Mol Cancer Ther 4, 996–1003. [DOI] [PubMed] [Google Scholar]
- Himeda CL, Barro MV and Emerson CP Jr., 2013. Pax3 synergizes with Gli2 and Zic1 in transactivating the Myf5 epaxial somite enhancer. Dev Biol 383, 7–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirai H, Verma M, Watanabe S, Tastad C, Asakura Y and Asakura A, 2010. MyoD regulates apoptosis of myoblasts through microRNA-mediated down-regulation of Pax3. J Cell Biol 191, 347–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hollenbach AD, McPherson CJ, Lagutina I and Grosveld G, 2002. The EF-hand calcium-binding protein calmyrin inhibits the transcriptional and DNA-binding activity of Pax3. Biochim Biophys Acta 1574, 321–8. [DOI] [PubMed] [Google Scholar]
- Hollenbach AD, Sublett JE, McPherson CJ and Grosveld G, 1999. The Pax3-FKHR oncoprotein is unresponsive to the Pax3-associated repressor hDaxx. EMBO J 18, 3702–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hornyak TJ, Hayes DJ, Chiu LY and Ziff EB, 2001. Transcription factors in melanocyte development: distinct roles for Pax-3 and Mitf. Mechanisms of Development 101, 47–59. [DOI] [PubMed] [Google Scholar]
- Hoth CF, Milunsky A, Lipsky N, Sheffer R, Clarren SK and Baldwin CT, 1993. Mutations in the paired domain of the human PAX3 gene cause Klein-Waardenburg syndrome (WS-III) as well as Waardenburg syndrome type I (WS-I). Am J Hum Genet 52, 455–62. [PMC free article] [PubMed] [Google Scholar]
- Hsieh MJ, Yao YL, Lai IL and Yang WM, 2006. Transcriptional repression activity of PAX3 is modulated by competition between corepressor KAP1 and heterochromatin protein 1. Biochem Biophys Res Commun 349, 573–81. [DOI] [PubMed] [Google Scholar]
- Huang SC, Ghossein RA, Bishop JA, Zhang L, Chen TC, Huang HY and Antonescu CR, 2016. Novel PAX3-NCOA1 Fusions in Biphenotypic Sinonasal Sarcoma With Focal Rhabdomyoblastic Differentiation. Am J Surg Pathol 40, 51–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hueber PA, Fukuzawa R, Elkares R, Chu L, Blumentkrantz M, He SJ, Anaka MR, Reeve AE, Eccles M, Jabado N, Iglesias DM and Goodyer PR, 2009. PAX3 is expressed in the stromal compartment of the developing kidney and in Wilms tumors with myogenic phenotype. Pediatr Dev Pathol 12, 347–54. [DOI] [PubMed] [Google Scholar]
- Ichi S, Boshnjaku V, Shen YW, Mania-Farnell B, Ahlgren S, Sapru S, Mansukhani N, McLone DG, Tomita T and Mayanil CS, 2011. Role of Pax3 acetylation in the regulation of Hes1 and Neurog2. Mol Biol Cell 22, 503–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iyengar AS, Loupe JM, Miller PJ and Hollenbach AD, 2012. Identification of CK2 as the kinase that phosphorylates Pax3 at Ser209 in early myogenic differentiation. Biochem Biophys Res Commun 428, 24–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iyengar AS, Miller PJ, Loupe JM and Hollenbach AD, 2014. Phosphorylation of PAX3 contributes to melanoma phenotypes by affecting proliferation, invasion, and transformation. Pigment Cell Melanoma Res 27, 846–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jalilian N, Tabatabaiefar MA, Farhadi M, Bahrami T and Noori-Daloii MR, 2015. A novel mutation in the PAX3 gene causes Waardenburg syndrome type I in an Iranian family. Int J Pediatr Otorhinolaryngol 79, 1736–40. [DOI] [PubMed] [Google Scholar]
- Jang MA, Lee T, Lee J, Cho EH and Ki CS, 2015. Identification of a Novel De Novo Variant in the PAX3 Gene in Waardenburg Syndrome by Diagnostic Exome Sequencing: The First Molecular Diagnosis in Korea. Ann Lab Med 35, 362–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keller C, Arenkiel BR, Coffin CM, El-Bardeesy N, DePinho RA and Capecchi MR, 2004. Alveolar rhabdomyosarcomas in conditional Pax3:Fkhr mice: cooperativity of Ink4a/ARF and Trp53 loss of function. Genes Dev 18, 2614–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kikuchi K, Tsuchiya K, Otabe O, Gotoh T, Tamura S, Katsumi Y, Yagyu S, Tsubai-Shimizu S, Miyachi M, Iehara T and Hosoi H, 2008. Effects of PAX3-FKHR on malignant phenotypes in alveolar rhabdomyosarcoma. Biochem Biophys Res Commun 365, 568–74. [DOI] [PubMed] [Google Scholar]
- Kim H, Ankamreddy H, Lee DJ, Kong KA, Ko HW, Kim MH and Bok J, 2014. Pax3 function is required specifically for inner ear structures with melanogenic fates. Biochemical and Biophysical Research Communications 445, 608–614. [DOI] [PubMed] [Google Scholar]
- Kioussi C, Gross MK and Gruss P, 1995. Pax3 - a Paired Domain Gene as a Regulator in Pns Myelination. Neuron 15, 553–562. [DOI] [PubMed] [Google Scholar]
- Knecht AK and Bronner-Fraser M, 2002. Induction of the neural crest: A multigene process. Nature Reviews Genetics 3, 453–461. [DOI] [PubMed] [Google Scholar]
- Kubic JD, Little EC, Lui JW, Iizuka T and Lang D, 2015a. PAX3 and ETS1 synergistically activate MET expression in melanoma cells. Oncogene 34, 4964–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubic JD, Lui JW, Little EC, Ludvik AE, Konda S, Salgia R, Aplin AE and Lang D, 2015b. PAX3 and FOXD3 Promote CXCR4 Expression in Melanoma. J Biol Chem 290, 21901–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubic JD, Mascarenhas JB, lizuka T, Wolfgeher D and Lang D, 2012. GSK-3 promotes cell survival, growth, and PAX3 levels in human melanoma cells. Mol Cancer Res 10, 1065–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kucharczuk KL, Love CM, Dougherty NM and Goldhamer DJ, 1999. Fine-scale transgenic mapping of the MyoD core enhancer: MyoD is regulated by distinct but overlapping mechanisms in myotomal and non-myotomal muscle lineages. Development 126, 1957–65. [DOI] [PubMed] [Google Scholar]
- Lagha M, Kormish JD, Rocancourt D, Manceau M, Epstein JA, Zaret KS, Relaix F and Buckingham ME, 2008. Pax3 regulation of FGF signaling affects the progression of embryonic progenitor cells into the myogenic program. Genes Dev 22, 1828–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lakkis MM, Golden JA, O’Shea KS and Epstein JA, 1999. Neurofibromin deficiency in mice causes exencephaly and is a modifier for Splotch neural tube defects. Dev Biol 212, 80–92. [DOI] [PubMed] [Google Scholar]
- Lam PY, Sublett JE, Hollenbach AD and Roussel MF, 1999. The oncogenic potential of the Pax3-FKHR fusion protein requires the Pax3 homeodomain recognition helix but not the Pax3 paired-box DNA binding domain. Mol Cell Biol 19, 594–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lang D and Epstein JA, 2003. Sox10 and Pax3 physically interact to mediate activation of a conserved c-RET enhancer. Hum Mol Genet 12, 937–45. [DOI] [PubMed] [Google Scholar]
- Lang D, Lu MM, Huang L, Engleka KA, Zhang M, Chu EY, Lipner S, Skoultchi A, Millar SE and Epstein JA, 2005. Pax3 functions at a nodal point in melanocyte stem cell differentiation. Nature 433, 884–7. [DOI] [PubMed] [Google Scholar]
- Lewis JT, Oliveira AM, Nascimento AG, Schembri-Wismayer D, Moore EA, Olsen KD, Garcia JG, Lonzo ML and Lewis JE, 2012. Low-grade sinonasal sarcoma with neural and myogenic features: a clinicopathologic analysis of 28 cases. Am J Surg Pathol 36, 517–25. [DOI] [PubMed] [Google Scholar]
- Libura J, Drukala J, Majka M, Tomescu O, Navenot JM, Kucia M, Marquez L, Peiper SC, Barr FG, Janowska-Wieczorek A and Ratajczak MZ, 2002. CXCR4-SDF-1 signaling is active in rhabdomyosarcoma cells and regulates locomotion, chemotaxis, and adhesion. Blood 100, 2597–606. [DOI] [PubMed] [Google Scholar]
- Liu Q, Yang G and Qian Y, 2017. Loss of MicroRNA-489–3p promotes osteosarcoma metastasis by activating PAX3-MET pathway. Mol Carcinog 56, 1312–1321. [DOI] [PubMed] [Google Scholar]
- Loupe JM, Miller PJ, Bonner BP, Maggi EC, Vijayaraghavan J, Crabtree JS, Taylor CM, Zabaleta J and Hollenbach AD, 2016a. Comparative transcriptomic analysis reveals the oncogenic fusion protein PAX3-FOXO1 globally alters mRNA and miRNA to enhance myoblast invasion. Oncogenesis 5, e246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loupe JM, Miller PJ, Bonner BP, Maggi EC, Vijayaraghavan J, Zabaleta J, Taylor CM, Tsien F, Crabtree JS and Hollenbach AD, 2016b. Acquisition of an oncogenic fusion protein serves as an initial driving mutation by inducing aneuploidy and overriding proliferative defects. Oncotarget 7, 62814–62835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loupe JM, Miller PJ, Crabtree JS, Zabaleta J and Hollenbach AD, 2017. Acquisition of an oncogenic fusion protein is sufficient to globally alter the landscape of miRNA expression to inhibit myogenic differentiation. Oncotarget 8, 87054–87072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loupe JM, Miller PJ, Ruffin DR, Stark MW and Hollenbach AD, 2015. Inhibiting phosphorylation of the oncogenic PAX3-FOXO1 reduces alveolar rhabdomyosarcoma phenotypes identifying novel therapy options. Oncogenesis 4, e145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lozano-Velasco E, Contreras A, Crist C, Hernandez-Torres F, Franco D and Aranega AE, 2011. Pitx2c modulates Pax3+/Pax7+ cell populations and regulates Pax3 expression by repressing miR27 expression during myogenesis. Dev Biol 357, 165–78. [DOI] [PubMed] [Google Scholar]
- Magnaghi P, Roberts C, Lorain S, Lipinski M and Scambler PJ, 1998. HIRA, a mammalian homologue of Saccharomyces cerevisiae transcriptional co-repressors, interacts with Pax3. Nat Genet 20, 74–7. [DOI] [PubMed] [Google Scholar]
- Manderfield LJ, Engleka KA, Aghajanian H, Gupta M, Yang S, Li L, Baggs JE, Hogenesch JB, Olson EN and Epstein JA, 2014. Pax3 and hippo signaling coordinate melanocyte gene expression in neural crest. Cell Rep 9, 1885–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marshall AD, Lagutina I and Grosveld GC, 2011. PAX3-FOXO1 induces cannabinoid receptor 1 to enhance cell invasion and metastasis. Cancer Res 71, 7471–80. [DOI] [PubMed] [Google Scholar]
- Mascarenhas JB, Littlejohn EL, Wolsky RJ, Young KP, Nelson M, Salgia R and Lang D, 2010. PAX3 and SOX10 activate MET receptor expression in melanoma. Pigment Cell Melanoma Res 23, 225–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuzaki Y, Hashimoto S, Fujita T, Suzuki T, Sakurai T, Matsushima K and Kawakami Y, 2005. Systematic identification of human melanoma antigens using serial analysis of gene expression (SAGE). J Immunother 28, 10–9. [DOI] [PubMed] [Google Scholar]
- Mayanil CS, George D, Freilich L, Miljan EJ, Mania-Farnell B, McLone DG and Bremer EG, 2001. Microarray analysis detects novel Pax3 downstream target genes. J Biol Chem 276, 49299–309. [DOI] [PubMed] [Google Scholar]
- Milet C and Monsoro-Burq AH, 2012. Neural crest induction at the neural plate border in vertebrates. Developmental Biology 366, 22–33. [DOI] [PubMed] [Google Scholar]
- Moase CE and Trasler DG, 1992. Splotch Locus Mouse Mutants - Models for Neural-Tube Defects and Waardenburg Syndrome Type-I in Humans. Journal of Medical Genetics 29, 145–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monsoro-Burq AH, Wang E and Harland R, 2005. Msx1 and Pax3 cooperate to mediate FGF8 and WNT signals during Xenopus neural crest induction. Developmental Cell 8, 167–178. [DOI] [PubMed] [Google Scholar]
- Moore S, Ribes V, Terriente J, Wilkinson D, Relaix F and Briscoe J, 2013. Distinct regulatory mechanisms act to establish and maintain Pax3 expression in the developing neural tube. PLoS Genet 9, e1003811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Motohashi N, Alexander MS, Casar JC and Kunkel LM, 2012. Identification of a novel microRNA that regulates the proliferation and differentiation in muscle side population cells. Stem Cells Dev 21, 3031–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murakami M, Tominaga J, Makita R, Uchijima Y, Kurihara Y, Nakagawa O, Asano T and Kurihara H, 2006. Transcriptional activity of Pax3 is co-activated by TAZ. Biochem Biophys Res Commun 339, 533–9. [DOI] [PubMed] [Google Scholar]
- Nabarro S, Himoudi N, Papanastasiou A, Gilmour K, Gibson S, Sebire N, Thrasher A, Blundell MP, Hubank M, Canderan G and Anderson J, 2005. Coordinated oncogenic transformation and inhibition of host immune responses by the PAX3-FKHR fusion oncoprotein. J Exp Med 202, 1399–410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naini S, Etheridge KT, Adam SJ, Qualman SJ, Bentley RC, Counter CM and Linardic CM, 2008. Defining the cooperative genetic changes that temporally drive alveolar rhabdomyosarcoma. Cancer Res 68, 9583–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Natoli TA, Ellsworth MK, Wu C, Gross KW and Pruitt SC, 1997. Positive and negative DNA sequence elements are required to establish the pattern of Pax3 expression. Development 124, 617–26. [DOI] [PubMed] [Google Scholar]
- Pandey PR, Chatterjee B, Olanich ME, Khan J, Miettinen MM, Hewitt SM and Barr FG, 2017. PAX3-FOXO1 is essential for tumour initiation and maintenance but not recurrence in a human myoblast model of rhabdomyosarcoma. J Pathol 241, 626–637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pani L, Horal M and Loeken MR, 2002. Rescue of neural tube defects in Pax-3-deficient embryos by p53 loss of function: implications for Pax-3-dependent development and tumorigenesis. Genes Dev 16, 676–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker CJ, Shawcross SG, Li H, Wang QY, Herrington CS, Kumar S, MacKie RM, Prime W, Rennie IG, Sisley K and Kumar P, 2004. Expression of PAX 3 alternatively spliced transcripts and identification of two new isoforms in human tumors of neural crest origin. Int J Cancer 108, 314–20. [DOI] [PubMed] [Google Scholar]
- Phelan ML, Rambaldi I and Featherstone MS, 1995. Cooperative interactions between HOX and PBX proteins mediated by a conserved peptide motif. Mol Cell Biol 15, 3989–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phelan SA and Loeken MR, 1998. Identification of a new binding motif for the paired domain of Pax-3 and unusual characteristics of spacing of bipartite recognition elements on binding and transcription activation. J Biol Chem 273, 19153–9. [DOI] [PubMed] [Google Scholar]
- Pingault V, Ente D, Dastot-Le Moal F, Goossens M, Marlin S and Bondurand N, 2010. Review and update of mutations causing Waardenburg syndrome. Hum Mutat 31, 391–406. [DOI] [PubMed] [Google Scholar]
- Pritchard C, Grosveld G and Hollenbach AD, 2003. Alternative splicing of Pax3 produces a transcriptionally inactive protein. Gene 305, 61–9. [DOI] [PubMed] [Google Scholar]
- Pruitt SC, Bussman A, Maslov AY, Natoli TA and Heinaman R, 2004. Hox/Pbx and Brn binding sites mediate Pax3 expression in vitro and in vivo. Gene Expr Patterns 4, 671–85. [DOI] [PubMed] [Google Scholar]
- Relaix F, Montarras D, Zaffran S, Gayraud-Morel B, Rocancourt D, Tajbakhsh S, Mansouri A, Cumano A and Buckingham M, 2006. Pax3 and Pax7 have distinct and overlapping functions in adult muscle progenitor cells. Journal of Cell Biology 172, 91–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Relaix F, Rocancourt D, Mansouri A and Buckingham M, 2004. Divergent functions of murine Pax3 and Pax7 in limb muscle development. Genes & Development 18, 1088–1105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russell WL, 1947. Splotch, a New Mutation in the House Mouse, Mus-Musculus. Genetics 32, 102–102. [Google Scholar]
- Sanchez-Ferras O, Coutaud B, Djavanbakht Samani T, Tremblay I, Souchkova O and Pilon N, 2012. Caudal-related homeobox (Cdx) protein-dependent integration of canonical Wnt signaling on paired-box 3 (Pax3) neural crest enhancer. J Biol Chem 287, 16623–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schafer BW, Czerny T, Bernasconi M, Genini M and Busslinger M, 1994. Molecular cloning and characterization of a human PAX-7 cDNA expressed in normal and neoplastic myocytes. Nucleic Acids Res 22, 4574–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scholl FA, Kamarashev J, Murmann OV, Geertsen R, Dummer R and Schafer BW, 2001. PAX3 is expressed in human melanomas and contributes to tumor cell survival. Cancer Res 61, 823–6. [PubMed] [Google Scholar]
- Schulte TW, Toretsky JA, Ress E, Helman L and Neckers LM, 1997. Expression of PAX3 in Ewing’s sarcoma family of tumors. Biochem Mol Med 60, 121–6. [DOI] [PubMed] [Google Scholar]
- Seidal T, Mark J, Hagmar B and Angervall L, 1982. Alveolar rhabdomyosarcoma: a cytogenetic and correlated cytological and histological study. Acta Pathol Microbiol Immunol Scand A 90, 345–54. [DOI] [PubMed] [Google Scholar]
- Shapiro DN, Sublett JE, Li B, Downing JR and Naeve CW, 1993. Fusion of PAX3 to a member of the forkhead family of transcription factors in human alveolar rhabdomyosarcoma. Cancer Res 53, 5108–12. [PubMed] [Google Scholar]
- Shern JF, Chen L, Chmielecki J, Wei JS, Patidar R, Rosenberg M, Ambrogio L, Auclair D, Wang J, Song YK, Tolman C, Hurd L, Liao H, Zhang S, Bogen D, Brohl AS, Sindiri S, Catchpoole D, Badgett T, Getz G, Mora J, Anderson JR, Skapek SX, Barr FG, Meyerson M, Hawkins DS and Khan J, 2014. Comprehensive genomic analysis of rhabdomyosarcoma reveals a landscape of alterations affecting a common genetic axis in fusion-positive and fusion-negative tumors. Cancer Discov 4, 216–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soleimani VD, Punch VG, Kawabe Y, Jones AE, Palidwor GA, Porter CJ, Cross JW, Carvajal JJ, Kockx CE, van IWF, Perkins TJ, Rigby PW, Grosveld F and Rudnicki MA, 2012. Transcriptional dominance of Pax7 in adult myogenesis is due to high-affinity recognition of homeodomain motifs. Dev Cell 22, 1208–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sommer A, Young-Wee T and Frye T, 1983. Previously undescribed syndrome of craniofacial, hand anomalies, and sensorineural deafness. Am J Med Genet 15, 71–7. [DOI] [PubMed] [Google Scholar]
- Stuart ET, Kioussi C and Gruss P, 1994. Mammalian Pax genes. Annu Rev Genet 28, 219–36. [DOI] [PubMed] [Google Scholar]
- Sumegi J, Streblow R, Frayer RW, Dal Cin P, Rosenberg A, Meloni-Ehrig A and Bridge JA, 2010. Recurrent t(2;2) and t(2;8) translocations in rhabdomyosarcoma without the canonical PAX-FOXO1 fuse PAX3 to members of the nuclear receptor transcriptional coactivator family. Genes Chromosomes Cancer 49, 224–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tajbakhsh S and Buckingham M, 2000. The birth of muscle progenitor cells in the mouse: spatiotemporal considerations. Curr Top Dev Biol 48, 225–68. [DOI] [PubMed] [Google Scholar]
- Tajbakhsh S, Rocancourt D, Cossu G and Buckingham M, 1997. Redefining the genetic hierarchies controlling skeletal myogenesis: Pax-3 and Myf-5 act upstream of MyoD. Cell 89, 127–138. [DOI] [PubMed] [Google Scholar]
- Tan WJ, Thike AA, Bay BH and Tan PH, 2014. Immunohistochemical expression of homeoproteins Six1 and Pax3 in breast phyllodes tumours correlates with histological grade and clinical outcome. Histopathology 64, 807–17. [DOI] [PubMed] [Google Scholar]
- Tanaka M and Herr W, 1990. Differential transcriptional activation by Oct-1 and Oct-2: interdependent activation domains induce Oct-2 phosphorylation. Cell 60, 375–86. [DOI] [PubMed] [Google Scholar]
- Tassabehji M, Newton VE, Liu XZ, Brady A, Donnai D, Krajewska-Walasek M, Murday V, Norman A, Obersztyn E, Reardon W and et al. , 1995. The mutational spectrum in Waardenburg syndrome. Hum Mol Genet 4, 2131–7. [DOI] [PubMed] [Google Scholar]
- Tomescu O, Xia SJ, Strezlecki D, Bennicelli JL, Ginsberg J, Pawel B and Barr FG, 2004. Inducible short-term and stable long-term cell culture systems reveal that the PAX3-FKHR fusion oncoprotein regulates CXCR4, PAX3, and PAX7 expression. Lab Invest 84, 1060–70. [DOI] [PubMed] [Google Scholar]
- Tremblay P, Dietrich S, Mericskay M, Schubert FR, Li ZL and Paulin D, 1998. A crucial role for Pax3 in the development of the hypaxial musculature and the long-range migration of muscle precursors. Developmental Biology 203, 49–61. [DOI] [PubMed] [Google Scholar]
- Tremblay P and Gruss P, 1994. Pax: genes for mice and men. Pharmacol Ther 61, 205–26. [DOI] [PubMed] [Google Scholar]
- Turc-Carel C, Lizard-Nacol S, Justrabo E, Favrot M, Philip T and Tabone E, 1986. Consistent chromosomal translocation in alveolar rhabdomyosarcoma. Cancer Genet Cytogenet 19, 361–2. [DOI] [PubMed] [Google Scholar]
- Underhill DA and Gros P, 1997. The paired-domain regulates DNA binding by the homeodomain within the intact Pax-3 protein. J Biol Chem 272, 14175–82. [DOI] [PubMed] [Google Scholar]
- Vogan KJ, Underhill DA and Gros P, 1996. An alternative splicing event in the Pax-3 paired domain identifies the linker region as a key determinant of paired domain DNA-binding activity. Mol Cell Biol 16, 6677–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vohra S and Biggin PC, 2013. Mutationmapper: a tool to aid the mapping of protein mutation data. PLoS One 8, e71711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waardenburg PJ, 1951. A new syndrome combining developmental anomalies of the eyelids, eyebrows and nose root with pigmentary defects of the iris and head hair and with congenital deafness. Am J Hum Genet 3, 195–253. [PMC free article] [PubMed] [Google Scholar]
- Walters ZS, Villarejo-Balcells B, Olmos D, Buist TW, Missiaglia E, Allen R, Al-Lazikani B, Garrett MD, Blagg J and Shipley J, 2014. JARID2 is a direct target of the PAX3-FOXO1 fusion protein and inhibits myogenic differentiation of rhabdomyosarcoma cells. Oncogene 33, 1148–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Li S, Xiao X, Wang P, Guo X and Zhang Q, 2010. PAX3 mutations and clinical characteristics in Chinese patients with Waardenburg syndrome type 1. Mol Vis 16, 1146–53. [PMC free article] [PubMed] [Google Scholar]
- Wang Q, Kumar S, Mitsios N, Slevin M and Kumar P, 2007. Investigation of downstream target genes of PAX3c, PAX3e and PAX3g isoforms in melanocytes by microarray analysis. Int J Cancer 120, 1223–31. [DOI] [PubMed] [Google Scholar]
- Wang Q, Kumar S, Slevin M and Kumar P, 2006. Functional analysis of alternative isoforms of the transcription factor PAX3 in melanocytes in vitro. Cancer Res 66, 8574–80. [DOI] [PubMed] [Google Scholar]
- Wang X, Bledsoe KL, Graham RP, Asmann YW, Viswanatha DS, Lewis JE, Lewis JT, Chou MM, Yaszemski MJ, Jen J, Westendorf JJ and Oliveira AM, 2014. Recurrent PAX3-MAML3 fusion in biphenotypic sinonasal sarcoma. Nat Genet 46, 666–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang XD, Morgan SC and Loeken MR, 2011. Pax3 stimulates p53 ubiquitination and degradation independent of transcription. PLoS One 6, e29379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang-Wuu S, Soukup S, Ballard E, Gotwals B and Lampkin B, 1988. Chromosomal analysis of sixteen human rhabdomyosarcomas. Cancer Res 48, 983–7. [PubMed] [Google Scholar]
- Watanabe A, Takeda K, Ploplis B and Tachibana M, 1998. Epistatic relationship between Waardenburg syndrome genes MITF and PAX3. Nat Genet 18, 283–6. [DOI] [PubMed] [Google Scholar]
- Wexler LH HL, 1997. Principles and Practices of Pediatric Oncology, Lippincott-Raven, Philadelphia. [Google Scholar]
- Wiggan O, Taniguchi-Sidle A and Hamel PA, 1998. Interaction of the pRB-family proteins with factors containing paired-like homeodomains. Oncogene 16, 227–36. [DOI] [PubMed] [Google Scholar]
- Williams BA and Ordahl CP, 1994. Pax-3 expression in segmental mesoderm marks early stages in myogenic cell specification. Development 120, 785–96. [DOI] [PubMed] [Google Scholar]
- Wilson D, Sheng G, Lecuit T, Dostatni N and Desplan C, 1993. Cooperative dimerization of paired class homeo domains on DNA. Genes Dev 7, 2120–34. [DOI] [PubMed] [Google Scholar]
- Wong WJ, Lauria A, Hornick JL, Xiao S, Fletcher JA and Marino-Enriquez A, 2016. Alternate PAX3-FOXO1 oncogenic fusion in biphenotypic sinonasal sarcoma. Genes Chromosomes Cancer 55, 25–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu TF, Yao YL, Lai IL, Lai CC, Lin PL and Yang WM, 2015. Loading of PAX3 to Mitotic Chromosomes Is Mediated by Arginine Methylation and Associated with Waardenburg Syndrome. J Biol Chem 290, 20556–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia L, Huang Q, Nie D, Shi J, Gong M, Wu B, Gong P, Zhao L, Zuo H, Ju S, Chen J and Shi W, 2013. PAX3 is overexpressed in human glioblastomas and critically regulates the tumorigenicity of glioma cells. Brain Res 1521, 68–78. [DOI] [PubMed] [Google Scholar]
- Xia SJ, Holder DD, Pawel BR, Zhang C and Barr FG, 2009. High expression of the PAX3-FKHR oncoprotein is required to promote tumorigenesis of human myoblasts. Am J Pathol 175, 2600–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao Y, Luo J, Zhang F, Li J, Han Y, Zhang D, Wang M, Ma Y, Xu L, Bai X and Wang H, 2016. A novel mutation in PAX3 associated with Waardenburg syndrome type I in a Chinese family. Acta Otolaryngol 136, 439–45. [DOI] [PubMed] [Google Scholar]
- Yang XM, Vogan K, Gros P and Park M, 1996. Expression of the met receptor tyrosine kinase in muscle progenitor cells in somites and limbs is absent in Splotch mice. Development 122, 2163–2171. [DOI] [PubMed] [Google Scholar]
- Zhang L, Xia L, Zhao L, Chen Z, Shang X, Xin J, Liu M, Guo X, Wu K, Pan Y and Fan D, 2015. Activation of PAX3-MET pathways due to miR-206 loss promotes gastric cancer metastasis. Carcinogenesis 36, 390–9. [DOI] [PubMed] [Google Scholar]