

Abstract
Background:
Measurement reliability and biological stability need to be considered when developing sampling protocols for population-based fecal microbiome studies.
Methods:
Stool samples were collected biannually over a two-year period and sequenced for the V1-V3 region of the 16S rRNA gene in 50 participants from the Multiethnic Cohort Study. We evaluated the temporal stability of the fecal microbiome on a community level with permutational multivariate analysis of variance (PERMANOVA), as well as on taxa and diversity measures with intraclass correlation coefficients.
Results:
Inter-individual differences were the predominant source of fecal microbiome variation, and variation within individual was driven more by changing abundances than the complete loss or introduction of taxa. Phyla and diversity measures were reliable over the two years. Most genera were stable over time, although those with low abundances tended to be more dynamic. Reliability was lower among participants who used antibiotics, with the greatest difference seen in samples taken within one month of reported use.
Conclusions:
The fecal microbiome as a whole is stable over a two-year period, although certain taxa may exhibit more temporal variability.
Impact:
When designing large epidemiologic studies, a single sample is sufficient to capture the majority of the variation in the fecal microbiome from 16S rRNA gene sequencing, while multiple samples may be needed for rare or less abundant taxa.
Keywords: fecal microbiome, temporal variability, antibiotics, community stability
Introduction
In the past decade, the gut microbiome has been of great interest in health research, with diseases such as colon cancer (1), inflammatory bowel disease (2), and cardiovascular disease (3) already linked to both community-wide shifts and changes in specific bacterial taxa. Our ability to identify associations between gut microbes and diseases will be greatly improved with the continued establishment of well-powered population-based longitudinal studies coupled with the decreasing costs of DNA sequencing. In order to conduct these large-scale studies, standardized methods that provide reliable estimates need to be implemented. Several studies have already investigated technical sources of variability due to different aspects of sample collection (4,5), processing (6,7), and sequencing (8–10).
Having reliable estimates that sufficiently capture temporal variation of the gut microbiome is also crucial. Microbial communities are complex and constantly changing in response to their environment. Factors such as diet (11–14), use of antibiotics and other medications (15,16), and exposure to pathogens (17) can have a pronounced impact on bacteria residing in the gut and other anatomical sites. In the context of epidemiologic research, a microbiome with dramatic fluctuations over time could require multiple sample collections or increased sample sizes for longitudinal studies. Previous studies have evaluated variation of the fecal microbiome over time, but have involved small numbers of participants (18,19), variable sampling periods (20), or only Caucasian populations (21), which may limit generalizability. Here, we assessed the temporal variability of the fecal microbiome in 50 older adults from a multiethnic population with biannual sampling over a two-year period.
Materials and methods
Study participants
The Multiethnic Cohort study (MEC) is a prospective cohort study conducted in Hawaii and Los Angeles County that was designed to investigate the association of lifestyle and genetic factors with the incidence of cancer and other chronic diseases. The study design, recruitment, and baseline characteristics have been described previously (22). Briefly, 215,251 men and women between the ages of 45-75 from primarily five racial/ethnic groups (African-American, Japanese-American, Latino, Native Hawaiian, and white) were enrolled into the study from 1993-1996 by completing a self-administered 26-page mailed questionnaire. Over 1800 of these participants (aged 60-77) were recruited in 2013-2017 as part of the MEC Adiposity Phenotype Study (APS) to investigate the relationships between the exposome, genome, microbiome, and metabolome with body fat distribution. Exclusion criteria for the MEC-APS included reported BMI outside the range of 18.5-40 kg/m2; oral or injection antibiotic use in the past 3 months; current or recent (<2 years) smoking; flu shot or other vaccinations in the past month; substantial weight change (>20 lbs) in the past 6 months; soft or metal implants; ileostomy or colectomy; dialysis; insulin or thyroid medication; and any of the following procedures or treatments in the past 6 months: chemotherapy, radiation therapy, corticosteroid hormones, prescription weight-loss drugs, endoscopy or irrigation of the large intestine. Percent body fat was measured by whole-body dual-energy X-ray absorptiometry (DXA) scans (23). Fifty individuals were randomly selected from the APS participants to have an equal distribution by sex (25 male and 25 female), the five main ethnic groups within the MEC (10 African American, 10 Japanese American, 10 Native Hawaiian, 10 Latino, and 10 whites), and BMI categories (within each sex-ethnic group, one from each of 22-24.9, 25-26.9, 27-29.9, 30-34.9 kg/m2 and one either from 18.5-21.9 or 35-40 kg/m2) in which to conduct our longitudinal fecal microbiome study. Institutional Review Board approval was obtained from all participating institutions and informed written consent was obtained from the study participants.
Sample collection
Over a two-year period, each participant was asked to collect a stool sample once every six months for a total of five samples. Stool samples were collected at home using a collection tube containing 5 mL RNAlater (Fisher Scientific) and sterile 5 mm glass beads (Ambion) to facilitate sample dispersion in RNAlater. Samples were then frozen overnight and either brought in or mailed to the study clinic the following morning. Collection materials and procedures have been described in detail previously (24). Along with each sample, participants were asked to fill out a stool collection questionnaire that included items on collection time, special diets, and consumption of probiotic foods in the past six months. The questionnaire also asked whether participants were treated with an oral, injection, or IV form of antibiotics in the past six months, and the most recent month antibiotics were taken. If, at baseline, the participants reported to have received antibiotic therapy during the past six months, collection was deferred by six months and the baseline eligibility questionnaire was re-administered.
Sample processing
Stool samples were shipped on dry ice from study centers in Honolulu, HI and Los Angeles, CA to the Fred Hutchinson Cancer Research Center (FHCRC) in Seattle, WA. Stool samples collected in RNAlater were thawed and homogenized at 10,000 RPM on ice for 30 seconds (Omni Tissue Homogenizer, Omni International, Kennesaw, GA). Homogenized sample (300 μL) was transferred into four FastPrep tubes (MP Biomedical, Santa Ana CA) along with 0.3 g zirconium beads (Biospec Products, Bartleville OK) which were previously sterilized in an oven (180°C for >2 hours), and stored at −80 °C. For DNA extraction, two FastPrep tubes from each sample were thawed on ice. Sterile phosphate buffered saline (300 μL) was added to each of the tubes, which were then centrifuged at 14,000 RPM for 10 minutes. The supernatant was removed and discarded. Preheated ASL buffer (50 °C; 700 μL; QIAGEN, Germantown MD) was added to the pellet in each sample tube. FastPrep tubes were placed in a FastPrep bead beater 24-5G (MP Biomedical) at 5.5 m/s for 45 seconds, followed by 95°C (Thermomixer, Eppendorf, Hauppauge NY) for 15 minutes at 15,000 RPM, and centrifuged for 3 minutes at 15,000 RPM. 520 μL of the supernatant was placed in a 1.5 mL tube containing an InhibitEX tablet (QIAGEN). Eppendorf tubes were centrifuged for 3 minutes at 15,000 RPM. The remaining DNA extraction procedures followed the standard QIAcube protocol for human stool (QIAGEN). Final elution of DNA was performed with 200 μL elution buffer (AE buffer; QIAGEN). DNA concentrations and purity were determined using the NanoDrop 8000 Spectrophotometer (ThermoFisher Scientific, Waltham, MA) and gel electrophoresis. Working stocks were diluted in AE buffer (QIAGEN) from genomic DNA and samples were stored at −20°C until shipped for sequencing.
Samples for sequencing were prepared using a working stock at final concentration of 20 ng/μL. Samples from the same participant were processed together in the same batch. FHC samples were used to assess variation in library preparation and sequencing batches. FHC samples were prepared by pooling stool from 6 participants outside the time-series study who had not used antibiotic in the past three months. From each participant, we collected five tubes of stool, with each tube containing 5 mL RNAlater and two scoops of stool that were stored at −80°C. All five tubes from each participant were thawed on ice, briefly homogenized individually, and then all combined into one container. Homogenized stools (400-500 μL) were distributed into multiple aliquots in FastPrep tubes and stored at −80°C. To assess DNA extraction, we used duplicate stool samples from three individuals outside of the time-series study who had not used antibiotics in the past three months. Two stool samples per individual were collected in RNAlater and frozen at −80 °C for one week. Samples were thawed on ice, homogenized and extracted using the protocol outlined above. Intraclass correlation coefficients (ICCs) for extraction duplicates were ≥0.93 for alpha diversity measures, ≥0.99 for the first PCoA axis for unweighted and weighted UniFrac, and ≥0.97 for the four most abundant phyla.
For paired-end sequencing of the V1-V3 region, the 27F mod forward PCR primer sequence was 5’-AGRGTTNGATCMTGGCTYAG-3’. The 519R reverse PCR primer sequence was 5’- GTNTTACNGCGGCKGCTG-3’. A 25-cycle PCR was performed using the HotStarTaq Plus Master Mix Kit (QIAGEN, USA) under the following conditions: 94°C for 3 minutes, followed by 28 cycles of 94°C for 30 seconds, 53°C for 40 seconds, and 72°C for 1 minute, after which a final elongation step at 72°C for 5 minutes was performed. After amplification, PCR products were checked in 2% agarose gel to determine the success of amplification and the relative intensity of bands. Multiple PCR products were pooled together in equal proportions based on their molecular weight and DNA concentrations. Pooled samples were purified using calibrated Ampure XP beads (Beckman Coulter, USA). The pooled and purified PCR products were used to prepare the Illumina DNA library using a ligation process (TruSeq Nano DNA LT, QIAGEN) which included Illumina adapters, pads, linkers and an 8 base pair (bp) barcode index. Sequencing was performed on the MiSeq using MiSeq Reagent Kit v3 following the manufacturer’s guidelines to obtain 2×300 bp paired-end reads (Illumina, San Diego, CA). FastQ files were exported and securely transferred (BaseSpace, Illumina) to FHCRC for bioinformatic analysis.
Microbiome bioinformatic data processing
To classify bacterial taxonomy, sequences were processed using QIIME v.1.8 (25). Sequences were joined with the fastq-join method, using min_overlap=15 and perc_max_diff=12. Sequences were filtered with split_libraries_fastq.py with q parameter set to 25, and defaults otherwise. The Nelson two-step method was used for operational taxonomic unit (OTU) generation at 97% similarity using the SILVA database (release 111, clustered at the 97% similarity level) for closed reference OTU picking following the UCLUST algorithm (26). The OTU table was filtered using the QIIME script filter_otus_from_otu_table.py with --min_count_fraction set to 0.00005 as recommended in Navas-Molina et al (27). Additional OTU entries were filtered out if they were detected as chimeras using QIIME’s identify_chimeric_seqs.py script with the blast_fragments method (28). The sequences were classified using the matching SILVA taxonomy for OTUs found in the first step of the Nelson method, and MOTHUR’s naive Bayesian Classifier (29,30) trained against the SILVA database (release 111, clustered at the 97% similarity level) for OTUs found in the second step. Sequences were aligned to the SILVA 16S rRNA gene reference alignment (31) using the NAST algorithm (32). Sequences that did not align to the appropriate 16S rRNA gene region were removed. The phylogenetic tree was constructed following the FastTree method (33). Sequence counts for each sample ranging from phylum to genus level were generated without rarefaction. Alpha diversity measures [phylogenetic diversity (34), Shannon index (35), Chao1 index (36)] and beta diversity matrices [unweighted and weighted UniFrac (37,38)] were calculated in QIIME based on the average of 10 sub-samples with rarefaction to 10,000 sequences per sample.
Statistical analysis
Differences in fecal microbiota composition were assessed using two phylogenetic beta diversity metrics, unweighted UniFrac and weighted UniFrac. Unweighted UniFrac is a qualitative measure that captures differences in the presence and absence of OTUs, while weighted UniFrac is a quantitative measure that additionally incorporates information on the relative abundance of OTUs (38). Principal coordinate analysis (PCoA) plots using the first two PCoA axes were generated for both unweighted and weighted UniFrac distances using the ‘cmdscale’ function in R. The variation in microbial community structure explained by individual, time point, sample receipt time, and antibiotic use was determined by PERMANOVA (999 permutations) for both unweighted and weighted UniFrac distances using the ‘adonis’ function from the R package ‘vegan’ (39). Due to the prevalence of use and impact of antibiotics, we stratified our analyses based on whether participants reported any antibiotic use during the 2-year study period.
To determine whether samples more closely resembled other samples from the same individual or samples from different individuals, we matched each non-baseline sample with the baseline sample it was most similar to as defined by the shortest distance using unweighted and weighted UniFrac metrics. We determined whether each pair of samples belonged to the same individual and then calculated the proportion of pairs that both belonged to the same person.
Taxon abundances are often normalized by converting raw counts into relative abundances per sample. Although this addresses the issue of varying sequencing depth, the subsequent data are constrained to a simplex due to the unit-sum constraint and, while useful for characterization, may not be appropriate for use with standard statistical approaches. Here, we applied the interquartile log-ratio transformation (IQLR) for all taxa abundances, which allows for analysis of compositional data by calculating log-ratios of abundances and has been shown to be effective in producing approximately multivariate normal data (40,41).
We used ICCs to assess the reliability of several commonly used microbiome measures, including the four most abundant phyla (Firmicutes, Bacteroidetes, Proteobacteria, Actinobacteria), the three alpha diversity measures described above, and three beta diversity measures (first PCoA axis for unweighted UniFrac, weighted UniFrac, Bray-Curtis, and Jaccard). ICCs were calculated by first fitting a linear mixed effects model with a random effect for participant using the ‘lmer’ function in the R package lme4 (42), and then dividing the between-individual variation by the total variation from the model using the ‘icc’ function in the R package sjstats. To assess the reliability of genera, we computed ICCs for abundances as well as presence/absence. ICCs for abundances were calculated using the approach described above. ICCs for presence/absence were calculated by first converting the genus abundance table to a presence/absence table by replacing all counts greater than 0 with 1. Any genus that was present in every sample was excluded as the ICC would be undefined due to no variation. Then, a generalized linear mixed effects model with a binomial distribution and a random effect for participant was fitted using the ‘glmer’ function in lme4 (43). ICCs were then computed using the ‘icc’ function in sjstats. For all ICC measures, reliability was considered excellent for ICC≥0.75, good for 0.74≥ICC≥0.60, fair for 0.59≥ICC≥0.40, and poor for ICC≤0.39 (44).
Next, we assessed whether one sample is sufficient or multiple samples over time are necessary when using the fecal microbiome in an association analysis with a health-related outcome. Since multiple studies have aimed to link the fecal microbiome with obesity (45–47), we selected baseline percent body fat as a benchmark for assessing stability of the association over time. The variation in fecal microbiome composition explained by baseline percent body fat was calculated using PERMANOVA R2 at baseline, as well as with the addition of subsequent samples (e.g. including baseline and the 6-month sample) using unweighted and weighted UniFrac to examine differences in the association when incorporating multiple time points.
We also explored recovery of the fecal microbiome from antibiotics among participants who reported antibiotic use during the two-year study period. Participants were excluded from this analysis if they did not provide the last date of antibiotic use, or had a baseline sample that failed laboratory quality control. We assessed recovery in samples that were taken after the first reported use of antibiotics only (i.e. samples were not included if they were collected after two or more courses of antibiotics), and categorized them based on time between last antibiotic use and date of sample collection (0-1 months, 1-3 months, 3-6 months, 6-12 months, 12-24 months). Percent change in alpha diversity (Shannon index and phylogenetic diversity) was calculated by dividing the difference in diversity between a sample and the baseline sample by the diversity of the baseline sample, and multiplying by 100. We also assessed changes in beta diversity by using unweighted and weighted UniFrac distances between each sample and the baseline sample corresponding to the same individual. Differences between the six-month and baseline samples for those not reporting antibiotic use were also included as a comparison. Changes in alpha diversity for each time interval was assessed using a one-sample t-test for a mean of zero. All analyses were conducted in R version 3.4.3.
Results
Participant characteristics
Participants (n=50) had a mean ± SD age of 68.6 ± 2.7 years, and were equally distributed by sex and the five race-ethnic groups of the MEC (Table 1) in accordance with our recruitment strategy. 23 (46%) participants reported using antibiotics at least once during the study period. Those who took antibiotics were more likely to be male and Latino. Samples were collected 186.8 ± 36.0 (mean ± SD) days apart. Alpha diversity and phyla abundances were comparable between antibiotics use group (Supplemental Table 1). The sequence data is available https://www.ncbi.nlm.nih.gov/sra under the accession number SRP153929.
Table 1.
Characteristics of MEC study participants by any reported antibiotic use.
| No antibiotic use (n=27) | Antibiotic use (n=23) | Total (n=50) | |
|---|---|---|---|
| Age, years | 68.2 ± 2.9 | 69.0 ± 2.3 | 68.6 ± 2.7 |
| Female | 15 (55.6) | 10 (43.5) | 25 (50) |
| Race/ethnicity | |||
| African American | 5 (18.5) | 5 (21.7) | 10 (20) |
| Japanese American | 6 (22.2) | 4 (17.4) | 10 (20) |
| Native Hawaiian | 7 (25.9) | 3 (13.0) | 10 (20) |
| Latino | 3 (11.1) | 7 (30.5) | 10 (20) |
| White | 6 (22.2) | 4 (17.4) | 10 (20) |
| Education, years | 15.0 ± 2.5 | 13.8 ± 3.4 | 14.4 ± 3.0 |
| Smoking status | |||
| Never | 19 (70.4) | 17 (73.9) | 36 (72.0) |
| Former | 8 (29.6) | 6 (26.1) | 14 (28.0) |
| Body fat % | 33.2 ± 6.8 | 32.2 ± 9.2 | 32.8 ± 7.9 |
Mean ± SD for continuous variables and n (%) for categorical variables
Temporal variation of the fecal microbiome
In our samples, we identified 10 phyla, 20 classes, 26 orders, 46 families, 93 genera, and 1220 OTUs. Three genera were present in every sample (Bacteroides and two belonging to Lachnospiraceae). There was 38,768 ± 11,596 (mean ± SD) sequences per sample and an average sequence length of 493 bp. ± 5 bp (mean ± SD). From the PCoA plots based on unweighted UniFrac, samples from the same individual generally clustered together (Figure 1A–C), particularly among those who did not take antibiotics (Figure 1A). There was greater overlap of samples between individuals when using weighted UniFrac (Figure 1D–F). The majority of microbiome variation was due to inter-individual differences (Table 2), accounting for 70% and 78% of the total unweighted UniFrac variation for those on and not on antibiotics, respectively. Inter-individual variation explained slightly less but remained the largest source of variation when using weighted UniFrac, accounting for 66% and 70% of the variation for those on and not on antibiotics, respectively. Variation was explained minimally by sample time point and days to receipt at study center (<1%). Inter-individual differences and antibiotic use were significant sources of microbiome variation while sample time point and days to receipt at study center were not.
Figure 1.
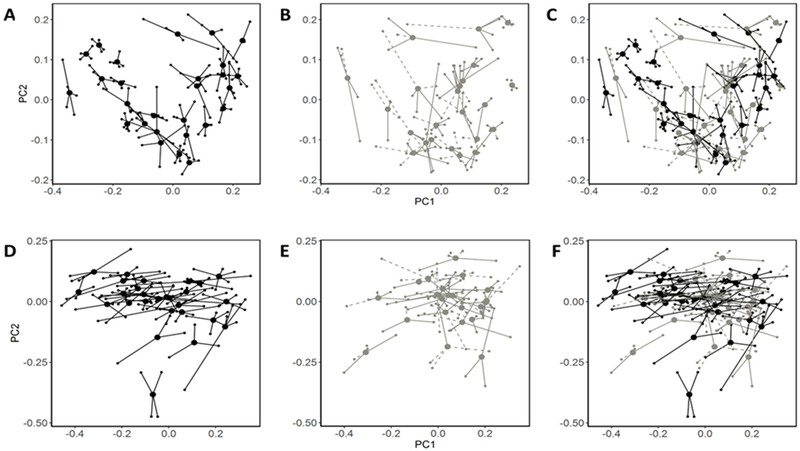
Variability of the gut microbiome. Principal coordinate plots based on unweighted UniFrac for no antibiotic use (A), antibiotic use (B), all participants (C), and weighted UniFrac for no antibiotic use (D), antibiotic use (E), all participants (F). Smaller dots indicate samples and are connected to larger dots which represent the mean PC1 and PC2 values for each individual. Dashed lines are connected to samples with reported antibiotic use.
Table 2.
Microbiome variation explained by inter-individual differences, time point of sample, days to sample receipt at study clinic, and antibiotic use calculated using a distance-based analysis of variance.
| No antibiotic use (n=27) |
Antibiotic use (n=23) |
Total (n=50) |
||||
|---|---|---|---|---|---|---|
| R2 | p | R2 | p | R2 | p | |
| Unweighted UNIFRAC | ||||||
| Individual | 0.777 | <0.001 | 0.696 | <0.001 | 0.743 | <0.001 |
| Time point | 0.006 | 0.797 | 0.008 | 0.643 | 0.004 | 0.424 |
| Days to receipt | 0.007 | 0.640 | 0.012 | 0.180 | 0.005 | 0.178 |
| Antibiotic use | 0.017 | 0.001 | ||||
| Weighted UNIFRAC | ||||||
| Individual | 0.701 | <0.001 | 0.655 | <0.001 | 0.687 | <0.001 |
| Time point | 0.003 | 0.909 | 0.009 | 0.454 | 0.004 | 0.434 |
| Days to receipt | 0.005 | 0.584 | 0.009 | 0.365 | 0.004 | 0.445 |
| Antibiotic use | 0.018 | 0.003 | ||||
To assess whether a single sample was representative of an individual’s microbiome over time, we matched each non-baseline sample to the baseline sample with the shortest UniFrac distance. The majority of non-baseline samples matched to the baseline sample of the same individual when using unweighted UniFrac distances (no antibiotics: 83%; antibiotics: 72%). Fewer samples matched correctly for weighted UniFrac, with about one-third of samples being most similar to the same person’s baseline sample (no antibiotics: 34%; antibiotics: 30%).
Reliability of microbiome measures
We next assessed fecal microbiome variability over time using ICCs of taxa and diversity measures. Among all participants, the four phyla had fair reliability, with ICCs between 0.56-0.59 (Table 3). Differences were seen when stratifying by antibiotic use, as ICCs were consistently higher among those not taking antibiotics compared to those who did. The majority of genera had at least fair reliability. For abundance measures, 79% of genera in the no-antibiotics group and 74% in the antibiotics group had ICC>0.40 (Figure 2A–C). For presence/absence, 86% in the no-antibiotics group and 84% in the antibiotics group had ICC>0.40 (Figure 2D–F). Genera with poor reliability were typically those with low abundance or low prevalence (Supplemental Table 2).
Table 3.
Temporal reliability of microbiome measures. Intraclass correlation coefficients for interquartile log-ratio transformed phyla, alpha diversity, and beta diversity measures.
| No antibiotic use (n=27) | Antibiotic use (n=23) | Total (n=50) | |
|---|---|---|---|
| Phylum | |||
| Firmicutes | 0.64 | 0.46 | 0.57 |
| Bacteroidetes | 0.62 | 0.59 | 0.56 |
| Proteobacteria | 0.65 | 0.44 | 0.56 |
| Actinobacteria | 0.67 | 0.49 | 0.59 |
| Alpha diversity | |||
| Phylogenetic diversity | 0.75 | 0.55 | 0.66 |
| Shannon index | 0.67 | 0.46 | 0.58 |
| Chao1 | 0.56 | 0.45 | 0.52 |
| Beta diversity | |||
| Unweighted UniFrac PC1 | 0.93 | 0.83 | 0.89 |
| Weighted UniFrac PC1 | 0.65 | 0.66 | 0.64 |
| Bray-Curtis PC1 | 0.95 | 0.88 | 0.90 |
| Jaccard PC1 | 0.95 | 0.90 | 0.90 |
Figure 2.
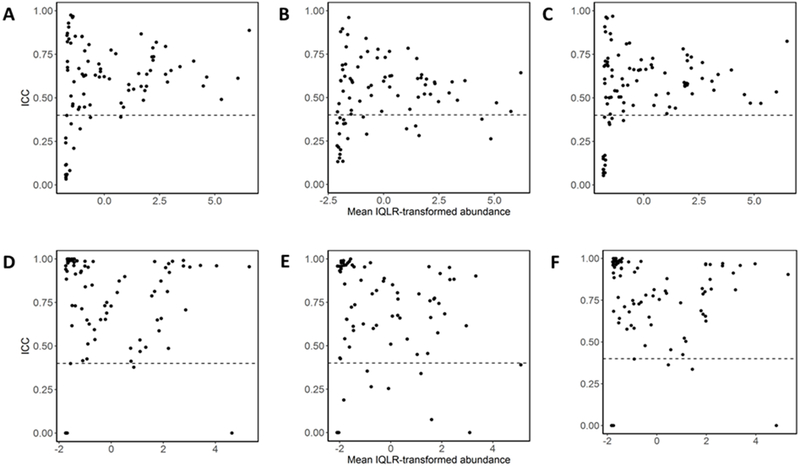
Reliability of genera. Intraclass correlations (ICC) genera were calculated for IQLR-transformed abundances for no antibiotic use (A), antibiotic use (B), all participants (C), and presence/absence for no antibiotic use (D), antibiotic use (E), all participants (F) and are plotted against mean abundance. Dotted line indicates ICC of 0.40.
Alpha diversity measures tended to have better reproducibility than individual phyla measures in the no-antibiotics group. Phylogenetic diversity had the highest reproducibility, followed by Shannon diversity and the Chao1 estimator. The ICCs of all three alpha diversity measures were greater in the no-antibiotics group than in the antibiotics group. We also assessed beta diversity reproducibility, finding unweighted UniFrac PC1, Bray-Curtis PC1, and Jaccard PC1 to have excellent stability over time regardless of antibiotic use, and weighted UniFrac PC1 to have good stability over time in both groups (Table 3).
Microbiome-body fat associations
To test the effect of variation in the microbiome over time on a relevant health outcome, we modeled the microbiome-body fat association starting with the baseline sample, followed by the addition of subsequent samples. Percent body fat had a wider range of values in the antibiotics group (11.9%-46.8%) than in the no-antibiotics group (21.4%-50.3%). The variation of the fecal microbiome explained by percent body fat did not fluctuate with the addition of subsequent samples, and remained relatively stable (Supplemental Table 3) whether using all participants (0.029-0.034), only participants not on antibiotics (0.035-0.056), or only participants on antibiotics (0.056-0.070) for unweighted UniFrac. Weighted UniFrac measures were slightly more variable but still consistent over time.
Recovery from antibiotics use
We also explored microbiome recovery from antibiotics by comparing post-antibiotic use samples to the pre-antibiotic baseline sample among participants who took antibiotics. Although none of the time intervals were significantly different from zero for Shannon index (Figure 3A), changes in the first month were the most variable. The percent change in phylogenetic diversity for samples taken in the first month were significant (Figure 3B). Box plots for beta diversity suggested little difference in unweighted UniFrac distance compared to baseline samples across time intervals, but differences were larger overall among antibiotic users than participants not taking antibiotics (Figure 3C). Recovery over time was more evident for weighted UniFrac, with distances from baseline after the first month post-antibiotics closely resembling those not taking antibiotics (Figure 3D)
Figure 3.
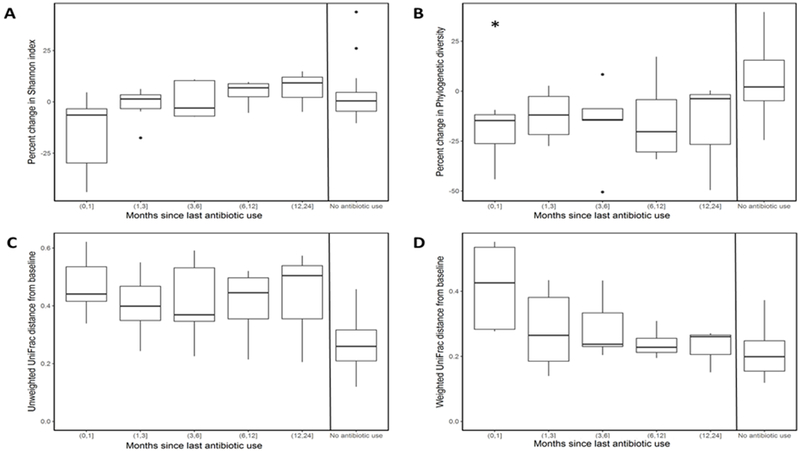
Microbiome recovery from antibiotics. Recovery was assessed among participants reporting use of antibiotics during the two years of collection. Percent change in Shannon index (A) and phylogenetic diversity (B) was calculated relative to the baseline sample and categorized on time since last reported antibiotic use. Changes in alpha diversity for each time interval were assessed using a one-sample t-test for a mean of zero (*p<0.05). Distances from baseline were computed for unweighted UniFrac (C) and weighted UniFrac (D) measures. Comparison of six-month to baseline samples among participants not taking antibiotics are also included.
Discussion
Using several approaches, we showed the fecal microbiome as a whole to be relatively stable over a two-year period. Samples from the same participant clustered together and an association analysis between the overall community structure and baseline body fat showed consistent results throughout the study period. Much of the variation was due to changes in taxa abundances rather than the complete loss or gain of taxa. Although reliability among participants who reported antibiotic use tended to be lower than among those who did not, the largest differences appeared to be among samples taken within a month of antibiotic use.
As with previous studies (18,19,21), inter-individual differences were the dominant source of variation, as evident in the wide phylum distribution of our samples, with relative abundances ranging from 15.8%-89.6% for Firmicutes and 6.7%-67.0% for Bacteroidetes. When matching non-baseline samples to the most similar baseline sample, we found that the majority matched to the same individual when using unweighted UniFrac, while fewer matched using weighted UniFrac, suggesting that changes over time were driven more by changing abundances of taxa rather than their presence or absence. Claesson et al. (21) conducted a similar analysis on stool samples collected 3 months apart. Although they found less discrepancy between weighted and unweighted UniFrac measures compared to our results, there was a similar pattern where fewer samples were matched when using weighted UniFrac.
There is also growing interest in studying the associations of specific taxa with disease, such as Fusobacterium and colon cancer (48,49) or Christensenellaceae and obesity (50). We found phyla measures, as well as the majority of genera, to be reliable. Temporal variation was more of a concern for genera with very low abundances or prevalence, some of which could be taxa that are transient and not representative of an individual’s microbiome over time or those that are near the detection limit and are thus not able to be consistently identified. Larger sample sizes may be necessary if these are of particular interest to a study. Less abundant taxa exhibited lower reproducibility in other methodologic studies as well (5).
In a set of exploratory analyses, we were able to assess the ability of the fecal microbiome to recover from antibiotics. Antibiotic use explained a small but significant proportion of the overall fecal microbiome variation. The strongest and most variable effect on diversity generally occurred in the weeks following use, with samples more closely resembling pre-antibiotic levels in the months that followed. Sampling the fecal microbiome one year (51), and even four weeks after antibiotics (52), has shown return in alpha diversity to pre-treatment levels, although recovery varied for different taxa. Similarly, the ELDERMET study reported that alpha diversity among those who reported antibiotic use within the past month was not significantly different from those who did not (53). However, nine genera were found to be different when using 16S rRNA gene sequencing, as were Bifidobacterium levels when measured by culture. With frequent sampling, Dethlefsen et al. was able to show that adults undergoing courses of ciprofloxacin saw decreases in OTU richness, phylogenetic diversity, and Shannon index within 3-4 days of administration (15). Participants began to recover within a week after taking the antibiotic, although the time needed to reach a stable level varied among participants and alterations in the abundances of certain taxa were apparent. While the gut microbial community as a whole may be able to recover from antibiotics, lingering effects on specific taxa highlight the need for the development of antibiotics with more targeted effects as an alternative to those that act on a broad range of bacteria.
Antibiotic use has also been associated with disease risk factors, including body weight, in animal models and epidemiologic studies, (54). We found that the fecal microbiome explained more variation in body fat among individuals who used antibiotics. Assuming that antibiotic use captured in our study reflects use before baseline (when percent body fat was measured), this finding might suggest a greater contribution by the altered microbial community structure to metabolic regulation and energy homeostasis. There is evidence that the type of antibiotic may have a different effect on overweight and obesity as well, since a longitudinal study by Bailey et al. (55) found an association between early-life exposure to broad-spectrum antibiotics with obesity, but not for narrow-spectrum antibiotics.
Our study design had several strengths. Among studies on temporal variation of the fecal microbiome, ours has one of the most ethnically diverse populations to date and one of few using elderly participants. We were able to collect samples on a consistent schedule over a longer period than other population-based studies, which typically collect samples for only a few months. The retention rate over the two years was also very high, with 49 participants sending all five samples and 1 participant only missing one.
A limitation of our study was that we were not able to assess other sources of microbiome variation, such as travel or recent health, as these were not included in the questionnaire that was filled out at each stool collection. Our study also used 16S rRNA gene data. Additional studies measuring temporal variation of other aspects of the gut microbiome, such as the metagenome and metatranscriptome (56), are crucial. Another limitation was that we did not have information on what types of antibiotics were used or reason for use. Antibiotics have varying mechanisms of action that include targeting bacterial cell walls or membranes, protein synthesis, and DNA or RNA synthesis (57). A two-center randomized controlled trial in the United Kingdom and Sweden reported different responses to the Shannon index from four different antibiotics, with effects on the gut microbiome ranging from no difference after one week to sustained reduction at one year (58). As antibiotics are frequently prescribed for treating a variety of infections, as well as for prophylaxis in preventing infections among high-risk patients, the disease state may also modify the effect of antibiotic treatment. However, the temporal trend we saw was comparable to studies conducted in participants who were healthy at the time of antibiotic administration (15,52).
In summary, we showed that a single assessment sufficiently captures the majority of fecal microbiome measurements in a population-based study, but special consideration should be taken with very rare or low abundant taxa. The assessment of methodologic issues, such as our test of the reliability of measurements, is an important step in designing robust, effective population-based studies to evaluate the role of the fecal microbiome in disease risk.
Supplementary Material
Acknowledgements
This work was supported in part by following grants from the National Institutes of Health: P01 CA168530,P30 CA015704, T32 CA009168, T32 CA094880,U01 CA164973.
We thank the Multiethnic Cohort Study participants who generously donated their time and effort to the Adiposity Phenotype Study.
We acknowledge the contribution of the study staff members whose excellent performance made this research possible: the Recruitment and Data Collection Core staff at USC (Adelaida Irimian, Chanthel Figueroa, Brenda Figueroa, Karla Soriano) and UH (Dr. Terrilea Burnett, Naomi Hee, Clara Richards, Cheryl Toyofuku, Hui Chang, Janice Nako-Piburn); the Data Management and Analysis Core staff at USC (Zhihan Huang) and UH (Maj Earle, Joel Julian, Anne Tome); the Project Administrative Core staff at UH (Eugene Okiyama); the Laboratory staff at FHCRC (Orsalem Kahsai, Wendy Thomas, Elizabeth Traylor, and Crystal Voyce); and Bioinformatics staff at FHCRC (Keith Curtis).
Footnotes
Conflict of interest: The following authors have no conflict of interest: B. C. Fu, T. W. Randolph, U. Lim, K. R. Monroe, I. Cheng, L. R. Wilkens, L. Le Marchand, J. W. Lampe, M. A.J. Hullar
References
- 1.Sears CL, Garrett WS. Microbes, microbiota, and colon cancer. Cell host & microbe 2014;15(3):317–28 doi 10.1016/j.chom.2014.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Morgan XC, Tickle TL, Sokol H, Gevers D, Devaney KL, Ward DV, et al. Dysfunction of the intestinal microbiome in inflammatory bowel disease and treatment. Genome biology 2012;13(9):R79 doi 10.1186/gb-2012-13-9-r79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wang Z, Klipfell E, Bennett BJ, Koeth R, Levison BS, Dugar B, et al. Gut flora metabolism of phosphatidylcholine promotes cardiovascular disease. Nature 2011;472(7341):57–63 doi 10.1038/nature09922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sinha R, Chen J, Amir A, Vogtmann E, Shi J, Inman KS, et al. Collecting Fecal Samples for Microbiome Analyses in Epidemiology Studies. Cancer epidemiology, biomarkers & prevention : a publication of the American Association for Cancer Research, cosponsored by the American Society of Preventive Oncology 2016;25(2):407–16 doi 10.1158/1055-9965.epi-15-0951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Flores R, Shi J, Yu G, Ma B, Ravel J, Goedert JJ, et al. Collection media and delayed freezing effects on microbial composition of human stool. Microbiome 2015;3:33 doi 10.1186/s40168-015-0092-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sinha R, Abu-Ali G, Vogtmann E, Fodor AA, Ren B, Amir A, et al. Assessment of variation in microbial community amplicon sequencing by the Microbiome Quality Control (MBQC) project consortium. Nature biotechnology 2017;35(11):1077–86 doi 10.1038/nbt.3981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Li F, Hullar MA, Lampe JW. Optimization of terminal restriction fragment polymorphism (TRFLP) analysis of human gut microbiota. Journal of microbiological methods 2007;68(2):303–11 doi 10.1016/j.mimet.2006.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Luo C, Tsementzi D, Kyrpides N, Read T, Konstantinidis KT. Direct comparisons of Illumina vs. Roche 454 sequencing technologies on the same microbial community DNA sample. PloS one 2012;7(2):e30087 doi 10.1371/journal.pone.0030087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schloss PD, Gevers D, Westcott SL. Reducing the effects of PCR amplification and sequencing artifacts on 16S rRNA-based studies. PloS one 2011;6(12):e27310 doi 10.1371/journal.pone.0027310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bokulich NA, Subramanian S, Faith JJ, Gevers D, Gordon JI, Knight R, et al. Quality-filtering vastly improves diversity estimates from Illumina amplicon sequencing. Nat Methods 2013;10(1):57–9 doi 10.1038/nmeth.2276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wu GD, Chen J, Hoffmann C, Bittinger K, Chen YY, Keilbaugh SA, et al. Linking long-term dietary patterns with gut microbial enterotypes. Science (New York, NY) 2011;334(6052):105–8 doi 10.1126/science.1208344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.David LA, Maurice CF, Carmody RN, Gootenberg DB, Button JE, Wolfe BE, et al. Diet rapidly and reproducibly alters the human gut microbiome. Nature 2014;505(7484):559–63 doi 10.1038/nature12820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Li F, Hullar MA, Schwarz Y, Lampe JW. Human gut bacterial communities are altered by addition of cruciferous vegetables to a controlled fruit- and vegetable-free diet. The Journal of nutrition 2009;139(9):1685–91 doi 10.3945/jn.109.108191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.O’Keefe SJ, Li JV, Lahti L, Ou J, Carbonero F, Mohammed K, et al. Fat, fibre and cancer risk in African Americans and rural Africans. Nature communications 2015;6:6342 doi 10.1038/ncomms7342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dethlefsen L, Relman DA. Incomplete recovery and individualized responses of the human distal gut microbiota to repeated antibiotic perturbation. Proceedings of the National Academy of Sciences of the United States of America 2011;108 Suppl 1:4554–61 doi 10.1073/pnas.1000087107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Falony G, Joossens M, Vieira-Silva S, Wang J, Darzi Y, Faust K, et al. Population-level analysis of gut microbiome variation. Science (New York, NY) 2016;352(6285):560–4 doi 10.1126/science.aad3503. [DOI] [PubMed] [Google Scholar]
- 17.Chang JY, Antonopoulos DA, Kalra A, Tonelli A, Khalife WT, Schmidt TM, et al. Decreased diversity of the fecal Microbiome in recurrent Clostridium difficile-associated diarrhea. The Journal of infectious diseases 2008;197(3):435–8 doi 10.1086/525047. [DOI] [PubMed] [Google Scholar]
- 18.Caporaso JG, Lauber CL, Costello EK, Berg-Lyons D, Gonzalez A, Stombaugh J, et al. Moving pictures of the human microbiome. Genome biology 2011;12(5):R50 doi 10.1186/gb-2011-12-5-r50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Costello EK, Lauber CL, Hamady M, Fierer N, Gordon JI, Knight R. Bacterial community variation in human body habitats across space and time. Science (New York, NY) 2009;326(5960):1694–7 doi 10.1126/science.1177486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Faith JJ, Guruge JL, Charbonneau M, Subramanian S, Seedorf H, Goodman AL, et al. The long-term stability of the human gut microbiota. Science (New York, NY) 2013;341(6141):1237439 doi 10.1126/science.1237439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Claesson MJ, Cusack S, O’Sullivan O, Greene-Diniz R, de Weerd H, Flannery E, et al. Composition, variability, and temporal stability of the intestinal microbiota of the elderly. Proceedings of the National Academy of Sciences of the United States of America 2011;108 Suppl 1:4586–91 doi 10.1073/pnas.1000097107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kolonel LN, Henderson BE, Hankin JH, Nomura AM, Wilkens LR, Pike MC, et al. A multiethnic cohort in Hawaii and Los Angeles: baseline characteristics. American journal of epidemiology 2000;151(4):346–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Maskarinec G, Lim U, Jacobs S, Monroe KR, Ernst T, Buchthal SD, et al. Diet Quality in Midadulthood Predicts Visceral Adiposity and Liver Fatness in Older Ages: The Multiethnic Cohort Study. Obesity (Silver Spring, Md) 2017;25(8):1442–50 doi 10.1002/oby.21868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fu BC, Randolph TW, Lim U, Monroe KR, Cheng I, Wilkens LR, et al. Characterization of the gut microbiome in epidemiologic studies: the multiethnic cohort experience. Annals of epidemiology 2016;26(5):373–9 doi 10.1016/j.annepidem.2016.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK, et al. QIIME allows analysis of high-throughput community sequencing data. Nat Methods 2010;7(5):335–6 doi 10.1038/nmeth.f.303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Edgar RC. Search and clustering orders of magnitude faster than BLAST. Bioinformatics (Oxford, England) 2010;26(19):2460–1 doi 10.1093/bioinformatics/btq461. [DOI] [PubMed] [Google Scholar]
- 27.Navas-Molina JA, Peralta-Sanchez JM, Gonzalez A, McMurdie PJ, Vazquez-Baeza Y, Xu Z, et al. Advancing our understanding of the human microbiome using QIIME. Methods in enzymology 2013;531:371–444 doi 10.1016/b978-0-12-407863-5.00019-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. Journal of molecular biology 1990;215(3):403–10 doi 10.1016/s0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 29.Schloss PD, Westcott SL, Ryabin T, Hall JR, Hartmann M, Hollister EB, et al. Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Applied and environmental microbiology 2009;75(23):7537–41 doi 10.1128/aem.01541-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wang Q, Garrity GM, Tiedje JM, Cole JR. Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Applied and environmental microbiology 2007;73(16):5261–7 doi 10.1128/aem.00062-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pruesse E, Quast C, Knittel K, Fuchs BM, Ludwig W, Peplies J, et al. SILVA: a comprehensive online resource for quality checked and aligned ribosomal RNA sequence data compatible with ARB. Nucleic acids research 2007;35(21):7188–96 doi 10.1093/nar/gkm864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Caporaso JG, Bittinger K, Bushman FD, DeSantis TZ, Andersen GL, Knight R. PyNAST: a flexible tool for aligning sequences to a template alignment. Bioinformatics (Oxford, England) 2010;26(2):266–7 doi 10.1093/bioinformatics/btp636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Price MN, Dehal PS, Arkin AP. FastTree 2--approximately maximum-likelihood trees for large alignments. PloS one 2010;5(3):e9490 doi 10.1371/journal.pone.0009490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Faith DP. Conservation evaluation and phylogenetic diversity. Biological conservation 1992;61(1):1–10. [Google Scholar]
- 35.Shannon CE, Weaver W. The mathematical theory of communication. University of Illinois press; 1998. [Google Scholar]
- 36.Chao A Nonparametric estimation of the number of classes in a population. Scandinavian Journal of statistics 1984:265–70. [Google Scholar]
- 37.Lozupone C, Knight R. UniFrac: a new phylogenetic method for comparing microbial communities. Applied and environmental microbiology 2005;71(12):8228–35 doi 10.1128/aem.71.12.8228-8235.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lozupone CA, Hamady M, Kelley ST, Knight R. Quantitative and qualitative beta diversity measures lead to different insights into factors that structure microbial communities. Applied and environmental microbiology 2007;73(5):1576–85 doi 10.1128/aem.01996-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dixon P VEGAN, a package of R functions for community ecology. Journal of Vegetation Science 2003;14(6):927–30. [Google Scholar]
- 40.Wu JR, Macklaim JM, Genge BL, Gloor GB. Finding the centre: corrections for asymmetry in high-throughput sequencing datasets. ArXiv e-prints. Volume 17042017. [Google Scholar]
- 41.Aitchison J The statistical analysis of compositional data. Journal of the Royal Statistical Society Series B (Methodological) 1982:139–77. [Google Scholar]
- 42.Bates D, Mächler M, Bolker B, Walker S. Fitting Linear Mixed-Effects Models Using lme4. 2015 2015;67(1):48 doi 10.18637/jss.v067.i01. [DOI] [Google Scholar]
- 43.Bates D, Mächler M, Bolker B, Walker S. Fitting linear mixed-effects models using lme4. arXiv preprint arXiv:14065823 2014. [Google Scholar]
- 44.Cicchetti DV. Guidelines, criteria, and rules of thumb for evaluating normed and standardized assessment instruments in psychology. Psychological assessment 1994;6(4):284. [Google Scholar]
- 45.Sze MA, Schloss PD. Looking for a Signal in the Noise: Revisiting Obesity and the Microbiome. mBio 2016;7(4) doi 10.1128/mBio.01018-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ley RE. Obesity and the human microbiome. Current opinion in gastroenterology 2010;26(1):5–11 doi 10.1097/MOG.0b013e328333d751. [DOI] [PubMed] [Google Scholar]
- 47.Turnbaugh PJ, Hamady M, Yatsunenko T, Cantarel BL, Duncan A, Ley RE, et al. A core gut microbiome in obese and lean twins. Nature 2009;457(7228):480–4 doi 10.1038/nature07540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Castellarin M, Warren RL, Freeman JD, Dreolini L, Krzywinski M, Strauss J, et al. Fusobacterium nucleatum infection is prevalent in human colorectal carcinoma. Genome research 2012;22(2):299–306 doi 10.1101/gr.126516.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kostic AD, Chun E, Robertson L, Glickman JN, Gallini CA, Michaud M, et al. Fusobacterium nucleatum potentiates intestinal tumorigenesis and modulates the tumor-immune microenvironment. Cell host & microbe 2013;14(2):207–15 doi 10.1016/j.chom.2013.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Goodrich JK, Waters JL, Poole AC, Sutter JL, Koren O, Blekhman R, et al. Human genetics shape the gut microbiome. Cell 2014;159(4):789–99 doi 10.1016/j.cell.2014.09.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Jakobsson HE, Jernberg C, Andersson AF, Sjolund-Karlsson M, Jansson JK, Engstrand L. Short-term antibiotic treatment has differing long-term impacts on the human throat and gut microbiome. PloS one 2010;5(3):e9836 doi 10.1371/journal.pone.0009836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Dethlefsen L, Huse S, Sogin ML, Relman DA. The pervasive effects of an antibiotic on the human gut microbiota, as revealed by deep 16S rRNA sequencing. PLoS biology 2008;6(11):e280 doi 10.1371/journal.pbio.0060280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.O’Sullivan O, Coakley M, Lakshminarayanan B, Conde S, Claesson MJ, Cusack S, et al. Alterations in intestinal microbiota of elderly Irish subjects post-antibiotic therapy. The Journal of antimicrobial chemotherapy 2013;68(1):214–21 doi 10.1093/jac/dks348. [DOI] [PubMed] [Google Scholar]
- 54.Cox LM, Blaser MJ. Antibiotics in early life and obesity. Nature reviews Endocrinology 2015;11(3):182–90 doi 10.1038/nrendo.2014.210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bailey LC, Forrest CB, Zhang P, Richards TM, Livshits A, DeRusso PA. Association of antibiotics in infancy with early childhood obesity. JAMA pediatrics 2014;168(11):1063–9 doi 10.1001/jamapediatrics.2014.1539. [DOI] [PubMed] [Google Scholar]
- 56.Mehta RS, Abu-Ali GS, Drew DA, Lloyd-Price J, Subramanian A, Lochhead P, et al. Stability of the human faecal microbiome in a cohort of adult men. Nature microbiology 2018;3(3):347–55 doi 10.1038/s41564-017-0096-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kohanski MA, Dwyer DJ, Collins JJ. How antibiotics kill bacteria: from targets to networks. Nature reviews Microbiology 2010;8(6):423–35 doi 10.1038/nrmicro2333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zaura E, Brandt BW, Teixeira de Mattos MJ, Buijs MJ, Caspers MP, Rashid MU, et al. Same Exposure but Two Radically Different Responses to Antibiotics: Resilience of the Salivary Microbiome versus Long-Term Microbial Shifts in Feces. mBio 2015;6(6):e01693–15 doi 10.1128/mBio.01693-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.