

Abstract
Genetically encoded fluorescent proteins or small-molecule probes that recognize specific protein binding partners can be used to label proteins to study their localization and function with fluorescence microscopy. However, these approaches are limited in signal-to-background resolution and the ability to temporally control labeling. Herein, we describe a covalent protein labeling technique using a fluorogenic malachite green probe functionalized with a photoreactive crosslinker. This enables a controlled covalent attachment to a genetically encodable fluorogen activating protein (FAP) with low background signal. We demonstrate covalent labeling of a protein in vitro as well as in live mammalian cells. This method is straightforward, displays high labeling specificity, and results in improved signal-to-background ratios in photoaffinity labeling of target proteins. Additionally, this probe provides temporal control over reactivity, enabling future applications in real-time monitoring of cellular events.
Graphical Abstract
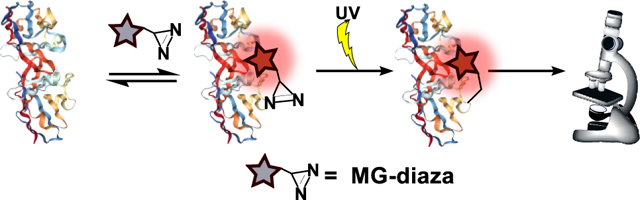
Fluorescent labeling of proteins in living systems has provided an invaluable window into cellular function.1,2 Protein expression,3 folding,4 post-translational modification,5 and localization6,7 can all now be monitored with fluorescent labels and microscopic analysis. The most common protein labeling technique employs genetic fusion of a POI with an intrinsically fluorescent protein (FP) such as green fluorescent protein or its variants.8 This process is typically accomplished through transfection of cells with a vector containing a promoter that induces the expression of the transgene. In most cases, overexpression of FP fusion proteins does not have an observable effect on the native properties and localization of the target protein.9,10 However, the intrinsic fluorescence of these FP labels often results in diffuse signals throughout the cell, making it difficult to monitor protein dynamics with high resolution. Additionally, promoters such as CMV and EF-1a, commonly used for expression of FP fusions, result in the production of large protein copy numbers, which in turn can compromise signal-to-noise ratios.11,12
Additional protein labeling techniques have been developed using non-covalent or covalent attachment of a fluorescent ligand to a peptide or protein sequence. One common strategy is the tetracysteine-biarsenical system to covalently attach a green or red fluorescent small molecule to a protein of interest.13,14 Despite the small size and high quantum yield of these systems, the arsenic-containing small-molecule dyes are shown to have elevated photobleaching properties, diminishing the clarity of subcellular tracking of proteins. Systems such as SNAP/Clip-tag,15,16 LAP,17,18 Halo-tag,15,19 and coiled-coil tag20 also employ a system where a small organic molecule covalently attaches to the POI. While these techniques are simple and enable covalent modification of target proteins, they still rely on constitutively active fluorophores, and hence background fluorescence is quite high in unreacted probes, requiring additional wash-out steps for effective visualization, rendering it difficult to use these systems for real time monitoring of cellular behavior.
Techniques that use fluorogenic molecules21–23 have significantly improved this resolution issue, overcoming the background signal generated from unbound molecules. However, these techniques lack either covalent attachment or temporal-control of target protein labeling, and hence their use is limited to non-covalent associations with short, diffusion-limited visualization lifetimes. The ability to temporally control protein labeling would enable pulse-chase experiments for studying time-dependent processes such as response to a therapeutic agent or flux in metabolic reactions.24–26 Temporally-controlled covalent protein labeling would also prove useful in pull-down experiments, as the protein of interest (POI) could be selectively labeled and isolated from a complex system.27,28 Thus, there remains a need for the development of a protein labeling strategy that can provide robust, stable, and temporally-controlled labeling with low background signal.
Herein, we address these needs using a photoaffinity labeling strategy, as light-induced reactions offer covalent attachment with precise temporal control, and can also offer spatial control of labeling if desired. Specifically, we use a fluorogen activating protein (FAP) that has been developed by Waggoner and coworkers and elaborated by Bruchez and coworkers. This FAP is composed of a single-chain variable fragment antibody that binds to and enhances the fluorescence of malachite green (MG).29 Non-covalent labeling using this protein-fluorophore pair has been used to image proteins in vitro and in live cells,30,31 however, this strategy has yet to be elaborated to enable covalent attachment of the fluorogenic label. Here we extend the functionality of this labeling system by appending a reactive diazirine group to the MG fluorogen, such that irradiation with light triggers covalent attachment to the FAP (Figure 1a). The diazirine reactive group is small in size and has been used as a photoactive cross-linker for proteomic profiling and fluorescent labeling of proteins,32–34 and the high photostability of MG35,36 minimizes challenges associated with photobleaching. The ability to form a covalent interaction will open the door to additional experimental possibilities such as immunoprecipitation and enhanced imaging contrast after wash-out.
Figure 1.

(a) Temporally-controlled covalent labeling of a protein of interest (POI). (b) Chemical structure of MG and MG-diazarine.
To generate our photoaffinity probe, we designed a malachite green (MG) fluorophore functionalized with a diazirine reactive group (MG-diazarine) (Figure 1b). Malachite green, a triphenylmethane dye, has very low fluorescence in solution, but can display up to a 20,000-fold increase in brightness when bound to a receptor such as the single-chain antibody-derived FAP. This property of MG provides low background signal for unbound and unreacted probe molecules, as well as for probes that may non-specifically react with biomolecules other than the FAP receptor. Another desirable feature of the MG-FAP system is its fluorescence emission maximum at 680 nm. This far red-shifted fluorescence emission further minimizes background signal by enabling imaging outside of the window where cellular autofluorescence is problematic. For the photoreactive group, we chose diazirine, which forms a highly reactive carbene species upon short bursts of UV irradiation at 355 nm, and is commonly used for photo-crosslinking of proteins in cells.32,37 Finally, we incorporated an arginine moiety in the linker connecting the reactive diazirine group and the MG, as this was found to improve solubility and membrane permeability of the fluorogenic probe.
The synthesis of MG-diazarine is outlined in Scheme 1. Amine-modified malachite green was generated using the method reported by Deng and coworkers38 and was coupled with Fmoc-Arg(Boc)2-OH. After deprotection of the Fmoc group using 20% piperidine in CH2Cl2, the resulting amine was coupled with succinimidyl 4,4’-azipentanoate. Oxidation of the triphenylmethane using chloranil, followed by deprotection of the Boc groups, yielded MG-diazarine.
Scheme 1.

Synthesis of MG-diazarine.
With the photoreactive fluorogen molecule in hand, we turned to testing the feasibility of our labeling approach in vitro. FAP protein was expressed and purified from bacteria, and this was incubated with 50 μM MG-diazarine for 10 min to allow for binding, then irradiated with 365 nm UV light for 5 min to initiate the diazirine group activation. The solution was then mixed 1:1 (v/v) with SDS denaturing loading dye and was heated to 90 °C for 10 min. Importantly, this denaturation step disrupts any non-covalent interactions of the fluorogen with the FAP, allowing for visualization of only the covalently bound FAP-MG-diazarine complexes. After cooling to room temperature, the samples were separated using SDS-PAGE followed by analysis using both Coomassie staining and fluorescence imaging. MG has very minimal fluorescence after denaturation of the FAP binding partner, which introduces a challenge for detection in SDS-PAGE. However, we have found that freezing the gel by incubation on dry ice provides a similar rotational confinement, producing sufficient fluorescence enhancement to enable visualization of the MG signal. As shown in Figure 2, labeling is only observed for the sample having both FAP protein and MG-diazarine, along with exposure to UV irradiation. No labeling is observed when the FAP is not expressed, no UV irradiation is used, or when MG-diazarine is replaced with MG lacking the photoreactive group. These control experiments demonstrate that covalent labeling of the FAP protein is attributable to binding of MG and subsequent light-induced reaction of the diazirine group.
Figure 2.

In vitro assessment of covalent photoaffinity labeling.
To quantify the rate of covalent labeling, we next conducted a time-course experiment. Solutions containing FAP protein and 50 μG-diazarine in 1× PBS were prepared and incubated at 37 °C to allow for noncovalent equilibration of the MG-diazarine with the FAP protein. After the initial incubation time, the samples were subjected to UV irradiation for varying quantities of time, and SDS-PAGE analysis was carried out using similar techniques as described above to determine the extent of covalent labeling (Figure 3a–b). Under these conditions, 50% labeling was observed within 10 min of UV activation (Figure 3b), which is comparable to previously reported photoaffinity labeling strategies.39,40
Figure 3.

Kinetics of labeling reaction. (a) SDS-PAGE analysis of labeling as a function of time. (b) Normalized fluorescence as a function of time.
While the MG probe only becomes fluorescent after binding to the FAP, we were still curious to know whether the diazirine could react non-specifically with other proteins. To investigate this question, we carried out the labeling reaction in a whole-cell lysate both with and without addition of FAP (Figure 4). The solutions were incubated for 30 min, then UV irradiated for 5 min and analyzed by SDS-PAGE. Encouragingly, the only bands visible in the fluorescence channel are those corresponding to the molecular weight of the FAP (major) and uncleaved FAP-His fusion (minor), and these bands are not observed in the no-FAP control. The absence of any other visible bands indicates that labeling is specific to the FAP and that the diazirine does not react with other proteins in a complex biological solution.
Figure 4.

Testing the specificity of FAP labeling in cell lysate.
After validating the ability of MG-diazarine to covalently label the FAP in vitro, we turned toward our ultimate goal of demonstrating protein labeling in live cells. HeLa cells were transfected with a plasmid encoding a FAP-mCerulean3 fusion protein. In this design, the blue fluorescent mCerulean3 protein serves as both a transfection reporter and as a model POI to evaluate the use of our FAP-MG-diazarine system. Importantly, the excitation and emission wavelengths of the mCerulean3 and MG-diazarine do not overlap, and thus there is no interference from fluorescence cross-talk or FRET signal. The cells were grown to 90% confluency and incubated with 50 μM MG-diazarine for 15 min. UV irradiation was then applied for 5 min, which we previously found to be sufficient for covalent labeling in vitro, and we verified that this irradiation procedure did not affect cell viability (Figure S7). UV photolabeling of cells was followed by exchange of the media and fluorescence imaging. The media was subsequently exchanged every 5 min and wash-out of unreacted probe followed by fluorescence imaging. While not necessary for using this imaging method in future applications, we performed these wash-out experiments to assess the stability of photolabeled versus non-covalent fluorogenic signal over time. Initially, the cells showed similar fluorescence intensity to control cells incubated with MG-diazarine but not exposed to UV irradiation. As expected, cells that were not transfected did not exhibit detectable MG-diazarine fluorescence (Figure 5b). This result indicates that the MG-diazarine fluorescence signal results from binding to the FAP and that UV activation of MG-diazarine in the presence of other cellular components does not result in unwanted background fluorescence. After each washing cycle, the transfected cells that were not UV irradiated showed a steady decrease in fluorescence, while the transfected cells that were irradiated with UV light maintained a consistent fluorescence intensity over 40 min of wash-out time (Figure 5b–c). This indicates that the covalent labeling reaction is also robust in the context of living cells, and that covalent labeling produced robust signal duration and stability. While the fluorescence of non-irradiated cells does not fully reach background levels, we hypothesize that this is due to the high binding affinity of the FAP for MG, making it difficult to fully deplete the non-covalently FAP-bound MG fraction in this control condition. However, the dramatic fluorescence decrease that we do observe suggests that unbound MG-diazarine is increasingly depleted from the cytoplasm, and thus our system would still meet this key requirement for pulse-chase labeling applications.
Figure 5.

Fluorescence imaging of HeLa cells transfected with mCerulean3-FAP. (a) Transfected cells and non-transfection control. (b) Wash-out of MG-diazarine from non-irradiated cells demonstrates robustness of covalent labeling approach. (c) Fluorescence signal as a function of washout time for cells irradiated and not irradiated with UV light.
In conclusion, we report a new approach for covalent fluorescent labeling of proteins of interest in vitro as well as in living cells. To accomplish this, we have synthesized a fluorogenic MG-diazarine probe that has an arginine unit to provide cell permeability and a diazirine reactive group for photoaffinity labeling. The MG-diazarine exhibits very low background signal, and has red-shifted fluorescence emission that is well outside of the cellular auto-fluorescence region. We show that this probe is capable of covalently labeling its FAP binding partner. The MG-diazarine probe is facile to synthesize, and plasmids encoding the FAP protein are widely available, making this approach very user friendly and broadly applicable. Unlike other covalent protein labeling approaches, the use of UV light to initiate probe attachment to the protein of interest provides temporal control over the labeling reaction to enable future applications such as pulse-chase imaging and studies of cellular behavior with enhanced signal stability and reduced background. Complimenting currently existing protein labeling strategies, this research provides a facile approach to covalent protein labeling in a format that is anticipated to open the door to new applications requiring temporal control.
Supplementary Material
ACKNOWLEDGMENT
This work was supported by the National Institutes of Health (R01GM116991 to J.M.H.).
Footnotes
Supporting Information. General experimental protocols, synthetic protocols and characterization data for MG-diazarine, and tabular data for cell wash-out experiment. This material is available free of charge via the Internet at http://pubs.acs.org.
REFERENCES
- (1).Toseland CP (2013) Fluorescent Labeling and Modification of Proteins. Journal of Chemical Biology 6 (3), 85–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Crivat G; Taraska JW (2011) Imaging Proteins inside Cells with Fluorescent Tags. Trends Biotechnol 30 (1), 8–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Chalfie M; Tu Y; Euskirchen G; Ward WW; Prasher D (1994) Green Fluorescent Protein as a Marker for Gene Expression. Science 263 (5148), 802–805. [DOI] [PubMed] [Google Scholar]
- (4).Waldo GS; Standish BM; Berendzen J; Terwilliger TC (1999) Rapid Protein-Folding Assay Using Green Fluorescent Protein. Nat. Biotechnol 17 (7), 691–695. [DOI] [PubMed] [Google Scholar]
- (5).Hertel F; Zhang J (2014) Monitoring of Post-Translational Modification Dynamics with Genetically Encoded Fluorescent Reporters. Biopolymers 101 (2), 180–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Lippincott-Schwartz J; Snapp EL; Phair RD (2018) The Development and Enhancement of FRAP as a Key Tool for Investigating Protein Dynamics. Biophysical Journal 115 (7), 1146–1155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Dangol S; Singh R; Chen Y; Jwa N (2017) Visualization of Multicolored in Vivo Organelle Markers for Co-Localization Studies in Oryza Sativa. Mol. Cells, 40 (11), 828–836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Tsien R (1998) The Green Fluorescent Protein. Annu. Rev. Biochem 67, 509–544. [DOI] [PubMed] [Google Scholar]
- (9).Das SC; Panda D; Nayak D; Pattnaik AK (2009) Biarsenical Labeling of Vesicular Stomatitis Virus Encoding Tetracysteine-Tagged M Protein Allows Dynamic Imaging of M Protein and Virus Uncoating in Infected Cells. J. Virol 83 (6), 2611–2622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Doyon JB; Zeitler B; Cheng J; Cheng AT; Cherone JM; Santiago Y; Lee AH; Vo TD; Doyon Y; Miller JC; Paschon DE; Zhang L; Rebar EJ; Gregory PD; Urnov FD; Drubin DG (2011) Rapid and Efficient Clathrin-Mediated Endocytosis Revealed in Genome-Edited Mammalian Cells. Nat. Cell Biol 13 (3), 331–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Qin JY; Zhang L; Clift KL; Hulur I; Xiang AP; Ren B; Lahn B (2010) Systematic Comparison of Constitutive Promoters and the Doxycycline-Inducible Promoter. PLoS One 5 (5), e10611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Running RJ; Allison DS (2004) High-Level Expression of Proteins in Mammalian Cells Using Transcription Regulatory Sequences from the Chinese Hamster EF-1 Gene. Biotechnol. Prog 20 (3), 880–889. [DOI] [PubMed] [Google Scholar]
- (13).Adams SR; Campbell RE; Gross LA; Martin BR; Walkup GK; Yao Y; O JL; Tsien RY (2002) New Biarsenical Ligands and Tetracysteine Motifs for Protein Labeling in Vitro and in Vivo: Synthesis and Biological Applications. J. Am. Chem. Soc 124 (21), 6063–6076. [DOI] [PubMed] [Google Scholar]
- (14).Griffin BA; Adams S; Tsien R (1998) Specific Covalent Labeling of Recombinant Protein Molecules Inside Live Cells. Science, 281 (5374), 269–272. [DOI] [PubMed] [Google Scholar]
- (15).Stagge F; Mitronova GY; Belov VN; Wurm CA; Jakobs S (2013) Snap-, CLIP-and Halo-Tag Labelling of Budding Yeast Cells. PLoS One 8 (10), e78745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Gautier A; Juillerat A; Heinis C; Reis Corre I; Kindermann M; Beaufils F; Johnsson K (2008) An Engineered Protein Tag for Multiprotein Labeling in Living Cells. Chemistry & Biology 15(2), 128–136. [DOI] [PubMed] [Google Scholar]
- (17).Yao JZ; Uttamapinant C; Poloukhtine A; Baskin JM; Codelli JA; Sletten EM; Bertozzi CR; Popik VV; Ting AY (2012) Fluorophore Targeting to Cellular Proteins via Enzyme-Mediated Azide Ligation and Strain-Promoted Cycloaddition. J. Am. Chem. Soc 134 (8), 3720–3728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Fernandez-Suarez M; Baruah H; Martinez-Hernandez L; Xie KT; Baskin JM; Bertozzi CR; Ting AY (2007) Redirecting Lipoic Acid Ligase for Cell Surface Protein Labeling with Small-Molecule Probes. Nat. Biotechnol 25 (12), 1483–1487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Los GV, Encell LP; Mcdougall MG; Hartzell DD; Karassina N; Zimprich C; Wood MG; Learish R; Friedman Ohana R; Urh M; Simpson D; Mendez J; Zimmerman K; Otto P; Vidugiris G; Zhu J; Darzins A; Klaubert DH; Bulleit RF; Wood KV HaloTag: (2018) A Novel Protein Labeling Technology for Cell Imaging and Protein Analysis. ACS Chem. Biol 3 (6), 373–382. [DOI] [PubMed] [Google Scholar]
- (20).Reinhardt U; Lotze J; Mo K; Beck-Sickinger AG; Seitz O (2015) Rapid Covalent Fluorescence Labeling of Membrane Proteins on Live Cells via Coiled-Coil Templated Acyl Transfer. Bioconjugate Chem 26 (10), 2106–2117. [DOI] [PubMed] [Google Scholar]
- (21).Bruchez MP (2015) Dark Dyes-Bright Complexes: Fluorogenic Protein Labeling. Curr. Opin. Chem. Biol 27, 18–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Chen Y; Clouthier CM; Tsao K; Strmiskova M; Lachance H; Keillor JW (2014) Coumarin-Based Fluorogenic Probes for No-Wash Protein Labeling. Angew. Chem 53 (50), 13785–13788. [DOI] [PubMed] [Google Scholar]
- (23).Telmer CA; Verma R; Teng H; Andreko S; Law L; Bruchez MP (2015) Rapid, Specific, No-Wash, Far-Red Fluorogen Activation in Subcellular Compartments by Targeted Fluorogen Activating Proteins. ACS Chem. Biol 10 (5), 1239–1246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Peraro L; Deprey KL; Moser MK; Zou Z; Ball HL; Levine B; Kritzer JA (2018) Cell Penetration Profiling Using the Chloroalkane Penetration Assay. J. Am. Chem. Soc 140, 11360–11369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Darabedian N; Gao J; Chuh KN; Woo CM; Pratt MR (2018) The Metabolic Chemical Reporter 6-Azido-6-Deoxy-Glucose Further Reveals the Substrate Promiscuity of O-GlcNAc Transferase and Catalyzes the Discovery of Intracellular Protein Modification by O-Glucose. J. Am. Chem. Soc 140 (23), 7092–7100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Schneider N; Ga C; Wiener J; Georgiev T; Gobet N; Weber W; Meier M (2018) Genetic Code Expansion Method for Temporal Labeling of Endogenously Expressed Proteins. ACS Chem. Biol 13 (11), 3049–3053. [DOI] [PubMed] [Google Scholar]
- (27).Titeca K; Lemmens I; Tavernier J; Eyckerman S (2019) Discovering Cellular Protein-protein Interactions: Technological Strategies and Opportunities. Mass Spectrom. Rev 38 (1), 79–111. [DOI] [PubMed] [Google Scholar]
- (28).Jain A; Liu R; Ramani B; Arauz E; Ishitsuka Y; Ragunathan K; Park J; Chen J; Xiang YK; Ha T (2011) Probing Cellular Protein Complexes Using Single-Molecule Pull-Down. Nature 473 (7348), 484–488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Szent-Gyorgyi C; Schmidt BAF; Schmidt BAF; Creeger Y; Fisher GW; Zakel KL; Adler S; Fitzpatrick JAJ; Woolford CA; Yan Q; Vasilev KV; Berget PB; Bruchez MP; Jarvik JW; Waggoner A (2008) Fluorogen-Activating Single-Chain Antibodies for Imaging Cell Surface Proteins. Nat. Biotechnol 26 (2), 235–240. [DOI] [PubMed] [Google Scholar]
- (30).Yan Q; Schmidt BF; Perkins LA; Naganbabu M; Saurabh S; Andreko SK; Bruchez MP (2015) Near-Instant Surface-Selective Fluorogenic Protein Quantification Using Sulfonated Triarylmethane Dyes and Fluorogen Activating Proteins. Org. Biomol. Chem 13, 2078–2086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Zeng G; Wang Y; Bruchez MP; Liang FS (2018) Self-Reporting Chemically Induced Protein Proximity System Based on a Malachite Green Derivative and the L5** Fluorogen Activating Protein. Bioconjugate Chem 29 (9), 3010–3015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Chan EWS; Chattopadhaya S; Panicker RC; Huang X and; Yao SQ (2004) Developing Photoactive Affinity Probes for Proteomic Profiling: Hydroxamate-Based Probes for Metalloproteases. J. Am. Chem. Soc 126 (44), 14435–14446. [DOI] [PubMed] [Google Scholar]
- (33).Tomohiro T; Kato K; Masuda S; Kishi H; Hatanaka Y (2011) Photochemical Construction of Coumarin Fluorophore on Affinity-Anchored Protein. Bioconjugate Chem 22 (3), 315–318. [DOI] [PubMed] [Google Scholar]
- (34).Bringmann G; Gampe CM; Reichert Y; Bruhn T; Faber JH; Mikyna M; Reichert M; Leippe M; Brun R; Gelhaus C (2007) Synthesis and Pharmacological Evaluation of Fluorescent and Photoactivatable Analogues of Antiplasmodial Naphthylisoquinolines. J. Med. Chem 50 (24), 6104–6115. [DOI] [PubMed] [Google Scholar]
- (35).Saurabh S; Zhang M; Mann VR; Costello AM (2015) Kinetically Tunable Photostability of Fluorogen-Activating Peptide - Fluorogen Complexes. Chemphyschem 16 (14), 2974–2980. [DOI] [PubMed] [Google Scholar]
- (36).Saurabh S; Perez AM; Comerci CJ; Shapiro L; Moerner WE (2016) Super-Resolution Imaging of Live Bacteria Cells Using a Genetically Directed, Highly Photostable Fluoromodule. J. Am. Chem. Soc 138 (33), 10398–10401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Hoffmann J-E; Dziuba D; Stein F; Schultz C (2018) A Bifunctional Noncanonical Amino Acid: Synthesis, Expression, and Residue-Specific Proteome-Wide Incorporation. Biochemistry 57, 4747–4752. [DOI] [PubMed] [Google Scholar]
- (38).Xing W; He L; Yang H; Sun C; Li D; Yang X; Li Y; Deng A (2009) Development of a Sensitive and Group-Specific Polyclonal Antibody-Based Enzyme-Linked Immunosorbent Assay (ELISA) for Detection of Malachite Green and Leucomalachite Green in Water and Fish Samples. J. Sci. Food Agric 89 (13), 2165–2173. [Google Scholar]
- (39).Uchinomiya S; Nonaka H; Wakayama S; Ojida A; Hamachi I (2013) In-Cell Covalent Labeling of Reactive His-Tag Fused Proteins. Chemical Communications 49 (44), 5022–5024. [DOI] [PubMed] [Google Scholar]
- (40).Chen Q; Hu X; Zhang D; Chen X; Wang J (2017) Selective Isolation of Myosin Subfragment-1 with a DNA-Polyoxovanadate Bioconjugate Chem 28 (12), 2976–2984. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.