

Abstract
Carboranes are boron-rich molecular clusters possessing electronic characteristics that allow for orthogonal approaches to vertex-selective modifications. We report improved functionalization methods utilizing orthogonal chemistry to achieve efficient substitution at electron-rich B-vertices and electron-poor C-vertices of carborane. Functionalization of B-vertices with alkyl and (hetero)aryl groups using the corresponding Grignard reagents has been improved through the use of a Pd-based precatalyst featuring an electron-rich biaryl phosphine ligand, resulting in reduced reaction times. Importantly, this method is tolerant towards alkyl-based Grignard reagents containing β-hydrogens. Furthermore, a transition metal-free approach to the substitution of carborane C-vertices with (hetero)aryl substrates has been developed under nucleophilic aromatic substitution (SNAr) conditions. The selective substitution of carboranes afforded by these methods holds potential for the rational synthesis of heterofunctionalized boron clusters with substituents on both boron and carbon-based vertices.
Keywords: carborane, boron clusters, cross-coupling, catalysis, nucleophilic aromatic substitution
Graphical Abstract
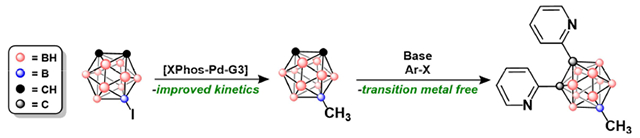
1. Introduction
Since the discovery of neutral carboranes (C2B10H12) more than 50 years ago, these icosahedral clusters have emerged as diverse building blocks for a variety of applications, including organic light emitting diodes,1 biomedicine,2 batteries,3 catalysis4, self-assembled materials5 and medicinal drug design.6 These compounds, which exist as three constitutional isomers (ortho, meta, and para) depending on relative positions of the two C-H vertices on the cage, feature several unique properties, such as three dimensional aromaticity, a large HOMO-LUMO gap, tunable dipole moments as a function of cluster symmetry, and thermal stability.7a The diversity of carborane applications, however, requires synthetic methods that can provide access to vertex-selective modification. Selectivity can be achieved, to a certain extent, by utilizing the inherent electronic non-uniformity of the cluster cage whereby certain B–H vertices are rendered more reactive than others towards electrophilic7a or organometallic7b-k reagents. Furthermore, under basic conditions the acidic protons bound to the carbon vertices can undergo facile deprotonation and subsequent selective functionalization with electrophiles.8 Nevertheless, current synthetic methods are still limited in their ability to achieve efficient substitution of carboranes. For example, current C-vertex substitution methods can install (hetero)aryl groups but often require the use of transition metal reagents (Figure 1A).8a-c Similarly, metal-catalyzed cross-coupling can be used to achieve B-vertex functionalization. However, the substitution of carbon-based nucleophiles is limited by substrate tolerance, long reaction times, and even poor reproducibility (Figure 1B).9a-d To improve on these procedures, we report a fast, high yielding B-vertex substitution method for B-substituted carboranes as well as a transition metal-free C-vertex substitution of (hetero)aryl substrates (Figure 1C). Importantly, these two methods can be performed sequentially, producing a convenient and straightforward strategy to form B- and C-functionalized carboranes.
Figure 1:
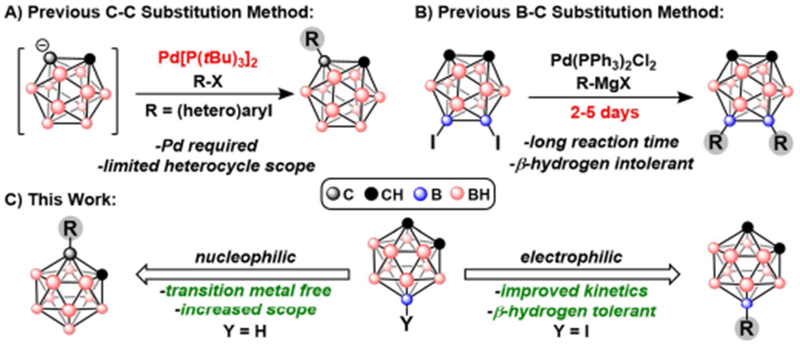
A) Previous substitution method for C-(hetero)arylation. B) Previous B-substitution method via Kumada coupling. C) Improved C- and B-vertex functionalization procedures.
2. B-Vertex Substitution
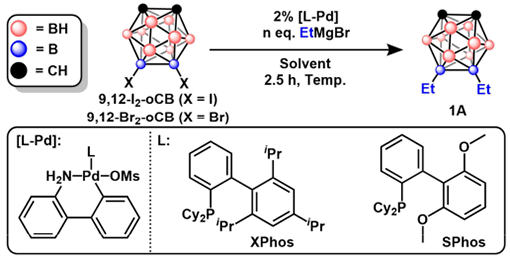
The boron vertices of carboranes have been especially difficult to substitute relative to traditional organic molecules. This is largely due to their hindered steric environment, variable electronic character, and strong non-hydridic B–H bonds.7a Friedel-Crafts halogenation, however, has been a vital tool for harnessing electrophilic reactivity at the electron-rich boron vertices of carboranes. The resulting B-halogenated carborane electrophiles provide access to B-C, B-O, or B-N substitution through metal-catalyzed cross-coupling. Zakharkin et al. was the first to report a Pd-mediated cross-coupling process with Pd(PPh3)2Cl2 between 9-I-ortho-carborane (9-I-oCB) or 9-I-meta-carborane (9-I-mCB) and several organomagnesium reagents, claiming to produce B-substituted clusters in a manner reminiscent to classical Kumada cross-coupling chemistry used for aryl halides.9a,b Subsequently, Li et al. reported a similar protocol for transformations between 9,12-I2-ortho-carborane (9,12-I2-oCB) and alkylmagnesium reagents.9c In both cases, authors claimed yields from 60-99% for coupling reactions that required 24-30 h for completion. Notably, a later report by Zheng et al. called the obtained yields and reaction times by the original authors into question, claiming that: “In our hands, the product yields of these reactions are not as high as been reported, and in most cases we have studied the yields are extremely low.”9d Additionally, Zheng et al. determined that reproducible cross-coupling could be obtained in 20-75% yield only after the introduction of a catalytic amount of CuI. While the method by Zheng et al. has improved the overall cross-coupling chemistry, several limitations, including long reaction times (2-5 days) and limited tolerance for Grignard reagents containing β-hydrogens, persisted. We hypothesize that these drawbacks can be further mitigated by replacing the Pd(PPh3)2Cl2 precatalyst with a system containing a biaryl phosphine ligand as was observed previously for B–O, B–N, and B–CN metal-catalyzed cross-coupling chemistry.9e-g
We initially targeted the coupling of 9,12-I2-oCB with EtMgBr in the presence of XPhos-Pd-G310 precatalyst under the reaction conditions reported by Zheng et al. to probe the tolerance of β-hydrogens presented by the Grignard nucleophile. Even with the inclusion of catalytic CuI, which has been reported9d to improve the yield of this reaction when using Pd(PPh3)2Cl2 as a precatalyst, very low conversion was observed (<5%) to the desired 9,12-diethyl-ortho-carborane (1A) was observed after 2.5 h (Entry 1, see SI for experimental details). A solvent (Entries 1-2) and temperature (Entries 2-3) screen revealed 1,4-dioxane as a promising solvent when employed at 75 °C with XPhos-Pd-G3. Notably, comparable yields were obtained with SPhos-Pd-G3 (Entry 4) under otherwise identical conditions. Furthermore, these conditions are selective for B–I bonds, with no desired product observed when using B–Br electrophiles (Entry 5). Under these optimized conditions, the issue of β-hydride elimination suggested in the previously reported system does not appear to be problematic.
With optimized conditions established, we assessed the scope of this method with ortho-carboranyl electrophiles (Figure 2). Starting with 9,12-I2-oCB, we observed nearly full conversions (>90%) to the disubstituted products in the presence of alkyl or arylmagnesium reagents. These compounds were purified via silica gel column chromatography providing products 1A, 1B, and 1C in 57-72% isolated yields. While these conditions allow for the isolation of disubstituted products (1A-C), we note that approximately 5-10% of the partially dehalogenated products (1D-E) are present in the reaction mixture of 1A-B. Interestingly, no observable dehalogenation is encountered when 9-I-oCB is used as a substrate and >95% conversions to the monosubstituted products 1D, 1E, and 1F are observed within 2 hours. Notably, a heterocycle compatible with magnesium reagents afforded >95% conversion to 1G, although slightly longer reaction time was needed (4 h).
Figure 2:
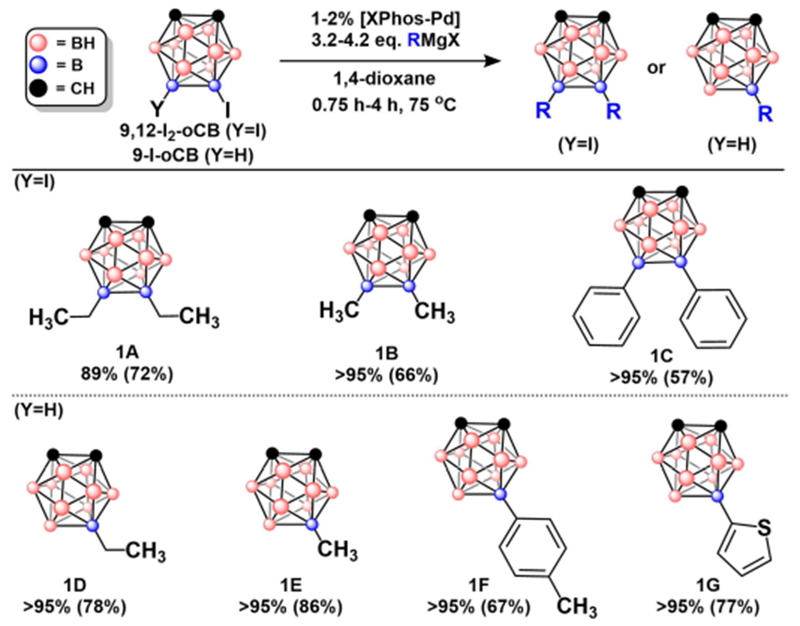
Reaction scheme for mono- and disubstitution of ortho-carborane. Ortho-carborane coupling products with GC-MS conversion and percent isolated yields in parentheses. See Table S2 in SI for exact reaction conditions.
To expand the utility of this method, we tested our conditions with 9-I-mCB and 9-Br-10-I-mCB to determine the tolerance of meta-carboranyl electrophiles and B-Br bonds (Figure 3). Slightly longer reaction times were required to reach >95% conversion for products 1H, 1I, and 1J. Despite >95% conversion observed for 1H, the low isolated yield (34%) likely resulted from the high volatility of the compound, as evidenced by observed sublimation in vacuo at room temperature when removing solvent. For products 1I and 1J, no substitution and/or reduction at the brominated vertex was observed, lending further evidence to the selectivity of this method for B–I bonds over B–Br bonds. This stands in contrast to the previously developed B–N and B–O cross-coupling methods using Pd-based catalysts with biaryl ligands that showed preferential functionalization of B–Br bonds over B–I congeners.9f
Figure 3:
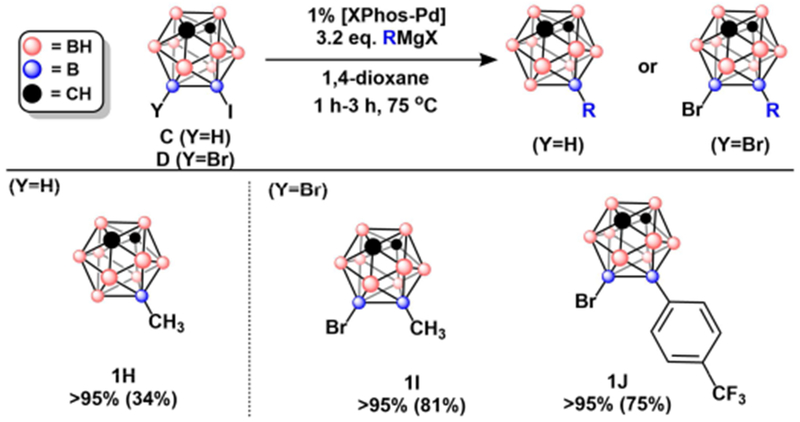
Reaction scheme for monocoupling of meta-carborane in the presence of B-Br bonds. Meta-carborane coupling products with GC-MS conversion and percent isolated yields in parentheses. See Table S2 in SI for exact reaction conditions.
3. C-vertex substitution
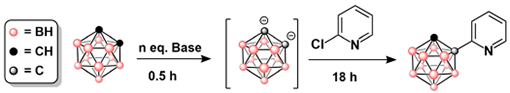
C-vertex substitution methods are perceived as more straightforward than the B-vertex functionalization: due to the acidic nature of the C–H bond, C-vertices can be deprotonated and treated with electrophiles or used as nucleophiles.8 While the metalation of carboranes and subsequent reactivity towards alkyl halides has produced a library of C-alkyl carboranes,7a C-vertex (hetero)arylation has been achieved through less straightforward procedures, such as metal-catalyzed cross-coupling that requires either stoichiometric or catalytic amounts of transition-metal reagents.8 For example, Cu-mediated coupling with (hetero)aryl iodides or bromides can yield C-pyridyl- and C-aryl-carboranes, but requires 25-90 h reaction times and can tolerate only a select group of functionalized (hetero)aryl substrates.8b More recent methods, such as Ni- and Pd-catalyzed cross coupling, demonstrate improved functional group tolerance for C-arylation, but are limited to attaching chloro- and methyl-pyridine.8a,c Transition metal-free C-substitution methods have been investigated, but require the reaction between a C-monosubstituted ortho-carborane (oCB) and a fluoroarene possessing a strong electron withdrawing group.11 This selectively produces 1,2-diaryl-ortho-carboranes, but this methodology was not applied to heterocyclic substrates. Overall, procedures for C-(hetero)arylation are currently limited by their use of transition metal reagents and small substrate scopes. Previous reports have demonstrated the possibility of C-vertex substitution under SNAr conditions,1a,11,12 prompting us to expand the number and type of substrates that can be appended to carborane using this approach.
For the initial optimization studies we tested 2-chloropyridine, estimating that it would be less reactive than fluoroheterocycles and more reactive than fluoroarenes. Typically, C–H deprotonation and successive metalation of carborane have been achieved with nBuLi in either diethyl ether or THF.7axref Nevertheless, we found that nBuLi was unable to facilitate the heteroarylation of carborane to 1-(2-pyridyl)-ortho-carborane (2A) under these conditions (Entry 1). We previously observed that KHMDS deprotonates 1,1’-bis-(ortho-carborane),13 indicating that it could potentially be applicable to our system. While 2 and 1 equivalents of KHMDS yielded sub-quantitative conversions (Entries 2 and 3, respectively), 3 equivalents of this base led to 99% conversion of oCB to product (Entry 4). Testing the effect of the disilazide counterion on the reaction, we observed 45% and 90% conversion with LiHMDS and NaHMDS, respectively (Entries 5 and 6), suggesting the cation significantly influences the reaction progression. High conversion efficiency with KHMDS led us to continue its use throughout the optimization studies. Next, we examined the effect of other ethereal solvents on the reaction progress. With conversions ranging from 86%-98% (Entries 7-9), we selected THF as our primary solvent.
Under the optimized conditions, 2A (Figure 4B) can be isolated in 61% yield after purification via silica gel column chromatography. This method tolerates bipyridine-based electrophiles, producing compounds 2B and 2C in 43% and 73% isolated yields, respectively. These bipyridine-based compounds would be difficult to synthesize using metal-catalyzed cross-coupling methods. The ability to generate 2C suggests that multiple C–C bond forming processes can be achieved at the electrophile under these conditions. Testing this method with (hetero)aryl fluorides resulted in the desired product formation under the optimized conditions (Figure 4, 2D-I see SI for experimental details). The lower room temperature reactivity of substrates originating from products 2D and 2E was circumvented by heating the reaction mixture to 80°C. This initially afforded 2E in 27% yield, and analysis of the 11B NMR data suggested the presence of deboronated products, which is consistent with previous reports of deboronation of oCB in the presence fluoride.14 To sequester fluoride ions and potentially prevent cage degradation, we used one equivalent of isopropoxytrimethylsilane under otherwise identical conditions, which increased the isolated yield of 2E to 44%. Similar observations were made with the 2,6-difluoropyridine substrate, but the yield remained nearly identical even with the addition of the fluoride scavenger. However, upon changing the solvent from THF to diethyl ether, the isolated yield for 2F increased to 75%. We attribute this observation to the likely lower solubility of the KF byproduct in diethyl ether, thereby decreasing its reactivity towards deboronation. The present SNAr method also proceeds with meta-carborane under similar conditions, as exemplified by the formation of 2G (Figure 4C). We also assessed the viability of conducting sequential C-vertex heterosubstitutions. Deprotonation of 2A and subsequent treatment with perfluorobenzene selectively produced the para-substituted product 2H in 29% isolated yield. Upon further optimization, we found that excess perfluorobenzene resulted in higher conversion to desired product. The best results were achieved with DME and 2 equivalents of KHMDS, resulting in 74% isolated product yield. Similar carborane-substituted phenylene molecules have been investigated for their luminescent properties, but are typically synthesized via cross coupling procedures that require multiple reagents such as CuI and Pd(PPh3)2Cl2.15 Overall, we demonstrate that SNAr methodology can be applied to both chloro- and fluoro(hetero)aryl substrates, however, in the latter case a competing reactivity of the fluoride byproduct needs to be mitigated by the judicious use of the additional reagents and solvents.
Figure 4:
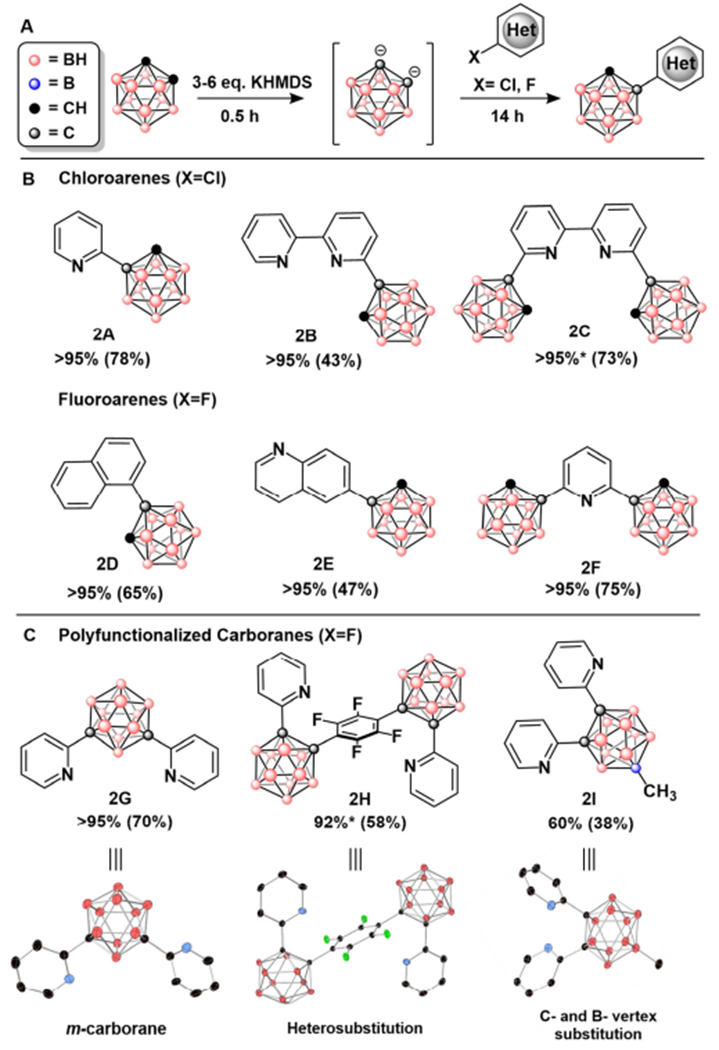
A) C-functionalization reaction scheme B) Products with GC-MS conversions and isolated yields in parentheses. C) Polyfunctionalized carboranes with associated single crystal X-ray structures obtained by extrapolating the general method in A. (*) indicates that conversion was determined through 1H NMR instead of GC-MS. See Table S4 in SI for exact reaction conditions.
a See SI for corresponding single crystal X-ray structures
Lastly, the described methods for forming B–C and C–C bonds in carboranes prompted us to explore whether these methods can be used in conjunction with each other to achieve the synthesis of heterofunctionalized clusters. Treatment of methylated carborane 1E with excess 2-fluoropyridine produced product 2I, demonstrating that both the B- and C-vertex functionalization methods can be used in a sequential manner. This compound was isolated in 38% yield. Single crystals of 2I were grown with hexanes and analyzed using X-ray crystallography, ultimately confirming its structural assignment (Figure 4C).
4. Conclusion
We have developed improved B- and C-vertex functionalization methods that can effectively afford previously inaccessible carborane derivatives. Specifically, by employing biaryl phosphine ligands, we have introduced an improved B-vertex substitution method that increases the rate of Pd-catalyzed Kumada cross-coupling conditions. Additionally, we have expanded the scope of C-vertex substitution while circumventing the requirement for transition metal-catalyzed cross-coupling. By utilizing the nucleophilic nature of the C-metalated carboranes, mild conditions can be used to achieve substitutions of heterocycles on carboranes. These improved methods represent a robust addition of transformations now available to the practitioners in boron cluster chemistry.
Supplementary Material
Table 1:
Optimization table for Kumada coupling with 9,12-I2-oCB or 9,12-Br2-oCB and EtMgBr. % Conv. indicates the % of 9,12-Et2-oCB present in the crude reaction mixture as determined by GC-MS.
 | ||||||
|---|---|---|---|---|---|---|
| Entry # | X | L | n | Solvent | T | % Conv. (% Yield) |
| 1a | I | XPhos | 5 | THF | 75 | 2 |
| 2 | I | XPhos | 4.2 | 1,4-dioxane | 65 | 30 |
| 3 | I | XPhos | 4.2 | 1,4-dioxane | 75 | 89b (77) |
| 4 | I | SPhos | 4.2 | 1,4-dioxane | 75 | 90 b (66) |
| 5 | Br | XPhos | 4.2 | 1,4-dioxane | 75 | 0 |
Included 2 mol% CuI additive.
Reaction mixture contained 5-10% dehalogenated product by GC-MS.
Table 2:
Optimization table of SNAr reaction between 2-chloropyridine and ortho-carborane. % Conv. indicates the % of 1-(2-pyridine)-oCB present in the crude reaction mixture as determined by GC-MS.
 | ||||
|---|---|---|---|---|
| Entry # | Base | Base eq. | Solvent | % Conv. |
| 1 | nBuLi | 2 | THF | 0 |
| 2 | KHMDS | 2 | THF | 92 |
| 3 | KHMDS | 1 | THF | 80 |
| 4 | KHMDS | 3 | THF | 99 |
| 5 | LiHMDS | 3 | THF | 45 |
| 6 | NaHMDS | 3 | THF | 90 |
| 7 | KHMDS | 3 | MTBEa | 86 |
| 8 | KHMDS | 3 | Etherb | 90 |
| 9 | KHMDS | 3 | DMEc | 98 |
Methyl-tert-butyl ether
Diethyl ether
1,2-Dimetheoxyethane
5. Acknowledgements
A.M.S. thanks the UCLA Department of Chemistry and Biochemistry for start-up funds, 3M for a Non-Tenured Faculty Award, the Alfred P. Sloan Foundation for a Fellowship in Chemistry, Research Corporation for Science Advancement (RCSA) for a Cottrell Scholar Award and the National Institutes of Health (NIH) for a Maximizing Investigators Research Award (MIRA, R35GM124746). C.M. thanks the UCLA Department of Chemistry and Biochemistry’s Daniel Kivelson Foundation for the Undergraduate Summer Research Fellowship in Chemistry.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.(a) Axtell JC; Kirlikovali KO; Djurovich PI; Jung D; Nguyen VT; Munekiyo B; Royappa AT; Rheingold AL; Spokoyny AM J. Am. Chem. Soc 2016, 138 (48), 15758. [DOI] [PubMed] [Google Scholar]; (b) Prokhorov AM; Hofbeck T; Czerwieniec R; Suleymanova AF; Kozhevnikov DN; Yersin H J. Am. Chem. Soc 2014, 136 (27), 9637. [DOI] [PubMed] [Google Scholar]
- 2.(a) Paxton RJ; Beatty BG; Hawthorne MF; Varadarajan A; Williams LE; Curtis FL; Knobler CB; Beatty JD; Shively JE Pro. Natl. Acad. Sci 1991, 88 (8), 3387. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Hawthorne MF; Maderna A Chem. Rev 1999, 99 (12), 3421. [DOI] [PubMed] [Google Scholar]; (c) Neirynck P; Schimer J; Jonkheijm P; Milroy LG; Cigler P; Brunsveld LJ Mater. Chem. B 2015, 3 (4), 539. [DOI] [PubMed] [Google Scholar]
- 3.McArthur SG; Geng L; Guo J; Lavallo V Inorg. Chem. Front 2015, 2 (12), 1101. [Google Scholar]
- 4.Selg C; Neumann W; Lönnecke P; Hey-Hawkins E; Zeitler K Chem. Eur. J 2017, 23 (13), 7932. [DOI] [PubMed] [Google Scholar]
- 5.(a) Farha OK; Spokoyny AM; Mulfort KL; Hawthorne MF; Mirkin CA; Hupp JT J. Am. Chem. Soc 2007, 129 (42), 12680. [DOI] [PubMed] [Google Scholar]; (b) Serino AC; Anderson ME; Saleh LMA; Dziedzic RM; Mills H; Heindreich LK; Spokoyny AM; Weiss PS ACS Appl. Mater. Interfaces 2017, 9, 40, 34592. [DOI] [PubMed] [Google Scholar]; (c) Kim J; Rim YS; Liu Y; Serino AC; Thomas JC; Chen H; Yang Y; Weiss PS Nano Lett. 2014, 14 (5), 2946. [DOI] [PubMed] [Google Scholar]; (d) Spokoyny AM; Farha OK; Mulfort KL; Hupp JT; Mirkin CA Inorg. Chim. Acta 2010, 364 (1), 266. [Google Scholar]
- 6.(a) Issa F; Kassiou M; Rendina LM Chem. Rev 2011, 111 (9), 5701. [DOI] [PubMed] [Google Scholar]; (b) Leśnikowski ZJ J. Med. Chem 2016, 59 (17), 7738. [DOI] [PubMed] [Google Scholar]; (c) Adamska A; Rumijowska,-Galewicz A; Ruszczynska A; Studzińska M; Jablońska A; Paradowska E; Bulska E; Munier-Lehman H; Dziadek J; Leśnikowski ZJ; Olejniczak AB Eur. J. Med. Chem 2016, 121 (4), 71. [DOI] [PubMed] [Google Scholar]
- 7.(a) For a general overview of carboranes: Grimes RN, Carboranes, 3rd edn, Elsevier, Oxford, 2016. [Google Scholar]; (b) For examples of B–H activation: Cheng R; Li B; Wu J; Zhang J; Qiu Z; Tang W; You S; Tang Y; Xie Z J. Am. Chem. Soc 2018, 140, 13, 4508–4511 [DOI] [PubMed] [Google Scholar]; (c) Quan Y; Qiu Z; Xie Z Chem. Eur. J 2018, 24, 2795–2805; [DOI] [PubMed] [Google Scholar]; (d) Cheng R; Qiu Z; Xie Z Nat. Comm 2017, 8, 14827. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Guo S; Cui P; Gao Y; Jin G Dalton Trans. 2018, 47, 13641–13646 [DOI] [PubMed] [Google Scholar]; (f) Eleazer BJ; Peryshkov DV Inorg. Chem 2018, 3, 79–109 [Google Scholar]; (g) Eleazer BJ; Smith MD; Popov AA; Peryshkov DV Chem. Sci 2018, 9, 2601–2608. [DOI] [PMC free article] [PubMed] [Google Scholar]; (h) Tang C; Zhang J; Xie Z Angew. Chem 2017, 129, 8768–8772. [Google Scholar]; (i) Lyu H; Quan Y; Xie Z Chem. Sci 2018, 9, 6390–6394. [DOI] [PMC free article] [PubMed] [Google Scholar]; (j) Mirabelli M; Sneddon L J. Am. Chem. Soc 1998, 110, 449–453. [Google Scholar]; (k) Estrada J; Lee S; McArthur S; El-Hellani A; Tham F; Lavallo V J. Organomet. Chem 2015, 798, 214–217. [Google Scholar]
- 8.(a) For a general overview of C-vertex substitution: Lu J-Y; Wan H; Zhang J; Wang Z; Li Y; Du Y; Li C; Liu Z-T; Liu Z-W; Lu J Chem. Eur. J 2016, 22 (49), 17542. [DOI] [PubMed] [Google Scholar]; (b) Coult R; Fox MA; Gill WR; Herbertson PL; MacBride JAH; Wade K J. Organomet. Chem 1993, 462 (1), 19 [Google Scholar]; (c) Tang C; Xie Z Angew. Chem. Int. Ed 2015, 54 (26), 7662. [DOI] [PubMed] [Google Scholar]; (d) Chan AL; Estrada J; Kefalidis CE; Lavallo V Organometallics 2016, 35 (19), 3257 [Google Scholar]; (e) Cabrera-González J; Viñas C; Haukka M; Bhattacharyya S; Gierschner J; Núñez R Chem. Eur. J 2016, 22 (38), 13588. [DOI] [PubMed] [Google Scholar]; (f) Powley SL; Schaefer L; Man WY; Ellis D; Rosair GM; Welch AJ Dalton Trans. 2016, 45, 3635. [DOI] [PubMed] [Google Scholar]
- 9.(a) Zakharkin LI, Kovredov AI, Ol’Shevskaya VA, Shaugumbekova Zh. S., Izv. Akad. Nauk SSSR, Ser. Khim 1980, 7. [Google Scholar]; (b) Zakharkin LI; Kovredov AI; Ol’shevskaya VA; Shaugumbekova ZS J. Organomet. Chem 1982, 226 (3), 217 [Google Scholar]; (c) Li J; Logan CF; Jones M Inorg. Chem 1991, 30 (25), 4866 [Google Scholar]; (d) Zheng Z; Jiang W; Zinn AA; Knobler CB; Hawthorne MF Inorg. Chem 1995, 34 (8), 2095 [Google Scholar]; (e) Sevryugina Y; Julius RL; Hawthorne MF Inorg. Chem 2010, 49 (22), 10627. [DOI] [PubMed] [Google Scholar]; (f) Dziedzic RM; Saleh LMA; Axtell JC; Martin JL; Stevens SL; Royappa AT; Rheingold AL; Spokoyny AM J. Am. Chem. Soc 2016, 138 (29), 9081. [DOI] [PubMed] [Google Scholar]; (g) Dziedzic RM; Martin JL; Axtell JC; Saleh LMA; Ong T-C; Yang Y-F; Messina MS; Rheingold AL; Houk KN; Spokoyny AM J. Am. Chem. Soc 2017, 139 (23), 7729. [DOI] [PubMed] [Google Scholar]
- 10.(a) For article on the synthesis of 3rd generation Pd-precatalysts: Bruno NC; Tudge MT; Buchwald SL Chem. Sci 2013, 4 (3), 916. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) For general literature on biaryl phosphine based precatalysts: Biscoe MR; Fors BP; Buchwald SL J. Am. Chem. Soc 2008, 130 (21), 6686. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Düfert MA; Billingsley KL; Buchwald SL J. Am. Chem. Soc 2013, 135 (34), 12877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ohta K; Goto T; Endo Y Tetrahedron Lett. 2005, 46 (3), 483. [Google Scholar]
- 12.(a) Henly TJ; Knobler CB; Hawthorne MF Organometallics 1992, 11, 2313 [Google Scholar]; (b) Islam MJ; Smith MD; Peryshkov DV J. Organomet. Chem 2018, 867, 208. [Google Scholar]
- 13.Kirlikovali KO; Axtell JC; Anderson K; Djurovich PI; Rheingold AL; Spokoyny AM Organometallics 2018, 37 (18), 3122. [Google Scholar]
- 14.(a) Getman TD Inorg. Chem 1998, 37, 3422–3423 [Google Scholar]; (b) Fox MA; MacBride JAH; Wade K Polyhedron 1996, 16 (14), 2499 [Google Scholar]; (c) Yoo J; Hwang J-W; Do Y Inorg. Chem 2001, 40 (3), 568. [DOI] [PubMed] [Google Scholar]
- 15.Bae HJ; Kim H; Lee KM; Kim T; Lee YS; Do Y; Lee MH Dalton Trans. 2014, 43 (13), 4978. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.