

Abstract
The MST1R (RON) kinase is overexpressed in >80% of human pancreatic cancers, but its role in pancreatic carcinogenesis is unknown. In this study, we examined the relevance of Mst1r kinase to Kras driven pancreatic carcinogenesis using genetically engineered mouse models. In the setting of mutant Kras, Mst1r overexpression increased acinar-ductal metaplasia (ADM), accelerated progression of pancreatic intraepithelial neoplasia (PanIN), and resulted in the accumulation of (mannose receptor C type 1) MRC1+, (arginase 1) Arg+ macrophages in the tumor microenvironment. Conversely, absence of a functional Mst1r kinase slowed PanIN initiation, resulted in smaller tumors, prolonged survival and a reduced tumor associated macrophage content. Mst1r expression was associated with increased production of its ligand Mst1, and in orthotopic models, suppression of Mst1 expression resulted in reduced tumor size, changes in macrophage polarization and enhanced T cell infiltration. This study demonstrates the functional significance of Mst1r during pancreatic cancer initiation and progression. Further, it provides proof of concept that targeting Mst1r can modulate pancreatic cancer growth and the microenvironment. This study provides further rationale for targeting Mst1r as a therapeutic strategy.
Keywords: Mst1r, MST1R, MST1, pancreatic cancer, macrophage
INTRODUCTION
Over the last several years, the genetic alterations that govern pancreatic carcinogenesis have been well-characterized and multiple genetically engineered mouse models of pancreatic cancer have been developed based on known driver mutations1–4. Disappointingly, however, our ability to translate these discoveries into impactful therapies remains slow and yet overall, less emphasis has been placed on downstream changes in gene and protein expression, often because these changes are dynamic, ill-defined, and therefore of unclear significance.
The upregulation of MST1R tyrosine kinase expression is a molecular event of particular interest in many epithelial neoplasms, as it can mediate oncogenic phenotypes and addiction to KRAS signaling5,6. Additionally, MST1R signaling has been shown to promote alternative activation of macrophages, which are potently immunosuppressive. Thus, MST1R on tumor cells and in tumor-associated macrophages is potentially targetable7. However, it remains unclear whether MST1R signaling actually promotes pancreatic cancer progression or if MST1R overexpression merely represents an epiphenomenon.
To address this question, we utilized mouse genetic gain and loss of function approaches. For Mst1r overexpression, we created a transgenic mouse strain to express Mst1r, in a pancreas specific manner while we used a Mst1r knock-in strain containing a targeted deletion of the tyrosine kinase signaling domain to study loss of Mst1r function. These studies reveal that in the setting of oncogenic Kras, Mst1r accelerates pancreatic duct neoplasia and progression to adenocarcinoma, though it is not absolutely required, and that functional Mst1r kinase results in increased tumor macrophage content and changes in polarization. Mst1r expression was associated with increased expression of its cognate ligand, Mst1. Additional studies in an orthotopic model demonstrate that loss of Mst1 expression, also resulted in reduced tumor growth associated with macrophage polarization toward the M1 state, suggesting that targeting Mst1/Mst1r signaling warrants continued investigation.
RESULTS
Mst1r Expression Accelerates Pancreatic Carcinogenesis
To study the effects of Mst1r expression on pancreatic duct carcinogenesis, we developed a transgenic mouse to direct pancreas-specific expression of the wild-type Mst1r protein using a Mst1r murine minigene driven by the Pdx-1 promoter utilized in our Pdx-1-cre strain (Pdx-1-Mst1r) (Supplemental 1A)4. Founder Pdx-1-Mst1r mice demonstrated 2-fold greater expression of Mst1r protein compared to normal pancreas (Supplemental 1B). A cohort of 32 Pdx-1-Mst1r mice were aged to 18 months. Fourteen animals were sacrificed at approximately 10 months secondary to severe dermatitis. Pdx-1 is expressed in terminally differentiated keratinocytes in the spinous layer of the epidermis, suggesting that Mst1r overexpression in the skin of these animals resulted in dysregulated inflammation11. No overt histopathologic changes were evident in the pancreata of these animals. Remaining mice were sacrificed at 18 months of age. While no overt pancreatic neoplasia developed in 17 mice, 1 animal developed a moderately-poorly differentiated primary pancreatic ductal adenocarcinoma with liver metastases (Supplemental 1C–I). Immunofluorescent staining for the epithelial marker CK-19 demonstrated ductal structures in both the primary pancreatic tumor and liver metastases (Supplemental 1H–I).
Since the majority of human pancreatic cancers harbor mutant Kras, we were most interested in studying the effects of Mst1r signaling in this setting. Thus, Pdx-1-Mst1r mice were bred to LSL-KrasG12D mice to yield mice (KR), which were then bred with Pdx-1-Cre to yield the compound strain termed, KRC, that overexpressed Mst1r in the pancreas in the presence of mutant Kras.
Animals were sacrificed at times ranging from 6 weeks to 15 months along with aged-matched controls (KC). Pancreatic tissue sections were analyzed for histology and immunostained to characterize Mst1r expression. Both KC and KRC strains exhibited acinar-ductal metaplasia (ADM) and PanIN formation (Figure 1A, B), though ADM was considerably more prominent in KRC mice (Figure 1C). In wild-type mice, basal Mst1r expression was restricted to islets (Supplemental 2A). In contrast, both KC and KRC strains demonstrated areas of Mst1r expression in ADM and PanIN lesions (Supplemental 2B), which were not present in wild-type pancreata. The presence of Mst1r expression in KC animals is likely secondary to known upregulation of Mst1r downstream of mutant KRAS12. In KRC mice, we observed an additional pattern of Mst1r expression, particularly in large lesions with extensive ADM. These areas of Mst1r expression were marked by concomitant expression of nuclear Pdx-1 consistent with presence of the Mst1r transgene (Figure 1D).
Figure 1. Mst1r Promotes Pancreatic Cancer Initiation and Progression.

(A) Low grade PanIN in a 6m KC mouse (B) Expansive PanIN formation in 6m KRC mouse. (C) Immunofluorescent staining for amylase (red, acini) and CK19 (green, ductal epithelium), confirming ADM within lesions of KRC pancreata (arrows). (D) Representative section from 6m KRC pancreas demonstrating sporadic areas of increased Mst1r expression (red) that correlate with nuclear expression of Pdx-1 (green), indicating the presence of the Pdx-1-Mst1r transgene. (E) Representative sections of KC, KRC and KTK−/−C pancreata demonstrates that KRC develop earlier onset of high grade PanIN2 and 3 compared to KC controls and KTK−/−C pancreata. (F) KRC animals have decreased survival compared with KC controls. Median survival 47 weeks (95% confidence interval 43–51 weeks) KC littermates failed to reach median survival in this cohort, p=0.004. (G) KTK−/−C animals have increased survival compared with KC controls. Median survival 63 weeks in KTK−/−C animals vs. 53 weeks in KC animals p=0.02 (Log-rank (Mantel-Cox) test).
We next compared age-matched KC, and KRC pancreata to determine the consequences gain of Mst1r function. Lesions that developed in KC mice up to 3 months were predominantly low grade PanIN (Figure 1E). In contrast, by 3 months, KRC mice developed large, abundant foci of ADM, many of which appeared to progress directly to high grade PanIN, a phenomenon we rarely observed in KC pancreata (Figure 1E). These lesions demonstrated a 2-fold increase in proliferation as assayed by Ki67 qPCR (Supplemental 2C). At later time points (6–9 months), KRC pancreata consistently contained more advanced PanIN than KC controls (Supplemental 2D, n=6, p <0.05). In fact, 9 month KRC pancreata were nearly replaced with high grade PanIN and invasive cancer, while lesions in KC pancreata remained predominantly low grade (Figure 1E). The survival of KRC mice was markedly reduced as compared to KC animals, in which median survival was not reached (Figure 1F, 95% CI 43–51 weeks). While we did observe liver and lung metastases in KRC mice as early as 3 months, it appeared that local disease progression was the most common cause of early lethality. Taken together, these results demonstrate that Mst1r accelerates both the initiation and progression of Kras-mediated pancreatic duct neoplasia in the mouse.
Loss of Functional Mst1R Kinase Slows Progression to Pancreatic Cancer
After finding that Mst1R kinase overexpression results in acceleration of pancreatic duct neoplasia, we sought to determine if Mst1r was required for progression to pancreatic cancer. Loss of function studies were conducted using mice with a targeted deletion of the tyrosine kinase signaling domain of the receptor, Mst1r TK−/− (referred to as TK−/−), created via a knock-in-out strategy8. Note that while, gain of function studies were conducted on a 129/B6 hybrid background, loss of function studies were conducted on a pure C57/B6 background with paired littermates. TK−/− mice were bred to LSL-KRASG12D and Pdx-1-Cre mice ultimately, yielding KTK−/−C, a strain bearing pancreas specific expression of mutant Kras with a functionally inactive Mst1r kinase. In stark contrast to KC mice, PanIN initiation and progression was delayed in KTK−/−C mice In these mice, sparse PanIN were apparent at 3 months of age, and those that were present were low grade (Figure 1E). In KTK−/−C mice, PanIN began to appear regularly at approximately 6 months, were predominantly low grade and these persisted well beyond 12 months with delayed progression to invasive cancer (Figure 1E). The survival of KTK−/−C mice significantly exceeded that of KC littermate controls (p=0.02) (Figure 1G). While we did observe eventual progression to carcinoma in some animals, total pancreas weight in KTK−/−C was reduced by 40% (Supplemental 2E) and tumor formation in KTK−/−C was much more focal as compared to KC in which multiple areas of the pancreas were involved by invasive carcinoma (Supplemental 2F). Thus, Mst1r function is not required for, but does significantly accelerate progression to pancreatic cancer.
Mst1r Signaling Alters Macrophage Polarization
In normal biology, Mst1r signaling results in attenuation of inflammation, by promoting changes in macrophage polarization from an “M1” and toward an “M2” state, as characterized by increased expression of M2 cell surface markers such as CD163 and MRC113,14. During our examinations of pancreatic histology from the gain and loss of function Mst1r strains, we noted clear differences in the immune cell infiltrates. We therefore characterized the number of F4/80+ macrophages and found a marked increase in KRC pancreata compared with KC at 6 months of age (Figure 2A). In comparing KC to KTK−/−C mice, the difference in macrophage accumulation was apparent even earlier, as macrophages are almost completely absent from KTK−/−C pancreata until 3m of age (Figure 2B). To more rigorously assess macrophage content, we examined macrophage content in the three strains using immunofluorescent double-labelling with F4/80+ and CD68+. This study confirmed the quantity of double labelled cells correlated with increasing expression of functional Mst1r kinase using strain-specific background controls (Supplemental 3A). We next examined the relative abundance of CD68+/CD163+ macrophages over time in the KC, KRC and KTK−/−C strains and noted that double-labelled cells were most abundant in KRC and least abundant in KTK−/−C pancreata, consistent with the relative activity of Mst1r kinase (Figure 2C–D and Supplemental Figure B).
Figure 2. Mst1r Effects Composition of the Immune Cell Infiltrate.

(A) Representative immunofluorescent staining of 6m KC and KRC mice revealing an increased infiltrate of F4/80+ cells (red) in KRC pancreata. (B) Immunohistochemistry from 6 week animals demonstrating the reduced F4/80+ cell infiltrate in KTK−/−C compared to KC pancreata. Quantitation revealed a significant reduction in F4/80 positive cells in KTK−/−C pancreata p=0.01 (C) Fluorescence double labelling for CD68/CD163 demonstrates an increase in KRC versus KC and KTK−/−C pancreata at multiple timepoints. (D) The average number of CD68/CD163 double labelled cells from KC, KRC and KTK−/−C were quantitated revealing an increasing number of double labelled cells with functional Mst1r overexpression at multiple timepoints, and a relation to strain by 2way ANOVA, p<0.0001.
(E) Pancreatic tissues were analyzed with flow cytometry. KRC and KC pancreata demonstrated a comparable infiltrate of CD11b+ Gr1low cells. (F) 12m pancreata from KRC, KC and KTK−/−C mice analyzed by flow cytometry. Note the predominance of CD45+ CD11b+ Gr1low CD11c− MRC1+ macrophages in KRC compared to the KC and KTK−/−C animals. The percentage of CD45+ CD11b+ Gr1low MRC1+ macrophages present in the 12m pancreata from the KRC, KC and
KTK−/−C animals analyzed by flow is represented (One-way ANOVA p=0.003). (G) 9m pancreata were analyzed for expression of Mst1r by qRT-PCR. Pancreata with high levels of Mst1r expression, regardless of strain, showed an increased percentage of CD45+ CD11b+ Gr1low CD11c+ MRC1+ cells compared to pancreatic tissues with low Mst1r expression (p=0.01). Data represents frequencies of gated cell populations, means were compared with non-parametric Kruskal-Wallis Test.
To further characterize the macrophage content, we examined pancreatic and splenic tissues from KC and KRC mice using multi-color flow cytometry15. We observed comparable infiltrates of CD45+CD11b+Gr1lo myeloid-lineage cells in KRC and KC animals, in both pancreatic and splenic cell suspensions (Figure 2E). After identifying the fraction of CD45+CD11b+Gr-1lo infiltrating cells, we then further characterized their expression of CD11c, a marker of inflammatory macrophages, and MRC1, another marker of immunosuppressive, tumor-associated macrophages16,17. Similar to our IHC studies, we observed the fraction of macrophages expressing cell surface markers indicative of an immunosuppressive phenotype (MRC1+) were significantly greater in KRC than in KC pancreata, while the fraction of CD11c+ macrophages was significantly greater in KC than in KRC pancreata (Figure 2F). Given our prior observations that KC pancreata develop increasing Mst1r expression associated with PanIN progression, we determined the relative quantity of Mst1r expression by Q-RT-PCR in order to characterize the relationship of macrophage phenotype to Mst1r expression. This analysis revealed that, independent of strain, high Mst1r expression was associated with a significant increase in the percentage of double positive MRC1+/CD11c+ macrophages as compared with tissues that had low Mst1r expression (p=0.01) (Figure 2G).
To further characterize macrophage phenotypes, we performed immunofluorescent staining of KRC pancreatic tissues using the markers F4/80, MRC1, MHC II, and Arginase I. Staining revealed that F4/80+/MRC1+ macrophages had a distinct morphology. These cells were small, round, and were frequently embedded in dense fibrous stroma surrounding the neoplastic cells (Supplemental Figure 4A). Additionally, these cells expressed Arginase I, a functional marker of the immunosuppressive tumor-promoting macrophages (Supplemental Figure 4B)18. We then stained these tissues for expression of MHCII, a marker of inflammatory macrophages with antigen presentation capacity17. In contrast to MRC1+ macrophages, MHCII+ cells were much larger with vacuolated cytoplasm suggestive of phagocytic activity and antigen-presenting capability (Supplemental Figure 4C). Multiple areas of F4/80+ cells were identified primarily in KRC pancreata, a result consistent with our flow cytometry data that demonstrated an increased number of double positive MRC1+/CD11c+ macrophages in tissues with high levels of Mst1r expression. Additionally, many of these macrophages co-stained with Ki67, a marker of active replication (Supplemental Figure 4D).
KPC mice have been shown to develop splenomegaly secondary to extramedullary hematopoiesis and accumulation of splenic CD11b+Gr1lo myeloid-derived suppressor cells18,19. We next analyzed the spleens of KRC and KC animals and observed a similar pattern of CD11b+Gr1lo expression. However, we did not observe any accumulation of either CD11c+ or MRC1+ macrophages in the spleen of KRC or KC animals, strongly suggesting that the differentiation of these macrophages is a result of soluble factors present within the tumor microenvironment (Supplemental Figure 4E).
Mst1-Mst1r Signaling Attenuates the M1 Macrophage State, But Does Not Independently Drive M2 Differentiation In Vitro
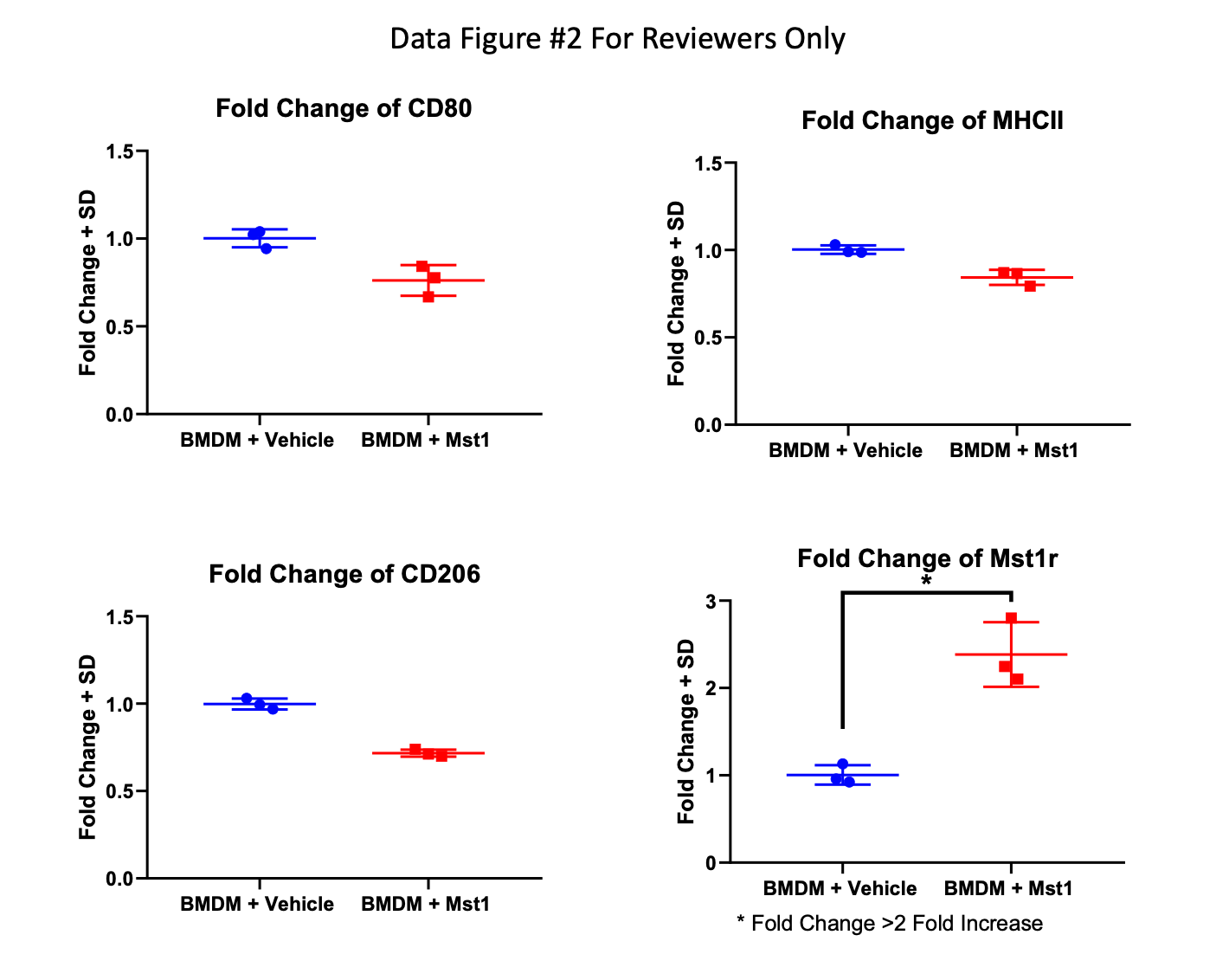
To further investigate if the alterations in macrophage phenotype observed were due to Mst1r signaling, we next examined if ligand-induced Mst1r activation was capable of altering macrophage polarization in vitro. Bone marrow-derived macrophages were treated with either Mst1r ligand, Mst1, or tumor conditioned media from a murine pancreatic cancer cell line, LMP, that expresses Mst1r20. Macrophages were treated with either IL-4 to polarize toward the immunosuppressive “M2” state (measured by expression of TGF-β and Arginase-1) or IFN-γ LPS to induce an inflammatory “M1” state (measured by expression of IFN-γ, IL-12β, and inducible nitric oxide synthase (iNOS)21. Mst1 stimulation inhibited the expression of IFN-γ IL-12 and iNOS but did not induce expression of Arginase I or TGF-β (Figure 3A, B). Thus, in this experimental setting, exposure to Mst1 strongly attenuated inflammatory M1 polarization, but did not fully induce an immunosuppressive M2 phenotype. LMP conditioned media, however, was capable of inducing M2 differentiation (Figure 3B), suggesting that signals in addition to Mst1r activation are required to fully drive polarization toward the M2 phenotype.
Figure 3. Mst1r-Mst1r Signaling Attenuates M1 Differentiation, but does not Independently Drive M2 Differentiation In Vitro.

Bone marrow derived myeloid cells were isolated and treated with either IL-4 or IFN-g to induce M1 or M2 macrophage polarization, respectively. Cells were treated with either Mst1, LMP conditioned media, or a combination of both Mst1 and LMP conditioned media. Cytokine expression was assayed by qRT-PCR (A) Mst1 attenuated M1 macrophage polarization, shown by decreased INFγ, IL-12b and iNOS. (B) Mst1 did not independently induce M2 polarization, as it failed to induce expression of Arginase-I or TGFβ. LMP-conditioned media was capable of inducing M2 macrophage differentiation. Data reflect mean fold change +/− SEM.
Mst1r Activates an Autocrine/Paracrine Positive Feedback Loop
We next sought to determine how Mst1r signaling in the epithelial tumor cell was effecting changes in macrophage state in the pancreatic tumor microenvironment. Given the effects of Mst1 on macrophage polarization, we hypothesized that Mst1r overexpression may in fact result in upregulation of Mst1 expression and secretion. To explore this, we derived multiple epithelial cell lines from tumors arising in KC and KRC pancreata and characterized their Mst1r and Mst1 expression by Q-RT-PCR. These studies revealed that Mst1r expressing lines demonstrated 10- to 60-fold increases in Mst1 expression as compared to Mst1r negative lines (Figure 4A). Additionally, we used a luciferase reporter assay to interrogate whether the Mst1r promoter was activated in response to Mst1 stimulation in multiple murine pancreatic cancer cell lines (PanIN 4313 and LMP). We found a significant increase in Mst1r transcription when stimulated with Mst1, with a positive regulatory response element located between −335 and −485 bp within the Mst1r promoter (Figure 4B, C). We similarly observed an increase in Mst1r transcript after bone marrow derived macrophages were exposed to Mst1 (Figure 4D). Immunostaining for Mst1 in pancreatic tissues confirmed Mst1 expression in epithelial cells with increased frequency/intensity in KRC vs. age-matched KC mice (Figure 4E). Finally, we screened tissue lysates for Mst1 protein levels and found significantly higher levels of Mst1 during disease progression (Supplemental 5A,B). Together, these data suggest that increased Mst1r expression results in increased expression of Mst1 by both pancreatic cancer cells and macrophages.
Figure 4. Mst1r Signaling Upregulates Mst1 Expression.

(A) KRC and CK epithelial cell lines derived from KC and KRC pancreata were screened for their relative expression of Mst1r and Mst1 by qRT-PCR. High levels of Mst1r expression correlated with similarly high levels of Mst1 expression in 3 out of 4 KRC epithelial cell lines. (B&C) Luciferase reporter assay results from murine pancreatic cell lines PanIN 4313 and 4964 LM. Cells were stimulated with 250ng/ml of Mst1 for 24–48h. A regulatory response element was located between −335bp and −485bp within the Mst1r promoter (p<0.0001, p =0.002). (D) Bone marrow-derived macrophages were exposed to 100 ng/mL Mst1 or vehicle. Q-RT-PCR demonstrated a significant increase in Mst1r transcript after Mst1 (p< 0.05). (E) Immunostaining in 9m pancreata demonstrate increased Mst1 expression in high grade PanIN lesions in KRC versus KC mice.
Mst1/Mst1r Knockdown Significantly Inhibits Tumor Growth In Vivo
To further test the hypothesis that Mst1r signaling from the epithelial tumor cell influences the tumor microenvironment, we utilized lentiviral shRNA to achieve knockdown of Mst1 expression in murine LMP cells (Figure 5A). The LMP pancreatic cancer cell line, derived from liver metastases of KPC mice, is highly tumorigenic when xenografted into the pancreata of syngeneic B6/129 F1 hybrid mice20. While Mst1 deficient cells were able to proliferate in vitro, they did proliferate more slowly than Mst1 intact cells (Supplemental 5C). Both lines were able to form tumors after orthotopic injection, however there was a profound reduction in tumor size from Mst1 deficient versus Mst1 intact lines (Figure 5B). We next performed flow cytometry on these tumors to determine the ratio of F4/80+/CD11c+ to F4/80+/MRC1+ expressing macrophages. Notably, the number of MRC1+ macrophages was markedly reduced in Mst1 deficient LMP tumors (p=0.03), suggesting that Mst1 secreted into the tumor microenvironment modulates macrophage polarization (Figure 5C). In orthotopic tumors, immunostaining for CD3+ T cells revealed marked infiltration throughout Mst1 deficient tumors, as compared to controls in which T cells were observed nearly exclusively at the periphery (Figure 5D). When we repeated this experiment in immunodeficient NSG mice, Mst-1 deficient tumors grew faster causing earlier lethality (data not shown), suggesting that an adaptive immune response was at least in part, responsible for growth suppression in the absence of Mst1-Mst1r signaling. Together, with our prior findings, these results suggest that Mst1r expression by pancreatic cancer cells results in secreted Mst1 protein that is then available to initiate autocrine and paracrine signaling within the tumor microenvironment (Figure 6A).
Figure 5. Mst1/Mst1r Regulates Orthotopic Pancreatic Cancer Growth.

(A) shRNA knockdown resulted in ~90% reduction in Mst1 expression in LMP cells compared to control. B) Mst1 KD tumors weighed significantly less than control tumors (p<0.001). (C) Flow cytometry was performed on single cell suspensions derived from LMP control or Mst1 knockdown tumors. Flow sorting demonstrated a reduction in MRC1+/CD11b+Grl− cells derived from Mst1 knockdown tumors, p=0.03. (D) Immunostaining for CD3 in LMP and LMP Mst1 KD tumors reveal an increase in CD3+ cells in the LMP Mst1 KD compared to the control LMP-derived tumors. (E) Multiplex ELISA assay of conditioned media from LMP control versus LMP Mst1 knockdown cell lines revealed an 8 fold reduction in Ccl2. (F) Q-RT-PCR for Ccl2 in 3 and 6 month old KRC and KTK−/−C pancreata demonstrated greater than 2 fold reduction in expression in the absence of functional Mst1r kinase.
Figure 6. Mst1r Activates a Feed-Forward Autocrine-Paracrine Signaling Loop that Effects Tumor Growth and Survival.

(A) Model suggesting that Mst1r overexpression results in increased Mst1 and increased Ccl2 within the tumor microenvironment, contributing to the attenuation of M1 differentiation and skewing macrophages towards an alternative tumor-promoting M2 state. (B&C) Analysis of the TCGA’s pancreatic cancer cohort revealed the association between high MST1R expression and both poor overall (p = 0.0009) and disease-free survival (p < 0.0001).
Mst1r Expression Correlates with Increased Ccl2 Secretion by Pancreatic Cancer Cells
Finally, we took advantage of the LMP Mst1/Mst1r knockdown cell lines to try and identify chemokines which may mediate the Mst1r related accumulation of macrophages in the pancreatic tumor microenvironment. We performed an ELISA to measure cytokine/chemokine levels on conditioned media from LMP control and knockdown cells. This revealed that an 8 fold reduction in the level of Ccl2 in the absence of Mst1/Mst1r expression (Figure 5E). To further evaluate this finding, we assayed for mRNA levels of Ccl2 in 3 month old pancreata from KRC and KTK−/−C mice. This confirmed a greater than 2 fold reduction in Ccl2 mRNA in KTK−/−C versus KRC mice (Figure 5F). As Ccl2 has been shown to mediate localization of bone marrow derived monocytes to the pancreatic tumor microenvironment, these findings suggest Mst1r mediated secretion of Ccl2 is at least partially responsible for our observations that macrophage content correlated with the level of functional Mst1r signaling22,23.
MST1R Expression Is Associated With Poor Outcomes in Human Pancreatic Cancer
To determine whether our findings regarding the relevance of Mst1r signaling in the mouse were relevant to human disease, we examined gene expression data from two independent cohorts. We first analyzed a publicly available dataset containing a gene expression profiling of 125 human primary pancreatic cancers and 46 normal pancreas (GSE71729). We confirmed MST1R overexpression in cancers compared with normal pancreas (data not shown). Interestingly we found that patients with a high expression of MST1R had a significantly worse overall survival (p = 0.0285). Analysis of the TCGA’s pancreatic cancer cohort confirmed the association between high MST1R expression and poor overall survival (p = 0.0009) and also revealed that high expression of MST1R was associated with poorer disease-free survival (p < 0.0001) (Figures 6B,C).
DISCUSSION
The MST1R kinase is overexpressed in human pancreatic cancer yet its contribution to pancreatic carcinogenesis remains unknown. In this study, we examined the consequences of gain and loss of Mst1r function in genetically engineered mouse models of pancreatic carcinogenesis initiated by mutant KRAS, widely accepted as a relevant model of human disease. Mst1r gain and loss of function each modulated pancreatic carcinogenesis, resulting in significant changes in survival. Mst1r associated carcinogenesis was marked by: 1) earlier onset of ADM that appears to progress rapidly to high-grade PanIN, 2) More rapid progression to invasive cancer, and 3) modulation of the immune microenvironment toward a tumor permissive phenotype.
Given the predominantly embryologic expression of the Pdx-1 promoter, the initial degree of Mst1r overexpression in the KRC compound strain is modest. As expected, we identified a progressive increase in Mst1r expression in KC mice, consistent with its putative role as a Kras effector6. The increasing expression of Mst1r in Kras mutant mice is important to highlight because they bias the experimental results towards the null hypothesis, when comparing KRC to KC mice, making the differences we observed more noteworthy. It is clear from our studies, however, that while Mst1r promotes disease progression in the setting of oncogenic Kras, it is not absolutely required for pancreatic carcinogenesis. In loss of function studies, we observed an obvious delay in disease progression and a modest, though statistically significant improvement in survival. Concordant with these findings, we observed a smaller tumor burden in the absence of Mst1r kinase activity. Importantly, mining of two independent databases revealed that high levels of Mst1r expression are a poor prognostic factor for survival in pancreatic cancer patients. These findings are in contrast to a prior report that failed to find MST1R prognostic for survival24. This disparity may be at least partially explained by some differences in methodology between the studies. The prior study initially characterized MST1R expression by immunohistochemistry using an antibody that we have found to bind off-target antigens. The IGCC mRNA data from this study, as the authors acknowledged, also had short follow-up at the time of their analysis.
Studies in other tumor types have similarly found that MST1R signaling impacts tumor progression9,25–27. One purported mechanism by which MST1R impacts tumor progression is via its expression on macrophages within the tumor microenvironment. MST1R signaling in peritoneal macrophages has been shown to drive macrophages away from immune-surveillance (the so-called “M1” state) towards an alternative, tumor-promoting “M2” phenotype via a mechanism that involves attenuation of iNOS and increased expression of Arginase-17,8,13,14. We therefore hypothesized that in addition to its effects on the epithelial tumor cell, MST1R signaling may affect pancreatic carcinogenesis indirectly by altering the polarization of infiltrating myeloid cells and/or tissue resident macrophages in the tumor microenvironment towards a tumor-promoting, immunosuppressive phenotype.
In keeping with this, we show that Mst1r overexpression in murine epithelial tumor cells is associated with upregulation of its own ligand, Mst1. These results are consistent with recent studies documenting tumor cell specific MST1 upregulation in both mouse and human prostate cancer28,29. We further found that Mst1 induces activation of the Mst1r promoter. To our knowledge, this reciprocal signaling between Mst1r expression and its own ligand, Mst1, has not been described. This finding that Mst1r signaling enhances Mst1 expression and that conversely, Mst1 knockdown is associated with decreased Mst1r expression, demonstrates the presence of a feed forward signaling loop leading both to an autocrine effect on Mst1r-expressing tumor cells, as well as a paracrine effect on Mst1r expressing macrophages within the tumor microenvironment. In the macrophage, Mst1-Mst1r signaling attenuates the expression of genes associated with tumor surveillance7. Since we observed that Mst1 stimulation is capable of attenuating the M1 phenotype, but not inducing the M2 state, this suggests that additional signals are necessary for this transition. Pancreatic cancer cell conditioned media was capable of inducing the M2 phenotype in vitro, also indicating that there are additional soluble factors secreted by tumor cells that influence macrophage state in the microenvironment. Among these, one report implicated lactate as a mediator of the M1-M2 transition30. The resultant skew towards the M2 phenotype is associated with increased expression of Arginase-I. Interestingly, increased metabolism of L-arginine is associated with the accumulation of ornithine and proline, precursors of collagen synthesis31. Histologically, we observed that most MRC1+ macrophages were embedded in dense stromal compartments characterized by abundant collagen deposition. In contrast, classically activated MHCII+ macrophages were not surrounded by stroma and maintained a large phagocytic phenotype consistent with their role as antigen-presenting cells. These results raise the possibility that the accumulation of alternatively activated macrophages within the tumor may result in increased arginine metabolism and collagen deposition, potentially contributing to the profound desmoplasia observed in the pancreatic cancer microenvironment. Our findings are consistent with those recently reported in a study that dissected the relative contribution of tissue resident versus bone marrow derived macrophages to pancreatic tumor progression. In this study, it was the F4/80+MHClow subset of macrophages which were most often tissue derived, that were associated with tumor progression and fibrosis32. Consistent with this study, we found that F4/80+/MRC1+ macrophages frequently co-express Ki67, and therefore acquire and maintain the ability to proliferate alongside cancer cells. Our studies also suggest that Mst1r signaling in pancreatic cancer cells results in secretion of Ccl2, a ligand for the Ccr2 receptor. Ccl2 has been shown to mediate trafficking of bone marrow derived monocytes to the pancreatic tumor microenvironment and influence tumor progression and prognosis22,23. Our findings are also consistent with the findings of Gardner et al. who noted a correlation of Ccl2 in a liver injury model33.
These results provide evidence of a positive feedback autocrine and paracrine Mst1-Mst1r signaling axis, and thereby suggest that targeting the Mst1-Mst1r axis may not only have a direct effect on the tumor cell but may have the added benefit of therapeutic immunomodulation within the tumor microenvironment. In the rapidly evolving field of cancer immunotherapy, targeting activities of the tumor-associated macrophage has emerged as one central therapeutic arm of investigation. The two primary strategies have been; 1) to modulate the activation state of the macrophage, and 2) exclusion of the macrophage from the tumor microenvironment. Successful modulation of macrophage phenotypes will require a thorough understanding of the signals driving activation states. Our data suggests Mst1r signaling may contribute to this modulation in the pancreatic cancer microenvironment and is therefore worthy of further study as an immunomodulatory strategy in this disease.
MATERIALS and METHODS
Mice
Animal protocols were approved by the UCSD Institutional Animal Care and Use Committee. PDX-1-cre, KrasLSLG12D and the Mst1r kinase deficient strain, (TK−/−) were described previously4,8. PDX-1-Mst1r animals were generated by insertion of a murine Mst1r mini-gene coding for wild type Mst1r protein under the control of the PDX-1 promoter9. PDX-1-Mst1r mice were screened for their pancreatic expression of Mst1r by western blot (Supplemental 1A,B). Mice with 2-fold increase in Mst1r were propagated forward. All strains were C57BL/6J except for the KrasLSLG12D, (B6/129S4). All experiments used littermate control mice and equal numbers of each gender. C57BL/6J/129S4 F1 hybrid mice were used for orthotopic implantation experiments. For animal studies group sizes were calculated by power analyses based on preliminary experiments using a significance level of 5% and a power of 80%. Animals were randomly assigned to experimental groups. The investigators were not blinded to allocation during experiments and outcome assessments. For genetically engineered mouse studies, equal numbers of male and female animals were used. Orthotopic experiments used male mice, aged 6–8 weeks.
Generating Epithelial Cell Lines
Pancreatic tissues were harvested from KrasCre (KC) and KrasRonCre (KRC) mice. Tissue fragments were embedded in Matrigel coated dishes and incubated at 37°C. Epithelial cells were isolated and propagated by clonal expansion. Mst1r knockdown and control lines were generated using shRNAs from Open Biosystems (Thermo Scientific) for mouse Mst1r, mouse Mst1 or scrambled control in the lenti pLKO.1 vector (Supplemental Table 1). Clones were isolated and analyzed for knockdown by Q-RT-PCR (Supplemental Table 2) and western blot (Anti-Mst1 Abcam, 1:3000 dilution).
Single Cell Suspensions
After sacrifice, intra-cardiac perfusion was performed with cold PBS. The body and tail of the pancreas were uniformly used for flow cytometry. The pancreatic head was placed in 10% saline-buffered formalin and paraffin embedded for histology. The body and tail were minced into 1–2mm pieces in dissociation buffer. Single cell suspensions were incubated at 37oC for 15 min and then passed through a 70uM filter into separation buffer containing EDTA. Cells were spun, counted and viability determined on a coulter counter.
Histology
Tissue sections were fixed overnight in 10% NBF and transferred to 70% ethanol. Paraffin embedding and cutting of tissue sections was performed by the UCSD Histology Core. Slides were baked overnight at 60°C and then either hematoxylin and eosin stained or processed for immunohistochemistry/immunofluorescence. Slides were cover-slipped with Permount (Fisher Scientific) and stored at room temperature. Antibodies/dilutions for are detailed in Supplemental Table 3.
Flow Cytometry
Cell pellets were re-suspended in 0.5% BSA in PBS. 1×106 cells were stained with fluorochrome conjugated antibodies at 4°C for 30 minutes. Cells were rinsed with 2mL cold PBS and spun at 1500 rpm to pellet. Supernatant was removed and the cells were re-suspended in 200uL of stabilizing fixative. Aqua Live/Dead Stain (Invitrogen) was used to exclude dead cells from the analysis. 2uL of BD Fc Block per tube was added and incubated at room temperature for 5 minutes prior to adding antibodies. Cells were analyzed on a Canto RUO Analyzer or FACS Aria Sorter equipped with 488 Argon, 633 Hene, and 405 Violet Lasers. Analyses were performed using FlowJo software in consultation with the UCSD/VA Research Flow Cytometry Core.
Bone marrow derived macrophage experiments
Primary murine macrophages were generated by flushing the bone marrow from the femur and tibia of C57BL/6 mice, followed by incubation for 5 days in DMEM containing 10% FBS and 10 ng/mL murine M-CSF (Peprotech). BMMs were subsequently cultured in DMEM containing 2% serum in the presence or absence of murine recombinant M-CSF (Peprotech) for 24 hours. Macrophages were then stimulated for 24h with relevant ligands and then cells were lysed and RNA prepared using the RNAeasy Kit (Qiagen).
Quantitative RT-PCR (Q-RT-PCR)
RNA was extracted from pancreata using Trizol (Invitrogen) and quality checked by gel electrophoresis. Cell lines were plated to achieve 90% confluency. Treatment groups included DMEM Complete alone or DMEM Complete with 100ng/ml recombinant murine Mst1 (R&D 6244-MS). Cells were incubated for the indicated times or overnight, after which time RNA was isolated using Trizol, then treated with DNAase1 (Invitrogen).
cDNA was synthesized from 1 μg of RNA using iScript cDNA Synthesis Kit (BioRad). Q-RT-PCR was performed using SoFast EvagGreen Supermix (BioRad) with cDNA corresponding to 50ng of total RNA and 500nM primers in a 20μl volume. Primers (sequences detailed in Supplemental Table 2) were examined by gel electrophoresis then sequenced. Relative fold change was calculated using the 2−ΔΔCt method with actin used to normalize. Either an untreated cell line, normal pancreas or KCE 329 cell line was used as the calibrator to generate the fold change10. Significance was set at 1.5-fold above or below the reference sample.
Orthotopic Experiments
6–8 month old F1 Hybrid male mice (B6129SF1/J mice, Jackson Laboratories, ME) were injected with 700,000 LMP cells into the pancreas using a 29G 1/2cc syringe. Tumors originating from the injection were allowed to grow for 3.5 weeks. Mice were then euthanized with the tumor removed and either fixed in 10% NBF, homogenized in Trizol (Life Technologies, CA) for RNA extraction or flash frozen in liquid Nitrogen and stored at −80oC for future processing.
Gene expression analyses
The GSE71729 dataset was downloaded from the Gene Expression Omnibus (GEO; http://www.ncbi.nlm.nih.gov/gds). The dataset corresponds to an Agilent human whole-genome 4×44K microarray (Agilent Technologies) and contains gene expression profiling for 46 normal pancreas specimens and 125 pancreatic cancers with matching clinical information. TCGA RNAseqV2 normalized expression values have been downloaded from the Genetic Data Commons Data Portal (https://portal.gdc.cancer.gov). 99 samples from the TCGA cohort with Mst1r expression values and clinical annotation were utilized for survival analysis. Selected samples show consistent OS time and status regarding clinical annotations retrieved from TCGA and Cbioportal websites (difference in median OS time <0.2 month). Survival curves and associated statistics were generated using GraphPad Prism. Patient stratification corresponding to high and low expression was performed using Cutoff Finder (http://molpath.charite.de/cutoff/). P-values refer to log-rank tests for survival curves and to unpaired t-test with Welch’s correction for boxplot.
Statistics
Data was analyzed using Student’s t test and ANOVA where appropriate. Survival analyses were performed using the Kaplan-Meier method. Flow cytometry data was analyzed using the nonparametric Kruskal-Wallis test. The Spearman’s rho coefficient was calculated for all correlations. All statistical tests were 2-sided, and p-values are reported where appropriate. Microsoft Excel, GraphPad Prism 5, and SPSS Statistical software were used for the analyses.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgments
Support: This work was supported by NIH CA155620 (AML), NIH T32CA121938 (MLB), Ride the Point (AML) and generous gifts to the Sally Ride Fund for Pancreatic Cancer Research (AML) and the Cure Pancreatic Cancer Fund (AML).
Abbreviations used in this manuscript
- MST1R
macrophage stimulating type 1 receptor
- MST1
macrophage stimulating type 1
- ADM
acinar-duct metaplasia
- Ccl2
C-C motif chemokine ligand 2
- PanIN
pancreatic intraepithelial neoplasia
- TCGA
the cancer genome atlas
- HGFL
hepatocyte growth factor-like protein
- GM-CSF
Granulocyte Macrophage Colony Stimulating Factor
- IL
interleukin
- iNOS
inducible nitric oxide synthase
Footnotes
Disclosures: The authors have no relevant disclosures or conflicts of interest.
REFERENCES
- 1.Ko AH. Progress in the treatment of metastatic pancreatic cancer and the search for next opportunities. J Clin Oncol 2015; 33:1779–86. [DOI] [PubMed] [Google Scholar]
- 2.Aguirre AJ, Bardeesy N, Sinha M, Lopez L, Tuveson DA, Horner J, et al. Activated Kras and Ink4a/Arf deficiency cooperate to produce metastatic pancreatic ductal adenocarcinoma. Genes Dev 2003;17:3112–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hingorani SR, Petricoin EF, Maitra A, Rajapakse V, King C, Jacobetz MA, et al. Preinvasive and invasive ductal pancreatic cancer and its early detection in the mouse. Cancer Cell 2003;4:437–50. [DOI] [PubMed] [Google Scholar]
- 4.Hingorani SR, Wang L, Multani AS, Combs C, Deramaudt TB, Hruban RH, et al. Trp53R172H and KrasG12D cooperate to promote chromosomal instability and widely metastatic pancreatic ductal adenocarcinoma in mice. Cancer Cell 2005;7:469–83. [DOI] [PubMed] [Google Scholar]
- 5.Logan-Collins J, Thomas RM, Yu P, Jaquish D, Mose E, French R, et al. Silencing of Mst1r receptor signaling promotes apoptosis and gemcitabine sensitivity in pancreatic cancers. Cancer Res 2010;70:1130–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Singh A, Greninger P, Rhodes D, Koopman L, Violette S, Bardeesy N, et al. A gene expression signature associated with “K-Ras addiction” reveals regulators of EMT and tumor cell survival. Cancer Cell 2009;15:489–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sharda DR, Yu S, Ray M, Squadrito ML, De Palma M, Wynn TA, et al. Regulation of macrophage arginase expression and tumor growth by the Ron receptor tyrosine kinase. J Immunol 2011;187:2181–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Waltz SE, Eaton L, Toney-Earley K, Hess KA, Peace BE, Ihlendorf JR, et al. Ron-mediated cytoplasmic signaling is dispensable for viability but is required to limit inflammatory responses. J. Clin. Invest J Clin Invest. 2001;108:567–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zinser GM, Leonis MA, Toney K, Pathrose P, Thobe M, Kader SA, et al. Mammary-specific Ron receptor overexpression induces highly metastatic mammary tumors associated with beta-catenin activation. Cancer Res 2006;66:11967–74. [DOI] [PubMed] [Google Scholar]
- 10.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001;25:402–8. [DOI] [PubMed] [Google Scholar]
- 11.Mazur PK, Gruner BM, Nakhai H, Sipos B, Zimber-Strobl U, Strobl LJ, et al. Identification of epidermal Pdx1 expression discloses different roles of Notch1 and Notch2 in murine Kras(G12D)-induced skin carcinogenesis in vivo. PLoS One 2010;5:e13578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Thomas RM, Toney K, Fenoglio-Preiser C, Revelo-Penafiel MP, Hingorani SR, Tuveson DA, et al. The Mst1r receptor tyrosine kinase mediates oncogenic phenotypes in pancreatic cancer cells and is increasingly expressed during pancreatic cancer progression. Cancer Res 2007;67:6075–82. [DOI] [PubMed] [Google Scholar]
- 13.Correll PH, Morrison AC, Lutz MA. Receptor tyrosine kinases and the regulation of macrophage activation. J Leukoc Biol 2004;75:731–7. [DOI] [PubMed] [Google Scholar]
- 14.Liu QP, Fuit K, Ward J, Correll PH. Negative regulation of macrophage activation in response to IFN-gamma and lipopolysaccharide by the STK/RON receptor tyrosine kinase. J Immunology 1999:163:6606–13. [PubMed] [Google Scholar]
- 15.Baumgarth N, Roederer M. A practical approach to multicolor flow cytometry for immunophenotyping. J Immunol Methods 2000;243:77–97. [DOI] [PubMed] [Google Scholar]
- 16.Mosser DM. The many faces of macrophage activation. J Leukoc Biol 2003;73:209–12. [DOI] [PubMed] [Google Scholar]
- 17.Martinez FO, Gordon S, Locati M, Mantovani A. Transcriptional profiling of the human monocyte-to-macrophage differentiation and polarization: new molecules and patterns of gene expression. J Immunol 2006;177:7303–11. [DOI] [PubMed] [Google Scholar]
- 18.Pylayeva-Gupta Y, Lee KE, Hajdu CH, Miller G, Bar-Sagi D. Oncogenic Kras-induced GM-CSF production promotes the development of pancreatic neoplasia. Cancer Cell 2012;21:836–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bayne LJ, Beatty GL, Jhala N, Clark CE, Rhim AD, Stanger BZ, et al. Tumor-derived granulocyte-macrophage colony-stimulating factor regulates myeloid inflammation and T cell immunity in pancreatic cancer. Cancer Cell 2012;21:822–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tseng WW, Winer D, Kenkel JA, Choi O, Shain AH, Pollack JR, et al. Development of an orthotopic model of invasive pancreatic cancer in an immunocompetent murine host. Clin Cancer Res 2010;16:3684–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Schmid MC, Varner JA. Myeloid cells in the tumor microenvironment: modulation of tumor angiogenesis and tumor inflammation. J Oncol 2010;2010:201026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Monti P, Leone BE, Marchesi F, Balzano G, Zerbi A, Scaltrini F, et al. The CC chemokine MCP-1/CCL2 in pancreatic cancer progression: regulation of expression and potential mechanisms of antimalignant activity. Cancer Res 2003;63:7451–61. [PubMed] [Google Scholar]
- 23.Sanford DE, Belt BA, Panni RZ, Mayer A, Deshpande AD, Carpenter D, et al. Inflammatory monocyte mobilization decreases patient survival in pancreatic cancer: a role for targeting the CCL2/CCR2 axis. Clin Cancer Res 2013;19:3404–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tactacan CM, Chang DK, Cowley MJ, Humphrey ES, Wu J, Gill AJ, et al. Mst1r is not a prognostic marker for resectable pancreatic cancer. BMC Cancer 2012;12:395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gurusamy D, Gray JK, Pathrose P, Kulkarni RM, Finkleman FD, Waltz SE. Myeloid-specific expression of Ron receptor kinase promotes prostate tumor growth. Cancer Res 2013;73:1752–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Eyob H, Ekiz HA, Welm AL. Mst1r promotes the metastatic spread of breast carcinomas by subverting antitumor immune responses. Oncoimmunology 2013;2:e25670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Thobe MN, Gurusamy D, Pathrose P, Waltz SE. The Ron receptor tyrosine kinase positively regulates angiogenic chemokine production in prostate cancer cells. Oncogene 2010;29:214–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Benight NM, Wagh PK, Zinser GM, Peace BE, Stuart WD, Vasiliauskas J, et al. HGFL supports mammary tumorigenesis by enhancing tumor cell intrinsic survival and influencing macrophage and T-cell responses. Oncotarget 2015;6:17445–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Vasiliauskas J, Nashu MA, Pathrose P, Starnes SL, Waltz SE. Hepatocyte growth factor-like protein is required for prostate tumor growth in the TRAMP mouse model. Oncotarget 2014;5:5547–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Colegio OR, Chu NQ, Szabo AL, Chu T, Rhebergen AM, Jairam V. Functional polarization of tumour-associated macrophages by tumour-derived lactic acid. Nature 2014;513:559–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bronte V, Zanovello P. Regulation of immune responses by L-arginine metabolism. Nat Rev Immunol 2005;5:641–54. [DOI] [PubMed] [Google Scholar]
- 32.Zhu Y, Herndon JM, Sojka DK, Kim KW, Knolhoff BL, Zuo C, et al. Tissue-Resident Macrophages in Pancreatic Ductal Adenocarcinoma Originate from Embryonic Hematopoiesis and Promote Tumor Progression. Immunity 2017;47:323–338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gardner CR, Hankey P, Mishin V, Yu S, Laskin JD, Laskin DL. Regulation of alternative macrophage activation in the liver following acetaminophen intoxication by stem cell-derived tyrosine kinase. Toxicol Appl Pharmacol 2012;262:139–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.