

Abstract

The constitutive androstane receptor (CAR) is a xenobiotic sensor governing the transcription of genes involved in drug disposition, energy homeostasis, and cell proliferation. However, currently available human CAR (hCAR) agonists are nonselective, which commonly activate hCAR along with other nuclear receptors, especially the closely related human pregnane X receptor (hPXR). Using a well-known hCAR agonist CITCO as a template, we report our efforts in the discovery of a potent and highly selective hCAR agonist. Two of the new compounds of the series, 18 and 19 (DL5050), demonstrated excellent potency and selectivity for hCAR over hPXR. DL5050 preferentially induced the expression of CYP2B6 (target of hCAR) over CYP3A4 (target of hPXR) on both the mRNA and protein levels. The selective hCAR agonist DL5050 represents a valuable tool molecule to further define the biological functions of hCAR, and may also be used as a new lead in the discovery of hCAR agonists for various therapeutic applications.
Keywords: Nuclear receptor, constitutive androstane receptor, structure−activity relationship, pregnane X receptor
The constitutive androstane receptor (CAR, NR1I3) belongs to the nuclear receptor (NR) subfamily 1I that also includes the pregnane X receptor (PXR, NR1I2) and vitamin D receptor (VDR, NR1I1).1,2 CAR plays an important role orchestrating the metabolism and detoxification of potentially toxic chemicals,3,4 by upregulating genes that are involved in the clearance of both xenobiotics and endobiotics.5,6 Although CAR-mediated metabolism of drugs (e.g., acetaminophen) can lead to the production of toxic metabolites,7 induction of CAR regulated genes has emerged as a therapeutic strategy for the treatment of various diseases. For instance, phenobarbital, a prototypical CAR activator, has been used to treat neonatal jaundice through its induction of UDP glucuronosyltransferase 1A1 (UGT1A1), the rate-limiting enzyme responsible for the metabolism of bilirubin.8 Maglich and co-workers have reported an important regulatory role of CAR in the metabolism of thyroid hormones during caloric restriction,9 while other studies revealed that activation of CAR ameliorates high-fat diet-induced obesity and diabetes in mice, suggesting that CAR modulators might be potential therapeutics for metabolic disorders.10,11 It is well-known that cyclophosphamide (CPA), a widely used chemotherapeutic prodrug, is converted to a therapeutically active metabolite 4-hydroxy-CPA primarily by CYP2B6 (target of hCAR) and to an inactive metabolite N-DCE-CPA predominantly by CYP3A4 (target of hPXR).12,13 Recent studies from our laboratory have shown that activation of CAR sensitizes cyclophosphamide (CPA)-based cancer treatment by enhancing biotransformation of CPA to its activate metabolite 4-hydroxy CPA.14,15
The activation mechanism of CAR begins with a ligand-triggered dissociation of CAR from its cochaperone partners, cytoplasmic CAR retention protein (CCRP), and heat shock protein 90 (Hsp90), in the cytosol (Figure 1).16 Dephosphorylation of CAR at Thr38 by protein phosphatase 2A (PP2A) generates free CAR,17 which then translocates into the nucleus and forms a heterodimer with the retinoid X receptor (RXR). The complex of RXR-CAR regulates its target genes upon binding to cis-acting responsive enhancer modules upstream of target genes.18 Different from most known NRs, CAR demonstrates constitutive and ligand-independent activity.19
Figure 1.
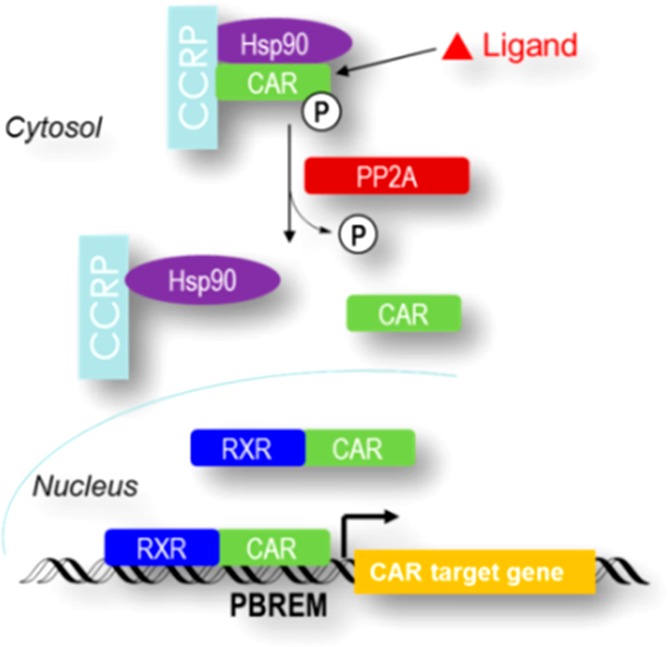
Activation mechanism of CAR.
Similar to most classical steroid receptors, CAR contains multiple functional domains including an N-terminal DNA-binding domain (DBD) that directly interacts with the promoters of its target genes, a hinge domain, and a multifunctional ligand-binding domain (LBD). The LBD contains a ligand binding pocket, a dimerization interface that binds to RXR, and a C-terminal ligand-dependent activation helix (AF2).20 The LBD of human CAR (hCAR) can bind to a wide range of structurally diversified ligands.21−23 Earning its name as an orphan nuclear receptor, physiologically relevant endogenous ligands of hCAR have not been identified as of yet.
Within the NR1I subfamily, hCAR shares a modestly conserved LBD and highly conserved DBD with human PXR (hPXR).24 As a result, these two nuclear receptors bind to multiple common ligands25 and regulate an overlapping set of genes (e.g., CYP2B6 and CYP3A4).24,26,27 Compared to hPXR, hCAR has a much smaller LBD (675 Å3 vs ∼1200 Å3 for hPXR).23 Unlike hPXR to which selective agonists (e.g., SR1281328) have been identified, no highly selective hCAR activator has been disclosed. The availability of potent and highly selective ligands for hCAR will not only benefit the confirmation of target genes for this important nuclear receptor but also verify whether hCAR is a viable therapeutic target.
In the past decade, efforts in both screens29−32 and medicinal chemistry33 have been devoted to the identification and development of agonists for hCAR. Among all known hCAR agonists, the GSK high-throughput screening hit CITCO is the most well-known for its good potency and selectivity.5,20 CITCO potentiates constitutive activity of hCAR by stabilizing the constitutive AF2-coactivator interaction.23 Although CITCO only indicates moderate selectivity for hCAR over hPXR, it is by far the most widely used chemical tool in elucidating the function of hCAR.34−37
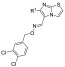
Herein, we report the development of hCAR agonists based on the structure of CITCO (Figure 2). Specifically, the 4-Cl-phenyl, imidazothiazole, and 3,4-diCl-phenyl groups of CITCO (1) were modified using compounds 2–29. Seven compounds were selected and further evaluated for their selectivity over hPXR. Two of the new compounds, 18 and 19 (DL5050), demonstrated excellent selectivity for hCAR over hPXR while maintaining similar hCAR agonistic potency to CITCO. In particular, compound DL5050 selectively induced the expression of cytochrome P450 CYP2B6 over CYP3A4 at both the mRNA and protein levels (Figure 4).
Figure 2.

Design of compounds 2–29 from CITCO.
Figure 4.
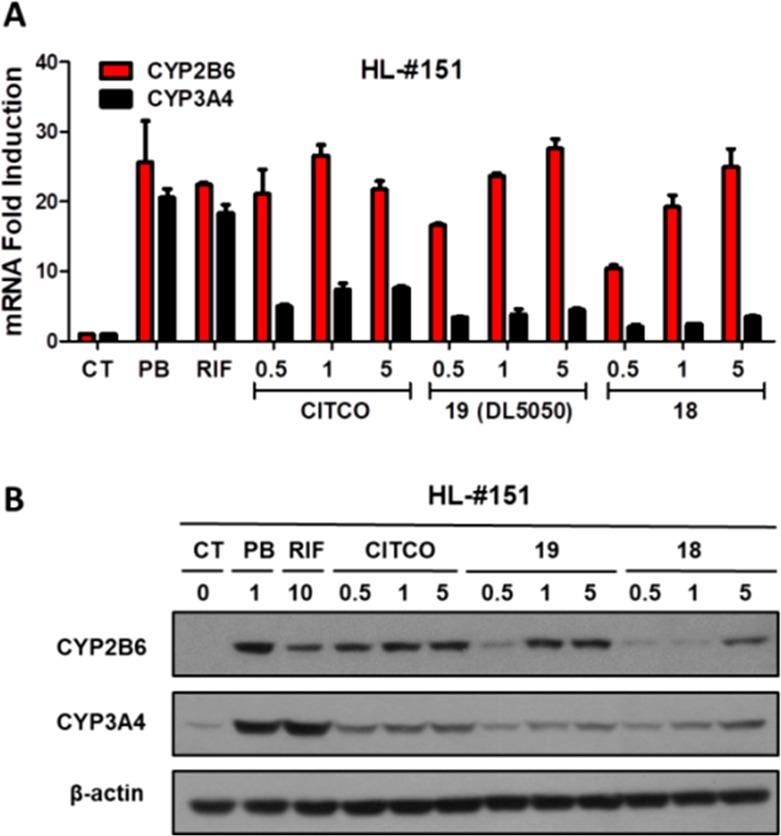
hCAR regulated induction of CYP2B6 and CYP3A4 in HPHs. (A) CYP2B6 and CYP3A4 mRNA levels were measured in HPHs prepared from human liver donors (HL-#151). The HPHs were treated with CITCO (0.5, 1, and 5 μM), PB (1 mM), RIF (10 μM), DL5050 (0.5, 1, and 5 μM), 18 (0.5, 1, and 5 μM), or vehicle control. (B) Representative immunoblots of CYP2B6 and CYP3A4 proteins in HPHs from liver donors #151. RT-PCR data obtained from three independent experiments were expressed as mean ± SD normalized against vehicle control.
The synthetic strategies for compounds 1–29 were outlined in Schemes 1 and 2. Condensation of bromomethyl ketone (30) with thiazol-2 amine provided imidazothiazole 31 (Scheme 1). Vilsmeier formylation of compound 31 gave aldehyde 32 in good yields. Treatment of aldehyde 32 with hydroxylamine 33 in EtOH led to the formation of final products 1–17.
Scheme 1. Synthesis of Compounds 1–17.

Scheme 2. Synthesis of Compounds 18–29.

As shown in Scheme 2, a modified four-step route was used to synthesize compounds 18–29. Condensation of bromomethyl ketone (30) with oxazol-2-amine in the mixture of THF and MeCN generated intermediate 34. TiCl4-mediated cyclization of compound 34 yielded imidazooxazole 35. Vilsmeier reaction of compound 35 using POCl3 in chloroform gave aldehyde 36 in excellent yields. Oximation of aldehyde 35 with substituted hydroxylamine 33 in the presence of AcOH led to the final compounds 18–29.
The new compounds were first evaluated for their potential modulation of hCAR activity using a cell-based quantitative high-throughput screening (qHTS) luciferase reporter assay on a double stable HepG2-CYP2B6-hCAR cell line.31 The potency was expressed as 50% effective concentration (EC50), and the maximum luciferase signal fold increase of a new compound over vehicle control as Emax. In addition, cytotoxicity of new compounds was evaluated in a luciferase-coupled ATP quantitation cell viability assay (Table S1).
In order to optimize R1, a collection of compounds (1–17) with a common (E)-imidazo[2,1-b]thiazole-5-carbaldehyde O-(3,4-dichlorobenzyl)oxime scaffold were tested (Table 1). The Emax values were shown as relative values compared to the basal value (negative control). Under our assay conditions, CITCO (1) indicated an EC50 of 0.62 μM and Emax of 2.8, which led to an Emax/EC50 value of 4.5. Removing the Cl atom, the analogue 2 showed a slightly increased potency. However, the Emax of compound 2 was also decreased. While substitution of the Cl atom with an F led to a new compound (3) with similar EC50 and Emax values, a larger Br group (4) at this position brought increased agonistic activity (Emax/EC50 = 5.0). Similarly improved activities were also observed when substituting the Cl with an Me group (5). Addition of a more polar CF3 group (6) yielded a weaker agonist. These results suggested that there was no demand for a hydrogen bond receptor at this position, instead, a small hydrophobic pocket might be available for new compound development. OMe (7) and i-Pr (8) groups at this position led to new compounds with slightly improved agonist activity. Large substituents, such as t-Bu (9), CN (10), Cy (11), and Ph (12), were also explored. Compounds 9 and 10 demonstrated weak agonistic activity for hCAR (Emax/EC50 < 1.0), and compounds 11 and 12 showed essentially no activity toward hCAR. Our results demonstrated that the hCAR agonist activity decreases while the size of substituents increase. Compound 13, with a bulky substitution on the 3,4-position also indicated no activity for hCAR, which is in agreement with our previous discovery. Shifting the Cl atom to the 3-position from the 4-position yielded compound 14 with a 2.6-fold decreased EC50 and slightly lower Emax value, implying that 3-substitution can also be tolerated by the agonist. Replacement of the 4-Cl-phenyl group with naphthalen-2-yl group led to compound 15 with an improved potency although its Emax was also decreased. The Emax/EC50 value of compound 15 was 1.4-fold better than that of CITCO. The regioisomer naphthalen-1-yl 16, however, indicated significantly lower EC50 and Emax values. Replacement of the aromatic group with an ethyl group led to compound 17 having a weak potency, highlighting the importance of an aromatic ring at this position. The cytotoxicity of new compounds was evaluated (Table S1). Similar to CITCO, our new compounds started to show toxicity at concentrations above 10 μM.
Table 1. EC50 and Emax of Compounds 1–17a.

EC50 and Emax values were calculated by nonlinear regression. Data are presented as mean ± SEM of at least three independent experiments in quadruplicate. ND (if the maximum tested concentration produced no effect).
Next, in an attempt to increase the Emax value of the compounds, we synthesized compounds 18–29 (Table 2). Replacing the imidazothiazole core with imidazoxazole, in CITCO and 15, yielded two new compounds, 18 and 19, with similar EC50 values but significantly larger Emax values (Emax/EC50 = 10 for both 18 and 19). These results indicated that the imidazoxazole core is preferred over the imidazothiazole for generating larger Emax values. Using compound 19 as a template, we studied the substitution effects on R2. When the 3-Cl atom was moved to the 2-position, the resulting 2,4-diCl-Bn analogue 20 indicated a 4.1-fold decreased EC50 value. After removal of one of the two Cl atoms from 20, the resulting compounds, 21 and 22, indicated 3.0- and 2.7-fold decreased EC50 values, respectively. When both Cl atoms of compound 19 were removed, the resulting compound 23 was 2.3-fold weaker; however, this compound also indicated a large Emax value of 4.4. Replacement of the ethylene linker with a structurally rigid five-membered ring generated a new compound (24) with diminished values for both EC50 and Emax. This result indicated the necessity of a flexible linker. The analogue with a pentafluorophenyl group (25) was less potent and efficacious in comparison to the unsubstituted phenyl analogue 23. In addition, different substitutions at the para-position of the Bn group, including CF3 (26), OMe (27), t-Bu (28), and PhO (29) were introduced to probe the depth of the binding pocket for R2. While compound 26 showed a comparable EC50 and Emax values to those of CITCO, compounds 27 and 28 indicated weak agonistic activities; in particular, the phenoxy analogue 29 demonstrated no agonistic activity toward hCAR. These results highlighted that large substitutions on the phenyl ring could not be tolerated in the pocket to which R2was bound. It is worth noting that the imidazoxazole-based compounds displayed less cytotoxicity compared to the ones listed in Table 1. In particular, compounds 18, 19, 23, and 26 showed no obvious cytotoxicity at the maximum tested concentration (92 μM) (Table S1).
Table 2. EC50 and Emax of Compounds 18–29a.
![]()

EC50 and Emax values were calculated by nonlinear regression. Data are presented as mean ± SEM of at least three independent experiments in quadruplicate. ND (if the maximum tested concentration produced no effect).
Based on the predicted hCAR activity, we selected seven new compounds including four potent hCAR agonists (18, 19 [DL5050], 23, and 26) and three weak hCAR agonists (12, 13, and 29) and tested their capacity in hPXR activation using a luciferase reporter assay (Figure 3B). The three weak hCAR agonists (12, 13, and 29) also indicated minimum agonistic activity for hPXR (Figure 3B). These results showed that CITCO analogs with large hydrophobic substitutions on either aromatic tail could abolish the potency in both hCAR and hPXR activation. These results also provided valuable information to define the size limitation of the LBD for both hCAR and hPXR based on the scaffold of CITCO. Among the four potent hCAR agonists, 23 exhibited potent hPXR activation at high concentrations with an Emax of 7.3 and EC50 of 7.3 μM (Figure 3B). Notably, compounds 18, 19, and 26 indicated negligible agonistic activity of hPXR and thereby represent selective agonists for hCAR over hPXR.
Figure 3.

Selectivity of eight compounds 12, 13, 18, 19 (DL5050), 23, 26, 29, and CITCO (1) for hCAR over hPXR. (A) Dose-dependent activation of hCAR by CITCO, 12, 13, 18, 19, 23, 26, and 29 in high throughput hCAR agonist luciferase gene reporter assays. (B) Dose-dependent activation of hPXR by CITCO, 12, 13, 18, 19, 23, 26, and 29 in high throughput hPXR agonist luciferase gene reporter assays.
Given that 26 demonstrated an hCAR activity that is markedly lower than that of 18 and 19 (DL5050), we next evaluated the effects of 18 and DL5050 on the induction of CYP2B6 and CYP3A4 genes in cultured human primary hepatocytes (HPH). As shown in Figure 4A, both 18 and DL5050 induced CYP2B6 mRNA expression in a concentration-dependent manner while only moderately altered the expression of CYP3A4. This was significantly different to the hCAR and hPXR dual activator (PB), the selective hPXR agonist (RIF), and even the moderately selective hCAR agonist (CITCO). At the protein level, the naphthyl analog DL5050 also demonstrated a similar concentration-dependent selective induction of CYP2B6 over CYP3A4 (Figure 4B). Conversely, compound 18 only showed marginal induction of CYP2B6 protein.
The binding mode of compound DL5050 in hCAR was also studied (Figure 5). The dichloro-phenyl ring of DL5050, which is the same as in CITCO, rests in the hydrophobic pocket flanked by Phe132, Tyr224, and Tyr 217. The imidazo[2,1-b]oxazole core of DL5050 fit deeper at the junction of helixes H5 and H3 than the imidazothiazole core of CITCO. Finally, the naphthyl ring of DL5050 establishes a π–π stacking interaction with Phe234, which is not present in CITCO, potentially providing a basis for the heightened activity of DL5050.
Figure 5.
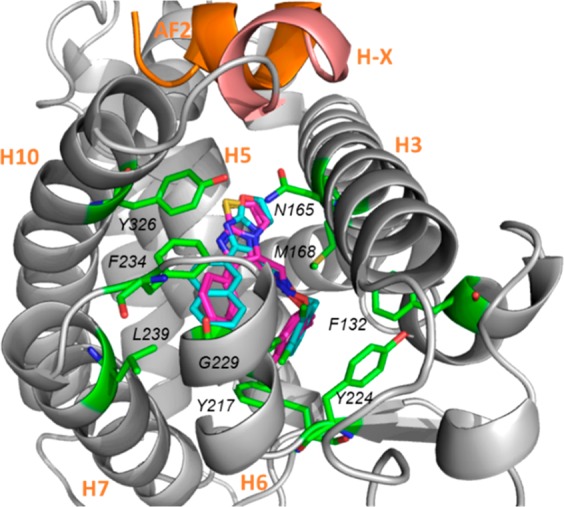
Docking result of compound DL5050 (orange) in the ligand-binding domain of hCAR in comparison with CITCO (magenta) using AutoDock Vina.38 Key hCAR residues that are expected to mediate interactions with ligands are highlighted in green. Helix X is showed in light pink. The activation function 2 helix (AF2) is shown in orange.
In summary, structure optimization of CITCO has yielded new hCAR agonists 18 and 19 (DL5050). While maintaining a similar hCAR activation potency as that of CITCO, these new compounds indicated a high specificity for hCAR over its closely related nuclear receptor, hPXR. One of the two new compounds, DL5050, exhibited potent induction of CYP2B6 gene at both the mRNA and protein levels while only resulted in minimal induction of CYP3A4. DL5050 displayed a strong induction preference in CYP2B6 over CYP3A4, which exceeds that of the known hCAR activator CITCO. The unique profile of compound DL5050 indicates this compound can be a tool for elucidating biological functions of hCAR with minimal hPXR interference. Additionally, the selectiveness of DL5050 for hCAR suggests it may be used as a therapeutic agent for various human diseases.
Acknowledgments
This work was supported in part by the Intramural Research Program of the National Center for Advancing Translational Sciences (NCATS).
Glossary
ABBREVIATIONS
- CAR
constitutive androstane receptor
- NR
nuclear receptors
- PXR
pregnane X receptor
- VDR
vitamin D receptor;
- CCRP
cytoplasmic CAR retention protein
- RXR
retinoid X receptor
- ER
estrogen receptor
- GR
glucocorticoid receptor
- DBD
DNA-binding domain
- LBD
ligand binding domain
- qHTS
quantitative high-throughput screening
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsmedchemlett.9b00079.
General experimental procedures, supplementary data, and copies of 1H and 13C NMR spectra and HPLC of compounds (PDF)
Author Contributions
§ D.L. and L.L. contribute equally to this work. All authors contributed to the writing of this manuscript. All authors have given approval to the final version of the manuscript.
The research was funded by the Maryland Innovation Initiative (MII) for the research grant to H.W. and F.X. This work was also supported in part by National Institutes of Health Grant GM107058 and GM121550 to H.W.
The authors declare no competing financial interest.
Supplementary Material
References
- Wei P.; Zhang J.; Egan-Hafley M.; Liang S.; Moore D. D. The nuclear receptor CAR mediates specific xenobiotic induction of drug metabolism. Nature 2000, 407, 920–923. 10.1038/35038112. [DOI] [PubMed] [Google Scholar]
- Timsit Y. E.; Negishi M. CAR and PXR: the xenobiotic-sensing receptors. Steroids 2007, 72, 231–246. 10.1016/j.steroids.2006.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie W.; Radominska-Pandya A.; Shi Y.; Simon C. M.; Nelson M. C.; Ong E. S.; Waxman D. J.; Evans R. M. An essential role for nuclear receptors SXR/PXR in detoxification of cholestatic bile acids. Proc. Natl. Acad. Sci. U. S. A. 2001, 98, 3375–3380. 10.1073/pnas.051014398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tzameli I.; Moore D. D. Role reversal: new insights from new ligands for the xenobiotic receptor CAR. Trends Endocrinol. Metab. 2001, 12, 7–10. 10.1016/S1043-2760(00)00332-5. [DOI] [PubMed] [Google Scholar]
- Maglich J. M.; Parks D. J.; Moore L. B.; Collins J. L.; Goodwin B.; Billin A. N.; Stoltz C. A.; Kliewer S. A.; Lambert M. H.; Willson T. M.; Moore J. T. Identification of a novel human constitutive androstane receptor (CAR) agonist and its use in the identification of CAR target genes. J. Biol. Chem. 2003, 278, 17277–17283. 10.1074/jbc.M300138200. [DOI] [PubMed] [Google Scholar]
- Stanley L. A.; Horsburgh B. C.; Ross J.; Scheer N.; Wolf C. R. Drug Metab. Rev. 2006, 38, 515–597. 10.1080/03602530600786232. [DOI] [PubMed] [Google Scholar]
- Zhang J.; Huang W.; Chua S. S.; Wei P.; Moore D. D. Modulation of acetaminophen-induced hepatotoxicity by the xenobiotic receptor CAR. Science 2002, 298, 422–424. 10.1126/science.1073502. [DOI] [PubMed] [Google Scholar]
- Huang W.; Zhang J.; Chua S. S.; Qatanani M.; Han Y.; Granata R.; Moore D. D. Induction of bilirubin clearance by the constitutive androstane receptor (CAR). Proc. Natl. Acad. Sci. U. S. A. 2003, 100, 4156–4161. 10.1073/pnas.0630614100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maglich J. M.; Watson J.; McMillen P. J.; Goodwin B.; Willson T. M.; Moore J. T. The nuclear receptor CAR is a regulator of thyroid hormone metabolism during caloric restriction. J. Biol. Chem. 2004, 279, 19832–19838. 10.1074/jbc.M313601200. [DOI] [PubMed] [Google Scholar]
- Gao J.; He J.; Zhai Y.; Wada T.; Xie W. The constitutive androstane receptor is an anti-obesity nuclear receptor that improves insulin sensitivity. J. Biol. Chem. 2009, 284, 25984–25992. 10.1074/jbc.M109.016808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong B.; Sara P. K.; Huang W.; Chen W.; Abu-Elheiga L. A.; Wakil S. J.; Stevens R. D.; Ilkayeva O.; Newgard C. B.; Chan L.; Moore D. D. Activation of nuclear receptor CAR ameliorates diabetes and fatty liver disease. Proc. Natl. Acad. Sci. U. S. A. 2009, 106, 18831–18836. 10.1073/pnas.0909731106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Z.; Roy P.; Waxman D. J. Role of human liver microsomal CYP3A4 and CYP2B6 in catalyzing N-dechloroethylation of cyclophosphamide and ifosfamide. Biochem. Pharmacol. 2000, 59, 961–972. 10.1016/S0006-2952(99)00410-4. [DOI] [PubMed] [Google Scholar]
- Code E. L.; Crespi C. L.; Penman B. W.; Gonzalez F. J.; Chang T. K.; Waxman D. J. Human cytochrome P4502B6: interindividual hepatic expression, substrate specificity, and role in procarcinogen activation. Drug Metab. Dispos. 1997, 25, 985–993. [PubMed] [Google Scholar]
- Wang D.; Li L.; Yang H.; Ferguson S. S.; Baer M. R.; Gartenhaus R. B.; Wang H. The constitutive androstane receptor is a novel therapeutic target facilitating cyclophosphamide-based treatment of hematopoietic malignancies. Blood 2013, 121, 329–338. 10.1182/blood-2012-06-436691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hedrich W. D.; Xiao J.; Heyward S.; Zhang Y.; Zhang J.; Baer M. R.; Hassan H. E.; Wang H. Activation of the constitutive androstane receptor increases the therapeutic index of CHOP in lymphoma treatment. Mol. Cancer Ther. 2016, 15, 392–401. 10.1158/1535-7163.MCT-15-0667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi K.; Sueyoshi T.; Inoue K.; Moore R.; Negishi M. Cytoplasmic accumulation of the nuclear receptor CAR by a tetratricopeptide repeat protein in HepG2 cells. Mol. Pharmacol. 2003, 64, 1069–1075. 10.1124/mol.64.5.1069. [DOI] [PubMed] [Google Scholar]
- Honkakoski P.; Negishi M. Protein serine/threonine phosphatase inhibitors suppress phenobarbital-induced Cyp2b10 gene transcription in mouse primary hepatocytes. Biochem. J. 1998, 330, 889–895. 10.1042/bj3300889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swales K.; Kakizaki S.; Yamamoto Y.; Inoue K.; Kobayashi K.; Negishi M. Novel CAR-mediated mechanism for synergistic activation of two distinct elements within the human cytochrome P450 2B6 gene in HepG2 cells. J. Biol. Chem. 2005, 280, 3458–3466. 10.1074/jbc.M411318200. [DOI] [PubMed] [Google Scholar]
- Mackowiak B.; Wang H. 2016. Mechanisms of xenobiotic receptor activation: Direct vs. indirect. Biochim. Biophys. Acta, Gene Regul. Mech. 2016, 1859, 1130–1140. 10.1016/j.bbagrm.2016.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu R. X.; Lambert M. H.; Wisely B. B.; Warren E. N.; Weinert E. E.; Waitt G. M.; Williams J. D.; Collins J. L.; Moore L. B.; Willson T. M.; Moore J. T. A structural basis for constitutive activity in the human CAR/RXRalpha heterodimer. Mol. Cell 2004, 16, 919–928. 10.1016/j.molcel.2004.11.042. [DOI] [PubMed] [Google Scholar]
- Xie W.; Barwick J. L.; Downes M.; Blumberg B.; Simon C. M.; Nelson M. C.; Neuschwander-Tetri B. A.; Brunt E. M.; Guzelian P. S.; Evans R. M. Humanized xenobiotic response in mice expressing nuclear receptor SXR. Nature 2000, 406, 435–439. 10.1038/35019116. [DOI] [PubMed] [Google Scholar]
- Moore L. B.; Parks D. J.; Jones S. A.; Bledsoe R. K.; Consler T. G.; Stimmel J. B.; Goodwin B.; Liddle C.; Blanchard S. G.; Willson T. M.; Collins J. L.; Kliewer S. A. Orphan nuclear receptors constitutive androstane receptor and pregnane X receptor share xenobiotic and steroid ligands. J. Biol. Chem. 2000, 275, 15122–15127. 10.1074/jbc.M001215200. [DOI] [PubMed] [Google Scholar]
- Moore L. B.; Maglich J. M.; McKee D. D.; Wisely B.; Willson T. M.; Kliewer S. A.; Lambert M. H.; Moore J. T. Pregnane X receptor (PXR), constitutive androstane receptor (CAR), and benzoate X receptor (BXR) define three pharmacologically distinct classes of nuclear receptors. Mol. Endocrinol. 2002, 16, 977–986. 10.1210/mend.16.5.0828. [DOI] [PubMed] [Google Scholar]
- Willson T. M.; Kliewer S. A. PXR, CAR and drug metabolism. Nat. Rev. Drug Discovery 2002, 1, 259–266. 10.1038/nrd753. [DOI] [PubMed] [Google Scholar]
- Cherian M. T.; Chai S. C.; Chen T. Small-molecule modulators of the constitutive androstane receptor. Expert Opin. Drug Metab. Toxicol. 2015, 11, 1099–1114. 10.1517/17425255.2015.1043887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faucette S. R.; Zhang T. C.; Moore R.; Sueyoshi T.; Omiecinski C. J.; LeCluyse E. L.; Negishi M.; Wang H. Relative activation of human pregnane X receptor versus constitutive androstane receptor defines distinct classes of CYP2B6 and CYP3A4 inducers. J. Pharmacol. Exp. Ther. 2006, 320, 72–80. 10.1124/jpet.106.112136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maglich J. M.; Stoltz C. M.; Goodwin B.; Hawkins-Brown D.; Moore J. T.; Kliewer S. A. Nuclear pregnane X receptor and constitutive androstane receptor regulate overlapping but distinct sets of genes involved in xenobiotic detoxification. Mol. Pharmacol. 2002, 62, 638–646. 10.1124/mol.62.3.638. [DOI] [PubMed] [Google Scholar]
- Kliewer S. A.; Goodwin B.; Willson T. M. The nuclear pregnane X receptor: a key regulator of xenobiotic metabolism. Endocr. Rev. 2002, 23, 687–702. 10.1210/er.2001-0038. [DOI] [PubMed] [Google Scholar]
- Molnar F.; Kublbeck J.; Jyrkkarinne J.; Prantner V.; Honkakoski P. An update on the constitutive androstane receptor (CAR). Drug Metabol. Drug Interact. 2013, 28, 79–93. 10.1515/dmdi-2013-0009. [DOI] [PubMed] [Google Scholar]
- Lynch C.; Pan Y.; Li L.; Ferguson S. S.; Xia M.; Swaan P. W.; Wang H. Identification of novel activators of constitutive androstane receptor from FDA-approved drugs by integrated computational and biological approaches. Pharm. Res. 2013, 30, 489–501. 10.1007/s11095-012-0895-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch C.; Zhao J.; Huang R.; Xiao J.; Li L.; Heyward S.; Xia M.; Wang H. Quantitative high-throughput identification of drugs as modulators of human constitutive androstane receptor. Sci. Rep. 2015, 5, 10405. 10.1038/srep10405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch C.; Mackowiak B.; Huang R.; Li L.; Heyward S.; Sakamuru S.; Wang H.; Xia M. Identification of modulators that activate the constitutive androstane receptor from the Tox21 10K compound library. Toxicol. Sci. 2019, 167, 282–292. 10.1093/toxsci/kfy242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smutny T.; Nova A.; Drechslerova M.; Carazo A.; Hyrsova L.; Hruskova Z. R.; Kunes J.; Pour M.; Spulak M.; Pavek P. 2-(3-Methoxyphenyl)quinazoline derivatives: a new class of direct constitutive androstane receptor (CAR) agonists. J. Med. Chem. 2016, 59, 4601–4610. 10.1021/acs.jmedchem.5b01891. [DOI] [PubMed] [Google Scholar]
- Tschuor C.; Kachaylo E.; Limani P.; Raptis D. A.; Linecker M.; Tian Y.; Herrmann U.; Grabliauskaite K.; Weber A.; Columbano A.; Graf R.; Humar B.; Clavien P. A. Constitutive androstane receptor (Car)-driven regeneration protects liver from failure following tissue loss. J. Hepatol. 2016, 65, 66–74. 10.1016/j.jhep.2016.02.040. [DOI] [PubMed] [Google Scholar]
- Shizu R.; Min J.; Sobhany M.; Pedersen L. C.; Mutoh S.; Negishi M. Interaction of the phosphorylated DNA-binding domain in nuclear receptor CAR with its ligand-binding domain regulates CAR activation. J. Biol. Chem. 2018, 293, 333–344. 10.1074/jbc.M117.806604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shizu R.; Osabe M.; Perera L.; Moore R.; Sueyoshi T.; Negishi M. Phosphorylated nuclear receptor CAR forms a homodimer to repress its constitutive activity for ligand activation. Mol. Cell. Biol. 2017, 37, e00649-16 10.1128/MCB.00649-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kandel B. A.; Thomas M.; Winter S.; Damm G.; Seehofer D.; Burk O.; Schwab M.; Zanger U. M. Genomewide comparison of the inducible transcriptomes of nuclear receptors CAR, PXR and PPARα in primary human hepatocytes. Biochim. Biophys. Acta, Gene Regul. Mech. 2016, 1859, 1218–1227. 10.1016/j.bbagrm.2016.03.007. [DOI] [PubMed] [Google Scholar]
- Trott O.; Olson A. J. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization and multithreading. J. Comput. Chem. 2009, 31, 455–461. 10.1002/jcc.21334. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.