

Abstract
Glucose-6-phosphate dehydrogenase (G6PDH) can initiate the glucose 6-phosphate (G6P) shunt around the Calvin-Benson cycle. In order to understand the regulation of flux through this pathway, we have characterized the biochemical parameters and redox regulation of the three functional plastidic isoforms of Arabidopsis G6PDH. When purified, recombinant proteins were measured, all three exhibited significant substrate inhibition by G6P but not NADP+, making the determination of enzyme kinetic parameters complex. We found that the half saturation concentration of G6PDH isoform 1 is increased under reducing conditions. The other two isoforms exhibit less redox regulation, however, isoform 2 is strongly inhibited by NADPH. Redox regulation of G6PDH1 can be partially reversed by hydrogen peroxide or protected against by presence of its substrate, G6P. Overall, our results support the conclusion that G6PDH can have significant activity throughout the day and can be dynamically regulated to allow or prevent flux through the glucose 6-phosphate shunt.
Keywords: Calvin-Benson cycle, glucose-6-phosphate dehydrogenase, glucose 6-phosphate, glucose 6-phosphate shunt, hydrogen peroxide, phosphoglucoisomerase, redox regulation
Introduction
Glucose 6-phosphate (G6P) is the first product out of the Calvin-Benson cycle in the starch synthesis pathway. However, it can also enter the oxidative pentose phosphate pathway creating a G6P shunt that bypasses the nonoxidative pentose phosphate pathway reactions that make up a significant part of the Calvin-Benson cycle. This pathway is generally considered to occur only in the dark because of the redox regulation of glucose-6-phosphate dehydrogenase (G6PDH) (1–6). In order to estimate flux through this alternative pathway and conditions where it may be important, it is critical to characterize regulation of G6PDH activity in the light.
The substrate of G6PDH, G6P, can be produced or consumed by three other reactions in the plastid: phosphoglucoisomerase (PGI), phosphoglucomutase (PGM), and glucose-6-phosphate/phosphate translocator 2 (GPT2). Phosphoglucoisomerase reversibly isomerizes fructose 6-phosphate (F6P) and G6P. Analysis of mutant lines of Clarkia xantiana indicated that PGI is not in great excess (7). There are two isoforms of PGI in Arabidopsis, one targeted to the plastid and the other found in the cytosol. The plastid PGI in particular is likely limiting given that G6P/F6P ratios in the plastid are significantly displaced from equilibrium and much lower than in the cytosol (8–12). Plants with loss-of-function mutations in the plastidic enzyme have 98.5% less starch in leaves (13). Loss-of-function mutants in the cytosolic enzyme results in increased starch and decreased sucrose (14). Second, PGM is an important reaction in starch synthesis that catalyzes the reversible reaction of G6P to glucose 1-phosphate (G1P). The Km for G6P is 47 μM and 8.5 μM for G1P with a Vmax of 115 and 328 μmol mg−1 min−1, respectively (15, 16). Hanson and McHale (17) showed that PGM had similar activity to PGI in Nicotiana sylvestris. Knockouts of PGM result in starchless plants (17–19). Finally, GPT2 is a glucose-6-phosphate/phosphate antiporter in the chloroplast membrane that is not normally present green tissue (20, 21). This is corroborated by the large concentration gradient in G6P between the chloroplast and cytosol (8–10). However, GPT2 is important in acclimation to light (22), is expressed in plants grown in high CO2 (23), and is increased when starch synthesis is repressed by knocking out starch synthesis genes (Kunz et al. 2010). When GPT2 is present, the gradient of G6P would result in G6P import into the plastid (8–10)
Here we focus on the characterization and biochemical regulation of the plastidic G6PDH isoforms due to its key role in the G6P shunt. There are six isoforms of G6PDH in Arabidopsis. Four of these are predicted to be targeted to the chloroplast and three of these are functional (24, 25). All three plastidic isoforms are expressed in leaf tissue. G6PDH1 and 2 have the highest relative expression (Wakao et al. 2005) It has been hypothesized that during the day, G6PDH initiates a G6P shunt around the Calvin-Benson cycle (26). The G6P shunt oxidizes and decarboxylates G6P to synthesize ribulose 5-phosphate (Ru5P). While the G6P shunt is a futile cycle, it has been proposed to play an important role in stabilization of photosynthesis.
Our goal was to characterize the kinetics and biochemical parameters of oxidized and reduced G6PDH isoforms and the key regulators of G6PDH. Novel findings indicate that G6PDH can remain fairly active during the day. We conclude that a G6P shunt is allowed and even likely in light of the kinetic parameters of G6PDH and that its activity could be modulated during the day to regulate flux through the G6P shunt.
Materials and Methods
Expression and purification of recombinant enzymes
C-terminal Strep-tagged (24, 27) plastidic G6PDH1, 2, and 3 genes were commercially synthesized by GenScript (https://www.genscript.com). All of the plasmid constructs were overexpressed in E. coli strain BL21. Cells were grown at 37ºC to an OD600 of 0.6 to1 and induced with 0.5 mM isopropyl β-D-1 thiogalactopyranoside (IPTG) at room temperature. Cells were grown overnight at room temperature after addition of IPTG. Cells were then centrifuged and resuspended in cold Buffer W (IBA, www.iba-lifesciences.com) with 1 mg ml−1 lysozyme, 1 μg ml−1 of DNAseI, and 1x protease inhibitor cocktail (Sigma, www.sigmaaldrich.com). Cells were then lysed by sonication (Branson Sonifier 250, us.vwr.com). The sonicator was set at 50% duty cycle and an output level of 1. The cells were sonicated using five steps where each step consisted of 15 s pulses and 15 s on ice. The lysate was centrifuged and supernatant collected. Protein was purified on a Strep-Tactin column (IBA) following the manufacturer’s instructions. For all purified proteins, SDS-PAGE was carried out and fractions containing >95% of total protein of interest were combined and concentrated using Amicon Ultra 0.5 ml centrifugal filters (molecular weight cut off 3 kDa). Glycerol was added to the concentrated protein to obtain a final protein solution with 15% glycerol. The glycerol stock of the proteins was aliquoted into small volumes, frozen in liquid nitrogen, and stored at −80˚C. The concentration of the proteins was determined using Pierce 660 nm protein assay reagent kit (ThermoFisher Scientific, www.thermofisher.com) using a bovine serum albumin standard. Final preparations of purified protein were run on an SDS-polyacrylamide gel and stained with Coomassie Blue to check the purity of the enzymes. Molecular weights were estimated from the protein construct using Vector NTI (ThermoFisher Scientific, www.thermofisher.com).
Coupled spectrophotometric assay for G6PDH
The activity of the purified G6PDH1, 2, and 3 was studied using coupled spectrophotometric assays. The concentration of G6P was determined using NADPH-linked assays measured spectrophotometrically. Purity of G6P was verified by untargeted LC-MS/MS. All assays were validated by demonstrating linear product formation, proportional to the time of the assay and amount of enzyme added. The assay was performed in 150 mM Hepes buffer pH 7.2 containing varying concentrations of NADP+, varying concentrations of G6P, and G6PDH. 8.3 ng of G6PDH1, 20 ng of G6PDH2, and 44 ng of G6PDH3 were used.
The concentrations used to study the half saturation concentration of G6PDH for G6P were 0 – 44.2 mM, and the concentrations used to study Km for NADP+ were 0 – 11 μM in a total volume of 800 μL. The concentrations were chosen to result in a number of data points spanning the range of activity up to enzyme saturation based on preliminary assays. When G6PDH was assayed with varied G6P, 0.6 mM NADP+ was added. The chosen concentration of NADP+ was in large excess and the concentration of NADP+ would change very little during the assay at this concentration. When G6PDH was assayed varying NADP+, 7.6 mM G6P was added for G6PDH1 and 3 and 15.4 mM for G6PDH2 since these were approximately the concentrations that gave maximal activity for each enzyme. Under these conditions, less than 5% of the substrates were consumed over the course of the assay. The assay mixtures were initally prepared by adding all the components except the enzyme and obtaining a stable baseline. The enzyme was added to start the reaction. Activity was recorded with a dual wavelength filter photometer (Sigma ZFP2) as the change in absorbance at 334 nm relative to 405 nm caused by NADP+ reduction to NADPH using an extinction coefficient of 6190 M−1 cm-1. These wavelengths were used because they correspond to mercury emission wavelengths of the lamp used in the filter photometer. By using two wavelengths, the sensitivity of the assay was greatly increased allowing measurements at low enzyme concentrations that caused very little change in substrate concentrations, even during long assays.
When assaying redox sensitivity, G6PDH was incubated with 10 mM DTT or 10 mM hydrogen peroxide at room temperature for 30 min before addition to the assay. The assay mixture was prepared with NADP+ and enzyme. After obtaining a stable baseline, 0.3 mM G6P for G6PDH1 and 3 and 1.6 mM G6P for G6PDH2 was added to initiate the reaction. For G6P protection assays, a stable baseline was obtained with G6PDH1 and 0.6 mM NADP+. The reactions were initiated by adding 10 mM DTT and 0.3 mM G6P. Activity was measured 30 or 60 min later to allow time for DTT deactivation. During an hour of incubation, less than 5% of added G6P and NADP+ was consumed and activity was still linear in response to time.
Kinetic characterization
Enzymes were assayed at varying concentrations of substrate while keeping the concentration of other substrates constant as described above. All G6PDH isoforms showed substrate inhibition for G6P, therefore we estimated regression lines and kinetic constants by finding the minimum of the sum of the squared residuals from the following equation using Solver in Excel, where v is the specific activity of the enzyme in μmol mg −1 min−1, X is the number bound inactivating substrate molecules, and H is the Hill cooperativity coefficient (28):
| Eq. 1 |
As recommended by LiCata and Allewell (29) X was constrained to be ≤ 2 in order to prevent unrealistic values of parameters giving good fits. However, because it is difficult to estimate Km in the presence of substrate inhibition, we determined the half saturation concentration from the maximum velocity seen in the assays (S0.5) of G6PDH. NADP+ kinetics were estimated using standard Michalis-Menten kinetics.
Inhibition studies
Different metabolites of the Calvin-Benson cycle were tested for their effect on G6PDH activity. All the metabolites were purchased from Sigma Aldrich. In metabolite screening assays, metabolites were assayed at a 1:1 ratio with the substrate. To determine the Ki of G6PDH for different metabolites, the assay was carried out in the presence of various concentrations of G6P and NADPH. Assay mixtures were prepared as described above with different concentrations of substrate. The G6P concentration was varied between 0 and 3.8 mM. The concentration of NADP+ in G6PDH assays was held constant at 600 μM. The concentration range used to study the Ki of G6PDH1 and 3 was 0–0.3 mM NADPH and 0–14.5 μM NADPH for the G6PDH2 assays based on preliminary assays. The mechanism of inhibition was determined from Hanes-Woolf plots. The Ki was determined from the non-linear least squares fitting of the activity vs. NADPH concentration plot using Solver in Excel using the standard equation for competitive inhibition as described below.
| Eq. 2 |
where Vmax is the maximum velocity, S is the G6P concentration, Km is the Michaelis constant, and Ki is the inhibition constant. Sum of least squares for Solver regressions for inhibition constants are shown in Table 1.
Table 1.
Kinetic constants and inhibition constants of G6PDH1, 2, and 3 as determined by NADPH-linked spectrophotometric assays. Km and kcat for NADP+ were determined by fitting the Michalis-Menten equation. Parameters for G6PDH were determined from a modified Michalis-Menten equation which includes substrate inhibition. Data points used in model fitting were n=3 different preparations. For inhibition constants, each number was determined from the fitted curves as described in the methods. Errors shown are S.E. (n=3). Sum of least-squares for inhibition parameters of G6PDH that were determined using Solver in Excel. RMSE- square root of mean squared error.
| G6PDH1 | G6PDH2 | G6PDH3 | ||
|---|---|---|---|---|
| G6P S0.5 (mM) | Oxidized | 0.3±0.0 | 1.6±0.4 | 0.3±0.1 |
| Reduced | 2.4±0.7 | 1.2±0.1 | 0.5±0.0 | |
| G6P Ki (mM) | Oxidized | 17.1±6.0 | 22.4±4.5 | 33.1±2.0 |
| Reduced | 56.2±6.4 | 3.7±1.1 | 44.9±11.9 | |
| kcat (s−1) | Oxidized | 51.8±6.1 | 14.2±5.3 | 11.3±0.6 |
| Reduced | 40.6±1.3 | 7.6±1.4 | 8.6±0.5 | |
| NADP Km (μM) | 0.6±0.2 | 1.3±0.03 | 0.6±0.06 | |
| Binding inhibitor substrate molecules (X) | 2.0±0.0 | 2.0±0.0 | 1.7±0.3 | |
| Hill cooperativity coefficient (H) | 1.0±0.0 | 1.0±0.0 | 1.0±0.0 | |
| Catalytic Efficiency G6P (mM−1 s−1) | 125.1±8.5. | 23.1±11.2 | 51.8±14.3 | |
| Catalytic Efficiency NADP (μM−1 s−1) | 109.7±38.8 | 21.0±2.5 | 20.1±1.6 | |
| NADPH Ki (μM) | 59 | 0.9 | 112 | |
| NADPH Ki RMSE | 15.8 | 3.3 | 2.1 | |
Midpoint potential of G6PDH1
As G6PDH1 was the most redox sensitive of the three enzymes under study, we performed a series of oxidation-reduction titrations with purified G6PDH1. Fully reduced DTT was prepared daily by combining 100 mM DTT with 200 mM sodium borohydride. The mixture was incubated on ice for 20 min and then neutralized by adding concentrated HCl to a final concentration of 0.2 M. The mixture was brought to a pH of 8 and diluted to a final concentration of 50 mM DTT. Oxidized DTT and buffers used in the assay were also pH 8. We used mixtures of oxidized and reduced DTT at different redox potentials, ranging from −420 to −124 mV in order to span the range from fully reduced enzyme to fully oxidized. The total concentration of DTT was 1–8.5 mM. 4.1 ng of G6PDH1 was incubated in the DTT mixture with 1 mg/ml BSA, pH 8 for 1 hr at 25˚C in an anaerobic environment. Activity of G6PDH was measured as described in ‘Coupled spectrophotometric assay for G6PDH’ using 0.3 mM G6P to initiate the reaction. The data were fit to the Nernst equation for a two-electron process. We used the Em of DTT as determined by Hutchison and Ort (30), −391 mV at pH 8. Oxidized and reduced DTT was quantified using modified protocols from Cho, Souitas (31) and Charrier and Anastasio (32) to calculate the potential.
| Eq. 3 |
Leaf extract assays
Arabidopsis Col-0 was grown on soil in a growth chamber with 12 h light at 120 μmol m−2 s−1, 23˚C and 12 h dark at 21˚C. Plants were treated at 0 or 200 μmol m−2 s−1 for one hour. Approximately 300 mg of leaf samples were collected in a 2 ml microfuge tube and immediately frozen by plunging in liquid nitrogen. Frozen samples were ground in a Retsch mill with 4 mm silicone carbide particles (BioSpec Products, www.biospec.com). One ml of cold extraction buffer (45 mM Hepes, pH 7.2, 30 mM NaCl, 10 mM mannitol, 2 mM EDTA, 0.5% Triton-X-100, 1% polyvinylpolypyrrolidone, 0.5% casein, 1% protease inhibitor cocktail) was added to the sample and vortexed for 30 s. The sample was centrifuged for 30 s at maximum speed and immediately placed on ice. G6PDH activity was assayed as described in “Coupled spectrophotometric assay for G6PDH”. Assays that used leaf extracts were normalized by amount of chlorophyll added to the assay mixture. Chlorophyll was quantified by lysing 50 μl of purified chloroplasts by sonication and adding supernatant to 1 ml of 95% ethanol. Chlorophyll concentration was calculated based on the OD654 (33):
| Eq. 4 |
Chloroplast isolation
Arabidopsis Col-0 was grown on soil in a growth chamber with 12 h light at 120 μmol m−2 s−1, 23˚C and 12 h dark at 21˚C. Chloroplasts were isolated using a Percoll gradient (34). Leaves were placed in a chilled blender with cold grinding buffer (330 mM mannitol, 50 mM Hepes, pH 7.6, 5 mM MgCl2, 1 mM MnCl2, 1 mM EDTA, 5 mM ascorbic acid, 0.25% BSA), blended, and then filtered through four layers of cheese cloth. Filtered liquid was centrifuged and the pellet was resuspended in resuspension buffer (330 mM mannitol, 50 mM Hepes, pH 7.6, 5 mM MgCl2, 1mM MnCl2, 1mM EDTA, 0.25% BSA). The resuspended pellet was layered on top of a 20–80% Percoll gradient which was centrifuged at 1200 g for 7 min. The bottom band in the gradient containing the intact chloroplasts was collected. One volume of resuspension buffer was added to collected chloroplasts and centrifuged at 1200 g for 2 min. The pellet was resuspended in 50 μl of water and vortexed to lyse the chloroplasts. One volume of 2x buffer (100 mM Hepes, pH 7.6, 10 mM MgCl2, 2 mM MnCl2, 2 mM EDTA, 2 mM EGTA, 60% glycerol, 0.2% Triton X-100, 0.2% PVPP) was added. Samples were stored at −80˚C until used for further analysis.
When chloroplast isolations were used to assess activity of fully oxidized and reduced plastidic G6PDH, isolated plastids were treated with 0 or 200 μmol m−2 s−1 of light for one hour, on ice to limit protease activity, to oxidize or reduce G6PDH before assaying activity. Assays that used isolated chloroplasts were normalized by the amount of chlorophyll added to the assay mixture as described in ‘Leaf extract assays’.
Results
Purification of recombinant G6PDH
The final concentration of G6PDH1 was 1.66 mg/ml, G6PDH2 was 1.90 mg/ml, and G6PDH3 was 0.177 mg/ml (Supplemental Fig. 1). The molecular weight of Strep-tagged recombinant G6PDH1 was ~65.2 kDa, G6PDH2 was ~70.2 kDa, and G6PDH3 was ~70.5 kDa. The maximum specific activity was ~55 μmol mg−1 protein min−1 for G6PDH1, ~22 μmol mg−1 protein min−1 for G6PDH2, and ~11 μmol mg−1 protein min−1 for G6PDH3. Maximum specific activities were determined from fitting varying NADP+ and 7.6 mM G6P for G6PDH1 and 3 and 15.4 mM for G6PDH2. One preparation of each recombinant enzyme was aliquoted and a fresh aliquot was used each day for all experiments that day. Each experiment described below consists of three technical replicates prepared from separate dilutions of enzymes aliquots.
Kinetic characterization of G6PDH isoforms
We determined the oxidized (fully active) biochemical parameters of G6PDH1, 2, and 3. Table 1 shows the Km and S0.5 (for both G6P and NADP+), G6P Ki, kcat, NADPH Ki, and other determined kinetic parameters of all three G6PDH isoforms. All three oxidized G6PDH isoforms showed substrate inhibition (Fig. 1a). G6PDH1 had a S0.5 of 0.3 mM and G6PDH3 had a S0.5 of 0.3 mM G6P while G6PDH1 had the highest kcat for G6P of 51.8 s-1. For NADP+ (Fig. 1b), G6PDH1 had the highest kcat.
Fig 1. Activity of G6PDH1, 2, and 3 at different G6P (a) and NADP+ (b) concentrations.
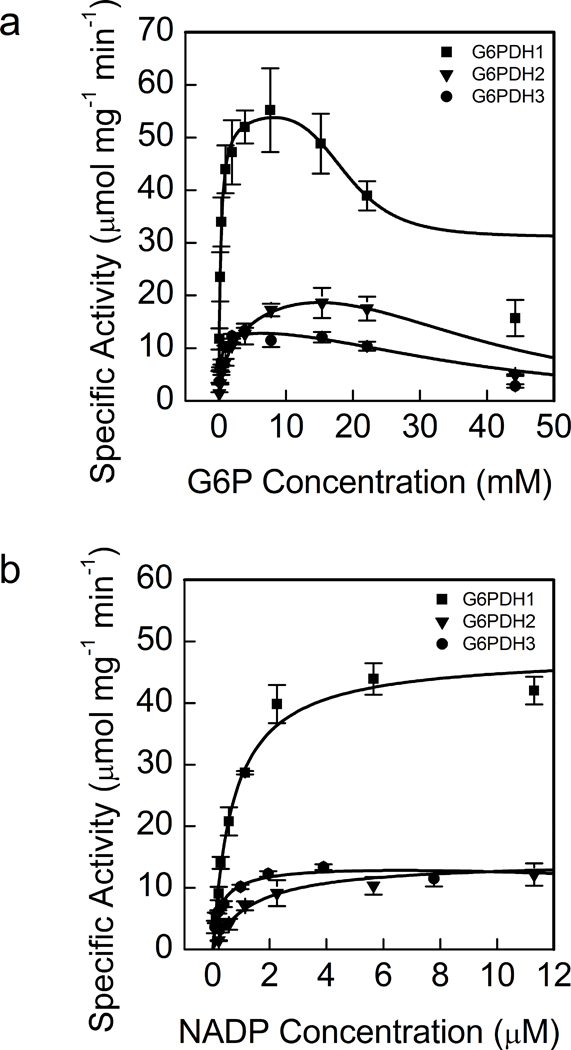
Each data point represents mean and error bars represent S.E. (n=3). All three isoforms of oxidized G6PDH showed substrate inhibition for G6P. G6PDH1 and 3 showed the greatest affinity for G6P and G6PDH3 had the greatest affinity for NADP+. During the G6P experiments NADP+ was 0.6 mM and during the NADP+ experiment assays were done 7.6 mM G6P for G6PDH1 and 3 and 15.4 mM for G6PDH2 since these were approximately the concentrations that gave maximal activity for each enzyme. In (a) lines represent data fit to Eq. 2 and in (b) lines represent data fit to the Michaelis-Menten equation.
G6PDH is inhibited by NADPH
Since the activity of an enzyme is also dependent on the presence of metabolic inhibitors, we tested ribulose 1,5-bisphosphate (RuBP), ribulose 5-phosphate (Ru5P), F6P, PGA, DHAP, E4P, NADPH, and 6PG for their effect on G6PDH activity. Only NADPH showed inhibition. While NADPH inhibited all three isoforms, G6PDH2 was the most inhibited. The calculated Ki values for NADPH are shown in Table 1. Sum of least squares for Solver regressions for inhibition constants are shown in Table 1. NADPH was found to be competitive for all isoforms based on the Hanes-Woolf plots, except above 14.5 μM for G6PDH2 and above 0.15 mM for G6PDH3 (Supplemental Fig. 2).
G6PDH1 is redox regulated
All isoforms of G6PDH were susceptible to deactivation by DTT, but G6PDH1 was the most deactivated after half an hour, losing approximately 90% of its activity (Fig. 2a). Kinetic characterization of G6PDH1 incubated with 10 mM DTT showed that decreased activity in G6PDH1 was due to both a decrease in kcat and an increase in S0.5. However, the kcat was less affected than the S0.5 (Table 1, Fig. 2b). Comparison of our results to those of Née, Zaffagnini (35), who used thioredoxins to deactivate G6PDH1, show that DTT is an acceptable mimic of thioredoxins to deactivate G6PDH1. Both results show that G6PDH1 will lose ~90% of activity when fully reduced. G6PDH2 and 3 did not significantly change the kcat or S0.5. G6PDH2 retained ~60% of activity and G6PDH3 retained ~80% of activity. Redox deactivation of G6PDH1 can be partially rescued by addition of hydrogen peroxide equimolar to DTT in vitro (Fig. 3a). However, G6PDH1 does not fully recover, returning to only ~65% of its original activity. Presumably there are additional mechanisms for fully reactivating G6PDH1 in vivo. G6PDH1 activity reached approximately 64% activity while 79% of the DTT was still reduced (Fig. 3b). The calculated Em at this time point was −407 mV. Based on our determined midpoint potential of G6PDH1 (see The midpoint potential of G6PDH1 is −378 mV at pH 8), we predict G6PDH1 would have < 5% activity at the redox potential in the assay. Therefore, we conclude the addition of hydrogen peroxide did not result in the re-activation of G6PDH1 by oxidizing DTT but that hydrogen peroxide was directly activating G6PDH1.
Fig 2. Activity of G6PDH 1, 2, and 3 with and without DTT treatment (a) and S0.5 shift with DTT in G6PDH 1 (b).
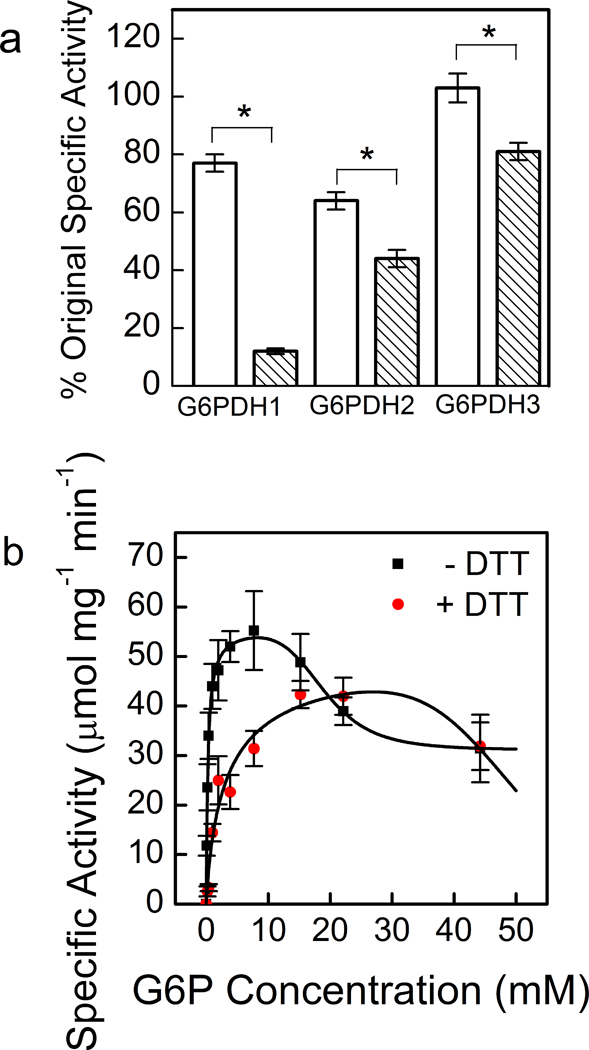
Each bar or data point represents mean and error bars represent S.E. (n=3). G6PDH1 was the most affected by DTT treatment. White bars indicate controls incubated without DTT for 30 min and shaded bars represent incubation with 10 mM DTT for 30 min. Single point assays were done S0.5 concentrations, 0.3 mM G6P for G6PDH1 and 3 and 1.6 mM for G6PDH2. Bars with an asterisk (*) are significantly different from corresponding controls as determined by Student’s t-test (P < 0.05). In (b), the lines represent data fit to Eq. 2, and there are two overlapping data points at the highest G6P concentration.
Fig 3. Partial re-activation of DTT-deactivated G6PDH1 with hydrogen peroxide.
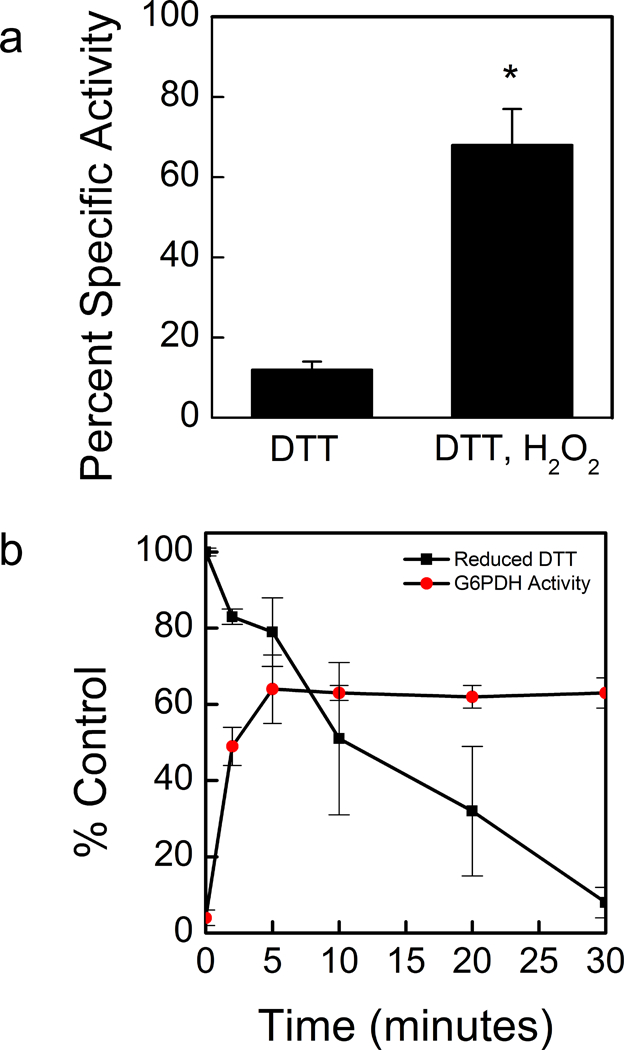
G6PDH1 deactivation by DTT could be partially recovered by addition of equimolar hydrogen peroxide. (a). Reactivation is not through DTT oxidation, but rather hydrogen peroxide directly effects G6PDH1 (b) In (b), at each time point G6PDH activity was assayed and DTT concentrations were quantified using protocols from Cho, Sioutas (68) and Charrier and Anastasio (32). Assays were done with 0.3 mM G6P. Each bar or data point represents mean and error bars represent S.E. (n=3). Bars with an asterisk (*) are significantly different as determined by two tailed Student’s t-test (P < 0.05).
Redox deactivation of G6PDH1 was decreased when G6P was present. When G6P was present at 0.3 mM during incubation with DTT, the activity of reduced G6PDH was higher than when G6P was not present (Fig. 4). Substrate interaction with other redox-regulated chloroplast enzymes has been reported before (36, 37).
Fig 4. G6PDH1 protection from deactivation by G6P.
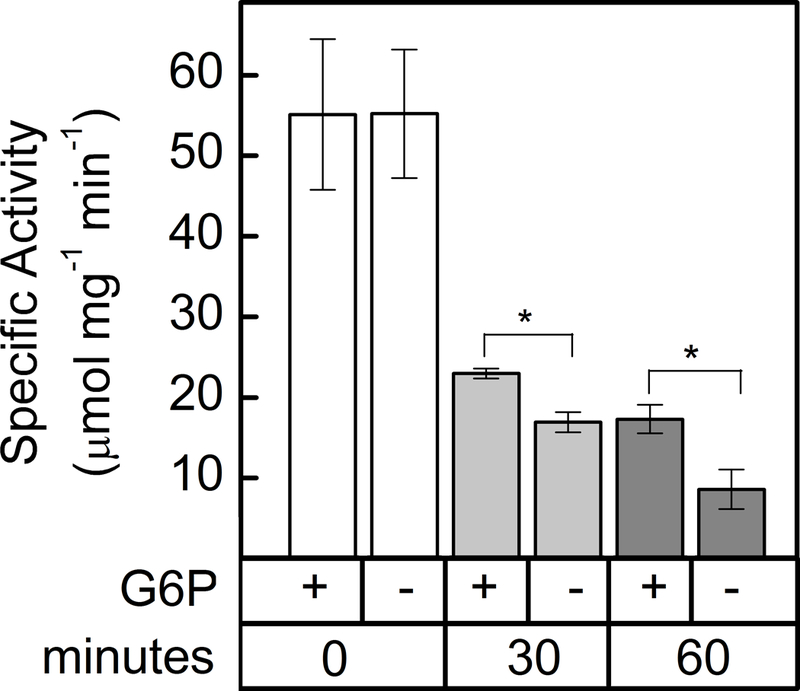
G6PDH1 is less deactivated by DTT after 30 and 60 min when G6P is present at 0.3 mM. Each bar represents the mean and error bars represent S.E. (n=3). Bars with asterisk (*) are significantly different as determined by two tailed Student’s t-test (P < 0.05).
The midpoint potential of G6PDH1 is −378 mV at pH 8
In addition to determining that G6PDH1 is susceptible to redox deactivation, we determined the midpoint potential of G6PDH1 (Fig. 5). The data was fit with the Nernst equation for a two-electron process. Incubation of G6PDH1 at higher redox potentials (−300 to −140 mV) did not increase activity any further. The midpoint potential of G6PDH1 at pH 8 was −378 mV. This corresponds to a midpoint potential of −318 mV at pH 7. We measured the amount of reduced DTT present at the beginning and end of an hour incubation with 3 mg/mL BSA. The measured amount of DTT did not change within the time of incubation. Additionally, the presence of 1 mg/mL or 3 mg/mL BSA did not change the measured amount of DTT, and hence the redox potential, within the first five min.
Fig 5. G6PDH1 midpoint potential.
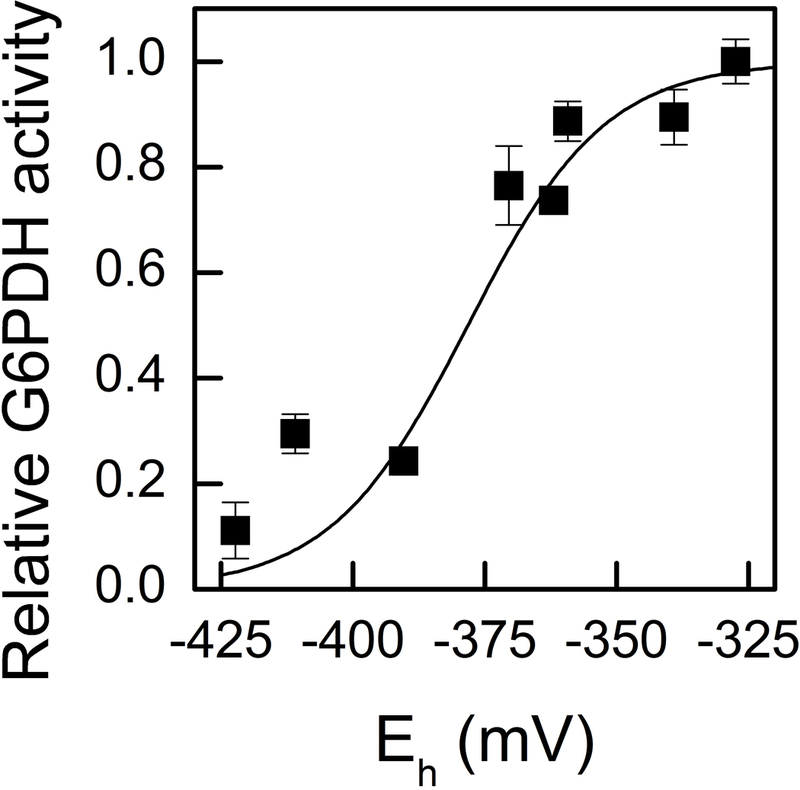
The midpoint potential of G6PDH1 was determined to be −378 mV at pH 8. Assays were done at the S0.5 concentration of G6P for oxidized G6PDH1, 0.3 mM. Each data point represents the mean and error bars represent S.E. (n=3). The dashed line represents the Nernst equation for one electron. The solid line represents the Nernst equation for two electrons.
G6PDH is active in isolated chloroplasts and leaf extracts
As well as performing in vitro characterization, we used rapid leaf extract assays and chloroplast isolations to determine the activity of redox-regulated G6PDH in the leaf. After illumination at 200 μmol m−2 s−1 for one hr, G6PDH activity in Arabidopsis leaf extracts decreased by about 30% (Fig. 6). When isolated chloroplasts were illuminated at 200 μmol m−2 s−1 for one hr, G6PDH activity decreased by 40%. Each chloroplast isolation and leaf extract had three biological replicates. The recombinant enzyme kinetic constants we determined indicate that a significant amount of the activity of G6PDH seen during the day can be caused by plastidial forms of the enzyme. Lendzian and Ziegler (38) showed similar results with spinach showing that this phenomenon is not restricted to Arabidopsis.
Fig 6. Whole leaf and chloroplast activity of G6PDH in Arabidopsis.
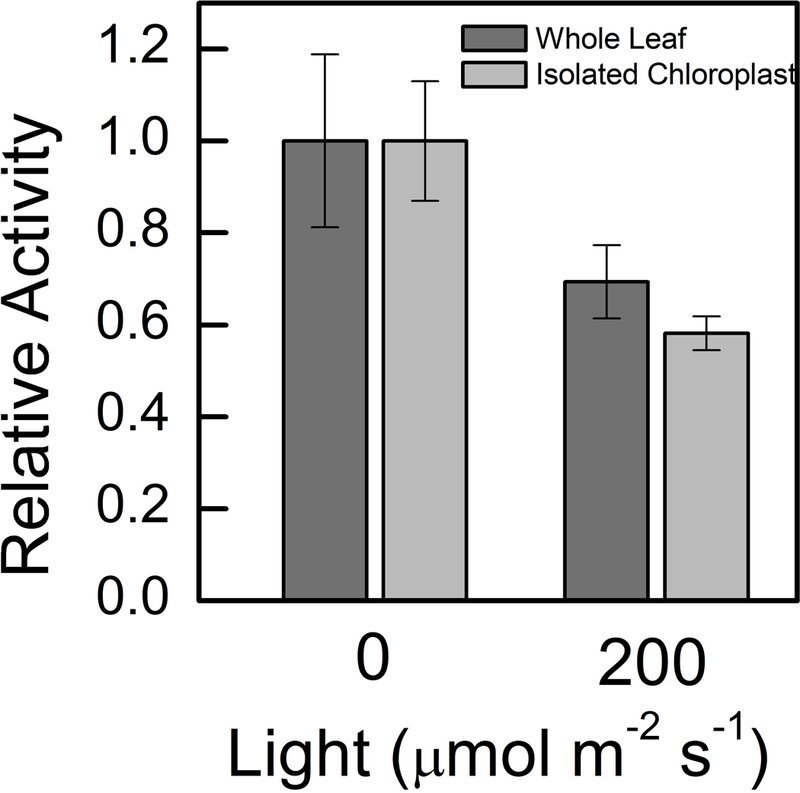
Whole leaf activity of G6PDH decreased 35% after illumination at 200 μmol m−2 s−1 for one hour. This represents the total redox sensitive G6PDH fraction in Arabidopsis leaves. Chloroplast G6PDH activity decreased 50% after illumination at 200 μmol m−2 s−1 for one hour. Samples were normalized by amount of chlorophyll added to the assay mixture. Assays were done with 5 mM G6P. Each bar represents the mean and error bars represent S.E. (n=3).
Discussion
Here we have investigated two questions: 1) what are the kinetic parameters of both oxidized and reduced G6PDH1 and 2) what are key regulators of plastidic G6PDH enzyme in order to understand how these factors impact G6PDH activity in the light and therefore flux through the G6P shunt.
G6PDH activity is highly sensitivity to G6P concentration in the plastid
The activity of G6PDH (and thus the G6P shunt) is sensitive to stromal G6P concentration by four mechanisms.
Deactivation of G6PDH in reducing conditions is primarily due to an increase in S0.5 (4).
G6P protects G6PDH from deactivation in reducing conditions (Fig 4) and catalytic site inhibition by diethyl decarbonate (6)
All isoforms of plastidic G6PDH show G6P substrate inhibition (Fig 1a).
G6P has been shown to relieve the inhibition of G6PDH by NADPH, as well as decrease the Km and increases the kcat of G6PDH in assays where NADP is varied (39, 40).
Our data corroborates previous research that shows G6PDH is in a reduced, less active form in the light, most likely to reduce futile cycling in leaves (4, 6, 24). However, while the enzyme is less active in the light (1–6) our results show that it can retain significant activity. While there is a slight decrease in kcat, G6PDH1 is predominately regulated by an increase in Km, agreeing with data from Scheibe, Geissler (4) in pea. Née, Aumont-Nicaise (6) have also characterized the kinetics of G6PDH1, but found a higher S0.5 and kcat than described here. Additionally, they propose changes in NADP+ binding play a role in G6PDH redox regulation.
The G6P concentration in the plastid is estimated to be approximately 1.4 mM (8, 9, 41). G6PDH regulation via a Km shift allows for up to ~30% maximal activity with increased substrate concentration, even in reducing conditions. Under some conditions the plastid G6P concentration might increase. For example, when plants are grown in high CO2, exposed to an increase in light intensity, or in plants deficient in starch synthesis (21–23), G6P concentrations may increase to the point where reduced G6PDH can readily consume G6P. However, the substrate inhibition of G6PDH limits the maximal activity of the enzyme at high substrate concentrations to possibly reduce futile cycling. While enzyme activity is generally dependent on the concentration of its substrate, because of the four ways that G6P can regulate G6PDH enzyme activity, G6PDH is particularly sensitive to the concentration of G6P.
G6PDH1 redox regulation allows modulation of G6PDH activity in the light
We determined the midpoint potential of G6PDH to be −378 mV at pH 8. Née, Zaffagnini (35) found a midpoint potential of 330 mV. Just over 10% of the difference may result from the difference in pH (8.0 in our study, 7.9 in Née, Zaffagnini (35)). We went to considerable lengths to ensure the DTT was fully reduced and to exclude oxygen during the incubations. In what follows we assume that the midpoint potential of G6PDH1 is −378 mV. This is close to the midpoint potential of other redox regulated enzymes in the Calvin-Benson cycle and electron transport (35, 42–47).
Assuming equilibrium, at the midpoint potential of G6PDH1 at pH 8 (−378 mV), using the Nernst equation, we calculate that all Calvin-Benson cycle enzymes and electron transport proteins are almost fully reduced and thus active while G6PDH maintains 50% of its activity (Table 2). Exceptions are ferredoxin and NADP+ dependent malate dehydrogenase (MDH), and NADP+ which are predicted to be oxidized at −378 mV. Ferredoxin should be oxidized during the day to accept electrons from PSI. MDH has maximial activity when fully reduced, but it has been shown that MDH activity is not fully active during the day (48, 49). Our prediction that NADPH would be 46% reduced is consistent with findings from Lendzian (50), which show the ratio of NADPH to NADP+ to be ~1 in a reconstituted spinach chloroplast. Although there may be deviations from redox equilibrium within the stroma, from these approximations we conclude that the midpoint potential of G6PDH1 is in a range to allow dynamic regulation of G6PDH and that it is theoretically possible to have flux through the Calvin-Benson cycle and the G6P shunt at the same time.
Table 2. Midpoint potentials and percent reduction of key Calvin-Benson cycle enzymes and electron transport proteins at −378 mV at pH 8, assuming equilibrium.
Calvin-Benson cycle enzymes are mostly active at the midpoint potential of G6PDH. One electron chemistry is assumed for ferredoxin and two electron chemistry for all others.
| Enzyme or metabolite | Midpoint potential, Em (mv) at pH 8 |
% reduced at −378 mV |
|---|---|---|
| G6PDH | −378 | 50.0 |
| Ferredoxin | −410 | 7.0 |
| NADPH | −380 | 46.0 |
| Thioredoxin f | −350 | 90.6 |
| Thioredoxin m | −360 | 81.1 |
| NADP−MDH | −390 | 27.5 |
| FBPase | −375 | 56.0 |
| PRK | −355 | 86.5 |
| Cyclic electron flow | −330 | 98.0 |
We have also shown that G6PDH can be activated upon addition of hydrogen peroxide. Brennan and Anderson (51) and Née, Zaffagnini (35) previously demonstrated a role for hydrogen peroxide regulation of G6PDH both in vivo and in vitro in the presence of thioredoxin. Additionally, in conditions where hydrogen peroxide can accumulate, such as high light, hydrogen peroxide can act on G6PDH directly to reverse G6PDH deactivation and modulate the consumption of G6P by the G6P shunt. It has been shown that hydrogen peroxide can be produced in situ in isolated chloroplasts and can transiently accumulate for signaling (52). The activity of G6PDH can be modulated by redox status of the plastid, G6P concentration, and hydrogen peroxide. Hydrogen peroxide can also stimulate cyclic electron flow (53), which could make up for the loss of ATP in the G6P shunt.
Redox regulation of dominant isoforms of G6PDH are found in many species, including Arabidopsis, pea, potato, spinach, and barley (4, 27, 54–57). We have shown that plastidic G6PDH from isolated Arabidopsis chloroplasts retains approximately 50% of its total activity, even in high light conditions. Additionally, cytosolic G6PDH could convert G6P in the cytosol to pentose phosphate and to be imported into the plastid by the xylulose-5-phosphate transporter (58).
Oxidative stress might also stimulate the G6P shunt. Drought or high light can result in an accumulation of hydrogen peroxide and other ROS products (see Suzuki, Koussevitzky (59) for a review). Based on current findings, we propose that, with accumulation of hydrogen peroxide, the S0.5 of G6PDH1 can decrease, increasing the flux through the G6P shunt. Sharkey and Weise (2016), proposed that the shunt can induce cyclic electron flow, which may help protect PSI. Photoprotective mechanisms of PSII, for example state transitions of the antenna complex or energy dependent quenching, are usually sufficient to safely dissipate excess excitation energy at PSII (60). However, with high light, in fluctuating light (61), and at low temperature (62), excess energy or electrons could still be passed on to PSI and result in PSI photoinhibition. Unlike PSII, the proteins of PSI have a low turnover rate and damage to PSI is considered more severe than damage to PSII (62–64). Coupling ATP consumption in the G6P shunt with cyclic electron flow would dissipate light energy at PSI (65–67).
Conclusion
Our data supports the conclusion that G6PDH can retain significant activity during the day and therefore allow flux through the G6P shunt in the light. This is particularly relevant in conditions where the redox status of the plastid or the plastidic concentration of G6P may be changing. G6PDH is still partially deactivated, reducing the loss of carbon while still maintaining regulatory flexibility to increase and decrease the G6P shunt (Fig. 7) as needed. However, in vivo flux measurments are not yet available for the G6P shunt. Characterization of of G6PDH mutants and quantification of flux through this pathway is essential to understand the physiological importance of the shunt.
Fig 7. The glucose 6-phosphate shunt.
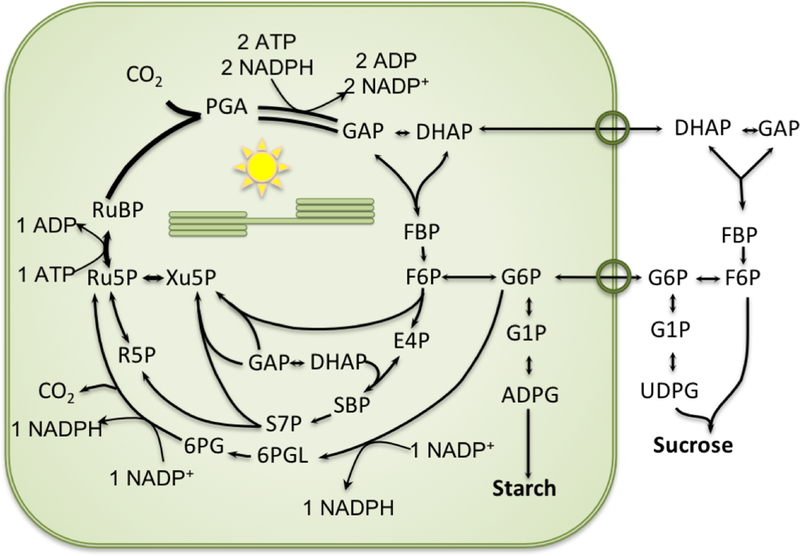
G6P is consumed by G6PDH to enter the G6PDH shunt. G6P re-enters the Calvin-Benson cycle as Ru5P. Overall, the shunt consumes three ATP and two NADP+ and produces two NAPDH. One CO2 molecule is lost for every G6P that enters the shunt.
Supplementary Material
Acknowledgements
We thank Michigan State University Research Technology Support Facility Mass Spectrometry Core for providing the facility for doing the LC-MS/MS work. This research was funded by U.S. Department of Energy Grant DE-FG02-91ER2002 (T.D.S. and A.L.P) and DE-FG02-11ER16220 (N.F.). A.L.P is partially supported by a fellowship from Michigan State University under the Training Program in Plant Biotechnology for Health and Sustainability (T32-GM110523). Partial salary support for T.D.S. came from Michigan AgBioResearch.
Abbreviations:
- 6PG
6-phosphogluconic acid
- At
Arabidopsis thaliana
- DHAP
dihydroxacetone phosphate
- E4P
erythrose 4-phosphate
- F6P
fructose 6-phosphate
- FBP
fructose 1,6-bisphosphate
- G6P
glucose 6-phosphate
- G6PDH
glucose-6-phosphate dehydrogenase
- PGA
3-phosphoglyceric acid
- PGI
phosphoglucoisomerase
- PGM
phosphoglucomutase
- So
Spinacia oleracea
- Xu5P
xylulose 5-phosphate
References
- 1.Buchanan BB, Gruissem W, Jones RL. Biochemistry & Molecular Biology of Plants. 2nd ed. Rockville: American Society of Plant Physiologists; 2015. [Google Scholar]
- 2.Buchanan BB. Role of light in the regulation of chloroplast enzymes. Annu Rev Plant Physiol. 1980; 31:341–74. [Google Scholar]
- 3.Anderson LE, Ng T-CL, Kyung-Eun Yoon P. Inactivation of pea leaf chloroplastic and cytoplasmic glucose 6-phosphate dehydrogenases by light and dithiothreitol. Plant Physiol. 1974; 53:835–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Scheibe R, Geissler A, Fickenscher K. Chloroplast glucose-6-phosphate dehydrogenase: Km shift upon light modulation and reduction. Archives of Biochemistry and Biophysics. 1989; 274:290–7. [DOI] [PubMed] [Google Scholar]
- 5.Heldt H-W, Piechulla B. Plant Biochemistry. Third ed Burlington MA: Elsevier Academic Press; 2005. [Google Scholar]
- 6.Née G, Aumont-Nicaise M, Zaffagnini M, Nessler S, Valerio-Lepiniec M, Bourguet-Issakidis E. Redox regulation of chloroplastic G6PDH activity by thioredoxin occurs through structural changes modifying substrate accessibility and cofactor binding. The Biochemical journal. 2014; 457:117–25. [DOI] [PubMed] [Google Scholar]
- 7.Kruckeberg AL, Neuhaus HE, Feil R, Gottlieb LD, Stitt M. Decreased-activity mutants of phosphoglucose isomerase in the cytosol and chloroplast of Clarkia xantiana. Impact on mass-action ratios and fluxes to sucrose and starch, and estimation of flux control coefficients and elasticity coefficients. Biochemical Journal. 1989; 261:457–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gerhardt R, Stitt M, Heldt HW. Subcellular metabolite levels in spinach leaves. Regulation of sucrose synthesis during diurnal alterations in photosynthetic partitioning. Plant Physiol. 1987; 83:399–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sharkey TD, Vassey TL. Low oxygen inhibition of photosynthesis is caused by inhibition of starch synthesis. Plant Physiol. 1989; 90:385–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Szecowka M, Heise R, Tohge T, Nunes-Nesi A, Vosloh D, Huege J, et al. Metabolic fluxes in an illuminated Arabidopsis rosette. Plant Cell. 2013; 25:694–714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schnarrenberger C, Oeser A. Two isoenzymes of glucosephosphate isomerase from spinach leaves and their intracellular compartmentation. Eur J Biochem. 1974; 45:77–82. [DOI] [PubMed] [Google Scholar]
- 12.Backhausen JE, Jöstingmeyer P, Scheibe R. Competitive inhibition of spinach leaf phosphoglucose isomerase isoenzymes by erythrose 4-phosphate. Plant Sci. 1997; 130:121–31. [Google Scholar]
- 13.Yu TS, Lue WL, Wang SM, Chen J. Mutation of Arabidopsis plastid phosphoglucose isomerase affects leaf starch synthesis and floral initiation. Plant Physiology. 2000; 123:319–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kunz HH, Zamani-Nour S, Hausler RE, Ludewig K, Schroeder JI, Malinova I, et al. Loss of cytosolic phosphoglucose isomerase affects carbohydrate metabolism in leaves and is essential for fertility of Arabidopsis. Plant Physiology. 2014; 166:753–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ray WJ, Roscelli GA. A kinetic study of the phosphoglucomutase pathway. Journal of Biological Chemistry. 1964; 239:1228–36. [PubMed] [Google Scholar]
- 16.Lowry OH, Passonneau JV. Phosphoglucomutase kinetics with the phosphates of fructose, glucose, mannose, ribose, and galactose. Journal of Biological Chemistry. 1969; 244. [PubMed] [Google Scholar]
- 17.Hanson KR, McHale NA. A starchless mutant of Nicotiana sylvestris containing a modified plastid phosphoglucomutase. Plant Physiol. 1988; 88:838–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Caspar T, Huber SC, Somerville C. Alterations in growth, photosynthesis, and respiration in a starchless mutant of Arabidopsis thaliana (L.) deficient in chloroplast phosphoglucomutase activity. Plant Physiology. 1985; 79:11–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kofler H, Häusler RE, Schulz B, Gröner F, Flügge UI, Weber A. Molecular characterisation of a new mutant allele of the plastid phosphoglucomutase in Arabidopsis, and complementation of the mutant with the wild-type cDNA. Molecular and General Genetics. 2000; 263:978–86. [DOI] [PubMed] [Google Scholar]
- 20.Kammerer B, Fischer K, Hilpert B, Schubert S, Gutensohn M, Weber A, et al. Molecular characterization of a carbon transporter in plastids from heterotrophic tissues: The glucose 6-phosphate phosphate antiporter. Plant Cell. 1998; 10:105–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kunz HH, Häusler RE, Fettke J, Herbst K, Niewiadomski P, Gierth M, et al. The role of plastidial glucose-6-phosphate/phosphate translocators in vegetative tissues of Arabidopsis thaliana mutants impaired in starch biosynthesis. Plant Biol. 2010; 12:115–28. [DOI] [PubMed] [Google Scholar]
- 22.Dyson BC, Allwood JW, Feil R, Xu YUN, Miller M, Bowsher CG, et al. Acclimation of metabolism to light in Arabidopsis thaliana: the glucose 6-phosphate/phosphate translocator GPT2 directs metabolic acclimation. Plant Cell Environ. 2015; 38:1404–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Leakey ADB, Xu F, Gillespie KM, McGrath JM, Ainsworth EA, Ort DR. Genomic basis for stimulated respiration by plants growing under elevated carbon dioxide. Proc Natl Acad Sci USA. 2009; 106:3597–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wakao S, Benning C. Genome-wide analysis of glucose-6-phosphate dehydrogenases in Arabidopsis. Plant J. 2005; 41:243–56. [DOI] [PubMed] [Google Scholar]
- 25.Meyer T, Hölscher C, Schwöppe C, von Schaewen A. Alternative tageting of Arabidopsis plastidic glucose-6-phosphate dehydrogenase G6PD1 involves cysteine-dependent interaction with G6PD4 in the cytosol. The Plant Journal. 2011; 66:745–58. [DOI] [PubMed] [Google Scholar]
- 26.Sharkey TD, Weise SE. The glucose 6-phosphate shunt around the Calvin-Benson Cycle. Journal of Experimental Botany. 2016; 67:4067–77. [DOI] [PubMed] [Google Scholar]
- 27.Wendt UK, Wenderoth I, Tegeler A, von Schaewen A. Molecular characterization of a novel glucose-6-phosphate dehydrogenase from potato (Solanum tuberosum L.). The Plant Journal. 2000; 23:723–33. [DOI] [PubMed] [Google Scholar]
- 28.Gray DW, Breneman SR, Topper LA, Sharkey TD. Biochemical characterization and homology modeling of methyl butenol synthase and implications for understanding hemiterpene synthase evolution in plants. J Biol Chem. 2011; 286:20582–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.LiCata VJ, Allewell NM. Is substrate inhibition a consequence of allostery in aspartate transcarbamylase? Biophysical Chemistry. 1997; 64:225–34. [DOI] [PubMed] [Google Scholar]
- 30.Hutchison RS, Ort DR. Measurment of equilibrium midpoint potentials of thiol/disulfide regulatory groups on thioredoxin activated chloroplast enzymes. Methods in Enzymology. 1995; 252:220–8. [DOI] [PubMed] [Google Scholar]
- 31.Cho AK, Souitas C, Miguel AH, Kumagai Y, Schmitz DA, Singh M, et al. Redox activity of airborne particulate matter at different sites in the Los Angeles Basin. Environ Res. 2005; 99:40–7. [DOI] [PubMed] [Google Scholar]
- 32.Charrier JG, Anastasio C. On dithiothreitol (DTT) as a measure of oxidative potential for ambient particles: evidence for the importance of soluble transition metals. Atmospheric Chemistry and Physics. 2013; 12:11317–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wintermans JGFM, DeMots A Spectrophotometric characteristics of chlorophylls a and b and their pheophytins in ethanol. Biochimica et Biophysica Acta. 1965; 109:448–53. [DOI] [PubMed] [Google Scholar]
- 34.Weise SE, Weber A, Sharkey TD. Maltose is the major form of carbon exported from the chloroplast at night. Planta. 2004; 218:474–82. [DOI] [PubMed] [Google Scholar]
- 35.Née G, Zaffagnini M, Trost P, Issakidis-Bourguet E. Redox regulation of chloroplastic glucose-6-phosphate dehydrogenase: A new role for f-type thioredoxin. Febs Lett. 2009; 583:2827–32. [DOI] [PubMed] [Google Scholar]
- 36.Woodrow IE, Walker DA. Regulation of stromal sedoheptulose-1,7-bisphosphatase activity and its role in controlling the reductive pentose phosphate pathway of photosynthesis. Biochimica et Biophysica Acta. 1983; 722:508–16. [Google Scholar]
- 37.Leegood RC, Kobayashi Y, Neimanis S, Walker DA, Heber U. Co-operative activation of chloroplast fructose-1,6-bisphosphatase by reductant, pH, and substrate. Biochimica et Biophysica Acta. 1982; 682:168–78. [Google Scholar]
- 38.Lendzian KJ, Ziegler H. Regulation of glucose-6-phosphate dehydrogenase in spinach chloroplasts by light. Planta. 1970; 94:27–36. [DOI] [PubMed] [Google Scholar]
- 39.Olavarría K, Valdés D, Cabrera R. The cofactor preference of glucose-6-phosphate dehydrogenase from Escherichia coli - modelling the physiological production of reduced cofactors. The FEBS journal. 2012; 279:2296–309. [DOI] [PubMed] [Google Scholar]
- 40.Shreve DS, Levy HR. Kinetic mechanism of glucose-6-phosphate dehydrogenase from the lactating rat mammary gland. Journal of Biological Chemistry. 1980; 255:2670–7. [PubMed] [Google Scholar]
- 41.Dietz KJ. A possible rate limiting function of chloroplast hexosemonophosphate isomerase in starch synthesis of leaves. Biochimica et Biophysica Acta. 1985; 839:240–8. [Google Scholar]
- 42.Cammack R, Rao KK, Bargeron CP, Hutson KG, Andrew PW, Rogers LJ. Midpoint redox potentials of plant and algal ferredoxins. Biochem J. 1977; 168:205–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Knaff DB. Oxidation-reduction properties of thioredoxins and thioredoxin-regulated enzymes. Physiologia Plantarum 2000; 110:309–13. [Google Scholar]
- 44.Strand DD, Fisher N, Davis GA, Kramer D. Redox regulation of the antimycin A sensitive pathway of cyclic electron flow around photosystem I in higher plant thylakoids. Biochimica et Biophysica Acta Bioenergetics. 2016; 1857:1–6. [DOI] [PubMed] [Google Scholar]
- 45.Hirasawa M, Brandes HK, Hartman FC, Knaff DB. Oxidation-reduction properties of the regulatory site of spinach phosphoribulokinase. Archives of Biochemistry and Biophysics. 1998; 350:127–31. [DOI] [PubMed] [Google Scholar]
- 46.Hirasawa M, Ruelland E, Schepens I, Issakidis-Bourguet E, Miginiac-Maslow M, Knaff DB. Oxidation-reduction properties of the regulatory disulfieds of sorghum chloroplast nicotinamide adenine dinucleotide phosphate-malate dehydrogenase. Biochemistry. 2000; 39:3344–50. [DOI] [PubMed] [Google Scholar]
- 47.Hirasawa M, Schürmann P, Jacquot JP, Manieri W, Jacquot P, Keryer E, et al. Oxidation-reduction properties of chlorplast thioredoxins, ferredoxin:thioredoxin reductase, and thioredoxin f-regulated enzymes. Biochemistry. 1999; 38:5200–5. [DOI] [PubMed] [Google Scholar]
- 48.Nakamoto H, Edwards GE. Dark activation of maize leaf NADP-malate dehydrogenase and pyruvate, orthophosphate dikinase in vivo under anaerobic conditions. Plant Science Letters. 1983; 32:139–46. [Google Scholar]
- 49.Nakamoto H, Edwards G. Influence of environmental factors on the light activation of pyruvate, Pi dikinase and NADP-malate dehydrogenase in maize. Functional Plant Biology. 1983; 10:279–89. [Google Scholar]
- 50.Lendzian KJ. Modulation of glucose-6-phosphate dehydrogenase by NADPH, NADP+ and dithiothreitol at variable NADPH/NADP+ ratios in an illuminated reconstituted Spinach (Spinacia oleracea L.) chloroplast system. Planta. 1980; 148:1–6. [DOI] [PubMed] [Google Scholar]
- 51.Brennan T, Anderson LE. Inhibition by catalase of dark-mediated glucose-6-phosphate dehydrogenase activation in pea chloroplasts. Plant Physiology. 1980; 66:815–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Smirnoff N, Arnaud D. Hydrogen peroxide metabolism and functions in plants. New Phytologist. 2019; 221. [DOI] [PubMed] [Google Scholar]
- 53.Strand DD, Livingston AK, Satoh-Cruz M, Froehlich JE, Maurino VG, Kramer DM. Activation of cyclic electron flow by hydrogen peroxide in vivo. Proc Natl Acad Sci USA. 2015; 112:5539–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wenderoth I, Scheibe R, von Schaewen A. Identification of the cysteine residues involved in redox modification of plant plastidic glucose-6-phosphate dehydrogenase. J Biol Chem. 1997; 272:26985–90. [DOI] [PubMed] [Google Scholar]
- 55.Schnarrenberger C, Oeser A, Tolbert NE. Two enzymes each of glucose-6-phosphate dehydrogenase and 6-phosphogluconate dehydrogenase in spinach leaves. Arch Biochem Biophys. 1973; 154:438–48. [DOI] [PubMed] [Google Scholar]
- 56.Semenikhina AV, Popova A, Matasova LV. Catalytic properties of glucose-6-phosphate dehydrogenase from pea leaves. Biochemistry. 1999; 64:863–6. [PubMed] [Google Scholar]
- 57.Wright DP, Huppe HC, Turpin DH. In vivo and in vitro studies of glucose-6-phosphate dehydrogenase from barley root plastids in relaiton to reductant supply for NO2- assimilation. Plant Physiology. 1997; 114:1413–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Eicks M, Maurino V, Knappe S, Flügge U-I, Fischer K. The plastidic pentose phosphate translocator represents a link between the cytosolic and the plastidic pentose phosphate pathways in plants. Plant Physiol. 2002; 128:512–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Suzuki N, Koussevitzky S, Mittler R, Miller G. ROS and redox signalling in the response of plants to abiotic stress. Plant Cell and Environment. 2011; 35:259–70. [DOI] [PubMed] [Google Scholar]
- 60.Derks A, Schaven K, Bruce D. Diverse mechanisms for photoprotection in photosynthesis. Dynamic regulation of photosystem II excitation in response to rapid environmental change. Biochimica et Biophysica Acta Bioenergetics. 2015; 1847:468–85. [DOI] [PubMed] [Google Scholar]
- 61.Allahverdiyeva Y, Suorsa M, Tikkanen M, Aro E-M. Photoprotection of photosystems in fluctuating light intensities. Journal of Experimental Botany. 2014; 66:2427–36. [DOI] [PubMed] [Google Scholar]
- 62.Sonoike K Photoinhibition of photosystem I. Physiologia Plantarum. 2011; 142:56–64. [DOI] [PubMed] [Google Scholar]
- 63.Scheller HV, Haldrup A. Photoinhibition of photosystem I. Planta. 2005; 221:5–8. [DOI] [PubMed] [Google Scholar]
- 64.Lima-Melo Y, Gollan PJ, Tikkanen M, Silveira JAG, Aro EM. Consequences of photosystem I damage and repair on photosynthesis and carbon utilization in Arabidopsis thaliana. Plant Journal. 2018. [DOI] [PubMed] [Google Scholar]
- 65.Munekage Y, Hashimoto M, Miyaka C, Tomizawa KI, Endo T, Tasaka M, et al. Cyclic electron flow around photosystem I is essential for photosynthesis. Nature. 2004; 429:579–82. [DOI] [PubMed] [Google Scholar]
- 66.Miyake C, Shinzaki Y, Miyata M, Tomizawa K. Enhancement of cyclic electron flow around PSI at high light and its contribution to the induction of non-photochemical quenching of chl fluorescence in intact leaves of tobacco plants. Plant Cell Physiol. 2004; 45:1426–33. [DOI] [PubMed] [Google Scholar]
- 67.Strand DD, Kramer D. Control of non-photochemical exciton quenching by the proton circuit of photosynthesis In: Demmig-Adams B, Garab G, Adams Iii WW, Gonvindjee, editors. Non-photochemical quenching and energy dissipation in plants, algae and cyanobacteria. Advances in photosynthesis and respiration including bioenergy and related processes. 40: Springer; 2014. p. 387–408. [Google Scholar]
- 68.Cho AK, Sioutas C, Miguel AH, Kumagai Y, Schmitz DA, Singh M, et al. Redox activity of airborne particlate matter at different sites in the Los Angeles Basin. Environmental Research. 2005; 99:40–7. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.