

SUMMARY
RLR-mediated Type-I IFN production plays a pivotal role in elevating host immunity for viral clearance and cancer immune-surveillance. Here we report that glycolysis, which is inactivated during RLR activation, serves as a barrier to impede type-I IFN production upon RLR activation. RLR-triggered MAVS-RIG-I recognition hijacks hexokinase binding to MAVS, leading to the impairment of hexokinase mitochondria localization and activation. Lactate serves as a key metabolite responsible for glycolysis-mediated RLR signaling inhibition by directly binding to MAVS transmembrane (TM) domain and preventing MAVS aggregation. Notably, lactate restoration reverses increased IFN production caused by lactate deficiency. Using pharmacological and genetic approaches, we show that lactate reduction by LDHA inactivation heightens type-I IFN production to protect mice from viral infection. Our study establishes a critical role of glycolysis-derived lactate in limiting RLR signaling and identifies MAVS as a direct sensor of lactate, which functions to connect energy metabolism and innate immunity.
Graphical Abstract
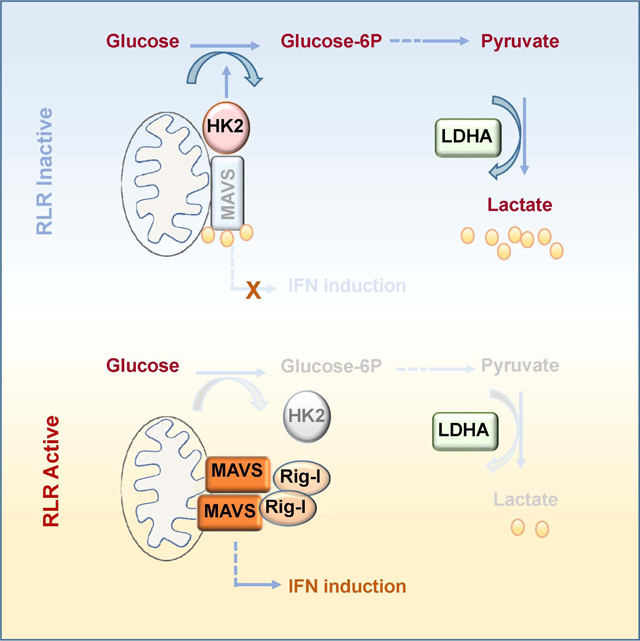
eTOC
Lactate acts as a regulator of the adaptor MAVS, allowing a cross-regulation between antiviral signaling and energy metabolism
INTRODUCTION
Type-I interferons (IFNs) produced by almost all type of cells play a vital role in host defense against viral infection and cancer immunosurveillance (Ivashkiv and Donlin, 2014; Zitvogel et al., 2015). Type-I IFNs composed of IFNα, IFNβ and other IFNs are induced following activation of cell-surface or intracellular pattern recognition receptors (PRRs), including retinoic acid-inducible gene I (RIG-I) like receptors (RLRs), stimulator of IFN genes protein (STING) and Toll-like receptors (TLRs) (Burdette et al., 2011; Kawai and Akira, 2010; Trinchieri, 2010). In response to microbial products (e.g. viral or bacterial components) or endogenous molecules (e.g. cytosolic and extracellular DNA and RNA), PRRs transmit distinct downstream signaling pathways to trigger type-I IFN production. Upon recognized by IFN receptors, type-I IFN signal activates the Janus kinase (JAK) signal transducer and activator of transcription (STAT) pathway, leading to the expression of IFN-stimulated genes (ISGs), which control innate and adaptive immunity and intracellular antimicrobial programs (MacMicking, 2012; Schoggins et al., 2011; Stark and Darnell, 2012).
RLRs like RIG-I and MDA5, as members of PRRs, are main cytosolic RNA sensors to trigger innate immune response by sequentially activating downstream axis for type-I IFN production (Schlee and Hartmann, 2016; Seth et al., 2005). Upon sensing diverse cytosolic dsRNAs, RIG-I undergoes conformational changes, oligomerization and exposure of the N-terminal CARD domains to recruit a signaling adaptor called mitochondrial antiviral-signaling protein (MAVS). The transmembrane domain (TM domain) at C-terminus of MAVS is necessary for its mitochondrial outer membrane localization. Once activated, MAVS develops a functional prion-like structure at mitochondria, which serves as a platform to form a MAVS signalosome for activating TBK1 and IKKe. Phosphorylation of IRF3 and NF-kB by TBK1 and IKKe drives their nuclear translocation, leading to subsequent transcription of type-I IFN and inflammatory cytokine (Honda et al., 2006; Hou et al., 2011; Ivashkiv and Donlin, 2014; Tamura et al., 2008). Although intracellular RLR-MAVS activation and type-I IFN induction are orchestrated by numerous mechanisms (Chen et al., 2013a; Feng et al., 2017; Gack et al., 2007; Jin et al., 2017; You et al., 2013; Zhu et al., 2014), little is known about whether energy metabolism, likely through the production of a certain metabolite, is crucially involved in the regulation of this key signaling node allowing for the host defense against virus.
As a major source of cellular energy and cell mass, glucose is metabolized via glycolysis into pyruvate, which can either be imported to mitochondria for tricarboxylic acid (TCA) cycle entry to generate acetyl-CoA, through pyruvate dehydrogenase complex (PDHc) in the presence of oxygen, or be catalyzed by lactate dehydrogenase (LDH) to generate lactate when oxygen is not available. Interestingly, most cancer cells are addicted to aerobic glycolysis known as “Warburg effect” for their survival and proliferation even in the presence of oxygen, accompanied by high lactate generation (DeBerardinis and Thompson, 2012; Parks et al., 2013). Although lactate is previously considered as a waste-product of glucose metabolism, accumulating evidence has underscored its pivotal role in regulating diverse biological processes, such as macrophage polarization, T helper cell differentiation as well as tumor immune-surveillance (Brand et al., 2016; Colegio et al., 2014; Peng et al., 2016). Till now, no direct protein target of lactate is identified to our knowledge. Whether lactate participates in other intracellular signal transduction and biological outcome through direct protein targeting remains to be determined.
RESULTS
Downregulation of glucose metabolism promotes RLR induced type-I IFN production.
To investigate whether energy metabolism participates in regulating type-I IFN production, we employed an unbiased, systemic metabolomics approach to examine the global metabolic changes during RLR-induced type I IFN production. Intriguingly, most metabolic intermediates downstream of glucose, such as phosphoenolpyruvate (PEP), pyruvate and lactate, exhibited decreased levels at the initial stage of type I IFN production (Figure 1A and 1B). The levels of TCA intermediates, such as aconitate, succinate, fumaric acid and malate, were also significantly downregulated, likely resulting from the reduced level of pyruvate, which shunts into the TCA cycle for the production of these intermediates. However, oxaloacetate (OAA) level was not significantly affected (Figure 1A). As the direct pyruvate downstream metabolite, Acetyl-CoA production would be expected to decrease due to the reduction in pyruvate level, leading to the slower consumption of OAA for citrate production and thus maintaining the OAA level. These results suggest that glucose metabolism is impaired during the RLR signaling activation.
Figure 1. Downregulation of glucose metabolism promotes RLR induced type-I IFN production.

A, HEK293 cells were transfected with Poly(I:C) and harvested for metabolomics analysis. Shown is the heatmap for dynamic changes of metabolites in glycolysis or oxidative phosphorylation. B, Quantification analysis of some intermediates in glycolysis and TCA cycle. C, Q-PCR analysis of IFN-β mRNA expression in HEK293 cells cultured with high glucose (25 mM) or low glucose (5 mM) and transfected with Poly(I:C)(1μg/ml). D and E, Q-PCR analysis of IFN- β mRNA expression in THP1 cells transfected with HTDNA (1 μg/ml) or stimulated with LPS (50 ng/ml). F and G, Q-PCR analysis of IFN-β or VSV mRNA expression from spleen tissue of mice fasted overnight and then treated with high glucose (1.5 g/kg) or low glucose (0.2 g/kg). H–J, Q-PCR analysis of IFN- β and Sendai viral specific gene expression in HEK293 cells pretreated with or without 2DG (2mM) and then transfected with Poly(I:C) (1 μg/ml) or infected with Sendai virus for indicated hours. Data are means±SD. * p < 0.05, **p < 0.01. See also Figure S1.
Systemic glucose metabolism changes during RLR-induced type-I IFN production raised the possibility that glucose catabolism is involved in the regulation of type-I IFN production. To test this possibility, we incubated cells with high or low glucose to monitor RLR induced cytokine production and found low glucose-treated cells showed much stronger induction of IFN-β and IL-6 mRNA upon poly(I:C) transfection, but not HTDNA or LPS treatment, which activates stimulator of interferon genes (STING) and Toll-like receptor (TLR) signaling, respectively, compared with those under high glucose treatment (Figure 1C–1E and Figure S1A–S1D), indicative of the specificity of glucose change in regulating RLR signaling. Importantly, higher IFN-β induction and lower virus replication were observed in fasted mice treated with low glucose compared to those treated with high glucose (Figure 1F and 1G). Similarly, 2-deoxy-glucose (2-DG), which blocks glucose metabolism by inhibiting hexokinase (HK) activity, markedly enhanced IFN-β and IL-6 production driven by different RLR stimuli including poly(I:C) transfection, Sendai virus (SEV) and Vesicular stomatitis virus (VSV) infection, leading to the reduction in virus replication (Figure 1H–1J and Figure S1E–S1G). These data suggest that downregulation of glucose metabolism promotes RLR induced type-I IFN production.
Mitochondria hexokinase activity is maintained by MAVS and inactivated during RLR activation.
Since most glucose intermediates downstream of hexokinase (HK) were down-regulated during RLR-mediated type-I IFN induction (Figure 2A), it is conceivable that HK activity might be affected during RLR activation. Indeed, HK activity decreased at early phase of RLR activation, correlated well with dynamic changes of glucose metabolites (Figure 2B and Figure S2A), indicating that a transient suppression of glucose metabolism through HK inhibition may be required for RLR-mediated type-I IFN induction. To further validate our notion, we knocked down HK2, the major isoform of HK in most human cell types, to mimic the suppression of glucose metabolism. Consistently, HK2 knockdown not only decreased glycolysis determined by reduced pyruvate and lactate levels, but also markedly enhanced TBK1-IRF3 signaling, cytokine production and SEV replication suppression upon RLR activation (Figure 2C and Figures S2B–S2E). Moreover, restoration of HK2 expression compromised the heightened effect of HK2 knockdown on IFN-β induction (Figures S2F).
Figure 2. Mitochondria hexokinase activity is maintained by MAVS and inactivated during RLR activation.

A, Scheme of selected reactions in glucose metabolic pathway. B, Analysis of hexokinase (HK) activity in purified mitochondria isolated from HEK293 cells transfected with poly(I:C). C, Q-PCR analysis of IFN-β mRNA expression in control or HK2 knockdown Hep3B cells transfected with or without Poly(I:C) (left panel). Immunoblot analysis of Hep3B cells with control or HK2 knockdown (right panel). D, Immunoblot analysis of HK2 level in the mitochondria fraction from THP1 cells transfected with Poly(I:C), HTDNA or stimulated with LPS. Tom20 was used as a mitochondria marker. E-F, Whole cell lysis of HEK293 cells infected with or without Sev were collected for immunoprecipitation (IP) with IgG or MAVS antibody, followed by Immunoblot (IB) analysis. G, HEK293 cells transfected with Flag-V or Flag-RIG-I(N) were immunoprecipitated with indicated antibodies and IP complexes were analyzed by IB analysis. H, HEK293 cells with control or RIG-I knockdown were infected with Sev for 4 hours and whole cell lysis were collected for IP with MAVS antibody, followed by IB analysis for indicated proteins. I and J, Measurement of mitochondria hexokinase activity, total pyruvate level, lactate level and ECAR in Hep3B cells with control or MAVS knockdown. Data are means±SD. * p < 0.05, **p < 0.01. See also Figure S2.
The mitochondria localization of HK2 is required for its activation and full glycolytic function in cell (DeWaal et al., 2018; Roberts and Miyamoto, 2015; Wolf et al., 2016). To gain insight into how HK activity was downregulated at early stage of RLR activation, we examined whether mitochondria HK2 level changes upon RLR activation. Of note, mitochondria HK2 decreased upon the Poly(I:C) transfection, but not HTDNA or LPS treatment (Figure 2D), which might account for the downregulation of HK activity during RLR activation. We next explored the mechanism underlying HK2 dissociation from mitochondria. Interestingly, MAVS, as a mitochondria localized protein, physiologically interacted with HK2, but not with other mitochondria proteins, like Tom20 (Figure 2E). Remarkably, the interaction between MAVS and HK2 declined during initial activation of RLR signaling triggered by Sev infection, but not STING signaling activation upon HTDNA transfection, correlated with the dissociation of HK2 from mitochondria (Figure 2F and Figure S2G). Moreover, a direct switch of MAVS binding from HK2 to RIG-I was observed in cells with ectopic expression of activated form of RIG-I (Figure 2G and Figure S2H). Furthermore, the dissociation of HK2 from MAVS upon RLR activation was not observed in RIG-I knockdown cells (Figure 2H and Figure S2I). These results suggested that RLR-triggered MAVS-RIG-I recognition hijacks HK2 binding to MAVS, leading to the impairment of HK mitochondria localization and activation.
As the downregulation of HK activity during RLR activation coincidences with RIG-I-MAVS recognition and subsequent MAVS activation upon RLR agonist stimulation (Schlee and Hartmann, 2016; Seth et al., 2005), we asked whether MAVS can regulate mitochondria HK activity. Interestingly, we found significant reduction of HK activity was observed in MAVS knockdown and knockout cells compared to control cells, accompanied by decreased pyruvate and lactate levels as well as extracellular acidification rate (ECAR) (Figure 2I-2J and Figure S2J–2K). These result suggest that MAVS maintains HK activity.
HK associates with the mitochondrial outer membrane through its interaction with the voltage-dependent anion channel (VDAC), which is involved in glycolysis regulation (Roberts and Miyamoto, 2015; Wolf et al., 2016). To examine whether VDAC binding to HK2 is necessary for the interaction between HK2 and MAVS, we applied a peptide that is known to disrupt HKs from VDAC (Prezma et al., 2013). Notably, disrupting VDAC1 and HK2 interaction by this peptide impaired the interaction of MAVS with HK2, indicating that binding to VDAC is required for HK2 interaction with MAVS (Figure S2L and S2M). Collectively, we conclude that MAVS is a previously unrecognized key regulator for HK, and switching MAVS binding from HK2 to RIG-I during RLR activation triggers HK2 release from mitochondria leading to impairing HK activity and subsequent glucose metabolism.
Anaerobic glycolysis inhibits RLR triggered MAVS-TBK1-IRF3 activation and type-I IFN production.
As the two major glucose catabolism pathways, oxidative phosphorylation (OXPHOS) and anaerobic glycolysis are tightly controlled in cells by a multistep mechanism to execute diverse biological processes (Lunt and Vander Heiden, 2011). We showed that downregulation of glucose metabolism is required for full activation of RLR signaling. To dissect the key step of glucose metabolism involved in regulating type-I IFN production, we adapted various strategies to challenge glucose metabolism either by triggering the switch from OXPHOS to anaerobic glycolysis or vice versa. Metabolic fate of pyruvate is mainly controlled by LDHA and PDHc, which convert pyruvate to lactate or acetyl-CoA for TCA cycle, respectively (Ganeshan and Chawla, 2014). As the crucial PDHc subunit, PDHA knockdown impaired mitochondria OXPHOS, thus promoting anaerobic glycolysis for lactate production. Interestingly, we found robust decrease of IFN-β production in PDHA knockdown cells compared with control cells upon RLR activation (Figure 3A and Figure S3A–S3D). Similarly, the treatment of cells with UK5099, a potent inhibitor of the mitochondria pyruvate transporter blocking pyruvate mitochondria entry for OXPHOS (Davies et al., 2017; Liu et al., 2018), elevated lactate level and led to decreased MAVS signaling and IFN-β induction (Figure S3E–S3H). By contrast, dichloroacetate (DCA), which shifts glucose metabolism from anaerobic glycolysis to OXPHOS by inhibiting pyruvate dehydrogenase kinase and thus activating PDHc (Baker et al., 2000; Michelakis et al., 2008), promoted IFN-β induction upon RLR activation, accompanied by the reduction of lactate level (Figure 3B and Figure S3I). Cells grown in galactose conditions are forced to respire by using OXPHOS instead of anaerobic glycolysis (Chang et al., 2013; Rossignol et al., 2004; Weinberg et al., 2010). While galactose expectedly impaired anaerobic glycolysis-mediated lactate production, it markedly enhanced IFN-β and IL-6 production upon poly(I:C) or Sev challenge (Figure 3C and Figure S3J–S3L). As a result, cells infected with GFP-labeled VSV showed a marked reduction in viral replication, as determined by GFP fluorescence, under galactose treatment (Figure 3D). These data suggest that anaerobic glycolysis serves as a negative signal to repress RLR-mediated MAVS signaling and cytokine production.
Figure 3. Anaerobic glycolysis inhibits RLR triggered MAVS-TBK1-IRF3 activation and type-I IFN production.

A–B, Q-PCR analysis of IFN-β mRNA expression in HEK293 cells with or without PDHA knockdown (A), or pretreated with or without DCA (10 mM) (B) and then transfected with Poly(I:C) (left panel) or infected with Sendai virus (right panel) for indicated times. C, Q-PCR analysis of IFN- β mRNA expression in immortalized bone marrow macrophage (iBMM) cells cultured in mediums containing glucose (25 mM) or galactose (25 mM) and then transfected with Poly(I:C) (left panel) or infected with Sendai virus (right panel) for indicated times. D, Microscopic images of VSV-GFP-infected iBMM cells pre-cultured in the same conditions as in C and then infected with VSV-GFP (MOI=0.1) as indicated times. Scale bar, 100 μm. E, Q-PCR analysis of IFN-β mRNA expression (left panel) and measurement of lactate secretion (right panel) in HEK293 cells exposed to normoxia (20% O2) or hypoxia (1% O2) and transfected with Poly(I:C). F, Immunoblot analysis of HEK293 cells with control or PDHA knockdown and transfected with Poly(I:C) for indicated times. G, Immunoblot analysis of HEK293 cells with control or PDHA knockdown. Cell mitochondria were isolated for SDD-AGE (upper panel) and whole cell lysates were used for SDS-PAGE (lower panel). Data are means±SD. **p < 0.01. See also Figure S3.
To further investigate the biological role of anaerobic glycolysis in RLR signaling activation, we treated cells with hypoxia, which physiologically promotes anaerobic glycolysis. While hypoxia challenge expectedly induced the expression of HIF1a targeted genes, such as VEGF and GLUT4, and promoted lactate generation, it markedly reduced IFN-β production upon RLR activation (Figure 3E and Figure S3M–S3P). Upon recognition of cytosolic RNA, RLR undergoes an ATP-dependent conformational change that facilitates MAVS oligomerization, which triggers TBK1 activation and subsequent IRF3 phosphorylation for IFN-β production. To understand how anaerobic glycolysis regulates type-I IFN production during RLR activation, we examined the phosphorylation of TBK1 and IRF3. Consistent with decreased IFN-β expression, the levels of TBK1 and IRF3 phosphorylation robustly declined in PDHA knockdown cells in response to poly(I:C) transfection (Figure 3F). Moreover, we demonstrated that PDHA knockdown attenuated MAVS aggregation upon Sev infection (Figure 3G). Taken together, these data suggest that anaerobic glycolysis suppresses RLR induced downstream activation by affecting MAVS or upstream function.
LDHA associated lactate negatively regulates RLR signaling.
Since LDHA catalyzing lactate generation from pyruvate is a key event for anaerobic glycolysis, we asked whether LDHA-associated lactate plays a role in negatively regulating type-I IFN by applying genetic and pharmacologic approaches. Indeed, knockdown of LDHA enhanced phosphorylation of TBK1 and IRF3 as well as IFN-β production upon RLR activation (Figure 4A–4C, Figure S4A and S4B). Importantly, LDHA restoration in these knockdown cells reversed this effect (Figure 4D). Similar to LDHA knockdown, treatment of sodium oxamate, a specific LDHA inhibitor (Crane et al., 2014; Zhao et al., 2014; Zhao et al., 2016), reduced lactate level and enhanced IFN-β production resulting in suppressing virus replication (Figure 4E–4H, Figure S4C and S4D). Sodium oxamate also promoted IRF3 and TBK1 phosphorylation and MAVS aggregation upon RLR activation (Figure S4E and S4F). Of note, the effect of LDHA inhibition on RLR signaling was not observed in cells treated by STING agonist (Figure S4G and S4H). Collectively, these results indicate that LDHA plays a crucial role in specifically suppressing RLR-MAVS signaling and subsequent type-I IFN production.
Figure 4. LDHA-associated lactate negatively regulates RLR signaling.

A–C, Measurement of lactate secretion (A), IFN-β mRNA expression (B) or protein levels (C) in Hep3B cells with control or LDHA knockdown and transfected with Poly (I:C) for 4 hours. D, Q-PCR analysis of IFN-β mRNA expression in Hep3B cells infected with control or LDHA shRNA along with or without Flag-LDHA expression and then transfected with Poly (I:C). E, Measurement of lactate secretion in HEK293 cells pretreated with or without Sodium Oxamate (20 mM) overnight. F-H, Q-PCR analysis of IFN- μ or Sev mRNA expression in HEK293 cells treated with or without sodium oxamate (20 mM) overnight and then transfected with Poly(I:C) for 2 hours or infected with Sev as indicated. I and J, Q-PCR analysis of IFN-β mRNA expression in Hep3B cells pretreated with or without sodium oxamate (20 mM) or 2-DG (2mM) and then added with or without Lactate (10 mM) before transfecting with Poly(I:C). K, Q-PCR analysis of IFN-β mRNA expression in Hep3B cells infected with control or HK2 shRNA and then added with or without Lactate (10 mM) before transfecting with Poly(I:C). L–M, Q-PCR analysis of IFN-β mRNA expression in iBMM cells cultured in mediums containing glucose (25 mM) or galactose (25 mM) and then added with or without Lactate before transfecting with Poly(I:C). N, Q-PCR analysis of IFN-β mRNA expression in Hep3B cells with control or MCT1 knockdown and then treated with or without sodium oxamate (20mM) overnight before lactate addition (10 mM) and poly(I:C) transfection. Data are means±SD. **p < 0.01. See also Figure S4 and S5.
We next explored whether LDHA-associated lactate is a direct metabolite meditating the inhibition of RLR signaling caused by anaerobic glycolysis. Of note, addition of lactate into cells pretreated with sodium oxamate reversed the effect of LDHA inhibitor on IFN-β promotion (Figure 4I and Figure S5A). Lactate add-back also compromised the effects of 2-DG on IFN-β induction and its upstream signaling (Figure 4J and Figure S5B). Similar results were also observed in HK2 knockdown cells (Figure 4K). Moreover, we showed that add-back of lactate rescued the effects of galactose on IFN-β induction and virus replication (Figure 4L and Figure S5C). To determine how much lactate is needed to inhibit IFN production, we added back various concentrations of exogenous lactate (0–10 mM) into cells treated with galactose, and found 5 mM lactate was required for inhibiting IFN-β production (Figure 4M and Figure S5D–S5E). To rule out the possibility that lactate affects IFN-β production by changing the acidity of the medium, acidic pH of medium was adjusted to equal level as cells without addition of lactate by using NaOH before Poly(I:C) transfection. Lactate could still inhibit IFN-β production to the similar extent as the condition where PH was not adjusted (Figure S5F and S5G). As cells utilize monocarboxylate transporter 1 (MCT1) for lactate uptake, we knocked down MCT1 in cells to determine whether lactate uptake is crucial for its effect. LDHA inhibitor-Oxamate or 2-DG treatment promoted IFN-β expression, but such effect was markedly impaired upon MCT1 knockdown (Figure 4N and Figure S5H–S5I), indicating that lactate transporting into cells is essential for inhibiting MAVS signaling. Collectively, we conclude that lactate serves as a key metabolite to mediate the inhibitory effect of anaerobic glycolysis on RLR signaling.
LDHA-associated lactate inhibits RLR signaling in vivo.
To further confirm glycolysis-derived lactate serves as a natural suppressor of RLR induced type-I IFN production in vivo, mice were fasted overnight to reduce the basal glucose and lactate level and then treated with high glucose or low glucose in the presence or absence of lactate addition. While the reduction of glucose level in mice enhanced type-I IFN and IL-6 induction triggered by VSV infection, lactate add-back reversed this effect (Figure 5A and Figure S6A), indicating that lactate plays a critical role in suppressing RLR signaling in vivo. Moreover, we used tamoxifen inducible LDHA knockout mice and primary macrophage cells, in which LDHA exon2 is flanked by loxP sites (Xie et al., 2014) and lactate generation is genetically inhibited, to monitor RLR-mediated cytokine production (Figure S6B). LDHA protein expression in diverse organs of Ldha−/− mice was abrogated upon tamoxifen treatment (Figure S6C). When challenged with VSV, Ldha−/− mice showed much higher cytokine induction in serum or tissue compared to that in Ldha+/+ mice (Figure 5B–5C and Figure S6D). Consistent with elevated IFN-β level, virus replication as determined by VSV-specific mRNA in different tissues was much lower in Ldha−/− mice than Ldha+/+ mice (Figure 5D). Pathological staining showed reduced infiltration of inflammatory cells in the lung of Ldha−/− mice compared with Ldha+/+ mice upon VSV infection (Figure 5E). In addition, peritoneal macrophages isolated from Ldha−/− mice exhibited reduced lactate secretion, but elevated IFN-β induction compared with those from Ldha+/+ mice upon poly (I:C) transfection, Sev or VSV infection, whereas no difference was observed between the groups when treated by HTDNA or HSV(Figure 5F–5G and Figure S6E–S6F). Moreover, lactate addition in Ldha−/− macrophage consistently reduced IFN-β production (Figure 5H). These data define the critical role of LDHA-associated lactate in suppressing RLR-mediated type-I IFN production for the host defense against viral infection in vivo.
Figure 5. LDHA-associated lactate inhibits RLR signaling in vivo.

A, Q-PCR analysis of IFN-β and IFN-α expression in lung from mice fasted overnight and then treated with high glucose (1.5 g/kg) or low glucose (0.2 g/kg) with or without following injection of sodium lactate (1 g/kg) and infected with VSV (2×107 pfu/g). B, ELISA analysis of IFN- β in sera of Ldha+/+ and Ldha−/− mice intraperitoneal injected with VSV (2×107 pfu/g). C and D, Q-PCR analysis of IFN- β expression in spleen (C) and VSV replication in different organs (D) from Ldha+/+ and Ldha−/− mice infected with VSV. E, Hematoxylin and eosin (HE) staining of lung sections in Ldha+/+ and Ldha−/− mice described in D. Scale bar, 100 mm. F and G, Analysis of lactate secretion (F) and IFN-β production (G) in supernatants of peritoneal macrophages generated from Ldha+/+ and Ldha−/− mice and treated with Poly(I:C) transfection, Sev or VSV infection. H, ELISA analysis of IFN-β production in supernatants of peritoneal macrophages generated from Ldha−/− mice and then added with or without lactate (10 mM) before VSV infection. I, Q-PCR analysis of type-I IFN expression in lung from mice injected with or without sodium oxamate (750 mg/kg) and then challenged by VSV (2×107 pfu/g). J, Q-PCR analysis of IFN-β mRNA expression in lung tissue from mice injected with or without sodium oxamate and infected with HSV. Data are means±SD. **p < 0.01. See also Figure S6.
Pharmacologically targeting LDHA-associated lactate enhances type-I IFN production and viral clearance in vivo.
Having shown that LDHA-mediated lactate serves as a nature inhibitor of RLR-mediated type-I IFN production and viral clearance in vivo, we then determined whether pharmacologically targeting LDHA by the LDHA inhibitor, sodium oxamate, in mice is an effective strategy to bolster IFN production for the host to defend against viral infection (Figure S6G). Remarkably, administration of sodium oxamate to mice reduced lactate production and promoted the induction of type-I IFN, IL-6 and virus clearance in various tissue samples after VSV infection (Figure 5I and Figure S6H–S6L). The effect of lactate on IFN production in vivo is specific for RLR, because no difference in IFN production was observed in mice injected with HSV that activates STING signaling (Figure 5J and Figure S6M). Thus, our results provide the proof of principle evidence that pharmacologically targeting LDHA by sodium oxamate is an effective strategy to enhance RLR triggered type-I IFN production for viral clearance.
LDHA-associated lactate negatively regulates RLR activation by targeting MAVS.
We next sought to identify the direct targets of lactate responsible for its action on RLR-signaling. To this end, we synthesized biotin-labeled lactate and performed biotin pulldown assay by mixing biotin-labeled lactate with the whole cell extracts. We focused on the key proteins that are involved in cytosolic RLR signaling activation including RIG-I, MDA5, TBK1, MAVS and IRF3 and found lactate specifically interacted with MAVS, but not with other components of cytosolic RLR signaling or other mitochondria proteins such as Tom20 (Figure 6A), indicative of the specificity of this interaction. Mass spectrometry analysis also identified a cluster of MAVS peptides in the lactate pulldown complex (Figure S7A). Of note, MAVS interacted with lactate, but not pyruvate, in cells under physiological conditions (Figure 6B and 6C). By using in vitro binding assay, we found MAVS binds to lactate in a dose-dependent manner. Pre-incubation with excessive lactate significantly attenuated the binding of MAVS to Biotin-lactate (Figure S7B and S7C). Collectively, these data suggest that lactate directly binds to MAVS.
Figure 6. LDHA-associated lactate negatively regulates RLR activation by targeting MAVS.

A, Immunoblot analysis of binding complexes isolated from HEK293 cell extracts incubated with biotin-labeled lactate or biotin control. B and C, Fluorescence analysis of lactate or pyruvate binding in complexes immunoprecipitated (IP) by IgG or anti-MAVS antibody from HEK293 cells. D and E, Immunoblot analysis of in vitro mapping assays among biotin-labeled lactate and various MAVS truncated protein translated in vitro by the TNT system. F, Schematic representation of the sequence for control or TM peptide of MAVS with TAT tag in N-terminal region. G, Immunoblot analysis of in vitro pulldown assays by incubating biotin-labeled lactate or biotin control with MAVS protein translated in vitro by the TNT system along with control or TM peptide. H, TM peptides identified by Mass Spectrometry through in vitro pulldown assay. I. Analysis of lactate binding with different doses of Tat-control or Tat-TM peptide. J, Immunofluorescence analysis of cellular localization of control or TM peptide in HeLa cells pretreated with each peptides, stained with MAVS or TAT antibody and imaged by confocal microscopy. Scale bar, 10 mm. K, Q-PCR analysis of IFN-β expression in Hep3B cells treated by control or TM peptide of MAVS and transfected with Poly(I:C). L, Q-PCR analysis of IFN-β expression in Hep3B cells pretreated with or without sodium oxamate overnight, then incubated with control or TM peptide of MAVS for 2 hours and addition of lactate, followed by transfection of Poly(I:C). Data are means±SD. *p < 0.05, **p < 0.01. See also Figure S7.
To explore the detailed interaction between lactate and MAVS, we carried out the domain mapping assay and found the transmembrane (TM) domain of MAVS that targets MAVS to the mitochondrial membrane is required for lactate-MAVS binding (Figure 6D and 6E). We next synthesized a peptide that constitutes MAVS TM domain (514–535aa) or control sequence with a trans-activator of transcription (TAT) tag placed in its N terminal region (Figure 6F and Figure S7D–S7E). In an in vitro binding assay, we found that TM peptide markedly interrupted the binding of lactate to MAVS in dose-dependent manner (Figure 6G and Figure S7F), indicating this peptide could compete with MAVS from binding to lactate. We next examined whether lactate could directly bind to TM domain of MAVS. In support of this notion, mass spectrometry analysis of in vitro biotin-lactate pulldown products revealed that the TM peptide of MAVS, but not control peptide, could bind to biotin-lactate (Figure 6H). Furthermore, we also used Tat antibody to perform immunoprecipitation assay and found that lactate could be specifically pulled down by Tat-TM peptide in a dose-dependent manner (Figure 6I). Thus, our data demonstrate that lactate binds to TM domain of MAVS.
We took the advantage of the cell penetration property (CPP) of the TAT peptide derived from the trans-activator of transcription of human immunodeficiency virus (Milletti, 2012). This CPP has been used to deliver large molecules or even small particles into the cells for testing their biological functions by overcoming the lipophilic barrier of the cell membranes. Using the immunofluorescence assay, we found both control and TM peptide could get into cells (Figure 6J). We then determined the effect of TM peptide on RLR induced type-I IFN production. Of note, adding TM peptide to the cells promoted RLR activation in a dose dependent fashion (Figure 6K and Figures S7G–S7H). Moreover, TM peptide also compromised the inhibitory effect of lactate on IFN-β and IL-6 production upon Poly (I:C) treatment (Figure 6L and Figures S7I–S7J). Collectively, these results suggest that lactate inhibits RLR signaling through its binding to the TM domain of MAVS.
Lactate inhibits MAVS mitochondria localization, RIG-I/MAVS association and MAVS aggregation.
In order to unravel the underlying mechanism by which lactate inhibits RIG-I-MAVS induced RLR activation, we explored whether lactate impacts the mitochondria localization of MAVS by binding to its TM domain, (Seth et al., 2005). Inhibition of lactate generation by oxamate potentiated MAVS mitochondria localization, but lactate add-back compromised this effect (Figure 7A and 7B). Mitochondria localization of MAVS provides the structural basis for RIG-I-MAVS recognition and subsequent MAVS activation. We sought to determine whether activated RIG-I could still bind to MAVS devoid of TM domain of MAVS. As expected, MAVS mutant defective of TM domain which could not localize in mitochondria failed to interact with activated RIG-I (Figures 7C). Notably, LDHA inhibitor treatment enhanced the interaction between activated RIG-I and MAVS in both basal and Sendai virus-infected conditions, whereas lactate treatment markedly reduced their binding (Figures 7D and 7E). Thus, lactate disrupts MAVS mitochondria localization and the interaction between RIG-I-MAVS interaction.
Figure 7. Lactate inhibits RIG-I/MAVS association and MAVS aggregation.

A, Immunofluorescence analysis of cellular localization of MAVS in Hep3B cells pretreated with or without Oxamate and added with lactate. Scale bar, 10mm. B, Immunoblot analysis of mitochondria fraction isolated from HEK293 cells treated as indicated. C-E, Cell lysates from HEK293 cells transfected or treated as indicated were immunoprecipitated with antibodies indicated, and IP complexes were analyzed by immunoblot analysis. F, Immunoblot analysis of in vitro MAVS aggregation. GST-RIG-I(N) was incubated with K63-Ub4 and then with mitochondria isolated from HEK293 cells preincubated with or without lactate (5 mM,10 mM) or pyruvate, followed by analysis of mitochondria extracts using SDD-AGE (left panel) and SDS-PAGE (right panel). GST-RIG-(N) was shown by Coomassie blue staining (CBB). G, Illustration of how glycolysis-derived lactate inhibits RLR signaling by targeting MAVS.
Recognition of MAVS by activated RIG-I is essential for MAVS aggregation and its downstream activation, we tested whether lactate could directly affect MAVS aggregation induced by activated RIG-I through performing in vitro MAVS aggregation assay. To this end, we incubated lactate with activated RIG-I protein (RIG-I(N)) along with mitochondria and ubiquitin chains as previously described (Hou et al., 2011). In the presence of mitochondria and unanchored K63-linked ubiquitin chains (K63-Ub4), we found that purified GST-RIG-I(N) triggered robust MAVS aggregation in vitro. Remarkably, lactate, but not pyruvate, impaired MAVS aggregation in a dose dependent manner (Figure 7F), indicative of direct effect of lactate on RIG-I-mediated MAVS aggregation. Collectively, these data suggest that lactate targets the TM domain of MAVS to obstruct its mitochondria localization and RIG-I-MAVS complex formation, thereby impairing MAVS aggregation and its downstream signaling activation.
DISCUSSION
Substantial evidence accumulated in recent years has highlighted the role of lactate not only in regulating tumor microenvironment and immune cell functions (Brand et al., 2016; Colegio et al., 2014), but also serving as a fuel for TCA cycle in cancer cells (Faubert et al., 2017). However, no direct targeting protein of lactate has been identified so far to help explain its mode of action. Our study identifies MAVS as a lactate sensor whose inactivation by direct lactate binding serves as a natural barrier to limit RLR signaling activation for type-I IFN production. We demonstrate that the binding of MAVS to lactate interrupts MAVS mitochondria localization, RIG-I and MAVS interaction and subsequent MAVS aggregation, thereby attenuating RLR signaling and downstream type-I IFN production (Fig. 7G).
Although metabolism has been linked to diverse biological processes, the connection between glucose metabolism and RLR-mediated innate immune activation remains puzzling. Through systematic approaches in conjunction with various biochemical assays, we unraveled that anaerobic glycolysis acts as a natural barrier to impede RLR signaling activation. Importantly, we demonstrated that glycolysis-derived lactate represents the first metabolite that directly binds to MAVS and suppresses its functions to orchestrate type-I IFN production. We rationalize that lactate accumulation by elevated LDH and/or glycolysis may be a potential mechanism for virus to evade host defense by inhibiting RLR-induced type-I IFN production. Notably, clinical studies reveal that elevated LDH or lactate levels are detected in patients under certain viral infection, especially in those with poor prognosis (Chen et al., 2013b; Hunt et al., 2015). Hence, pharmacologically targeting LDHA-dependent lactate production is a promising strategy to strengthen host innate immune response through heightening type-I IFN production for viral clearance and cancer immune-surveillance.
TM domain of MAVS is not only crucial for the mitochondria localization of MAVS, but also required for RIG-I recognition. Interestingly, our findings that TM peptide competes with MAVS from binding to lactate and lactate directly binds to TM domain of MAVS may provide the molecular basis of how lactate disrupts MAVS mitochondria localization and MAVS-RIG-I interaction, thereby impairing MAVS aggregation and RLR-mediated IFN production for viral clearance. Of note, applying TM peptide from MAVS to the cells not only enhances IFN production under normal conditions, but also relieves the inhibitory effect of lactate on RLR-mediated IFN production. Thus, our findings suggest that targeting lactate-MAVS interaction may represent another promising strategy for clinical drug development based on the knowledge learned from TM peptide to boost our body immunity for viral and cancer clearance. This strategy may offer a superior way than directly targeting LDHA, as it maintains the level of lactate for normal cellular functions.
Type-I IFNs are critical cell-intrinsic antimicrobial factors that limit the spread of infectious agents, such as viral pathogens. (MacMicking, 2012; Schoggins et al., 2011; Stark and Darnell, 2012). Beyond their important role in host defense against viruses, type-I IFNs also play a critical role in regulating the functions of diverse innate and adaptive immune cells including dendritic cell, regulatory T (TReg) cells and cytotoxic T lymphocytes (CTLs), thereby contributing to immune-surveillance of cancer (Zitvogel et al., 2015). While energy metabolism has emerged to regulate diverse biological processes, its role in RLR signaling activation and type-I IFN production has not been defined. Our study reveals that glycolysis serves as physiological barriers to limit type-I IFN production by promoting lactate generation, both in vitro and in vivo. Hence, in order to achieve optimal type-I IFN production, it is imperative for the host to develop a strategy to inactivate glycolysis and thereby reducing lactate production during RLR challenge. In this respect, we demonstrate the RLR challenge switches the recognition of MAVS from HK2 to RIG-I, resulting in HK2 mitochondria dissociation and HK inactivation. Our study therefore uncovers that MAVS is a previously unrecognized player that maintains HK2 activity and glycolysis. Thus, MAVS possess dual roles in regulating glycolysis and innate immune response, and viral infection hijacks MAVS’s function from glycolysis to innate immune regulation.
Since cancer cells often overexpressed HK2 (Patra et al., 2013), exposed to hypoxia microenvironment and favored glycolysis for energy metabolism known as Warburg effect (Nakazawa et al., 2016; Pavlova and Thompson, 2016), it is likely that limiting type-I IFN production by these conditions through generating more lactate may serve as another important mechanism for cancer cells to evade immune surveillance. In summary, our study provides not only the crucial molecular insight into how energy metabolism and type-I IFN cross-talk to regulate diverse biological processes, but also offers an important paradigm and strategy for the management of various human diseases, such as viral infection and cancer.
STAR Methods
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for reagents may be directed to, and will be fulfilled by the Lead Contact, Hui-Kuan Lin (hulin@wakehealth.edu).
EXPERIMENTAL MODELS AND SUBJECT DETAILS
Mice.
Ldha+/+ and Ldha−/− mice on a C57BL/6J background were previously described (Xie et al., 2014). All animal procedures were approved by Institutional Animal Care and Use Committee in Wake Forest University School of Medicine. Littermates of the same sex were randomly assigned to experimental groups. 6–8 weeks old mice were treated with tamoxifen (20 mg/ml in corn oil) or coin oil by intraperitoneal injection. According to protocols provided by Jackson lab for inducible Cre driver lines, LDHA exon2 deletion was confirmed by genotyping 10 days after final injection. Mice were then used for peritonea macrophage isolation or virus infection by intraperitoneal injection. IFN-β concentration in supernatants of peritoneal macrophage or sera from Ldha+/+ and Ldha−/− mice were determined by using the mouse IFN-β ELISA kit (Thermo Fisher 424001) according to the manufacturer’s protocol. All the analyses were performed blindly. Ldha−/− mice were randomly allocated into experimental groups for further treatment and cell samples were allocated based on the genotype of interest.
Cells.
HEK293, Hep3B, Raw264.7 cells and immortalized bone marrow macrophage (iBMM) cells were cultured in DMEM medium supplied with 10% FBS, 2 mM glutamine, penicillin (100 U/mL) and streptomycin (100 mg/mL). THP-1 cells were cultured in RPMI-1640 medium supplied with 10% FBS, 2 mM glutamine, penicillin (100 U/mL) and streptomycin (100 mg/mL). Primary peritoneal macrophages were isolated from mice 14 days after tamoxifen intraperitoneal injection as previously described (Chen et al., 2013a). Cells were plated 24 hours before transfection with Poly(I:C) (1 μg/ml), HTDNA (1 μg/ml) or siRNA (10 nM) by Lipofectamine 2000 (Thermo Fisher Scientific). THP-1 cells were pretreated by PMA (100 ng/mL) overnight for differentiation before the next manipulation. To generate shRNA knockdown cells, HEK293T cells were prepared and co-transfected with either luciferase (shLuc) or target gene shRNA with packaging plasmid (pHelper) and envelop plasmid (pEnv) by using the calcium phosphate transfection method. Medium was changed 6 hours later and virus particles were harvested after another 48 hours to infect parental cells, then selected by puromycin.
Viruses.
VSV and HSV-1 were propagated and tittered by plague assay on Vero cells. For in vivo cytokine production studies, age and sex-matched groups of littermate mice were intraperitoneally injected with VSV or HSV-1 (2×107 pfu/g). For cell based assays, cells were infected with VSV-GFP (0.1 MOI) or SeV (1 MOI) for the indicated hours and cytokine production was analyzed by Q-PCR or ELISA.
METHODS DETAILS.
Immunoblotting (IB), Immunoprecipitation (IP) and Pull down assays.
For Immunoblotting, whole cell lysates were prepared in E1A buffer (50 mM Hepes, pH 7.6, 250 mM NaCl, 0.1% Nonidet P-40, 5mM EDTA) supplemented with complete protease inhibitor cocktail (Roche, 04693132001). Cell lysates were separated by SDS-PAGE and proteins were visualized by enhanced chemiluminescence according to the manufacturer’s instructions (ThermoFisher Scientific). For protein-protein interactions, cells were lysed by RIPA lysis buffer (150 mM NaCl, 0.5% NP-40, 5 mM EDTA) containing a mixture of protease inhibitor cocktail (Roche). Primary antibodies were incubated with magic protein agarose A/G beads for 30 min at room temperature, followed by incubating with cell lysates for 3 hours with rotating at RT. The beads were washed four times with lysis buffer and analyzed by IB. For Biotin-lactate pull down assays, magic Dynabeads MyOne Streptavidin T1 was preincubated with free biotin or biotin-labeled lactate in PBS for 1 hour at RT, and then incubated with cell lysates overnight with rotating at 4°C. The beads were washed 3–4 times and analyzed by Immunoblotting.
Lactate treatment.
For lactate addiction assay, we used two forms of lactate in the paper, one is the acid form of lactate (L-lactic acid), which was used for in vitro cell based study and in vitro MAVS aggregation assay, while the other one is the basic form of lactate (sodium lactate), which was used for in vivo animal experiments.
Immunofluorescent Confocal Microscopy.
Cells were fixed with 3% paraformaldehyde for 20 min at 25°C and permeabilized for 20 min with 0.5% Tween-20. Samples were blocked with 2% goat serum in phosphate-buffered saline (PBS) for 30 min. Anti-MAVS (1:500, Santa Cruz) and anti-TAT (1:500, Abcam) antibodies were used to detect the MAVS protein and TM peptides, respectively. Staining was visualized with secondary antibodies and the images were captured with a digital camera under a confocal microscope.
RNA extraction and quantitative RT-PCR
Total RNA was extracted via TRIzol reagent (Ambion 15596–018). Total RNAs (0.5–1mg) were subjected to reverse transcription with PrimeScript RT Master Mix (Takara, DRR036A). To determine relative mRNA level, Q-PCR was performed using universal SYBR Green Supermix (Bio-Rad 172–5125) and gene expression was normalized to that of GAPDH or HPRT. Primers used for Q-PCR were listed in Table S1.
Measurement of hexokinase activity and Lactate level.
For hexokinase activity detection, mitochondria were isolated from cells and pellet was lysed and subjected to Hexokinase activity measurement by using Hexokinase Colorimetric assay kit (Biovision K789–100). Secreted lactate level was measured by lactate Plus Test Strips (Nova Biomedical/fisher), whereas intracellular lactate level was measured by using lactate Colorimetric/Fluorometric assay kit (Biovision K607–100) according to manufacturer’s protocol. For Seahorse analysis, cells with control or MAVS knockdown were prepared, the XF24 Extracellular Flux analyzer (Seahorse Biosciences, Billerica, MA) was used to measure extracellular acidification rate (ECAR).
In vivo and in vitro MAVS aggregation assays.
In vivo MAVS aggregation was performed according to published protocol (Hou et al., 2011). In brief, mitochondria were isolated by using the Mitochondria isolation kit (Thermo 89874), and mitochondria pellet was suspended in 1 × sample buffer (0.5 × TBE, 10% glycerol, 2% SDS, and 0.0025% bromophenol blue) and subjected to Semi-denaturing detergent agarose gel electrophoresis (SDD-AGE). Samples were loaded onto a vertical 1.5% agarose gel (Bio-Rad). After electrophoresis in the running buffer (1 × TBE and 0.1% SDS) for 40 min with a constant voltage of 80–100 V at 4°C, the proteins were transferred to Immobilon membrane (Millipore) for immunoblotting.
For in vitro MAVS aggregation, crude mitochondria were isolated and RIG-I activation was detected as previously described (Zeng et al., 2009). Briefly, each ml mixture contained 100 ng GST-RIG-I(N) and 50–100 ng ubiquitin chains (K63–Ub4 from Boston Biochem UC-310B) in buffer containing 20 mM HEPES-KOH (pH 7.0) and 10% (v/v) glycerol. After incubation at RT for 10 min in total 10 μl reaction system, 1 μl of reaction mixture was mixed with 10 μg of mitochondrial fraction in 10 μl Buffer B (20 mM HEPES-KOH [pH 7.0], 5 mM MgCl2, and 0.25 M D-mannitol) at 30°C for 30 min. Mitochondria fraction was then pelleted at 10,000 g for 10 min and washed twice with Buffer C (20 mM HEPES-KOH at pH 7.4, 0.5 mM EGTA, 0.25 MD-mannitol, and EDTA-free protease inhibitor cocktail) and then subjected SDD-AGE.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistics.
Statistical significance was identified by Student’s t test. p values of less than 0.05 were considered statistically significant; * p < 0.05, **p < 0.01.
Supplementary Material
Figure S1. Inhibition of glucose metabolism promotes RLR-induced type-I IFN production, Related to Figure 1. A, Q-PCR analysis of IFN-β mRNA expression in RAW264.7 macrophage cells cultured in mediums with high glucose (25 mM) or low glucose (5 mM) and transfected with HMW Poly(I:C) for indicated hours. B–D, Q-PCR analysis of IFN-β or IL-6 mRNA expression in THP1 cells transfected with Poly(I:C) (1μg/ml), HTDNA (1 μg/ml) or treated with LPS (50 ng/ml). E, Q-PCR analysis of IFN-β mRNA expression in Hep3B cells pretreated with or without 2-DG (2mM) overnight and then transfected with Poly(I:C) for indicated hours. F, Q-PCR analysis of IL-6 mRNA expression in HEK293 cells pretreated with or without 2-DG (2mM) and then transfected with Poly(I:C) for indicated hours. G, Q-PCR analysis of IFN-β mRNA expression in HEK293 cells pretreated with or without 2-DG (2mM) overnight and then infected with VSV for indicated hours. Data are means±SD. *p < 0.05, **p < 0.01.
Figure S2. Reciprocal regulation between hexokinase and MAVS, Related to Figure 2. A, Analysis of Hexokinase (HK) activity in purified mitochondria isolated from HEK293 cells infected with Sev for indicated hours. B and C, Analysis of pyruvate, lactate level (B) and HK2 expression (C) in Hep3B cells with control or HK2 knockdown by using Colorimetric assay kit or immunoblotting. D, Q-PCR analysis of IL-6 mRNA expression in Hep3B cells with control or HK2 knockdown and transfected with Poly (I: C). E, Q-PCR analysis of IFN-β or Sev specific mRNA expression in Hep3B cells with control or HK2 knockdown and infected with Sev. F, Q-PCR analysis of IFN-β mRNA expression in Hep3B cells infected with control or HK2 shRNA with or without Flag-HK2 expression and then transfected with Poly(I:C). G, Whole cell lysates of THP1 cells transfected with HTDNA for indicated hours were collected for IP with MAVS antibody, followed by IB analysis for indicated proteins. H, HEK293 cells transfected with Flag-Vector or Flag-RIG-I(N) together HA-MAVS were immunoprecipitated with IgG or anti-HA antibody, and IP complexes were analyzed by immunoblot analysis. I, Q-PCR analysis of IFN-β mRNA expression in HEK293 cells with control or RIG-I knockdown and infected with Sev as described in Figure. 2H. J, Immunoblot analysis of MAVS expression in Hep3B cells with control or MAVS knockdown. K, Analysis of hexokinase (HK) activity in purified mitochondria isolated from wild-type and MAVS knockout MEF cells (upper panel). Immunoblot analysis of MAVS protein level in wild type or MAVS knockout MEFs (lower panel). L, HEK293 cells were pre-incubated with control or VDAC competitive peptide (0.5μM) for 90 min and then immunoprecipitated with IgG or anti-MAVS antibody, and IP complexes were analyzed by immunoblot analysis. M, HEK293 cells transfected with Myc-VDAC were treated with the same conditions as in L and immunoprecipitated with IgG or anti-Myc antibody, IP complexes were analyzed by immunoblot analysis. Data are means±SD. **p < 0.01.
Figure S3. Anaerobic glycolysis affects RLR triggered type-I IFN production, Related to Figure 3. A and B, Q-PCR analysis of PDHA mRNA expression (A) and measurement of lactate secretion (B) for HEK293 cells with control and PDHA knockdown as described in Fig. 3A. C and D, Q-PCR analysis of IFN-β (C) and PDHA (D) mRNA expression HEK293 cells transfected with control or PDHA siRNA. E, Analysis of lactate secretion in supernatant of HEK293 cells treated with or without UK5099 (10 μM) overnight. F and G, Q-PCR analysis of IFN-β mRNA expression in HEK293 cells pretreated with UK5099 overnight and then transfected with Poly(I:C) (F) or infected with Sev (G). H. Immunoblot analysis of HEK293 cells treated with or without UK5099 (10 μM) overnight and then transfected with Poly(I:C) as indicated. I. Analysis of lactate secretion in HEK293 cells treated with DCA as described in Figure 3B. J, Detection of lactate secretion in immortalized bone marrow macrophage cells cultured in mediums containing glucose (25 mM) or galactose (25 mM) as described in Figure 3C. K and L, Q-PCR analysis of IFN-β (Raw264.7 cells) or IL-6 (iBMM cells) expression treated the same as in J and transfected with Poly(I:C) for indicated hours. M, Q-PCR analysis of VEGF mRNA expression in HEK293 cells exposed to normoxia (20% O2) or hypoxia (1% O2) and transfected with Poly(I:C). N, Q-PCR analysis of IFN-β expression in RAW264.7 macrophage cells treated with the same conditions as in M and transfected with Poly(I:C) for indicated hours. O and P, Q-PCR analysis of Glut4 mRNA expression (O) and measurement of lactate secretion (P) in RAW264.7 macrophage cells as indicated. Data are means±SD. **p < 0.01.
Figure S4. LDHA inhibits RLR induced type-I IFN production, Related to Figure 4. A, Immunoblot analysis of LDHA expression in Hep3B cells infected with control or two independent LDHA shRNAs. B, Q-PCR determination of IFN-β mRNA expression in Hep3B cells with control or two independent LDHA shRNAs infection and then transfected with Poly(I:C) for 2 hours. C, Q-PCR determination of IFN-β mRNA expression in Hep3B cells pretreated with or without sodium oxamate (20 mM) overnight and then transfected with Poly(I:C) for 4 hours. D, Q-PCR analysis of IFN-β expression in HEK293 cells pretreated with or without Sodium Oxamate (20 mM) overnight and then infected with VSV for indicated hours. E and F, Immunoblot analysis of Hep3B cells treated with or without sodium oxamate (20 mM) and then infected with Sendai virus for indicated times. Whole cell lysates were subjected to SDS-PAGE (E) and mitochondria were isolated for SDD-AGE (F). G and H, Q-PCR analysis of IFN-β expression (G) and immunoblot analysis (H) in THP1 cells pretreated with or without Sodium Oxamate (20 mM) overnight and then transfected with HTDNA as indicated. Data are means±SD. *p < 0.05, **p < 0.01.
Figure S5. LDHA-associated lactate inhibits RLR induced type-I IFN production, Related to Figure 4. A, Q-PCR analysis of IFN-β mRNA expression in HEK293 cells pretreated with or without sodium oxamate (20 mM) overnight and then cultured in medium in the presence or absence of lactate (10 mM) 2 hours before infection with Sev for 8 hours. B, Immunoblot analysis of Hep3B cells treated with or without 2-DG and then cultured in medium with or without lactate (10 mM) before transfecting with Poly(I:C). Cell mitochondria were isolated for SDD-AGE (upper panel) and whole cell lysates were used for SDS-PAGE (lower panel). C, Microscopic images of VSV-GFP-infected immortalized bone marrow macrophage cultured in medium containing glucose (25 mM) or galactose (25 mM) with or without addition of lactate (10 mM) 2 hours before infecting with VSV-GFP (MOI=0.1) for 16 hours. Scale bar, 100 mm. D, The total ion current (TIC) chromatogram and Mass spectrum of intracellular lactate for Figure 4M. E, List of intracellular lactate concentration for each group in Figure 4M. F and G, Q-PCR analysis of IFN-β in Hep3B cells treated with or without Oxamate (F) or 2-DG (G) overnight, then added with lactate (10 mM) along with or without NaOH to correct PH level before Poly(I:C) transfection. H, Q-PCR analysis of IFN-β mRNA expression in Hep3B cells with control or MCT1 knockdown and then treated with or without 2-DG overnight before lactate addition (10 mM) and Poly(I:C) transfection. I, Q-PCR analysis of MCT1 expression in Hep3B cells with control or MCT1 knockdown. Data are means±SD. *p < 0.05, **p < 0.01.
Figure S6. In vivo targeting LDHA enhances RLR signaling, Related to Figure 5. A, Q-PCR analysis of IL-6 expression in lung from mice fasted overnight and then treated with high glucose (1.5 g/kg) or low glucose (0.2 g/kg) with or without injection of sodium lactate (1 g/kg) and VSV infection. B, Schematic representation of the procedures for LDHA deletion in mice and peritoneal macrophage isolation. C, Immunoblot analysis of LDHA protein level in different organs from mice with intraperitoneal injection of tamoxifen (20 mg/ml) or vehicle. D, Q-PCR analysis of IFN- α or IL-6 expression in spleen from Ldha+/+ and Ldha−/− mice treated as in Figure 5. E, Immunoblot analysis of LDHA expression in peritoneal macrophages isolated from Ldha+/+ or Ldha−/− mice. F, ELISA analysis of IFN-β expression in supernatant of Ldha+/+ or Ldha−/− macrophages transfected with HTDNA or infected with HSV. G, Schematic representation of the procedures for mice intraperitoneal injection of sodium oxamate and VSV infection. H, Analysis of lactate secretion in peripheral blood of mice intraperitoneally injected with or without sodium oxamate (750 mg/kg). I-L, Q-PCR analysis of IL-6 (I) or VSV specific gene expression (J-L) in different organs from mice treated with or without sodium oxamate and then challenged with VSV. M, Q-PCR analysis of IFN-α mRNA expression in lung tissue from mice injected with or without sodium oxamate and infected with HSV. Data are means±SD. **p < 0.01.
Figure S7. Lactate negatively regulates RLR activation by targeting MAVS, Related to Figure 6. A, MAVS peptides identified by Mass Spectrometry through in vitro pulldown. B, Immunoblot analysis of in vitro pulldown assays by incubating biotin-labeled lactate with increased doses of MAVS protein translated by the TNT system. C, Immunoblot analysis of in vitro pulldown assays by incubating biotin-labeled lactate and MAVS protein together with or without lactate. D-E, Base peak ion Chromatogram (left panel) and mass spectrum (right panel) of the ion Chromatography peak for control peptide (D) or TM peptide (E). F, Schematic representation of the procedures for in vitro biotin pulldown assay indicated in Figure 6G. G, Q-PCR analysis of IFN-β expression in HEK293 cells treated by control or TM peptide and transfected with Poly(I:C). H, Q-PCR analysis of IL-6 expression in Hep3B cells treated by control or TM peptide and transfected with Poly(I:C). I and J, Q-PCR analysis of IFN-β or IL-6 expression in Hep3B cells pretreated with or without 2-DG overnight, then treated by control or TM peptide for 2 hours, addition of lactate (5 mM) and transfected with Poly(I:C). Data are means±SD. *p < 0.05, **p < 0.01.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse monoclonal anti-MAVS(E-3) | Santa Cruz Biotechnology | Cat# sc-166583; RRID:AB_2012300 |
| Mouse monoclonal anti-HK2(B-8) | Santa Cruz Biotechnology | Cat# sc-374091; RRID:AB_10917915 |
| Mouse monoclonal anti-β-Actin(C4) | Santa Cruz Biotechnology | Cat# sc-47778; RRID: AB_626632 |
| Mouse monoclonal anti-c-Myc(9E10) | Santa Cruz Biotechnology | Cat# sc-40; AB_627268 |
| Rabbit polyclonal anti-HA-probe | Santa Cruz Biotechnology | Cat# sc-805; RRID: AB_631618 |
| Rabbit monoclonal anti-Rig-l(D14G6) | Cell Signaling Technology | Cat# 3743; RRID:AB_2269233 |
| Rabbit monoclonal anti-TBK1(D1B4) | Cell Signaling Technology | Cat# 3504; RRID:AB_2255663 |
| Rabbit monoclonal anti-IRF3(D6l4C) | Cell Signaling Technology | Cat# 11904; RRID:AB_2722521 |
| Rabbit monoclonal anti-TBK1(phosS172) (D52C2) | Cell Signaling Technology | Cat# 5483; RRID:AB_10693472 |
| Rabbit monoclonal anti-IRF3(phosS396) (4D4G) | Cell Signaling Technology | Cat# 4947; RRID:AB_823547 |
| Rabbit monoclonal anti-PDHA(C54G1) | Cell Signaling Technology | Cat# 3205; RRID:AB_2162926 |
| Rabbit monoclonal anti-MDA5(D74E4) | Cell Signaling Technology | Cat# 5321; RRID:AB_10694490 |
| Rabbit polyclonal anti-MAVS | Cell Signaling Technology | Cat# 3993; RRID:AB_823565 |
| Rabbit polyclonal anti-LDHA | Cell Signaling Technology | Cat# 2012; RRID:AB_2137173 |
| Mouse monoclonal anti-HIV-TAT(N3) | Abcam | Cat# ab63957; RRID:AB_1139536 |
| Bacterial and Virus Strains | ||
| VSV-GFP | Prof. Zhijian J. Chen (UT Southwestern Medical Center, USA). | N/A |
| HSV-1 | Prof. Jiahuai Han (Xiamen U. China) | N/A |
| SEV | ATCC | Cat# VR907 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| D-(+)-Glucose | Sigma-Aldrich | Cat# G7021 |
| D-(+)-Galactose | Sigma-Aldrich | Cat# G0750 |
| L(+)-Lactic acid | Sigma-Aldrich | Cat# L1750 |
| Sodium L-lactate | Sigma-Aldrich | Cat# L7022 |
| Sodium Oxamate | Sigma-Aldrich | Cat# 02751 |
| 2-Deoxy-D-glucose(2-DG) | Sigma-Aldrich | Cat# D8375 |
| Sodium dichloroacetate(DCA) | Sigma-Aldrich | Cat# 347795 |
| UK5099 | Sigma-Aldrich | Cat# PZ0160 |
| Poly(l:C) | Sigma-Aldrich | Cat# P1530 |
| HT-DNA | Sigma-Aldrich | Cat# D6898 |
| Tamoxifen | Sigma-Aldrich | Cat# 10540-29-1 |
| TRIzol | Thermo Fisher Scientific | Cat# 15596018 |
| Dynabeads MyOne Streptavidin T1 | Thermo Fisher Scientific | Cat# 65601 |
| Human Tetra Ubiquitin/Ub4 WT Chains (K63linked) | R&D | Cat# UC310B |
| Con-peptide | This paper | N/A |
| TM-peptide | This paper | N/A |
| VDAC competitive peptide | (Prezma et al., 2013) | N/A |
| Critical Commercial Assays | ||
| Mouse IFN-β ELISA kit | Thermo Fisher | Cat# 424001 |
| Hexokinase Colorimetric assay kit | Biovision | Cat# K789–100 |
| Lactate Colorimetric/Fluorometric assay kit | Biovision | Cat# K607–100 |
| Mitochondria isolation kit | Thermo Fisher | Cat# 89874 |
| Experimental Models: Cell Lines | ||
| Human: HEK293 | ATCC | Cat# CRL-11268 |
| Human: THP-1 | ATCC | Cat# TIB-202 |
| Human: Hep3B | ATCC | Cat# HB-8064 |
| Mouse: Raw264.7 | ATCC | ATCC® TIB-71™ |
| Mouse: iBMM | Prof. Feng Shao (National Institute of Biological Sciences, China) | N/A |
| Mouse: primary peritonea macrophages | This paper | N/A |
| Experimental Models: Organisms/Strains | ||
| Mouse: Cretm-LDHAfl/fl C57BL/6J | (Xie et al., 2014) | N/A |
| Oligonucleotides | ||
| siRNA: human MCT1-#1 | RiboBio | Cat# stB0007914A |
| siRNA: human MCT1-#2 | RiboBio | Cat# stB0007914B |
| siRNA: human PDHA | Thermo Fisher | Cat# 4427038 |
| ShRNA: human HK2-#1 | Sigma-Aldrich | Cat# TRCN0000037670 |
| ShRNA: human HK2-#2 | Sigma-Aldrich | Cat# TRCN0000195582 |
| ShRNA:human MAVS-#1 | Sigma-Aldrich | Cat# TRCN0000236029 |
| ShRNA: human MAVS-#2 | Sigma-Aldrich | Cat# TRCN0000236030 |
| ShRNA: human Rig-I | Sigma-Aldrich | Cat# TRCN0000153712 |
| ShRNA: human PDHA | Sigma-Aldrich | Cat# TRCN0000028630 |
| ShRNA: human LDHA#1 | Sigma-Aldrich | Cat# TRCN0000164922 |
| ShRNA: human LDHA#2 | Sigma-Aldrich | Cat# TRCN0000026541 |
| qPCR pimers, see Table S1 | This paper | N/A |
| Recombinant DNA | ||
| PCDNA3-MAVS | Prof. Zhijian J. Chen (UT Southwestern Medical Center, USA). | N/A |
| pEF-BOS-Flag-RIG-l | Prof. Zhijian J. Chen (UT Southwestern Medical Center, USA). | N/A |
| Human HK2 cDNA Construct | Prof. Qinxi Li(Xiamen U. China)(Zhang et al.,2017) | N/A |
| Human LDHA cDNA Construct | Prof. Ping Gao(Xiamen U. China)(Zhong et al.,2017) | N/A |
| Human VDAC cDNA Construct | Prof. Hui Zhong (Molecular Genetics Department Beijing Institute of Biotechnology, China)(Guanetal., 2013) | N/A |
Highlights.
Lactate inhibits RLR-mediated interferon production.
This regulation occurs through direct sensing of lactate by MAVS
MAVS associates with hexokinase but this association is disrupted by RIG-I
Targeting LDHA enhances type I IFN production and viral clearance
Acknowledgements.
We thank Drs. Zhijian Chen (UT South Western Medical Center), Qinxi Li (Xiamen University, China), Ping Gao (Xiamen University, China) and Hui Zhong (Beijing Institute of Biotechnology, China) for sharing with various plasmids. We are indebted to Dr. Steve Kridel (Wake Forest Baptist Comprehensive Cancer Center) for providing the Seahorse device for our study. We acknowledge the support of the Wake Forest Baptist Comprehensive Cancer Center Cell Engineering Shared Resource, supported by the National Cancer Institute’s Cancer Center Support Grant (P30CA012197). This work is supported by Start-ups and Anderson Endowed Professorship fund from Wake Forest School of Medicine and NIH grants (R01CA182424 and R01CA193813) to H.-K.L as well as NIH grants (R01CA194094 and R01CA197178) to H.-Y.L. We also acknowledge the support by the National key research and development program (2017YFA0503900), National Natural Science Foundation of China (31671413) and Beijing Nova program (Z161100004916147) to W.-N.Z. and National Natural Science Foundation of China (81790252) to H.-Y.L.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interests: The authors declare no competing interests.
References
- Baker JC, Yan X, Peng T, Kasten S, and Roche TE (2000). Marked differences between two isoforms of human pyruvate dehydrogenase kinase. J Biol Chem 275, 15773–15781. [DOI] [PubMed] [Google Scholar]
- Brand A, Singer K, Koehl GE, Kolitzus M, Schoenhammer G, Thiel A, Matos C, Bruss C, Klobuch S, Peter K, et al. (2016). LDHA-Associated Lactic Acid Production Blunts Tumor Immunosurveillance by T and NK Cells. Cell Metab 24, 657–671. [DOI] [PubMed] [Google Scholar]
- Burdette DL, Monroe KM, Sotelo-Troha K, Iwig JS, Eckert B, Hyodo M, Hayakawa Y, and Vance RE (2011). STING is a direct innate immune sensor of cyclic di-GMP. Nature 478, 515–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang CH, Curtis JD, Maggi LB Jr., Faubert B, Villarino AV, O’Sullivan D, Huang SC, van der Windt GJ, Blagih J, Qiu J, et al. (2013). Posttranscriptional control of T cell effector function by aerobic glycolysis. Cell 153, 1239–1251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen W, Han C, Xie B, Hu X, Yu Q, Shi L, Wang Q, Li D, Wang J, Zheng P, et al. (2013a). Induction of Siglec-G by RNA viruses inhibits the innate immune response by promoting RIG-I degradation. Cell 152, 467–478. [DOI] [PubMed] [Google Scholar]
- Chen Y, Liang W, Yang S, Wu N, Gao H, Sheng J, Yao H, Wo J, Fang Q, Cui D, et al. (2013b). Human infections with the emerging avian influenza A H7N9 virus from wet market poultry: clinical analysis and characterisation of viral genome. Lancet 381, 1916–1925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colegio OR, Chu NQ, Szabo AL, Chu T, Rhebergen AM, Jairam V, Cyrus N, Brokowski CE, Eisenbarth SC, Phillips GM, et al. (2014). Functional polarization of tumour-associated macrophages by tumour-derived lactic acid. Nature 513, 559–563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crane CA, Austgen K, Haberthur K, Hofmann C, Moyes KW, Avanesyan L, Fong L, Campbell MJ, Cooper S, Oakes SA, et al. (2014). Immune evasion mediated by tumor-derived lactate dehydrogenase induction of NKG2D ligands on myeloid cells in glioblastoma patients. Proc Natl Acad Sci U S A 111, 12823–12828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies LC, Rice CM, Palmieri EM, Taylor PR, Kuhns DB, and McVicar DW (2017). Peritoneal tissue-resident macrophages are metabolically poised to engage microbes using tissue-niche fuels. Nat Commun 8, 2074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeBerardinis RJ, and Thompson CB (2012). Cellular metabolism and disease: what do metabolic outliers teach us? Cell 148, 1132–1144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeWaal D, Nogueira V, Terry AR, Patra KC, Jeon SM, Guzman G, Au J, Long CP, Antoniewicz MR, and Hay N (2018). Hexokinase-2 depletion inhibits glycolysis and induces oxidative phosphorylation in hepatocellular carcinoma and sensitizes to metformin. Nat Commun 9, 446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faubert B, Li KY, Cai L, Hensley CT, Kim J, Zacharias LG, Yang C, Do QN, Doucette S, Burguete D, et al. (2017). Lactate Metabolism in Human Lung Tumors. Cell 171, 358–371e359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng H, Lenarcic EM, Yamane D, Wauthier E, Mo J, Guo H, McGivern DR, Gonzalez-Lopez O, Misumi I, Reid LM, et al. (2017). NLRX1 promotes immediate IRF1-directed antiviral responses by limiting dsRNA-activated translational inhibition mediated by PKR. Nat Immunol 18, 1299–1309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gack MU, Shin YC, Joo CH, Urano T, Liang C, Sun L, Takeuchi O, Akira S, Chen Z, Inoue S, et al. (2007). TRIM25 RING-finger E3 ubiquitin ligase is essential for RIG-I-mediated antiviral activity. Nature 446, 916–920. [DOI] [PubMed] [Google Scholar]
- Ganeshan K, and Chawla A (2014). Metabolic regulation of immune responses. Annu Rev Immunol 32, 609–634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guan K, Zheng Z, Song T, He X, Xu C, Zhang Y, Ma S, Wang Y, Xu Q, Cao Y, et al. (2013). MAVS regulates apoptotic cell death by decreasing K48-linked ubiquitination of voltage-dependent anion channel 1. Mol Cell Biol 33, 3137–3149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honda K, Takaoka A, and Taniguchi T (2006). Type I interferon [corrected] gene induction by the interferon regulatory factor family of transcription factors. Immunity 25, 349–360. [DOI] [PubMed] [Google Scholar]
- Hou F, Sun L, Zheng H, Skaug B, Jiang QX, and Chen ZJ (2011). MAVS forms functional prion-like aggregates to activate and propagate antiviral innate immune response. Cell 146, 448–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunt L, Gupta-Wright A, Simms V, Tamba F, Knott V, Tamba K, Heisenberg-Mansaray S, Tamba E, Sheriff A, Conteh S, et al. (2015). Clinical presentation, biochemical, and haematological parameters and their association with outcome in patients with Ebola virus disease: an observational cohort study. Lancet Infect Dis 15, 1292–1299. [DOI] [PubMed] [Google Scholar]
- Ivashkiv LB, and Donlin LT (2014). Regulation of type I interferon responses. Nat Rev Immunol 14, 36–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin S, Tian S, Luo M, Xie W, Liu T, Duan T, Wu Y, and Cui J (2017). Tetherin Suppresses Type I Interferon Signaling by Targeting MAVS for NDP52-Mediated Selective Autophagic Degradation in Human Cells. Mol Cell 68, 308–322e304. [DOI] [PubMed] [Google Scholar]
- Kawai T, and Akira S (2010). The role of pattern-recognition receptors in innate immunity: update on Toll-like receptors. Nat Immunol 11, 373–384. [DOI] [PubMed] [Google Scholar]
- Liu X, Cooper DE, Cluntun AA, Warmoes MO, Zhao S, Reid MA, Liu J, Lund PJ, Lopes M, Garcia BA, et al. (2018). Acetate Production from Glucose and Coupling to Mitochondrial Metabolism in Mammals. Cell 175, 502–513e513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lunt SY, and Vander Heiden MG (2011). Aerobic glycolysis: meeting the metabolic requirements of cell proliferation. Annu Rev Cell Dev Biol 27, 441–464. [DOI] [PubMed] [Google Scholar]
- MacMicking JD (2012). Interferon-inducible effector mechanisms in cell-autonomous immunity. Nat Rev Immunol 12, 367–382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michelakis ED, Webster L, and Mackey JR (2008). Dichloroacetate (DCA) as a potential metabolic-targeting therapy for cancer. Br J Cancer 99, 989–994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milletti F (2012). Cell-penetrating peptides: classes, origin, and current landscape. Drug Discov Today 17, 850–860. [DOI] [PubMed] [Google Scholar]
- Nakazawa MS, Keith B, and Simon MC (2016). Oxygen availability and metabolic adaptations. Nat Rev Cancer 16, 663–673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parks SK, Chiche J, and Pouyssegur J (2013). Disrupting proton dynamics and energy metabolism for cancer therapy. Nat Rev Cancer 13, 611–623. [DOI] [PubMed] [Google Scholar]
- Patra KC, Wang Q, Bhaskar PT, Miller L, Wang Z, Wheaton W, Chandel N, Laakso M, Muller WJ, Allen EL, et al. (2013). Hexokinase 2 is required for tumor initiation and maintenance and its systemic deletion is therapeutic in mouse models of cancer. Cancer Cell 24, 213–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavlova NN, and Thompson CB (2016). The Emerging Hallmarks of Cancer Metabolism. Cell Metab 23, 27–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng M, Yin N, Chhangawala S, Xu K, Leslie CS, and Li MO (2016). Aerobic glycolysis promotes T helper 1 cell differentiation through an epigenetic mechanism. Science 354, 481–484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prezma T, Shteinfer A, Admoni L, Raviv Z, Sela I, Levi I, and Shoshan-Barmatz V (2013). VDAC1-based peptides: novel pro-apoptotic agents and potential therapeutics for B-cell chronic lymphocytic leukemia. Cell Death Dis 4, e809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts DJ, and Miyamoto S (2015). Hexokinase II integrates energy metabolism and cellular protection: Akting on mitochondria and TORCing to autophagy. Cell Death Differ 22, 364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossignol R, Gilkerson R, Aggeler R, Yamagata K, Remington SJ, and Capaldi RA (2004). Energy substrate modulates mitochondrial structure and oxidative capacity in cancer cells. Cancer Res 64, 985–993. [DOI] [PubMed] [Google Scholar]
- Schlee M, and Hartmann G (2016). Discriminating self from non-self in nucleic acid sensing. Nat Rev Immunol 16, 566–580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoggins JW, Wilson SJ, Panis M, Murphy MY, Jones CT, Bieniasz P, and Rice CM (2011). A diverse range of gene products are effectors of the type I interferon antiviral response. Nature 472, 481–485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seth RB, Sun L, Ea CK, and Chen ZJ (2005). Identification and characterization of MAVS, a mitochondrial antiviral signaling protein that activates NF-kappaB and IRF 3. Cell 122, 669–682. [DOI] [PubMed] [Google Scholar]
- Stark GR, and Darnell JE Jr. (2012). The JAK-STAT pathway at twenty. Immunity 36, 503–514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamura T, Yanai H, Savitsky D, and Taniguchi T (2008). The IRF family transcription factors in immunity and oncogenesis. Annu Rev Immunol 26, 535–584. [DOI] [PubMed] [Google Scholar]
- Trinchieri G (2010). Type I interferon: friend or foe? J Exp Med 207, 2053–2063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinberg F, Hamanaka R, Wheaton WW, Weinberg S, Joseph J, Lopez M, Kalyanaraman B, Mutlu GM, Budinger GR, and Chandel NS (2010). Mitochondrial metabolism and ROS generation are essential for Kras-mediated tumorigenicity. Proc Natl Acad Sci U S A 107, 8788–8793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf AJ, Reyes CN, Liang W, Becker C, Shimada K, Wheeler ML, Cho HC, Popescu NI, Coggeshall KM, Arditi M, et al. (2016). Hexokinase Is an Innate Immune Receptor for the Detection of Bacterial Peptidoglycan. Cell 166, 624–636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie H, Hanai J, Ren JG, Kats L, Burgess K, Bhargava P, Signoretti S, Billiard J, Duffy KJ, Grant A, et al. (2014). Targeting lactate dehydrogenase--a inhibits tumorigenesis and tumor progression in mouse models of lung cancer and impacts tumor-initiating cells. Cell Metab 19, 795–809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- You F, Wang P, Yang L, Yang G, Zhao YO, Qian F, Walker W, Sutton R, Montgomery R, Lin R, et al. (2013). ELF4 is critical for induction of type I interferon and the host antiviral response. Nat Immunol 14, 1237–1246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng W, Xu M, Liu S, Sun L, and Chen ZJ (2009). Key role of Ubc5 and lysine-63 polyubiquitination in viral activation of IRF3. Mol Cell 36, 315–325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Wang S, Jiang B, Huang L, Ji Z, Li X, Zhou H, Han A, Chen A, Wu Y, et al. (2017). c-Src phosphorylation and activation of hexokinase promotes tumorigenesis and metastasis. Nat Commun 8, 13732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao W, Chang C, Cui Y, Zhao X, Yang J, Shen L, Zhou J, Hou Z, Zhang Z, Ye C, et al. (2014). Steroid receptor coactivator-3 regulates glucose metabolism in bladder cancer cells through coactivation of hypoxia inducible factor 1alpha. J Biol Chem 289, 11219–11229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y, Wang A, Zou Y, Su N, Loscalzo J, and Yang Y (2016). In vivo monitoring of cellular energy metabolism using SoNar, a highly responsive sensor for NAD(+)/NADH redox state. Nat Protoc 11, 1345–1359. [DOI] [PubMed] [Google Scholar]
- Zhong X, Tian S, Zhang X, Diao X, Dong F, Yang J, Li Z, Sun L, Wang L, He X, et al. (2017). CUE domain-containing protein 2 promotes the Warburg effect and tumorigenesis. EMBO Rep 18, 809–825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu J, Zhang Y, Ghosh A, Cuevas RA, Forero A, Dhar J, Ibsen MS, Schmid-Burgk JL, Schmidt T, Ganapathiraju MK, et al. (2014). Antiviral activity of human OASL protein is mediated by enhancing signaling of the RIG-I RNA sensor. Immunity 40, 936–948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zitvogel L, Galluzzi L, Kepp O, Smyth MJ, and Kroemer G (2015). Type I interferons in anticancer immunity. Nat Rev Immunol 15, 405–414. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Inhibition of glucose metabolism promotes RLR-induced type-I IFN production, Related to Figure 1. A, Q-PCR analysis of IFN-β mRNA expression in RAW264.7 macrophage cells cultured in mediums with high glucose (25 mM) or low glucose (5 mM) and transfected with HMW Poly(I:C) for indicated hours. B–D, Q-PCR analysis of IFN-β or IL-6 mRNA expression in THP1 cells transfected with Poly(I:C) (1μg/ml), HTDNA (1 μg/ml) or treated with LPS (50 ng/ml). E, Q-PCR analysis of IFN-β mRNA expression in Hep3B cells pretreated with or without 2-DG (2mM) overnight and then transfected with Poly(I:C) for indicated hours. F, Q-PCR analysis of IL-6 mRNA expression in HEK293 cells pretreated with or without 2-DG (2mM) and then transfected with Poly(I:C) for indicated hours. G, Q-PCR analysis of IFN-β mRNA expression in HEK293 cells pretreated with or without 2-DG (2mM) overnight and then infected with VSV for indicated hours. Data are means±SD. *p < 0.05, **p < 0.01.
Figure S2. Reciprocal regulation between hexokinase and MAVS, Related to Figure 2. A, Analysis of Hexokinase (HK) activity in purified mitochondria isolated from HEK293 cells infected with Sev for indicated hours. B and C, Analysis of pyruvate, lactate level (B) and HK2 expression (C) in Hep3B cells with control or HK2 knockdown by using Colorimetric assay kit or immunoblotting. D, Q-PCR analysis of IL-6 mRNA expression in Hep3B cells with control or HK2 knockdown and transfected with Poly (I: C). E, Q-PCR analysis of IFN-β or Sev specific mRNA expression in Hep3B cells with control or HK2 knockdown and infected with Sev. F, Q-PCR analysis of IFN-β mRNA expression in Hep3B cells infected with control or HK2 shRNA with or without Flag-HK2 expression and then transfected with Poly(I:C). G, Whole cell lysates of THP1 cells transfected with HTDNA for indicated hours were collected for IP with MAVS antibody, followed by IB analysis for indicated proteins. H, HEK293 cells transfected with Flag-Vector or Flag-RIG-I(N) together HA-MAVS were immunoprecipitated with IgG or anti-HA antibody, and IP complexes were analyzed by immunoblot analysis. I, Q-PCR analysis of IFN-β mRNA expression in HEK293 cells with control or RIG-I knockdown and infected with Sev as described in Figure. 2H. J, Immunoblot analysis of MAVS expression in Hep3B cells with control or MAVS knockdown. K, Analysis of hexokinase (HK) activity in purified mitochondria isolated from wild-type and MAVS knockout MEF cells (upper panel). Immunoblot analysis of MAVS protein level in wild type or MAVS knockout MEFs (lower panel). L, HEK293 cells were pre-incubated with control or VDAC competitive peptide (0.5μM) for 90 min and then immunoprecipitated with IgG or anti-MAVS antibody, and IP complexes were analyzed by immunoblot analysis. M, HEK293 cells transfected with Myc-VDAC were treated with the same conditions as in L and immunoprecipitated with IgG or anti-Myc antibody, IP complexes were analyzed by immunoblot analysis. Data are means±SD. **p < 0.01.
Figure S3. Anaerobic glycolysis affects RLR triggered type-I IFN production, Related to Figure 3. A and B, Q-PCR analysis of PDHA mRNA expression (A) and measurement of lactate secretion (B) for HEK293 cells with control and PDHA knockdown as described in Fig. 3A. C and D, Q-PCR analysis of IFN-β (C) and PDHA (D) mRNA expression HEK293 cells transfected with control or PDHA siRNA. E, Analysis of lactate secretion in supernatant of HEK293 cells treated with or without UK5099 (10 μM) overnight. F and G, Q-PCR analysis of IFN-β mRNA expression in HEK293 cells pretreated with UK5099 overnight and then transfected with Poly(I:C) (F) or infected with Sev (G). H. Immunoblot analysis of HEK293 cells treated with or without UK5099 (10 μM) overnight and then transfected with Poly(I:C) as indicated. I. Analysis of lactate secretion in HEK293 cells treated with DCA as described in Figure 3B. J, Detection of lactate secretion in immortalized bone marrow macrophage cells cultured in mediums containing glucose (25 mM) or galactose (25 mM) as described in Figure 3C. K and L, Q-PCR analysis of IFN-β (Raw264.7 cells) or IL-6 (iBMM cells) expression treated the same as in J and transfected with Poly(I:C) for indicated hours. M, Q-PCR analysis of VEGF mRNA expression in HEK293 cells exposed to normoxia (20% O2) or hypoxia (1% O2) and transfected with Poly(I:C). N, Q-PCR analysis of IFN-β expression in RAW264.7 macrophage cells treated with the same conditions as in M and transfected with Poly(I:C) for indicated hours. O and P, Q-PCR analysis of Glut4 mRNA expression (O) and measurement of lactate secretion (P) in RAW264.7 macrophage cells as indicated. Data are means±SD. **p < 0.01.
Figure S4. LDHA inhibits RLR induced type-I IFN production, Related to Figure 4. A, Immunoblot analysis of LDHA expression in Hep3B cells infected with control or two independent LDHA shRNAs. B, Q-PCR determination of IFN-β mRNA expression in Hep3B cells with control or two independent LDHA shRNAs infection and then transfected with Poly(I:C) for 2 hours. C, Q-PCR determination of IFN-β mRNA expression in Hep3B cells pretreated with or without sodium oxamate (20 mM) overnight and then transfected with Poly(I:C) for 4 hours. D, Q-PCR analysis of IFN-β expression in HEK293 cells pretreated with or without Sodium Oxamate (20 mM) overnight and then infected with VSV for indicated hours. E and F, Immunoblot analysis of Hep3B cells treated with or without sodium oxamate (20 mM) and then infected with Sendai virus for indicated times. Whole cell lysates were subjected to SDS-PAGE (E) and mitochondria were isolated for SDD-AGE (F). G and H, Q-PCR analysis of IFN-β expression (G) and immunoblot analysis (H) in THP1 cells pretreated with or without Sodium Oxamate (20 mM) overnight and then transfected with HTDNA as indicated. Data are means±SD. *p < 0.05, **p < 0.01.
Figure S5. LDHA-associated lactate inhibits RLR induced type-I IFN production, Related to Figure 4. A, Q-PCR analysis of IFN-β mRNA expression in HEK293 cells pretreated with or without sodium oxamate (20 mM) overnight and then cultured in medium in the presence or absence of lactate (10 mM) 2 hours before infection with Sev for 8 hours. B, Immunoblot analysis of Hep3B cells treated with or without 2-DG and then cultured in medium with or without lactate (10 mM) before transfecting with Poly(I:C). Cell mitochondria were isolated for SDD-AGE (upper panel) and whole cell lysates were used for SDS-PAGE (lower panel). C, Microscopic images of VSV-GFP-infected immortalized bone marrow macrophage cultured in medium containing glucose (25 mM) or galactose (25 mM) with or without addition of lactate (10 mM) 2 hours before infecting with VSV-GFP (MOI=0.1) for 16 hours. Scale bar, 100 mm. D, The total ion current (TIC) chromatogram and Mass spectrum of intracellular lactate for Figure 4M. E, List of intracellular lactate concentration for each group in Figure 4M. F and G, Q-PCR analysis of IFN-β in Hep3B cells treated with or without Oxamate (F) or 2-DG (G) overnight, then added with lactate (10 mM) along with or without NaOH to correct PH level before Poly(I:C) transfection. H, Q-PCR analysis of IFN-β mRNA expression in Hep3B cells with control or MCT1 knockdown and then treated with or without 2-DG overnight before lactate addition (10 mM) and Poly(I:C) transfection. I, Q-PCR analysis of MCT1 expression in Hep3B cells with control or MCT1 knockdown. Data are means±SD. *p < 0.05, **p < 0.01.
Figure S6. In vivo targeting LDHA enhances RLR signaling, Related to Figure 5. A, Q-PCR analysis of IL-6 expression in lung from mice fasted overnight and then treated with high glucose (1.5 g/kg) or low glucose (0.2 g/kg) with or without injection of sodium lactate (1 g/kg) and VSV infection. B, Schematic representation of the procedures for LDHA deletion in mice and peritoneal macrophage isolation. C, Immunoblot analysis of LDHA protein level in different organs from mice with intraperitoneal injection of tamoxifen (20 mg/ml) or vehicle. D, Q-PCR analysis of IFN- α or IL-6 expression in spleen from Ldha+/+ and Ldha−/− mice treated as in Figure 5. E, Immunoblot analysis of LDHA expression in peritoneal macrophages isolated from Ldha+/+ or Ldha−/− mice. F, ELISA analysis of IFN-β expression in supernatant of Ldha+/+ or Ldha−/− macrophages transfected with HTDNA or infected with HSV. G, Schematic representation of the procedures for mice intraperitoneal injection of sodium oxamate and VSV infection. H, Analysis of lactate secretion in peripheral blood of mice intraperitoneally injected with or without sodium oxamate (750 mg/kg). I-L, Q-PCR analysis of IL-6 (I) or VSV specific gene expression (J-L) in different organs from mice treated with or without sodium oxamate and then challenged with VSV. M, Q-PCR analysis of IFN-α mRNA expression in lung tissue from mice injected with or without sodium oxamate and infected with HSV. Data are means±SD. **p < 0.01.
Figure S7. Lactate negatively regulates RLR activation by targeting MAVS, Related to Figure 6. A, MAVS peptides identified by Mass Spectrometry through in vitro pulldown. B, Immunoblot analysis of in vitro pulldown assays by incubating biotin-labeled lactate with increased doses of MAVS protein translated by the TNT system. C, Immunoblot analysis of in vitro pulldown assays by incubating biotin-labeled lactate and MAVS protein together with or without lactate. D-E, Base peak ion Chromatogram (left panel) and mass spectrum (right panel) of the ion Chromatography peak for control peptide (D) or TM peptide (E). F, Schematic representation of the procedures for in vitro biotin pulldown assay indicated in Figure 6G. G, Q-PCR analysis of IFN-β expression in HEK293 cells treated by control or TM peptide and transfected with Poly(I:C). H, Q-PCR analysis of IL-6 expression in Hep3B cells treated by control or TM peptide and transfected with Poly(I:C). I and J, Q-PCR analysis of IFN-β or IL-6 expression in Hep3B cells pretreated with or without 2-DG overnight, then treated by control or TM peptide for 2 hours, addition of lactate (5 mM) and transfected with Poly(I:C). Data are means±SD. *p < 0.05, **p < 0.01.