

Abstract
Enrichment of modified peptides from global peptides is inevitable in mass spectrometric analysis protein modifications because of their importance in the study of cellular functions and low abundance in the global proteomic analysis. Recent advances in enrichment methods for modified peptides such as phosphopeptides and intact glycopeptides (IGPs) show that the methods for proteomic analyses of both protein modifications are robust. We have recently observed and reported a large number of IGPs from phosphoproteomic analysis using IMAC-based phosphopeptides enrichment procedure. To determine whether phosphorylated peptides could be specifically isolated from coenriched IGPs in IMAC experiments with different pH, IMAC procedures were performed at different pH conditions, and we found that the enrichment of phosphopeptides at pH 2.0 was the optimal condition for having the highest number of phosphopeptide identifications; however, coenrichment of phosphopeptides and glycopeptides was inevitable in the entire pH range. The hydrophilic enrichments of IGPs performed before or after IMAC enrichment were evaluated subsequently to determine the optimal workflow for simultaneous analyses of phosphopeptides and glycopeptides, and IMAC enrichment followed by hydrophilic enrichment was chosen as the optimized workflow. Applying the workflow to the TMT-labeled peptides from luminal and basal-like type of breast cancer patient-derived xenograft (PDX) models allowed quantitative analyses of phospho- and glycoproteomics with 17582 phosphopeptides and 3468 glycopeptides identified, and 1237 phosphopeptides and 236 glycopeptides showed significant expression differences between luminal and basal-like, respectively. This method allows simultaneous analyses of phosphoprotein and glycoprotein modifications, extending our understanding of roles of glycosylation and phosphorylation in biology and diseases.
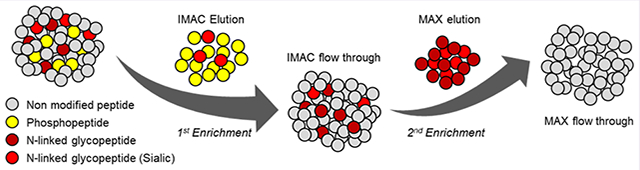
Graphical Abstract
INTRODUCTION
Phosphorylation and glycosylation are among the most common protein modifications occurring in a cellular system. In a mammalian cell, especially, about 30% and 50% of all proteins can have phosphorylation and glycosylation.1 These modifications play an important role in regulating signal transduction, protein stability, and protein−protein interaction. For example, N-linked glycosylation of membrane protein regulates the activity of the extracellular domain, and phosphorylation of tyrosine kinase mediates signaling pathways. Furthermore, it has been found that aberrant glycosylation and phosphorylation is related to various cancer such as breast, ovarian, and so on.2−4 Therefore, achieving knowledge of phosphorylation and glycosylation in cancer is not only a key to clarifying the pathogenesis but also potentially a great help in the treatment and diagnosis of cancer.
Even though the mass spectrometry is one of the most advanced technologies developed to date to analyze protein medications, there are challenges to analyze phosphorylated and glycosylated peptides because of their low abundance and ion suppression in the phospho- and glyco- proteomic analysis.5,6 Therefore, it is necessary for the specific enrichments of modified peptides prior to MS analysis. Various enrichment methods have been developed to isolate phosphopeptides such as metal oxide affnity chromatography (MOAC),7 immobilized metal ion affnity chromatography (IMAC),8,9 ion-exchange chromatography,10 and immunoprecipitation.11−13 Among these methods, IMAC technique is widely used due to its simple process, independent of the phosphopeptide type, and easy for MS analysis. This method is based on negatively charged molecules that bind to a metal ion (Fe3+) and enrich the phosphate group (-PO3−) by neutralizing the carboxyl groups of aspartic acid, glutamic acid, and the C-terminus at low pH and high organic solvent conditions. However, the carboxyl groups present in sialic acid has a pKa value different from that of the carboxyl groups present in the amino acid due to electronegativity and has a pKa value similar to that of a phosphate group.14 To this end, several studies have attempted to separate sialylated glycopeptides and phosphopeptides.15,16 Lasen et al. reported the novel enrichment method for specific sialylated peptides using titanium oxide (TiO2). In this method, enrichment was performed at highly acidic condition after removing the phosphopeptides by the enzyme, and then 192 and 97 of sialylated peptides were identified with high specificity effciency in human plasma and salvia, respectively.17 In our recently published paper, we also confirmed that a large number of glycopeptides, especially sialylated glycopeptides, were identified in the phosphoproteomic analysis using IMAC process.18 Therefore, we hypothesized that that phosphoproteomic analysis using IMAC enrichment also coenriched sialylated glycopeptides at the specific pH condition using for phosphopeptide enrichment, but there is no definite result yet to investigate this coenrichment phenomenon.
Compared with phosphoproteomic analysis, intact glycopeptide analysis is challenging because of the high complexity of assignment of glycopeptides from complicated LC-MS/MS spectra and enrichment process.19 Glycosidases (such as PNGase F) release glycans from peptide/proteins, making it easy to identify peptides backbone for glycosylated sites and glycans, but the glycosylation site-specific glycan information is lost. In recent study, we reported the development of novel normal approach and search algorithms for identification of intact-glycopeptides, called N-linked glycan and glycosite analysis (NGAG) and GPQuest tool based on the identified glycans and glycosites. This approach and search tool combined with analysis of glycans, glycosites, and intact glycopeptides expanded the identification of IGPs.20−23 To enrich intact glycopeptides, hydrophilic interaction chromatography (HILIC);24,25 electrostatic repulsion hydrophilic interaction (ERLIC),26,27 a mixed-mode with hydrophilic, reversed-phase, and anion-exchange capture (such as MAX);28 hydrazine modification;29 lectin affinity chromatography;30 and boronic acid affinity31 have been used to enrich glycosylated peptides. HILIC-based methods for glycopeptide enrichment, particularly N-linked glycopeptides, are a widely used method because of their high reproducibility, convenient experiment process, and absent bias toward glycan species.28
Recently, simultaneous enrichment methods have been investigated for glyco- and phosphopeptides enrichment. Zhang et al.26 optimized the ERLIC method to simultaneously identify both glyco- and phosphopeptides with the identification of 544 and 922 of unique glycopeptides and glycosylated site as well as 383 and 915 of phosphoproteins and phosphorylation sites, respectively. Several multifunctional micromaterials also have been developed that simultaneously select for glyco- and phosphor-peptides. For example, Zou et al. have developed material Ti4 + ions, which is selective for phosphorylation, by coupling it to the hydrophilic material CS @ PGMA @ IDA (chitosan; poly glycidyl methacrylate; iminodiacetic acid).32 The new material was designed to be selective for glycopeptides as well as for phosphopeptides and identified 235 and 256 of glyco- and phosphoproteins. Here we investigated enrichment efficiency of phosphopeptides and IGPs according to pH condition in phosphopeptide enrichment and confirmed the characteristic of coeluted IGPs and ideal pH condition for identification of phosphopeptides. By the addition of glycopeptide enrichment, we also developed and optimized workflow for both phosphopeptides and IGPs, and then optimization workflow has been applied to PDX samples, which were labeled with TMT and fractionated, and identified huge IGPs and phosphopeptides. The optimized method, simultaneous enrichment, is a useful tool to identify and quantify IGPs and phosphopeptides.
RESULTS AND DISCUSSION
Characterization for Identified Phosphopeptides at Different pH Conditions.
To evaluate the characteristics for phosphopeptide enrichment by IMAC at different pH conditions, 0.3 mg of peptide samples was bound to Fe3+- NTA beads and washed at different pH conditions (pH 1−6 and 0.5−5.5), respectively (Figure 1A). Eluents from IMAC were analyzed by LC-MS/MS and searched by SEQUEST algorithm. In results at pH from 1 to 6, the number of phosphopeptides identified and specificity of enrichment at different pH conditions were determined (Figure 1B). The number of peptide spectra matches (PSMs) and unique peptides of phosphopeptides, and phosphopeptide specificity (number of phosphopeptide PSMs/total PSMs) at pH 2 were 14 308, 4519, and 99%, respectively, which were the highest values compared with other conditions. Even though at both pH 1 and 3 conditions also have high specificity value, over 95%, but the number of PSMs and number of peptides of phosphopeptides were about 10% less than those identified at pH 2 condition. Interestingly, at pH 5 and 6, both the number of PSMs of phosphopeptides and specificity were decreased dramatically, but instead, the number of PSMs ssof nonphosphopeptides was increased. Similar to the above results, pH conditions from 0.5 to 5.5, both pH 1.5 and 2.5 have highest PSM number and highest specificity (Supplementary Figure 1A). It was supposed that the iron ions (Fe3+) might no longer have a positive charge in conditions with pH over 4 because pKa value of iron ions on NTA is about 4.3. For this reason, nonspecific binding was increased at from pH 4.5 to 6 conditions.
Figure 1.

Phosphopeptide enrichment at different pH conditions. (A) Workflow of IMAC experiments at different pH conditions. The pH conditions were used from 1 to 6 and from 0.5 to 5.5, respectively. (B) The numbers of identified phospho-PSMs, phosphopeptides and specificity. Specificity is calculated as phospho-PSMs/total PSMs. (C) The percentages of single or multiple phosphorylations in a tryptic peptide. (D) The percentages of phospho-serine, -threonine, and -tyrosine identified.
Multiphosphorylation events per peptide occurred approximately 20% at pH 2 and 3 conditions, but the percentage was increased to 45% at pH 1 condition (Figure 1C). At the pH 1 condition, especially, doubly phosphorylated peptides were increased significantly to 35%. Triple and quadruple phosphorylation peptides were 8% and 2% at pH 1 condition, and these percentages were the highest among the pH conditions tested. In spite of the sustained high specificity at pH 1, the decrease in the total number of phosphopeptides and/or singly phosphopeptides means that it is hard for singly phosphorylated peptide to get the negative ion at pH 1 condition. On the other hand, in the case of multiply phosphorylated peptides, it is considered that the number is increased because there is more opportunity to get negative charge than single phosphorylation. As a result of pH ranging from 0.5 to 5.5, multiply phosphorylated peptides at pH 0.5 were confirmed to be about 25% and about 17% at pH 1.5 to 3.5 (Supplementary Figure 1B). The portion of phosphothreonine and -tyrosine residues changed slightly at high pH condition (Figure 1D). The percentage of threonine and tyrosine residues, which were about 17% and 2% at pH 1−4, were increased to ~20% and ~4% at pH 5 and 6, respectively. In addition, the increasing portion of phospho-threonine and -tyrosine residues at the high pH value can be also confirmed in Supplementary Figure 1C.
Coeluted Intact-Glycopeptides by IMAC.
Phosphopep-tide isolation by IMAC is based on charge interaction between the phosphate group (negative charge) and iron ion group (positive charge). The pKa value of phosphate group is known about 2.1 and carboxylic group of sialic acid, one of the sugar classes, also has a similar pKa value of 2.5. It means that the IGP containing sialic acid group can be cocaptured and eluted with phosphopeptides by IMAC. To determine the IGPs coeluted by IMAC, the spectra containing oxonium ion (204.8 of MS2 ion) were extracted and counted. The 204.8 of oxonium ion, which was HexNAc fragment ion produced by HCD, was used for prediction to spectrum from a glycopeptide.22,33,34 Some oxonium ion spectra which have sufficient MS2 ions were assigned to IGP by GPQuest.22,35 Numbers of oxonium ion spectra and identified glycopeptides were different at different pH conditions (Figure 2A). Unlike enrichment of phosphopeptides, IGPs have the highest number of oxonium ion spectra and glyco-PSM at pH 3 condition, not at pH 2. An average of 8722 of oxonium ion spectra have been detected at pH 3 condition, and an average of 6578 of oxonium ion spectra was detected at pH 2 condition. Even though the number of glycopeptide spectra detected from IMAC enrichment at pH 2 condition was not the highest, a large number of glycopeptides were codetected at pH 2, and this number was ~50% of the number of phospho-PSMs at pH2. Likewise, the number of oxonium ion spectra and the number of glyco-PSMs was highest at pH 3. About 430 of glyco-PSMs were identified at pH 3 condition, and this result showed the IGPs could be coenriched with phosphopeptides at pH 2 and 3. To determine the glycans from the identified intact-glycopeptides, glycopeptides were classified to contain high-mannose, fucosylated without sialylated, sialylated, and others (Figure 2B). Overall, about 54.2%, 31%, or 9.8% of total glycans were identified as glycopeptides containing sialylated, fucosylated without sialylated glycans, or other hybrid or complex glycans, while just 5% of intact glycopeptides containing high-mannose. Especially, about 61.7% of sialylated glycopeptides were identified at pH 2 condition, indicating that selective IGPs, especially sialylated IGPs were coenriched in phosphopeptide enrichment by IMAC. The large percentage of complex glycans (~40%) identified without sialic acids could be possible due to the loss of sialic acids during the sample preparation and/or mass spectrometry analysis.34,36,37 The fucosylated glycopeptide, which is inevitably present in sialic acid glycosylation synthesis mechanism, can be indirectly deduced from the broken form of sialic acid detected on the mass spectrometer. For this reason, it is presumed that a considerable number of fucosylated glycopeptide coeluted in IMAC are from sialylated glycopeptides.
Figure 2.

Characteristics of IGPs according to IMAC experiments at different pH conditions. (A) The numbers of spectra containing oxonium ions (left y-axis) and identified glycopeptides (right y-axis).(B) The numbers of glycopeptide-PSMs and percentages according to the type of glycans in identified glycopeptides.
Workflow for Enrichment of Both Phosphopeptides and Glycopeptides by IMAC and MAX.
According to the above results, it was confirmed that enrichment efficiency of phosphopeptide at IMAC is affected by pH condition because of the negative ion capability of phosphate and competition by other negative ion molecules, especially sialylated glycopeptides. At pH 2 condition, even though a large number of IGPs are coenriched, it is obvious that pH 2 is the best condition to enrich phosphopeptides because of the optimal balance in these factors. To determine the workflow for identification of both phosphopeptides and IGPs, we performed glycopeptide enrichment process using MAX cartridge. The sequential enrichment was performed in two different order: one is to enrich the phosphopeptides with IMAC followed by enrichment with MAX using IMAC flow through or the IMAC and MAX can be performed in opposite order (Figure 3A). The numbers of phosphopeptides and IGPs identified in each method were compared (Figure 3B, supplementary Table 1). In total, 12 433 and 8331 of phospho-PSMs and -peptides were identified, and 192 and 146 of glyco-PSMs and -peptides were identified in IMAC enrichment. In MAX enrichment following IMAC, only about a hundred phosphopeptides were identified, but 611 and 506 of glyco-PSMs and -peptides were identified. Since the phosphopeptides were depleted in the first IMAC, the number of phosphopeptides in the second MAX was almost not identified. In the second MAX, however, a large number of IGPs were identified. A total of 12 563 and 8442 of phospho-PSMs and -peptides and 803 and 652 of glyco-PSMs and -peptides were identified through a sequential enrichment method. In the MAX followed by IMAC enrichment method, approximately 400 of phosphopeptides and 823 and 652 of glyco-PSMs and -peptides were identified in the first MAX experiments. As in the previous experiment, not many phosphopeptides were not identified with IGP enrichment by MAX, but a large number of phospho-PSMs and -peptides were identified at 7941 and 5476, respectively, in IMAC enrichment followed MAX enrichment (Figure 3B). A total of 8592 and 5916 of phospho-PSM and peptides and 824 and 653 of glyco-PSMs and -peptides were identified from IMAC after MAX experiment, which was a number of approximately 4000 and 2400 phospho-PSMs and -peptides decreased compared with the MAX after IMAC experiment. Comparisons of two independent results show that IMAC followed by MAX workflow identified more phosphopeptides and similar IGPs.
Figure 3.

Performances of the two workflows for phosphoproteomic and glycoproteomic analyses. (A) Illustration of the workflow for phosphopeptide and IGPs enrichments using IMAC and MAX. The second enrichment was performed using the flow through from the first one.(B) The numbers of identified phosphopeptides (left y-axis) and IGPs (right y-axis) across two experimental workflows. (C) Glycan profile distribution for identified IGPs. “Sialyl” means sialylated glycopeptide with or without fucosylated form.
In the case of phosphopeptides identified in the first MAX, there were more peptides with relatively lower hydrophobic values and single phosphorylation peptides compared with IMAC (Supplementary Figure 2). Hydrophilic peptides, which include not only IGP but also unmodified and phosphopeptides, were enriched by MAX column and the eluted hydrophilic peptides from MAX are high enough to bury the coeluted hydrophilic phosphopeptides. In addition, the decreasing ratio of multiple phosphorylation peptides shows that the phosphate group still remains a negative charge in the MAX column. The relatively low specificity in MAX enrichment results in a mixture of hydrophilic peptides and increases the sample complexity. This could be a possible reason why hydrophilic phosphopeptides were lost when the MAX experiment was performed first. The number of identified glycol-PSMs and peptides are similar between two experimental workflows. The types of glycans identified in these two experimental workflows showed that sialylated glycopeptides were largely identified in IMAC followed by MAX than MAX followed by IMAC (Figure 3C). The high abundance of hydrophilic peptides eluted from MAX might interfere with the identification of sialic-acid-containing peptides as well as the phosphopeptides eluted from MAX enrichment.
Quantitative Analysis of Phosphopeptides and Glycopeptides from Two Breast PDX Tumors.
The phosphopeptides and glycopeptides of two subtypes of breast cancer PDX model, which are a basal-like and a luminal type, were analyzed quantitatively through sequentially enrichment worklow using IMAC followed by MAX enrichment. The five samples in each subtype were labeled with TMT10-plex and then fractionated to 13 before enrichment (Supplementary Figure 3). From the first IMAC and subsequent MAX enrichment, a large number of phosphopeptides and IGPs were identified (Figure 4A, supplementary Table 2). The total of 17 852 and 5286 of phospho-peptides and -proteins, as well as 3468 and 660 of glycopeptides and proteins were identified. A large number of IGPs were identified in IMAC as well as in MAX, whereas the phosphorylated peptides were mostly identified in the IMAC. These results demonstrate that phosphopeptides can achieve sufficient recovery by IMAC enrichment, whereas IGPs can be increased identification coverage through IMAC as well as MAX. Identified peptides from PDX models were derived from Homo sapiens as well as Mus musculus, and some of the peptides were not distinguishable due to share the peptide sequences between the two species. The numbers of different types of glycans according to origins showed that most of the identified sialylated and fucosylated IGPs were derived from mus-musculus, while IGPs from homosapiens were mainly enriched in high-mannose (Figure 4B). Almost high-mannose were enriched by MAX and 55% of total high-mannose peptides were from human, and only 18% are from the mouse. Initial IMAC enrichment helped to increase the coverage of sialylated and fucosylated peptides, and about 60% and 43% of identified sialylated and fucosylated peptides were from mouse (only about 30% and 34% from human). Compared with the unfractioned result, which was shown in Figure 3C, the high-mannose peptides increased about 3-fold, whereas the sialylated and fucosylated peptides increased about 6- and 7-fold, respectively. Reduced complexity of sample through fractionation can improve sensitivity for low abundance peptides by decreasing the dynamic range of coeluting peptides, and the increased identifications of a sialylated and fucosylated peptide are expected to be due to the fractionation.
Figure 4.

Characteristics of simultaneously enriched phosphopeptides and IGPs. (A) Numbers of identified phosphopeptides (left y-axis) and IGPs (right y-axis) using workflow by IMAC followed by MAX enrichments from 13 fractions. (B) Glycan profile distribution. Pie charts show the spectral distribution according to their glycan class. “Homo+Mus” indicate indistinguishable species of the identified peptides.
Isobaric-tag-based (TMT, iTRAQ, demethylation, etc.) proteomic approaches are widely used in the quantitative method for multiple protein modifications with multiple enrichment processes because the isobaric labeling can reduce the experimental variation.38,39 Huang et al.40 used 24 breast cancer PDX models with various subtypes to quantitatively analyze not only phosphorylated proteins but also normal proteins and demonstrated the correlation of proteomics with transcriptomic. The phosphoproteomic analysis for the basallike (WHIM2) and luminal (WHIM16) subtypes of PDX models we used showed a high level of correlation coefficient (r = 0.75) when compared with the quantitative results of the Huang et al., and indicating similar results were observed with the published data (Supplementary Figure 4). A total of 1237 of differentially expressed phosphopeptides derived from human were selected by abundance ratio (>2-fold, luminal/basal) and p-value (<0.01), and 753 of proteins were determined on the basis of those peptides as differentially expressed phosphoproteins (DEPhos, Supplementary Table 3). The 405 and 832 phosphopeptides were up- and down-regulated in luminal subtype, respectively. At the protein level, 261 and 499 phosphoproteins were up- and down-regulated, and then 7 proteins had both up- and down-regulated phosphorylated peptides. From analyzing the gene ontology of DEPhos, up-regulated proteins were enriched in biological processes such as cell death, apoptosis, and biosynthesis in the luminal subtype compared with those of basal, whereas down-regulated proteins were enriched in DNA replication, transcription, and cell division (Figure 5A). A total 236 of differentially expressed IGPs were selected from human proteins and confirmed as membrane proteins, and those IGPs represented 26 up- and 46 down-regulated glycoproteins, respectively, in the luminal subtype (three glycoproteins had IGPs with increased and decreased level within the same proteins) (Supplementary Table 3). Overexpression of glycosylated proteins in were enriched in immune response and cell proliferation, whereas reduced proteins were enriched in cell adhesion and enzyme-linked receptor protein signaling pathway as well as cell proliferation (Figure 5B). Among the differentially expressed proteins identified by glycopeptides or phosphopeptides, 171 proteins were identified by both glycoproteomics and phosphoproteomics. In the case of CD276 protein, which was known important immune checkpoint and regulating of T cell function in breast cancer, the phosphorylation site at 525S41 as well as glycosylation site at 322N42 were simultaneously identified.
Figure 5.

Enriched biological processes for differentially expressed phosphorylated proteins and glycosylated proteins. Gene ontology classification of phosphoproteins (A) and glycoproteins (B) was performed according to significantly up- or down-regulated peptides.
METHODS
Tryptic Digestion, TMT Labeling, and Basic Reverse-Phase Fractionation.
Breast cancer patient-derived xenograft (PDX) models (basal-like and luminal type of cancer) were lysed using 8 M urea lysis buffer (8 M urea, 75 mM NaCl, 50 mM Tris-HCl, pH8), and protein concentration was determined using BCA assay kit (23225, Pierce Biotechnology). Enzymatic tryptic digestion was performed as previous described.43 Digested samples were desalted by C18 (WAT036820, Sep-Pak tC18 cartridge, Waters) and dried using Speed-Vac.
Tandem Mass Tag (TMT) labeling and basic reverse-phase liquid chromatography fractionation (bRPLC). The same amount (0.3 mg) of digested peptides for basal-like and luminal type were labeled with TMT-10 plex (90406, Pierce Biotechnology) as the manufacturing process. Basal-like samples were labeled with 126, 127C, 128C, 129C, and 130 and luminal samples were labeled with 127N, 128N, 129N, 130N, and 131 of TMT channel, respectively. Pooled TMT-labeled peptides were fractionated to 96 well and combined to 13 fractions for the enrichment process.43,44 TMT 10plex labeled peptides were separated on a reversed-phase Zorbax extend-C-18 column (4.6 × 100 mm, 1.8 μm particles, Agilent Technology) using an Agilent 1220 Infinity HPLC System. HPLC gradient condition was set as follows with solvent A (10 mM ammonium formate, pH 10) and solvent B (10 mM ammonium formate in 90% (v/v) ACN, pH 10): 2% (v/v) B for 10 min, from 2 to 8% B for 5 min, from 8 to 35% B for 85 min, from 35 to 95% B for 5 min, and 95% B for 25 min. Eluents were collected on the 96-well plate with 1 mL/min and concatenated into 12 fractions by 15, 27, 39, 51, 63, 75, 87 and plus one more fraction by pooled from 3 to 14. The samples were dried in a Speed-Vac.
IMAC Enrichment.
IMAC procedure was performed using Fe3+- NTA agarose beads that were prepared freshly using Ni2+-NTA agarose beads (QIAGEN, cat no. 30210) as previously described.45 To conjugate and wash the peptides at different pH condition, IMAC bind/wash buffer of from pH 1 to 6 were made by 1% (v/v), 0.15%,0.05%, 0.03% TFA and nonacid, and 0.005% (v/v) NH4OH in 80% acetonitrile (ACN), respectively. Likewise bind/wash buffer of from pH 0.5 to 5.5 were prepared by 3%, 0.3%, 0.1%, 0.04%, 0.01% TFA, and 0.001% NH4OH in 80% ACN, respectively. Each sample for IMAC was reconstituted in 200 μL of bind/wash buffers respectively and incubated with 50 μL of 5% (v/v) Fe3+-NTA agarose beads to conjugate for 30 min at RT. The supernatant after conjugation was collected (nonbinding) by centrifuge. The beads were put on the Stage Tip and were washed three times each with 200 μL of bind/wash buffer by centrifugation. The washed out buffer from beads were collected together with nonbinding buffer to be used to subsequently enrichment process. The peptides conjugated to beads were eluted with 75 μL potassium phosphate buffer (500 mM KH2PO4, pH7) and 50% (v/v) ACN in 0.1% (v/v) formic acid. Eluted peptides were dried and stored at −80 °C until LC-MS/MS analysis.
MAX Enrichment.
The IGPs enrichment process was performed using MAX cartridge (Oasis MAX cartridge 30 mg, Waters, U.S.A.) as previously described.28 Peptide mixture was reconstituted with 1 mL of 95% (v/v) ACN in 1% (v/v) TFA. The MAX cartridge was conditioned with 2 mL of methanol, 50% ACN in 1% TFA, and 95% ACN in 1% TFA, respectively, and the peptide mixture was subsequently loaded onto the cartridge. The flow through after peptide loading and the washing with 4 mL of 95% ACN in 1% TFA were combined for the subsequent enrichment process. Elution buffer (50% ACN in 0.1% TFA) allows elution of the IGPs from the cartridge, and eluted samples were dried using Speed-Vac.
LC-MS/MS Analysis.
All samples were analyzed by a Q-exactive mass spectrometer (Thermo Fisher Scientific, U.S.A.) combined with the Ultimate 3000 UPLC system (Thermo Fisher Scientific, U.S.A.). The peptides (≈ 1ug) were loaded onto trap column (5 μm, 2 cm × 75 μm ID, Thermo Fisher Scientific, U.S.A.) and separated on analytic column (3 μm, 50 cm × 75 μm ID, Thermo Fisher) with a linear gradient of 5−35% buffer B (95% ACN and 0.1% FA) for 60 min at a flow rate of 250 nL/min controlled by Chromelon software. MS scans were acquired using a data-dependent top 10 modes which were choosing the most abundant intensity from precursor ion scan survey (300−1800 of mass range). For MS1 scan, resolution and automatic gain control (AGC) values were set at 70 K at 200 m/z and 3 × 106. MS/MS spectra were acquired by HCD fragmentation with 30 of normalized collision energy (NCE) and 35 K of a resolving power at 200 m/z.
Database Search.
For the same.raw data files, SEQUEST (Proteome discoverer 2.0, Thermo Fisher Scientific) and GPQuest (v2.0, in-house platform) were performed for identification of phosphopeptides and IGPs, respectively. SEQUEST search algorithm was performed against the combined database (UniProt human and mouse, both released from Sep. 2015). Tolerance of precursor ion and product ion was set at 10 ppm and 0.06 Da, respectively. The phosphorylation (+79.998 on serine, threonine, and tyrosine) and oxidation (+15.998 on methionine) modifications were set as variable and carbamidomethylation (+57.105 on cysteine) was set as fixed parameters. TMT-10plex (+273.121 on lysine and n-terminus) was set as fixed modification only in TMT labeled sample set. Percolator was set to 0.01 for a peptide which is allowed a maximum of two miss cleavages and over six peptide lengths. For quantifying of phosphopeptides, intensities of reporter ion were summed depending on the phosphopeptide group and comparison between the two groups, luminal and basal of PDXs, is expressed as the median value of the fold change ratio for luminal/basal-like intensities in every five replicates. Prior GPQuest search, the.raw data file format was converted to mzXML with the centroid all scans option using MS-Convert (Trans-Proteomic Pipeline platform). MS1 and MS2 tolerance were set as 10 and 20 ppm, respectively. GPQuest search was performed against in-house glycosite database containing 20 421 peptides with human N-glycan database (in-house database, 277 of N-glycan composition). The glycopeptide-spectral matches were determined with 0.01 of false discovery rate (FDR) and minimum Morpheus score of five and intensity coverage above 10%. The oxonium ion-containing spectra, which is having 204.8 of fragmentation ion with above 10% of highest intensity, were counted using Python platform.
Data Analysis.
Consensus process in Proteome Discoverer 2.0 is used to roll up the reporter ion intensity from individual peptidespectrum matches (PSMs) to phosphopeptide groups and calculate the adjusted p-value and fold-change ratio between PDX samples (luminal/basal-like) with five replicates. The reporter ion intensities from GPQuest results also were summed as a median value depending on the IGP, meaning the same peptide backbone with the same glycan species, and the p-value and fold-change ratio for basal-like/luminal were calculated using two t-test analyses. Differentially expressed peptides were selected with under 0.01 of p-value and over 2-fold changes. Gene ontology analysis is performed by DAVID (Bioinformatics Resource 6.7v)46,47 and filtered significant biological process as p-value < 0.01.
CONCLUSIONS
Developments of MS/MS spectral search algorithm and instrument have led to the discovery of unmatched spectra. GPQuest 2.0 identified IGPs that have not been seen from global and phosphoproteomic data by identifying IGPs through fragmentation obtained by HCD.22,35 A significant number of IGPs have been reported to coelute in IMAC experiments for phosphopeptide isolation, and we expected this is caused by the pKa value of the phosphopeptide and glycopeptide functional groups. The coelution efficiency of phosphopeptides and IGPs were investigated according to the pH conditions. We confirmed pH 2 was the optimal pH condition for the phosphopeptide enrichment by IMAC and the coelution of IGPs, especially sialylated glycopeptides at the IMAC enrichment. IGPs that were not caught in the IMAC process were enriched through additional MAX procedures and resulted in identifying a large number of IGPs. Identifications of ~18 000 of phosphopeptides and ~3500 IGPs in the experiment through fractionation as well as identification of 8400 and 650 phosphopeptides and glycopeptides in single LC-MS/MS run demonstrate sufficient improvement for simultaneous proteomic analyses of phosphopeptides and glycopeptides. Using the isobaric tag approach shows the potentially quantitatively analysis for IGPs as well as phosphopeptides and allows us to find modified peptides and biological processes between luminal and basal subtypes.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by Funding: National Cancer Institute, the Clinical Proteomic Tumor Analysis Consortium (CPTAC,U24CA210985).
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acschembio.8b00902.
Supplementary figures (PDF)
Data set of identification of phosphopeptides and IGPs in optimization workflow (Supplementary Table 1) (XLSX)
Data set of identification of phosphopeptides and IGPs in quantitative analysis (Supplementary Table 2) (XLSX)
Data set of differentially expressed phosphopeptides and IGPs (Supplementary Table 3) (XLSX)
The authors declare no competing financial interest.
REFERENCES
- (1).Khoury GA, Baliban RC, and Floudas CA (2011) Proteome-wide post-translational modification statistics: frequency analysis and curation of the swiss-prot database. Sci. Rep 1, 90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Pinho SS, and Reis CA (2015) Glycosylation in cancer: mechanisms and clinical implications. Nat. Rev. Cancer 15, 540–555. [DOI] [PubMed] [Google Scholar]
- (3).Magalhaes A, Duarte HO, and Reis CA (2017) Aberrant Glycosylation in Cancer: A Novel Molecular Mechanism Controlling Metastasis. Cancer Cell 31, 733–735. [DOI] [PubMed] [Google Scholar]
- (4).Yu L, Lu M, Jia D, Ma J, Ben-Jacob E, Levine H, Kaipparettu BA, and Onuchic JN (2017) Modeling the Genetic Regulation of Cancer Metabolism: Interplay between Glycolysis and Oxidative Phosphorylation. Cancer Res. 77, 1564–1574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Zhao Y, and Jensen ON (2009) Modification-specific proteomics: strategies for characterization of post-translational modifications using enrichment techniques. Proteomics 9, 4632–4641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Macek B, Mann M, and Olsen JV (2009) Global and sitespecific quantitative phosphoproteomics: principles and applications. Annu. Rev. Pharmacol. Toxicol 49, 199–221. [DOI] [PubMed] [Google Scholar]
- (7).Wolschin F, Wienkoop S, and Weckwerth W (2005) Enrichment of phosphorylated proteins and peptides from complex mixtures using metal oxide/hydroxide affinity chromatography (MOAC). Proteomics 5, 4389–4397. [DOI] [PubMed] [Google Scholar]
- (8).Andersson L, and Porath J (1986) Isolation of phosphoproteins by immobilized metal (Fe3+) affinity chromatography. Anal. Biochem 154, 250–254. [DOI] [PubMed] [Google Scholar]
- (9).Posewitz MC, and Tempst P (1999) Immobilized gallium(III) affinity chromatography of phosphopeptides. Anal. Chem 71, 2883–2892. [DOI] [PubMed] [Google Scholar]
- (10).Miquel E, Alegria A, Barbera R, and Farre R (2005) Speciation analysis of calcium, iron, and zinc in casein phosphopeptide fractions from toddler milk-based formula by anion exchange and reversed-phase high-performance liquid chromatography-mass spectrometry/flame atomic-absorption spectroscopy. Anal. Bioanal. Chem 381, 1082–1088. [DOI] [PubMed] [Google Scholar]
- (11).Zhang G, and Neubert TA (2006) Use of detergents to increase selectivity of immunoprecipitation of tyrosine phosphorylated peptides prior to identification by MALDI quadrupole-TOF MS. Proteomics 6, 571–578. [DOI] [PubMed] [Google Scholar]
- (12).Zhang H, Zha X, Tan Y, Hornbeck PV, Mastrangelo AJ, Alessi DR, Polakiewicz RD, and Comb MJ (2002) Phosphoprotein analysis using antibodies broadly reactive against phosphorylated motifs. J. Biol. Chem 277, 39379–39387. [DOI] [PubMed] [Google Scholar]
- (13).Rush J, Moritz A, Lee KA, Guo A, Goss VL, Spek EJ, Zhang H, Zha XM, Polakiewicz RD, and Comb MJ (2005) Immunoaffinity profiling of tyrosine phosphorylation in cancer cells. Nat. Biotechnol 23, 94–101. [DOI] [PubMed] [Google Scholar]
- (14).Ficarro SB, McCleland ML, Stukenberg PT, Burke DJ, Ross MM, Shabanowitz J, Hunt DF, and White FM (2002) Phosphoproteome analysis by mass spectrometry and its application to Saccharomyces cerevisiae. Nat. Biotechnol 20, 301–305. [DOI] [PubMed] [Google Scholar]
- (15).Palmisano G, Lendal SE, and Larsen MR (2011) Titanium dioxide enrichment of sialic acid-containing glycopeptides. Methods Mol. Biol 753, 309–322. [DOI] [PubMed] [Google Scholar]
- (16).Palmisano G, Lendal SE, Engholm-Keller K, Leth-Larsen R, Parker BL, and Larsen MR (2010) Selective enrichment of sialic acid-containing glycopeptides using titanium dioxide chroma tography with analysis by HILIC and mass spectrometry. Nat. Protoc 5, 1974–1982. [DOI] [PubMed] [Google Scholar]
- (17).Larsen MR, Jensen SS, Jakobsen LA, and Heegaard NH (2007) Exploring the sialiome using titanium dioxide chromatography and mass spectrometry. Mol. Cell. Proteomics 6, 1778–1787. [DOI] [PubMed] [Google Scholar]
- (18).Hu Y, Shah P, Clark DJ, Ao M, and Zhang H (2018) Reanalysis of Global Proteomic and Phosphoproteomic Data Identified a Large Number of Glycopeptides, Anal. Chem.908065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Wuhrer M, Catalina MI, Deelder AM, and Hokke CH (2007) Glycoproteomics based on tandem mass spectrometry of glycopeptides Chromatogr J. B: Anal. Technol. Biomed. Life Sci 849, 115–128. [DOI] [PubMed] [Google Scholar]
- (20).Sun S, Shah P, Eshghi ST, Yang W, Trikannad N, Yang S, Chen L, Aiyetan P, Hoti N, Zhang Z, Chan DW, and Zhang H (2016) Comprehensive analysis of protein glycosylation by solidphase extraction of N-linked glycans and glycosite-containing peptides. Nat. Biotechnol 34, 84–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Sun S, Hu Y, Jia L, Eshghi ST, Liu Y, Shah P, and Zhang H (2018) Site-Specific Profiling of Serum Glycoproteins Using N-Linked Glycan and Glycosite Analysis Revealing Atypical NGlycosylation Sites on Albumin and alpha-1B-Glycoprotein. Anal. Chem 90, 6292–6299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Toghi Eshghi S, Shah P, Yang W, Li X, and Zhang H (2015) GPQuest: A Spectral Library Matching Algorithm for Site-Specific Assignment of Tandem Mass Spectra to Intact N-glycopeptides. Anal. Chem 87, 5181–5188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Shah P, Wang X, Yang W, Toghi Eshghi S, Sun S, Hoti N, Chen L, Yang S, Pasay J, Rubin A, and Zhang H (2015) Integrated Proteomic and Glycoproteomic Analyses of Prostate Cancer Cells Reveal Glycoprotein Alteration in Protein Abundance and Glycosylation. Mol. Cell. Proteomics 14, 2753–2763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Ruhaak LR, Huhn C, Waterreus WJ, de Boer AR, Neususs C, Hokke CH, Deelder AM, and Wuhrer M (2008) Hydrophilic interaction chromatography-based high-throughput sample preparation method for N-glycan analysis from total human plasma glycoproteins. Anal. Chem 80, 6119–6126. [DOI] [PubMed] [Google Scholar]
- (25).Mysling S, Palmisano G, Hojrup P, and Thaysen-Andersen M (2010) Utilizing ion-pairing hydrophilic interaction chromatography solid phase extraction for efficient glycopeptide enrichment in glycoproteomics. Anal. Chem 82, 5598–5609. [DOI] [PubMed] [Google Scholar]
- (26).Zhang H, Guo T, Li X, Datta A, Park JE, Yang J, Lim SK, Tam JP, and Sze SK (2010) Simultaneous characterization of glyco- and phosphoproteomes of mouse brain membrane proteome with electrostatic repulsion hydrophilic interaction chromatography. Mol. Cell. Proteomics 9, 635–647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Alpert AJ (2008) Electrostatic repulsion hydrophilic interaction chromatography for isocratic separation of charged solutes and selective isolation of phosphopeptides. Anal. Chem 80, 62–76. [DOI] [PubMed] [Google Scholar]
- (28).Yang W, Shah P, Hu Y, Toghi Eshghi S, Sun S, Liu Y, and Zhang H (2017) Comparison of Enrichment Methods for Intact N- and O-Linked Glycopeptides Using Strong Anion Exchange and Hydrophilic Interaction Liquid Chromatography. Anal. Chem 89, 11193–11197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Zhang H, Li XJ, Martin DB, and Aebersold R (2003) Identification and quantification of N-linked glycoproteins using hydrazide chemistry, stable isotope labeling and mass spectrometry. Nat. Biotechnol 21, 660–666. [DOI] [PubMed] [Google Scholar]
- (30).Kaji H, Saito H, Yamauchi Y, Shinkawa T, Taoka M, Hirabayashi J, Kasai K, Takahashi N, and Isobe T (2003) Lectin affinity capture, isotope-coded tagging and mass spectrometry to identify N-linked glycoproteins. Nat. Biotechnol 21, 667–672. [DOI] [PubMed] [Google Scholar]
- (31).Xu Y, Wu Z, Zhang L, Lu H, Yang P, Webley PA, and Zhao D (2009) Highly specific enrichment of glycopeptides using boronic acid-functionalized mesoporous silica. Anal. Chem 81, 503–508. [DOI] [PubMed] [Google Scholar]
- (32).Zou X, Jie J, and Yang B (2017) Single-Step Enrichment of N-Glycopeptides and Phosphopeptides with Novel Multifunctional Ti(4+)-Immobilized Dendritic Polyglycerol Coated Chitosan Nano-materials. Anal. Chem 89, 7520–7526. [DOI] [PubMed] [Google Scholar]
- (33).Carr SA, Huddleston MJ, and Bean MF (1993) Selective identification and differentiation of N- and O-linked oligosaccharides in glycoproteins by liquid chromatography-mass spectrometry. Protein Sci. 2, 183–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Yang S, Zhang L, Thomas S, Hu Y, Li S, Cipollo J, and Zhang H (2017) Modification of Sialic Acids on Solid Phase: Accurate Characterization of Protein Sialylation. Anal. Chem 89 (12), 6330–6335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Hu YW, Yang WM, Shah P, Sun SS, Ao MH, and Zhang H (2017) GPQuest 3: A Tool for Large-scale and Comprehensive Glycosylation Analysis on MS data. Glycobiology 27, 1218–1218. [Google Scholar]
- (36).Toyoda M, Ito H, Matsuno YK, Narimatsu H, and Kameyama A (2008) Quantitative derivatization of sialic acids for the detection of sialoglycans by MALDI MS. Anal. Chem 80, 5211–5218. [DOI] [PubMed] [Google Scholar]
- (37).Sekiya S, Wada Y, and Tanaka K (2005) Derivatization for stabilizing sialic acids in MALDI-MS. Anal. Chem 77, 4962–4968. [DOI] [PubMed] [Google Scholar]
- (38).Rauniyar N, and Yates JR 3rd. (2014) Isobaric labeling-based relative quantification in shotgun proteomics. J. Proteome Res 13, 5293–5309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Cheng L, Pisitkun T, Knepper MA, and Hoffert JD (2016) Peptide Labeling Using Isobaric Tagging Reagents for Quantitative Phosphoproteomics. Methods Mol. Biol 1355, 53–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Huang KL, Li S, Mertins P, Cao S, Gunawardena HP, Ruggles KV, Mani DR, Clauser KR, Tanioka M, Usary J, Kavuri SM, Xie L, Yoon C, Qiao JW, Wrobel J, Wyczalkowski MA, Erdmann-Gilmore P, Snider JE, Hoog J, Singh P, Niu B, Guo Z, Sun SQ, Sanati S, Kawaler E, Wang X, Scott A, Ye K, McLellan MD, Wendl MC, Malovannaya A, Held JM, Gillette MA, Fenyo D, Kinsinger CR, Mesri M, Rodriguez H, Davies SR, Perou CM, Ma C, Reid Townsend R, Chen X, Carr SA, Ellis MJ, and Ding L (2017) Proteogenomic integration reveals therapeutic targets in breast cancer xenografts. Nat. Commun 8, 14864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Dephoure N, Zhou C, Villen J, Beausoleil SA, Bakalarski CE, Elledge SJ, and Gygi SP (2008) A quantitative atlas of mitotic phosphorylation. Proc. Natl. Acad. Sci. U. S. A 105, 10762–10767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Chen R, Jiang X, Sun D, Han G, Wang F, Ye M, Wang L, and Zou H (2009) Glycoproteomics analysis of human liver tissue by combination of multiple enzyme digestion and hydrazide chemistry. J. Proteome Res 8, 651–661. [DOI] [PubMed] [Google Scholar]
- (43).Zhou JY, Chen L, Zhang B, Tian Y, Liu T, Thomas SN, Chen L, Schnaubelt M, Boja E, Hiltke T, Kinsinger CR, Rodriguez H, Davies SR, Li S, Snider JE, Erdmann-Gilmore P, Tabb DL, Townsend RR, Ellis MJ, Rodland KD, Smith RD, Carr SA, Zhang Z, Chan DW, and Zhang H (2017) Quality Assessments of Long-Term Quantitative Proteomic Analysis of Breast Cancer Xenograft Tissues. J. Proteome Res 16, 4523–4530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Mertins P, Tang LC, Krug K, Clark DJ, Gritsenko MA, Chen L, Clauser KR, Clauss TR, Shah P, Gillette MA, Petyuk VA, Thomas SN, Mani DR, Mundt F, Moore RJ, Hu Y, Zhao R, Schnaubelt M, Keshishian H, Monroe ME, Zhang Z, Udeshi ND, Mani D, Davies SR, Townsend RR, Chan DW, Smith RD, Zhang H, Liu T, and Carr SA (2018) Reproducible workflow for multiplexed deep-scale proteome and phosphoproteome analysis of tumor tissues by liquid chromatography-mass spectrometry. Nat. Protoc 13, 1632–1661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Zhang H, Liu T, Zhang Z, Payne SH, Zhang B, McDermott JE, Zhou JY, Petyuk VA, Chen L, Ray D, Sun S, Yang F, Chen L, Wang J, Shah P, Cha SW, Aiyetan P, Woo S, Tian Y, Gritsenko MA, Clauss TR, Choi C, Monroe ME, Thomas S, Nie S, Wu C, Moore RJ, Yu KH, Tabb DL, Fenyo D, Bafna V, Wang Y, Rodriguez H, Boja ES, Hiltke T, Rivers RC, Sokoll L, Zhu H, Shih IM, Cope L, Pandey A, Zhang B, Snyder MP, Levine DA, Smith RD, Chan DW, Rodland KD, Investigators C, et al. (2016) Integrated Proteogenomic Characterization of Human High-Grade Serous Ovarian Cancer. Cell 166, 755–765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Huang DW, Sherman BT, and Lempicki RA (2009) Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc 4, 44–57. [DOI] [PubMed] [Google Scholar]
- (47).Huang DW, Sherman BT, and Lempicki RA (2009) Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 37, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.