

Abstract
We examined activin receptor type IIA (ActRIIA) activation in chronic kidney disease (CKD) by signal analysis and inhibition in mice with Alport syndrome, using the ActRIIA ligand trap RAP-011 initiated in 75 day old Alport mice. At 200 days of age there was severe CKD and associated Mineral and Bone Disorder (CKD-MBD), consisting of osteodystrophy, vascular calcification, cardiac hypertrophy, hyperphosphatemia, hyperparathyroidism, elevated FGF23 and reduced klotho. The CKD-induced bone resorption and osteoblast dysfunction was reversed, and bone formation was increased by RAP-011. ActRIIA inhibition prevented formation of calcium apatite deposits in aortic adventitia and tunica media and significantly decreased mean aortic calcium concentration from 0.59 in untreated to 0.36 mg/g in treated Alport mice. Aortic ActRIIA stimulation in untreated mice increased p-Smad2 levels and transcription of sm22α and αSMA. ActRIIA inhibition reversed aortic expression of the osteoblast transition markers Runx2 and osterix. Heart weight was significantly increased by 26% in untreated mice but remained normal during RAP-011 treatment. In 150 day old mice, the GFR was significantly reduced by 55%, but only by 30% in the RAP-011 treated group. In 200 day old mice, the mean BUN was 100 mg/dl in untreated mice compared to 60 mg/dl in the treated group. In the kidneys of 200 day old mice, ActRIIA and p-Smad2 were induced and MCP-1, fibronectin and interstitial fibrosis were stimulated; all were attenuated by RAP-011 treatment. Hence, activation of ActRIIA signaling during early CKD contributes to the CKD-MBD components of osteodystrophy and cardiovascular disease, and to renal fibrosis. Thus, inhibition of ActRIIA signaling is efficacious in improving and delaying CKD-MBD in this model of Alports syndrome.
Keywords: Chronic kidney disease, vascular calcification, bone, mineral metabolism, fibrosis, cardiovascular disease
INTRODUCTION:
The chronic kidney disease – mineral bone disorder (CKD-MBD) is a major contributor to the excess mortality associated with the CKD pandemic. (1–5) The CKD-MBD begins early in the course of kidney disease,(6–8) and at its inception consists of skeletal,(7, 9) vascular (8) and cardiac diseases,(10) elevations of FGF23 (6) and reductions in klotho. (11) (12) The early CKD-MBD precedes the development of secondary hyperparathyroidism, calcitriol deficiency, and hyperphosphatemia, which are CKD-MBD components associated with more advanced CKD. Recent kidney disease progression clinical trials demonstrate that cardiovascular morbidity may be worsened at the same time kidney function is improving, separating cardiovascular disease progression from efficacy in the progression of CKD, (13, 14) making coordinate therapy of the CKD-MBD and intrinsic CKD a requisite. Other recent studies targeting the CKD-MBD demonstrate that CKD-stimulated atherosclerotic vascular calcification (VC) was inhibited by an Activin receptor type IIA (ActRIIA) ligand trap. (15) In the ldlr−/− high fat fed atherosclerotic diabetic mice, CKD stimulated VC through dedifferentiation of vascular smooth muscle and osteoblastic transition, which was reversed by the ligand trap. (15) A putative ligand for the trap, Activin A, was increased in the kidney and the circulation, (15) suggesting that ActRIIA signaling might be a critical systemic mechanism of both renal disease progression and CKD-MBD production. To examine this hypothesis in a murine homologue of human kidney disease, we have used a model of Alport Syndrome.
Activins are widely expressed factors involved in cell differentiation, proliferation, and inflammation.(16, 17) They belong to the TGFβ superfamily and activate signal transduction by forming and ligating receptor heteromultimers. Type II receptors (activin receptor type IIA and IIB (ActRIIA and B)) are the ligand binding components, which are active plasma membrane serine/threonine kinases that upon ligand binding dimerize, attract and phosphorylate type I receptors (the ALK kinases of which there are seven). ActRIIA and B are receptors for a diverse group of TGFβ superfamily member ligands. The activated receptor complexes attract and phosphorylate regulatory SMADs (portmanteau of C. Elegans SMA, and Drosphilia Mothers against decaplentaplegic), in the case of activins, SMADs 2 and 3 (supplemental Fig. S1) The activated regulatory SMADs associate with co-SMAD 4 and translocate to the nucleus with other transcription factors, which in the case of osteoclasts critically includes cfos (supplemental Fig. S1).
Alport syndrome is most often caused by mutations in the COL4A5 gene on the X chromosome.(18) The syndrome is characterized by progressive kidney disease caused by an abnormal glomerular basement membrane unable to withstand the mechanical forces of post-natal glomerular perfusion. The disease begins in childhood as hematuria (stage 0), progresses through microalbuminuria (stage 1), proteinuria (stage 2), and progressive renal impairment and tubulointerstitial nephritis (stage 3) to end stage kidney disease (stage 4) during the second to fourth decades depending on the mutation type.(18–20) Autosomal recessive and dominant forms of Alport syndrome are less common and are due to mutations in the COL4A3 or COL4A4 genes.(21–23)
The murine X-linked Alport syndrome (XLAS) homologue, Col4a5y/- on the C57Bl/6J background, is relatively faithful to the human Alport kidney disease.(24) Hematuria (stage 0) begins early in life and proteinuria is established by day 100 (stage 2), with progression to CKD with reductions of GFR to 10–20% of normal by day 200 of life (stage 4).(24)(Fang Y and Hruska KA, unpublished observations). Significant mortality begins thereafter. Treatment of murine and human Alport syndrome with angiotensin converting enzyme inhibitors has delayed progression of CKD, but only if started early in the course of disease.(20, 25–27)
The CKD-MBD has been studied in murine Alport syndrome using Col4a3 deficiency in the background strain 129X1/Sv. (28) The progression to the late stages of the CKD-MBD syndrome with hyperphosphatemia, calcitriol deficiency and hyperparathyroidism was rapid not allowing for full development of CKD-MBD with its attendant vascular calcification/cardiac hypertrophy. The shortened lifespan was 8–14 weeks due to kidney failure, and an observed early elevation of FGF23 was not attributable to increased osteocyte secretion, which only occurred late in the disease course.(28) Here we examine renal osteodystrophy, vascular calcification, heart weights and tubulointerstitial nephritis in an Alport model with an extended lifespan and demonstrate that ActRIIA signaling was stimulated in the skeleton, vasculature and the kidney, and that a ligand trap for the receptor (RAP-011) started early in the course of disease was efficacious in preventing osteodystrophy, vascular calcification, vascular osteoblastic transition, cardiomegaly and delaying progression of Alport nephritis.
RESULTS:
These studies were performed in a cohort of Alport mice and their wild type littermates as described in Methods that were maintained on a standard mouse chow until euthanasia at 200 days of life (do). Alport mice were treated with vehicle or RAP-011, 10mg/kg, subcutaneous twice weekly beginning at day 75 of life and continued until euthanasia.
The Chronic Kidney Disease – Mineral Bone Disorder (CKD-MBD) in Alport mice
Osteodystrophy
A critical component of the CKD-MBD is renal osteodystrophy. 200 day old Alport mice had an osteodystrophy produced by high resorption, but without change in bone formation compared to WT. Alport distal femurs had significantly increased eroded trabecular perimeters and increased osteoclast numbers along with osteoclast perimeters (Figure 1A–C). Osteoblast numbers and perimeter were also increased. (Figure 1D–E) The latter did not lead to increased mineral apposition or bone formation rate (Figure 1F–G) due to a decrease in the bone formation rate per osteoblast (Figure 1H), produced by a delay in mineralization. As a result, there was an increase in osteoid area and osteoid perimeter (Figure 1J–K).
Figure one.
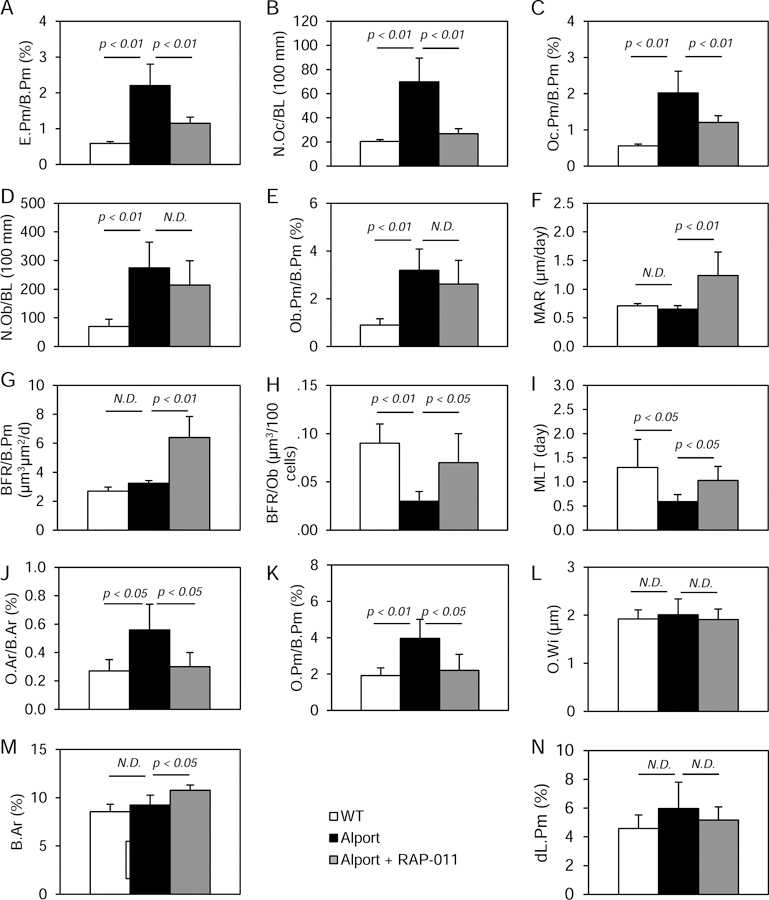
Bone histomorphometry in the groups of mice: Wild-type (white bars), Alport vehicle-treated (black bars), Alport RAP-011 treated (gray bars). A, Eroded perimeter/bone perimeter (E.Pm/B.Pm). B, Osteoclast number (N. Oc/BL(100 mm)). C, Osteoclast perimeter/bone perimeter (Oc.Pm/B.Pm). D, Osteoblast number (N.Ob/BL (110mm)). E, Osteoblast perimeter/bone perimeter (Ob.Pm/B.Pm). F, Mineral apposition rate (MAR). G, Bone formation rate/bone perimeter (BFR/B.Pm). H, Bone formation rate/osteoblast (BFR/Ob). I, Mineralization lag time (MLT). J, Osteoid area/bone area (O.Ar/B.Ar). K, Osteoid perimeter/bone perimeter (O.Pm/B.Pm). L, Osteoid width (OW). M, Bone Area (B.Ar.). N, Double label perimeter (dL.Pm). Alport mice had increased bone resorption and osteoclasts prevented by RAP-011 treatment. Alport mice had increased osteoblast number and surface but MAR and BFR/B.Pm were not increased. RAP-011 did not further increase osteoblast number but increased MAR and BFR/B.Pm. Alport mice had increased osteoid perimeter which was corrected by RAP-011 treatment. N.D., not different. n=13 for WT, 8 for Alport and 12 for RAP-01, see Methods for histomorphometry techniques. Data are mean ± SEM.
RAP-011 treatment, which suggests the role of ActRIIA signaling in the osteodystrophy of the Alport mice, produced a correction of eroded trabecular perimeters (Figure 1A) and osteoclast numbers along with osteoclast perimeters (Figure 1B–C). Osteoblast numbers and perimeter were not decreased by RAP-011 (Figure 1D–E), and RAP-011 treatment increased mineral apposition rate, bone formation rate and corrected bone formation rate per osteoblast (Figure 1F–H). As a result, osteoid area and osteoid perimeter were corrected (Figure 1J–K), and structurally, bone area was increased (Figure 1M).
Vascular Calcification
One of the critical components of the CKD-MBD is vascular calcification. Our Alport model is in the C57Bl/6J strain, which is a resistance strain for vascular calcification.(29) However, as shown in (Figure 2A–B), by 200 do Alport mice have calcium deposits in the tunica media adjacent to the adventitia of the vessels (Figure 2B). Tissue calcium levels were increased in the aorta compared to wild-type littermates (Figure 2C). In the Alport aortas, RAP-011 treatment blocked the increase in aortic calcium levels (Figure 2C).
Figure two.
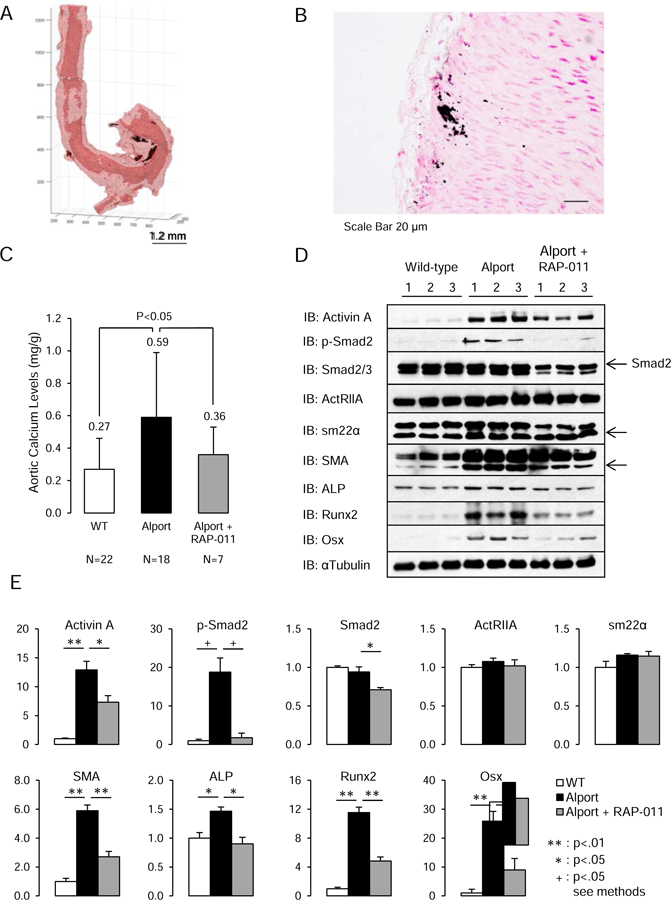
Vascular calcification in Alport mice and aortic ActRIIA signaling. A, Deposits of calcium phosphate (black patches) were detected by micro-CT in the aortic adventitia and media adjacent to the adventitia of Alport mice. B, Deposits of calcium phosphate were detected by von Kossa staining in the aortic adventitia and media adjacent to the adventitia of Alport mice (scale bar 20µm). C, Aortic calcium levels in the groups of mice. Alport mice had significantly elevated aortic Ca levels, which were reduced by RAP-011 treatment. D, Analysis of ActRIIA signaling in aortic homogenates. Westerns of aortic homogenates from 3 WT, 3 Alport vehicle-treated, and 3 Alport RAP-011 treated mice. E, quantitation of the Westerns in D. Activin A and p-Smad2 levels were increased in homogenates from Alport vehicle-treated mice, and they were reduced in homogenates from Alport RAP-011 treated mice. SM22α (Transgelin) and SMA (smooth muscle actin) were increased in the homogenates from the Alport vehicle-treated mice aortas. SMA was decreased in the homogenates from the Alport RAP-011 treated mice aortas compared to vehicle-treated Alport mice. Runx2 and osterix (Osx) were induced in the aortic homogenates from the Alport vehicle-treated mice and reduced by RAP-011 treatment. n = 6 for each group in E.
Serum and Plasma Chemistries
Also, as seen in other murine CKD models different from human CKD, there was an increase in the serum calcium in the Alport mice; this was not affected by RAP-011 treatment (Table 1). The 200 day old Alport mice were hyperphosphatemic, and RAP-011 did not affect the serum phosphate (Pi) levels (Table 1). PTH levels were increased in Alport mice to 805±493 pg/ml from 181±39 in wild-type littermates (Table 1 and Supplementary Figure S2A). ActRIIA inhibition with RAP-011 had no effect on PTH levels. FGF23 levels were increased in Alport mice to 2624±2540 pg/ml from 213±88 in wild-type littermates (Table 1 and Supplementary Figure S2B). ActRIIA inhibition with RAP-011 had no effect on FGF23 levels. These results were similar to CKD produced by renal ablation in C56BL/6J mice (Table 1).
Table 1.
Serum biochemical parameters in the various groups of animals
| Group 1 | Group 2 | Group 3 | |
|---|---|---|---|
| Mouse | wildtype | Alport | Alport |
| Sex | Male | Male | Male |
| Days postnatal | 200 | 200 | 200 |
| Treatment | None | Vehicle | RAP-011 |
| N | 15 | 21 | 10 |
| Ca (mg/dl) | 8.5 ± .5 | 9.62 ± 1.0 * | 9.8 ± 0.8 * |
| Phosphorus (mg/dl) | 9.2 ± 0.9 | 10.5 ± 2.9 | 10.8 ± 2.0 |
| Parathyroid Hormone (pg/ml) | 181 ± 39 | 805 ± 493 * | 700 ± 750 |
| FGF 23 (pg/ml) | 213 ± 88 | 2624 ± 2540 * | 1639 ± 1050 |
| Mouse C57Bl6J # | Sham operated | Ablative CKD | Ablative CKD |
| Sex | Male | Male | Male |
| Weeks postnatal | 28 | 28 | 28 |
| Treatment | None | Vehicle | RAP-011 |
| N | 6 | 6 | 6 |
| Ca (mg/dl) | 7.9 | 9.0 | 7.5 |
| Phosphorus (mg/dl) | 11.1 | 10.4 | 10.9 |
| Parathyroid Hormone (pg/ml) | 181 | 382 | 392 |
| FGF23 (pg/ml) | 221 | 561 | 1745 |
P < 0.05, groups 2 and 3 compared to group 1
ActRIIA signaling in the aorta
To examine the mechanism of RAP-011’s effect on aortic calcium levels, we analyzed ActRIIA signaling in the aortas of the Alport mice. As shown in Figure 2D, ActRIIA levels were not decreased in aortas from 200 day old Alport mice with severe CKD as they were by induction of CKD in the diabetic ldlr−/− high fat fed mouse,(15), and RAP-011 treatment had no effect on ActRIIA levels. The primary ActRIIA ligand, activin A (inhibin βA homodimer), which was decreased in the aortas of ldlr−/− high fat mice,(15) was induced in the Alport aortas, and decreased by RAP-011 treatment (Figure 2D and E). Alport mice had increased aortic p-Smad2 levels consistent with increased ActRIIA signaling, and they also had increased aortic sm22α and α-smooth muscle actin (SMA) levels (Figure 2D). RAP-011 treatment decreased p-Smad2 levels, but total Smad 2/3 levels were also decreased. RAP-011 treatment did not affect sm22α levels, but it did decrease SMA levels (Figure 2D and E). Despite the different distribution of aortic calcium deposits in the Alport aortas compared to other models of vascular calcification, and the increase not decrease in vascular smooth muscle proteins, CKD (200 do) in Alport mice induced osteoblastic transition in the aortas detected by Runx2 and osterix levels (Figure 2D). Runx2 and osterix levels were reduced by RAP-011 treatment. The effects of CKD in the aortas of Alport mice were similar to the effects of CKD produced by renal ablation in C57BL/6J mice (supplemental Figs. S3).
Cardiac Hypertrophy
A third critical component of the CKD-MBD is its effects on the heart and cardiac hypertrophy. Left ventricular hypertrophy (LVH) is a near-validated surrogate of cardiac outcomes in human CKD, and cardiac hypertrophy develops in our Alport mice by 200 days (Figure 3). RAP-011 treatment prevented development of cardiac hypertrophy (Figure 3).
Figure three.
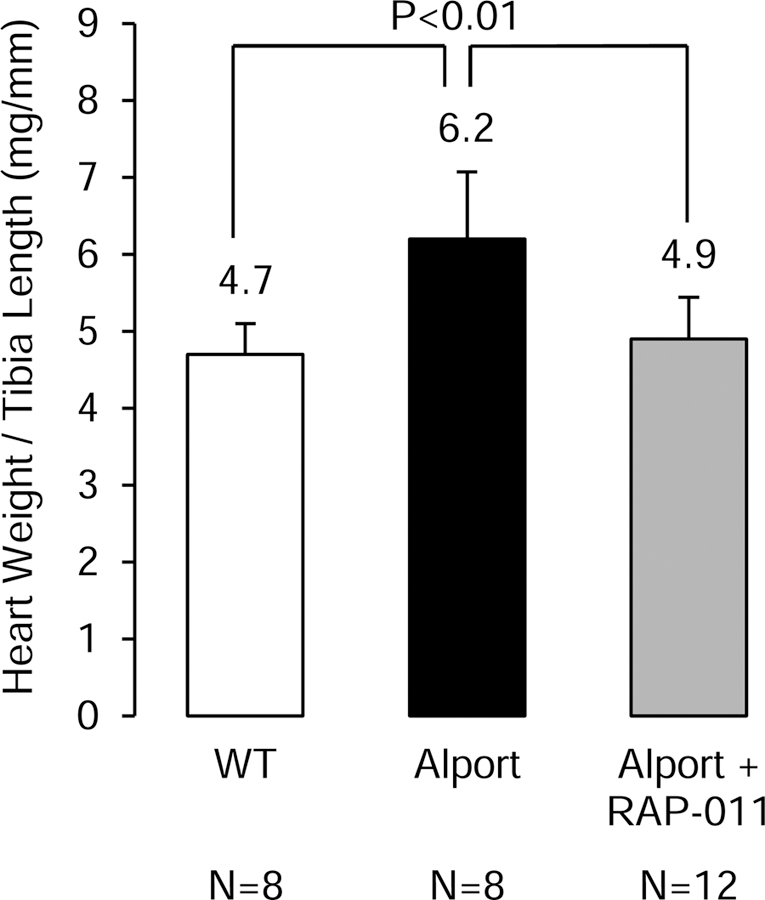
Cardiac weights in the groups of mice. Cardiac hypertrophy developed in the Alport vehicle-treated mice, and this was prevented in the Alport mice treated with RAP-011.
Renal Klotho
A fourth critical component of the CKD-MBD is renal klotho expression, as most of systemic klotho derives from the kidney.(30) Loss of klotho in CKD is associated with vascular calcification, cardiac hypertrophy and osteodystrophy, and its replacement has been shown to be efficacious.(11, 31, 32) Homogenates of Alport kidneys demonstrated major reductions in klotho expression (Figure 4). Treatment of Alport mice with RAP-011 did not significantly increase klotho expression. Thus, increasing klotho was not the mechanism of RAP-011 effects on the skeleton, vasculature or heart.
Figure four.
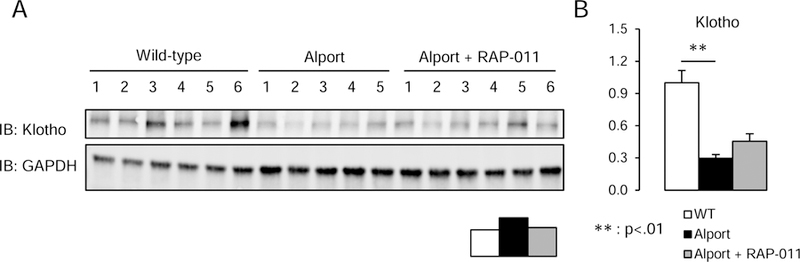
Western analysis of renal klotho. A, Klotho levels in renal homogenates from 6 wild-type mice, 5 Alport vehicle-treated mice, and 6 Alport RAP-011 treated mice. B, quantitation of the klotho levels in A. Data are reported as mean ± SEM.
Alport Nephritis
Renal Function
The BUN of 150 day old (do) Alport mice was 43±22 mg/dl (n=8) compared to 30±2 mg/dl in 150 do wild type (WT) littermates. Inulin clearances in 150 do Alport mice revealed that RAP-011 treatment preserved renal function compared to the Alport vehicle-treated mice whose GFR was significantly depressed (46% of WT) (Figure 5A). By day of life 200, serum BUN of the three groups of mice (Figure 5B), revealed severe azotemia in the 200 do Alport mice, BUN 99±39 mg/dl, p<0.05, consistent with severe CKD and GFR ≤ 10–15% of WT (stage 4 Alport Syndrome). There was a 40% reduction in the BUN as a result of RAP-011 treatment, (Stage 3 Alport syndrome). The urinary albumin-to-creatinine ratio (UACR) at 200 days was threefold elevated in the Alport mice compared to wild type littermates (Figure 5C), and RAP-011 did not significantly decrease the UACR at this time point consistent with no change in proteinuria between stages 3 and 4 in Alport syndrome.
Figure five.
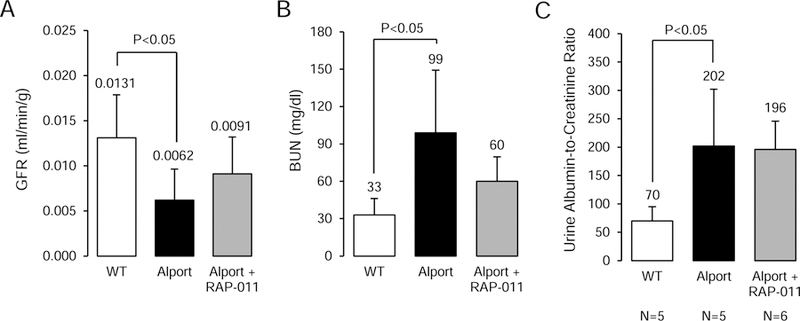
Kidney function in Alport mice. A, Inulin clearances in 150 day old groups of mice. The 150 do Alport vehicle-treated mice had significantly reduced GFR compared to WT littermates. RAP-011 had better preserved GFR than the vehicle-treated mice. B, Serum BUN in 200 day old groups of mice. The BUN of 200 do Alport mice were consistent with severe CKD and GFR ≤ 10–15% of WT. RAP-011 treatment decreased the BUN at 200 do. C, Urine albumin-to-creatinine ratios in the groups of mice. Consistent with delayed progression of the RAP-011 treated Alport mice compared to vehicle-treated Alport, the UACR was not different between the two groups of mice. The “n” for the groups in A-C were as follows: WT = 6, Alport vehicle = 7, and Alport RAP-011 = 7; p<0.05.
Renal Pathology
The kidneys from 200 day old Alport mice revealed a consistent severe interstitial fibrosis (Figure 6B) producing a major increase in interstitial volume from 12 to 59% (Figure 6D). There was a significant reduction in interstitial fibrosis at 200 do in the kidneys from RAP-011 treated mice compared to Alport mice vehicle-treated (Figure 6C and D).
Figure six.
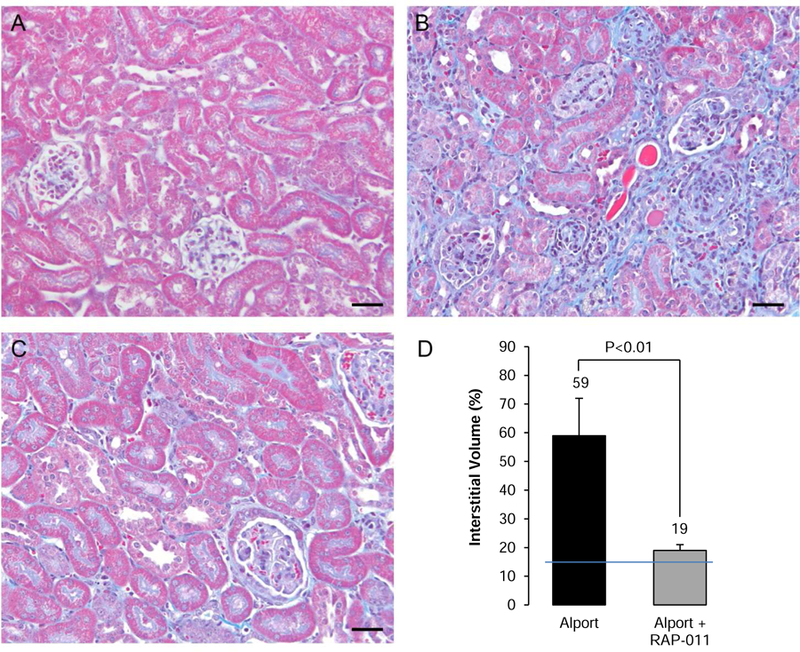
Photomicrographs of trichrome stained cortical sections (scale bars 50µm) from: A, 200 do wild-type littermate mouse; B, 200 do vehicle-treated Alport mouse showing severe interstitial fibrosis and glomerulosclerosis; C, 200 do RAP-011 treated mouse showing less interstitial fibrosis than in the vehicle-treated mice and retention of more normal glomerular morphology; D, Quantification of interstitial inflammation and fibrosis: The results are mean ±SD, n=6 kidneys per group. Five fields from each kidney were measured to get the interstitial volume of that kidney. The line through the bars is the upper limit of normal interstitial volume.
ActRIIA signaling in the kidney
To examine the mechanism of RAP-011’s effect on kidney function, and tubulointerstitial fibrosis, we analyzed ActRIIA signaling in kidney homogenates of the Alport mice. As shown in Figure 7A and B, p-Smad 2 and total smad 2/3 levels (the transcription regulators traveling to the nucleus produced by the activated ActRIIA receptor complex) were increased in renal homogenates from vehicle-treated Alport mice compared to WT littermates. (Figure 7A and B). ActRIIA expression was higher in Alport kidneys, and fibronectin and MCP-1 expression were stimulated. The primary ActRIIA ligand, activin A, was decreased by RAP-011 treatment along with smad 2/3 expression. RAP-011 treatment also decreased fibronectin and MCP-1 levels (Figure 7 A and B).
Figure seven.
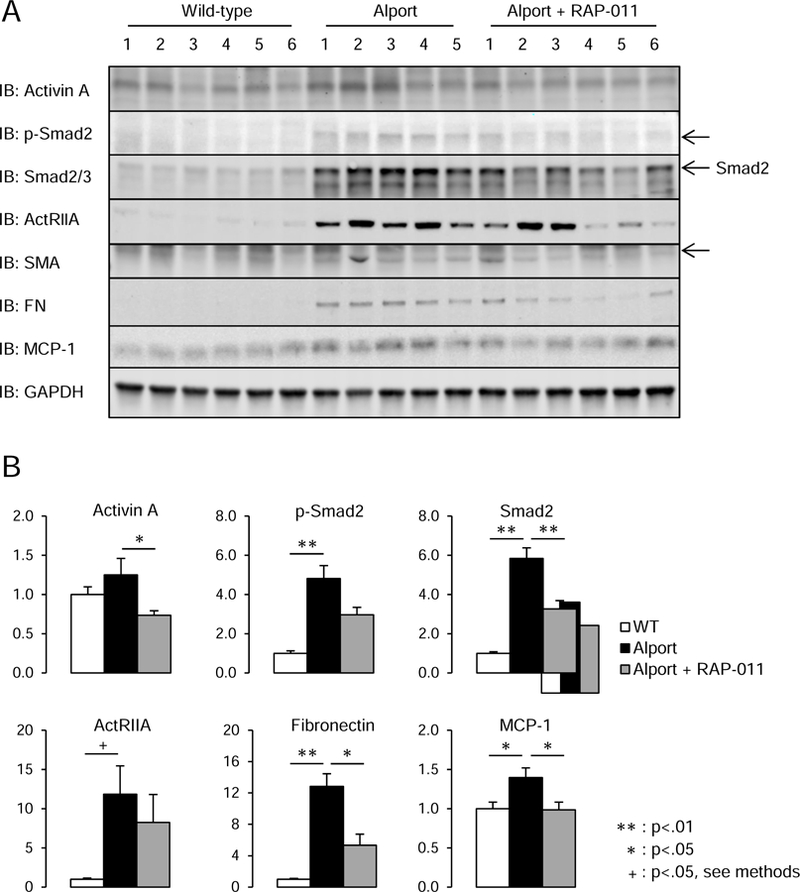
Analysis of ActRIIA signaling in renal homogenates. A, Westerns of kidney homogenates from 6 WT, 5 Alport vehicle-treated, and 6 Alport RAP-011 treated mice. p-Smad2 and Smad 2/3 levels were increased in homogenates from Alport vehicle-treated mice, and they were reduced in homogenates from Alport RAP-011 treated mice. ActRIIA was induced in Alport mice, and the targets fibronectin and MCP-1 were also induced in Alport mice. RAP-011 treatment lowered fibronectin and MCP-1 levels. B, quantitation of the Westerns in A. Data are reported as mean ± SEM.
DISCUSSION:
The results of the studies presented here establish that ActRIIA signaling is activated by CKD throughout the body in the murine homologue of X-linked Alport syndrome. CKD activation of ActRIIA signaling was observed in two other models of CKD, thus, we do not think these findings are specific to Alport syndrome. We analyzed the skeleton, the vasculature, the heart and the kidney. The indicators of ActRIIA activation were the studies of signal transduction in the vasculature and the kidney, and in addition, the effects of its inhibition in all four tissues by RAP-011. Although TGFβ and bone morphogenetic protein 7 have been extensively studied in CKD, (33–39) other members of the TGFβ superfamily have not. The studies reported here and our recent studies are the first to examine ActRIIA activation in CKD. (15) They are indicated from the results of translational profiles of myofibroblasts during renal fibrosis, a common pathway of many kidney diseases, which demonstrate that inhibin βA is one of the major proteins expressed. (40) Inhibin βA homodimers form activin A, a known hormone, raising the possibility that renal fibrosis may produce direct systemic effects through activation of its receptors, type II activin receptors (ActRII) and type I activin receptor-like kinase (ALK) receptors.
Our results demonstrate that CKD in the Alport mice produces an osteodystrophy due to bone resorption by osteoclast stimulation (Fig. 1A–C) and a decrease in osteoblast function (Fig. 1H) and mineralization (Fig 1I–K). ActRIIA inhibition prevented osteoclast stimulation and increased osteoblast function and bone formation (Fig. 1G). These results demonstrate the surprising finding that ActRIIA signaling is able to cause features of renal osteodystrophy heretofore considered to be due only to secondary hyperparathyroidism. They are consistent with the effects of activin A on osteoclastogenesis and function (41, 42) and on osteoblast function. (43) They are also consistent with the effects of RAP-011 on CKD-stimulated osteoclast function in the high fat fed diabetic ldlr−/− mice.(44) Also consistent with the high fat fed diabetic ldlr−/− mice.(44) was the correction of the BFR per osteoblast by RAP-011. This implies that osteoblast ActRIIA signaling delays mineralization in agreement with the effects of activin A described by Chantry et al. (43) The effects of ActRIIA inhibition were despite no change in the elevated PTH and FGF23 levels of the 200 day old Alport mice. The effect of PTH to stimulate remodeling and osteoclastogenesis is through RANK ligand (RANKL) production by osteoblasts and osteocytes (supplemental Fig. 1). (45, 46) The role of activin A in osteoclastogenesis is distal to RANKL and thus, ActRIIA inhibition blocked osteoclast stimulation in the presence of PTH. The effects of ActRIIA inhibition in the skeleton are the strongest indication that the ligand responsible for its stimulation during CKD is activin A, since the other potential ligands, GDF11, BMP9 and 10, and myostatin are not likely critical in osteoclastogenesis. Myostatin does stimulate osteoclastogenesis but through the activin A/ActRII pathway, (47) and myostatin is not produced in the kidney stimulated by CKD. Whereas, activin A is produced by renal myofibroblasts and is elevated in the circulation of Alport mice. (15)
In the vasculature, ActRIIA activation by CKD surprisingly stimulated vascular smooth muscle specific protein expression. Although ActRIIA function in the vasculature has not been extensively studied, this result is consistent with the effects of Smad 2/3 transcription regulators on vascular differentiation and vascular smooth muscle function.(48, 49) However, in CKD the thought is that vascular smooth muscle cell (VSMC) dedifferentiation contributes to osteoblastic transition leading to vascular calcification. (50–52) In diabetic atherosclerotic ldlr−/− high fat fed mice, we have shown that CKD indeed decreases VSMC specific protein expression, and also decreases expression and signaling of ActRIIA, (8, 15) and Lin et al have shown that Runx2 deletion in VSMC of ldlr−/− mice prevents high fat induced vascular calcification. (53) Yet here in the context absent diabetes and atherosclerosis, ActRIIA signaling is stimulated by CKD leading to increased expression of smooth muscle proteins, sm22α and αSMA. This is in agreement with studies showing that activin A promotes the contractile phenotype of smooth muscle cells. (54) Despite its effects on VSMC biomarkers, CKD stimulated osteoblastic transition in the aorta (Figure 2D). This could be consistent with a second cell source contributing to the osteoblastic phenotype. This concept is consistent with recent findings of aortic adventitial mesenchymal stem cell like progenitors differentiating towards vascular smooth muscle cells and migrating to the arterial neointima, but expressing osteoblastic transcription factors. (55) Our finding of patchy aortic calcification mainly in the aortic arch adventitia and media adjacent to the adventitia is consistent with a precursor state to diffuse medial VC, and is distinct from CKD stimulated atherosclerotic vascular calcification where it resides mainly in atheroma deposits,(8) and from the diffuse medial calcification induced by MGP deficiency, osteoprotegerin deficiency, adenine feeding or high phosphorus diet. (56–59) Thus, the data are consistent with recent reports in ldlr−/− high fat fed mice with CKD in which elevated activin A produced by renal myofibroblasts stimulated osteoblastic transition and VC, which was inhibited by administration of a ActRIIA ligand trap. (15) They are also consistent with recent studies in vascular smooth muscle cells implicating ALK1 in CKD-stimulated vascular calcification. (60) ALK1 is a type 1 TGFβ family receptor that is a binding partner with ActRIIA, and recently implicated in cardiovascular remodeling and fibrosis. (61) They are also consistent with the effect of the fibrodysplasia ossificans progressive mutation in ALK2 (ACVR1), another ActRIIA binding partner, which imparts responsiveness to activin A, which then stimulates resident muscle satellite cells to undergo osteoblastic differentiation.(62)
Treatment with RAP-011 was effective in blocking the CKD stimulation of cardiac hypertrophy despite no change in FGF23 levels suggesting another mechanism of left ventricular hypertrophy stimulation in CKD was inhibited by the ligand trap or improved kidney function limited cardiac hypertrophy. The role of ActRIIA in cardiac development is related to the roles of bone morphogenetic proteins (BMPs), see Morrell et al (63) for review. Recent studies of cardiac hypertrophy during aging further implicate ActRIIA signaling as having an important role in cardiac function. (64, 65) Further study is required to distinguish the mechanism of ActRIIA stimulation by CKD and the cardiac effects of RAP-011.
Our results demonstrate that the progressive kidney disease of Alport syndrome in the Col4a5 deficient mouse was delayed by inhibition of ActRII signaling through treatment with RAP-011, such that while vehicle-treated mice reached the equivalent of stage 4 Alport syndrome (severe CKD), the RAP-011 treated mice were in the equivalent of stage 3 Alport syndrome (proteinuria and early to moderate decrease in GFR). (66) The treatment regimen utilized a design that began therapy in early kidney disease (75 do) when the mice manifest only hematuria. This treatment plan was devised for two reasons. The first was that our experience using BMP-7 as a therapeutic for Alport was not successful when began at day of life 125, but was effective when begun earlier in the course of disease (unpublished observations of Yifu Fang and Keith Hruska). These results are consistent with the salutary effects of knocking down a BMP antagonist, USAG-1 or Wise, during development and post-natal life. (67) Secondly, this strategy mirrors the human experience with angiotensin converting enzyme inhibition in Alport, wherein, late institution of therapy when fibrosis is most active was not effective in delaying disease progression, but institution early in the course of disease was more effective (EARLY PRO-TECT Alport trial of early ACEI, NCT01485978). Our results with ActRIIA inhibition in Alport nephritis are in agreement with recent studies implicating activin in cyst formation in murine PKD models and decrease of PKD progression with a ligand trap for the ActRIIB receptor.(68) Activin A binds both ActRIIA and ActRIIB and in the kidney the receptors may be redundant though they are not in the skeleton.
The efficacy of RAP-011 treatment in the skeletal, vascular, and cardiac components of the CKD-MBD in the Alport mice could have derived from direct tissue inhibition of ActRIIA or from less stimulation of ActRIIA due to improved kidney function. We favor both possibilities, and our previous studies (15, 44) favor direct tissue inhibition of ActRIIA, because the CKD in the ldlr−/− high fat fed mice was ablative and RAP-011 could not have improved kidney function preventing development of the CKD-MBD. To further investigate this issue more directly related to the Alport findings, we performed renal ablation produced CKD in C57BL/6J mice, the background strain of our Alport mice. As shown in supplemental Fig. 3, CKD increased aortic Ca levels in C57BL/6J mice, stimulated osteoblastic transition in the aorta shown by expression of Runx2, and increased activin A and pSmad2. These findings are similar to the results in Alport mice shown in Fig. 2. RAP-011 decreased aortic Ca, pSmad2, and Runx2, again without affecting the elevated PTH and FGF23 levels (supplemental Fig. 4) This data also suggests that the findings in 200 do Alport mice with CKD were due to CKD and not specific to Alport syndrome, since the CKD-MBD phenotype is similar in C57BL/6J and Alport mice.
In conclusion, the study of murine Alport syndrome demonstrates activation of ActRIIA signaling in the kidney, skeleton, vasculature and the heart during CKD. The studies support the concept that an endocrine factor produced in the kidney during disease disrupts organ homeostasis outside of the kidney and participates in the pathogenesis of the CKD-MBD. The studies provide support for the hypothesis that inhibition of ActRIIA signaling will be efficacious in the CKD-MBD and CKD related to the upregulation of activin in the kidney during disease and its effects to stimulate fibrosis and systemic ActRIIA.
CONCISE METHODS:
Animal Model:
The model of CKD we used was the murine homolog of X-linked Alport syndrome, which is a deficiency in the gene for the α5 chain of type IV collagen, Col4a5.(24) The mice develop spontaneous kidney disease, and they were used to confirm the effects of renal ablation induced CKD on circulating activin levels, the CKD-MBD and renal fibrosis. Breeding pairs were obtained from Yoav Segal, Univ. of Minnesota and were bred for experiments. All mice received a standard chow diet throughout. Hemizygote males spontaneously developed kidney disease comparable with human CKD stage 3–4 at 150 days after birth, stage 4–5 equivalent disease by 200 days and developed hematuria by day 75 of life.
The study design utilizes three groups of mice, wild type littermates, Alport mice treated with vehicle, and Alport mice receiving subcutaneous injections of RAP-011, a ligand trap for the ActRIIA receptor (Celgene, Summit, NJ) given 10 mg/kg twice a week beginning at day 75 of life until euthanasia at 200 days. The dose used was previously shown in PK/PD studies to be an efficacious dose for stimulation of bone formation,(69) and in Agapova et al. (15). The rest of the methods are in the supplemental materials.
Supplementary Material
Acknowledgements:
The studies reported here were supported by NIH grants DK070790 and DK 089137 (KAH) and DK080770 (HHM) and by the musculoskeletal research center core NIH P30 AR057235.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Disclosure:
KAH and HHM have been Celgene consultants. The other authors have no competing financial interests.
REFERENCES:
- 1.Bansal N, Katz R, Robinson-Cohen C, et al. Absolute rates of heart failure, coronary heart disease, and stroke in chronic kidney disease: An analysis of 3 community-based cohort studies. JAMA Cardiology 2016. [DOI] [PMC free article] [PubMed]
- 2.Block GA, Hulbert-Shearon TE, Levin NW, Port FK. Association of serum phosphorus and calcium X phosphate product with mortality risk in chronic hemodialysis patients: a national study. Am J Kidney Dis 1998;31(4):607–17. [DOI] [PubMed] [Google Scholar]
- 3.Go AS, Chertow GM, Fan D, McCulloch CE, Hsu Cy. Chronic kidney disease and the risks of death, cardiovascular events, and hospitalization. New Engl J Med 2004;351:1296–305. [DOI] [PubMed] [Google Scholar]
- 4.Gutierrez OM, Mannstadt M, Isakova T, Rauh-Hain JA, Tamez H, Shah A, et al. Fibroblast Growth Factor 23 and Mortality among Patients Undergoing Hemodialysis. New Engl J Med 2008;359:584–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Blacher J, Guerin AP, Pannier B, Marchais SJ, London GM. Arterial calcifications, arterial stiffness, and cardiovascular risk in end-stage renal disease. Hypertension 2001;38:938–42. [DOI] [PubMed] [Google Scholar]
- 6.Isakova T, Wahl P, Vargas GS, Gutierrez OM, Scialla J, Xie H, et al. Fibroblast growth factor 23 is elevated before parathyroid hormone and phosphate in chronic kidney disease. Kidney Int 2011;79(12):1370–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pereira RC, Juppner H, Azucena-Serrano CE, Yadin O, Salusky IB, Wesseling-Perry K. Patterns of FGF-23, DMP1 and MEPE expression in patients with chronic kidney disease. Bone 2009;45(6):1161–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fang Y, Ginsberg C, Sugatani T, Monier-Faugere MC, Malluche H, Hruska KA. Early chronic kidney disease-mineral bone disorder stimulates vascular calcification. Kidney Int 2014;85(1):142–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Malluche HH, Ritz E, Lange HP. Bone histology in incipient and advanced renal failure. Kidney Int 1976;9(4):355–62. [DOI] [PubMed] [Google Scholar]
- 10.Kottgen A, Russell SD, Loehr LR, Crainiceanu CM, Rosamond WD, Chang PP, et al. Reduced Kidney Function as a Risk Factor for Incident Heart Failure: The Atherosclerosis Risk in Communities (ARIC) Study. Journal of the American Society of Nephrology 2007;18(4):1307–15 [DOI] [PubMed] [Google Scholar]
- 11.Hu MC, Shi M, Zhang J, Qui+Ýones H, Griffith C, Kuro-o M, et al. Klotho Deficiency Causes Vascular Calcification in Chronic Kidney Disease. J Am Soc Nephrol 2011;22:124–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hu M-C, Shi M, Zhang J, Quinones H, Kuro-o M, Moe OW. Klotho deficiency is an early biomarker of renal ischemia-reperfusion injury and its replacement is protective. Kidney Int 2010;78(12):1240–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.de Zeeuw D, Akizawa T, Audhya P, Bakris GL, Chin M, Christ-Schmidt H, et al. Bardoxolone Methyl in Type 2 Diabetes and Stage 4 Chronic Kidney Disease. New England Journal of Medicine 2013;369:2492–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chin MP, Reisman SA, Bakris GL, O’Grady M, Linde PG, McCullough PA, et al. Mechanisms Contributing to Adverse Cardiovascular Events in Patients with Type 2 Diabetes Mellitus and Stage 4 Chronic Kidney Disease Treated with Bardoxolone Methyl. American Journal of Nephrology 2014;39(6):499–508. [DOI] [PubMed] [Google Scholar]
- 15.Agapova OA, Fang Y, Sugatani T, Seifert ME, Hruska KA. Ligand trap for the activin type IIA receptor protects against vascular disease and renal fibrosis in mice with chronic kidney disease. Kidney International 2016; 89(6):1231–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Xia Y, Schneyer AL. The biology of activin: recent advances in structure, regulation and function. Journal of Endocrinology 2009;202(1):1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tsuchida K, Nakatani M, Hitachi K, Uezumi A, Sunada Y, Ageta H, et al. Activin signaling as an emerging target for therapeutic interventions. Cell Communication and Signaling 2009;7(6):15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kruegel J, Rubel D, Gross O. Alport syndrome[mdash]insights from basic and clinical research. Nat Rev Nephrol 2013;9(3):170–8. [DOI] [PubMed] [Google Scholar]
- 19.Jais JP, Knebelmann B, Giatras I, DeMarchi M, Rizzoni G, Renieri A, et al. X-linked Alport syndrome: Natural history in 195 families and genotype-phenotype correlations in males. Journal of the American Society of Nephrology 2000;11:649–57. [DOI] [PubMed] [Google Scholar]
- 20.Kashtan CE. Long-term management of Alport syndrome in pediatric patients. Pediatr Health, Medicine and Therapeutics 2013;4:41–5. [Google Scholar]
- 21.Van Der Loop FTL, Heidet L, Timmer EDJ, Van Den Bosch BJC, Leinonen A, Antignac C, et al. Autosomal dominant Alport syndrome caused by a COL4A3 splice site mutation. Kidney Int 2000;58:1870–5. [DOI] [PubMed] [Google Scholar]
- 22.Miner JH. Pathology vs. molecular genetics: (re)defining the spectrum of Alport syndrome. Kidney Int 2014;86(6):1081–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Longo I, Porcedda P, Mari F, Giachino D, Meloni I, Deplano C, et al. COL4A3/COL4A4 mutations: From familial hematuria to autosomal-dominant or recessive Alport syndrome. Kidney Int 2002;61:1947–56. [DOI] [PubMed] [Google Scholar]
- 24.Rheault MN, Kren SM, Thielen BK, Mesa HA, Crosson JT, Thomas W, et al. Mouse Model of X-Linked Alport Syndrome. J Am Soc Nephrol 2004;15:1466–74. [DOI] [PubMed] [Google Scholar]
- 25.Gross O, Beirowski B, Koepke M-L, Kuck J, Reiner M, Addicks K, et al. Preemptive ramipril therapy delays renal failure and reduces renal fibrosis in COL4A3-knockout mice with Alport syndrome1. Kidney Int 2003;63(2):438–46. [DOI] [PubMed] [Google Scholar]
- 26.Katayama K, Nomura S, Tryggvason K, Ito M. Searching for a treatment for Alport syndrome using mouse models. World Journal of Nephrology 2014;3(4):230–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Savige J, Gregory M, Gross O, Kashtan C, Ding J, Flinter F. Expert Guidelines for the Management of Alport Syndrome and Thin Basement Membrane Nephropathy. Journal of the American Society of Nephrology 2013;24(3):364–75. [DOI] [PubMed] [Google Scholar]
- 28.Stubbs JR, He N, Idiculla A, Gillihan R, Liu S, David V, et al. Longitudinal evaluation of FGF23 changes and mineral metabolism abnormalities in a mouse model of chronic kidney disease. J Bone Miner Res 2012;27(1):38–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Demer LL, Tintut Y. Vascular Calcification: Pathobiology of a Multifaceted Disease. Circulation 2001;104(16):1881–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lindberg K, Amin R, Moe OW, Hu M-C, Erben RG, Östman Wernerson A, et al. The Kidney Is the Principal Organ Mediating Klotho Effects. J Am Soc Nephrol 2014;25(10):2169–75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hu MC, Shi M, Cho HJ, Adams-Huet B, Paek J, Hill K, et al. Klotho and Phosphate Are Modulators of Pathologic Uremic Cardiac Remodeling. J Am Soc Nephrol 2015;26(6):1290–302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Xie J, Yoon J, An S-W, Kuro-o M, Huang C-L. Soluble Klotho Protects against Uremic Cardiomyopathy Independently of Fibroblast Growth Factor 23 and Phosphate. Journal of the American Society of Nephrology 2015;26(5):1150–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Morrissey J, Hruska K, Guo G, Wang S, Chen Q, Klahr S. Bone morphogenetic protein-7 improves renal fibrosis and accelerates the return of renal function. Journal of the American Society of Nephrology 2002;13:S14–S21. [PubMed] [Google Scholar]
- 34.Hruska KA, Guo G, Wozniak M, Martin D, Miller S, Liapis H, et al. Osteogenic protein-1 (OP-1) prevents renal fibrogenesis associated with ureteral obstruction. Am J Phys (Renal) 2000;279:F130–F43. [DOI] [PubMed] [Google Scholar]
- 35.Wang SN, Lapage J, Hirschberg R. Loss of tubular bone morphogenetic protein-7 in diabetic nephropathy. J Am Soc Nephrol 2001;12:2392–9. [DOI] [PubMed] [Google Scholar]
- 36.Mitu GM, Wang S, Hirschberg R. BMP7 is a podocyte survival factor and rescues podocytes from diabetic injury. AJP - Renal Physiology 2007;293:F1641–F8. [DOI] [PubMed] [Google Scholar]
- 37.Wang S, Hirschberg R. BMP7 antagonizes TGF-beta-dependent fibrogenesis in mesangial cells. AJP - Renal Physiology 2003;284:F1006–F13. [DOI] [PubMed] [Google Scholar]
- 38.Meng X-m, Nikolic-Paterson DJ, Lan HY. TGF-[beta]: the master regulator of fibrosis. Nat Rev Nephrol 2016;12(6):325–38. [DOI] [PubMed] [Google Scholar]
- 39.Liu Y. Renal fibrosis: New insights into the pathogenesis and therapeutics. Kidney International 69(2):213–7. [DOI] [PubMed] [Google Scholar]
- 40.Grgic I, Krautzberger AM, Hofmeister A, Lalli M, DiRocco DP, Fleig SV, et al. Translational Profiles of Medullary Myofibroblasts during Kidney Fibrosis. Journal of the American Society of Nephrology 2014;25(9):1979–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sugatani T, Alvarez UM, Hruska KA. Activin A stimulates I kappaB-alpha/NFkappaB and RANK expression for osteoclast differentiation, but not AKT survival pathway in osteoclast precursors. J Cell Biochem 2003;90(1):59–67. [DOI] [PubMed] [Google Scholar]
- 42.Gaddy-Kurten D, Coker JK, Abe E, Jilka RL, Manolagas SC. Inhibin suppresses and activin stimulates osteoblastogenesis and osteoclastogenesis in murine bone marrow cultures. Endocrinology 2002;143:74–83. [DOI] [PubMed] [Google Scholar]
- 43.Chantry AD, Heath D, Mulivor AW, Pearsall S, Baud’huin M, Coulton L, et al. Inhibiting activin-A signaling stimulates bone formation and prevents cancer-induced bone destruction in vivo. Journal of Bone and Mineral Research 2010;25(12):2633–46. [DOI] [PubMed] [Google Scholar]
- 44.Sugatani T, Agapova OA, Fang Y, Berman AG, Wallace JM, Malluche HH, et al. Ligand trap of the activin receptor type IIA inhibits osteoclast stimulation of bone remodeling in diabetic mice with chronic kidney disease. Kidney International 2017;91(1):86–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.O’Brien CA, Plotkin LI, Galli C, Goellner JJ, Gortazar AR, Allen MR, et al. Control of Bone Mass and Remodeling by PTH Receptor Signaling in Osteocytes. PLOS ONE 2008;3(8):e2942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Xiong J, O’Brien CA. Osteocyte RANKL: New insights into the control of bone remodeling. Journal of Bone and Mineral Research 2012;27(3):499–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Dankbar B, Fennen M, Brunert D, Hayer S, Frank S, Wehmeyer C, et al. Myostatin is a direct regulator of osteoclast differentiation and its inhibition reduces inflammatory joint destruction in mice. Nat Med 2015;21(9):1085–90. [DOI] [PubMed] [Google Scholar]
- 48.Goumans M-J, Liu Z, ten Dijke P. TGF-[beta] signaling in vascular biology and dysfunction. Cell Res 2009;19(1):116–27. [DOI] [PubMed] [Google Scholar]
- 49.Guo X, Chen S-Y. Transforming growth factor-β and smooth muscle differentiation. World Journal of Biological Chemistry 2012;3(3):41–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Moe SM, Chen NX. Pathophysiology of Vascular Calcification in Chronic Kidney Disease. Circulation Research 2004;95(6):560–7. [DOI] [PubMed] [Google Scholar]
- 51.Tyson KL, Reynolds JL, McNair R, Zhang Q, Weissberg PL, Shanahan CM. Osteo/chondrocytic transcription factors and their target genes exhibit distinct patterns of expression in human arterial calcification. Arterioscler Thromb Vasc Biol 2003;23:489–94. [DOI] [PubMed] [Google Scholar]
- 52.Speer MY, Yang HY, Brabb T, Leaf E, Look A, Lin WL, et al. Smooth Muscle Cells Give Rise to Osteochondrogenic Precursors and Chondrocytes in Calcifying Arteries. Circulation Research 2009;104:733–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lin M-E, Chen TM, Wallingford MC, Nguyen NB, Yamada S, Sawangmake C, et al. Runx2 deletion in smooth muscle cells inhibits vascular osteochondrogenesis and calcification but not atherosclerotic lesion formation. Cardiovascular Research 2016;112(2):606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Engelse MA, Neele JM, van Achterberg TAE, van Aken BE, van Schaik RHN, Pannekoek H, et al. Human Activin-A Is Expressed in the Atherosclerotic Lesion and Promotes the Contractile Phenotype of Smooth Muscle Cells. Circulation Research 1999;85(10):931–9. [DOI] [PubMed] [Google Scholar]
- 55.Kramann R, Goettsch C, Wongboonsin J, Iwata H, Schneider Rebekka K, Kuppe C, et al. Adventitial MSC-like Cells Are Progenitors of Vascular Smooth Muscle Cells and Drive Vascular Calcification in Chronic Kidney Disease. Cell Stem Cell 2016;19(5):628–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Luo G, Ducy P, McKee MD, Pinero GJ, Loyer E, Behringer RR, et al. Spontaneous calcification of arteries and cartilage in mice lacking matrix GLA protein. Nature 1997;386:78–81 [DOI] [PubMed] [Google Scholar]
- 57.Bucay N, Sarosi I, Dunstan CR, Morony S, Tarpley J, Capparelli C, et al. Osteoprotegerin-deficient mice develop early onset osteoporosis and arterial calcification. Genes and Development 1998;12:1260–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.El-Abbadi MM, Pai AS, Leaf EM, Yang HY, Bartley BA, Quan KK, et al. Phosphate feeding induces arterial medial calcification in uremic mice: role of serum phosphorus, fibroblast growth factor-23, and osteopontin. Kidney Int 2009;75:1297–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Shobeiri N, Adams MA, Holden RM. Vascular Calcification in Animal Models of CKD: A Review. American Journal of Nephrology 2010;31(6):471–81. [DOI] [PubMed] [Google Scholar]
- 60.Zhu D, Mackenzie NCW, Shanahan CM, Shroff RC, Farquharson C, MacRae VE. BMP-9 regulates the osteoblastic differentiation and calcification of vascular smooth muscle cells through an ALK1 mediated pathway. Journal of Cellular and Molecular Medicine 2015;19(1):165–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.González-Núñez M, Muñoz-Félix JM, López-Novoa JM. The ALK-1/Smad1 pathway in cardiovascular physiopathology. A new target for therapy? Biochimica et Biophysica Acta (BBA) - Molecular Basis of Disease 2013;1832(10):1492–510. [DOI] [PubMed] [Google Scholar]
- 62.Hatsell SJ, Idone V, Wolken DMA, Huang L, Kim HJ, Wang L, et al. ACVRIR206H receptor mutation causes fibrodysplasia ossificans progressiva by imparting responsiveness to activin A. Science Translational Medicine 2015;7(303):303ra137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Morrell NW, Bloch DB, ten Dijke P, Goumans M-JTH, Hata A, Smith J, et al. Targeting BMP signalling in cardiovascular disease and anaemia. Nat Rev Cardiol 2016;13(2):106–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Loffredo Francesco S, Steinhauser Matthew L, Jay Steven M, Gannon J, Pancoast James R, Yalamanchi P, et al. Growth Differentiation Factor 11 Is a Circulating Factor that Reverses Age-Related Cardiac Hypertrophy. Cell 2013;153(4):828–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Garber K No longer going to waste. Nat Biotech 2016;34(5):458–61. [DOI] [PubMed] [Google Scholar]
- 66.Gross O, Perin L, Deltas C. Alport syndrome from bench to bedside: the potential of current treatment beyond RAAS blockade and the horizon of future therapies. Nephrology Dialysis Transplantation 2014;29(suppl 4):iv124–iv30. [DOI] [PubMed] [Google Scholar]
- 67.Tanaka M, Asada M, Higashi AY, Nakamura J, Oguchi A, Tomita M, et al. Loss of the BMP antagonist USAG-1 ameliorates disease in a mouse model of the progressive hereditary kidney disease Alport syndrome. J Clin Invest 2010;120:768–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Leonhard WN, Kunnen SJ, Plugge AJ, Pasternack A, Jianu SBT, Veraar K, et al. Inhibition of Activin Signaling Slows Progression of Polycystic Kidney Disease. Journal of the American Society of Nephrology 2016;27(12):3589–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Lotinun S, Pearsall RS, Davies MV, Marvell TH, Monnell TE, Ucran J, et al. A soluble activin receptor Type IIA fusion protein (ACE-011) increases bone mass via a dual anabolic-antiresorptive effect in Cynomolgus monkeys. Bone 2010;46(4):1082–8. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.