

Abstract
Background
The LysM receptor-like kinases (LysM-RLKs) are important to both plant defense and symbiosis. Previous studies described three clades of LysM-RLKs: LysM-I/LYKs (10+ exons per gene and containing conserved kinase residues), LysM-II/LYRs (1–5 exons per gene, lacking conserved kinase residues), and LysM-III (two exons per gene, with a kinase unlike other LysM-RLK kinases and restricted to legumes). LysM-II gene products are presumably not functional as conventional receptor kinases, but several are known to operate in complexes with other LysM-RLKs. One aim of our study was to take advantage of recently mapped wild tomato transcriptomes to evaluate the evolutionary history of LysM-RLKs within and between species. The second aim was to place these results into a broader phylogenetic context by integrating them into a sequence analysis of LysM-RLKs from other functionally well-characterized model plant species. Furthermore, we sought to assess whether the Group III LysM-RLKs were restricted to the legumes or found more broadly across Angiosperms.
Results
Purifying selection was found to be the prevailing form of natural selection within species at LysM-RLKs. No signatures of balancing selection were found in species-wide samples of two wild tomato species. Most genes showed a greater extent of purifying selection in their intracellular domains, with the exception of SlLYK3 which showed strong purifying selection in both the extracellular and intracellular domains in wild tomato species. The phylogenetic analysis did not reveal a clustering of microbe/functional specificity to groups of closely related proteins. We also discovered new putative LysM-III genes in a range of Rosid species, including Eucalyptus grandis.
Conclusions
The LysM-III genes likely originated before the divergence of E. grandis from other Rosids via a fusion of a Group II LysM triplet and a kinase from another RLK family. SlLYK3 emerges as an especially interesting candidate for further study due to the high protein sequence conservation within species, its position in a clade of LysM-RLKs with distinct LysM domains, and its close evolutionary relationship with LYK3 from Arabidopsis thaliana.
Electronic supplementary material
The online version of this article (10.1186/s12862-019-1467-3) contains supplementary material, which is available to authorized users.
Keywords: Phylogenetics, Population genetics, Solanum, Plant immunity, Symbiosis
Background
Plants are regularly targeted for colonization by organisms ranging from pathogenic to beneficial [1]. Pathogenic organisms infect the plant and use it for nourishment, eventually reducing host fitness [1]. Pathogens can induce changes in the host plant’s genetic regulation to maximize the amount of nutrients that can be accessed and to avoid detection and subsequent host defense responses [1]. Beneficial symbiotic organisms also use the host plant for nourishment but offer benefits to the plant in exchange (e.g. better uptake of water and nutrients) [1]. Plants that can differentiate between pathogenic and beneficial symbiotic organisms improve their chances of survival and reproduction [1]. Detection of the presence of these organisms involves extracellular receptors, often receptor-like kinases (RLKs), which recognize proteins or other molecular “patterns” produced by the colonizing organisms and trigger phosphorylation and downstream signaling cascades within the cell [1, 2]. The signaling cascades then activate the appropriate defensive or symbiotic responses [2].
Genes containing the LysM motif, including the family of LysM-RLKs, have been implicated in the detection of both plant-symbiotic and -pathogenic organisms [3]. In the case of beneficial symbiotic organisms, some LysM-RLKs are the key coordinators of the plant’s cooperative response with the symbiont [3]. Other LysM-RLKs are necessary for detecting pathogens and activating defense responses. These responses can be initiated by the recognition of pathogen-derived molecules such as chitin as it is shed by the invading pathogen [3]. LysM-RLKs sometimes function together as heterodimers, with the extracellular region of one LysM-RLK detecting the presence of the colonizing pathogen or symbiont while the kinase domain of another LysM-RLK mediates the symbiotic or defense response [3, 4]. Some LysM-RLKs, such as Oryza sativa CERK1 (OsCERK1), also function as dual-purpose detectors of both pathogenic and symbiotic organisms [5]. It is not clear whether genes that play similar functional roles are more closely related to one another or if convergent evolution in microbe discrimination is widespread in this protein family.
LysM-RLKs, with three repetitions of the LysM domain, are ubiquitous among land plants [6] and may have evolved as part of a signaling module prior to colonization of land and the origin of symbiosis with mycorrhiza [7]. Shiu et al. describe two main clades of LysM-RLKs: LysM-I and LysM-II [8], with LysM-RLKs in Group II lacking conserved kinase residues; for example, the glycine-rich loop is missing from the kinase domains of all Group II LysM-RLKs from Arabidopsis thaliana and Solanum lycopersicum [6, 9]. LysM-I RLKs have ten or more exons, while LysM-II RLKs typically have one or two [6]. Another group consisting of four LysM-RLK genes (two from Medicago truncatula and two from Lotus japonicus) contain classically conserved kinase residues (like LysM-I RLKs) and two exons (like LysM-II RLKs). However, due to their structural similarities to both groups, these genes cannot be classified unambiguously into either Group I or Group II, and their clade has been named Group III [10, 11]. Arrighi et al. have suggested that the Group III LysM-RLKs MtLYR5 and MtLYR6 are of chimeric origin, arising from the fusion of one gene encoding a LysM triplet and a second gene encoding a kinase domain, unlike those found in other LysM genes [10]. Lohmann et al. came to the same conclusion based on their analyses of the only other characterized Group III genes (found in L. japonicus) and further suggested that Group III arose within the dicot lineage [11]. Until now, Group III genes have not been reported from outside the Leguminosae. Furthermore, although phylogenetic analyses of LysM-RLKs from a variety of plant species have been conducted, a comprehensive phylogenetic analysis of this family across several species (including tomato) is lacking. Simultaneously, the availability of newly-described functions of individual LysM-RLKs from well-studied species provided an opportunity to evaluate the distribution of functional specificity in a phylogenetic context.
Here we synthesize the currently available information about function (i.e. microbe specificity) and phylogenetic relationships of LysM-RLKs, including those from tomato, and show a close relationship between Group II and Group III LysM-RLKs. Newly discovered putative Group III LysM-RLKs are present in a wide variety of Rosid species. However, they were not detected outside the Rosid clade. Orthologs of SlLYK3 show evidence of strong purifying selection in wild tomatoes, and although the kinase domain of SlLYK8 is truncated in cultivated tomato, we find that this is not the case for orthologs in wild tomato species.
Results
Distribution of microbe specificity across the phylogenetic tree
The goal of this study was to understand the evolutionary history of the LysM-RLKs on two levels: at the microevolutionary scale (within the clade of wild tomatoes) and at the macroevolutionary scale. At the larger evolutionary time scale, understanding the broader evolutionary patterns, especially in terms of microbe recognition, will allow us to place the insights gained from the microevolutionary analysis in context. We aimed to test if LysM-RLKs with similar microbial recognition specificity clustered phylogenetically. We assembled a large set of all previously defined (canonical) LysM-RLKs reported from species for which the greatest amount of functional data was available: A. thaliana, L. japonicus, M. truncatula, O. sativa, and S. lycopersicum [4, 9, 11, 17, 33–45]. We then aligned the LysM-RLK protein sequences using MUSCLE, inferred the phylogenetic relationships and combined it with the known functions of the proteins (Fig. 1). The maximum-likelihood phylogeny was rooted using MAD [27], a modified midpoint rooting method that takes lineage-specific heterotachy into account. This makes this method robust to variation in evolutionary rates among lineages and allows rooting without a priori determination of an outgroup. We differentiated between functions inferred by correlated gene expression/regulation patterns and those established by analyses of mutant phenotypes. This was an important distinction to make, because it is possible for a gene to be co-regulated upon microbe exposure without the gene necessarily playing a role in symbiosis or defense (e.g. LjLYS11) [43]. While we do recover multiple small clades of closely related sequences reported to fulfill similar functions, in most cases, major clades do not appear to be strictly associated with a specific form of microbe recognition. This suggests that microbe recognition can evolve convergently throughout the family and that orthologous genes can encode for different recognition specificities. This agrees with the results of de Mita et al. who showed neofunctionalization among LysM-RLK gene duplicates [46]. However, it should be noted that most of the genes have not necessarily been tested for each of the functions listed, and functions in the best-studied functional process - Rhizobia symbiosis – dominate the tree. This bias in functional characterization may limit our power to detect a strong correlation between the type of microbe recognition and phylogenetic position.
Fig. 1.
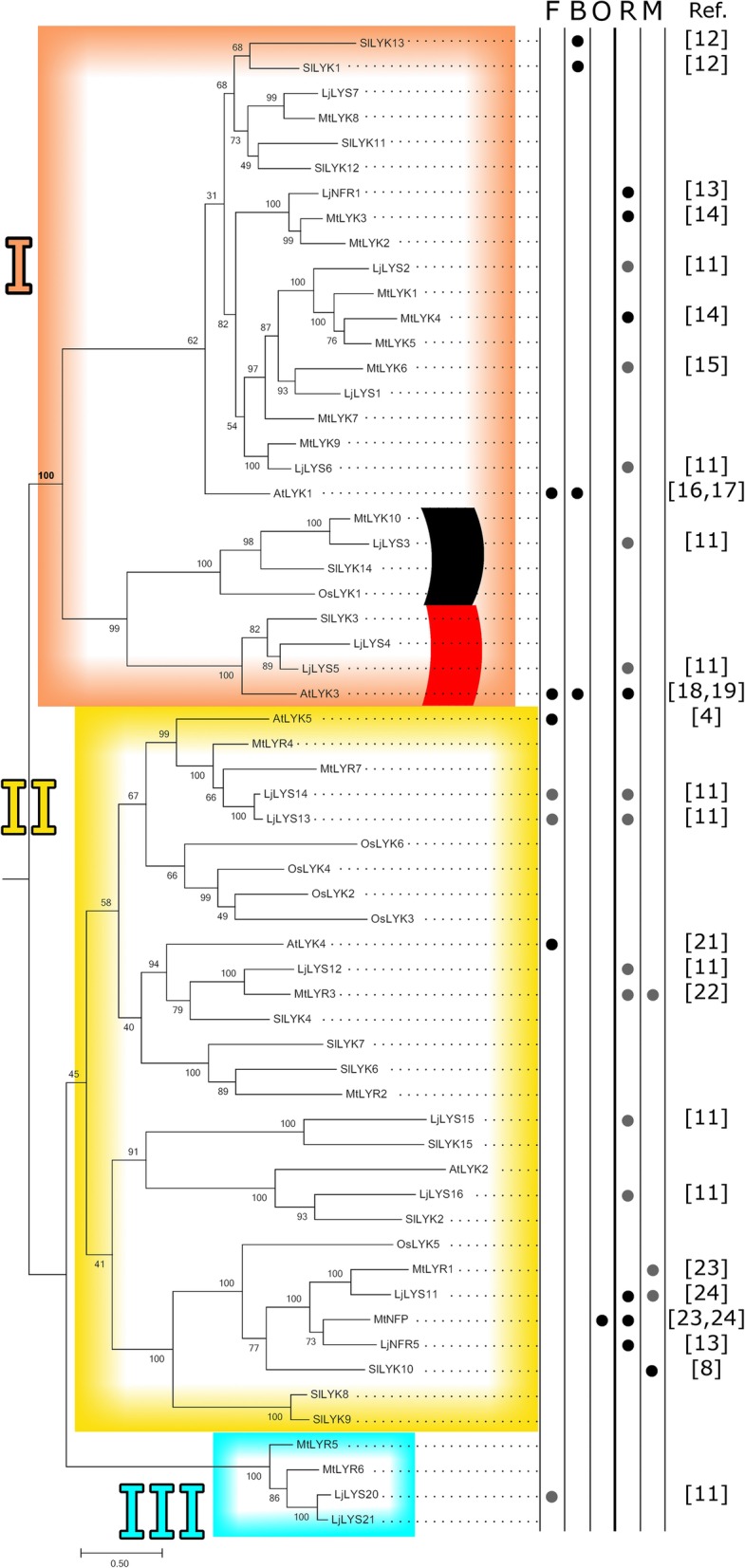
LysM-RLK phylogeny and functions. Maximum likelihood phylogeny and functions of canonical LysM-RLKs from Solanum lycopersicum (Sl), Arabidopsis thaliana (At), Lotus japonicus (Lj), Oryza sativa (Os), and Medicago truncatula (Mt). The phylogeny and 500 bootstrap replicates were inferred using RAxML under the WAG model with empirical frequencies and seed values of 100. The species of gene origin is given by the first two letters of the name given on the phylogeny. The phylogeny was rooted using the method of Minimal Ancestor Deviation [27]. The scale bar indicates amino acid substitutions per site. Gene functions are indicated: defense against fungi (F), bacteria (B), and oomycetes (O) and symbiosis with rhizobia (R) and mycorrhiza (M). LysM-RLKs form three clades. Red and black arcs indicate groups of proteins with distinct LysM domain sequences. Functions verified by mutation phenotypes are indicated by black circles. Functions inferred by differential expression are indicated by gray circles. Citations for sources of functional information are shown in brackets
Group III LysM-RLKs cluster with group II LysM-RLKs
The relationships between the major gene groups are generally consistent with those of previously published phylogenies based on entire coding regions or kinase domains of a subset of these LysM-RLKs (Fig. 1; references [9, 12]). We observe a separation of the LysM-RLKs into two major clades: Group I sequences in one clade and Group II and Group III sequences in a second clade. The monophyly of these clades is well-supported (100% bootstrap support, bold on Fig. 1), assuming correct rooting of the phylogeny. Furthermore, the genes of Group I are further differentiated into two subclades. These genes differ by the presence of a microexon in one group and the absence of this exon in the other group, as previously reported by Lohmann and colleagues [11]. According to our analysis, Group II and Group III share a more recent common ancestor than either does with Group I.
Group III LysM-RLKs exist in diverse Rosid species
Group III LysM-RLKs were previously reported only from L. japonicus and M. truncatula. To determine whether Group III LysM-RLKs are present outside of the Leguminosae and specifically in tomato, we searched NCBI’s non-redundant protein database for homologs of LjLYS20 using BlastP with a limit of 250 hits. This produced as many putative Group III LysM-RLKs as obtained by increasing the search limit to 500 hits or by using each of the four known Group III LysM-RLKs (LjLYS20, LjLYS21, MtLYR5 and MtLYR6) and merging the results. The 250 hits aligned to the query by BLAST were at least 42% identical to the query sequence (679 amino acids). The majority of candidate sequences identified in the search originated from legume species, but some came from additional species across the Rosid clade. No sequences were found in the Asterids, the clade containing the tomatoes. To determine if these new sequences were bona fide Group III LysM-RLKs, we determined whether they contained the highly-conserved CXC motif between the 1st and 2nd LysM domains. From the GUIDANCE alignment of these sequences, we determined that 117 of the 250 hits had the defining CXC motif.
To determine whether additional putative Group III LysM-RLKs may have been missed in the BlastP search, an online psiBLAST [47] with LjLYS20 as query was performed. Five iterations of psiBLAST [47] were run, under standard settings and using an E-value cut-off of 0.05. Only the portions of the sequences which aligned to the query were downloaded. Since the CXC motif occurs at positions 109–111 of LjLYS20, the first 130 amino acids of the hits were aligned with the canonical Group III LysM-RLKs using MUSCLE to check for the presence of the CXC motif. The second psiBLAST iteration produced some sequences with CXC motifs, but alignment with the canonical LysM-RLKs revealed that they did not have LysM domains and were not part of the LysM-RLK family. The third iteration produced a sequence with CXC motif which closely resembled another sequence from the same species in the original BLASTp search. The fourth and fifth iterations produced no sequences with CXC motifs. We concluded that the psiBLAST search did not result in additional putative Group III LysM-RLKs and that the BlastP search was sufficiently exhaustive. We included the 117 unique putative Group III sequences from the BlastP search in our phylogenetic analysis of the canonical LysM-RLKs.
In the combined analysis, the canonical Group III members (LjLYS20, LjLYS21, MtLYR5 and MtLYR6) and new putative Group III LysM-RLK genes form a clade together (Additional files 3 and 4). In agreement with our initial analysis of the canonical LysM-RLKs (Fig. 1), we observe that Group II and Group III share a more recent common ancestor with one another than either does with Group I. However, all Group II genes lack conserved kinase residues, while Group III genes encode a kinase unlike other LysM-RLK kinases [10, 11]. Since the extracellular LysM region and intracellular kinase region of Group III genes may have different ancestries due their putative chimeric origin [10, 11], we inferred the phylogenetic history of these two regions separately (Fig. 2, Additional files 5 and 6).
Fig. 2.

Phylogeny of canonical LysM-RLKs and CXC-motif-containing BlastP hits of LjLYS20. a Phylogeny of the LysM domain sequences only shows Group III and putative Group III sequences forming a clade with Group II. The phylogeny and 500 bootstrap replicates were inferred using RAxML under the WAG model with empirical frequencies and seed values of 100 and rooted using the method of Minimal Ancestor Deviation [27]. b Phylogeny of the kinase domains only shows Group I and Group II forming a clade together, while Group III and putative Group III sequences form another clade. The phylogeny and 500 bootstrap replicates were inferred using RAxML under the JTT model with empirical frequencies and seed values of 100 and rooted using the method of Minimal Ancestor Deviation [27]. The scale bar indicates amino acid substitutions per site
If these newly identified sequences were true homologs of the four original Group III sequences, we expected Group II sequences to form a clade together with the original and new Group III sequences in analyses based on the LysM domain region. As before, we used the MAD method to root these phylogenies. We recovered a clade containing the Group II genes along with the new and canonical Group III genes with 91% bootstrap support in our analysis of the LysM domain region of these genes (bold in Fig. 2a, Additional file 5). In contrast, a phylogeny based on the kinase domain would be expected to include both the new and previously described Group III genes in a separate monophyletic clade from Group I and II genes and this is what we observe (Fig. 2b, Additional file 6).
The new Group III LysM-RLKs in our analysis indicate a wider taxonomic distribution of Group III genes in species outside the Leguminosae. These new gene sequences are found in species throughout the Rosids, but not from the Asterids (which includes tomatoes) or other Eudicots. This is consistent with Group III genes arising relatively early in the Rosid lineage. Nevertheless, their presence exclusively in the Rosid lineage implies that they arose after the monocot/dicot split. In contrast, since Group I and Group II genes are found in both monocots and dicots, these genes likely originated and began to diverge prior to the monocot/dicot split. The sequence similarity in the LysM domain region between Group III and Group II genes implies that a Group II ancestral gene was the likely donor of the LysM triplet at the time of origin of Group III genes in Rosids.
The LysM domains of SlLYK3, AtLYK3, LjLYS4, and LjLYS5 are distinct from other LysM-RLKs
The ability of LysM-RLK proteins to detect and distinguish between ligands depends on their three extracellular LysM domains. Therefore, the evolutionary history of this region is especially relevant for understanding how functional differences arise and are maintained. Based on previous functional studies, the LysM2 domain of certain LysM-RLKs has emerged as the most critical for ligand recognition and discrimination [48]. Therefore, we were interested in uncovering the evolutionary history of each LysM domain in isolation and whether individual domains showed differential patterns of association with functional recognition. To this end, we sought to reconstruct the evolutionary history of the LysM1, LysM2, and LysM3 domains from the set of canonical LysM-RLK genes of S. lycopersicum, A. thaliana, L. japonicus, M. truncatula, and O. sativa.
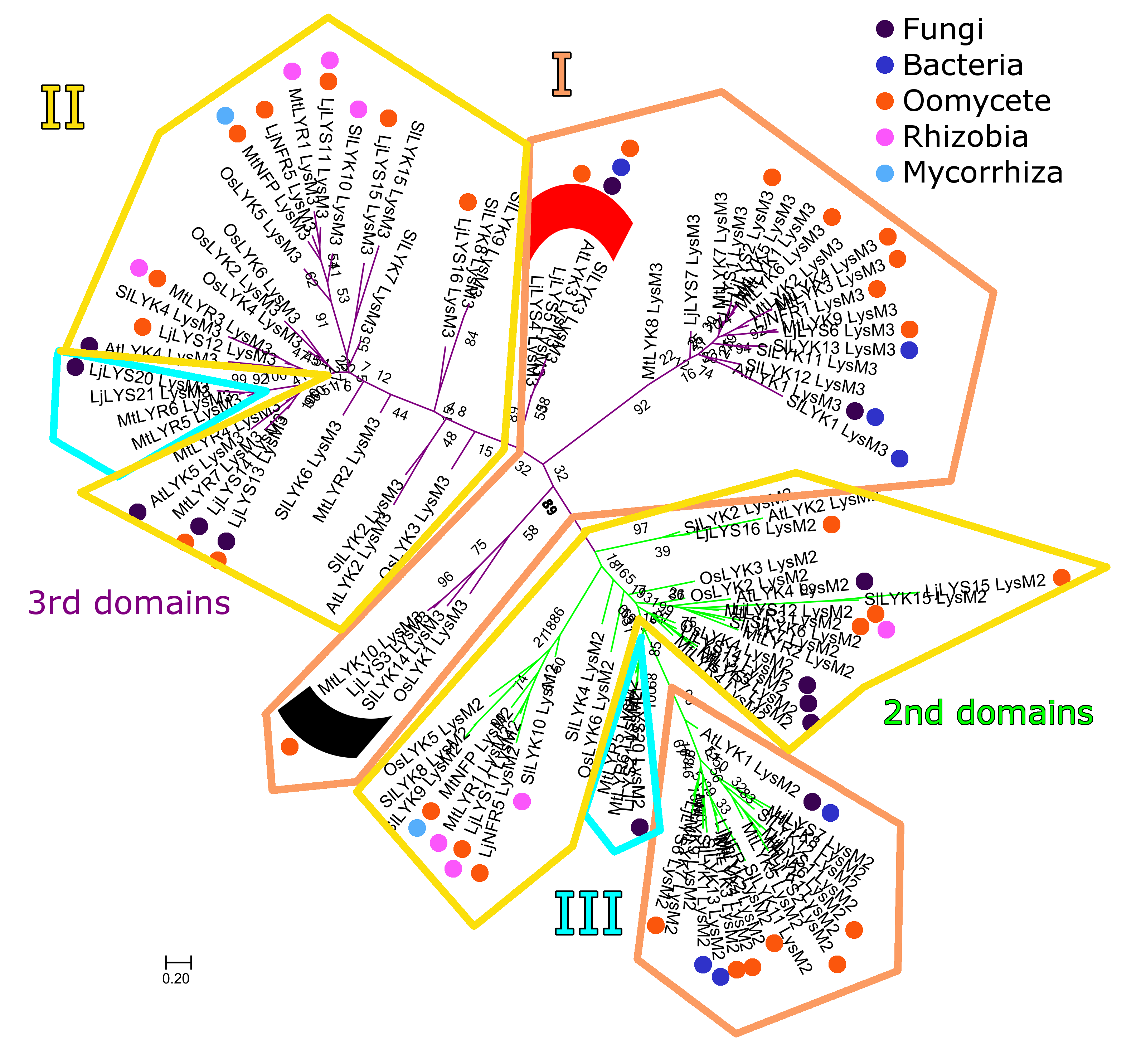
The short sequence lengths (about 60 amino acids) and substantial variation between the individual LysM domain sequences led to poor phylogenetic resolution. Since the three-LysM-domain structure is ancient [3], we expected to recover a tree with three distinct clades consisting of sequences from the first, second and third domains, respectively. However, our phylogenetic analysis did not show three monophyletic clades according to domain order (Fig. 3, Additional file 7). Instead resolution, especially between the first and third domain sequences, was exceptionally low. Furthermore, branches subtending some lineages were substantially longer than others. The lineages containing sequences from four genes in particular, SlLYK3, AtLYK3, LjLYS4, and LjLYS5 (indicated in red swatches in Figs. 1 and 3 and referred to subsequently as the “red clade”), stood out for each LysM domain and consistently formed clades supported by bootstrap values near 90%.
Fig. 3.

Unrooted phylogeny of individual LysM-RLK domains. Phylogeny of amino acid sequences of each of the three LysM-RLK protein domains from each of the canonical LysM-RLKs of Solanum lycopersicum (Sl), Arabidopsis thaliana (At), Lotus japonicus (Lj), Oryza sativa (Os), and Medicago truncatula (Mt). Each gene is represented three times in the tree, once for each individual LysM domain (see emphasis on SlLYK3 in phylogeny), with color-coding by domain position. The phylogeny and 500 bootstrap replicates were inferred using RAxML under the WAG model with empirical frequencies and seed values of 100. The Log-likelihood was −13,081. The scale bar indicates amino acid substitutions per site. Sequences from the first, second, and third LysM domains generally cluster in clades with others of the corresponding LysM domain, but the first and third domains do not form separate clades. Especially long branches subtend the groups of the first and second domains of genes of interest, referred to as the red clade and the black clade. Domain sequences from the red clade are highlighted in red: AtLYK3, SlLYK3, LjLYS4, and LjLYS5. Those from the black clade are highlighted in black: OsLYK1, MtLYK10, SlLYK14, and LjLYS3. The third domain of OsLYK1 and first domain of LjLYS3 are separated from the corresponding domains of the second group
We suspected that the long branches subtending these clades could indicate a problem with the alignment of these sequences. A GUIDANCE [25] analysis indicated that for the third domain, similar alignments resulted independent of alignment method. For the first and second domains, different alignments were recovered depending upon which alignment method was used.
In particular, the GUIDANCE sequence score was less than 0.8 for the alignments of the first domains of all of the proteins, as well as the second domains of the previously described group SlLYK3, AtLYK3, LjLys4, and LjLYS5 (the red clade) and another group OsLYK1, SlLYK14, LjLYS3, and MtLYK10 (indicated by a black swatch in Figs. 1 and 3 and referred to as the “black clade”). We conclude that the phylogenetic signal in these data is very weak due to extensive sequence divergence.
This analysis prompted us to inspect the degree to which residues of the individual LysM domains were conserved. Also we investigated whether the LysM domain sequences from the genes belonging to the red and black clades had substantially diverged from the other genes, especially at residues otherwise conserved across genes. The WebLogo [49] analysis of the sequences for the three individual LysM domains, excluding the sequences belonging to the red and black clades, showed that the conservation of individual residues varied across all three LysM domains (Fig. 4). A moderate number of conserved residues (four) were shared between both groups of LysM-RLKs in the third LysM domain (Fig. 4). However, the amino acid logos for the LysM domain sequences from the red and black clades show nearly no overlap with the conserved residues in LysM domains 1 and 2. In summary, the representative genes of the red and black clades are evolutionarily divergent compared to those from other LysM-RLK genes, showing nearly no overlap in conserved residues in LysM domains 1 and 2. This is further indicated by their position on the phylogeny based on the entire protein sequences (Fig. 1): There it is noted that genes from the red and black clades belong to Group I and form a well-supported monophyletic sister clade to the other members of the Group I LysM-RLKs.
Fig. 4.

Amino acid logos of LysM-RLK domains. Logos of LysM-RLK LysM domains, with those of the red and black clades (AtLYK3, SlLYK3, LjLYS4, LjLYS5, MtLYK10, OsLYK1, LjLYS3, and SlLYK14) computed separately. The third domains of both sets of sequences share conserved amino acids, while first and second domains of the two sequence sets share few conserved amino acids
Based upon the outcome of the Logo analysis and GUIDANCE, we reanalyzed the individual LysM domains for the gene and domain combinations that could be confidently aligned (Additional files 8 and 9). For this analysis, red and black clade domain 2 sequences and all sequences from domain 1 were excluded due to low confidence in the alignments (as described above). The resulting phylogeny shows good resolution between domain 2 and 3 sequences (bootstrap support of 89%, bold on Additional file 8), consistent with the maintenance of this domain structure tracing back at least to the split of monocots and dicots.
Purifying selection is the prevalent form of natural selection in LysM-RLKs from wild tomatoes
To evaluate the evolutionary history of the LysM-RLKs on a more recent microevolutionary timescale, we investigated the patterns of polymorphism and divergence in LysM-RLK genes in a young pair of wild tomato species, S. chilense and S. peruvianum. We first calculated standard population genetic summary statistics for these genes (Table 1) in the species of interest. LysM-RLK sequences from S. ochranthum and S. lycopersicoides were used as outgroups. These are ideal outgroups. Their evolutionary position is outside our clade of interest, which itself also includes the cultivated tomato, S. lycopersicum [20]; these two outgroup species are evolutionarily closer to the wild tomatoes than is S. tuberosum (potato); and both outgroup species are diploids, rather than polyploids.
Table 1.
Summary of allelic diversity and tests of neutrality of LysM-RLKs in wild tomatoes
| No. Seqs | No. Sites | Haplotypes | π all sites (non,syn,silent) | πa/πs | S | MK p-value (uncorrected) | |
|---|---|---|---|---|---|---|---|
| LYK1 | 1881 | ||||||
| chil v ochr | 17 v 1 | 13 | 0.0027 (0.0016, 0.0059, 0.0059) | 0.28 | 19 | 0.062 | |
| chil v lyco | 17 v 1 | 0.0027 (0.0016, 0.0059, 0.0059) | 0.28 | 0.026 | |||
| peru v ochr | 17 v 1 | 15 | 0.0049 (0.0026, 0.0118, 0.0117) | 0.22 | 43 | 0.185 | |
| peru v lyco | 17 v 1 | 0.0049 (0.0026, 0.0118, 0.0117) | 0.22 | 0.146 | |||
| LYK3 | 1992 | ||||||
| chil v ochr | 17 v 1 | 16 | 0.0046 (0.0011, 0.0160, 0.0159) | 0.07 | 37 | 0.036 | |
| chil v lyco | 17 v 1 | 0.0046 (0.0011, 0.0152, 0.0151) | 0.07 | 0.718 | |||
| peru v ochr | 16 v 1 | 15 | 0.0056 (0.0008, 0.0211, 0.0209) | 0.04 | 51 | 0.005 | |
| peru v lyco | 16 v 1 | 0.0056 (0.0008, 0.0215, 0.0214) | 0.04 | 0.822 | |||
| LYK4 | 1938 | ||||||
| chil v ochr | 15 v 1 | 15 | 0.0080 (0.0061, 0.0138, 0.0137) | 0.44 | 51 | 0.510 | |
| chil v lyco | 15 v 1 | 0.0080 (0.0061, 0.0138, 0.0137) | 0.44 | 0.760 | |||
| peru v ochr | 16 v 1 | 16 | 0.0089 (0.0054, 0.0202, 0.0201) | 0.26 | 80 | 0.275 | |
| peru v lyco | 16 v 1 | 0.0089 (0.0054, 0.0202, 0.0201) | 0.26 | 0.336 | |||
| LYK6 | 1599 | ||||||
| chil v ochr | 15 v 1 | 13 | 0.0151 (0.0119, 0.0246, 0.0246) | 0.48 | 108 | 0.108 | |
| chil v lyco | 15 v 1 | 0.0151 (0.0119, 0.0245, 0.0245) | 0.48 | 0.069 | |||
| peru v ochr | 8 v 1 | 8 | 0.0161 (0.0135, 0.0244, 0.0244) | 0.55 | 87 | 0.118 | |
| peru v lyco | 8 v 1 | 0.0161 (0.0135, 0.0244, 0.0244) | 0.55 | 0.064 | |||
| LYK8 | 1149 | ||||||
| chil v ochr | 13 v 1 | 12 | 0.0048 (0.0044, 0.0064, 0.0064) | 0.69 | 34 | 0.025 | |
| peru v ochr | 17 v 1 | 16 | 0.0052 (0.0042, 0.0087, 0.0087) | 0.47 | 55 | 0.026 | |
| LYK9 | 1890 | ||||||
| chil v ochr | 17 v 1 | 17 | 0.0073 (0.0042, 0.0174, 0.0176) | 0.24 | 74 | 0.818 | |
| chil v lyco | 17 v 1 | 0.0073 (0.0042, 0.0174, 0.0176) | 0.24 | 73 | 0.604 | ||
| peru v ochr | 17 v 1 | 17 | 0.0068 (0.0045, 0.0135, 0.0146) | 0.33 | 79 | 0.813 | |
| peru v lyco | 17 v 1 | 0.0068 (0.0045, 0.0174, 0.0146) | 0.33 | 77 | 0.649 |
Population samples were available for Solanum chilense (chil) and Solanum peruvianum (peru). Single allelic sequences from Solanum ochranthum (ochr) and Solanum lycopersicoides (lyco) were used for outgroup comparisons in the tests of neutrality. Haplotypes, the number of unique alleles in each population sample, apply to the first species listed in column 1, as do values for S, the number of segregating sites. The ratio of non-synonymous to synonymous average pairwise differences (πa/πs) was calculated for S. chilense and S. peruvianum. The McDonald-Kreitman test was applied to each pair of species listed in column 1. Uncorrected p-values for the McDonald-Kreitman analyses are reported, but following correction for multiple testing, none of the corrected p-values were below 5%
We identified six LysM-RLKs for which our criterium for a minimum sample size for complete gene sequences from 8 different individuals for both S. chilense and S. peruvianum was met. This corresponded to the following set of genes: LYK1, LYK3, LYK4, LYK6, LYK8 and LYK9. The polymorphism at non-synonymous sites at the LysM-RLK genes ranged from 0.11% (LYK3) to 1.19% (LYK6) in S. chilense and 0.08% (LYK3) to 1.4% (LYK6) in S. peruvianum (Table 1). The polymorphism at synonymous sites at the LysM-RLK genes ranged from 0.64% (LYK8) to 2.5% (LYK6) in S. chilense and 0.87% (LYK8) to 2.4% (LYK6) in S. peruvianum. These values are consistent with the species-wide mean polymorphism at non-synonymous (0.18%) and synonymous (1.27%) sites in S. chilense and non-synonymous (0.22%) and synonymous (1.69%) sites in S. peruvianum as reported in [19].
To determine whether selection had differential effects on the pattern of sequence polymorphism in the intracellular or extracellular domains, we calculated the summary statistics for each of these domains separately (Table 2). The ratio of non-synonymous (πa) and synonymous (πs) pairwise differences is often used to gauge the impact of natural selection on the distribution of sequence variation [50]. For most genes, the ratio of πa/πs was higher in the extracellular domain than in the intracellular/kinase domain. The distribution of variation at LYK3 gene stands out because the ratios of πa/πs are extremely low (≤ 0.06) and are nearly equivalent in the extracellular and intracellular domains (Table 2). This indicates that a similar degree of purifying selection may be acting on the intracellular and extracellular domains of LYK3 in wild tomatoes.
Table 2.
Summary of allelic diversity applied separately to extracellular and intracellular domains of LysM-RLKs from wild tomatoes
| Gene region | No. Seqs | No. Sites | Haplotypes | π all sites (non,syn,silent) | πa/πs | S | |
|---|---|---|---|---|---|---|---|
| LYK1 | |||||||
| chil v ochr | Extracellular | 17 v 1 | 699 | 8 | 0.0026 (0.0025, 0.0032, 0.0032) | 0.79 | 8 |
| Intracellular | 1101 | 9 | 0.0028 (0.0011, 0.0079, 0.0078) | 0.14 | 10 | ||
| chil v lyco | Extracellular | 17 v 1 | 699 | 8 | 0.0026 (0.0025, 0.0032, 0.0032) | 0.80 | 8 |
| Intracellular | 1101 | 9 | 0.0028 (0.0011, 0.0079, 0.0078) | 0.14 | 10 | ||
| peru v ochr | Extracellular | 17 v 1 | 699 | 12 | 0.0063 (0.0042, 0.0129, 0.0129) | 0.33 | 21 |
| Intracellular | 1101 | 15 | 0.0043 (0.0018, 0.0117, 0.0116) | 0.16 | 21 | ||
| peru v lyco | Extracellular | 17 v 1 | 699 | 12 | 0.0063 (0.0043, 0.0130, 0.0130) | 0.33 | 21 |
| Intracellular | 1101 | 15 | 0.0043 (0.0018, 0.0117, 0.0116) | 0.16 | 21 | ||
| LYK3 | |||||||
| chil v ochr | Extracellular | 17 v 1 | 705 | 12 | 0.0046 (0.0005, 0.0176, 0.0176) | 0.03 | 13 |
| Intracellular | 1203 | 16 | 0.0044 (0.0009, 0.0158, 0.0156) | 0.06 | 21 | ||
| chil v lyco | Extracellular | 17 v 1 | 705 | 12 | 0.0046 (0.0005, 0.0155, 0.0155) | 0.03 | 13 |
| Intracellular | 1203 | 16 | 0.0044 (0.0009, 0.0158, 0.0156) | 0.06 | 21 | ||
| peru v ochr | Extracellular | 16 v 1 | 705 | 13 | 0.0086 (0.0008, 0.0328, 0.0328) | 0.03 | 24 |
| Intracellular | 1203 | 14 | 0.0042 (0.0008, 0.0155, 0.0153) | 0.05 | 25 | ||
| peru v lyco | Extracellular | 16 v 1 | 705 | 13 | 0.0088 (0.0009, 0.0349, 0.0349) | 0.02 | 24 |
| Intracellular | 1203 | 14 | 0.0042 (0.0008, 0.0155, 0.0153) | 0.05 | 25 | ||
| LYK4 | |||||||
| chil v ochr | Extracellular | 15 v 1 | 801 | 15 | 0.0089 (0.0074, 0.0136, 0.0136) | 0.54 | 18 |
| Intracellular | 1053 | 14 | 0.0074 (0.0053, 0.0139, 0.0137) | 0.38 | 31 | ||
| chil v lyco | Extracellular | 15 v 1 | 801 | 15 | 0.0089 (0.0074, 0.0136, 0.0136) | 0.54 | 18 |
| Intracellular | 1053 | 14 | 0.0074 (0.0053, 0.0139, 0.0137) | 0.38 | 31 | ||
| peru v ochr | Extracellular | 16 v 1 | 801 | 16 | 0.0106 (0.0087, 0.0161, 0.0089) | 0.54 | 41 |
| Intracellular | 1053 | 11 | 0.0075 (0.0029, 0.0236, 0.0233) | 0.12 | 33 | ||
| peru v lyco | Extracellular | 16 v 1 | 801 | 16 | 0.0106 (0.0087, 0.0161, 0.0161) | 0.54 | 41 |
| Intracellular | 1053 | 11 | 0.0075 (0.0029, 0.0236, 0.0233) | 0.12 | 33 | ||
| LYK6 | |||||||
| chil v ochr | Extracellular | 15 v 1 | 780 | 12 | 0.0146 (0.0132, 0.0198, 0.0198) | 0.66 | 29 |
| Intracellular | 741 | 13 | 0.0150 (0.0105, 0.0265, 0.0265) | 0.39 | 31 | ||
| chil v lyco | Extracellular | 15 v 1 | 780 | 12 | 0.0146 (0.0132, 0.0198, 0.0198) | 0.66 | 29 |
| Intracellular | 741 | 13 | 0.0150 (0.0105, 0.0265, 0.0265) | 0.39 | 31 | ||
| peru v ochr | Extracellular | 8 v 1 | 780 | 8 | 0.0152 (0.0149, 0.0166, 0.0166) | 0.90 | 33 |
| Intracellular | 741 | 8 | 0.0188 (0.0127, 0.0397, 0.0397) | 0.31 | 30 | ||
| peru v lyco | Extracellular | 8 v 1 | 780 | 8 | 0.0152 (0.0149, 0.0165, 0.0165) | 0.90 | 33 |
| Intracellular | 741 | 8 | 0.0188 (0.0127, 0.0397, 0.0397) | 0.31 | 30 | ||
| LYK8 | |||||||
| chil v ochr | Extracellular | 13 v 1 | 771 | 9 | 0.0042 (0.0053, 0.0006, 0.0006) | 9.1 | 9 |
| Intracellular* | 300 | 3 | 0.0022 (0.0000, 0.0116, 0.0116) | 0.00 | 3 | ||
| peru v ochr | Extracellular | 17 v 1 | 771 | 13 | 0.0038 (0.0032, 0.0058, 0.0058) | 0.55 | 16 |
| Intracellular* | 300 | 9 | 0.0047 (0.0035, 0.0101, 0.0101) | 0.34 | 11 | ||
| LYK9 | |||||||
| chil v ochr | Extracellular | 17 v 1 | 774 | 14 | 0.0086 (0.0068, 0.0145, 0.0145) | 0.47 | 22 |
| Intracellular | 1038 | 14 | 0.0060 (0.0020, 0.0199, 0.0202) | 0.10 | 22 | ||
| chil v lyco | Extracellular | 17 v 1 | 774 | 14 | 0.0086 (0.0068, 0.0145, 0.0145) | 0.47 | 22 |
| Intracellular | 1038 | 14 | 0.0060 (0.0020, 0.0199, 0.0202) | 0.10 | 22 | ||
| peru v ochr | Extracellular | 17 v 1 | 774 | 14 | 0.0053 (0.0040, 0.0092, 0.0092) | 0.44 | 25 |
| Intracellular | 1038 | 17 | 0.0082 (0.0051, 0.0168, 0.0188) | 0.30 | 32 | ||
| peru v lyco | Extracellular | 17 v 1 | 774 | 14 | 0.0053 (0.0040, 0.0092, 0.0092) | 0.44 | 25 |
| Intracellular | 1038 | 17 | 0.0082 (0.0052, 0.0169, 0.0189) | 0.30 | 32 | ||
Population samples were available for Solanum chilense (chil) and Solanum peruvianum (peru). Single allelic sequences from Solanum ochranthum (ochr) and Solanum lycopersicoides (lyco) were used for outgroup comparisons. Haplotypes, the number of unique alleles in each population sample, apply to the first species listed in column 1, as do values for S, the number of segregating sites. The ratio of non-synonymous to synonymous average pairwise differences (πa/πs) was calculated for S. chilense and S. peruvianum. *The kinase domain of LYK8 is truncated
We then applied two standard tests of neutrality to determine if the pattern of genetic variation was consistent with a recent history of directional or balancing selection. According to Tajima’s D, no evidence for selection at these six genes could be detected. According to the McDonald-Kreitman test, three genes may have experienced recent selection: SlLYK1, SlLYK3, and SlLYK8 (Table 1). In the analysis of alleles of SlLYK3 from S. peruvianum (with S. ochranthum as the outgroup), an excess of non-synonymous fixed differences between species was observed. This is consistent with a recent bout of directional selection at this locus. However, after correcting for multiple testing, the corrected p-value exceeded a significance threshold of 0.05. After 22 tests, the Šidák correction [51] requires a p-value of 0.0023 or less for a 5% significance threshold. The lowest uncorrected p-value for an individual test – SlLYK3 with p = 0.00507 – is equivalent to a p-value of 10–11% after correction. Therefore, following correction for multiple testing, we fail to reject the null hypothesis of equal ratios of replacement to silent changes for fixed differences compared to segregating sites. In summary, the intraspecific and interspecific ratios of non-synonymous and synonymous variation are consistently less than 1 for all the genes analyzed. This indicates the action of purifying selection operating at these loci. Furthermore, the tests of neutrality did not indicate strong balancing or directional selection operating at these loci, except possibly with the case of SlLYK3, a member of the red clade.
Purifying selection extends to some LysM-RLKs in A. thaliana
To see if similar evolutionary patterns were present at LysM-RLKs in other plants species, we extended our population genetic analysis to the model plant species, A. thaliana. We analyzed the sequence variation from 68 accessions for the following genes: AtLYK1, AtLYK2, AtLYK3, AtLYK4 and AtLYK5 (Table 3). These genomic sequences were retrieved from Cao et al. [52]. In particular, we focused on the distribution of variation at AtLYK3, since this gene is a member of the red clade (Fig. 1) and is phylogenetically closely related to SlLYK3, which showed evidence for strong purifying selection in wild tomatoes (Tables 1 and 2). The patterns of variation at AtLYK3 are also consistent with the action of purifying selection (Table 3: πa/πs = 0.178). However, the ratio of πa/πs is not as low as it is for LYK3 in wild tomatoes (Table 1; πa/πs ≤ 0.06).
Table 3.
Summary of allelic diversity at LysM-RLK from A. thaliana
| π (silent) | π (non-coding) | π (syn) | π (non-syn) | πa/πs | π (all sites) | |
|---|---|---|---|---|---|---|
| AtLYK1 | 0.01519 | 0.00745 | 0.04017 | 0.00285 | 0.069 | 0.00996 |
| -extracellular | 0.01687 | 0.00490 | 0.04678 | 0.00273 | 0.057 | 0.01017 |
| -intracellular | 0.01469 | 0.00847 | 0.03933 | 0.00177 | 0.044 | 0.00970 |
| AtLYK2 | 0.00134 | 0.00290 | 0.00098 | 0.00081 | 0.820 | 0.00094 |
| -extracellular | 0.00242 | 0.00296 | 0.00211 | 0.00026 | 0.123 | 0.00097 |
| -intracellular | 0.00080 | 0.00000 | 0.00081 | 0.00082 | 1.015 | 0.00082 |
| AtLYK3 | 0.01090 | 0.00932 | 0.01570 | 0.00281 | 0.178 | 0.00730 |
| -extracellular | 0.01258 | 0.00875 | 0.02217 | 0.00587 | 0.262 | 0.00939 |
| -intracellular | 0.01032 | 0.00956 | 0.01307 | 0.00121 | 0.092 | 0.00650 |
| AtLYK4 | 0.00258 | 0.00273 | 0.00236 | 0.00188 | 0.794 | 0.00219 |
| -extracellular | 0.00236 | 0.00326 | 0.00057 | 0.00265 | 4.629 | 0.00251 |
| -intracellular | 0.00299 | 0.00195 | 0.00433 | 0.00075 | 0.173 | 0.00164 |
| AtLYK5 | 0.00258 | 0.00325 | 0.00150 | 0.00061 | 0.403 | 0.00151 |
| -extracellular | 0.00153 | 0.00162 | 0.00136 | 0.00000 | 0.000 | 0.00077 |
| -intracellular | 0.00366 | 0.00507 | 0.00163 | 0.00110 | 0.672 | 0.00220 |
Wild tomatoes encode orthologs of SlLYK8 with intact kinase domains
In the annotated genome of the cultivated tomato, S. lycopersicum, the SlLYK8 gene is predicted to encode an extracellular LysM region and a truncated intracellular kinase domain. The SlLYK8 gene is lacking more than half of the kinase region present in the closely-related SlLYK9 gene. The missing region includes the catalytic loop and activation segment of the kinase. However, we observed strong protein sequence conservation in the intracellular regions of the orthologs of SlLYK8 in S. peruvianum and S. chilense (Table 2: πa/πs = 0.34 and πa/πs = 0.00 respectively). This was counter to our intuition that natural selection would be relaxed in the intracellular region, if these genes no longer encoded functional kinases. Alternatively, we reasoned that the SlLYK8 gene may be intact in the wild species and only recently truncated (or mis-annotated) in the cultivated species, S. lycopersicum. To test this hypothesis, we evaluated the de novo assembled transcriptomes of these wild species (assembled without read-mapping to the annotated genome of S. lycopersicum). This was necessary because coding regions not annotated in the original S. lycopersicum reference genome would fail to be mapped from the wild species, even if they were present in these wild species.
We screened the de novo assemblies to check whether SlLYK8 orthologous sequences were linked to kinase-encoding sequences [19]. Due to its high sequence similarity to SlLYK8, we also included SlLYK9 in our analyses. The genomic sequences of SlLYK8 and SlLYK9 were used as query sequences in a BlastN search against the de novo assembled transcriptomes. After filtering by sequence length and percent identity to SlLYK8 and SlLYK9, no single sequence was assigned to both queries. Amino acid translations of the sequences which extended beyond the original position of SlLYK8 truncation in S. lycopersicum were aligned with MUSCLE, and this alignment was used to infer a maximum likelihood tree rooted using the method of MAD [27] (Fig. 5). Three sequences had higher sequence similarity to SlLYK8 than to SlLYK9 and extended beyond the position of SlLYK8 truncation. Further inspection of the sequences revealed that each has an intact kinase, including all essential kinase residues. One of the full-length sequences was found in S. chilense and two were from S. peruvianum, which suggests that the truncation of the kinase in SlLYK8 happened after the divergence between S. lycopersicum and the wild tomato species included here.
Fig. 5.
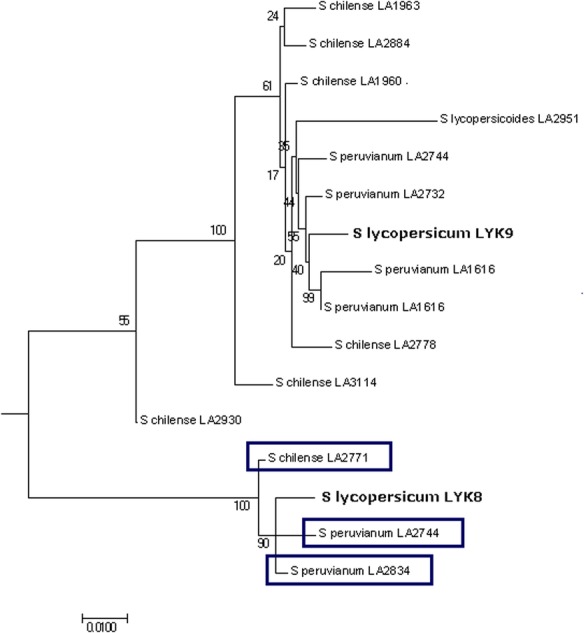
Phylogeny of wild tomato SlLYK8 and SlLYK9 orthologs with intact kinases. This phylogeny includes BLAST hits for Solanum lycopersicum LysM-RLKs SlLYK8 and SlLYK9 which extend past the point of SlLYK8 truncation. The phylogeny and 500 bootstrap replicates were inferred using RAxML under the GTRGAMMA model and seed values of 100 and rooted using the method of Minimal Ancestor Deviation [27]. The scale bar indicates nucleotide substitutions per site. Three sequences from Solanum peruvianum and Solanum chilense with intact kinase domains share more recent ancestry with SlLYK8 than with SlLYK9 and show the greatest percent identity with SlLYK8
Discussion
Here we investigated the evolutionary history of LysM-RLKs, with a special focus on wild tomatoes. These species show evidence of strong purifying selection at these genes, as opposed to directional or balancing selection observed at other well-known pathogenic recognition proteins in wild tomatoes [53–55]. In particular, the orthologs of SlLYK3 have been subject to especially strong purifying selection, specifically in their extracellular domains. This gene belongs to the red clade of LysM-RLKs, which has distinct first and second LysM domains compared to other members of group I (Figs. 1 and 4).
There are currently no data on the function and/or specificity of SlLYK3 or its orthologs from wild tomatoes, but its homolog, AtLYK3, from A. thaliana negatively regulates fungal and bacterial defense [38] and is essential for suppressing the flg22-triggered immune responses in the presence of Nod factors [39]. The presence of strong protein conservation of SlLYK3 orthologs within wild tomatoes, coupled with the distinctness of its LysM domains, makes it an interesting candidate for further functional characterization. Like AtLYK3, orthologs in tomato may fulfill multiple roles. It may do this by detecting multiple microbial ligands with partner receptors or by coordinating signals to control immune response after microbe detection.
Our analyses also uncovered additional novel aspects of LysM-RLK evolution in tomatoes. For example, we detected complete kinase domains in orthologs of SlLYK8 from S. chilense and S. peruvianum. While it is still unclear whether all wild tomatoes have orthologs of SlLYK8 with intact kinase domains, its presence in both S. peruvianum and S. chilense shows that some wild tomatoes do. Sequences from SlLYK8 alleles with intact kinases from other individuals may have been below our cutoffs for percent identity or sequence length or not sufficiently expressed at the time samples were taken. If intact kinase domains are found in additional wild tomato species, it would suggest that the truncation of the kinase may be unique to S. lycopersicum and could have been a fairly recent evolutionary change. Broader taxonomic sampling and analysis of genomic sequences will help to resolve the timing and direction of these evolutionary changes.
It was previously postulated that Group III originated after the split between Group I and Group II, because Group III is only found in dicots [11]. Our analysis indicates that the extracellular (LysM triplet) domain regions of Group II and Group III genes are more closely related to each other than either is to genes from Group I, and that the kinase domains of Group III genes differ from those of Groups II and III (as previously postulated [10, 11]). Despite expanding the known number of species containing LysM-III genes, we still did not find any outside of the Rosids, a clade of the Eudicots. Zhang et al. noted that LysM-II genes in both M. truncatula and O. sativa lacked activation loops and conservation at residues necessary for activity [6], and we note that the same is true for the Glycine-rich loop; this sequence is missing in every LysM-II gene in our analysis, and multiple representatives of this clade are present in each species. Taken together, this implies that the initial divergence in this gene family was between Group I and Group II LysM-RLKs, and that Group III LysM-RLKs originated subsequently (but prior to the divergence of E. grandis from the other Rosids) via a fusion of a Group II LysM triplet with a kinase domain from another protein family.
Conclusions
The LysM-RLKs belong to a diverse family of proteins with many functions in plant symbiosis and defense and little correspondence between function and phylogenetic relationships. Here we provided an overview of the functions and phylogenetic relationships and found that the Group III LysM-RLKs share a closer relationship with those in Group II than those in Group I. Newly identified Group III LysM-RLKs were found in a variety of Rosid species. The kinase domain of SlLYK8 homologs is intact in at least some wild tomato species. We suggest that SlLYK3 is a prime candidate for future functional analysis, owing to its close relationship to the multi-functional AtLYK3, its distinct LysM domains, and signs that the extracellular domains experience strong protein conservation in wild tomatoes.
Methods
Sources for protein sequences
Amino acid sequences from A. thaliana, S. lycopersicum, L. japonicus, O. sativa, and M. truncatula were obtained from the sources listed in Additional file 1: Table S1 [9–18]. To determine the positions of the three LysM domains, we identified the Cysteine residues (including CXC motifs) which occur prior to the start of the first LysM domain and between the proceeding LysM domains. The conserved Proline at the end of the third LysM domain marked the end of the LysM triplet.
Transcriptome data and coverage selection criteria
DNA sequences of LysM-RLKs from Solanum chilense, Solanum peruvianum, Solanum ochranthum, and Solanum lycopersicoides were obtained from Beddows et al. [19], with the exception of the SlLYK8 sequences, which were compiled from the same source data but with minimum read depth of 5 (Additional file 2). The study by Beddows et al. [19] provides an extensive transcriptome dataset for individuals of 18 different populations of S. chilense and S. peruvianum, respectively. In both studies, the two species, S. ochranthum and S. lycopersicoides, were used as outgroups. These two species are closely related to wild tomatoes, but lie outside the wild tomato clade. Because the cultivated tomato (S. lycopersicum) is nested within the clade of wild tomatoes [20], it could not be used as an outgroup. Sequences were included in the population genetic analysis, provided they met the following conditions for sequence completeness and sample size:
Individual sequences had < 10% N-content (undetermined nucleotides) in the coding region
A minimum sample size of 8 complete sequences (alleles) for both S. chilense and S. peruvianum. Alleles were sampled from different individuals independent of their gene sequence, i.e. alleles from different individuals may be identical in sequence.
Accession LA0752 was included in the S. chilense sequence set. Sequences from LA1274, an accession described as Solanum corneliomulleri, were included in the S. peruvianum data set (see Beddows et al. [19] for justification).
For the population genetic analysis of SlLYK8, four genotypes (LA2750, LA2884, LA0752, and LA2930) were excluded because the alleles of SlLYK8 could not be unambiguously inferred for these genotypes. De novo assembled transcriptomes were obtained from the same reads available from Beddows et al. [19]. They were assembled using Trinity [21] version 2014-07-17 with standard settings and mapped to S. lycopersicum (cultivar Heinz 1706) with GMAP [22] version 2017-05-08 with standard settings.
Alignments
For data sets with fewer than 100 sequences or shorter than 200 amino acids, protein sequence alignments were done with the MUSCLE algorithm [24] implemented in MEGA7 [23]. For larger datasets, GUIDANCE [25] (with the MAFFT option) was used.
Phylogenetic analyses
Phylogenies based on protein sequences were inferred in RAxML [26] using the protein substitution model that best fit the data (found using the PROTGAMMAAUTO function). For DNA sequences, the GTRGAMMA function was used. Branch support was assessed by performing 500 non-parametric bootstrap replicates. Seed values of 100 were chosen for reproducibility. Phylogenies were rooted with Minimal Ancestor Deviation (MAD) [27].
Population genetic analysis
All population genetic tests were performed in DnaSP [28] on a randomly selected haplotype from each sampled individual. Significance for the McDonald-Kreitman test [29] was determined by the G-test when the assumptions of this test were met; otherwise, Fisher’s exact test was used. Tajima’s D [30] was calculated using the total number of mutations.
The identification of SlLYK8 from wild tomatoes
Two BlastN searches [31] were performed against the de novo transcriptomes described above: one with the SlLYK8 sequence (1149 nucleotides in length) as the query and one with the corresponding positions of the SlLYK9 sequence (1890 nucleotides in length) as the query. Percent identity was used to measure the similarity of the hits to SlLYK8 and SlLYK9. All hits at least 1000 nucleotides long and showing 97% or greater identity to either SlLYK8 or SlLYK9 (as reported in the BlastN results table) were included in further analyses. The sequences were translated in six frames and aligned together with the amino acid sequences from SlLYK8 and SlLYK9. Translations which spanned at least 40% of the SlLYK8 gene and extended beyond the end of SlLYK8 were included in the alignment.
Identification of group III LysM-RLK homologs
The LjLYS20 amino acid sequence was used as a query in an online BlastP search (with max 250 hits and standard settings) [31] against NCBI’s non-redundant protein sequences database [32]. The full-length hits were aligned with the canonical LysM-RLKs and filtered according to the presence of the CXC motif between the first and second LysM domains. Exact duplicate sequences from the same species were removed before phylogenetic analysis.
Additional files
Table S1. Amino acid sequence sources. These are the sources of sequences used to infer the protein phylogenies [9–18]. (PDF 200 kb)
SlLYK8 ortholog sequences generated for this study. This is a Fasta-formatted text file containing the sequences generated from reads from several wild tomato species (Solanum peruvianum: peru, Solanum chilense: chil, Solanum lycopersicoides: lyco, and Solanum ochranthum: ochr) which were mapped to the region corresponding to SlLYK8 in Solanum lycopersicum. Unlike the rest of the LysM-RLK orthologs obtained from [19], these sequences were assessed with a minimum read depth of five sequences. The rest of the mapping procedure was the same as that used for the other sequences. (TXT 35 kb)
{kind=link}
Phylogeny of new putative Group III LysM-RLKs and canonical LysM-RLKs. The maximum likelihood phylogeny and 500 bootstrap replicates were inferred using RAxML assuming the JTT model and seed values of 100. (PNG 987 kb)
Phylogeny from Additional file 3 in Newick format. (NWK 7 kb)
Phylogeny from Fig. 2a in Newick Format. (NWK 6 kb)
Phylogeny from Fig. 2b in Newick Format. (NWK 6 kb)
Phylogeny from Fig. 3 in Newick format. (NWK 6 kb)
{kind=link}
Phylogeny of reliably aligned individual LysM-RLK domains. Phylogeny of amino acid sequences of the LysM-RLK LysM domains which scored 0.80 or higher when evaluated with GUIDANCE. Each of the first domains scored below this cutoff, and all were omitted. All third domains were included. Second domains of genes highlighted in red and black were omitted. The maximum likelihood phylogeny and 500 bootstrap replicates were inferred using RAxML under the WAG model with empirical frequencies and seed values of 100. The Log-likelihood was − 9529. The second and third domains of the sequences included form distinct clades. Known functions of the proteins (Fig. 1) were mapped onto the individual domains in the tree. (PNG 1616 kb)
Phylogeny from Additional file 8 in Newick format. (NWK 7 kb)
Acknowledgements
We would like to thank Alisandra Denton for help with the assembly of the Arabidopsis population genetic dataset, Thorsten Klösges for bioinformatic assistance, the Rose lab for helpful suggestions and insights during the analysis, and Christopher Blum, Sophie de Vries, Jan de Vries, Ingo Ebersberger, Ovidiu Popa and two anonymous reviewers for helpful comments on earlier versions of this paper.
Declarations
A portion of the work described here was presented as part of the doctoral dissertation of Dr. Sarah Richards.
Abbreviations
- At
Arabidopsis thaliana
- CERK1
Chitin elicitor receptor-like kinase 1
- Eg
Eucalyptus grandis
- Lj
Lotus japonicus
- LYK
LysM-RLK with classically conserved kinase domain
- LYR
LYK-related, or LysM-RLK without classically conserved kinase domain
- LYS
LysM-RLKs in Lotus japonicus
- LysM-RLK
lysin motif RLK
- Mt
Medicago truncatula
- Os
Oryza sativa
- RLK
receptor-like kinase
- Sl
Solanum lycopersicum
Authors’ contributions
LR and SR developed the hypotheses. SR tested the hypotheses and wrote the manuscript. LR edited the manuscript. Both authors approved of the final version of the manuscript.
Funding
Financial support for the collection, analysis and storage of the data was provided by Deutsche Forschungsgemeinschaft (DFG) grants RO 2491/4–1 and RO 2491/5–2.
Availability of data and materials
The SlLYK8 ortholog sequences generated for this study can be found in the Supplemental Material for this paper. All other sequences analyzed in this study can be found in public repositories as indicated in the citations.
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
Laura E. Rose is a member of the editorial board (Associate Editor) of this journal.
Footnotes
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Sarah Richards, Email: sarah.richards@hhu.de.
Laura E. Rose, Email: laura.rose@hhu.de
References
- 1.Oldroyd GED, Robatzek S. The broad spectrum of plant associations with other organisms. Curr Opin Plant Biol. 2011;14(4):347–350. doi: 10.1016/j.pbi.2011.06.001. [DOI] [PubMed] [Google Scholar]
- 2.Rajamuthiah R, Mylonakis E. Effector triggered immunity. Virulence. 2014;5(7):697–702. doi: 10.4161/viru.29091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gust AA, Willmann R, Desaki Y, Grabherr HM, Nürnberger T. Plant LysM proteins: modules mediating symbiosis and immunity. Trends Plant Sci. 2012;17(8):495–502. doi: 10.1016/j.tplants.2012.04.003. [DOI] [PubMed] [Google Scholar]
- 4.Cao Y, Liang Y, Tanaka K, Nguyen C, Jedrzejczak R, Joachimiak A, et al. The kinase LYK5 is a major chitin receptor in Arabidopsis and forms a chitin-induced complex with related kinase CERK1. Elife. 2014;3:e03766. doi: 10.7554/eLife.03766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhang X, Dong W, Sun J, Feng F, Deng Y, He Z, et al. The receptor kinase CERK1 has dual functions in symbiosis and immunity signaling. Plant J. 2014;81(2):258–267. doi: 10.1111/tpj.12723. [DOI] [PubMed] [Google Scholar]
- 6.Zhang XC, Cannon SB, Stacey G. Evolutionary genomics of LysM genes in land plants. BMC Evol Biol. 2009;9(1):183. doi: 10.1186/1471-2148-9-183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Delaux PM, Radhakrishnan GV, Jayaraman D, Cheema J, Malbreil M, Volkening JD, et al. Algal ancestor of land plants was preadapted for symbiosis. Proc Natl Acad Sci USA. 2015;112(43):13390–13395. doi: 10.1073/pnas.1515426112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shiu SH, Karlowski WM, Pan R, Tzeng YH, Mayer KFX, Li WH. Comparative analysis of the receptor-like kinase family in Arabidopsis and Rice. Plant Cell. 2004;16(5):1220–1234. doi: 10.1105/tpc.020834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Buendia L, Wang T, Girardin A, Lefebvre B. The LysM receptor-like kinase SlLYK10 regulates the arbuscular mycorrhizal symbiosis in tomato. New Phytol. 2016;210(1):184–195. doi: 10.1111/nph.13753. [DOI] [PubMed] [Google Scholar]
- 10.Arrighi JF, Barre A, Amor BB, Bersoult A, Soriano LC, Mirabella R, et al. The Medicago truncatula lysine motif-receptor-like kinase gene family includes NFP and new nodule-expressed genes. Plant Physiol. 2006;142(1):265–279. doi: 10.1104/pp.106.084657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lohmann GV, Shimoda Y, Nielsen MW, Jørgensen FG, Grossmann C, Sandal N, et al. Evolution and regulation of the Lotus japonicus LysM receptor gene family. Mol Plant-Microbe Interact. 2010;23(4):510–521. doi: 10.1094/MPMI-23-4-0510. [DOI] [PubMed] [Google Scholar]
- 12.Zhang XC, Wu X, Findley S, Wan J, Libault M, Nguyen HT, et al. Molecular evolution of Lysin motif-type receptor-like kinases in plants. Plant Physiol. 2007;144(2):623–636. doi: 10.1104/pp.107.097097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rhee SY, Beavis W, Berardini TZ, Chen G, Dixon D, Doyle A, et al. The Arabidopsis information resource (TAIR): a model organism database providing a centralized, curated gateway to Arabidopsis biology, research materials and community. Nucleic Acids Res. 2003;31(1):224. doi: 10.1093/nar/gkg076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.National Center for Biotechnology Information (NCBI). Bethesda, MD, USA. https://www.ncbi.nlm.nih.gov/. Accessed 21 Oct 2016.
- 15.J. Craig Venter Institute MedicMine (JCVI MedicMine). Rockville, MD, USA. https://medicmine.jcvi.org/. Accessed 21 Oct 2016.
- 16.Kawahara Y, de la Bastide M, Hamilton JP, Kanamori H, McCombie WR, Ouyang S, et al. Improvement of the Oryza sativa Nipponbare reference genome using next generation sequence and optical map data. Rice. 2013;6.1:4. doi: 10.1186/1939-8433-6-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zeng L, Velásquez AC, Munkvold KR, Zhang J, Martin GB. A tomato LysM receptor-like kinase promotes immunity and its kinase activity is inhibited by AvrPtoB. Plant J. 2012;69(1):92–103. doi: 10.1111/j.1365-313X.2011.04773.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fernando-Pozo N, Menda N, Edwards JD, Saha S, Tecle IY, Strickler SR, et al. The sol genomics network (SGN) – from genotype to phenotype breeding. Nucleic Acids Res. 2015;43.D1:D1036–41. [DOI] [PMC free article] [PubMed]
- 19.Beddows I, Reddy A, Kloesges T, Rose LE. Population genomics in wild tomatoes—the interplay of divergence and admixture. Genome Biol Evol. 2017;9(11):3023–3038. doi: 10.1093/gbe/evx224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bedinger PA, Chetelat RT, McClure B, Moyle LC, Rose JK, Stack SM, et al. Interspecific reproductive barriers in the tomato clade: opportunities to decipher mechanisms of reproductive isolation. Sex Plant Reprod. 2011;24(3):171–187. doi: 10.1007/s00497-010-0155-7. [DOI] [PubMed] [Google Scholar]
- 21.Grabherr MG, Haas BJ, Yassour M, Levin JZ, Thompson DA, Amit I, Adiconis X, Fan L, Raychowdhury R, Zeng Q, Chen Z, Mauceli E, Hacohen N, Gnirke A, Rhind N, di Palma F, Birren BW, Nusbaum C, Lindblad-Toh K, Friedman N, Regev A. Full-length transcriptome assembly from RNA-seq data without a reference genome. Nat Biotechnol. 2011;29(7):644–652. doi: 10.1038/nbt.1883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wu TD, Watanabe CK. GMAP: a genomic mapping and alignment program for mRNA and EST sequences. Bioinformatics. 2005;21:1859–1875. doi: 10.1093/bioinformatics/bti310. [DOI] [PubMed] [Google Scholar]
- 23.Kumar S, Stecher G, Tamura K. MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol Biology Evol. 2016;33(7):1870–1874. doi: 10.1093/molbev/msw054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Edgar RC. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004;32(5):1792–1797. doi: 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sela I, Ashkenazy H, Katoh K, Pupko T. GUIDANCE2: accurate detection of unreliable alignment regions accounting for the uncertainty of multiple parameters. Nucleic Acids Res. 2015;43(W1):W7–14. doi: 10.1093/nar/gkv318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Stamatakis A. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics. 2014;30(9):1312–1313. doi: 10.1093/bioinformatics/btu033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tria FDK, Landan G, Dagan T. Phylogenetic rooting using minimal ancestor deviation. Nat Ecol Evol. 2017;1:s41559–s41017. doi: 10.1038/s41559-017-0193. [DOI] [PubMed] [Google Scholar]
- 28.Librado P, Rozas J. DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics. 2017;15(11):1451–1452. doi: 10.1093/bioinformatics/btp187. [DOI] [PubMed] [Google Scholar]
- 29.McDonald JH, Kreitman M. Adaptive protein evolution at the Adh locus in drosophila. Nature. 1991;351(6328):652. doi: 10.1038/351652a0. [DOI] [PubMed] [Google Scholar]
- 30.Tajima F. Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics. 1989;123(3):585–595. doi: 10.1093/genetics/123.3.585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215(3):403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 32.O'Leary NA, Wright MW, Brister JR, Ciufo S, Haddad D, McVeigh R, Rajput B, Robbertse B, Smith-White B, Ako-Adjei D, Astashyn A. Reference sequence (RefSeq) database at NCBI: current status, taxonomic expansion, and functional annotation. Nucleic Acids Res. 2015;44(D1):D733–D745. doi: 10.1093/nar/gkv1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Radutoiu S, Madsen LH, Madsen EB, Felle HH, Umehara Y, Grønlund M, Sato S, Nakamura Y, Tabata S, Sandal N, Stougaard J. Plant recognition of symbiotic bacteria requires two LysM receptor-like kinases. Nature. 2003;425(6958):585. doi: 10.1038/nature02039. [DOI] [PubMed] [Google Scholar]
- 34.Limpens E, Franken C, Smit P, Willemse J, Bisseling T, Geurts R. LysM domain receptor kinases regulating rhizobial nod factor-induced infection. Science. 2003;302(5645):630–633. doi: 10.1126/science.1090074. [DOI] [PubMed] [Google Scholar]
- 35.Jones KM, Sharopova N, Lohar DP, Zhang JQ, VandenBosch KA, Walker GC. Differential response of the plant Medicago truncatula to its symbiont Sinorhizobium meliloti or an exopolysaccharide-deficient mutant. Proc Natl Acad Sci U S A. 2008;105(2):704–709. doi: 10.1073/pnas.0709338105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Miya A, Albert P, Shinya T, Desaki Y, Ichimura K, Shirasu K, Narusaka Y, Kawakami N, Kaku H, Shibuya N. CERK1, a LysM receptor kinase, is essential for chitin elicitor signaling in Arabidopsis. Proc Natl Acad Sci U S A. 2007;104(49):19613–19618. doi: 10.1073/pnas.0705147104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Willmann R, Lajunen HM, Erbs G, Newman MA, Kolb D, Tsuda K, Katagiri F, Fliegmann J, Bono JJ, Cullimore JV, Jehle AK. Arabidopsis lysin-motif proteins LYM1 LYM3 CERK1 mediate bacterial peptidoglycan sensing and immunity to bacterial infection. Proc Natl Acad Sci U S A. 2011;108(49):19824–19829. doi: 10.1073/pnas.1112862108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Paparella C, Savatin DV, Marti L, De Lorenzo G, Ferrari S. The Arabidopsis LYSIN MOTIF-CONTAINING RECEPTOR-LIKE KINASE3 regulates the cross talk between immunity and abscisic acid responses. Plant Physiol. 2014;165(1):262–267. doi: 10.1104/pp.113.233759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Liang Y, Cao Y, Tanaka K, Thibivilliers S, Wan J, Choi J, ho Kang C, Qiu J, Stacey G. Nonlegumes respond to rhizobial nod factors by suppressing the innate immune response. Science. 2013;341(6152):1384–1387. doi: 10.1126/science.1242736. [DOI] [PubMed] [Google Scholar]
- 40.Wan J, Tanaka K, Zhang XC, Son GH, Brechenmacher L, Nguyen TH, Stacey G. LYK4, a lysin motif receptor-like kinase, is important for chitin signaling and plant innate immunity in Arabidopsis. Plant Physiol. 2012;160(1):396–406. doi: 10.1104/pp.112.201699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Fliegmann J, Canova S, Lachaud C, Uhlenbroich S, Gasciolli V, Pichereaux C, Rossignol M, Rosenberg C, Cumener M, Pitorre D, Lefebvre B. Lipo-chitooligosaccharidic symbiotic signals are recognized by LysM receptor-like kinase LYR3 in the legume Medicago truncatula. ACS Chem Biol. 2013;8(9):1900–1906. doi: 10.1021/cb400369u. [DOI] [PubMed] [Google Scholar]
- 42.Hogekamp C, Arndt D, Pereira PA, Becker JD, Hohnjec N, Küster H. Laser microdissection unravels cell-type-specific transcription in arbuscular mycorrhizal roots, including CAAT-box transcription factor gene expression correlating with fungal contact and spread. Plant Physiol. 2011;157(4):2023–2043. doi: 10.1104/pp.111.186635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rasmussen SR, Füchtbauer W, Novero M, Volpe V, Malkov N, Genre A, Bonfante P, Stougaard J, Radutoiu S. Intraradical colonization by arbuscular mycorrhizal fungi triggers induction of a lipochitooligosaccharide receptor. Sci Rep. 2016;6:29733. doi: 10.1038/srep29733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rey T, Nars A, Bonhomme M, Bottin A, Huguet S, Balzergue S, Jardinaud MF, Bono JJ, Cullimore J, Dumas B, Gough C. NFP, a LysM protein controlling nod factor perception, also intervenes in Medicago truncatula resistance to pathogens. New Phytol. 2013;198(3):875–886. doi: 10.1111/nph.12198. [DOI] [PubMed] [Google Scholar]
- 45.Amor BB, Shaw SL, Oldroyd GE, Maillet F, Penmetsa RV, Cook D, Long SR, Dénarié J, Gough C. The NFP locus of Medicago truncatula controls an early step of nod factor signal transduction upstream of a rapid calcium flux and root hair deformation. Plant J. 2003;34(4):495–506. doi: 10.1046/j.1365-313X.2003.01743.x. [DOI] [PubMed] [Google Scholar]
- 46.De Mita S, Streng A, Bisseling T, Geurts R. Evolution of a symbiotic receptor through gene duplications in the legume–rhizobium mutualism. New Phytol. 2014;201(3):961–972. doi: 10.1111/nph.12549. [DOI] [PubMed] [Google Scholar]
- 47.Altschul SF, Madden TL, Schäffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Radutoiu S, Madsen LH, Madsen EB, Jurkiewicz A, Fukai E, Quistgaard EM, et al. LysM domains mediate lipochitin–oligosaccharide recognition and Nfr genes extend the symbiotic host range. EMBO J. 2007;26(17):3923–3935. doi: 10.1038/sj.emboj.7601826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Crooks GE, Hon G, Chandonia JM, Brenner SE. WebLogo: A sequence logo generator. Genome Res. 2004;14:1188–1190. doi: 10.1101/gr.849004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Greenman C, Stephens P, Smith R, Dalgliesh GL, Hunter C, Bignell G, et al. Patterns of somatic mutation in human cancer genomes. Nature. 2007;446(7132):153. doi: 10.1038/nature05610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Šidák Z. Rectangular confidence regions for the means of multivariate normal distributions. J Am Stat Assoc. 1967;62(318):626–633. [Google Scholar]
- 52.Cao J, Schneeberger K, Ossowski S, Günther T, Bender S, Fitz J, et al. Whole-genome sequencing of multiple Arabidopsis thaliana populations. Nat Genet. 2011;43(10):956. doi: 10.1038/ng.911. [DOI] [PubMed] [Google Scholar]
- 53.Rose LE, Michelmore RW, Langley CH. Natural variation in the Pto pathogen resistance gene within species of wild tomato (Lycopersicon): II. Population genetics of Pto. Genetics. 2007;175:1307–1319. doi: 10.1534/genetics.106.063602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Rose LE, Grzeskowiak L, Hörger A, Groth M, Stephan W. Targets of selection in a disease resistance network in wild tomatoes. Mol Plant Pathol. 2011;12:921–927. doi: 10.1111/j.1364-3703.2011.00720.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hörger AC, Ilyas M, Stephan W, Tellier A, van der Hoorn RAL, Rose LE. Balancing selection at the tomato RCR3 Guardee gene family maintains variation in strength of pathogen defense. PLoS Genet. 2012;8:e1002813. doi: 10.1371/journal.pgen.1002813. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Amino acid sequence sources. These are the sources of sequences used to infer the protein phylogenies [9–18]. (PDF 200 kb)
SlLYK8 ortholog sequences generated for this study. This is a Fasta-formatted text file containing the sequences generated from reads from several wild tomato species (Solanum peruvianum: peru, Solanum chilense: chil, Solanum lycopersicoides: lyco, and Solanum ochranthum: ochr) which were mapped to the region corresponding to SlLYK8 in Solanum lycopersicum. Unlike the rest of the LysM-RLK orthologs obtained from [19], these sequences were assessed with a minimum read depth of five sequences. The rest of the mapping procedure was the same as that used for the other sequences. (TXT 35 kb)
Phylogeny of new putative Group III LysM-RLKs and canonical LysM-RLKs. The maximum likelihood phylogeny and 500 bootstrap replicates were inferred using RAxML assuming the JTT model and seed values of 100. (PNG 987 kb)
Phylogeny from Additional file 3 in Newick format. (NWK 7 kb)
Phylogeny from Fig. 2a in Newick Format. (NWK 6 kb)
Phylogeny from Fig. 2b in Newick Format. (NWK 6 kb)
Phylogeny from Fig. 3 in Newick format. (NWK 6 kb)
Phylogeny of reliably aligned individual LysM-RLK domains. Phylogeny of amino acid sequences of the LysM-RLK LysM domains which scored 0.80 or higher when evaluated with GUIDANCE. Each of the first domains scored below this cutoff, and all were omitted. All third domains were included. Second domains of genes highlighted in red and black were omitted. The maximum likelihood phylogeny and 500 bootstrap replicates were inferred using RAxML under the WAG model with empirical frequencies and seed values of 100. The Log-likelihood was − 9529. The second and third domains of the sequences included form distinct clades. Known functions of the proteins (Fig. 1) were mapped onto the individual domains in the tree. (PNG 1616 kb)
Phylogeny from Additional file 8 in Newick format. (NWK 7 kb)
Data Availability Statement
The SlLYK8 ortholog sequences generated for this study can be found in the Supplemental Material for this paper. All other sequences analyzed in this study can be found in public repositories as indicated in the citations.