

Abstract
The pulmonary vascular endothelium is involved in the pathogenesis of acute and chronic lung diseases. Endothelial cell (EC)-derived products such as extracellular vesicles (EVs) serve as EC messengers that mediate inflammatory as well as cytoprotective effects. EC-EVs is a broad term, which encompasses exosomes and microvesicles of endothelial origin. EVs are comprised of lipids, nucleic acids, and proteins that reflect not only the cellular origin but also the stimulus that triggered their biogenesis and secretion. This chapter presents an overview of the biology of EC-EVs and summarizes key findings regarding their characteristics, components, and functions. The role of EC-EVs is specifically delineated in pulmonary diseases characterized by endothelial dysfunction, including pulmonary hypertension, acute respiratory distress syndrome and associated conditions, chronic obstructive pulmonary disease, and obstructive sleep apnea.
INTRODUCTION
Extracellular vesicles (EVs) are small membrane-derived vesicles shed by cells into the extracellular space (Gyorgy et al. 2011). Over the last two decades, EVs have attracted significant interest mainly because they are major players in cellular communication and regulate diverse cellular responses; EVs, upon their release extracellularly, deliver their bioactive cargo to target cells modulating processes such as coagulation, inflammation, apoptosis, angiogenesis, and proliferation. In addition, EVs not only are present in biological fluids (e.g. plasma, urine, bronchoalveolar lavage, saliva, sputum, pleural effusions), but also their levels, cellular source, and molecular cargo are altered in various diseases, strongly suggesting that they could serve as biomarkers, injurious stimuli, and a source of therapeutic targets (Yanez-Mo et al. 2015).
The presence of biologically active vesicles in human plasma was first documented by Wolf, almost 50 years ago (Wolf 1967). These vesicles were described to be shed from the plasma membrane of platelets and were named microparticles, while later, other groups discovered another category of small vesicles released after fusion of multivesicular bodies (MVBs) with the cell membrane, which were named exosomes (Johnstone et al. 1987). Almost all cell types spontaneously or upon activation release vesicles that could originate from either the plasma membrane or the MVBs. EVs derived from endothelial cells (EC) have been of particular interest as they shed directly to the bloodstream and can interact with circulating blood cells or travel to distant sites (Hromada et al. 2017). The endothelial cell-derived EV (EC-EV) plasma levels are low in healthy individuals, but elevated in acute or chronic pathologic conditions characterized by endothelial dysfunction, such as sepsis, atherosclerosis, acute coronary syndrome, diabetes mellitus, and others (Dignat-George and Boulanger 2011; Chironi et al. 2009; Jansen, Nickenig, and Werner 2017).
The vascular endothelium lines the interior surface of blood vessels and acts as a semipermeable barrier that separates the blood from the surrounding tissues. ECs are the major regulators of vascular homeostasis, they mediate immune responses, and participate in processes such as inflammation, coagulation, and angiogenesis (Orfanos et al. 2004). Normal endothelial function is disrupted in lung diseases including pulmonary hypertension, acute respiratory distress syndrome (ARDS), chronic obstructive pulmonary disease (COPD), obstructive sleep apnea (OSA), and bronchopulmonary dysplasia (BPD) (Maniatis et al. 2008; Green and Turner 2017; Mourani and Abman 2015; Ranchoux et al. 2018; Lurie 2011). Interestingly, ECs are highly heterogeneous and depending on their location can have distinct morphology and function; especially in the lung, distinct ECs, for example, macrovascular vs microvascular, appear to have specific roles and behaviors in response to physiologic or inflammatory stimuli (Stevens et al. 2001). As expected, the heterogeneous ECs can produce diverse EVs in response to various mechanical, chemical, and molecular stimuli; however, the specific role and characteristics of these EC-EVs in mediating lung disease pathogenesis are only beginning to be explored.
The goal of this chapter is to present an overview of the EV biology, with emphasis on exosomes and microvesicles, and summarize recent findings and knowledge gaps on the mechanisms that regulate the generation of EC-EVs, their characteristics (e.g. molecular cargo), and their possible role as biomarkers, mediators of lung vascular diseases, and their potential as therapeutic delivery vehicles.
OVERVIEW OF ENDOTHELIAL EXTRACELLULAR VESICLES (EVs)
Classification of EVs
Eukaryotic EVs can be classified according to their size and the process of their biogenesis. The main EV categories are: a) exosomes (~30–150 nm), b) microvesicles (MVs), also named microparticles (MPs) or ectosomes (~0.1 −1 μm), and c) apoptotic bodies (ABs) (~2–5 μm) (Xu et al. 2016; Yanez-Mo et al. 2015; Kalra et al. 2012).
Exosomes:
Exosomes are generated through a process that involves first the inward budding of an endosomal membrane and subsequent formation of MVBs, and second the release of exosomes into the extracellular space after fusion of the MVBs with the plasma membrane. Basic cargo carried by exosomes includes molecules associated with the plasma membrane, the cytosol, and the endosomal compartments (Thery, Zitvogel, and Amigorena 2002).
Microvesicles:
MVs shed directly from the plasma membrane and are enriched in phosphatidylserine (PS). A well-described mechanism that underlies MV formation involves increased calcium influx, activation of the enzymes scramblase and floppase, and inhibition of flippase, all enzymes responsible for maintaining plasma membrane asymmetry. These events lead to loss of phospholipid asymmetry and the exposure of PS in the outer membrane (Hugel et al. 2005; Piccin, Murphy, and Smith 2007; Morel et al. 2011). MV shedding is then completed by the actions of the calcium-dependent enzymes calpain and gelsolin. MV basic cargo includes membrane and cytosolic components (Hargett and Bauer 2013).
Apoptotic bodies:
ABs are generated during the characteristic membrane blebbing occurring in apoptotic cell death (van Niel, D’Angelo, and Raposo 2018).
Despite extensive research on EVs, their isolation, identification, and characterization remain challenging and diverse among research groups. Recently, ISEV (International Society for Extracellular Vesicles) published guidelines for EV analyses (Lotvall et al. 2014; Witwer et al. 2013) in order to promote the standardization of EV protocols. Briefly, based on the ISEV recommendations, EVs of interest should be isolated from biological fluids or conditioned media and should be characterized using multiple methodologies for a) EV common markers; analytic approaches such as immunoblotting, flow cytometry (FACS), and mass spectrometry could be employed to identify EV-associated molecules, b) size and morphology; transmission electron microscopy, atomic force microscopy, nanoparticle tracking analysis (NTA), and dynamic light scattering can be used to characterize the size and morphology of isolated EVs, and c) enumeration; NTA, FACS, and imaging flow cytometry can be used to provide an estimation of EV concentrations. Currently, there is no consensus on a “gold standard” method to isolate the EV subpopulations; for exosome isolation, there are various protocols that employ ultracentrifugation (≥100,000 × g), ultrafiltration, and density gradient separation (Thery et al. 2006; Raimondo et al. 2011), while for MV isolation, a high-speed centrifugation (~14,000– 21,000 × g) after eliminating dead cells, ABs, and cell debris is a standard isolation method. However, due to the significant overlap in the EV physical characteristics, most of the isolation techniques can result in mixed EV populations and this need to be considered when interpreting EV-relevant data.
Upon isolation, subtypes of EVs can be identified based on their content. Even though, all EVs carry common proteins and lipids, there are molecules such as the tetraspanins CD9, CD81, CD83, and CD63, and the MVB synthesis proteins, ALG-2 interacting protein X (Alix) and tumor susceptibility gene 101 protein (TSG101) that are mainly enriched in exosomes (Kreimer et al. 2015; Thery, Zitvogel, and Amigorena 2002) (Figure 1). MVs are enriched in PS (Kreimer et al. 2015) and FACS analysis after staining with annexin V is a usual method to quantify/identify MVs. However, there are studies that have also identified MVs lacking PS in their outer membrane (Connor et al. 2010).
Figure 1. Schematic representation of endothelial cell-derived extracellular vesicles (EC-EVs).
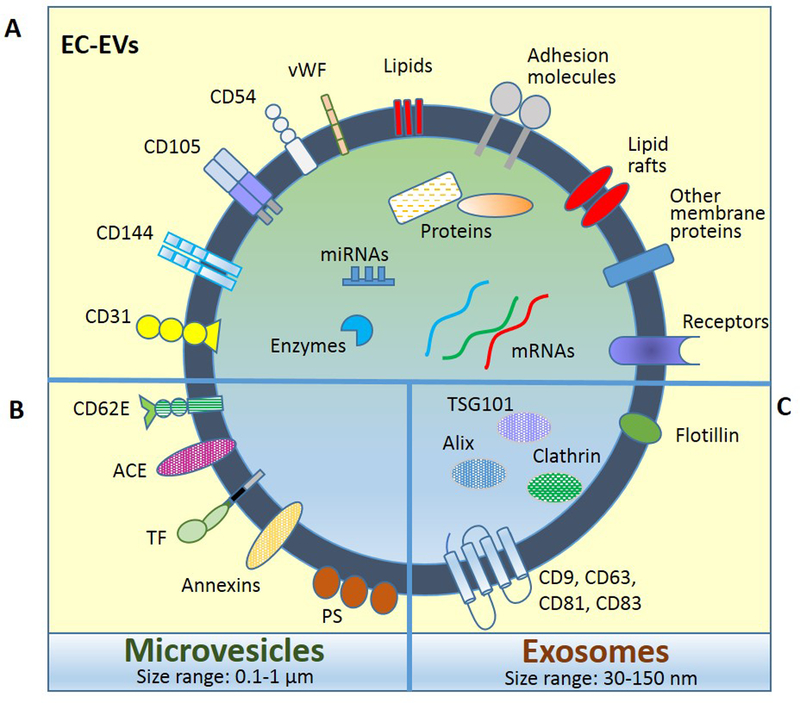
EV main categories, microvesicles and exosomes, carry common components (A) but also are enriched in specific molecules (B and C). EC markers that identify EVs of endothelial origin include CD62E, CD31, CD144, CD105, CD54, and vWF. Abbreviations: CD62E; E-selectin, CD31; PECAM-1, CD144; VE-cadherin, CD105; endoglin, CD54; ICAM-1, vWF; von Willebrand factor, ACE; angiotensin converting enzyme, TF; tissue factor, PS; phosphatidylserine, TSG101; tumor susceptibility gene 101 protein, CD9, CD63, CD81, CD82; tetraspanins.
Based on their cell type of origin, EVs can further be classified in other categories as platelet EVs, epithelial EVs, stem cell-derived EVs, endothelial EVs, etc. For this classification, there are cell-specific markers that can be used to distinguish the source of EVs. To identify EC- EVs several markers exist (Figure 1). E-selectin (CD62E) and vascular endothelial (VE)-cadherin (CD144) are selective markers for EVs of endothelial origin. Angiotensin-converting enzyme (ACE) is abundantly expressed in pulmonary capillary endothelium and could serve to identify specific EC-EV subpopulations. Other common endothelial markers such as platelet endothelial cell adhesion molecule-1, PECAM-1 (CD31), intercellular adhesion molecule 1, ICAM-1 (CD54), endoglin (CD105), vascular cell adhesion molecule 1, VCAM-1 (CD106), melanoma cell adhesion molecule, MCAM (CD146), integrin aV (CD51), and von Willebrand factor (vWf) can also be expressed in other cell types, and should be used to identify EC-EVs (in mixed samples such as plasma) only if combined with other markers in order to exclude EVs originating from non-EC sources (Dignat-George and Boulanger 2011).
EV molecular cargo
EVs carry a variety of biological molecules that can be delivered to target cells and their cargo is defined by the cellular origin and the specific conditions that stimulated their production and release. Overall, EVs are composed of proteins, nucleic acids, and lipids (Figure 1). In addition, as it has recently been shown, EVs are also capable of carrying whole organelles, such as mitochondria (Boudreau et al. 2014; Morrison et al. 2017). To date, numerous high-throughput studies have been performed to uncover the cargo of EVs derived from various cell types and body fluids, and three data repositories have been established for EV proteomic, lipidomic, and transcriptomic data: ExoCarta, Vesiclepedia, and EVpedia (Keerthikumar et al. 2016; Kalra et al. 2012; Kim et al. 2013) (Choi et al. 2013). The goals of these studies were to identify: a) a common set of EV molecules, in order to advance our understanding of the mechanisms triggering the vesicle biogenesis and release, and b) the cell type- and stimulus-specific EV components, in order to provide important information on the biological function of EVs, the activation status of the cell of origin, and the EV potential fate. Very importantly, the identification of unique molecules in EVs that are cell- and stimulus-specific can potentially be used as biomarkers and therapeutic targets.
EV protein cargo:
Among the common proteins found in EVs are: adhesion proteins (e.g. integrins, PECAMs, EPCAMs, ICAMs), cytoskeletal proteins (e.g. actin, tubulins, profilin, cofilin), receptors (e.g. PDGFR, EGFR), annexins and tetraspanins, and enzymes (e.g. enolases, glyceraldehyde 3-phosphate dehydrogenase). Common EV proteins associate with the following main subcellular localization categories: plasma membrane, cytoskeleton, cytosol, and vesicles. Cell motion, vesicle-mediated transport, translation, protein localization are among the main biological processes associated with the common EV proteins (Choi et al. 2013, 2015; Pocsfalvi et al. 2016).
EV nucleic acid cargo:
According to a growing number of studies, a variety of nucleic acids exist into the extracellular space within vesicles; mRNA, miRNA, long noncoding RNA, chromosomal and mitochondrial DNA are some of the nucleic acids identified in EVs (Kim et al. 2017; Nerlich et al. 2018; Cai et al. 2016; Hu, Drescher, and Chen 2012). These molecules can be transferred within vesicles from the origin cell to the recipient cell and can induce or alter gene and protein expression processes.
EV lipid cargo:
Lipids are essential components of EVs that not only play a role in their formation, but also have major biological functions. The lipid membrane of EVs resembles the plasma membrane and consists mainly of phospholipids, glycolipids, and cholesterol. Several lipidomic studies have determined the lipid composition of different types of EVs. Common EV lipids include phosphatidylcholine (PC), phosphatidylserine (PS), phosphatidylethanolamine (PE), sphingomyelin (SM), and cholesterol (Choi et al. 2013; Kreimer et al. 2015; Pocsfalvi et al. 2016; Dignat-George and Boulanger 2011). EV-associated lipids regulate numerous cellular responses including coagulation, angiogenesis, and death (Tripisciano et al. 2017; Kim et al. 2002; Beloribi et al. 2012).
Endothelial EV cargo:
EVs in biological fluids are an important source of biological information, however, these fluids contain mixed populations of vesicles derived from a variety of cellular sources and generated under diverse conditions. Thus, the characterization of the composition of cell type- and stimulus-specific EVs can only be achieved by performing in vitro studies using primary cell cultures or cell lines. Examples of unique proteome cargo have been found in EVs derived from human umbilical vein endothelial cells (HUVEC) after stimulation with TNF-α or plasminogen activator inhibitor-1 (PAI-1) (Banfi et al. 2005; Peterson et al. 2008; Sander et al. 2008), in EVs derived from human pulmonary artery endothelial cells (HPAEC) after stimulation with mechanical cyclic stretch or lipopolysaccharide (LPS) (Letsiou et al. 2015), and in human microvascular endothelial cells (HMEC-1) exposed to hypoxia or TNF-α (de Jong et al. 2012). A proteomic analysis of MVs derived from young HUVEC revealed that these MVs carry functional peptide antioxidant components that can act as ROS scavengers, as well as NAPDH- synthesizing enzymes (Bodega et al. 2017). Interestingly, the MV-associated antioxidant machinery is potentiated in senescent HUVEC. Several other studies have addressed how the status of the cell of origin affects the EC-EV protein content. For example, Jimenez et al., performed an EC-MV antigenic characterization in activated versus apoptotic endothelial cells (Jimenez et al. 2003). In this study, renal and brain microvascular EC and coronary macrovascular EC were treated with TNF-α or were deprived of growth factors to induce activation or apoptosis, respectively. The authors found that in apoptosis, EC-MVs positive for CD31 and CD105 were greatly increased, while EC-expression of CD31 and CD105 was decreased. In contrast, activation leads to increased release of EC-MVs positive for CD62E, CD54, and CD106, markers that are also induced in the activated EC. This study further demonstrated that macrovascular EC produce much fewer EVs compared to microvascular EC during activation and apoptosis. This phenomenon could be explained by the fact that macrovascular EC are overall less responsive to growth factor deprivation or activation. Given the special characteristics of ECs located in different vascular beds, Scruggs et al. investigated whether MVs isolated from vascular cells would carry the unique glycocalyx properties of their parent cell. This study showed that pulmonary microvascular and artery endothelial cells express unique glycocalyx linkages, while EVs released spontaneously or after injury did not recapitulate the glycocalyx of the parent cell (Scruggs et al. 2015).
Several studies have analyzed the nucleic acid content of EC-EVs. Van Balcom et al. performed a semi-quantitative and qualitative analysis of small RNAs in both endothelial exosomes and the parent cells, HMEC-1. They demonstrated that different RNA classes were distributed differentially between cells and exosomes, and that endothelial exosomes contain miRNAs that can regulate angiogenesis, proliferation, and differentiation (miR-10b, LET71, miR-126, miR-100, miR-27b, miR-30a, miR-25, miR-221) (van Balkom et al. 2015). The authors also observed that fragments of small non-coding RNAs such as snoRNAS, scaRNAs, vRNAs, and yRNAs are more abundant in exosomes compared to cells, which is especially important given the biological role that has recently been attributed to these molecules. This study not only emphasizes the targeted packaging of small RNAs in exosomes but also proposes a new cellular mechanism that involves localized degradation of RNAs in the proximity of MVBs and subsequent elimination of these RNA fragments through exosomes. miRNA profiling has also been performed in EVs-derived from mouse brain endothelial cells (b.End5) after stimulation with inflammatory stimuli. This study identified several miRNAs upregulated in EVs derived from stimulated EC (including the miR-328–3p, miR-211–5p, let-7d-3p, miR-698–3p, miR204–5p, miR-615–3p) (Yamamoto et al. 2015). Similarly, TNF-α triggers specific packaging of miRNAs in microvesicles produced from human aortic endothelial cells (HAEC) (Alexy et al. 2014). De Jong et al. analyzed the mRNA content of endothelial EVs (exosomes) and identified a unique mRNA profile in exosomes produced from HMEC-1 exposed to hypoxia or TNF-α that clearly distinguishes them from control exosomes (de Jong et al. 2012).
Regarding the lipid cargo of EC-EVs, Hussain et al. performed an analysis of the phospholipid composition of endothelial MVs derived from IL-1alpha-stimulated HUVEC (Abid Hussein et al. 2008). EC-MVs from activated EC were enriched in both PS and PE and could trigger thrombin formation in a tissue factor (TF)-dependent mechanism. All together, these studies clearly demonstrate that EC-EV cargo is primarily defined by the cell of origin and the existing cellular conditions.
Numerous studies have identified EVs in biological fluids (plasma, bronchoalveolar lavage, sputum, saliva, etc) of patients with lung diseases, however, a few of them attempted to characterize the composition of these EVs. Nevertheless, it is evident that specific subsets of molecules are packaged in EVs in a disease-specific manner (Alipoor et al. 2016; Vykoukal et al. 2017; Sun et al. 2018; Qazi et al. 2010; Ramacciotti et al. 2010).
Endothelial EV production and downstream effects
In a previous section, we described the general mechanisms by which different types of EVs are generated and released extracellularly. In this section, we will review specific mechanisms involved in endothelial EV secretion and present some of the EV biological effects with our main focus remaining on in vitro observations (Summarized in Table 1).
Table 1.
Endothelial-cell derived extracellular vesicle production
| EV source | Stimulus | EV Function | Reference |
|---|---|---|---|
| HUVEC | Activated protein C | Anti-coagulant | (Perez-Casal et al. 2005) |
| Angiogenic factors | Regulate angiogenesis | (Taraboletti et al. 2002) | |
| Angiotensin II receptor 1 autoantibody | Impair endothelial function • Induce ROS increase • Reduce NO synthesis |
(Yang et al. 2014) | |
| Complement proteins | Exert prothrombinase activity | (Hamilton et al. 1990) | |
| C-reactive protein | Possibly contribute to vascular injury | (Wang et al. 2007) | |
| High glucose | Induce oxidative stress in EC and promote platelet/EC interactions | (Terrisse et al. 2010) | |
| Hypoxia/ reoxygenation |
Induce oxidative stress and apoptosis in cardiomyocytes | (Zhang, Shang, et al. 2016) | |
| IL-1α | Pro-coagulant | (Abid Hussein et al. 2008) | |
| PAI-1 | Pro-coagulant | (Brodsky et al. 2002) | |
| PAI-1 | Impair endothelial function • Inhibit NO production • Increase pulmonary capillary permeability and induce neutrophil recruitment to the lungs |
(Densmore et al. 2006; Buesing et al. 2011) | |
| Shear stress | Not assessed | (Vion, Ramkhelawon, et al. 2013) | |
| Spontaneously | • Impair angiogenesis* • Increase apoptosis* *In high EV doses |
(Mezentsev et al. 2005) | |
| Spontaneously | Suppress monocyte activation • Reduce NF-kB, TNF-α, and IL-1β • Increase IL-10, TGF-β, and MRC1 |
(Njock et al. 2015) | |
| TNF-α | Pro-coagulant | (Combes et al. 1999; Szotowski et al. 2005) | |
| TNF-α/ Ionizing radiation | Pro-coagulant | (Szotowski et al. 2007) | |
|
HUVEC & HDMEC |
Ethanol | Promote angiogenesis | (Lamichhane et al. 2017) |
| HMEC-1 | Hypoxia | Mediate extracellular matrix remodeling | (de Jong et al. 2016) |
| Thrombin | Not assessed | (Sapet et al. 2006) | |
| TNF-α | Induce maturation of plasmacytoid dendritic cells | (Angelot et al. 2009) | |
| HLMVEC | Intracellular LPS | Not assessed | (Cheng et al. 2017) |
| TNF-α | Pro-inflammatory • NF-kB activation in EC • Induce CXCL-10, IL-6, sICAM-1, C5a, and TNF-α release from EC |
(Liu et al. 2017) | |
| TNF-α, H2O2, Cigarette smoke |
Not assessed | (Takahashi et al. 2013) | |
|
HLMVEC & HPAEC |
Cigarette smoke | Suppress efferocytosis | (Serban et al. 2016) |
| Hypoxia, LPS | Promote overproliferation and apoptosis resistance in PASMC |
(Zhao et al. 2017) | |
| LPS | Induce lung EC barrier disruption | (Sun et al. 2012) | |
| Mechanical stretch | Pro-inflammatory Induce alveolar-capillary barrier disruption and neutrophil recruitment to the lungs |
(Vion, Birukova, et al. 2013; Letsiou et al. 2015) | |
| HBMEC | Mechanical stretch | Not assessed | (Andrews et al. 2016) |
| HAEC | TNF-α | Pro-inflammatory Induce sICAM-1 release |
(Curtis et al. 2009) |
| HCAEC | Serum-starved induced apoptosis | Anti-inflammatory • Reduce TNF-α-induced endothelial ICAM-1 expression and monocyte adhesion to EC Mediate EC repair |
(Jansen et al. 2015; Jansen et al. 2013) |
| Human EPC | Spontaneously | Promote angiogenesis | (Deregibus et al. 2007) |
| Spontaneously | Anti-apoptotic | (Burger et al. 2015) | |
| Spontaneously | Promote angiogenesis | (Zhang, Chen, et al. 2016) | |
| Spontaneously | Anti-inflammatory • Reduce LPS-induced HMGB-1 and VCAM-1 in EC |
(Zhou et al. 2018) | |
| RMVEC | Spontaneously | Impair endothelial function • Reduce NO levels • Impair vasorelaxation • Increase superoxide production |
(Brodsky et al. 2004) |
| MLEC | TNF-α | Pro-inflammatory • NF-kB activation and ICAM-1 upregulation in EC |
(Andrews and Rizzo 2016) |
| MAEC | Angiotensin II | Pro-inflammatory • VCAM-1 upregulation and ROS increase in EC • Macrophage adhesion |
(Burger et al. 2011) |
| bEnd5 | Cytokine Combo/ LPS |
Pro-inflammatory • VEGF-B upregulation in pericytes |
(Yamamoto et al. 2015) |
Abbreviations: HMEC-1 or HDMEC; Human dermal microvascular EC, HLMVEC; Human lung microvascular EC, HBMEC; human brain microvascular EC, HAEC; Human aortic EC, HPAEC; human pulmonary artery EC, HCAEC; human coronary artery EC, EPC; endothelial progenitor cells, MLEC; mouse lung EC, RMVEC; rat renal microvascular EC, MAEC; mouse aortic EC, bEND5; Mouse brain EC, PASMC; pulmonary artery smooth muscle cells.
In 1990, Hamilton et al. observed that assembly of the complement proteins C5b-9 on human endothelial cells results in increased vesiculation of membrane particles from the endothelial surface (Hamilton et al. 1990). These vesicles expressed binding sites for factor Va suggesting an EV-prothrombinase activity. Subsequent studies demonstrated that TNF-α stimulates different types of endothelial cells to release EVs with unique properties. In HUVEC, TNF-α-derived MVs trigger coagulation in vitro, in a TF-dependent manner (Combes et al. 1999; Szotowski et al. 2005). HMEC-1 in response to TNF-α produce MVs, which can induce the maturation of freshly isolated plasmacytoid dendritic cells (PDCs) (Angelot et al. 2009). Interestingly, EVs derived from quiescent HMEC-1 could also stimulate PDC maturation, however, PDC response was dose-dependent suggesting that PDCs are likely to be activated in pathological conditions in which there is an increased production of EVs. In human aortic endothelial cells (HAECs), TNF-α, through a pathway that involves p38 activation, increases the release of EVs, which then induce the increased production of the pro-inflammatory mediator, soluble endothelial ICAM-1 (sICAM-1) (Curtis et al. 2009). Similarly, MVs from human lung microvascular (HLMVEC) and mouse lung endothelial cells (MLEC) stimulated with TNF-α activate the NF-kB pathway and lead to pro-inflammatory signaling (Liu et al. 2017; Andrews and Rizzo 2016).
Several other endothelial injurious stimuli have been linked to EV release. PAI-1 dose-dependently increases the number of EVs released from HUVEC. PAI-1- EC-EVs express uPAR and αvβ3 integrin and have procoagulant activities (Brodsky et al. 2002). Thrombin stimulation of HMEC-1 induces a dose- and time-dependent release of MVs through the caspase-2-RhoA/ROCK pathway. Gene expression profiling and gene silencing assays revealed that ROCK- II and not ROCK-I was the critical mediator for thrombin-induced EC-MVs (Sapet et al. 2006). C- reactive protein (CRP) also stimulates EV release from HUVEC, an effect that is related to impaired tetrahydro-L-biopterin (BH4)-dependent NO production. Treatment with exogenous BH4 reduces EC-MV shedding and restores NO levels, suggesting that the released EVs could contribute to CRP-induced vascular injury (Wang et al. 2007). Angiotensin-II (Ang-II) triggers EV formation through pathways that involve Ang II receptor I binding and NADPH oxidase and Rho kinase activation (Burger et al. 2011). Ang-II-EC-EVs are enriched in lipid rafts/caveolae components and can stimulate ROS production and pro-inflammatory responses in ECs in an EGFR-dependent manner. Angiotensin II receptor type 1 autoantibody (AT1-AA), which is found in patients with hypertension, activates the Ang I receptor similar to Ang II and promotes MV generation from HUVEC in a dose- and time-dependent manner through p38 activation. AT1-AA-EVs increase ROS and reduce NO synthesis in ECs (Yang et al. 2014). Yamamoto et al. showed that inflammatory cytokines and LPS induce a dose-dependent release of EVs from b.End5 (Yamamoto et al. 2015). These EVs transfer their miRNA content to pericytes and enhance their VEGF-B expression, an effect that could result in pathological neovascularization and/or vascular leakage (Yamamoto et al. 2015). HMEC-1 produce exosomes spontaneously and their numbers do not change after TNF-α or hypoxia (de Jong et al. 2012), whereas LPS or hypoxia enhance exosome release from HPAEC (Zhao et al. 2017). HPAEC-derived exosomes could be internalized by pulmonary artery smooth muscle cells (PASMC), promoting their proliferation and inducing apoptosis resistance. HUVEC exposed to ethanol produce high amounts of exosomes, enriched in the lncRNAs, HOTAIR, and MALAT1, which can mediate the pro-angiogenic effects of EVs in the presence of ethanol (Lamichhane et al. 2017). Toxic substances such as cigarette smoke (CS) can reach circulation following CS inhalation and activate ECs to produce EVs through the activity of the acid sphingomyelinase (aSMase). CS-induced EVs, which are enriched in the miRNAs, let-7d, miR-191, miR126, miR125a, and in ceramide, found to inhibit efferocytosis of peripheral blood monocyte-derived macrophages (Serban et al. 2016). On the contrary, cytoprotective molecules can stimulate the release of EVs with protective properties. For example, exposure of HUVEC to activated protein C (APC) triggers the release of EPCR-containing MVs that can bind to APC and promote anti-coagulant effects (Perez-Casal et al. 2005).
Non-molecular stimuli have also been found to stimulate EC-EV production. Ionizing radiation (IR) of HUVEC causes the release of TF-containing EC-MVs and this effect is potentiated in the presence of TNF-α. EV production and IR-induced ROS formation and apoptosis are inhibited in the presence of antioxidants (Szotowski et al. 2007), suggesting again a potential role of EC-MVs in mediating vascular injury. HUVEC exposed to a hypoxia/reoxygenation (H/R) injury model secrete EC-MVs, which contribute to oxidative stress and apoptosis in cardiomyocytes, (Zhang, Shang, et al. 2016). Another study reported that sustained atheroprone low shear stress stimulates EC-MV release through activation of Rho and ERK½ kinases and cytoskeletal reorganization, while atheroprotective high shear stress limits EC-MV release by regulating NO-dependent pathways (Vion, Ramkhelawon, et al. 2013). Moreover, excessive mechanical forces that injure the endothelium result in increased EC-MV production in vitro (HPAEC and human brain microvascular endothelial cells; HBMVEC) and in vivo (Letsiou et al. 2015; Vion, Birukova, et al. 2013; Andrews et al. 2016). Endothelial cells release exosomes that carry in their outer membrane the lysyl oxidase-like 2 (LOXL2), through which they can interact with the extracellular matrix (ECM). The expression and activity of LOX2 are significantly increased in exosomes derived from hypoxia-treated EC, suggesting that hypoxia-derived exosomes could play a potential role in focal ECM remodeling, a key process involved in both fibrosis and wound healing (de Jong et al. 2016).
An important recent finding with therapeutic significance is that endothelial progenitor cells (EPC) produce EVs with cytoprotective properties. EPC-derived EVs are incorporated into HMEC through integrin binding and promote proliferative, antiapoptotic, and proangiogenic effects (Deregibus et al. 2007). EPC-EVs exert these effects by delivering their mRNA content to the target ECs to induce eNOS phosphorylation and activate the PI3K/Akt pathway. Intravenous injection of EPC-EVs in rats protects against ischemia-reperfusion-induced acute kidney injury, by enhancing tubular cell proliferation, and reducing apoptosis and leucocyte infiltration (Cantaluppi et al. 2012). This effect was mediated by the pro-angiogenic miR-126 and miR-296 carried within EVs. Similar results were obtained in a study from Burger et al., which demonstrated that exosomes from human cord blood-derived endothelial colony-forming cells (ECFC) mediate protective effects in a mouse model of ischemic acute kidney injury and attenuate hypoxia-induced EC apoptosis in vitro (Burger et al. 2015). Other studies show that EPC-derived exosomes exert proangiogenic effects and promote proliferation and migration in HMEC by activating the ERK½ signaling pathway (Zhang, Chen, et al. 2016). The therapeutic potential of EPC-exosomes was recently evaluated in a sepsis model; intravenous injection of EPC-exosomes in septic mice improves the survival, suppresses lung and renal vascular leakage, and reduces multi-organ failure (Zhou et al. 2018). These exosomes were enriched in miR-126–3p and 5p, targeting the LPS-induced high mobility group box 1(HMGB1) and VCAM-1 levels in HMVEC.
The observation that spontaneously released EC-EVs appear to have homeostasis restoring properties rather than injury-inducing is not limited to EPC. Unstimulated HUVEC-derived EVs suppress the activation of pro-inflammatory genes (e.g. TNF-α, IL-1β) and enhance markers of alternative activation or immunomodulatory responses (e.g. IL-10, MRC1, TGF-β) in monocytes challenged with LPS. In vivo, EVs isolated from normal mouse plasma were also capable of suppressing monocyte activation and inflammation. These effects could be mediated by anti-inflammatory miRNAs carried by the EVs, such as the miR-10a (Njock et al. 2015). Along the same line, MVs derived from serum-starved human coronary artery endothelial cells (HCAEC) protect against TNF-α-induced ICAM-1 expression on adjacent ECs and prevent subsequent monocyte adhesion (Jansen et al. 2015), while they contribute to endothelial repair processes by promoting EC proliferation and migration (Jansen, Yang, Hoelscher, et al. 2013). These EVs are highly enriched in miR-126, which is transferred to adjacent ECs to regulate SPRED1 expression, a protein involved in migration and proliferation. Interestingly, EC-MVs derived from glucose-treated HCAEC carry less miR-126 and their capacity to promote endothelial repair is suppressed (Jansen, Yang, Hoelscher, et al. 2013). However, the level of EVs is important since pathologic concentrations of EVs derived from quiescent EC induce apoptosis and impair angiogenesis (Mezentsev et al. 2005), and can directly affect acetylcholine-induced vasorelaxation and NO production by aortic rings (Brodsky et al. 2004).
Taken together, it is evident that endothelial cells can release EVs with unique properties via specific mechanisms dependent on the environment in which they are produced. EVs target neighbor or distant cells or exert autocrine actions on the endothelial cells themselves. Moreover, the effects of EC-EVs appear to be dose-dependent and when their concentration exceeds the normal range might lead to deleterious effects on target cells contributing to disease pathophysiology.
Mechanisms by which EVs interact with target cells
Upon their release, EVs target neighboring or distant cells to mediate their effects or to be cleared. To date, several mechanisms have been proposed to describe the interaction of EVs with the recipient cell, and these include a) ligand/receptor signaling, b) fusion of the vesicle with the plasma membrane of the cell recipient, c) clathrin-mediated endocytosis, d) caveolin-mediated endocytosis, e) phagocytosis, and f) macropinocytosis (Mulcahy, Pink, and Carter 2014; Maas, Breakefield, and Weaver 2017). In this section, we will present a few examples of microvesicle and exosome uptake mechanisms.
Jansen et al. demonstrated that endothelial microvesicles are incorporated by HCAEC in an annexin I/phosphatidylserine receptor-dependent manner and protect against apoptosis (Jansen et al. 2012). In another study, EC-MVs induced by TNF-α were internalized through a mechanism that involves caveolin-1 in MLEC to activate pro-inflammatory signaling (Andrews and Rizzo 2016). Caveolae are constitutively expressed in abundance along the pulmonary vascular bed and lipid rafts may represent a predominant EV uptake mechanism in the lung. Another EV internalization mechanism involves the interaction of PS in the EC-EV membrane with lactadherin (also known as milk fat globule-EGF factor 8 protein; MFGE8), which is present in ECs and serves as a bridge between the PS and the αvß3 integrin (Terrisse et al. 2010). This interaction enables the EC-MVs to attach to the cell membrane and enter the cell target without fusion. This uptake can be inhibited in the presence of annexin V, suggesting that PS in the EC-EV membrane plays a major role in mediating the EV-EC interactions. Upon internalization, these EC-EVs activate the endothelium (HUVEC) and increase its association to unstimulated platelets (Terrisse et al. 2010). Lactadherin not only promotes endocytosis but it appears to play a central role in the clearance of circulating PS-expressing MVs by macrophages (Dasgupta et al. 2009). Interestingly, lactadherin-deficient mice have an increased number of platelet-derived MVs compared to wild-type littermates. Another mechanism that describes the clearance of MVs by the endothelium involves the developmental endothelial locus-1 (Del-1), an endothelial-derived opsonin (Dasgupta et al. 2012). MV uptake by HUVEC or HMVEC can be inhibited in the presence of an antibody against Del-1 in vitro, while Del-1 deficient mice show decreased uptake of MVs in lung endothelial cells. To track the path of EVs upon internalization, Faille et al. showed that platelet MVs upon endocytosis in the endothelium do not stay along the plasma membrane but move into large intracellular vesicles. Then, the EVs co-localize with early endosomes and acidic lysosomes, while they did not colocalize with the ER (Faille et al. 2012).
Numerous studies have been performed to track the route of exosomes upon their release and to investigate their internalization mechanisms (McKelvey et al. 2015). A real-time study demonstrated that exosomes attach to the membrane of the target cell before they get internalized by endocytosis and not fusion. After uptake, exosomes were diffused in the cytoplasm or actively transported along the cytoskeleton and targeted the lysosomes (Tian et al. 2013). Similarly, another study showed that exosomes upon internalization through macropinocytosis, colocalize with Lamp1, a marker of late endosomes or lysosomes (Fitzner et al. 2011). Exosomes derived from EPCs could be incorporated into endothelial cells to enhance their proliferation, migration, and angiogenic properties (Zhang, Chen, et al. 2016). This study did not explore the mechanism by which exosomes were internalized, however, they showed that the vesicles were visible in the perinuclear region of HMEC-1, within two hours.
ENDOTHELIAL EVs AND LUNG DISEASES
Vascular endothelial dysfunction in lung diseases
Endothelial dysfunction in the pulmonary circulation is broadly described as a loss of the ability of the endothelium to maintain vascular tone, limit coagulation, and provide a semi-permeable barrier to infiltrates and fluid. Multiple pulmonary vascular diseases exhibit some or all of the traits of endothelial dysfunction including pulmonary hypertension, acute respiratory distress syndrome, and COPD. The role of EC-EVs in endothelial dysfunction and repair are described.
Endothelial EVs in pulmonary hypertension
Pulmonary hypertension is a complex disease involving proliferation, inflammation, and increased vasocontractility within the lung vasculature. Incorporated into each aspect of this fatal disease is a role for EVs. All EVs are not injurious and in some instances, they are beneficial for restoring homeostasis, however the larger body of work is focused on the detrimental role of EVs in propagating PH and the use of EVs as biomarkers in PH progression.
Increased tone and vasocontractility is a recognized component of all forms of pulmonary hypertension, in particular hypoxia-induced hypertension. MVs from the circulation of rats exposed to chronic hypoxia to then injected into normal rat inhibit endothelium dependent vasorelaxation (Tual-Chalot et al. 2010). When pulmonary artery endothelial cells and aortic endothelial cells were treated with the PH MVs, decreased nitric oxide production was observed. Intriguingly, the increased oxidative stress also occurred in the pulmonary artery endothelium, but not the aortic, suggesting that the components of the MVs have differential effects (Tual-Chalot et al. 2010). This brings to light a question beginning to advance in the field as to whether EVs are capable of specifically targeting select vascular beds, or whether the cells themselves recognize components of the EVs after interactions. This question will require a great deal more investigation.
In patients and all animal models of pulmonary arterial hypertension there is significant intravascular diapedesis of inflammatory cells such as macrophages and neutrophils (Nicolls and Voelkel 2017; Rabinovitch et al. 2014; Tuder et al. 1994). These inflammatory cells release cytokines responsible for stimulation of pulmonary endothelial and smooth muscle hypercontractility, proliferation and apoptotic resistance. Data from animal models suggest these events are sentinel in the progression of complex vascular lesions that ultimately occlude vessels. The culmination of this hypercontractility and vascular occlusion is right heart failure. Thus, our understanding of the events perpetuating this injury is vital.
In order for inflammatory cells to escape the circulation they must directly interact with the pulmonary endothelium (Bevilacqua et al. 1994). Increased expression of intercellular adhesion molecules on an activated endothelium induces the attachment and migration of inflammatory cells. Extracellular vesicles, including MVs and exosomes, increase ICAM-1 and endothelial-leukocyte adhesion molecule (E-selectin) on endothelium in vascular diseases (Wang et al. 2011; Jansen, Yang, Franklin, et al. 2013; Barry et al. 1997). In pulmonary arterial hypertension there are significant increases in total circulating MVs. Circulating MVs from a model of PAH, when applied to naïve pulmonary artery endothelium, induce increased ICAM-1 expression and function, suggesting that MVs likely play a role in the recruitment of inflammatory infiltrates and formation of the obstructive lesions (Blair, Haven, and Bauer 2016). These MVs only induced ICAM-1 expression in the pulmonary artery and not the pulmonary microcirculation (Blair, Haven, and Bauer 2016). Again, this implicates either a selective targeting mechanism or cellular sorting and use of EV populations.
Formation of vascular lesions requires proliferation and apoptosis resistance of pulmonary artery endothelium and pulmonary artery smooth muscle cells (Cool et al. 1999; Machado 2012; Mitani et al. 2001). Extracellular vesicles isolated from the plasma of the monocrotaline-induced mouse model stimulate right ventricular hypertrophy and remodeling of pulmonary vessels when injected into normal healthy mice. Injection of EV-depleted plasma from monocrotaline mice did not induce any observable PH traits in normal mice, supporting the idea that the EVs are responsible for injury. When these same EVs are used to treat pulmonary vein endothelial cells, the cells became apoptotic resistant. Apoptosis resistance and proliferation of pulmonary artery smooth muscle cells is also induced, in part, by exosomes derived from stimulated pulmonary endothelium. However, the agent used to stimulate exosome release in these studies by Zhao, et al. was LPS, not a usual PH stimulus such as hypoxia. This raises the question as to whether the observed apoptosis resistance would occur under PH (Peterson et al. 2008; Shai et al. 2012; Kucharzewska et al. 2013). More recent work on translationally controlled tumor protein (TCTP) reveals it is expressed in exosomes from blood outgrowth endothelial cells derived from PAH patients. The protein can be transferred via exosomes between endothelial and vascular smooth muscle cells. TCTP confers apoptosis resistance in tumor cells, and this data suggests a potential mechanism dependent on exosome delivery in PAH (Ferrer et al. 2018).
One of the confounding issues surrounding PAH is that it is most frequently a diagnosis of exclusion. The gold standard for diagnosis is right heart catheterization, which does not come without significant patient risk. For a number of years investigators have worked toward identifying circulating, easily accessible biomarkers. The use of brain-type natriuretic peptide (BNP or pro-NT-BNP) has increased significantly as an indicator of pathophysiologic changes in the ventricle. However, this marker is not apparent until late-stage damage has occurred. These insights are pushing investigators to search for dynamic biomarkers reflective of the ongoing pulmonary arteriopathy and indicators of PAH severity.
As mentioned before, one characteristic of a subset of MVs is that they are pro-coagulant expressing tissue factor and phosphatidylserine on their external membranes. Using samples from 20 patients with PAH, Bakouboula and others found increased pro-coagulant MVs in blood drawn from the pulmonary artery during right heart catheterization. The highest expression of tissue factor on MVs was found in patients with severe disease in World Health Organization functional class 3 or greater. They also identified a population of CD105-positive MVs that were significantly higher than controls (Bakouboula et al. 2008). CD105 is endoglin, an accessory receptor transforming growth factor-β, which may play a role in excessive endothelial cell proliferation during vascular lesion formation in PAH (Harrison et al. 2003). However, CD105 is not specific for endothelial cells and in these investigations the authors report there is no significant change in other markers of endothelial MVs such as PECAM or E-selectin. This may be due to a technical issue limiting the study to analysis of only pro-coagulant MVs bound to annexin-V. More recent work has indicated that not all MVs or extracellular vesicles exhibit phosphatidylserine on their membrane. While the focus on procoagulant MVs began to yield some insight into the pulmonary vascular disease, this limitation may have overlooked other vital microvesicle populations.
Using centrifugation and flow cytometry, not selective to the procoagulant activities, Amabile et al. examined microvesicles in the systemic circulation of pulmonary hypertension patients. PH patients in their study had increased circulating E-selectin, VE-cadherin, and PECAM positive MVs, all markers of endothelium. The PECAM and VE-cadherin positive MVs in this study correlated with the mean pulmonary artery pressure and with BNP, suggesting that these EC-MVs are likely indicators of the hemodynamic severity of PH. E-selectin microvesicles didn’t correlate with hemodynamics, but did correlate with high-sensitivity C reactive protein, a marker of inflammation in vascular disease (Amabile et al. 2008). These data suggest subpopulations of EC-MVs may be indicative of the multiple injuries that occur in a multifactorial disease such as pulmonary hypertension. In a follow up study, this group analyzed EC-MV populations from patients undergoing right heart catheterization for diagnosis of PH to determine whether any of the EC-MV populations could be predictive of worse outcomes. These patients were not receiving any PH-specific therapy at the time of the catheterization. Increased E- selectin-positive EC-MVs, above a threshold level of 353 events/μL, correlated with poor outcomes including hospital admission for right heart failure or death (Amabile et al. 2009). They also determined, in follow-up visits, that none of the EC-MVs were reflective of the addition of PH therapy. Interestingly, this group did not examine endoglin-positive microvesicles in either study, but these data support the notion that pre-selecting for procoagulant MVs did limit the populations that can be analyzed and may be beneficial as biomarkers. These observations show why it is vital to report specific methods and for proper comparisons, use the same method of isolation and characterization. While the field is in flux as to the definition of terms for the different extracellular vesicles and even isolation protocols, evidence is clearly mounting that their value as biomarkers is high.
While EVs that are upregulated during pulmonary vascular diseases are frequently detrimental and indicative of injury, some studies highlight their role in repair of the vasculature. For many years stem cell therapies were touted for being the magic bullet for vascular diseases. However, studies utilizing stem cell, or more appropriately mesenchymal stromal cell (MSC), conditioned media revealed that the media itself proffered the same repair effects as the cells themselves. Recently, EVs isolated from MSC media have been tested in pulmonary hypertension. In the hypoxic model of PH, pre-treatment of mice with injections of MSC-EVs (exosomes) decreased circulating markers of inflammation, prevented inflammatory cell infiltration, and blocked the pulmonary vascular remodeling. Further, media cleared of MSC-EVs did not have these effects, nor did exosomes from cultured mouse lung fibroblasts (Lee et al.2012). MSC extracellular vesicles (microvesicles) have also been injected in established PAH in rats and reduced the mean pulmonary artery pressure and vascular remodeling(Chen et al. 2014). These data are supported in work from Aliotta et al. that found that MSC-exosomes prevented and reversed monocrotaline-induced PAH and this may be dependent on the microRNA cargo of the exosomes. Several miRNAs identified in the MSC-exosomes are considered anti-inflammatory and anti-proliferative (Aliotta et al. 2016). While these miRNAs have been vetted in cell culture systems, it remains to be seen whether exosomes can deliver miRNAs in vivo. Studies of extracellular vesicles as drug delivery tools are in their early stages, but to truly capitalize on the usefulness of these particles we have a great deal about cellular processing to understand.
Endothelial EVs in ARDS and associated conditions
Acute Respiratory Distress Syndrome (ARDS) is a common devastating complication of clinical disorders such as sepsis, pneumonia, and trauma (Matthay, Ware, and Zimmerman 2012). ARDS is defined as an acute syndrome of lung inflammation and increased alveolar-capillary permeability associated with severe hypoxia and bilateral infiltrates on chest radiographs with no evidence of left heart failure (studied in preclinical models as acute lung injury) (Force et al. 2012). Despite recent advances in the management of these critically ill patients, the mortality rate remains unacceptably high (>30%). To date, there is no effective pharmacologic treatment, and the only intervention proven to reduce mortality in ARDS is mechanical ventilation, which can also aggravate lung injury, a process termed ventilator-induced lung injury (VILI) (Slutsky and Ranieri 2013).
A key event in the pathogenesis of ARDS is dysfunction of the lung endothelium caused by a variety of pulmonary or systemic inflammatory stimuli, including bacteria and their associated molecules (e.g. LPS and exotoxins), thrombin, mechanical stretch, and cytokines (Maniatis et al. 2008; Maniatis and Orfanos 2008). These stimuli lead to a number of responses including endothelial adhesion molecule expression, release of pro-inflammatory mediators, EC cytoskeleton rearrangement, apoptosis, etc. These events contribute to lung injury by promoting EC barrier disruption and excessive translocation of inflammatory immune cells into the lungs.
An increasing number of studies are demonstrating that extracellular vesicles are linked to ARDS and acute lung injury (ALI) pathogenesis. Densmore et al. were the first to investigate the role of endothelial EVs in ALI (Densmore et al. 2006). In their studies, EC-EVs induce endothelial dysfunction by causing impairment of endothelium vasodilation in both mouse and human ex vivo isolated vessels. In vivo, EC-EVs derived from PAI-1-stimulated EC, when injected intravenously into Brown Norway rats lead to disruption of the alveolar-capillary barrier, induce pulmonary edema, and stimulate interstitial neutrophil accumulation, all characteristics of ALI. In mice, EC-EVs caused increased pulmonary capillary permeability that was significantly potentiated in LPS-treated mice, suggesting that EC-EVs could enhance the inflammatory response induced by other primary insults. Mechanistically, the vesicles caused a decrease in NO production in stimulated EC by targeting the eNOs activity and its association with Hsp90 (Densmore et al. 2006). These observations were complemented with subsequent studies from the same group demonstrating that EC-EVs exert additional pro-inflammatory properties. Specifically, EC-EVs when injected intravenously into mice trigger pulmonary and systemic release of IL-1 β and TNF-α, recruitment and activation of neutrophils, and similar to their previous study, exacerbation of pre-existing lung injury (induced by LPS) (Buesing et al. 2011).
Several other studies aimed to characterize the levels and biological roles of EC-EVs in ALI. In a mouse model of endotoxemia-induced ALI, LPS triggers the release of different subpopulations of endothelial MVs (CD144+, CD62E+, CD54+, CD31+), while treatment with simvastatin, which protects against ALI, reduced significantly the levels of all shed EC-EVs (Yu et al. 2017). In a rat model of ALI, there was an increase in the number of circulating CD54 (ICAM- 1) positive MVs in LPS-treated animals compared to controls (Li et al. 2015). MVs isolated from the blood of LPS-treated rats when injected intratracheally or intravenously to naïve rats induce both systemic and local pulmonary inflammation (Li et al. 2015). A candidate protein that could mediate the inflammatory responses of LPS-induced EC-EVs is sphingosine-1-phosphate receptor 3 (S1PR3) (Sun et al. 2012). Nitrated S1PR3 has been detected in the plasma of murine ALI and in humans with severe sepsis-induced ARDS. EC barrier disrupting agents, such as LPS, induce S1PR3 nitration and shedding of EC-EVs that contain S1PR3. These EVs, in a S1PR3-dependent manner, induce EC permeability, suggesting again a critical role for EVs in ALI pathogenesis (Sun et al. 2012). In a mouse model of LPS-induced direct ALI, Soni et al. found that microvesicles derived from alveolar macrophages and epithelial cells are rapidly released after LPS into the alveolar space, however, in this study, the authors did not assess the EC-EVs (Soni et al. 2016). Lung endothelial cell apoptosis, an important feature of ALI, is significantly induced in the absence of intersectin-1 (ITSN-1), a highly abundant lung EC membrane- associated protein. Interestingly, mice lacking ITSN-1 release into the circulation increased levels of EVs that carry Alk5 (or transforming growth factor ß receptor I), which they transfer to dysfunctional EC to enhance their survival (Bardita et al. 2015). Apart from apoptosis, other forms of cell death can result in severe dysfunction of the endothelial barrier. Intracellular LPS that enters the cell via bacterial vesicles or by bacterial breaching of the plasma membrane triggers pyroptosis in ECs by binding to caspases 4/5 in humans and caspase-11 in mice (Cheng et al.2017). EC pyroptosis contributes to ALI by causing lytic cell death and release of the pro- inflammatory cytokine IL-1 ß. The importance of this pathway was highlighted by data showing that mice deficient for endothelial caspase-11 are protected against ALI induced by endotoxemia. Interestingly, pyroptotic ECs release EVs (CD31+) in vitro through a mechanism that requires LPS internalization (Cheng et al. 2017). In vivo, endotoxemia in wild-type mice is associated with a significant increase in circulating endothelial EVs, an effect that is almost completely abolished in endothelial-specific caspase-11 deficient mice (Cheng et al. 2017), suggesting that caspase-11 is required for EC-EV production. In an experimental rat model of acute pancreatitis-induced lung injury, an increase in circulating exosomes with unique proteome was observed. These exosomes could reach the alveolar compartment, where they could be internalized by macrophages. In vitro experiments showed that plasma exosomes induced under inflammatory conditions polarize the alveolar macrophages towards a pro-inflammatory phenotype (Bonjoch et al. 2016). However, the source of these exosomes was not determined. In another model of pulmonary inflammation, serum exosomes from rats that inhaled zinc-oxide nanoparticles found to carry a unique miRNA cargo compared to healthy rats. These EV-associated miRNAs may contribute directly or indirectly to lung inflammation (Qiao et al. 2018).
Excessive mechanical forces, generated during injurious mechanical ventilation (i.e. VILI), can damage the lung vascular endothelial cells or induce pro-inflammatory cascades (Wang et al. 2017). Similar to barrier disruptive agents, such as LPS, pathologic mechanical cyclic stretch (18% cyclic stretch; mimics stresses generated in vivo during VILI) stimulates human lung endothelial cells to release increased numbers of EC-MVs (Letsiou et al. 2015; Vion, Birukova, et al. 2013). Vion et al. explored the mechanisms that regulate EC-MV generation in vitro and found that a pan-caspase inhibitor limits the 18% cyclic stretch-induced EC-MV generation, while Rho kinase or calpain inhibitors had no effect (Vion, Birukova, et al. 2013). Proteomic analysis of MVs isolated from the supernatant of HPAEC revealed unique proteins in MVs-induced by 18% cyclic stretch compared to MVs derived from control or LPS-treated cells (Letsiou et al. 2015). 18% cyclic stretch-EC-MVs, similar to PAI-1-EVs, induce lung injury in mice as assessed by increased alveolar-capillary permeability and pulmonary neutrophil accumulation (Letsiou et al. 2015). In the same study, it was shown that VILI mice had increased levels of CD62E positive MVs in plasma and bronchoalveolar lavage (BAL) compared to control mice (Letsiou et al. 2015). Other studies later confirmed the hypothesis that injurious mechanical ventilation is associated with EC-MV production. In a murine model of VILI, different subpopulations of endothelial MVs (CD144+, CD62E+, CD31+, CD54+) were significantly elevated. Treatment of mice with the protective agent ligustrazine (Tetramethylpyrazine; TMP) reduced VILI-induced pulmonary edema, markers of endothelial dysfunction (serum and lung tissue E-selectin and VCAM-1), and levels of circulating EC-MVs (Pan et al. 2017). Along that same line, high levels of circulating CD31+MVs were detected in rats exposed to high tidal volume mechanical ventilation (Cabrera-Benitez et al. 2015). Taken together, these studies suggest that endothelial EVs could serve not only as markers of endothelial dysfunction in ALI, but also as therapeutic targets since they appear to mediate numerous inflammatory responses.
Only a few studies have characterized the cellular origin of EVs in ARDS patients. Guervilly et al. found that leukocyte MVs are highly increased in the alveolar space of ARDS patients, while endothelial microvesicles (CD31+/CD41-) were detectable in the BAL of 6 patients (total 52), in 1 patient in the ventilated control group (ICU patients ventilated for non-pulmonary disorders, total 10), but in none of the control group (healthy individuals) (Guervilly et al. 2011). In a more recent study, endothelial EVs (CD31+/CD41-) were found elevated in the plasma of ARDS patients compared to healthy controls (Cheng et al. 2017). In the same study, EC-MVs from patients with cardiogenic pulmonary edema were also assessed and compared to healthy controls. No significant difference was observed between these two groups, suggesting that EC-MV increase in ARDS is a specific indicator of endothelial destruction occurring during endotoxemia (Cheng et al. 2017). Although these data are exciting and support in vivo and in vitro observations, due to the small number of patients tested, their importance should be evaluated in follow up larger-scale studies. Clearly, however, EV levels are altered in ARDS. Bastarache et al. reported increased levels of pro-coagulant EVs in the edema fluid of ARDS patients compared to patients with hydrostatic pulmonary edema (Bastarache et al. 2009). Interestingly, a more recent study demonstrated that the levels of PS+MVs were lower in the plasma of ICU patients who developed ARDS compared to those who did not, however, data characterizing MV phenotypes was not included (Shaver et al. 2017). An important observation of this study was that the MV levels differed according to the risk factors for ARDS, and pneumonia patients had the highest amount of PS+MVs compared to patients with sepsis, trauma, aspiration, or those receiving multiple transfusions.
Pneumonia, the most frequent cause of ARDS and sepsis, is a severe infection of the lung. A variety of organisms, including bacteria, viruses, or fungi can cause pneumonia through mechanisms that involve complex pathogen-host interactions. Similar to ALI pathogenesis, the lung endothelium is a primary target in severe lung infections (Muller-Redetzky, Suttorp, and Witzenrath 2014). Despite antibiotic or other appropriate treatments, pneumonia can progress to life-threatening conditions through mechanisms that are not fully understood. Moreover, the role of EVs in mediating the pathogenic mechanisms of pneumonia is just beginning to be explored and our knowledge is limited to vesicles originating from pathogens and epithelial or immune cells. For example, administration of influenza virus (IAV) in mice upregulates IL-36α gene and protein expression levels in the lungs, and in vitro, IAV induces IL-36α secretion from alveolar epithelial cells through MVs (Aoyagi et al. 2017). IL-36γ can also be secreted within MVs and exosomes by lung macrophages in response to Gram-negative (Klebsiella pneumoniae) and -positive (Streptococcus pneumoniae) bacteria (Kovach et al. 2016). Alveolar epithelial cells upon stimulation with pneumolysin, a Streptococcus pneumoniae exotoxin, release MVs that contain mitochondrial DNA (Nerlich et al. 2018). A role for endothelial MVs in pneumonia was suggested in a recent study by Lashin et al. who analyzed MVs from plasma of septic patients due to community-acquired pneumonia (CAP) and healthy volunteers (Lashin et al. 2018). The authors found that MVs (Annexin V/CD51+) potentially derived from endothelial cells were significantly elevated in CAP patients compared to healthy individuals. More importantly, in the same study, it was demonstrated that in patients with CAP, the numbers of MVs originating from endothelial and not other sources were higher in non-survivors suggesting that the EC-MVs could serve as a prognostic marker for survival. These observations clearly indicate that the role of endothelial EVs in pneumonia warrants further investigation.
Sepsis, a systemic inflammatory response to infection, is a critical illness that can progress to ARDS, multi-organ failure, and even death. It is characterized by excessive endothelial activation, failure of several EC functions, and microvascular leakage (Ince et al. 2016). Since the endothelium is primarily affected in sepsis, it is with no surprise that endothelial EVs are also involved in the pathogenesis and progression of the disease (Reid and Webster 2012). Data from clinical studies, however, seem conflicting. One of the first studies in the field that was published almost 20 years ago showed that endothelial CD62E+ MVs and platelets MVs were decreased in patients with multi-organ dysfunction syndrome (MODS) and sepsis compared to healthy controls, while only the granulocyte MV subset was increased in patients (Joop et al. 2001). Ogura et al. then measured the EC-MV levels in patients with severe systemic inflammatory response syndrome (SIRS) and found that endothelial MVs were significantly increased in these patients compared to healthy individuals (Ogura et al. 2004). These observations were confirmed in a cohort of septic shock patients. This latter study showed that platelet- and endothelial-derived MVs (CD146+) were increased in septic patients when compared to nonseptic subjects (Mostefai et al. 2008). In a more recent study, endothelial dysfunction measurements in severe sepsis correlated with lower numbers and reduced function of circulating cells implicated in endothelial repair, while EC-MV (CD31+/CD42b-) numbers did not differ between patients and healthy controls (van Ierssel et al. 2013). Using high-sensitivity flow cytometry, Lehner et al. analyzed EC-MV subpopulations in septic shock patients (Lehner et al. 2016) and demonstrated that CD31+/CD41- MVs were increased by a 3-fold in the plasma of patients compared to healthy volunteers. CD144+, CD62E+, and CD106+ endothelial MVs were low in both groups. Interestingly, the authors argued that CD31 is also present in some leukocyte subsets and hypothesized that CD31+/CD41-MVs are mostly not endothelium derived. Extensive disease heterogeneity in sepsis as well as the non-standardized EC-EV analysis may partially explain these variable results, and additional studies are needed to clarify the role of EC-EVs in sepsis.
Additional studies have addressed the functional role of sepsis-derived MVs. Mostefai et al. demonstrated that isolated MVs from septic patients when injected intravenously into mice enhance the contraction of mouse aorta in response to LPS and serotonin, and increase the production of the vasoconstrictor thromboxane A2. These observations suggest that these MVs might have protective effects against vascular hyporeactivity in sepsis (Mostefai et al. 2008). Another study showed that active caspase-1 is released within microvesicles in sepsis and these MVs could induce apoptosis in healthy lymphocytes (Exline et al. 2014). This MV effect was reproduced in vitro after stimulation of whole blood with LPS. LPS-MVs, similar to sepsis-derived MVs, could induce lymphocyte cell death, a response, which was blocked after inhibition of either caspase-1 or disruption of MVs (Exline et al. 2014). Herrmann et al. then explored the pro-inflammatory and procoagulant properties of MVs isolated from septic patients (Herrmann et al. 2015). Exposure of EC to plasma (from septic patients)-derived MVs did not lead to significant inflammatory responses. In contrast, these MVs had increased procoagulant activity compared to control patients.
Very recently, a new study presented promising data on the therapeutic effects of EPC- derived exosomes against sepsis (Zhou et al. 2018). For this study, the authors employed a cecal ligation and puncture (CLP) murine model and EPC-exosomes were administrated intravenously 4 hours after CLP. Mice treated with EPC-exosomes had improved survival, reduced lung and renal vascular leakage, and diminished liver and kidney dysfunction. In order to elucidate the underlying protective mechanisms, the investigators analyzed the exosomal miRNA content. This analysis revealed that the exosomal miR-126–3p and 5pcould suppress LPS-induced HMGB1 and VCAM-1 in HMEC. These data suggest that EPC-exosomes can prevent endothelial dysfunction and could potentially improve sepsis outcomes (Zhou et al. 2018).
Endothelial EVs in COPD, asthma, and BPD
Chronic obstructive pulmonary disease (COPD) and asthma are chronic lung inflammatory conditions associated with high morbidity (Green and Turner 2017). COPD is characterized by lung destruction, which normally is not fully reversible and progresses over time and results in airflow obstruction and reduction of lung capacity. COPD can be induced by chronic exposure of the airway to environmental toxic substances such as cigarette smoke (CS) that cause epithelial injury, accelerate epithelial senescence, and induce pulmonary vascular impairment (Green and Turner 2017). In addition, a small percentage of patients develop COPD as a result of alpha 1 anti-trypsin (A1AT) deficiency. Asthma is characterized by variable airflow obstruction and airway hyperresponsiveness (AHR) and can be triggered mainly by allergens (Fujita et al. 2014).
Lung EC contribute to the pathogenesis of COPD and asthma in different ways (Huertas et al. 2018; Green and Turner 2017). In COPD, a) EC activation and up-regulation of EC adhesion molecules mediate the transendothelial migration (TEM) of neutrophils to the lung tissues, b) there is an excessive apoptosis of pulmonary endothelial cells, which contributes to the destruction of the alveoli, c) there is an increased percentage of senescent endothelial cells, and d) several endothelial functions, such as NO production, are impaired (Green and Turner 2017). Moreover, the endothelial progenitor cells that are critical mediators in the repair process of the injured endothelium appear to malfunction in both COPD and asthma (Green and Turner 2017). The role of the lung endothelium in asthma has been studied toa lesser extent, but there is enough evidence to support that its functions are altered in the course of the disease. For instance, migration of eosinophils, which are critical players in asthma, is mediated by the upregulation of EC adhesion molecules and EC-derived chemokines that are present in asthmatic subjects and in ECs exposed to allergens (Green and Turner 2017).
It is well documented that EVs originating from different cell types play an important role in COPD pathogenesis and progression (Takahashi and Kubo 2014; Kadota et al. 2016; Kubo 2018). Moreover, several clinical studies have demonstrated that EVs of endothelial origin are altered in COPD. Smokers with signs of early emphysema had increased plasma levels of EC-EVs (CD42b-/CD31+), 76% of which were ACE positive suggesting that they originate from the pulmonary capillaries (Gordon et al. 2011). Increased EC-MV (CD144+, CD31+/CD41-, CD62E+ MVs) release was also evident in patients with stable COPD compared to non-COPD volunteers, while patients with exacerbated COPD had also higher EC-MV levels compared to stable COPD (Takahashi et al. 2012). The same study identified a subset of EC-MVs (CD62E+) to be significantly higher in COPD patients with frequent exacerbations than in those with fewer exacerbations. This could be explained by the fact that during exacerbations, there is an increase in endothelial activation (CD62E upregulation) and damage that triggers the release of these vesicles. This suggests that measuring the specific EC-MV subpopulations could serve as an indicator of patient’s susceptibility to exacerbation. The same research group, later, studied the association between EC-MV numbers and changes in forced expiratory volume in 1s (FEV1) in patients with COPD (Takahashi et al. 2014). They found that the number of CD62E+ and CD144+ (VE-cadherin) MVs showed significant negative correlation with annual FEV1 changes and that high CD62E+ MV levels in stable COPD patients are predictive of rapid FEV1 decline. More evidence about the role of EC-MVs in COPD was provided by Thomashow et al., who reported that EC-MVs (CD31+/CD42-) were increased in mild and severe COPD and emphysema. In this study, CD62E+ MV levels were increased only in severe COPD patients (Thomashow et al. 2013). To assess the role of cigarette smoke in regulating the endothelial function in COPD, Strulovici-Bare et al. compared the plasma EC-MV levels in non-smokers and healthy or COPD smokers, and they found that smokers had elevated levels of circulating EC-MVs (CD31+/CD42b-) (Strulovici-Barel et al. 2016). The EC-MV levels remained stable over a period of 1 year in subjects who did not change their smoking habits, while smoking cessation resulted in a decrease of EC-MVs in healthy smokers but not in COPD smokers. Very interestingly, Heiss et al. showed that secondhand smoke (SHS) can also have an effect on endothelial MV release. For these studies, they exposed healthy non-smokers to SHS and measured endothelial responses and EC-MV levels. They found that brief exposure to SHS results in sustained endothelial activation and injury as assessed by mobilization of dysfunctional EPCs and elevated EC-MV (CD144+, CD31+/CD41-, CD62E+) and VEGF levels (Heiss et al. 2008). Endothelial MVs in COPD patients are not only elevated in the circulation but also locally. A recent study demonstrated that patients with COPD had high CD31+ MV levels in their sputum, and a correlation was found between MV levels and lung damage (Lacedonia et al. 2016). Taken together, these data suggest that there is a persistent endothelial cell activation and damage in patients with COPD, which results in a stable release of MVs.
Data obtained from in vitro and in vivo studies correlate well with the clinical observations. Serban et al. demonstrated a direct effect of cigarette smoke on EV release (Serban et al. 2016). In this study, CS exposure resulted in increased production of EC-EVs in mouse plasma and in the supernatants of HLMVEC. In a previous study, the responses of pulmonary versus systemic (aortic) endothelial cells to CS were investigated. MVs positive for CD31 and CD146, but not for VE-cadherin and CD62E, were released from HLMVEC upon CS exposure, while in aortic endothelial cells, only the CD146+ MV subtypes were slightly induced (Takahashi et al. 2013). In vivo, Wistar rats exposed to CS for a period of 2–6 months had increased circulating levels of CD31+/CD42b- MVs compared to unchallenged rats, and this was accompanied by a decline in pulmonary function (Liu et al. 2014). Finally, an important finding for the field of COPD was that EVs derived from healthy pulmonary endothelial cells carry and deliver the cytoprotective A1AT to lung epithelial cells, unveiling a new pathway in A1AT trafficking (Lockett et al. 2014).
In asthma, bronchial epithelial cells, mast cells, T or B cells, and dendritic cells are the major sources of extracellular vesicles (Fujita et al. 2014; Wahlund et al. 2017), while very limited information exists on endothelial EVs. Duarte et al. investigated the phenotype of circulating EVs in asthmatic patients and they found that platelet MV levels were significantly increased in asthma compared to non-asthmatics (Duarte et al. 2013). In this study, endothelial MVs (CD31+/CD42b) were also increased in asthma by almost 50%, however, the difference was not statistically significant. Since lung ECs are activated in asthma, additional studies are needed to fill the knowledge gap about the role of EC-EVs in this disease.
Bronchopulmonary dysplasia (BPD) occurs in low birth weight and premature infants and is characterized by pathological evidence of large oversimplified alveoli and dysmorphic pulmonary vasculature (Hwang and Rehan 2018). BPD usually occurs when infants are born during the time when lungs are developing from the canalicular stage to the saccular stage, which is differentiated by the development and function of type II pneumocytes that begin to produce surfactant (Collaco and McGrath-Morrow 2018; Hwang and Rehan 2018). Structurally, this coincides with capillary invasion into the mesenchyma and establishment of the pulmonary microcirculation. Thus, the loss of proper development of the pulmonary circulation also increases the incidence of pulmonary hypertension in BPD infants. There are numerous proposed causes of BPD, including intrauterine growth restriction, pre-eclampsia, pre- and postnatal inflammation, blood transfusion, and maternal smoking (Malleske, Chorna, and Maitre 2018). A number of these causes of BPD including pre-eclampsia, inflammation, blood transfusion and smoking are all stimuli for the release of deleterious extracellular vesicles (Ling et al. 2014; Redman and Sargent 2007; Kim et al. 2018; Aung et al. 2017; Chang et al. 2017; Liu et al. 2014; Serban et al. 2016).
Treatment or support of infants with BPD often includes steroid therapy, however the results are highly controversial due to long-been studied
term risks associated with steroid use (Doyle, Ehrenkranz, and Halliday 2014b, 2014a; Doyle et al. 2014). Along with steroid use, replacement of surfactant has been used for a number of years and although use of surfactant decreases the need for ventilation, it does not decrease incidence of BPD (Stevens et al. 2007). There are newer versions of artificial surfactant that are more resistant to inactivation that may prove beneficial (Sato and Ikegami 2012). Mechanical ventilation strategies have been adapted over the previous decade to accommodate lung capacity of newborns and although mechanical stress still induces injury, more recent work suggests a lower incidence of BPD using volume-targeted ventilation (Wheeler et al. 2011). However, ventilator use also increases potentially dangerous extracellular vesicles as mentioned in a previous section.
A limited number of studies have examined the potential roles for extracellular vesicles in BPD. Exosomal miRNA content was recently tested for potential as a biomarker in extremely premature infants. Lal et al. utilized bronchoalveolar lavage fluid from infants with severe BPD, hyperoxia-exposed mice, and hyperoxia-exposed bronchial epithelial cells, the latter serve as models of the disease, and found that exosomes were increased in total number and had increased expression of miR-876–3p (Lal et al. 2018). In a prospective cohort study of infant aspirates, miR-876–3p was reduced in BPD-susceptible compared to BPD-resistant infants. The majority of exosomes in this study were identified as epithelial in origin, which is not surprising considering the source material. Much of the remainder of the exosome population was neutrophilic, suggesting a bronchial inflammatory response in the preterm infants. In work from Veerappan and others, mast cell exosomes were also identified in the aspirates of preterm infants (Veerappan et al. 2016). These studies clearly indicate that exosomes have the potential to be a powerful predictor of disease.
Exosomes, in particular those that are derived from mesenchymal stromal cells, are touted as the effector agent in MSC therapy for a variety of vascular diseases and BPD is no exception. Exosomes carefully characterized from media conditioned by human MSC cultures were used to treat newborn mice exposed to hyperoxia, a common model of BPD. MSC-exosomes delivered intravenously during postnatal exposure to hyperoxia ameliorated classic histologic features of BPD such as alveolar simplification, septal fibrosis, and loss of peripheral pulmonary vessels. Further, the exosomes improved lung function including decreased pulmonary hypertension and improved pressure volume loops. MSC-exosomes frequently modulate inflammatory cell differentiation and infiltration (Willis et al. 2018). In the mouse model of BPD, the exosomes induced signaling consistent with the cytokine profile of anti-inflammatory macrophages and regulated alveolar macrophage phenotype in an in vitro model.
Overall, while a role for exosomes as a biomarker of the severity of BPD or the potential for MSC-exosomes as a treatment are clear, the mechanistic studies for either remain incomplete. There is no currently available literature specifically on endothelial EVs in BPD. While BPD is clearly a disease involving poor development of the pulmonary vasculature and endothelial dysfunction, this area shows a significant knowledge gap to be investigated in the future.
Endothelial EVs in obstructive sleep apnea
Obstructive sleep apnea (OSA) is a respiratory disorder characterized by recurrent episodes of partial or complete obstruction of the upper airways during sleep that leads to episodes of intermittent hypoxia. OSA is associated with other conditions such as hypertension and cardiovascular disorders and affects a big portion of the population. Interestingly, several studies have demonstrated that OSA syndrome is characterized by endothelial dysfunction (Stiefel et al. 2013; Lurie 2011; Feng, Zhang, and Chen 2012). Reactive oxygen species (ROS) and other pro-inflammatory mediators generated in OSA can lead to EC activation and injury, and markers of endothelial injury such as ICAM-1, VCAM-1, and E-selectin (CD62E) are elevated in the circulation of patients with OSA (El-Solh et al. 2002). OSA directly induces vascular endothelial dysfunction by reducing the endothelial repair capacity, promoting endothelial apoptosis, and decreasing endothelial NO production (Jelic et al. 2008; Jelic et al. 2009).
Numerous studies have demonstrated an important role for EVs in the OSA syndrome and EVs originating from platelets, leukocytes, and endothelial cells are the most studied ones (Trzepizur et al. 2014; Khalyfa, Kheirandish-Gozal, and Gozal 2017). Jelic et al. explored the levels of endothelial EVs in a cohort of newly diagnosed patients with OSA and no other cofounding factors including obesity. They found that EC-MV (CD31+/CD42b-) levels were increased in patients compared to healthy controls and that the EC-MV levels were negatively correlated with the number of circulating EPCs and baseline brachial artery flow mediated dilation (FMD) values (FMD represents indirect maker of endothelial nitric oxide-mediated reactivity) (Jelic et al. 2009). Treatment of these patients with CPAP (continuous positive airway pressure) normalized the levels of EPCs and tended to decrease the EC-MV levels and the increase of FMD. Concurrently, another group published a study measuring the circulating levels of MVs in patients with minimally symptomatic OSA (Ayers et al. 2009). In this study, MVs derived from platelets and leukocytes, but not endothelial cells (measured as CD31+/CD41-) were significantly increased in OSA patients compared to healthy controls. The same group, in a follow-up study, performed a randomized controlled trial, which aimed to examine the effect of a 2-week withdrawal of CPAP treatment on levels of circulating MVs (Ayers et al. 2013). CPAP withdrawal led to a recurrence of OSA and an increase in the levels of endothelial (CD62E+) MVs compared to patients who remained in CPAP. An important finding was that CPAP withdrawal did not cause significant changes in CD31+/CD41-MVs, as well as in granulocyte, leukocyte, and platelet- derived MVs.
In another cohort of patients, different endothelial MV subpopulations (CD31+/CD42-, CD31+/Annexin V+, CD62E+) were found higher in OSA patients compared to controls, while CPAP therapy decreased the levels of the CD62E+ subset of EC-MVs but had no effect on the other EC-MV subtypes (Yun et al. 2010). This study also examined how OSA severity influences EC-MV levels. Interestingly, only the levels of CD31+/CD42- subset of EC-MVs differed among mild, moderate, and severe OSA. Priou et al. then compared the plasma MV phenotypes in OSA patients with ODI (oxyhemoglobin desaturation index≥3%) ≥10 events per hour of sleep (desaturator group) and ODI<10 events per hour (nondesaturator group) and also tested the biological functions of OSA-associated MVs (Priou et al. 2010). In these settings, granulocyte- derived MVs (CD66B+ and CD62L+ MVs) were increased in the desaturator group compared to the nondesaturator. CD146 or MCAM+, platelet, annexin V+, and erythrocyte MVs did not differ between groups. It has to be noted that for this study, CD146 was used to identify MVs of endothelial origin, and therefore the data obtained here regarding the EC-MV levels cannot be directly compared to other studies. Another finding of this study was that MVs from desaturators could decrease NO production in endothelial cells in vitro and increase the expression of endothelial adhesion molecules and cyclooxygenase-2. Ex vivo, desaturators-derived MVs impair the endothelium-dependent relaxation in response to acetylcholine (Priou et al. 2010). In a recent study, Bikov et al. investigated the diurnal changes in circulating MVs in OSA and found that CD41+ and annexin V+ MVs increase significantly at 5 o’clock in the evening and that this increase was more pronounced in patients with greater severity (Bikov et al. 2017). Even though the endothelial MVs were not measured in this study, these data highlight the diurnal pattern of MV production and the importance of being consistent when collecting MVs. On the biological role of OSA-MVs, Tual-Chalot et al. showed that circulating MVs from OSA patients carry VEGF and stimulate endothelial cells to secrete endothelin-1, which triggers angiogenesis (Tual-Chalot et al. 2014).
Khalyfa et al. have been investigating the role of exosomes in OSA (Khalyfa, Kheirandish-Gozal, and Gozal 2017). Their studies employed a human model of intermittent hypoxia (IH) and they demonstrated that exosomes isolated from plasma samples (from individuals who have been exposed to IH) were primarily derived from endothelial sources. IH-induced exosomes were biologically active and could induce endothelial dysfunction; IH-exosomes could disrupt the EC barrier, reduce eNOs expression and phosphorylation, increase ICAM-1 expression and monocyte adhesion to EC (Khalyfa et al. 2017; Khalyfa et al. 2016). Moreover, these studies used microarray approaches to reveal an altered exosomal miRNA cargo in exosomes isolated after IH exposure compared to baseline (before exposure) and an altered mRNA profile in ECs treated with IH-exosomes. These findings clearly demonstrate that endothelial exosomes also play a major role in the pathogenesis of OSA.
Endothelial EVs in other lung diseases
Lung endothelial dysfunction and injury characterizes other severe lung diseases including idiopathic pulmonary fibrosis, cystic fibrosis, and pulmonary thromboembolism.
Idiopathic pulmonary fibrosis (IPF) is an interstitial lung disease characterized by aberrant accumulation of fibrotic tissue in the lungs parenchyma associated with poor prognosis. Multiple mechanisms have been proposed to underlie IPF pathogenesis, which include abnormal vascular repair and remodeling, and decreased levels of EPCs (Strieter and Mehrad 2009; Malli et al.2013). A very recent study by Bacha et al. was the first to establish a role for circulating MVs in IPF (Bacha et al. 2018). In this study, circulating MVs were phenotyped in recently diagnosed IPF patients (<9 months) and healthy controls. Endothelial CD62E+ MVs and platelet MVs were significantly increased in IPF patients, while the CD62E+ MVs were also associated with disease severity. Other MV subtypes that were measured (CD31+, CD144+, CD66b+) did not differ significantly between the two groups. Another important finding of this study was that endothelial microvesicles isolated from EPCs that were extracted from IPF patients induce fibroblast migration and show increased ability to generate plasmin. These observations suggest that fibrotic processes are associated with endothelial activation and that the IPF-EC-MVs may have a major role in the pathogenesis of the disease.
Cystic fibrosis (CF) is a genetic disease, caused by mutations in the cystic fibrosis transmembrane conductance regulator (CFTR) gene and is characterized by the buildup of thick mucus in the lungs and other organs. The role of MVs in CF is just beginning to be explored. Porro et al. demonstrated that MVs, mainly derived from granulocytes, were detectable in sputa obtained from CF patients (Porro et al. 2010) and that MVs derived from CF sputum are pro-inflammatory when injected into the murine lung (Porro et al. 2013). CFTR is a membrane protein present at the apical surface of epithelial cells (localized in lipid rafts), which acts as a chloride channel and regulates the transepithelial movement of water and electrolyte in exocrine tissues. In CF, there is a malfunction of this channel, because of mutations in the cftr gene (Hwang et al.2018). Interestingly, EVs are capable of carrying the protein and mRNA of CFTR, which they deliver to recipient cells to restore the CFTR chloride channel activity (Vituret et al. 2016; Gonzalez et al. 2012). Recent reports suggest that endothelial dysfunction can also be a feature of cystic fibrosis (Totani et al. 2017; Romano et al. 2001; Poore et al. 2013), however, to date, there are no studies on endothelial EVs and CF, and new studies exploring this axis could provide important insight into the pathophysiology and diagnosis of this devastating disease.
Pulmonary thromboembolism (PTE) is an acute complication of deep vein thrombosis (DVT) caused by a blood clot that blocks the blood flow in the lungs (Yan et al. 2017). EVs play a major role in promoting coagulation and therefore are considered important mediators in venous thromboembolism (VTE) conditions (Yuana, Sturk, and Nieuwland 2013; Rautou and Mackman. On the other hand, the vascular endothelium is also involved in thrombotic processes by regulating coagulation and fibrinolysis (Blann and Lip 2001) and can be damaged in severe cases of PTE (Lehnert et al. 2017). Chirinos et al demonstrated that in VTE patients, there is a marked activation of endothelial cells, platelets, and leukocytes, and an increased release of circulating EC-MVs (CD31+/CD42b-) and formation of EC-MV-leukocyte conjugates (Chirinos et al. 2005). 44% of patients recruited were diagnosed with PTE, however, a specific association of EC-MVs and PTE was not reported. In further studies, platelet and endothelial MVs were measured in plasma of patients with acute pulmonary embolism, and the data indicated that only the platelet MVs were higher in PTE patients compared to healthy controls (Bal et al. 2010). Interestingly, this difference was not statistically significant when for the analysis, PTE patients were compared to control patients (no VTE) with cardiovascular risk factors. Another study investigated the levels of endothelial MVs (CD144+) in chronic thromboembolic pulmonary hypertension (CTEPH) and PTE (Belik et al. 2016). CTPEH, which is a complication of PTE, is a chronic disease characterized by obstruction of pulmonary arteries by unresolved thromboemboli. CTEPH patients have increased levels of CD144+ EVs compared to PTE and healthy controls. Moreover, CTPEH-EVs were enriched in endoglin, which they could transfer to pulmonary endothelial cells to promote angiogenesis and inhibit apoptosis. The authors conclude that endoglin/CD144+ EVs could serve as protective mediators in CTEPH, by promoting endothelial survival and angiogenesis to repair the effects of vascular occlusion and endothelial damage. In a rat model of experimental pulmonary embolism, the protein composition of circulating EVs changes as a result of pulmonary vascular occlusion (Watts et al. 2012). Specifically, PTE-EVs carry proteins that have prothrombotic characteristics (fibronectin, fibrinogen, vWF) or can inhibit fibrinolysis (alpha 2 macroglobulin).
CONCLUSIONS AND FUTURE DIRECTIONS
The pulmonary endothelium was once considered an inert barrier. However, investigations into both health and disease of the lung have revealed that the endothelium is an active contributor to pulmonary homeostasis. The pulmonary vascular endothelium is exposed to a variety of toxic and inflammatory stimuli that can alter its normal function. It is therefore not surprising that endothelial dysfunction is a primary component in most acute and chronic lung diseases. Understanding how the lung endothelium is being affected in pathological conditions allows us to develop new endothelial-targeted therapeutics. Towards this end, it is now evident that endothelial cells under both normal and pathological conditions secrete extracellularly functional membrane vesicles. These EVs work as messengers transferring signals to recipient cells. In many cases these signals are detrimental and enhance the endothelial dysfunction, but in others the “message” is repair driven. Clinical and basic science studies have demonstrated that the numbers of endothelium-derived EVs are low under normal conditions, however, their levels, composition, and functions change dramatically in pathology-related to vascular injury. Therefore, EC-EVs not only serve as markers of endothelial dysfunction but they likely also contribute to disease pathogenesis or progression by regulating diverse cellular responses (Figure 2).
Figure 2. Endothelial EV-Cell interactions in homeostasis and lung disease.
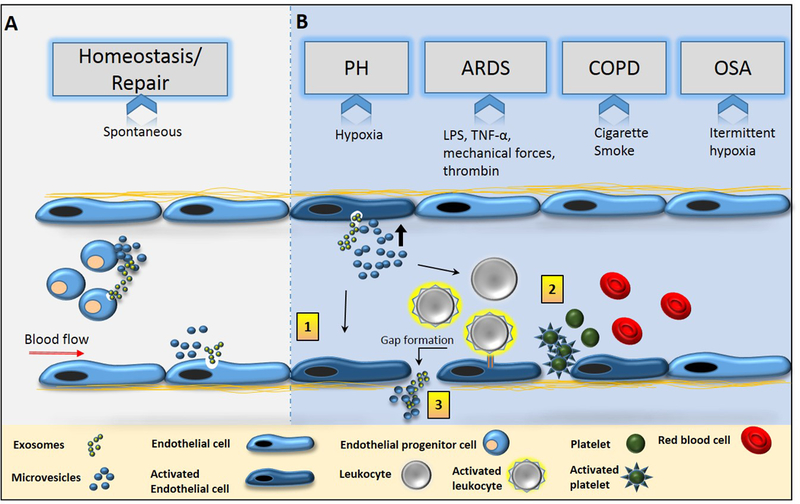
(A) Vascular or progenitor endothelial cells secrete EVs spontaneously into the circulating blood where they contribute to primary homeostatic functions and repair by promoting angiogenesis, proliferation, and other anti-inflammatory effects. (B) Increased numbers of microvesicles and exosomes are released from EC after stimulation with lung disease-relevant stimuli such as hypoxia, mechanical stretch, bacterial toxins (e.g. LPS), inflammatory cytokines (e.g. TNF-α) and other endogenous mediators (e.g. thrombin), or toxic substances (e.g. cigarette smoke). Upon their release, EC-EVs interact with the vascular endothelium (1) or modulate the activities of circulating blood cells (2) to promote lung damage by inducing endothelial dysfunction, pro-inflammatory signaling, coagulation, etc. EVs can also be released from the basolateral EC surface or escape the circulation after EC barrier disruption and target other lung cells such as smooth muscle cells and macrophages (3). Abbreviations: PH; Pulmonary hypertension, ARDS; Acute respiratory distress syndrome, COPD; chronic obstructive pulmonary disease, OSA; obstructive sleep apnea.
Numerous studies have demonstrated that EC-EV levels are altered in lung vascular diseases (as summarized in Table 2), yet there is much to be understood and evaluated. Based on the data presented here some areas of future work include: 1) advancing the current technology towards achieving reproducible data in regards to the isolation and quantification of EC-EV in clinical and basic science studies, 2) understanding the distinct characteristics of EVs generated from discrete phenotypic foci of the lung endothelium, 3) expand studies evaluating if EC-EVs can serve as markers of early endothelial dysfunction in lung diseases, 4) advance our knowledge on the functional role of EC-EVs derived during acute and chronic lung endothelial dysfunction, 5) perform thorough investigations of the mechanisms by which EC-EVs interact with recipient cells including endocytosis or direct membrane interactions under in vitro and in vivo conditions 6) evaluate the signaling mechanisms directly involved in EC-EV stimulated endothelial dysfunction, and 7) continue to characterize the lung EC-EV cargo.
Table 2.
Endothelial cell-derived extracellular vesicle (EC-EV) levels in lung diseases.
| Disease or Condition |
Model | EC-EV phenotypes | Levels |
EV
medium |
Other major findings | Reference |
|---|---|---|---|---|---|---|
| PH | Patients |
CD62E+ CD144+ CD31+/CD41- |
↑ ↑ ↑ |
Plasma | • EC-MVs (CD31+/CD41- and CD144+) predict hemodynamic severity • CD45+ increased, no changes in CD41+ |
(Amabile et al. 2008) |
| Patients |
CD62E+ CD144+ CD31+/CD41- |
↑ ↑ |
Plasma | • CD62E+ predict poor outcomes • CD45+ increased |
(Amabile et al. 2009) | |
| Patients | CD105+ | ↑ | Plasma | • TF+ increased • TF+ correlate with severity of PH |
(Bakouboula et al. 2008) | |
| Patients (CTEPH) |
CD105+ CD144+ |
↑ | Plasma | CD105+ from patients stimulate increased CD105 expression in vitro, promote angiogenesis, and inhibit apoptosis in ECs | (Belik et al. 2016) | |
| Patients | CD62E+ | ↑ | Plasma | • CD31/CD61+ (PMVs) and CD11b+ increased • Patients with thrombo-embolic PH had increased CD62E+ compared to non-embolic PH |
(Diehl et al. 2011) | |
| Patients (IPAH) |
CD31+/CD42b- | ↑ | Plasma | CD39 ectonucleotidase expression was increased on all EV populations | (Visovatti et al. 2012) | |
| Monocrotaline PH rat model | CD31+/CD42b- | ↑ | Plasma | CD31+/CD42b- increased during disease but treatment with hepatocyte growth factor reduced EVs and reversed PAH | (Chen et al. 2014) | |
| Hypoxic PH rat model | CD54+ | ─ |
Plasma | • CD61+ (PMVs) and erythrocyte MVs increased • No changes in CD45+ • Total EVs increased in hypoxia and total EVs impaired endothelial-dependent relaxation |
(Tual-Chalot et al. 2010) | |
| Su5416/Hypoxia PAH rat model | Total EVs | ↑ | Plasma | EVs from PAH stimulate adhesion molecule expression | (Blair, Haven, and Bauer 2016) | |
| Monocrotaline PH murine model | Total EVs | ↑ | Plasma | EVs from monocrotaline-PH mice injected into healthy mice induced PH; exosomes found to be the injury-inducing population | (Aliotta et al. 2016; Aliotta et al. 2013) | |
| ARDS | Patients | EC-EVs not assessed | Pulmonary edema fluid | Procoagulant alveolar EVs are increased in ARDS | (Bastarache et al. 2009) | |
| Patients | CD31+/CD41- | ↑ | BAL | • EC-EVs were detectable in 6/52 ARDS patients • Leukocyte EVs are increased in ARDS |
(Guervilly et al. 2011) | |
| Patients | EC-EVs not assessed | Plasma | Phosphatidylserine+ EVs are decreased in ICU patients who develop ARDS | (Shaver et al. 2017) | ||
| Patients & ALI murine model- LPS (IP) |
CD31+/CD41- | ↑ | Plasma | EC pyroptosis induces MV release in a caspase-dependent manner | (Cheng et al. 2017) | |
| ALI murine model-LPS (IP) |
CD144+ CD62E+ CD31+ CD54+ |
↑ ↑ ↑ ↑ |
Plasma | Simvastatin protects against ALI and reduces plasma EC-MV levels | (Yu et al. 2017) | |
| ALI rat model- LPS (IT) | CD54+ | ↑ | Plasma | Plasma MVs from LPS-treated rats induce pulmonary inflammation | (Li et al. 2015) | |
| ALI murine model-LPS (IT) | EC-EVs not assessed | BAL | Alveolar macrophage and epithelial EVs are increased in ALI | (Soni et al. 2016) | ||
| VILI murine model | CD62E+ | ↑ | Plasma, BAL | Pathologic mechanical stretch-induced EC-MVs cause lung injury | (Letsiou et al. 2015) | |
| VILI murine model |
CD144+ CD62E+ CD31+/CD41- CD54+ |
↑ ↑ ↑ ↑ |
Plasma | Treatment of mice with a protective agent (Tetramethylpyrazine) reduces VILI and EC-MV levels | (Pan et al. 2017) | |
| VILI rat model | CD31+ | ↑ | Plasma | Lung tissue PECAM −1 (CD31) levels are reduced in VILI | (Cabrera-Benitez et al. 2015) | |
| COPD | Patients (smokers with signs of early emphysema) | CD31+/CD42b- | ↑ | Plasma | 76% of EC-MVs were angiotensin converting enzyme positive, indicating the pulmonary capillaries as their main source | (Gordon et al. 2011) |
| Patients (stable COPD) |
CD144+ CD31+/CD41- CD62E+ CD146+ |
↑ ↑ ↑ ─ |
Plasma | • CD62E+ MVs are higher in patients with frequent exacerbations | (Takahashi et al. 2012) | |
| Patients |
CD31+/CD42-
CD62E+ |
↑ ↑ |
Plasma | • CD31+/CD42- were increased in mild and severe COPD • CD62E+ were increased in severe COPD |
(Thomashow et al. 2013) | |
| Patients | CD31+ | ↑ | Sputum | Positive correlation of MV levels and lung damage | (Lacedonia et al. 2016) | |
| Patients (Smokers) |
CD31+/CD42b- | ↑ | Plasma | EC-MV levels are not altered in COPD smokers following smoking cessation | (Strulovici-Barel et al. 2016) | |
| Patients (Secondhand smoke) |
CD144+ CD31+/CD41- CD62E+ |
↑ ↑ ↑ |
Plasma | Secondhand smoke results in dysfunctional endothelial progenitor cell mobilization | (Heiss et al. 2008) | |
| Patients & cigarette smoke murine model | CD31+/CD42b- | ↑ | Plasma | EC-MV levels are higher in mild COPD compared to severe COPD | (Serban et al. 2016) | |
| Cigarette smoke (Rat model) |
CD31+/CD42b- | ↑ | Plasma | Negative correlation between EC-MVs and dynamic compliance | (Liu et al. 2014) | |
| OSA | Patients | CD31+/CD42b- | ↑ | Blood | EMVs negatively correlate with circulating EPCs | (Jelic et al. 2009) |
| Patients (Minimally symptomatic OSA) | CD31+/CD41- | ─ | Plasma | Annexin V+, PMPs+, and CD45+ increased | (Ayers et al. 2009) | |
| Patients |
CD31+/CD42- CD31+/Annexin V+ CD62E+ |
↑ ↑ ↑ |
Plasma | CPAP decreased CD62E+ | (Yun et al. 2010) | |
| Patients (desaturators vs nondesaturators) |
CD146+ | ─ | Plasma | • CD66B+ and CD62L+ increased in OSA (desaturators) • OSA (desaturators)-MVs induce endothelial dysfunction |
(Priou et al. 2010) | |
| Patients (2 week CPAP withdrawal) |
CD62E+
CD31+/CD41- |
↑ ─ |
Plasma | No changes in CD66B+, CD45+, and PMPs after a 2 week CPAP withdrawal | (Ayers et al. 2013) | |
| Patients | CD146+ | ─ | Plasma | OSA-MVs carry VEGF and stimulate angiogenesis | (Tual-Chalot et al. 2014) | |
| Patients | EC-EVs not assessed | Plasma | CD41+ and annexin V+ increase in the evening and with severity | (Bikov et al. 2017) | ||
| Healthy volunteers exposed to human intermittent hypoxia (IH) | Endothelial exosomes | ↑ | Plasma | IH-induced exosomes cause endothelial dysfunction | (Khalyfa et al. 2016) |
Description of symbols: + indicates EVs positive, - indicates EVs negative, ↑ indicates increased EV levels, ─ indicates no change in EV levels.
Abbreviations: MVs; microvesicles, PMVs; platelet microvesicles, PH; Pulmonary hypertension, IPAH; Idiopathic pulmonary arterial hypertension, CTEPH; chronic thromboembolic pulmonary hypertension, ARDS; Acute respiratory distress syndrome, ALI; Acute Lung injury, VILI; ventilator-induced lung injury, BAL; Bronchoalveolar lavage, COPD; chronic obstructive pulmonary disease, OSA; obstructive sleep apnea, CPAP; continuous positive airway pressure.
A new exciting field of interest is the use of EVs as therapeutics. Accumulating evidence indicates that EVs derived from mesenchymal stem cells protect against chronic or acute lung disorders (Monsel et al. 2016; Willis, Kourembanas, and Mitsialis 2017; Chen, Li, and Chen 2015), while endothelial progenitor cells are also attracting attention as a source of EVs with potent cytoprotective properties. While these data have driven the field in novel directions, many of the issues raised in understanding EV interactions and signaling remain. So, while it is evident that EC-EVs are no longer cellular trash, much work remains in understanding their role in homeostasis and disease.
ACKNOWLEDGMENTS
Dr Eleftheria Letsiou is a recipient of an ERS-EU RESPIRE2 Marie Sktodowska-Curie Research Fellowship - Number MCF 9019–2015. Dr. Natalie Bauer is funded by the National Institutes of Health-NIH R01HL133066.
REFERENCES
- Abid Hussein M. N., Boing AN, Biro E, Hoek FJ, Vogel GM, Meuleman DG, Sturk A, and Nieuwland R. 2008. ‘Phospholipid composition of in vitro endothelial microparticles and their in vivo thrombogenic properties’, Thromb Res, 121: 865–71. [DOI] [PubMed] [Google Scholar]
- Alexy T, Rooney K, Weber M, Gray WD, and Searles CD. 2014. ‘TNF-alpha alters the release and transfer of microparticle-encapsulated miRNAs from endothelial cells’, Physiol Genomics, 46: 833–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aliotta JM, Pereira M, Wen S, Dooner MS, Del Tatto M., Papa E, Goldberg LR, Baird GL, Ventetuolo CE, Quesenberry PJ, and Klinger JR. 2016. ‘Exosomes induce and reverse monocrotaline-induced pulmonary hypertension in mice’, Cardiovasc Res, 110: 319–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alipoor SD, Mortaz E, Garssen J, Movassaghi M, Mirsaeidi M, and Adcock IM. 2016. ‘Exosomes and Exosomal miRNA in Respiratory Diseases’, Mediators Inflamm, 2016: 5628404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amabile N, Heiss C, Chang V, Angeli FS, Damon L, Rame EJ, McGlothlin D, Grossman W, De Marco T, and Yeghiazarians Y. 2009. ‘Increased CD62e(+) endothelial microparticle levels predict poor outcome in pulmonary hypertension patients’, J Heart Lung Transplant, 28: 1081–6. [DOI] [PubMed] [Google Scholar]
- Amabile N, Heiss C, Real WM, Minasi P, McGlothlin D, Rame EJ, Grossman W, De Marco T, and Yeghiazarians Y. 2008. ‘Circulating endothelial microparticle levels predict hemodynamic severity of pulmonary hypertension’, Am J Respir Crit Care Med, 177: 1268–75. [DOI] [PubMed] [Google Scholar]
- Andrews AM, Lutton EM, Merkel SF, Razmpour R, and Ramirez SH. 2016. ‘Mechanical Injury Induces Brain Endothelial-Derived Microvesicle Release: Implications for Cerebral Vascular Injury during Traumatic Brain Injury’, Front Cell Neurosci, 10: 43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrews AM, and Rizzo V. 2016. ‘Microparticle-Induced Activation of the Vascular Endothelium Requires Caveolin-1/Caveolae’, PLoS One, 11: e0149272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Angelot F, Seilles E, Biichle S, Berda Y, Gaugler B, Plumas J, Chaperot L, Dignat-George F, Tiberghien P, Saas P, and Garnache-Ottou F. 2009. ‘Endothelial cell-derived microparticles induce plasmacytoid dendritic cell maturation: potential implications in inflammatory diseases’, Haematologica, 94: 1502–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aoyagi T, Newstead MW, Zeng X, Kunkel SL, Kaku M, and Standiford TJ. 2017. ‘IL-36 receptor deletion attenuates lung injury and decreases mortality in murine influenza pneumonia’, Mucosal Immunol, 10: 1043–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aung HH, Tung JP, Dean MM, Flower RL, and Pecheniuk NM. 2017. ‘Procoagulant role of microparticles in routine storage of packed red blood cells: potential risk for prothrombotic post-transfusion complications’, Pathology, 49: 62–69. [DOI] [PubMed] [Google Scholar]
- Ayers L, Ferry B, Craig S, Nicoll D, Stradling JR, and Kohler M. 2009. ‘Circulating cell-derived microparticles in patients with minimally symptomatic obstructive sleep apnoea’, Eur Respir J, 33: 574–80. [DOI] [PubMed] [Google Scholar]
- Ayers L, Stoewhas AC, Ferry B, Stradling J, and Kohler M. 2013. ‘Elevated levels of endothelial cell-derived microparticles following short-term withdrawal of continuous positive airway pressure in patients with obstructive sleep apnea: data from a randomized controlled trial’, Respiration, 85: 478–85. [DOI] [PubMed] [Google Scholar]
- Bacha NC, Blandinieres A, Rossi E, Gendron N, Nevo N, Lecourt S, Guerin CL, Renard JM, Gaussem P, Angles-Cano E, Boulanger CM, Israel-Biet D, and Smadja DM. 2018. ‘Endothelial Microparticles are Associated to Pathogenesis of Idiopathic Pulmonary Fibrosis’, Stem Cell Rev, 14: 223–35. [DOI] [PubMed] [Google Scholar]
- Bakouboula B, Morel O, Faure A, Zobairi F, Jesel L, Trinh A, Zupan M, Canuet M, Grunebaum L, Brunette A, Desprez D, Chabot F, Weitzenblum E, Freyssinet JM, Chaouat A, and Toti F. 2008. ‘Procoagulant membrane microparticles correlate with the severity of pulmonary arterial hypertension’, Am J Respir Crit Care Med, 177: 536–43. [DOI] [PubMed] [Google Scholar]
- Bal L, Ederhy S, Di Angelantonio E, Toti F, Zobairi F, Dufaitre G, Meuleman C, Mallat Z, Boccara F, Tedgui A, Freyssinet JM, and Cohen A. 2010. ‘Factors influencing the level of circulating procoagulant microparticles in acute pulmonary embolism’, Arch Cardiovasc Dis, 103: 394–403. [DOI] [PubMed] [Google Scholar]
- Banfi C, Brioschi M, Wait R, Begum S, Gianazza E, Pirillo A, Mussoni L, and Tremoli E. 2005. ‘Proteome of endothelial cell-derived procoagulant microparticles’, Proteomics, 5: 4443–55. [DOI] [PubMed] [Google Scholar]
- Bardita C, Predescu DN, Sha F, Patel M, Balaji G, and Predescu SA. 2015. ‘Endocytic deficiency induced by ITSN-1s knockdown alters the Smad2/3-Erk½ signaling balance downstream of Alk5’, J Cell Sci, 128: 1528–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barry OP, Pratico D, Lawson JA, and FitzGerald GA. 1997. ‘Transcellular activation of platelets and endothelial cells by bioactive lipids in platelet microparticles’, J Clin Invest, 99: 2118–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bastarache JA, Fremont RD, Kropski JA, Bossert FR, and Ware LB. 2009. ‘Procoagulant alveolar microparticles in the lungs of patients with acute respiratory distress syndrome’, Am J Physiol Lung Cell Mol Physiol, 297: L1035–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belik D, Tsang H, Wharton J, Howard L, Bernabeu C, and Wojciak-Stothard B. 2016. ‘Endothelium-derived microparticles from chronically thromboembolic pulmonary hypertensive patients facilitate endothelial angiogenesis’, J Biomed Sci, 23: 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beloribi S, Ristorcelli E, Breuzard G, Silvy F, Bertrand-Michel J, Beraud E, Verine A, and Lombardo D. 2012. ‘Exosomal lipids impact notch signaling and induce death of human pancreatic tumoral SOJ-6 cells’, PLoS One, 7: e47480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bevilacqua MP, Nelson RM, Mannori G, and Cecconi O. 1994. ‘Endothelial-leukocyte adhesion molecules in human disease’, Annu Rev Med, 45: 361–78. [DOI] [PubMed] [Google Scholar]
- Bikov A, Kunos L, Pallinger E, Lazar Z, Kis A, Horvath G, Losonczy G, and Komlosi ZI.’Diurnal variation of circulating microvesicles is associated with the severity of obstructive sleep apnoea’, Sleep Breath, 21: 595–600. [DOI] [PubMed] [Google Scholar]
- Blair LA, Haven AK, and Bauer NN. 2016. ‘Circulating microparticles in severe pulmonary arterial hypertension increase intercellular adhesion molecule-1 expression selectively in pulmonary artery endothelium’, Respir Res, 17: 133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blann AD, and Lip GY. 2001. ‘Virchow’s triad revisited: the importance of soluble coagulation factors, the endothelium, and platelets’, Thromb Res, 101: 321–7. [DOI] [PubMed] [Google Scholar]
- Bodega G, Alique M, Bohorquez L, Ciordia S, Mena MC, and Ramirez MR. 2017. ‘The Antioxidant Machinery of Young and Senescent Human Umbilical Vein Endothelial Cells and Their Microvesicles’, Oxid Med Cell Longev, 2017: 7094781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonjoch L, Casas V, Carrascal M, and Closa D. 2016. ‘Involvement of exosomes in lung inflammation associated with experimental acute pancreatitis’, J Pathol, 240: 235–45. [DOI] [PubMed] [Google Scholar]
- Boudreau LH, Duchez AC, Cloutier N, Soulet D, Martin N, Bollinger J, Pare A, Rousseau M, Naika GS, Levesque T, Laflamme C, Marcoux G, Lambeau G, Farndale RW, Pouliot M, Hamzeh-Cognasse H, Cognasse F, Garraud O, Nigrovic PA, Guderley H, Lacroix S, Thibault L, Semple JW, Gelb MH, and Boilard E. 2014. ‘Platelets release mitochondria serving as substrate for bactericidal group IIA-secreted phospholipase A2 to promote inflammation’, Blood, 124: 2173–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brodsky SV, Malinowski K, Golightly M, Jesty J, and Goligorsky MS. 2002. ‘Plasminogen activator inhibitor-1 promotes formation of endothelial microparticles with procoagulant potential’, Circulation, 106: 2372–8. [DOI] [PubMed] [Google Scholar]
- Brodsky SV, Zhang F, Nasjletti A, and Goligorsky MS. 2004. ‘Endothelium-derived microparticles impair endothelial function in vitro’, Am J Physiol Heart Circ Physiol, 286: H1910–5. [DOI] [PubMed] [Google Scholar]
- Buesing KL, Densmore JC, Kaul S, Pritchard KA Jr., Jarzembowski JA, Gourlay DM, and Oldham KT. 2011. ‘Endothelial microparticles induce inflammation in acute lung injury’, J Surg Res, 166: 32–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burger D, Montezano AC, Nishigaki N, He Y, Carter A, and Touyz RM. 2011. ‘Endothelial microparticle formation by angiotensin II is mediated via Ang II receptor type I/NADPH oxidase/ Rho kinase pathways targeted to lipid rafts’, Arterioscler Thromb Vasc Biol, 31 : 1898–907. [DOI] [PubMed] [Google Scholar]
- Burger D, Vinas JL, Akbari S, Dehak H, Knoll W, Gutsol A, Carter A, Touyz RM, Allan DS, and Burns KD. 2015. ‘Human endothelial colony-forming cells protect against acute kidney injury: role of exosomes’, Am J Pathol, 185: 2309–23. [DOI] [PubMed] [Google Scholar]
- Cabrera-Benitez NE, Valladares F, Garcia-Hernandez S, Ramos-Nuez A, Martin-Barrasa JL, Martinez-Saavedra MT, Rodriguez-Gallego C, Muros M, Flores C, Liu M, Slutsky AS, and Villar J. 2015. ‘Altered Profile of Circulating Endothelial-Derived Microparticles in Ventilator-Induced Lung Injury’, Crit Care Med, 43: e551–9. [DOI] [PubMed] [Google Scholar]
- Cai J, Wu G, Jose PA, and Zeng C. 2016. ‘Functional transferred DNA within extracellular vesicles’, Exp Cell Res, 349: 179–83. [DOI] [PubMed] [Google Scholar]
- Cantaluppi V, Gatti S, Medica D, Figliolini F, Bruno S, Deregibus MC, Sordi A, Biancone L, Tetta C, and Camussi G. 2012. ‘Microvesicles derived from endothelial progenitor cells protect the kidney from ischemia-reperfusion injury by microRNA-dependent reprogramming of resident renal cells’, Kidney Int, 82: 412–27. [DOI] [PubMed] [Google Scholar]
- Chang AL, Kim Y, Seitz AP, Schuster RM, Lentsch AB, and Pritts TA. 2017‘Erythrocyte-Derived Microparticles Activate Pulmonary Endothelial Cells in a Murine Model of Transfusion’, Shock, 47: 632–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, Li C, and Chen L. 2015. ‘The Role of Microvesicles Derived from Mesenchymal Stem Cells in Lung Diseases’, Biomed Res Int, 2015: 985814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen JY, An R, Liu ZJ, Wang JJ, Chen SZ, Hong MM, Liu JH, Xiao MY, and Chen YF. 2014. ‘Therapeutic effects of mesenchymal stem cell-derived microvesicles on pulmonary arterial hypertension in rats’, Acta Pharmacol Sin, 35: 1121–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng KT, Xiong S, Ye Z, Hong Z, Di A, Tsang KM, Gao X, An S, Mittal M, Vogel SM, Miao EA, Rehman J, and Malik AB. 2017. ‘Caspase-11-mediated endothelial pyroptosis underlies endotoxemia-induced lung injury’, J Clin Invest, 127: 4124–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chirinos JA, Heresi GA, Velasquez H, Jy W, Jimenez JJ, Ahn E, Horstman LL, Soriano AO, Zambrano JP, and Ahn YS. 2005. ‘Elevation of endothelial microparticles, platelets, and leukocyte activation in patients with venous thromboembolism’, J Am Coll Cardiol, 45: 1467–71. [DOI] [PubMed] [Google Scholar]
- Chironi GN, Boulanger CM, Simon A, Dignat-George F, Freyssinet JM, and Tedgui A. 2009. ‘Endothelial microparticles in diseases’, Cell Tissue Res, 335: 143–51. [DOI] [PubMed] [Google Scholar]
- Choi DS, Kim DK, Kim YK, and Gho YS. 2013. ‘Proteomics, transcriptomics and lipidomics of exosomes and ectosomes’, Proteomics, 13: 1554–71. [DOI] [PubMed] [Google Scholar]
- Choi DS, Kim DK, Kim YK, and Gho YS. 2015. ‘Proteomics of extracellular vesicles: Exosomes and ectosomes’, Mass Spectrom Rev, 34: 474–90. [DOI] [PubMed] [Google Scholar]
- Collaco JM, and McGrath-Morrow SA. 2018. ‘Respiratory Phenotypes for Preterm Infants, Children, and Adults: Bronchopulmonary Dysplasia and More’, Ann Am Thorac Soc. [DOI] [PubMed] [Google Scholar]
- Combes V, Simon AC, Grau GE, Arnoux D, Camoin L, Sabatier F, Mutin M, Sanmarco M, Sampol J, and Dignat-George F. 1999. ‘In vitro generation of endothelial microparticles and possible prothrombotic activity in patients with lupus anticoagulant’, J Clin Invest, 104: 93–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connor DE, Exner T, Ma DD, and Joseph JE. 2010. ‘The majority of circulating platelet- derived microparticles fail to bind annexin V, lack phospholipid-dependent procoagulant activity and demonstrate greater expression of glycoprotein Ib’, Thromb Haemost, 103: 1044–52. [DOI] [PubMed] [Google Scholar]
- Cool CD, Stewart JS, Werahera P, Miller GJ, Williams RL, Voelkel NF, and Tuder RM. 1999. ‘Three-dimensional reconstruction of pulmonary arteries in plexiform pulmonary hypertension using cell-specific markers. Evidence for a dynamic and heterogeneous process of pulmonary endothelial cell growth’, Am J Pathol, 155: 411–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curtis AM, Wilkinson PF, Gui M, Gales TL, Hu E, and Edelberg JM. 2009. ‘p38 mitogen-activated protein kinase targets the production of proinflammatory endothelial microparticles’, J Thromb Haemost, 7: 701–9. [DOI] [PubMed] [Google Scholar]
- Dasgupta SK, Abdel-Monem H, Niravath P, Le A, Bellera RV, Langlois K, Nagata S, Rumbaut RE, and Thiagarajan P. 2009. ‘Lactadherin and clearance of platelet-derived microvesicles’, Blood, 113: 1332–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dasgupta SK, Le A, Chavakis T, Rumbaut RE, and Thiagarajan P. 2012. ‘Developmental endothelial locus-1 (Del-1) mediates clearance of platelet microparticles by the endothelium’, Circulation, 125: 1664–72. [DOI] [PubMed] [Google Scholar]
- de Jong OG, van Balkom BW, Gremmels H, and Verhaar MC. 2016. ‘Exosomes from hypoxic endothelial cells have increased collagen crosslinking activity through up-regulation of lysyl oxidase-like 2’, J Cell Mol Med, 20: 342–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Jong OG, Verhaar MC, Chen Y, Vader P, Gremmels H, Posthuma G, Schiffelers RM, Gucek M, and van Balkom BW. 2012. ‘Cellular stress conditions are reflected in the protein and RNA content of endothelial cell-derived exosomes’, J Extracell Vesicles, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Densmore JC, Signorino PR, Ou J, Hatoum OA, Rowe JJ, Shi Y, Kaul S, Jones DW, Sabina RE, Pritchard KA Jr., Guice KS, and Oldham KT. 2006. ‘Endothelium-derived microparticles induce endothelial dysfunction and acute lung injury’, Shock, 26: 464–71. [DOI] [PubMed] [Google Scholar]
- Deregibus MC, Cantaluppi V, Calogero R, Lo Iacono M, Tetta C, Biancone L, Bruno S, Bussolati B, and Camussi G. 2007. ‘Endothelial progenitor cell derived microvesicles activate an angiogenic program in endothelial cells by a horizontal transfer of mRNA’, Blood, 110: 2440–8. [DOI] [PubMed] [Google Scholar]
- Dignat-George F, and Boulanger CM. 2011. ‘The many faces of endothelial microparticles’, Arterioscler Thromb Vasc Biol, 31: 27–33. [DOI] [PubMed] [Google Scholar]
- Doyle LW, Ehrenkranz RA, and Halliday HL. 2014a. ‘Early (< 8 days) postnatal corticosteroids for preventing chronic lung disease in preterm infants’, Cochrane Database Syst Rev: CD001146. [DOI] [PubMed] [Google Scholar]
- Doyle LW, Ehrenkranz RA, and Halliday HL. 2014b. ‘Late (> 7 days) postnatal corticosteroids for chronic lung disease in preterm infants’, Cochrane Database Syst Rev: CD001145. [DOI] [PubMed] [Google Scholar]
- Doyle LW, Halliday HL, Ehrenkranz RA, Davis PG, and Sinclair JC. 2014. ‘An update on the impact of postnatal systemic corticosteroids on mortality and cerebral palsy in preterm infants: effect modification by risk of bronchopulmonary dysplasia’, J Pediatr, 165: 1258–60. [DOI] [PubMed] [Google Scholar]
- Duarte D, Taveira-Gomes T, Sokhatska O, Palmares C, Costa R, Negrao R, Guimaraes JT, Delgado L, Soares R, and Moreira A. 2013. ‘Increased circulating platelet microparticles as a potential biomarker in asthma’, Allergy, 68: 1073–5. [DOI] [PubMed] [Google Scholar]
- El-Solh AA, Mador MJ, Sikka P, Dhillon RS, Amsterdam D, and Grant BJ. 2002. ‘Adhesion molecules in patients with coronary artery disease and moderate-to-severe obstructive sleep apnea’, Chest, 121: 1541–7. [DOI] [PubMed] [Google Scholar]
- Exline MC, Justiniano S, Hollyfield JL, Berhe F, Besecker BY, Das S, Wewers MD, and Sarkar A. 2014. ‘Microvesicular caspase-1 mediates lymphocyte apoptosis in sepsis’, PLoS One, 9: e90968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faille D, El-Assaad F, Mitchell AJ, Alessi MC, Chimini G, Fusai T, Grau GE, and Combes V. 2012. ‘Endocytosis and intracellular processing of platelet microparticles by brain endothelial cells’, J Cell Mol Med, 16: 1731–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng J, Zhang D, and Chen B. 2012. ‘Endothelial mechanisms of endothelial dysfunction in patients with obstructive sleep apnea’, Sleep Breath, 16: 283–94. [DOI] [PubMed] [Google Scholar]
- Ferrer E, Dunmore BJ, Hassan D, Ormiston ML, Moore S, Deighton J, Long L, Yang XD, Stewart DJ, and Morrell NW. 2018. ‘A Potential Role for Exosomal TCTP Export in Vascular Remodeling in Pulmonary Arterial Hypertension’, Am J Respir Cell Mol Biol. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitzner D, Schnaars M, van Rossum D, Krishnamoorthy G, Dibaj P, Bakhti M, Regen T, Hanisch UK, and Simons M. 2011. ‘Selective transfer of exosomes from oligodendrocytes to microglia by macropinocytosis’, J Cell Sci, 124: 447–58. [DOI] [PubMed] [Google Scholar]
- Force, Ards Definition Task, Ranieri VM, Rubenfeld GD, Thompson BT, Ferguson ND, Caldwell E, Fan E, Camporota L, and Slutsky AS. 2012. ‘Acute respiratory distress syndrome: the Berlin Definition’, JAMA, 307: 2526–33. [DOI] [PubMed] [Google Scholar]
- Fujita Y, Yoshioka Y, Ito S, Araya J, Kuwano K, and Ochiya T. 2014. ‘Intercellular communication by extracellular vesicles and their microRNAs in asthma’, Clin Ther, 36: 873–81. [DOI] [PubMed] [Google Scholar]
- Gonzalez G, Vituret C, Di Pietro A, Chanson M, Boulanger P, and Hong SS. 2012. ‘Microparticle-mediated transfer of the viral receptors CAR and CD46, and the CFTR channel in a CHO cell model confers new functions to target cells’, PLoS One, 7: e52326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon C, Gudi K, Krause A, Sackrowitz R, Harvey BG, Strulovici-Barel Y, Mezey JG, and Crystal RG. 2011. ‘Circulating endothelial microparticles as a measure of early lung destruction in cigarette smokers’, Am J Respir Crit Care Med, 184: 224–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green CE, and Turner AM. 2017. ‘The role of the endothelium in asthma and chronic obstructive pulmonary disease (COPD)’, Respir Res, 18: 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guervilly C, Lacroix R, Forel JM, Roch A, Camoin-Jau L, Papazian L, and Dignat- George F. 2011. ‘High levels of circulating leukocyte microparticles are associated with better outcome in acute respiratory distress syndrome’, Crit Care, 15: R31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gyorgy B, Szabo TG, Pasztoi M, Pal Z, Misjak P, Aradi B, Laszlo V, Pallinger E, Pap E, Kittel A, Nagy G, Falus A, and Buzas EI. 2011. ‘Membrane vesicles, current state-of- the-art: emerging role of extracellular vesicles’, Cell Mol Life Sci, 68: 2667–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamilton KK, Hattori R, Esmon CT, and Sims PJ. 1990. ‘Complement proteins C5b-9 induce vesiculation of the endothelial plasma membrane and expose catalytic surface for assembly of the prothrombinase enzyme complex’, J Biol Chem, 265: 3809–14. [PubMed] [Google Scholar]
- Hargett LA, and Bauer NN. 2013. ‘On the origin of microparticles: From “platelet dust” to mediators of intercellular communication’, Pulm Circ, 3: 329–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrison RE, Flanagan JA, Sankelo M, Abdalla SA, Rowell J, Machado RD, Elliott CG, Robbins IM, Olschewski H, McLaughlin V, Gruenig E, Kermeen F, Halme M, Raisanen-Sokolowski A, Laitinen T, Morrell NW, and Trembath RC. 2003‘Molecular and functional analysis identifies ALK-1 as the predominant cause of pulmonary hypertension related to hereditary haemorrhagic telangiectasia’, J Med Genet, 40: 865–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heiss C, Amabile N, Lee AC, Real WM, Schick SF, Lao D, Wong ML, Jahn S, Angeli FS, Minasi P, Springer ML, Hammond SK, Glantz SA, Grossman W, Balmes JR, and Yeghiazarians Y. 2008. ‘Brief secondhand smoke exposure depresses endothelial progenitor cells activity and endothelial function: sustained vascular injury and blunted nitric oxide production’, J Am Coll Cardiol, 51: 1760–71. [DOI] [PubMed] [Google Scholar]
- Herrmann IK, Bertazzo S, O’Callaghan DJ, Schlegel AA, Kallepitis C, Antcliffe DB, Gordon AC, and Stevens MM. 2015. ‘Differentiating sepsis from non-infectious systemic inflammation based on microvesicle-bacteria aggregation’, Nanoscale, 7: 13511–20. [DOI] [PubMed] [Google Scholar]
- Hromada C, Muhleder S, Grillari J, Redl H, and Holnthoner W. 2017. ‘Endothelial Extracellular Vesicles-Promises and Challenges’, Front Physiol, 8: 275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu G, Drescher KM, and Chen XM. 2012. ‘Exosomal miRNAs: Biological Properties and Therapeutic Potential’, Front Genet, 3: 56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huertas A, Guignabert C, Barbera JA, Bartsch P, Bhattacharya J, Bhattacharya S, Bonsignore MR, Dewachter L, Dinh-Xuan AT, Dorfmuller P, Gladwin MT, Humbert M, Kotsimbos T, Vassilakopoulos T, Sanchez O, Savale L, Testa U, and Wilkins MR. 2018. ‘Pulmonary vascular endothelium: the orchestra conductor in respiratory diseases: Highlights from basic research to therapy’, Eur Respir J, 51. [DOI] [PubMed] [Google Scholar]
- Hugel B, Martinez MC, Kunzelmann C, and Freyssinet JM. 2005. ‘Membrane microparticles: two sides of the coin’, Physiology (Bethesda), 20: 22–7. [DOI] [PubMed] [Google Scholar]
- Hwang JS, and Rehan VK. 2018. ‘Recent Advances in Bronchopulmonary Dysplasia: Pathophysiology, Prevention, and Treatment’, Lung, 196: 129–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang TC, Yeh JT, Zhang J, Yu YC, Yeh HI, and Destefano S. 2018. ‘Structural mechanisms of CFTR function and dysfunction’, J Gen Physiol, 150: 539–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ince C, Mayeux PR, Nguyen T, Gomez H, Kellum JA, Ospina-Tascon GA, Hernandez G, Murray P, De Backer D, and Adqi Xiv Workgroup. 2016. ‘The Endothelium in Sepsis’, Shock, 45: 259–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jansen F, Nickenig G, and Werner N. 2017. ‘Extracellular Vesicles in Cardiovascular Disease: Potential Applications in Diagnosis, Prognosis, and Epidemiology’, Circ Res, 120: 1649–57. [DOI] [PubMed] [Google Scholar]
- Jansen F, Yang X, Baumann K, Przybilla D, Schmitz T, Flender A, Paul K, Alhusseiny A, Nickenig G, and Werner N. 2015. ‘Endothelial microparticles reduce ICAM-1 expression in a microRNA-222-dependent mechanism’, J Cell Mol Med, 19: 2202–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jansen F, Yang X, Franklin BS, Hoelscher M, Schmitz T, Bedorf J, Nickenig G, and Werner N. 2013. ‘High glucose condition increases NADPH oxidase activity in endothelial microparticles that promote vascular inflammation’, Cardiovasc Res, 98: 94–106. [DOI] [PubMed] [Google Scholar]
- Jansen F, Yang X, Hoelscher M, Cattelan A, Schmitz T, Proebsting S, Wenzel D, Vosen S, Franklin BS, Fleischmann BK, Nickenig G, and Werner N. 2013. ‘Endothelial microparticle-mediated transfer of MicroRNA-126 promotes vascular endothelial cell repair via SPRED1 and is abrogated in glucose-damaged endothelial microparticles’, Circulation, 128: 2026–38. [DOI] [PubMed] [Google Scholar]
- Jansen F, Yang X, Hoyer FF, Paul K, Heiermann N, Becher MU, Abu Hussein N., Kebschull M, Bedorf J, Franklin BS, Latz E, Nickenig G, and Werner N. 2012. ‘Endothelial microparticle uptake in target cells is annexin I/phosphatidylserine receptor dependent and prevents apoptosis’, Arterioscler Thromb Vasc Biol, 32: 1925–35. [DOI] [PubMed] [Google Scholar]
- Jelic S, Lederer DJ, Adams T, Padeletti M, Colombo PC, Factor P, and Le Jemtel TH. 2009. ‘Endothelial repair capacity and apoptosis are inversely related in obstructive sleep apnea’, Vasc Health Risk Manag, 5: 909–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jelic S, Padeletti M, Kawut SM, Higgins C, Canfield SM, Onat D, Colombo PC, Basner RC, Factor P, and LeJemtel TH. 2008. ‘Inflammation, oxidative stress, and repair capacity of the vascular endothelium in obstructive sleep apnea’, Circulation, 117: 2270–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jimenez JJ, Jy W, Mauro LM, Soderland C, Horstman LL, and Ahn YS. 2003. ‘Endothelial cells release phenotypically and quantitatively distinct microparticles in activation and apoptosis’, Thromb Res, 109: 175–80. [DOI] [PubMed] [Google Scholar]
- Johnstone RM, Adam M, Hammond JR, Orr L, and Turbide C. 1987. ‘Vesicle formation during reticulocyte maturation. Association of plasma membrane activities with released vesicles (exosomes)’, J Biol Chem, 262: 9412–20. [PubMed] [Google Scholar]
- Joop K, Berckmans RJ, Nieuwland R, Berkhout J, Romijn FP, Hack CE, and Sturk A. 2001. ‘Microparticles from patients with multiple organ dysfunction syndrome and sepsis support coagulation through multiple mechanisms’, Thromb Haemost, 85: 810–20. [PubMed] [Google Scholar]
- Kadota T, Fujita Y, Yoshioka Y, Araya J, Kuwano K, and Ochiya T. 2016. ‘Extracellular Vesicles in Chronic Obstructive Pulmonary Disease’, Int J Mol Sci, 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalra H, Simpson RJ, Ji H, Aikawa E, Altevogt P, Askenase P, Bond VC, Borras FE, Breakefield X, Budnik V, Buzas E, Camussi G, Clayton A, Cocucci E, Falcon-Perez JM, Gabrielsson S, Gho YS, Gupta D, Harsha HC, Hendrix A, Hill AF, Inal JM, Jenster G, Kramer-Albers EM, Lim SK, Llorente A, Lotvall J, Marcilla A, Mincheva-Nilsson L, Nazarenko I, Nieuwland R, Nolte-’t Hoen EN, Pandey A, Patel T, Piper MG, Pluchino S, Prasad TS, Rajendran L, Raposo G, Record M, Reid GE, Sanchez-Madrid F, Schiffelers RM, Siljander P, Stensballe A, Stoorvogel W, Taylor D, Thery C, Valadi H, van Balkom BW, Vazquez J, Vidal M, Wauben MH, Yanez-Mo M, Zoeller M, and Mathivanan S. 2012. ‘Vesiclepedia: a compendium for extracellular vesicles with continuous community annotation’, PLoS Biol, 10: e1001450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keerthikumar S, Chisanga D, Ariyaratne D, Al Saffar H, Anand S, Zhao K, Samuel M, Pathan M, Jois M, Chilamkurti N, Gangoda L, and Mathivanan S. 2016. ‘ExoCarta: A Web-Based Compendium of Exosomal Cargo’, J Mol Biol, 428: 688–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khalyfa A, Kheirandish-Gozal L, and Gozal D. 2017. ‘Circulating exosomes in obstructive sleep apnea as phenotypic biomarkers and mechanistic messengers of end-organ morbidity’, Respir Physiol Neurobiol. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khalyfa A, Youssefnia N, Foster GE, Beaudin AE, Qiao Z, Pialoux V, Pun M, Hanly PJ, Kheirandish-Gozal L, Poulin MJ, and Gozal D. 2017. ‘Plasma Exosomes and Improvements in Endothelial Function by Angiotensin 2 Type 1 Receptor or Cyclooxygenase 2 Blockade following Intermittent Hypoxia’, Front Neurol, 8: 709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khalyfa A, Zhang C, Khalyfa AA, Foster GE, Beaudin AE, Andrade J, Hanly PJ, Poulin MJ, and Gozal D. 2016. ‘Effect on Intermittent Hypoxia on Plasma Exosomal Micro RNA Signature and Endothelial Function in Healthy Adults’, Sleep, 39: 2077–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim CW, Lee HM, Lee TH, Kang C, Kleinman HK, and Gho YS. 2002. ‘Extracellular membrane vesicles from tumor cells promote angiogenesis via sphingomyelin’, Cancer Res, 62: 6312–7. [PubMed] [Google Scholar]
- Kim DK, Kang B, Kim OY, Choi DS, Lee J, Kim SR, Go G, Yoon YJ, Kim JH, Jang SC, Park KS, Choi EJ, Kim KP, Desiderio DM, Kim YK, Lotvall J, Hwang D, and Gho YS. 2013. ‘EVpedia: an integrated database of high-throughput data for systemic analyses of extracellular vesicles’, J Extracell Vesicles, 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim KM, Abdelmohsen K, Mustapic M, Kapogiannis D, and Gorospe M. 2017. ‘RNA in extracellular vesicles’, Wiley Interdiscip Rev RNA, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim Y, Xia BT, Jung AD, Chang AL, Abplanalp WA, Caldwell CC, Goodman MD, and Pritts TA. 2018. ‘Microparticles from stored red blood cells promote a hypercoagulable state in a murine model of transfusion’, Surgery, 163: 423–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovach MA, Singer BH, Newstead MW, Zeng X, Moore TA, White ES, Kunkel SL, Peters-Golden M, and Standiford TJ. 2016. ‘IL-36gamma is secreted in microparticles and exosomes by lung macrophages in response to bacteria and bacterial components’, J Leukoc Biol, 100: 413–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreimer S, Belov AM, Ghiran I, Murthy SK, Frank DA, and Ivanov AR. 2015. ‘Mass-spectrometry-based molecular characterization of extracellular vesicles: lipidomics and proteomics’, J Proteome Res, 14: 2367–84. [DOI] [PubMed] [Google Scholar]
- Kubo H 2018. ‘Extracellular Vesicles in Lung Disease’, Chest, 153: 210–16. [DOI] [PubMed] [Google Scholar]
- Kucharzewska P, Christianson HC, Welch JE, Svensson KJ, Fredlund E, Ringner M, Morgelin M, Bourseau-Guilmain E, Bengzon J, and Belting M. 2013. ‘Exosomes reflect the hypoxic status of glioma cells and mediate hypoxia-dependent activation of vascular cells during tumor development’, Proc Natl Acad Sci U S A, 110: 7312–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lacedonia D, Carpagnano GE, Trotta T, Palladino GP, Panaro MA, Zoppo LD, Foschino Barbaro M. P., and Porro C. 2016. ‘Microparticles in sputum of COPD patients: a potential biomarker of the disease?’, Int J Chron Obstruct Pulmon Dis, 11: 527–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lal CV, Olave N, Travers C, Rezonzew G, Dolma K, Simpson A, Halloran B, Aghai Z, Das P, Sharma N, Xu X, Genschmer K, Russell D, Szul T, Yi N, Blalock JE, Gaggar A, Bhandari V, and Ambalavanan N. 2018. ‘Exosomal microRNA predicts and protects against severe bronchopulmonary dysplasia in extremely premature infants’,JCI Insight , 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamichhane TN, Leung CA, Douti LY, and Jay SM. 2017. ‘Ethanol Induces Enhanced Vascularization Bioactivity of Endothelial Cell-Derived Extracellular Vesicles via Regulation of MicroRNAs and Long Non-Coding RNAs’, Sci Rep, 7: 13794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lashin HMS, Nadkarni S, Oggero S, Jones HR, Knight JC, Hinds CJ, and Perretti M. 2018. ‘Microvesicle Subsets in Sepsis Due to Community Acquired Pneumonia Compared to Faecal Peritonitis’, Shock, 49: 393–401. [DOI] [PubMed] [Google Scholar]
- Lee C, Mitsialis SA, Aslam M, Vitali SH, Vergadi E, Konstantinou G, Sdrimas K, Fernandez-Gonzalez A, and Kourembanas S. 2012. ‘Exosomes mediate the cytoprotective action of mesenchymal stromal cells on hypoxia-induced pulmonary hypertension’, Circulation, 126: 2601–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehner GF, Harler U, Haller VM, Feistritzer C, Hasslacher J, Dunzendorfer S, Bellmann R, and Joannidis M. 2016. ‘Characterization of Microvesicles in Septic Shock Using High-Sensitivity Flow Cytometry’, Shock, 46: 373–81. [DOI] [PubMed] [Google Scholar]
- Lehnert P, Johansson PI, Ostrowski SR, Moller CH, Bang LE, Olsen PS, and Carlsen J. 2017. ‘Coagulopathy in patients with acute pulmonary embolism: a pilot study of whole blood coagulation and markers of endothelial damage’, Scand J Clin Lab Invest, 77: 19–26. [DOI] [PubMed] [Google Scholar]
- Letsiou E, Sammani S, Zhang W, Zhou T, Quijada H, Moreno-Vinasco L, Dudek SM, and Garcia JG. 2015. ‘Pathologic mechanical stress and endotoxin exposure increases lung endothelial microparticle shedding’, Am J Respir Cell Mol Biol, 52: 193–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Meng X, Liang X, Gao Y, and Cai S. 2015. ‘Administration of microparticles from blood of the lipopolysaccharide-treated rats serves to induce pathologic changes of acute respiratory distress syndrome’, Exp Biol Med (Maywood), 240: 1735–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ling L, Huang H, Zhu L, Mao T, Shen Q, and Zhang H. 2014. ‘Evaluation of plasma endothelial microparticles in pre-eclampsia’, J Int Med Res, 42: 42–51. [DOI] [PubMed] [Google Scholar]
- Liu H, Ding L, Zhang Y, and Ni S. 2014. ‘Circulating endothelial microparticles involved in lung function decline in a rat exposed in cigarette smoke maybe from apoptotic pulmonary capillary endothelial cells’, J Thorac Dis, 6: 649–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Zhang R, Qu H, Wu J, Li L, and Tang Y. 2017. ‘Endothelial microparticles activate endothelial cells to facilitate the inflammatory response’, Mol Med Rep, 15: 1291–96. [DOI] [PubMed] [Google Scholar]
- Lockett AD, Brown MB, Santos-Falcon N, Rush NI, Oueini H, Oberle AJ, Bolanis E, Fragoso MA, Petrusca DN, Serban KA, Schweitzer KS, Presson RG Jr., Campos M, and Petrache I. 2014. ‘Active trafficking of alpha 1 antitrypsin across the lung endothelium’, PLoS One, 9: e93979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lotvall J, Hill AF, Hochberg F, Buzas EI, Di Vizio D, Gardiner C, Gho YS, Kurochkin IV, Mathivanan S, Quesenberry P, Sahoo S, Tahara H, Wauben MH, Witwer KW, and Thery C. 2014. ‘Minimal experimental requirements for definition of extracellular vesicles and their functions: a position statement from the International Society for Extracellular Vesicles’, J Extracell Vesicles, 3: 26913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lurie A 2011. ‘Endothelial dysfunction in adults with obstructive sleep apnea’, Adv Cardiol, 46: 139–70. [DOI] [PubMed] [Google Scholar]
- Maas SLN, Breakefield XO, and Weaver AM. 2017. ‘Extracellular Vesicles: Unique Intercellular Delivery Vehicles’, Trends Cell Biol, 27: 172–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Machado RD 2012. ‘The molecular genetics and cellular mechanisms underlying pulmonary arterial hypertension’, Scientifica (Cairo), 2012: 106576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malleske DT, Chorna O, and Maitre NL. 2018. ‘Pulmonary sequelae and functional limitations in children and adults with bronchopulmonary dysplasia’, Paediatr Respir Rev, 26: 55–59. [DOI] [PubMed] [Google Scholar]
- Malli F, Koutsokera A, Paraskeva E, Zakynthinos E, Papagianni M, Makris D, Tsilioni I, Molyvdas PA, Gourgoulianis KI, and Daniil Z. 2013. ‘Endothelial progenitor cells in the pathogenesis of idiopathic pulmonary fibrosis: an evolving concept’, PLoS One, 8: e53658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maniatis NA, Kotanidou A, Catravas JD, and Orfanos SE. 2008. ‘Endothelial pathomechanisms in acute lung injury’, Vascul Pharmacol, 49: 119–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maniatis NA, and Orfanos SE. 2008. ‘The endothelium in acute lung injury/acute respiratory distress syndrome’, Curr Opin Crit Care, 14: 22–30. [DOI] [PubMed] [Google Scholar]
- Matthay MA, Ware LB, and Zimmerman GA. 2012. ‘The acute respiratory distress syndrome’, J Clin Invest, 122: 2731–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKelvey KJ, Powell KL, Ashton AW, Morris JM, and McCracken SA. 2015. ‘Exosomes: Mechanisms of Uptake’, J Circ Biomark, 4: 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mezentsev A, Merks RM, O’Riordan E, Chen J, Mendelev N, Goligorsky MS, and Brodsky SV. 2005. ‘Endothelial microparticles affect angiogenesis in vitro: role of oxidative stress’, Am J Physiol Heart Circ Physiol, 289: H1106–14. [DOI] [PubMed] [Google Scholar]
- Mitani Y, Ueda M, Komatsu R, Maruyama K, Nagai R, Matsumura M, and Sakurai M. 2001. ‘Vascular smooth muscle cell phenotypes in primary pulmonary hypertension’, Eur Respir J, 17: 316–20. [DOI] [PubMed] [Google Scholar]
- Monsel A, Zhu YG, Gudapati V, Lim H, and Lee JW. 2016. ‘Mesenchymal stem cell derived secretome and extracellular vesicles for acute lung injury and other inflammatory lung diseases’, Expert Opin Biol Ther, 16: 859–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morel O, Jesel L, Freyssinet JM, and Toti F. 2011. ‘Cellular mechanisms underlying the formation of circulating microparticles’, Arterioscler Thromb Vasc Biol, 31: 15–26. [DOI] [PubMed] [Google Scholar]
- Morrison TJ, Jackson MV, Cunningham EK, Kissenpfennig A, McAuley DF, O’Kane CM, and Krasnodembskaya AD. 2017. ‘Mesenchymal Stromal Cells Modulate Macrophages in Clinically Relevant Lung Injury Models by Extracellular Vesicle Mitochondrial Transfer’, Am J Respir Crit Care Med, 196: 1275–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mostefai HA, Meziani F, Mastronardi ML, Agouni A, Heymes C, Sargentini C, Asfar P, Martinez MC, and Andriantsitohaina R. 2008. ‘Circulating microparticles from patients with septic shock exert protective role in vascular function’, Am J Respir Crit Care Med, 178: 1148–55. [DOI] [PubMed] [Google Scholar]
- Mourani PM, and Abman SH. 2015. ‘Pulmonary Hypertension and Vascular Abnormalities in Bronchopulmonary Dysplasia’, Clin Perinatol, 42: 839–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulcahy LA, Pink RC, and Carter DR. 2014. ‘Routes and mechanisms of extracellular vesicle uptake’, J Extracell Vesicles, 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller-Redetzky HC, Suttorp N, and Witzenrath M. 2014. ‘Dynamics of pulmonary endothelial barrier function in acute inflammation: mechanisms and therapeutic perspectives’, Cell Tissue Res, 355: 657–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nerlich A, Mieth M, Letsiou E, Fatykhova D, Zscheppang K, Imai-Matsushima A, Meyer TF, Paasch L, Mitchell TJ, Tonnies M, Bauer TT, Schneider P, Neudecker J, Ruckert JC, Eggeling S, Schimek M, Witzenrath M, Suttorp N, Hippenstiel S, and Hocke AC. 2018. ‘Pneumolysin induced mitochondrial dysfunction leads to release of mitochondrial DNA’, Sci Rep, 8: 182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicolls MR, and Voelkel NF. 2017. ‘The Roles of Immunity in the Prevention and Evolution of Pulmonary Arterial Hypertension’, Am J Respir Crit Care Med, 195: 1292–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Njock MS, Cheng HS, Dang LT, Nazari-Jahantigh M, Lau AC, Boudreau E, Roufaiel M, Cybulsky MI, Schober A, and Fish JE. 2015. ‘Endothelial cells suppress monocyte activation through secretion of extracellular vesicles containing antiinflammatory microRNAs’, Blood, 125: 3202–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogura H, Tanaka H, Koh T, Fujita K, Fujimi S, Nakamori Y, Hosotsubo H, Kuwagata Y, Shimazu T, and Sugimoto H. 2004. ‘Enhanced production of endothelial microparticles with increased binding to leukocytes in patients with severe systemic inflammatory response syndrome’, J Trauma, 56: 823–30; discussion 30–1. [DOI] [PubMed] [Google Scholar]
- Orfanos SE, Mavrommati I, Korovesi I, and Roussos C. 2004. ‘Pulmonary endothelium in acute lung injury: from basic science to the critically ill’, Intensive Care Med, 30: 1702–14. [DOI] [PubMed] [Google Scholar]
- Pan S, Fei A, Jing L, Zhang X, and Gao C. 2017. ‘Increased Circulating Endothelial Microparticles Associated with PAK4 Play a Key Role in Ventilation-Induced Lung Injury Process’, Biomed Res Int, 2017: 4902084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez-Casal M, Downey C, Fukudome K, Marx G, and Toh CH. 2005. ‘Activated protein C induces the release of microparticle-associated endothelial protein C receptor’, Blood, 105: 1515–22. [DOI] [PubMed] [Google Scholar]
- Peterson DB, Sander T, Kaul S, Wakim BT, Halligan B, Twigger S, Pritchard KA Jr., Oldham KT, and Ou JS. 2008. ‘Comparative proteomic analysis of PAI-1 and TNF- alpha-derived endothelial microparticles’, Proteomics, 8: 2430–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piccin A, Murphy WG, and Smith OP. 2007. ‘Circulating microparticles: pathophysiology and clinical implications’, Blood Rev, 21: 157–71. [DOI] [PubMed] [Google Scholar]
- Pocsfalvi G, Stanly C, Vilasi A, Fiume I, Capasso G, Turiak L, Buzas EI, and Vekey K. 2016. ‘Mass spectrometry of extracellular vesicles’, Mass Spectrom Rev, 35: 3–21. [DOI] [PubMed] [Google Scholar]
- Poore S, Berry B, Eidson D, McKie KT, and Harris RA. 2013. ‘Evidence of vascular endothelial dysfunction in young patients with cystic fibrosis’, Chest, 143: 939–45. [DOI] [PubMed] [Google Scholar]
- Porro C, Di Gioia S, Trotta T, Lepore S, Panaro MA, Battaglino A, Ratclif L, Castellani S, Bufo P, Martinez MC, and Conese M. 2013. ‘Pro-inflammatory effect of cystic fibrosis sputum microparticles in the murine lung’, J Cyst Fibros, 12: 721–8. [DOI] [PubMed] [Google Scholar]
- Porro C, Lepore S, Trotta T, Castellani S, Ratclif L, Battaglino A, Di Gioia S, Martinez MC, Conese M, and Maffione AB. 2010. ‘Isolation and characterization of microparticles in sputum from cystic fibrosis patients’, Respir Res, 11: 94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Priou P, Gagnadoux F, Tesse A, Mastronardi ML, Agouni A, Meslier N, Racineux JL, Martinez MC, Trzepizur W, and Andriantsitohaina R. 2010. ‘Endothelial dysfunction and circulating microparticles from patients with obstructive sleep apnea’, Am J Pathol, 177: 974–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qazi KR, Torregrosa Paredes P, Dahlberg B, Grunewald J, Eklund A, and Gabrielsson S. ‘Proinflammatory exosomes in bronchoalveolar lavage fluid of patients with sarcoidosis’, Thorax, 65: 1016–24. [DOI] [PubMed] [Google Scholar]
- Qiao Y, Liang X, Yan Y, Lu Y, Zhang D, Yao W, Wu W, and Yan Z. 2018. ‘Identification of Exosomal miRNAs in Rats With Pulmonary Neutrophilic Inflammation Induced by Zinc Oxide Nanoparticles’, Front Physiol, 9: 217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabinovitch M, Guignabert C, Humbert M, and Nicolls MR. 2014. ‘Inflammation and immunity in the pathogenesis of pulmonary arterial hypertension’, Circ Res, 115: 165–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raimondo F, Morosi L, Chinello C, Magni F, and Pitto M. 2011. ‘Advances in membranous vesicle and exosome proteomics improving biological understanding and biomarker discovery’, Proteomics, 11: 709–20. [DOI] [PubMed] [Google Scholar]
- Ramacciotti E, Hawley AE, Wrobleski SK, Myers DD Jr., Strahler JR, Andrews PC, Guire KE, Henke PK, and Wakefield TW. 2010. ‘Proteomics of microparticles after deep venous thrombosis’, Thromb Res, 125: e269–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ranchoux B, Harvey LD, Ayon RJ, Babicheva A, Bonnet S, Chan SY, Yuan JX, and Perez VJ. 2018. ‘Endothelial dysfunction in pulmonary arterial hypertension: an evolving landscape (2017 Grover Conference Series)’, Pulm Circ, 8: 2045893217752912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rautou PE, and Mackman N. 2013. ‘Microvesicles as risk markers for venous thrombosis’, Expert Rev Hematol, 6: 91–101. [DOI] [PubMed] [Google Scholar]
- Redman CW, and Sargent IL. 2007. ‘Microparticles and immunomodulation in pregnancy and pre-eclampsia’, J Reprod Immunol, 76: 61–7. [DOI] [PubMed] [Google Scholar]
- Reid VL, and Webster NR. 2012. ‘Role of microparticles in sepsis’, Br J Anaesth, 109: 503–13. [DOI] [PubMed] [Google Scholar]
- Romano M, Collura M, Lapichino L, Pardo F, Falco A, Chiesa PL, Caimi G, and Davi G. 2001. ‘Endothelial perturbation in cystic fibrosis’, Thromb Haemost, 86: 1363–7. [PubMed] [Google Scholar]
- Sander TL, Ou JS, Densmore JC, Kaul S, Matus I, Twigger S, Halligan B, Greene AS, Pritchard KA Jr., and Oldham KT. 2008. ‘Protein composition of plasminogen activator inhibitor type 1-derived endothelial microparticles’, Shock, 29: 504–11. [DOI] [PubMed] [Google Scholar]
- Sapet C, Simoncini S, Loriod B, Puthier D, Sampol J, Nguyen C, Dignat-George F, and Anfosso F. 2006. ‘Thrombin-induced endothelial microparticle generation: identification of a novel pathway involving ROCK-II activation by caspase-2’, Blood, 108: 1868–76. [DOI] [PubMed] [Google Scholar]
- Sato A, and Ikegami M. 2012. ‘SP-B and SP-C containing new synthetic surfactant for treatment of extremely immature lamb lung’, PLoS One, 7: e39392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scruggs AK, Cioffi EA, Cioffi DL, King JA, and Bauer NN. 2015. ‘Lectin-Based Characterization of Vascular Cell Microparticle Glycocalyx’, PLoS One, 10: e0135533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serban KA, Rezania S, Petrusca DN, Poirier C, Cao D, Justice MJ, Patel M, Tsvetkova I, Kamocki K, Mikosz A, Schweitzer KS, Jacobson S, Cardoso A, Carlesso N, Hubbard WC, Kechris K, Dragnea B, Berdyshev EV, McClintock J, and Petrache I. 2016. ‘Structural and functional characterization of endothelial microparticles released by cigarette smoke’, Sci Rep, 6: 31596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shai E, Rosa I, Parguina AF, Motahedeh S, Varon D, and Garcia A. 2012. ‘Comparative analysis of platelet-derived microparticles reveals differences in their amount and proteome depending on the platelet stimulus’, J Proteomics, 76 Spec No.: 287–96. [DOI] [PubMed] [Google Scholar]
- Shaver CM, Woods J, Clune JK, Grove BS, Wickersham NE, McNeil JB, Shemancik G, Ware LB, and Bastarache JA. 2017. ‘Circulating microparticle levels are reduced in patients with ARDS’, Crit Care, 21: 120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slutsky AS, and Ranieri VM. 2013. ‘Ventilator-induced lung injury’, N Engl J Med, 369: 2126–36. [DOI] [PubMed] [Google Scholar]
- Soni S, Wilson MR, O’Dea KP, Yoshida M, Katbeh U, Woods SJ, and Takata M. 2016. ‘Alveolar macrophage-derived microvesicles mediate acute lung injury’, Thorax, 71: 1020–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevens TP, Harrington EW, Blennow M, and Soll RF. 2007. ‘Early surfactant administration with brief ventilation vs. selective surfactant and continued mechanical ventilation for preterm infants with or at risk for respiratory distress syndrome’, Cochrane Database Syst Rev: CD003063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevens T, Rosenberg R, Aird W, Quertermous T, Johnson FL, Garcia JG, Hebbel RP, Tuder RM, and Garfinkel S. 2001. ‘NHLBI workshop report: endothelial cell phenotypes in heart, lung, and blood diseases’, Am J Physiol Cell Physiol, 281: C1422–33. [DOI] [PubMed] [Google Scholar]
- Stiefel P, Sanchez-Armengol MA, Villar J, Vallejo-Vaz A, Moreno-Luna R, and Capote F. 2013. ‘Obstructive sleep apnea syndrome, vascular pathology, endothelial function and endothelial cells and circulating microparticles’, Arch Med Res, 44: 409–14. [DOI] [PubMed] [Google Scholar]
- Strieter RM, and Mehrad B. 2009. ‘New mechanisms of pulmonary fibrosis’, Chest, 136: 1364–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strulovici-Barel Y, Staudt MR, Krause A, Gordon C, Tilley AE, Harvey BG, Kaner RJ, Hollmann C, Mezey JG, Bitter H, Pillai SG, Hilton H, Wolff G, Stevenson CS, Visvanathan S, Fine JS, and Crystal RG. 2016. ‘Persistence of circulating endothelial microparticles in COPD despite smoking cessation’, Thorax, 71: 1137–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun X, Singleton PA, Letsiou E, Zhao J, Belvitch P, Sammani S, Chiang ET, Moreno-Vinasco L, Wade MS, Zhou T, Liu B, Parastatidis I, Thomson L, Ischiropoulos H, Natarajan V, Jacobson JR, Machado RF, Dudek SM, and Garcia JG. 2012. ‘Sphingosine-1-phosphate receptor-3 is a novel biomarker in acute lung injury’, Am J Respir Cell Mol Biol, 47: 628–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Y, Huo C, Qiao Z, Shang Z, Uzzaman A, Liu S, Jiang X, Fan LY, Ji L, Guan X, Cao CX, and Xiao H. 2018. ‘Comparative Proteomic Analysis of Exosomes and Microvesicles in Human Saliva for Lung Cancer’, J Proteome Res. [DOI] [PubMed] [Google Scholar]
- Szotowski B, Antoniak S, Goldin-Lang P, Tran QV, Pels K, Rosenthal P, Bogdanov VY, Borchert H, Schultheiss HP, and Rauch U. 2007. ‘Antioxidative treatment inhibits the release of thrombogenic tissue factor from irradiation- and cytokine-induced endothelial cells’, Cardiovasc Res, 73: 806–12. [DOI] [PubMed] [Google Scholar]
- Szotowski B, Antoniak S, Poller W, Schultheiss HP, and Rauch U. 2005. ‘Procoagulant soluble tissue factor is released from endothelial cells in response to inflammatory cytokines’, Circ Res, 96: 1233–9. [DOI] [PubMed] [Google Scholar]
- Takahashi T, Kobayashi S, Fujino N, Suzuki T, Ota C, He M, Yamada M, Suzuki S, Yanai M, Kurosawa S, Yamaya M, and Kubo H. 2012. ‘Increased circulating endothelial microparticles in COPD patients: a potential biomarker for COPD exacerbation susceptibility’, Thorax, 67: 1067–74. [DOI] [PubMed] [Google Scholar]
- Takahashi T, Kobayashi S, Fujino N, Suzuki T, Ota C, Tando Y, He M, Yamada M, Kurosawa S, Yamaya M, and Kubo H. 2013. ‘Differences in the released endothelial microparticle subtypes between human pulmonary microvascular endothelial cells and aortic endothelial cells in vitro’, Exp Lung Res, 39: 155–61. [DOI] [PubMed] [Google Scholar]
- Takahashi T, Kobayashi S, Fujino N, Suzuki T, Ota C, Tando Y, Yamada M, Yanai M, Yamaya M, Kurosawa S, Yamauchi M, and Kubo H. 2014. ‘Annual FEV1 changes and numbers of circulating endothelial microparticles in patients with COPD: a prospective study’, BMJ Open, 4: e004571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi T, and Kubo H. 2014. ‘The role of microparticles in chronic obstructive pulmonary disease’, Int J Chron Obstruct Pulmon Dis, 9: 303–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terrisse AD, Puech N, Allart S, Gourdy P, Xuereb JM, Payrastre B, and Sie P. 2010. ‘Internalization of microparticles by endothelial cells promotes platelet/endothelial cell interaction under flow’, J Thromb Haemost, 8: 2810–9. [DOI] [PubMed] [Google Scholar]
- Thery C, Amigorena S, Raposo G, and Clayton A. 2006. ‘Isolation and characterization of exosomes from cell culture supernatants and biological fluids’, Curr Protoc Cell Biol, Chapter 3: Unit 3 22. [DOI] [PubMed] [Google Scholar]
- Thery C, Zitvogel L, and Amigorena S. 2002. ‘Exosomes: composition, biogenesis and function’, Nat Rev Immunol, 2: 569–79. [DOI] [PubMed] [Google Scholar]
- Thomashow MA, Shimbo D, Parikh MA, Hoffman EA, Vogel-Claussen J, Hueper K, Fu J, Liu CY, Bluemke DA, Ventetuolo CE, Doyle MF, and Barr RG. 2013. ‘Endothelial microparticles in mild chronic obstructive pulmonary disease and emphysema. The Multi-Ethnic Study of Atherosclerosis Chronic Obstructive Pulmonary Disease study’, Am J Respir Crit Care Med, 188: 60–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian T, Zhu YL, Hu FH, Wang YY, Huang NP, and Xiao ZD. 2013. ‘Dynamics of exosome internalization and trafficking’, J Cell Physiol, 228: 1487–95. [DOI] [PubMed] [Google Scholar]
- Totani L, Plebani R, Piccoli A, Di Silvestre S, Lanuti P, Recchiuti A, Cianci E, Dell’Elba G, Sacchetti S, Patruno S, Guarnieri S, Mariggio MA, Mari VC, Anile M, Venuta F, Del Porto P, Moretti P, Prioletta M, Mucilli F, Marchisio M, Pandolfi A, Evangelista V, and Romano M. 2017. ‘Mechanisms of endothelial cell dysfunction in cystic fibrosis’, Biochim Biophys Acta, 1863: 3243–53. [DOI] [PubMed] [Google Scholar]
- Tripisciano C, Weiss R, Eichhorn T, Spittler A, Heuser T, Fischer MB, and Weber V. 2017. ‘Different Potential of Extracellular Vesicles to Support Thrombin Generation: Contributions of Phosphatidylserine, Tissue Factor, and Cellular Origin’, Sci Rep, 7:6522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trzepizur W, Martinez MC, Priou P, Andriantsitohaina R, and Gagnadoux F. 2014. ‘Microparticles and vascular dysfunction in obstructive sleep apnoea’, Eur Respir J, 44: 207–16. [DOI] [PubMed] [Google Scholar]
- Tual-Chalot S, Gagnadoux F, Trzepizur W, Priou P, Andriantsitohaina R, and Martinez MC. 2014. ‘Circulating microparticles from obstructive sleep apnea syndrome patients induce endothelin-mediated angiogenesis’, Biochim Biophys Acta, 1842: 202–7. [DOI] [PubMed] [Google Scholar]
- Tual-Chalot S, Guibert C, Muller B, Savineau JP, Andriantsitohaina R, and Martinez MC. ‘Circulating microparticles from pulmonary hypertensive rats induce endothelial dysfunction’, Am J Respir Crit Care Med, 182: 261–8. [DOI] [PubMed] [Google Scholar]
- Tuder RM, Groves B, Badesch DB, and Voelkel NF. 1994. ‘Exuberant endothelial cell growth and elements of inflammation are present in plexiform lesions of pulmonary hypertension’, Am J Pathol, 144: 275–85. [PMC free article] [PubMed] [Google Scholar]
- van Balkom BW, Eisele AS, Pegtel DM, Bervoets S, and Verhaar MC. 2015. ‘Quantitative and qualitative analysis of small RNAs in human endothelial cells and exosomes provides insights into localized RNA processing, degradation and sorting’, J Extracell Vesicles, 4: 26760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Ierssel SH, Van Craenenbroeck EM, Hoymans VY, Vrints CJ, Conraads VM, and Jorens PG. 2013. ‘Endothelium dependent vasomotion and in vitro markers of endothelial repair in patients with severe sepsis: an observational study’, PLoS One, 8: e69499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Niel G, D’Angelo G, and Raposo G. 2018. ‘Shedding light on the cell biology of extracellular vesicles’, Nat Rev Mol Cell Biol. [DOI] [PubMed] [Google Scholar]
- Veerappan A, Thompson M, Savage AR, Silverman ML, Chan WS, Sung B, Summers B, Montelione KC, Benedict P, Groh B, Vicencio AG, Peinado H, Worgall S, and Silver RB. 2016. ‘Mast cells and exosomes in hyperoxia-induced neonatal lung disease’, Am J Physiol Lung Cell Mol Physiol, 310: L1218–32. [DOI] [PubMed] [Google Scholar]
- Vion AC, Birukova AA, Boulanger CM, and Birukov KG. 2013. ‘Mechanical forces stimulate endothelial microparticle generation via caspase-dependent apoptosis- independent mechanism’, Pulm Circ, 3: 95–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vion AC, Ramkhelawon B, Loyer X, Chironi G, Devue C, Loirand G, Tedgui A, Lehoux S, and Boulanger CM. 2013. ‘Shear stress regulates endothelial microparticle release’, Circ Res, 112: 1323–33. [DOI] [PubMed] [Google Scholar]
- Vituret C, Gallay K, Confort MP, Ftaich N, Matei CI, Archer F, Ronfort C, Mornex JF, Chanson M, Di Pietro A, Boulanger P, and Hong SS. 2016. ‘Transfer of the Cystic Fibrosis Transmembrane Conductance Regulator to Human Cystic Fibrosis Cells Mediated by Extracellular Vesicles’, Hum Gene Ther, 27: 166–83. [DOI] [PubMed] [Google Scholar]
- Vykoukal J, Sun N, Aguilar-Bonavides C, Katayama H, Tanaka I, Fahrmann JF, Capello M, Fujimoto J, Aguilar M, Wistuba II, Taguchi A, Ostrin EJ, and Hanash SM.‘Plasma-derived extracellular vesicle proteins as a source of biomarkers for lung adenocarcinoma’, Oncotarget, 8: 95466–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wahlund CJE, Eklund A, Grunewald J, and Gabrielsson S. 2017. ‘Pulmonary Extracellular Vesicles as Mediators of Local and Systemic Inflammation’, Front Cell Dev Biol, 5: 39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang JG, Williams JC, Davis BK, Jacobson K, Doerschuk CM, Ting JP, and Mackman N. 2011. ‘Monocytic microparticles activate endothelial cells in an IL-1beta- dependent manner’, Blood, 118: 2366–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang JM, Wang Y, Huang JY, Yang Z, Chen L, Wang LC, Tang AL, Lou ZF, and Tao J. 2007. ‘C-Reactive protein-induced endothelial microparticle generation in HUVECs is related to BH4-dependent NO formation’, J Vasc Res, 44: 241–8. [DOI] [PubMed] [Google Scholar]
- Wang T, Gross C, Desai AA, Zemskov E, Wu X, Garcia AN, Jacobson JR, Yuan JX, Garcia JG, and Black SM. 2017. ‘Endothelial cell signaling and ventilator-induced lung injury: molecular mechanisms, genomic analyses, and therapeutic targets’, Am J Physiol Lung Cell Mol Physiol, 312: L452–L76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watts JA, Lee YY, Gellar MA, Fulkerson MB, Hwang SI, and Kline JA. 2012. ‘Proteomics of microparticles after experimental pulmonary embolism’, Thromb Res,130: 122–8. [DOI] [PubMed] [Google Scholar]
- Wheeler KI, Klingenberg C, Morley CJ, and Davis PG. 2011. ‘Volume-targeted versus pressure-limited ventilation for preterm infants: a systematic review and meta-analysis’, Neonatology, 100: 219–27. [DOI] [PubMed] [Google Scholar]
- Willis GR, Fernandez-Gonzalez A, Anastas J, Vitali SH, Liu X, Ericsson M, Kwong A, Mitsialis SA, and Kourembanas S. 2018. ‘Mesenchymal Stromal Cell Exosomes Ameliorate Experimental Bronchopulmonary Dysplasia and Restore Lung Function through Macrophage Immunomodulation’, Am J Respir Crit Care Med, 197: 104–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willis GR, Kourembanas S, and Mitsialis SA. 2017. ‘Therapeutic Applications of Extracellular Vesicles: Perspectives from Newborn Medicine’, Methods Mol Biol, 1660: 409–32. [DOI] [PubMed] [Google Scholar]
- Witwer KW, Buzas EI, Bemis LT, Bora A, Lasser C, Lotvall J, Nolte-’t Hoen EN, Piper MG, Sivaraman S, Skog J, Thery C, Wauben MH, and Hochberg F. 2013. ‘Standardization of sample collection, isolation and analysis methods in extracellular vesicle research’, J Extracell Vesicles, 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf P 1967. ‘The nature and significance of platelet products in human plasma’, Br J Haematol, 13: 269–88. [DOI] [PubMed] [Google Scholar]
- Xu R, Greening DW, Zhu HJ, Takahashi N, and Simpson RJ. 2016. ‘Extracellular vesicle isolation and characterization: toward clinical application’, J Clin Invest, 126: 1152–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto S, Niida S, Azuma E, Yanagibashi T, Muramatsu M, Huang TT, Sagara H, Higaki S, Ikutani M, Nagai Y, Takatsu K, Miyazaki K, Hamashima T, Mori H, Matsuda N, Ishii Y, and Sasahara M. 2015. ‘Inflammation-induced endothelial cell-derived extracellular vesicles modulate the cellular status of pericytes’, Sci Rep, 5: 8505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan C, Wang X, Su H, and Ying K. 2017. ‘Recent Progress in Research on the Pathogenesis of Pulmonary Thromboembolism: An Old Story with New Perspectives’, Biomed Res Int, 2017:6516791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yanez-Mo M, Siljander PR, Andreu Z, Zavec AB, Borras FE, Buzas EI, Buzas K, Casal E, Cappello F, Carvalho J, Colas E, Cordeiro-da Silva A., Fais S, Falcon-Perez JM, Ghobrial IM, Giebel B, Gimona M, Graner M, Gursel I, Gursel M, Heegaard NH, Hendrix A, Kierulf P, Kokubun K, Kosanovic M, Kralj-Iglic V, Kramer- Albers EM, Laitinen S, Lasser C, Lener T, Ligeti E, Line A, Lipps G, Llorente A, Lotvall J, Mancek-Keber M, Marcilla A, Mittelbrunn M, Nazarenko I, Nolte-’t Hoen EN, Nyman TA, O’Driscoll L, Olivan M, Oliveira C, Pallinger E, Del Portillo HA, Reventos J, Rigau M, Rohde E, Sammar M, Sanchez-Madrid F, Santarem N, Schallmoser K, Ostenfeld MS, Stoorvogel W, Stukelj R, Van der Grein SG, Vasconcelos MH, Wauben MH, and De Wever O. 2015. ‘Biological properties of extracellular vesicles and their physiological functions’, J Extracell Vesicles, 4: 27066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang S, Zhong Q, Qiu Z, Chen X, Chen F, Mustafa K, Ding D, Zhou Y, Lin J, Yan S, Deng Y, Wang M, Zhou Y, Liao Y, and Zhou Z. 2014. ‘Angiotensin II receptor type 1 autoantibodies promote endothelial microparticles formation through activating p38 MAPK pathway’, J Hypertens, 32: 762–70. [DOI] [PubMed] [Google Scholar]
- Yu Y, Jing L, Zhang X, and Gao C. 2017. ‘Simvastatin Attenuates Acute Lung Injury via Regulating CDC42-PAK4 and Endothelial Microparticles’, Shock, 47: 378–84. [DOI] [PubMed] [Google Scholar]
- Yuana Y, Sturk A, and Nieuwland R. 2013. ‘Extracellular vesicles in physiological and pathological conditions’, Blood Rev, 27: 31–9. [DOI] [PubMed] [Google Scholar]
- Yun CH, Jung KH, Chu K, Kim SH, Ji KH, Park HK, Kim HC, Lee ST, Lee SK, and Roh JK. 2010. ‘Increased circulating endothelial microparticles and carotid atherosclerosis in obstructive sleep apnea’, J Clin Neurol, 6: 89–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Chen C, Hu B, Niu X, Liu X, Zhang G, Zhang C, Li Q, and Wang Y. 2016. ‘Exosomes Derived from Human Endothelial Progenitor Cells Accelerate Cutaneous Wound Healing by Promoting Angiogenesis Through Erk½ Signaling’, Int J Biol Sci, 12: 1472–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Q, Shang M, Zhang M, Wang Y, Chen Y, Wu Y, Liu M, Song J, and Liu Y. 2016. ‘Microvesicles derived from hypoxia/reoxygenation-treated human umbilical vein endothelial cells promote apoptosis and oxidative stress in H9c2 cardiomyocytes’, BMC Cell Biol, 17: 25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao L, Luo H, Li X, Li T, He J, Qi Q, Liu Y, and Yu Z. 2017. ‘Exosomes Derived from Human Pulmonary Artery Endothelial Cells Shift the Balance between Proliferation and Apoptosis of Smooth Muscle Cells’, Cardiology, 137: 43–53. [DOI] [PubMed] [Google Scholar]
- Zhou Y, Li P, Goodwin AJ, Cook JA, Halushka PV, Chang E, and Fan H. 2018. ‘Exosomes from Endothelial Progenitor Cells Improve the Outcome of a Murine Model of Sepsis’, Mol Ther. [DOI] [PMC free article] [PubMed] [Google Scholar]