

Abstract
Ubiquitin-conjugating enzymes (E2) enable protein ubiquitination by conjugating ubiquitin to their catalytic cysteine for subsequent transfer to a target lysine sidechain. Deprotonation of the incoming lysine enables its nucleophilicity, but determinants of lysine activation remain poorly understood. We report a novel pathogenic mutation in the E2 UBE2A, identified in two brothers presenting mild intellectual disability. The pathogenic Q93E mutation yields UBE2A with impaired aminolysis activity but no loss of ability to be conjugated with ubiquitin. Importantly, the low intrinsic reactivity of Q93E-UBE2A is not overcome by a cognate ubiquitin E3 ligase, Rad18, with the UBE2A target, PCNA. However, Q93E-UBE2A is reactive at high pH or with a low pKa amine as the nucleophile, providing the first evidence of reversion of a defective UBE2A mutation. We propose that Q93E substitution perturbs the UBE2A catalytic microenvironment essential for lysine deprotonation during ubiquitin transfer, generating an enzyme that is disabled but not dead.
INTRODUCTION
Protein ubiquitination is a post-translational modification that regulates both protein function and levels1. A trio of enzymes, ubiquitin-activating enzyme (E1), ubiquitin-conjugating enzyme (E2), and ubiquitin-ligating enzyme (E3), work in series to conjugate ubiquitin to a lysine residue on a target protein through an isopeptide bond2. The modification has myriad cellular effects, and as a consequence, mutations in components of the ubiquitination machinery are linked to human disease, including cancer, neurodevelopmental, and neurodegenerative disorders3. Mutations in ubiquitination enzymes are documented contributors to neurodevelopmental pathophysiology in intellectual disability (ID) and autism spectrum disorders4.
A prototypic disorder involving an altered ubiquitination enzyme is X-linked intellectual disability (XLID)-type Nascimento5, related to abnormalities in the UBE2A gene (Gene ID 7319). The human locus for the UBE2A gene is at Xq24 and the transcript is ubiquitously expressed, with highest mRNA levels in heart, testis, and brain6. Individuals identified with this syndrome carry intragenic mutations (point mutations or small deletions) or larger Xq24 deletions encompassing the UBE2A gene5,7–19. Common clinical features include moderate-to-severe intellectual disability, prominent dysmorphic features, impaired speech, urogenital malformations, skin abnormalities, and epilepsy10. Larger deletions encompassing UBE2A and adjacent genes appear to result in severe forms of the clinical syndrome with a higher prevalence of white matter changes, heart defects, and urogenital malformations compared to patients with intragenic UBE2A pathogenic variants.
UBE2A shares 95% amino acid identity to UBE2B (Gene ID 7320) at 5q31.1 but UBE2B variants have only been associated with male infertility20. Molecular mechanisms linking mutations in UBE2A to neurodevelopmental disorders are still poorly understood. Emerging data implicate defective mobilization of the E3 Parkin as a source of neuronal vesicle trafficking and clearance of dysfunctional mitochondria abnormalities, both suspected to contribute to the neurodevelopmental phenotype in patients with UBE2A deficiency syndrome11. Previously reported pathogenic mutations are distributed along the six exons of the UBE2A gene5,7–19. Limited functional and structural information on mutant forms of UBE2A limit current understanding of the contribution of specific UBE2A variants to the pathogenesis of ID.
We identified a novel missense mutation c.277C>G (p.Q93E) in UBE2A in two brothers presenting atypical phenotype with only mild ID and impaired speech. The impact of this novel mutation on UBE2A function and structure was characterized to reveal molecular mechanisms underlying the human disorder. We found that the Q93E mutation yields an enzyme with impaired ability to transfer ubiquitin to lysine, thereby inhibiting product formation. The defect is not rescued by the presence of an E3 ligase (RAD18) toward a specific UBE2A target, PCNA. Our results implicate Q93 of UBE2A in the deprotonation of an incoming lysine, a crucial step for ubiquitin transfer from an E2~ubiquitin conjugate to substrate. Strikingly, the activity of UBE2A Q93E mutant is partially recovered by increased pH, providing the first report of a potential reversion of a defective mutation in UBE2A related to XLID-type Nascimento.
RESULTS
Identification of UBE2A Q93E mutation in XLID patients
Two brothers from a Brazilian family (Figure 1A) were diagnosed with idiopathic mild intellectual disability and speech impairment. Extensive clinical genetic investigation, including fra(X) screening, karyotype and chromosome microarray analysis, failed to identify the aetiology of the condition. Recent whole exome sequencing revealed that the affected individuals carry a missense variant in UBE2A exon 5 (chrX:1187165 c.277C>G). This variant has not been previously reported. Sanger sequencing confirmed the mutation in the probands and was used to determine familial segregation (Figure 1B). The mother and three unaffected daughters are heterozygous carriers of the UBE2A mutation (Supplementary Figure 1). All female carriers showed extreme skewed X-inactivation in peripheral blood (>90%). The variant is therefore considered as the cause of the phenotype. The mutation results in an amino acid change of Gln to Glu at position 93 near the active site of UBE2A. Position 93 harbors glutamine in both UBE2A and UBE2B but is not highly conserved among E2s (Supplementary Figure 2).
Figure 1: Identification and functional characterization of Q93E in UBE2A.
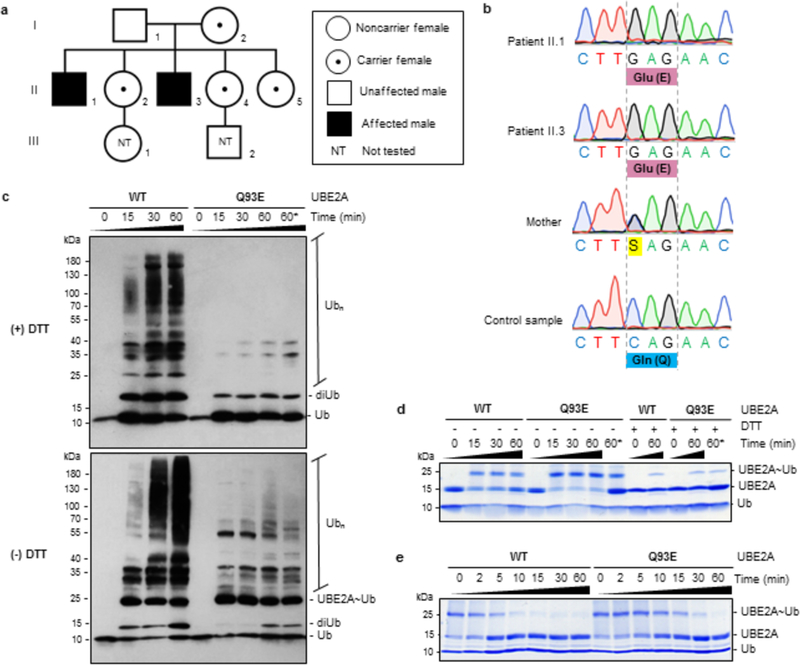
(A) Three-generation heredogram (I, II and Ill) representing the family of patients with ID, with probands shaded in black. Carrier females are indicated by a black dot within unblackened circle. Individuals in generation III were not tested (NT). (B) Sanger sequencing analysis of exon five of UBE2A showing the c.277C->G (p.Q93E) mutation in the two siblings (Patients II.1 and II.3). The mother is a heterozygous carrier. (C) Western blot analysis of in vitro ubiquitination assay of wild-type (WT) UBE2A and Q93E mutant enzyme using anti-ubiquitin antibody, in the presence (+) or absence (-) of reducing agent (DTT). (D) Thioester bond formation monitored by SDS-PAGE, using non-reducing (-DTT) or reducing (+DTT) sample loading buffer. (E) Evaluation of UBE2A~Ub conjugate ability in transferring ubiquitin to free lysine, visualized by SDS-PAGE in non-reducing conditions. Ub - ubiquitin; diUb - diubiquitin; Ubn - polyubiquitin chain; UBE2A~Ub - thioester conjugate of ubiquitin with UBE2A; * - double concentration of Q93E-UBE2A mutant on reaction.
The Q93E mutation impairs UBE2A activity
Given the proximity of Q93 to the catalytic cysteine, we tested whether the mutation alters general UBE2A activity. Purified recombinant wild-type (WT) and Q93E-UBE2A were assayed for their ability to generate polyubiquitin chains21. Each E2 was incubated with E1, ATP/Mg2+, and ubiquitin, in the absence of an E3 or a substrate. WT-UBE2A generated high molecular-weight products as detected by western blotting against ubiquitin, but there was a marked lack of products generated by the mutant E2 (Figure 1C). To test whether loss of activity is due to impaired conjugation of ubiquitin to the mutant E2 by E1, assays in which the E2~Ub conjugate is visualized directly were compared (Figure 1D). Both WT- and Q93E-UBE2A are robustly conjugated with ubiquitin, implying that the defect is in the second (transfer) step of product formation. In the assay shown in Figure 1C, products are generated when a lysine on an acceptor ubiquitin (Ubacceptor) attacks the thioester of E2~Ubdonor to carry out an aminolysis reaction. To test if the Q93E mutation affects the intrinsic aminolysis activity of UBE2A, we carried out lysine discharge assays. Activity towards the ε-amino group of free lysine reflects the ability of E2~Ub conjugates to transfer ubiquitin without complications associated with protein substrates22,23. Upon addition of free lysine to E2~Ub, WT-UBE2A~Ub is depleted within ~10 minutes while depletion of Q93E-UBE2A~Ub took over 30 minutes (Figure 1E, Supplementary Figure 3A). Hence, Q93E-UBE2A is compromised in aminolysis activity towards free lysine and lysine attached to ubiquitin.
The activity of other UBE2A mutations previously reported to be involved in XLID Nascimento-type, namely R7W, R11Q and G23R7,11 were compared. Each XLID mutant showed a detectable decrease in polyubiquitin chain formation in vitro (Supplementary Figure 4A), although the reductions were more modest than those observed for Q93E. Ubiquitin conjugation to the E2 active site was not impaired in these mutants (Supplementary Figure 4B). These mutation sites are far from the E2 active site: R7 and R11 are in the putative E1/E3 binding surface and G23 is on the “backside” surface of UBE2A that binds the E3 enzyme RAD1821.
XLID Q93E mutation affects the UBE2A catalytic site
We determined crystal structures of WT- and Q93E-UBE2A at 1.85 and 2.20 Å resolution, respectively (Figure 2A and Supplementary Table 1) and compared the structures. Superposition of the two structures revealed high similarity (RMSD of 0.17 Å) and a typical E2 fold, featuring a 4-stranded antiparallel β-sheet, four α-helices, and a short 310-helix24. The most notable difference was the loss of a hydrogen bond between the Q93 sidechain and the backbone carbonyl of L89 — the residue adjacent to active-site C88 (Figure 2B, Supplementary Figure 5).
Figure 2: Structural comparison between wild-type and Q93E human UBE2A.
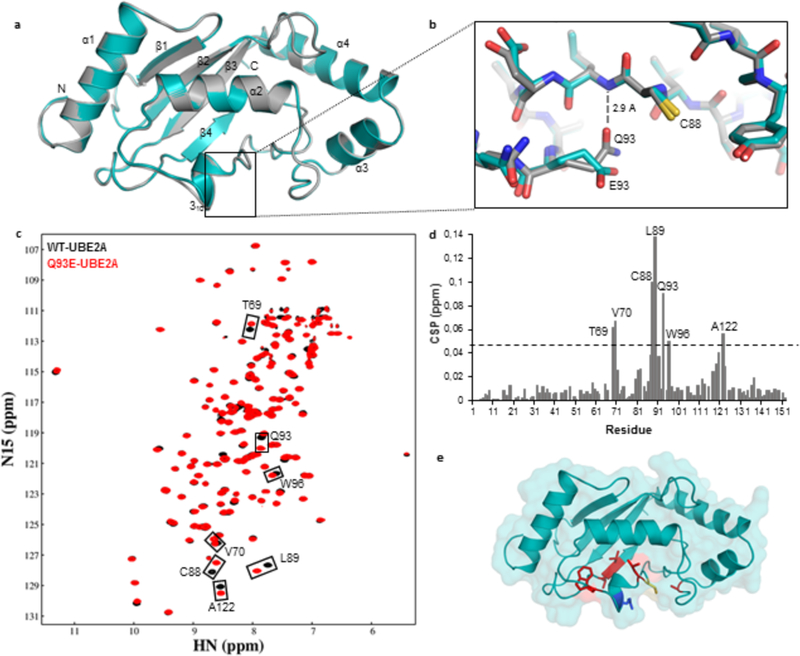
(A) Ribbon representation of wild-type UBE2A (grey) and Q93E mutant (cyan) crystal structures, superposed. (B) Close up view of Q93E mutation from (A) highlighting the interactions made by Q93 sidechain. The nearby catalytic residue C88 is also indicated. (C) Overlay of (1H,15N)-HSQC spectra of 15N-labeled wild-type UBE2A (black) and Q93E mutant (red). Residues showing significant chemical shift perturbation (CSP) are indicated. (D) Graph showing the weighted CSP for each residue when introducing the Q93E mutation into UBE2A. The dashed line indicates twice the standard deviation above average. (E) Residues presenting the most significant CSPs are mapped in red on Q93E-UBE2A crystal structure. Catalytic Cys88 is colored in yellow and Q93E mutation is shown in blue.
We used NMR spectroscopy to gain more insight into the basis for the reduced activity of Q93E-UBE2A. Assignment of backbone resonances of Q93E-UBE2A was carried out using standard triple resonance approaches; backbone assignments for WT-UBE2A were based on previously reported UBE2B NMR spectra (BMRB code17443). Overlay of the (1H,15N)-HSQC spectra of wild-type and Q93E-UBE2A are highly similar, with residues near the catalytic site (T69, V70, L89, W96, and A122) and the catalytic C88 (Figure 2C–E) exhibiting small chemical shift perturbations (CSPs). Consistent with the loss of hydrogen bonding between L89 and Q93 implied from the mutant crystal structure, the two largest CSPs are for L89 and C88, confirming that the Q93E mutation detectably alters the chemical environment of the catalytic residue.
XLID Q93E mutation preserves the UBE2A~Ub conformation
The orientation and interactions of ubiquitin within the E2 active site play a central role in its transfer to substrate25. In E2~Ub conjugates characterized structurally, the conjugated ubiquitin is flexible and assumes a range of conformations relative to the E2 domain26. Binding of an E2~Ub to some RING-type E3s promotes a “closed” state of the conjugate, in which ubiquitin is positioned against the E2 helix, α226,27. The “closed” state has enhanced reactivity, presumably by positioning the thioester bond for nucleophilic attack by incoming lysine28. We investigated if the ability of UBE2A~Ub to adopt a “closed” conformation is altered in the mutant by monitoring formation of an oxyester conjugate of C88S-UBE2A variant, which is more stable than the thioester conjugate25. 15N-C88S-UBE2A or 15N-C88S/Q93E-UBE2A was conjugated with unlabeled ubiquitin directly in the NMR tube, in the presence of E1 and Mg2+. After starting the reaction by addition of ATP, spectral changes were monitored through acquisition of sequential (1H,15N)-HSQC spectra. The spectrum of oxyester-linked 15N-C88S-UBE2A-O~Ub showed substantial CSPs, especially for peaks corresponding to residues surrounding the UBE2A catalytic site (Figure 3A and B). There are perturbations to residues on helix-α2 (residues D101, S103, I105, S111, L112 and L113) (Figure 3B), the E2 region that interacts with ubiquitin in the closed conformation26. The oxyester of the mutant 15N-C88S/Q93E-UBE2A (UBE2AQ93E-O~Ub) showed similar perturbations to those observed with UBE2A-O~Ub conjugate (Figure 3C).
Figure 3: Comparison of wild-type and Q93E mutant UBE2A-O~Ub oxyester conjugates.
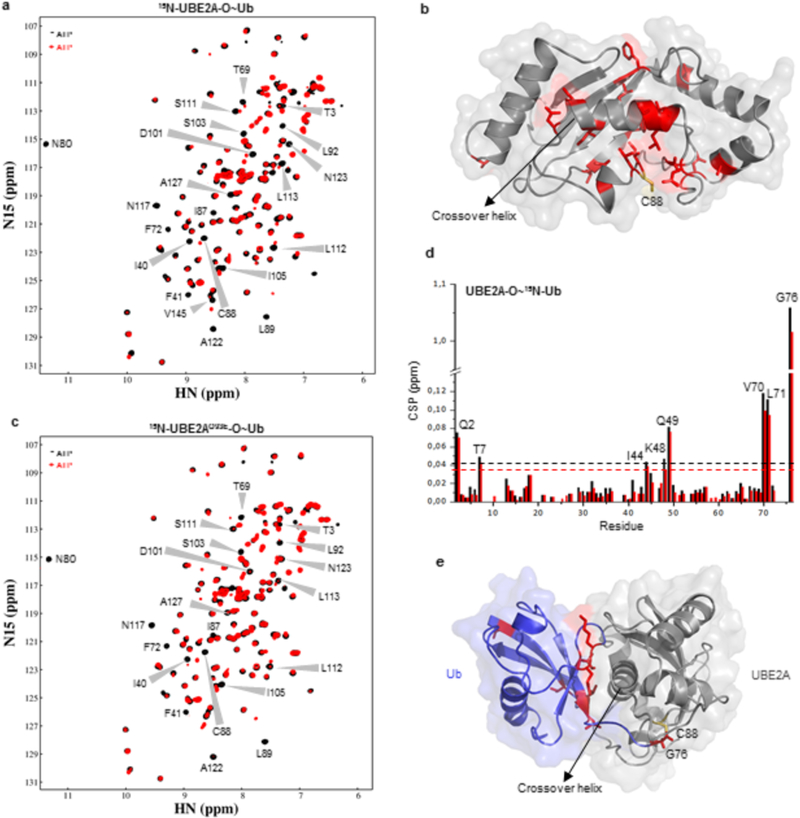
(A) (1H,15N)-HSQC spectra of 15N-C88S-UBE2A before (black) and after (red) addition of ATP, in the presence of E1, ubiquitin and Mg2+. Residues presenting CSPs larger than one standard deviation above the average after oxyester formation are indicated on spectra. (B) Mapping of residues perturbed in (A) upon conjugation with ubiquitin on WT UBE2A structure. Catalytic residue C88 is shown in yellow. (C) (1H,15N)-HSQC spectra of ubiquitin conjugation with 15N-C88S/Q93E-UBE2A. Black - before ATP addition; Red - after ATP addition. (D) CSP chart of 15N-Ub upon conjugation with C88S-UBE2A (black) or C88S/Q93E-UBE2A (red) (corresponding to spectra in Supplementary Figure 6A). Dashed lines indicate one standard deviation above the average. (E) CSP mapping onto the structural model of UBE2A~Ub showing Ub residues that experienced significant NMR chemical shift changes. Catalytic cysteine C88 is indicated in yellow.
A reciprocal setup allowed changes to the 15N-Ub spectra upon oxyester conjugation to be observed (Supplementary Figure 6A). CSPs were highly similar for both conjugates (Figure 3D, Supplementary Figure 6A). A crystal structure of the yeast homolog of UBE2A, Rad6~Ub, is in an open state29, so to assess whether the UBE2A oxyester conjugates adopts a closed state in solution, we built a molecular model of UBE2A~Ub based on a Ubc13~Ub crystal structure (PDB code 5aiu)30 that is in a closed conformation (Figure 3E). In the model of UBE2A~Ub, NMR-perturbed Ub residues I44, K48, Q49, V70 and L71 are in the interface with E2 helix-α2 31,32. Thus, the NMR results indicate that UBE2A-O~Ub visits closed conformations in solution, consistent with UBE2A’s ability to assemble polyubiquitin chains in the absence of an E3. Furthermore, the results indicate that UBE2AQ93E-O~Ub conjugate adopts a similar conformation. The main spectral difference between UBE2A-O~Ub and UBE2AQ93E-O~Ub conjugates was for ubiquitin Gly76, the residue that forms the oxyester bond with UBE2A (Supplementary Figure 6A). The difference indicates that the chemical environment of the oxyester bond (the mimic of the reactive bond of E2~Ub) is altered in the UBE2AQ93E-O~Ub conjugate.
The human E2, UBE2D2 (UbcH5b), has arginine at the position homologous to UBE2A Q93. Substitution of R90 in UBE2D2 with Glu (R90E-UBE2D2) generates a mutant with impaired polyubiquitination activity33. An interaction between the substituted Glu residue in R90E-UBE2D2 and R74 in the C-terminal tail of Ub was proposed to explain the loss of function. If a similar interaction is in play in Q93E-UBE2A, we would predict a difference in the chemical shift of the Ub R74 peak between wild-type and Q93E- UBE2A~Ub, but none is observed (Supplementary Figure 6B). Furthermore, the peak corresponding to E93 is not perturbed between the apo- and conjugate form of the E2 (Supplementary Figure 6C). Together, the observations argue against a similar interaction as the source of the activity defect in Q93E-UBE2A~Ub.
Lastly, considering that an interaction between ubiquitin and the backside of some E2 enzymes is critical for polyubiquitin chain formation21,33,34, we tested whether the Q93E mutation affects the UBE2A backside interaction with ubiquitin. Comparison of (1H,15N)-HSQC spectra of wild-type and Q93E-UBE2A in the presence of a 10-fold molar excess of ubiquitin were highly similar (Supplementary Figure 7), confirming that the weak backside interaction is not detectably impaired.
Overall, the data demonstrate that the activity defects of Q93E-UBE2A are not due to altered conformation of either the apo- or conjugate form. Instead, the evidence points to a change in the chemical microenvironment of the active site.
UBE2A Q93 residue facilitates the ubiquitin transfer
The ability to observe sidechain NH2 resonances for Gln and Asn sidechains by NMR allowed us to follow the fate of the Q93 sidechain during C88S-UBE2A oxyester formation (Figure 4A). Although the Q93 backbone amide resonance is unperturbed upon ubiquitin conjugation (Figure 4A, left panel), its sidechain NH2 resonances are considerably perturbed (Figure 4A, right panel). To further investigate the Q93 sidechain, we generated the thioester in the NMR tube with wild-type UBE2A. The Q93 sidechain resonances have different resonance positions in (uncharged) wild-type and C88S-UBE2A, consistent with the notion that Q93 senses the chemical environment of the active site (-SH versus -OH; Supplementary Figure 8). In the first spectrum after ATP addition (collected for 80 minutes post-ATP addition), we observe a substantial CSP in the C88 peak (Supplementary Figure 9A) and for the Q93 sidechain peaks (Figure 4B; red spectrum). At five hours post-ATP addition, a second set of Q93 sidechain resonances that are further shifted appear (Figure 4B; cyan spectrum). The observation of two sets of peaks indicates that the Q93 sidechain exists in two slowly-interconverting conformations at this point in the reaction. Importantly, neither of the Q93 resonances overlays with the original peak (Figure 4B; black spectrum), so neither correspond to discharged, unmodified UBE2A. In the same spectrum, there is substantial loss of intensity for most peaks. A set of specific UBE2A residues exhibit an even larger reduction in peak intensity (Supplementary Figure 9A and B), essentially the same residues that were perturbed in UBE2A-O~Ub oxyester spectra (Supplementary Figure 9C and Figure 3B), indicating the effect is linked to formation of the thioester bond between UBE2A and ubiquitin. We propose that the Q93 sidechain participates in a new interaction during the process. To test this hypothesis, we examined the impact of Q93 substitution with alanine on polyubiquitin chain assembly activity. As predicted, the Q93A substitution reduced UBE2A activity compared to wild- type-UBE2A (Figure 5A), consistent with involvement of the Q93 sidechain in the facilitation of ubiquitin transfer. We note that most E2 family members have residues capable of forming hydrogen bonds at the Q93-equivalent position of UBE2A (Supplementary Figure 2).
Figure 4: Monitoring of the Q93 residue during UBE2A catalysis.
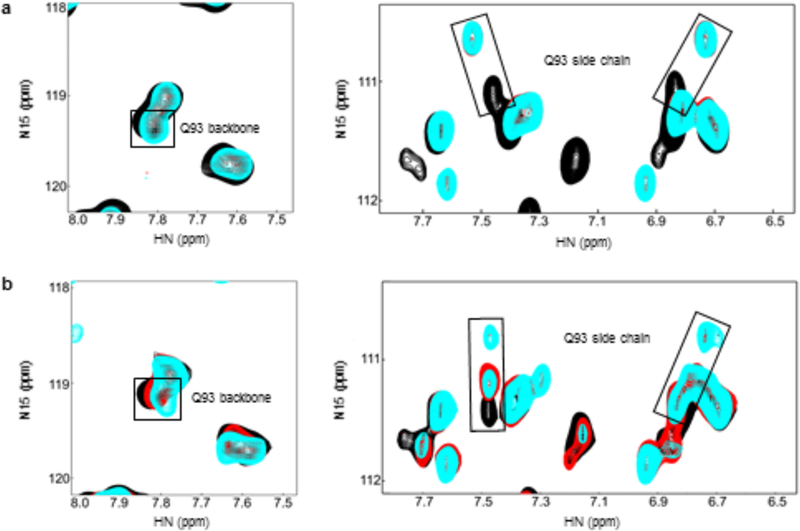
(1H,15N)-HSQC spectra of (A) 15N-C88S-UBE2A during oxyester linkage formation and (B) 15N-WT- UBE2A during thioester linkage formation, showing the peaks correspondent to Q93 backbone and Q93 sidechain. Black - before ATP addition, red - right after ATP addition, cyan - 5 hours after ATP addition.
Figure 5: Wild-type and mutants UBE2A enzymatic activity.
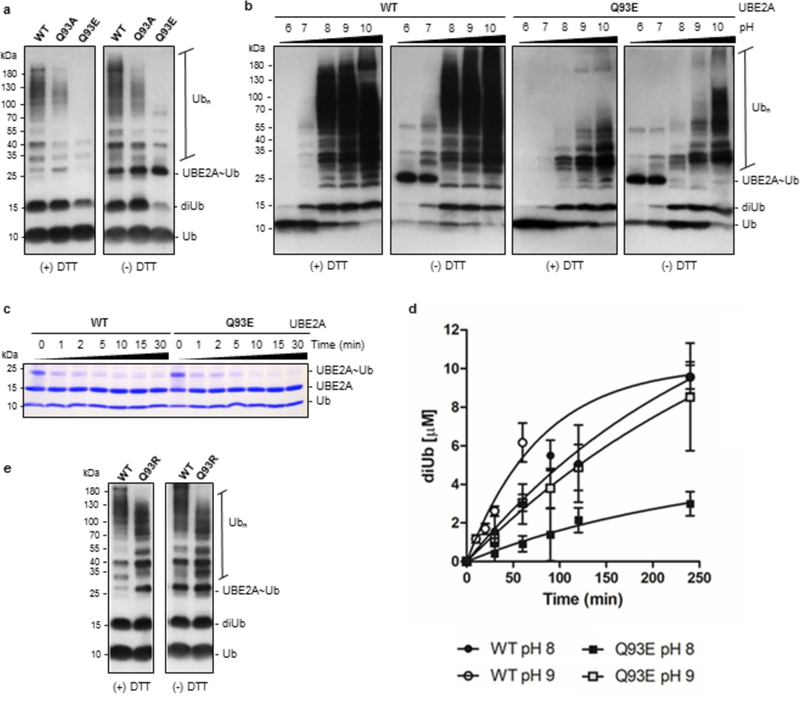
(A) In vitro ubiquitination assay analyzed by western blot using anti-ubiquitin antibody comparing WT-UBE2A, Q93A and Q93E mutants at pH 8 (30 minutes), under reducing (+ DTT) and non-reducing (- DTT) conditions. (B) The ability of WT- and Q93E-UBE2A in assemble polyubiquitin chains was assayed at different pHs (6–10) (2 hours). (C) Discharge of UBE2A~Ub in the presence of the low pKa nucleophile hydroxylamine, verified through non-reducing coomassie stained SDS-PAGE. (D) Graph showing the formation of diubiquitin by WT- and Q93E-UBE2A at pH 8 and 9. Data represent the mean ± standard deviation of three independent experiments. The respective rates were determined from a linear regression (Supplementary Figure 10, C and D). (E) Comparison between WT-UBE2A and a mutant enzyme presenting a basic residue in position 93 (Q93R), through polyubiquitination assay at pH 8 (30 minutes). Ub - ubiquitin; diUb - diubiquitin; Ubn - polyubiquitin chain; UBE2A~Ub - thioester conjugate of ubiquitin with UBE2A.
Defective Q93E mutant is active in alkaline conditions
The Q93E mutant is more severely impaired than Q93A-UBE2A in its polyubiquitination activity, suggesting that the presence of the carboxylate group of E93, as opposed to the loss of the amide group of Q93, is particularly detrimental to activity (Figure 5A). In assays performed as a function of pH, the ability of Q93E-UBE2A to assemble polyubiquitin chains is partially restored at pH 9 or above (Figure 5B). An essential step in ubiquitin transfer via aminolysis is deprotonation of the incoming lysine that acts as a nucleophile to attack the E2~Ub conjugate. Given the high pKa of lysine (about 10.5), residues near the E2 active site must create a microenvironment that lowers the effective pKa of the attacking lysine to enable formation of the isopeptide bond35. The calculated pKa value for a substrate lysine near the catalytic cysteine of the SUMO-specific E2, Ubc9, is lowered almost 3 pH units compared with the pKa of free lysine35. The increase in Q93E-UBE2A activity at high pH implies that the mutant E2 has lost its ability to suppress the pKa of the incoming lysine, thereby slowing the rate of ubiquitin transfer to a lysine amino group. To test this hypothesis, we performed reactions with hydroxylamine (pKa ~ 6.0)36 as the nucleophile, as it does not require deprotonation to act as a nucleophile at neutral pH. In contrast to reactions with free lysine (Figure 1E), the initial rates of discharge using hydroxylamine were indistinguishable for wild-type and Q93E-UBE2A (Figure 5C, Supplementary Figure 3B). Both enzymes exhibit considerable activity, indicating that when deprotonation is not necessary, Q93E-UBE2A is active. Together, the results strongly imply a primary impact of the Q93E-UBE2A mutation is its inability to lower the pKa of the attacking lysine sidechain.
To calculate the pKa value of lysine from an Ubacceptor molecule that is proximal to the catalytic site of UBE2A, we incorporated a Ub molecule into our UBE2A~Ubdonor model based on the crystal structure of Ubc13~Ub presenting an acceptor ubiquitin near the thioester linkage (PDB code 2gmi)37. Using this model, multi-conformational continuum electrostatics38 predict an increase in lysine pKa from 9.3 in the WT complex to 13.7 in the Q93E-UBE2A mutant complex. The calculations support the notion that substitution of Glu at position 93 leads to impaired lysine deprotonation caused by an increase in the pKa of the attacking lysine.
To gain insight into the activation of Q93E-UBE2A at high pH, we measured the rate of diubiquitin formation by WT and Q93E enzymes at pH 8 and 9 (Figure 5D, Supplementary Figure 10). We conducted single-turnover assays in which the charging reactions were done using lysine-free ubiquitin (K0-Ub) to hinder polyubiquitin formation (Supplementary Figure 10A) and the discharge reactions were performed using equimolar concentration of wild-type ubiquitin (after addition of EDTA to inhibit E1 enzyme). Diubiquitin formation was measured for both enzymes at the two pH conditions (Supplementary Figure 10B), quantified, and the rates of product formation were determined from linear regression analysis (Supplementary Figure 10C). The rate of diubiquitin formation by Q93E-UBE2A at pH 8 was about four-fold slower than that of WT-UBE2A (0.013 ± 0.001 μM/min versus 0.047 ± 0.008 μM/min, respectively; Supplementary Figure 10D). Increasing the reaction pH from 8 to 9 led to a reduction in the gap between rates (0.041 ± 0.003 μM/min for mutant versus 0.101 ± 0.006 μM/min for WT-UBE2A). The rate observed for Q93E-UBE2A at pH 9 is quite similar to that of WT-UBE2A at pH 8, corroborating the idea that the impairment caused by Q93E substitution can be overcome by increased pH.
To further explore this hypothesis, we substituted Q93 in UBE2A with Arg, the residue found in numerous E2s including UBE2D2. As predicted by our model, the Q93R mutation yields increased UBE2A activity compared to the WT enzyme (Figure 5E). Thus, we conclude that the nature of the amino acid sidechain at position 93 residue in UBE2A contributes directly to enzyme activity via modulation of lysine activation during ubiquitin transfer.
UBE2A Q93E mutation diminishes monoubiquitination of PCNA
To assess the Ub transfer activity of Q93E-UBE2A in the presence of an E3 and a substrate we performed reactions that contained either wild-type or Q93E-UBE2A, the RING E3 Rad18, and the substrate PCNA. This pairing of E2 and E3 is known to transfer monoubiquitin to a specific lysine on PCNA39. Both forms of the E2 generate monoubiquitinated PCNA (Figure 6), but both the rate and level of product formation is reduced with Q93E-UBE2A (t1/2 of 22.8 min versus 6.9 min for WT). The results indicate that the aminolysis defect in Q93E-UBE2A is not compensated by the E3 ligase Rad18, implying that the presence of mutant E2 could lead to decreased levels of ubiquitinated product. To assess whether a positively-charged residue facilitates lysine activation for ubiquitin transfer to substrate, we conducted the PCNA ubiquitination experiment with Q93R-UBE2A (Figure 6A). The variant is highly active in this reaction, producing more product than WT-UBE2A at an increased rate (t1/2 of 2.9 min; Figure 6B). In combination, the data show that the more active the E2 is intrinsically, the more product is generated in E3-dependent reactions.
Figure 6: PCNA monoubiquitination assay.
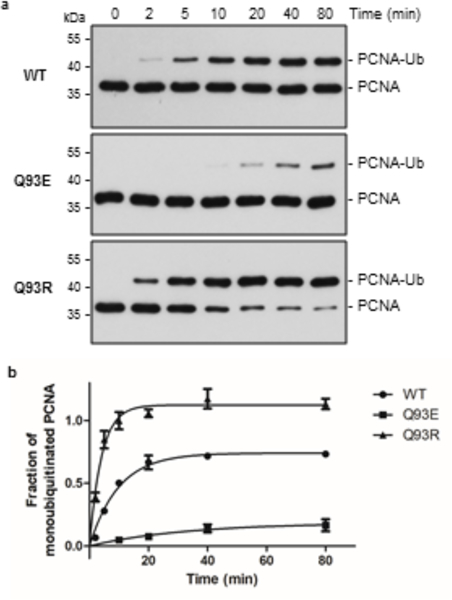
Comparison between WT-, Q93E- and Q93R-UBE2A enzymes in the ability of monoubiquitinate PCNA. (A) Anti-PCNA western blot (uncropped blots are shown in Supplementary Figure 11). PCNA-Ub - monoubiquitinated PCNA. (B) Graph showing the formation of ubiquitinated PCNA by each enzyme, quantified by ImageJ. Three independent experiments were conducted and mean ± standard deviations are indicated.
DISCUSSION
Mutations in the UBE2A gene are known to cause X-linked intellectual disability5,7–19. Uncovering the molecular mechanisms that underlie UBE2A-type ID can expand understanding of ID pathophysiology and clarify fundamental catalytic mechanisms of this important class of enzymes. As central players of protein ubiquitination, E2s carry out two reactions: transthiolation that conjugates ubiquitin to the E2 active site, and aminolysis that transfers ubiquitin to an amino group on a substrate23. Here, we demonstrate that a novel pathogenic missense UBE2A mutation Q93E impairs the enzyme’s ability to carry out aminolysis, while its transthiolation activity is preserved. Q93E-UBE2A is defective not only with ubiquitin serving as the nucleophile, inhibiting polyubiquitin chain formation, but also with free lysine, indicating that the defect is in the aminolysis reaction itself. Furthermore, the mutant E2 is defective in its ability to monoubiquitinate PCNA in the presence of its cognate E3 ligase (RAD18), indicating that the intrinsic defect cannot be fully overcome by an activating E3. These observations strongly suggest that Q93E-UBE2A generates its bona fide ubiquitinated products more slowly and at lower levels than wild-type UBE2A, even in the presence of cognate E3 ligases.
For lysine to act as a nucleophile, its ε-amino group must be deprotonated 35,40. Two mechanisms are proposed for lysine deprotonation by E2s: 1) direct abstraction of a proton by a proton acceptor and 2) reduction of the pKa of the incoming lysine by the microenvironment near the E2 active site35. In the former case, D117 of UBE2D141 and H94 of UBE2G242 are proposed to serve as proton acceptors for an incoming lysine. But neither of the corresponding residues in UBE2A (S120 and Q93, respectively) can serve as proton acceptors and indeed, none of the residues observed to be perturbed in Q93E- UBE2A can receive a proton. Furthermore, the high similarity of ubiquitin resonances in WT and Q93E-UBE2A~Ub conjugates, other than for Ub G76, argue against the existence of a proton acceptor in the ubiquitin moiety. Therefore, our work supports a mechanism in which lysine deprotonation is not carried out directly by a specific amino acid residue from UBE2A or Ubdonor serving as proton acceptor.
Despite the conserved architecture of the active site and position of the catalytic cysteine among E2s, the identity and location of residues that participate in catalysis differ, suggesting that positioning and deprotonation of an incoming lysine may involve distinct sets of residues43. In the SUMO-specific E2 Ubc9, residues N85, Y87, and D127 near the active-site cysteine provide a microenvironment that lowers the incoming lysine pKa to enhance deprotonation and nucleophilicity at physiological pH and help position the incoming lysine35. Although the NMR data indicate that substitution of Glu for Gln at position 93 perturbs the environment around the active site, the UBE2A residues that correspond to Ubc9 N85, Y87, and D127 (N80, Y82, and S120) exhibit no or very minor perturbations in Q93E-UBE2A spectra compared to WT-UBE2A spectra. Our study identifies a previously unappreciated contribution of the Q93 sidechain to the aminolysis reaction. Previous studies on UBE2B indicated an involvement of Q93 residue in formation of the thioester with ubiquitin21. In contrast, the defect in Q93- UBE2A is in discharge of the ubiquitin from the thioester and not in thioester formation. Residues at the Q93-equivalent position are not strictly conserved in the E2 family, although almost half of human E2s have Arg or Lys at this position and only UBE2A and UBE2B have Gln (Supplementary Figure 2). Divergent effects of mutations at this E2 position have been reported. Substitutions with alanine inhibit formation of the E2~Ub conjugate in human UBE2C (UbcH10) 44, restricts polyubiquitin chain formation by UBE2G245, and has no apparent effect on UBE2D2 and UBE2K activity33,46. In UBE2D2, UBE2K, and UBE2A, glutamic acid substitution at this position results in an E2 with impaired activity33,46.
The combined observations indicate that the “Q93” position, which sits at the opening to the active site of E2s may modulate both the thioester formation and discharge activities of an E2. A positively-charged residue at this position appears to enhance activity, while substitution with a negatively-charged Glu impairs activity. Three human E2s, UBE2J2, UBE2U, and Ubc9, contain a negatively-charged residue at this position, suggesting that different residues are responsible for modulating the aminolysis reaction carried out by these E2s.
Studies conducted with cells from a mouse Ube2A knockout or containing patient-derived UBE2A mutations (R7W, I87MfsX14 and Q128X) have shown common defects in mitochondria, suspected to be caused by defective polyubiquitination of mitochondrial proteins11. These studies associated XLID mutations in UBE2A with disruption in mitophagy11. R7W and R11Q UBE2A mutations result in a loss of interaction with UBR4, a protein linked to lysosome-mediated degradation and autophagy47. R11Q and G23R pathogenic mutations are defective in polyubiquitin chain assembly21,47. Here, we show similar reductions in the polyubiquitin chain assembly by the mutants R7W, R11Q and G23R and even more pronounced reductions by the novel Q93E mutation in UBE2A. Therefore, defects in polyubiquitin chain formation and disruption of the mitophagy/autophagy processes seem to be a common feature related to different XLID UBE2A mutations.
Altogether, our data support a model in which glutamic acid at position 93 in UBE2A markedly reduces the enzyme’s ability to transfer ubiquitin from its active site to the ε-amino group of a lysine. The defect is caused by changes in the active site microenvironment that impair the reduction of pKa of an incoming lysine needed for its nucleophilicity. The results may have important implications for strategies to specifically inhibit UBE2A, recognized as a potential target for melanoma, breast, and ovarian cancer48–50. Moreover, our finding that increased pH can partially rescue Q93E- UBE2A activity could present possibilities to modulate the activity of the mutant enzyme with compounds that restore its ability to reduce the pKa of an incoming lysine.
Supplementary Material
ACKNOWLEDGEMENTS
We thank LNBio/CNPEM for accessibility to core facilities as well as for financial support. We also thank the access to MX2 beamline at the Brazilian Synchrotron Light Laboratory (LNLS) and the MX2 staff for technical assistance. We are very grateful to H. Powell for his assistance in X-ray data processing. And we would also like to thank T. Sixma and C. Hill for Addgene plasmids (#63571 and #61937, respectively). This work was supported by the Brazilian National Council for Scientific and Technological Development (CNPq C.R. -306879/2014-0; K.G.F - 310536/2014-6 and 422790/2016-8) and grants from Sâo Paulo Research Foundation (FAPESP C.R. - 2012/50981-5 and 2013/08028-1; M.M. - 2015/06281-7) and NIH/NIGMS (R.E.K. - R01 GM088055).
Footnotes
COMPETING FINANCIAL INTERESTS STATEMENT
The authors declare no competing financial interests.
REFERENCES
- 1.Komander D & Rape M The Ubiquitin Code. Annu. Rev. Biochem. 81, 203–229 (2012). [DOI] [PubMed] [Google Scholar]
- 2.Pickart CM Mechanisms Underlying Ubiquitination. Annu. Rev. Biochem. 70, 503–533 (2001). [DOI] [PubMed] [Google Scholar]
- 3.Popovic D, Vucic D & Dikic I Ubiquitination in disease pathogenesis and treatment. Nat. Med. 20, 1242–1253 (2014). [DOI] [PubMed] [Google Scholar]
- 4.Louros SR & Osterweil EK Perturbed proteostasis in autism spectrum disorders. J. Neurochem. 139, 1081–1092 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nascimento RMP, Otto PA, de Brouwer APM & Vianna-Morgante AM UBE2A, which encodes a ubiquitin-conjugating enzyme, is mutated in a novel X- linked mental retardation syndrome. Am. J. Hum. Genet. 79, 549–55 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Koken MHM et al. Expression of the Ubiquitin-Conjugating DNA Repair Enzymes HHR6A and B Suggests a Role in Spermatogenesis and Chromatin Modification. Dev. Biol. 173, 119–132 (1996). [DOI] [PubMed] [Google Scholar]
- 7.Budny B et al. Novel missense mutations in the ubiquitination-related gene UBE2A cause a recognizable X-linked mental retardation syndrome. Clin. Genet. 77, 541–551 (2010). [DOI] [PubMed] [Google Scholar]
- 8.De Leeuw N et al. UBE2A deficiency syndrome: Mild to severe intellectual disability accompanied by seizures, absent speech, urogenital, and skin anomalies in male patients. Am. J. Med. Genet. Part A 152 A, 3084–3090 (2010). [DOI] [PubMed] [Google Scholar]
- 9.Honda S et al. Novel deletion at Xq24 including the UBE2A gene in a patient with X-linked mental retardation. J. Hum. Genet. 55, 244–247 (2010). [DOI] [PubMed] [Google Scholar]
- 10.Czeschik JC et al. X-linked intellectual disability type Nascimento is a clinically distinct, probably underdiagnosed entity. OrphanetJ. Rare Dis. 8, 146 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Haddad DM et al. Mutations in the Intellectual Disability Gene Ube2a Cause Neuronal Dysfunction and Impair Parkin-Dependent Mitophagy. Mol. Cell 50, 831–843 (2013). [DOI] [PubMed] [Google Scholar]
- 12.Tucker T et al. Single exon-resolution targeted chromosomal microarray analysis of known and candidate intellectual disability genes. Eur. J. Hum. Genet. 22, 792–800 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Utine GE et al. Etiological yield of SNP microarrays in idiopathic intellectual disability. Eur. J. Paediatr. Neurol. 18, 327–337 (2014). [DOI] [PubMed] [Google Scholar]
- 14.Niranjan TS et al. Affected kindred analysis of human X chromosome exomes to identify novel X-linked intellectual disability genes. PLoS One 10, 1–22 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Thunstrom S, Sodermark L, Ivarsson L, Samuelsson L & Stefanova M UBE2A deficiency syndrome: A report of two unrelated cases with large Xq24 deletions encompassing UBE2A gene. Am. J. Med. Genet. Part A 167, 204–210 (2015). [DOI] [PubMed] [Google Scholar]
- 16.Tzschach A et al. Next-generation sequencing in X-linked intellectual disability. Eur. J. Hum. Genet. 23, 1513–1518 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tsurusaki Y et al. A novel UBE2A mutation causes X-linked intellectual disability type Nascimento. Hum. Genome Var. 4, 17019 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Xiao B et al. Marked yield of re-evaluating phenotype and exome/target sequencing data in 33 individuals with intellectual disabilities. Am. J. Med. Genet. Part A 176, 107–115 (2018). [DOI] [PubMed] [Google Scholar]
- 19.Giugliano T et al. UBE2A deficiency in two siblings: A novel splicing variant inherited from a maternal germline mosaicism. Am. J. Med. Genet. Part A 176, 722–726 (2018). [DOI] [PubMed] [Google Scholar]
- 20.Suryavathi V et al. Novel Variants in UBE2B Gene and Idiopathic Male Infertility. J. Androl. 29, 564–571 (2008). [DOI] [PubMed] [Google Scholar]
- 21.Hibbert RG, Huang A, Boelens R & Sixma TK E3 ligase Rad18 promotes monoubiquitination rather than ubiquitin chain formation by E2 enzyme Rad6. Proc. Natl. Acad. Sci. U. S. A. 108, 5590–5595 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pickart CM & Rose IA Functional heterogeneity of ubiquitin carrier proteins. J. Biol. Chem. 260, 1573–1581 (1985). [PubMed] [Google Scholar]
- 23.Stewart MD, Ritterhoff T, Klevit RE & Brzovic PS E2 enzymes: more than just middle men. Cell Res. 26, 423–440 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.van Wijk SJL & Timmers HTM The family of ubiquitin-conjugating enzymes (E2s): deciding between life and death of proteins. FASEBJ. 24, 981–993 (2010). [DOI] [PubMed] [Google Scholar]
- 25.Page RC, Pruneda JN, Amick J, Klevit RE & Misra S Structural Insights into the Conformation and Oligomerization of E2~Ubiquitin Conjugates. Biochemistry 51, 4175–4187 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Middleton AJ, Wright JD & Day CL Regulation of E2s: A Role for Additional Ubiquitin Binding Sites? J. Mol. Biol. 429, 3430–3440 (2017). [DOI] [PubMed] [Google Scholar]
- 27.Wickliffe KE, Lorenz S, Wemmer DE, Kuriyan J & Rape M The mechanism of linkage-specific ubiquitin chain elongation by a single-subunit E2. Cell 144, 769–781 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pruneda JN et al. Structure of an E3:E2~Ub Complex Reveals an Allosteric Mechanism Shared among RING/U-box Ligases. Mol. Cell 47, 933–942 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kumar P et al. Role of a non-canonical surface of Rad6 in ubiquitin conjugating activity. Nucleic Acids Res. 43, 9039–9050 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Branigan E, Plechanovová A, Jaffray EG, Naismith JH & Hay RT Structural basis for the RING-catalyzed synthesis of K63-linked ubiquitin chains. Nat. Struct. Mol. Biol. 22, 597–602 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pruneda JN, Stoll KE, Bolton LJ, Brzovic PS & Klevit RE Ubiquitin in Motion: Structural Studies of the Ubiquitin-Conjugating Enzyme~Ubiquitin Conjugate. Biochemistry 50, 1624–1633 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Plechanovov A, Jaffray EG, Tatham MH, Naismith JH & Hay RT Structure of a RING E3 ligase and ubiquitin-loaded E2 primed for catalysis. Nature 489, 115–120 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sakata E et al. Crystal Structure of UbcH5b~Ubiquitin Intermediate: Insight into the Formation of the Self-Assembled E2~Ub Conjugates. Structure 18, 138–147 (2010). [DOI] [PubMed] [Google Scholar]
- 34.Brzovic PS, Lissounov A, Christensen DE, Hoyt DW & Klevit RE A UbcH5/ubiquitin noncovalent complex is required for processive BRCA1-directed ubiquitination. Mol. Cell 21, 873–880 (2006). [DOI] [PubMed] [Google Scholar]
- 35.Yunus A. a & Lima CD Lysine activation and functional analysis of E2- mediated conjugation in the SUMO pathway. Nat. Struct. Mol. Biol. 13, 491–499 (2006). [DOI] [PubMed] [Google Scholar]
- 36.Mollin J, Kasparek F & Lasovsky J On the basicity of hydroxylamine and it derivatives. Chem. zresli 29, 39–43 (1975). [Google Scholar]
- 37.Eddins MJ, Carlile CM, Gomez KM, Pickart CM & Wolberger C Mms2- Ubc13 covalently bound to ubiquitin reveals the structural basis of linkage- specific polyubiquitin chain formation. Nat. Struct. Mol. Biol. 13, 915–920 (2006). [DOI] [PubMed] [Google Scholar]
- 38.Georgescu RE, Alexov EG & Gunner MR Combining Conformational Flexibility and Continuum Electrostatics for Calculating pKas in Proteins. Biophys. J. 83, 1731–1748 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hoege C, Pfänder B, Moldovan G-L, Pyrowolakis G & Jentsch S RAD6- dependent DNA repair is linked to modification of PCNA by ubiquitin and SUMO. Nature 419, 135–141 (2002). [DOI] [PubMed] [Google Scholar]
- 40.Saha A, Lewis S, Kleiger G, Kuhlman B & Deshaies RJ Essential Role for Ubiquitin-Ubiquitin-Conjugating Enzyme Interaction in Ubiquitin Discharge from Cdc34 to Substrate. Mol. Cell 42, 75–83 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhen Y et al. Exploring the RING-catalyzed ubiquitin transfer mechanism by MD and QM/MM calculations. PLoS One 9, (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ju T, Bocik W, Majumdar A & Tolman JR Solution structure and dynamics of human ubiquitin conjugating enzyme Ube2g2. Proteins Struct. Funct. Bioinforma. 78, 1291–1301 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cappadocia L & Lima CD Ubiquitin-like Protein Conjugation: Structures, Chemistry, and Mechanism. Chem. Rev. 118, 889–918 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lin Y, Hwang WC & Basavappa R Structural and functional analysis of the human mitotic-specific ubiquitin-conjugating enzyme, UbcH10. J Biol Chem 277, 21913–21921 (2002). [DOI] [PubMed] [Google Scholar]
- 45.Li W, Tu D, Brunger AT & Ye Y A ubiquitin ligase transfers preformed polyubiquitin chains from a conjugating enzyme to a substrate. Nature 446, 333–337 (2007). [DOI] [PubMed] [Google Scholar]
- 46.Middleton AJ & Day CL The molecular basis of lysine 48 ubiquitin chain synthesis by Ube2K. Sci. Rep. 5, 16793 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hong JH et al. KCMF1 (potassium channel modulatory factor 1) Links RAD6 to UBR4 (ubiquitin N-recognin domain-containing E3 ligase 4) and lysosome- mediated degradation. Mol. Cell. Proteomics 14, 674–85 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sanders MA et al. Novel Inhibitors of Rad6 Ubiquitin Conjugating Enzyme: Design, Synthesis, Identification, and Functional Characterization. Mol. Cancer Ther. 12, 373–383 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Rosner K et al. Rad6 is a Potential Early Marker of Melanoma Development. Transl. Oncol. 7, 384–392 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Somasagara RR et al. Rad6 upregulation promotes stem cell-like characteristics and platinum resistance in ovarian cancer. Biochem. Biophys. Res. Commun. 469, 449–455 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.