

Abstract
The introduction of fluorine into bioactive molecules is a matter of importance in medicinal chemistry. In this study, representatives of various chemical entities of fluoroaromatic compounds were synthesized. Depending on the reaction conditions, either tetrafluorophthalimides or ammonium tetrafluorophthalamates are accessible from tetrafluorophthalic anhydride and primary amines. Tetrafluorophthalamic acids undergo thermal decarboxylation to yield tetrafluorobenzamides. These could be successfully converted upon treatment with primary amines, in the course of an aromatic nucleophilic substitution, to 2,3,5-trifluorobenzamides with respective amino substituents at the 4-position. The five structure types were characterized by means of spectroscopic and crystallographic methods. The synthesized compounds were evaluated as inhibitors of angiogenesis by measuring microvessel outgrowth in a rat aortic ring assay. The biological activity was maintained throughout these different polyfluorinated chemotypes.
Keywords: angiogenesis, fluorine, tetrafluorobenzamides, tetrafluorophthalimides, trifluorobenzamides
Introduction
Angiogenesis, the formation of new blood vessels from existing ones, is driven by a finely tuned balance between factors which constitute a functional network. The process of angiogenesis is important for several physiological and pathological conditions, including cancer, where functional vessels are required to supply nutrients and oxygen, and to sustain tumor growth and metastatic dissemination. Angiogenesis can be inhibited by blocking pro-angiogenic or raising antiangiogenic factors or by killing tumor-activated endothelial cells. Antiangiogenic drugs have been shown to slow tumor progression and metastasis.[1] Accordingly, therapies based on such drugs represent a promising approach for the treatment of cancer diseases and there remains a need for novel agents against angiogenesis.[2]
The importance of tetrafluorination of simple phthalimides to achieve antiangiogenic activity has been shown.[3] Similarly, compound I (Figure 1), a polyfluorinated phthalimide derivative, leads to a strongly enhanced inhibition of the microvessel outgrowth in an angiogenesis model,[4] and of the TNF-a pro-duction by monocytes.[5] It possesses antimyeloma capacity, activates the stress kinase p38 pathway, affects blood vessels depending on their state of maturity,[6] and was active in a mouse xenograft model at low doses in combination with flavopiridol.[7] Whereas phthalimidobarbituric acids of type II failed to act as antiangiogenic agents, the fluorinated analogues III showed a strong decrease in microvessel outgrowth in rat aortic ring explants.[8,9] The intact fused five-membered ring was not essential, as the activity was retained in case of the tetrafluorobenzamide derivatives IV.[8,9]
Figure 1.

Structures of (fluorinated) phthalimide and benzamide derivatives as antiangiogenic agents.
Covalently bound fluorine has attracted much attention in drug development, although it is an unusual element in natural products. The substitution of hydrogen by fluorine in bioactive molecules produces only minor changes in the 3D structure due to the similar van der Waals radii of fluorine and hydrogen. Strong dipole moments of the carbon–fluorine bond result from the large electronegativity of fluorine and may contribute to interand intramolecular interactions.[10]
The property of covalently bound fluorine in organic molecules as a hydrogen-bond acceptor has been a controversial issue. Meanwhile, a number of spectroscopic, crystallographic and computational studies provided several lines of evidence for weak C–F⋯H–X hydrogen bonds. Hydrogen bonds to fluo-rine have the potential to effectively alter the binding events of fluorinated compounds to biological target structures.[11] In addition to an enhanced target affinity, increased metabolic stability can be attained; in particular, labile sites of metabolic oxidation can be blocked by fluorination.[10]
The high electronegativity of fluorine and the resulting polarization of the C–F bond lead to a remarkable alteration of the physicochemical properties, such as acidity or basicity of adjacent functional groups. Moreover, the addition of fluorine or trifluoromethyl groups on aryl or heteroaryl groups can increase the lipophilicity and enhance the membrane permeability in comparison with the non-fluorinated parent compounds.[10]
In the course of the determination of favorable (fluorophilic) and unfavorable (fluorophobic) environments within target proteins, inhibitors of serine and metalloproteases,[12] cysteine proteases,[13] and protein kinases[14] have been established.
In this study, it was intended to incorporate several simplified amino components into tetrafluorophthalimides III and tetrafluorobenzamides IV and to extend the series of test compounds by two types of related polysubstituted fluorobenzene derivatives decorated with the same amino components. The resulting compounds were investigated on the basis of spectroscopic and crystallographic data. Finally, the entire library of fluoro compounds was subjected to the examination of antiangiogenic properties.
Results and Discussion
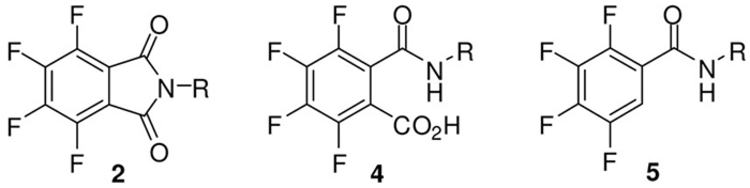
Six representatives of the chemotype of tetrafluorophthalimides (2a–f) were synthesized by condensation of tetrafluorophthalic anhydride (1) with different primary amines. The ring-opening/recyclization reactions were performed in AcOH at 1188C (Scheme 1). Under different conditions, when treating 1 with primary amines in toluene at reflux, the corresponding ammonium tetrafluorophthalamates (3a–f) were obtained. In the absence of AcOH, the recyclization step is prevented because the products accumulate as salts without carbonyl reactivity and precipitate. Tetrafluorophthalamic acids 4a–f were easily accessible by protonation of 3 in an aqueous medium. Chemotypes 3 and 4 constitute intermediate structures of a previously reported tandem ring opening/decarboxylation sequence.[9] In the present study, we have isolated and characterized these compounds and provided those of type 4 to a biological evaluation.
Scheme 1.

Synthesis of polyfluorinated compounds 2–7: i) RNH2, AcOH, reflux, 3 h; ii) RNH2, dry toluene, reflux, 3 h; iii) H2O, pH 1–2, 4°C; iv) 1. dry DMSO, 153°C, 4 h, 2. H2O, RT, 3 days; v) Δ, 4 h.
Thermal decarboxylation of tetrafluorophthalamic acids 4 was achieved in DMSO at 1538C to successfully produce 5a–f. These tetrafluorobenzamides represent structurally simplified analogues of the aforementioned antiangiogenic barbituric acids IV (Figure 1).
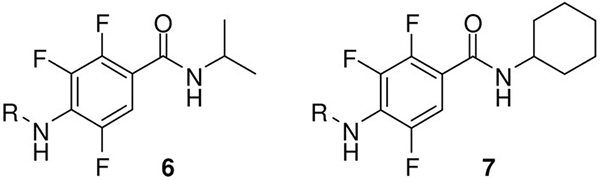
To introduce a further point of diversity, we considered to subject tetrafluoro derivatives to a nucleophilic substitution with amines. The reaction of certain phthalimides of type 2 with primary amines led to mixtures of 4- and 5-substituted regioisomers, which influenced cellular vesicular systems, in particular the generation of lipids droplets.[15] Mixtures of even four isomeric products might be formed from tetrafluoro-phthalamic acids 4 and amines. However, the transformation of tetrafluorobenzamides 5 is expected to proceed regioselectively. A corresponding conversion has been reported, even though in rare cases.[16]
We selected two tetrafluorobenzamides (5b and 5d) for the reaction with primary amines, the isopropyl derivative 5b because of its promising antiangiogenic activity (Table 1) and 5d because the bulky cyclohexyl moiety was expected to be advantageous. The nucleophilic substitution occurred unsurprisingly at the para position. With the so substituted products 6a–d and 7a–e, we received nine highly functionalized benzene derivatives. This transformation could also be accomplished under microwave assisted conditions with similar yields, as exemplified for 7c and 7d.
Table 1.
Inhibitory effects of tetrafluoro derivatives 2, 4, and 5 in the rat aortic ring assay of angiogenesis.
 | ||||
|---|---|---|---|---|
| Substitution pattern | R | Inhibition [%][a] | ||
| 2 | 4 | 5 | ||
| a | nPr | 59 | 55 | n.i.[b] |
| b | iPr | 81 | n.i. | 58 |
| c | iBu | 67 | 50 | 31 |
| d | cHex | 59 | 30 | 35 |
| e | Bn | 10 | 50 | 31 |
| f | tBu | n.d.[c] | 46 | 44 |
| Positive controls[d] | Inhibition [%][a] | |||
| CAI[e] | TNP-470 | III[f] | IV[f] | |
| R1 = Et, R2 = iPr | ||||
| 73 | 47 | >95 | 90 | |
Compounds were administered as a single dose at a concentration of 50 mm and incubated for 5 days. Outgrowth was compared with microvessel outgrowth from rings treated with the control (0.5% DMSO). Control outgrowth is classed at 100% outgrowth.
No inhibitory activity.
Not determined.
Incubation was conducted for 4 days.
A concentration of 30 μM was used.
Data from ref. [9].
The 13C NMR spectra of tetrafluorophthalimides 2 exhibited three distinct shifts of the carbon resonances of the fused benzene ring, C3a/7a, C4/7, and C5/6. Our assignments are in agreement with previous work[9] and a detailed NMR investigation including N-phenyl-tetrafluorophthalimide.[17] The magni-tude of the C–F coupling of carbons C3a/7a varied from 7 to 10 Hz, although larger constants could be assumed for twobond fluorine couplings, for example, as described for C3a/7a in N-phenyl-tetrafluorophthalimide (2J(C,F)=13.5 Hz).[17] However, smaller values (2J(C,F) ≈7 Hz) have been reported for four derivatives of N-phenyl-tetrafluorophthalimide.[18] The 2J(C,F) coupling constants of 4-fluoroisoindoline (8, Figure 2) of ≈%19 Hz for both C3a and C5 indicate that carbons which are engaged in ring-fusion do not necessarily produce small 2J(C,F) constants. Thus, the fixed orientation of the carbonyl groups relative to the C–F bonds at positions 4 and 7 attributes to the small coupling constants in case of 2a–f.
Figure 2.

Reference compounds.
The C1 and C6 signals of ammonium tetrafluorophthalamates 3 derived from 2J(C,F) coupling constants of either around 22 Hz or 15 Hz could initially not be distinguished. In order to assign the chemical shifts and the corresponding coupling constants to carbons C1 and C6, we have recorded a 13C NMR spectrum of 2-fluorobenzamide (9, Figure 2).[19] An X-ray crystal structure of 9 revealed the orientation of the NH2 moiety to the fluorine substituent, while in other ortho-halo-benzamides the oxygen pointed in the direction of the halogen.[20] As in 9, a preferred intramolecular F⋯H–N interaction was also postulated for our compounds 3. We have now considered the 2J(C1,F) coupling constant in 9 of 14.2 Hz as a reference for the 2J(C6,F) coupling constants in compounds 3. Accordingly, we could assign the 13C NMR signals with 2J(C,F) values of around 15 Hz to C6 (δ=120 ppm) and those with 22 Hz to C1 (δ=128 ppm). In 3-fluorobenzamide (10, Figure 2), the 2J(C2,F) coupling constant of 22.6 Hz was determined. Thus, the magnitude of the heteronuclear spin-spin coupling depended not only on the distance between the carbon and fluorine nuclei, but also on the orientation of their orbitals and intramolecular interactions.
Tetrafluorophthalamic acids 4 exhibited 2J(C,F) values for the heteronuclear coupling of C6 but also of C1 were between 13 and 19 Hz, indicating a different pattern of intramolecular interactions in comparison with the corresponding salts 3. The assignment of the 13C NMR signals was based on the chemical shifts and in relation to compounds 3 and 5 (see below); hence C6 resonates at 123 ppm, C1 at 118 ppm. A single crystal X-ray analysis of 4a as a representative compound was carried out (Figure 3). As in case of 2-fluorobenzamide (9), the NH is oriented toward the 5-fluorine substituent to form an intramolecular H⋯F hydrogen bond. It produces, together with an intermolecular contact to a carbonyl oxygen, a bifurcated hydrogen bond network and accounts for a dihedral angle of 37 between the tetrafluorinated benzene ring and the plane through the carboxamide moiety. The carboxylate plane is twisted relative to the aromatic plane by 70. A sandwich π–π stacking is created with alternating orientation of the substituents and a 3.7 Å centroid-distance of the benzene rings.
Figure 3.

Left: Molecular plot of 2,3,4,5-tetrafluoro-6-(propylcarbamoyl)benzoic acid (4a) showing the displacement ellipsoids at the 50% probability level for the non-H atoms. Right: Packing diagram of 4a. The N–H⋯F and N–H⋯O contacts are shown as broken lines.[21]
The regioselectivity of the nucleophilic substitution was confirmed by the chemical shifts in the 13C NMR spectra. Compounds 6 and 7 exhibited 2J(C1,F) values of ≈13 Hz, similar to the coupling constants of the carboxamide-substituted aromatic carbons in 3 and 4. The unsubstituted aromatic carbons of 5, 6 and 7 showed 2J(C6,F) constants in the usual range of 20–23 Hz. An X-ray refraction of 7e (Figure 4) revealed a sandwich π–π stacking (4.5 Å centroid-distance) and that the residues are pointing in the same direction. The amidic and every other anilinic NH moiety make intramolecular N–H⋯F bonds. Due to further intermolecular N–H⋯O contact, bifurcated Hbotif is formed. The geometrical analysis demonstrated a chair conformation of the cyclohexane ring.
Figure 4.

Top: Molecular plot of 4-(benzylamino)-N-cyclohexyl-2,3,5-trifluorobenzamide (7e) showing the displacement ellipsoids at the 50% probability level for the non-H atoms. Bottom: Packing diagram of 7e. The N–H⋯F and N–H⋯O contacts are shown as broken lines.[21]
To determine the antiangiogenic activity of the synthesized compounds, the rat aortic ring assay was performed and the compounds were administered at a concentration of 50 μm (Table 1). 5-Amino-1-{[3,5-dichloro-4-(4-chlorobenzoyl)phenyl]methyl}−1H-1,2,3-triazole-4-carboxamide (CAI), a synthetic, cytostatic inhibitor of non-voltage-operated calcium channels and calcium channel-mediated signaling pathways, as well as O-chloroacetyl-carbamoyl fumagillol (TNP-470), a methionine aminopeptidase-2 and neovascularization inhibitor, were used as positive controls. Moreover, data of two particularly potent representatives of tetrafluorophthalimido and benzamido barbituric acids (types III and IV) were included in Table 1.
In comparison with the DMSO-treated ring explants, most of the test compounds of chemotypes 2, 4, and 5 showed moderate inhibitory activities, with the N-isopropyl-tetrafluorophthalimide 2b as the most potent derivative. Thus, the isopropyl residue was advantageous for antiangiogenic activity. However, the strong activity of tetrafluorophthalimides with a barbituric acid moiety as the N-substituent, as reported previously,[9] was not reached with compound 2b. The corresponding derivatives of chemotypes 4 and 5 bearing the isopropyl moiety, that is, 4b and 5b, showed lower activity than 2b. The antiangiogenic potential of tetrafluorobenzamides has already been elucidated,[9] and was confirmed for chemotype 5 in this study. Tetrafluorophthalamic acids have first been shown as antiangiogenic agents herein. The exchange of hydrogen by a carboxyl group did not have a strong impact on angiogenesis inhibition, as can be seen by a comparison of representatives 5a–f with the analogues 4a–f. Chemotypes 4 and 5 are considered to be more appropriate for further structural optimization because these ring-opened derivatives are more conformationally flexible than phthalimides of type 2. Moreover, phthalimides 2 are inferior in stability because of the easy opening of the five-membered ring.
We selected two tetrafluorobenzamides, 5b and 5d, and introduced a further point of diversity, that is, an amino substituent at position 4. The antiangiogenesis data in response to trifluorobenzamides 6 and 7 at an inhibitor concentration of 50 μm are presented in Table 2. Among the N-isopropylcarboxamides 6, the cyclohexylamino derivative 6d exhibited an improved activity (80% inhibition), when compared with its precursor 5b (58% inhibition). In case of 5d (35% inhibition), the exchange of fluorine by all four different amino groups led to slightly improved inhibitors 7 (>50% inhibition). Thus, trifluorobenzamides have been identified as a new chemotype of antiangiogenetic agents.
Table 2.
Inhibitory effects of trifluoro derivatives 6 and 7 in the rat aortic ring assay of angiogenesis.
 | |||
|---|---|---|---|
| Substitution pattern | R | Inhibition [%][a] | |
| 6 | 7 | ||
| a | nPr | 51 | 54 |
| b | iPr | 32 | 67 |
| c | iBu | 52 | 51 |
| d | cHex | 80 | 54 |
| e | Bn | n.a.[b] | n.a.[c] |
Compounds were administered as a single dose at a concentration of 50 μM and incubated for 5 days. Outgrowth was compared with micro-vessel outgrowth from rings treated with the control (0.5% DMSO). Control outgrowth is classed at 100% outgrowth.
Not applicable.
No inhibitory activity.
The antiangiogenic activity of the two best compounds in the rat aortic ring model was complemented by a preliminary in vitro human umbilical vein endothelial cell (HUVEC) cytotoxicity assay, in which 2b and 6d, at a concentration of 10 mm, inhibited the survival of HUVECs 74% and 78%, respectively.
Conclusions
In this study, different molecular entities of fluoroaromatic compounds were synthesized and subjected to detailed spectroscopic and crystallographic analyses. Such compounds are potentially bioactive.
We have determined the antiangiogenic activity of tetrafluorophthalimides with ordinary N-alkyl residues. Although these compounds 2 were less potent than tetrafluorophthalimides with a barbituric acid moiety as N-substituent, they provide a starting point for tailored structural modifications and a comparative analysis of the resulting chemotypes. With respect to antiangiogenic activity, ring opening as realized in 4–7 was tolerated. In particular, we found that one fluorine substituent can be replaced by an amino group leading to 6/7 without an essential loss of bioactivity. Thus, the repertoire of structural frameworks to develop angiogenesis inhibitors was expanded. Further biological studies are ongoing to understand the clinical utility of selected compounds from this cohort.
Future structural modifications are intended as follows. The carboxamide moiety of 6/7 can be revised and re-equipped with a barbituric acid unit. The scopes and limitations of the aromatic amino substitution need to be further explored. It can also be envisaged to use this substitution for the introduction of linkers which bear a second functionality. Thus, our study might pave the way for the design of bifunctional molecules with fluorescent or biotin labels as tool compounds for studying angiogenesis.
Experimental Section
Melting points were determined on a Gallenkamp capillary melting point apparatus or on a Rapido Boetius apparatus, and are uncorrected. The purity of the obtained substances and the composition of the reaction mixtures were controlled by thin-layer chromatography (TLC) using Merck aluminum silica gel plates with 60 F254 indicator. Preparative column chromatography was performed using Merck silica gel 60 (63–200 mesh). Petroleum ether used was a mixture of alkanes boiling between 40–60°C, according to the supplier’s declaration. NMR spectra were recorded on a Bruker Avance DRX 500 spectrometer at 500 MHz (1H NMR) and 125 MHz (13C NMR) in [D6]DMSO. Chemical shifts (δ) are reported in ppm. Spin multiplicities are indicated by the following symbols: s (singlet), br s (broad singlet), d (doublet), t (triplet), q (quartet), sext (sextet), sept (septet), oct (octet), non (nonet), m (multiplet). All multiplets related with J(C, F) couplings in 13C NMR spectra are centered. Mass spectra were recorded on an MS-50 A.E.I. spectrometer under electron impact ionization (EI) at 70 eV. Elemental analyses were performed with a Vario EL apparatus.
Chemistry
Synthesis of tetrafluorophthalimides 2a–f: General Procedure. A mixture of the primary amine (1.5 mmol) and tetrafluorophthalic anhydride (1, 0.40 g, 1.8 mmol) in glacial AcOH (11 mL) was stirred under reflux for 3 h. The resulting solution was then evaporated to dryness under reduced pressure. The oily residue was recrystallized from EtOH.
4,5,6,7-Tetrafluoro-2-propylisoindoline-1,3-dione (2a) was prepared from 1 and propylamine (89 mg, 0.12 mL); yield 49%(0.19 g); mp: 106–108°C; 1H NMR ([D6]DMSO): δ=0.86 (t, J=7.4 Hz, 3H, CH3), 1.59 (sext, J=7.3 Hz, 2H, CH2CH3) 3.50 (t, J=7.1 Hz, 2H, NCH2); 13C NMR ([D6]DMSO): δ=11.22 (CH3), 21.12 (CH2CH3), 39.91 (NCH2), 114.30 (m, 3J(C,F)=9.4 Hz, C3a, C7a), 142.50 (m, 1J(C,F)=264.2 Hz, C4, C7), 144.09 (m, 1J(C,F)=260.9 Hz, C5, C6), 162.86 (CO). Elemental analysis calcd (%) for C11H7F4NO2: C 50.59, H 2.70, N 5.36, found: C 50.65, H 2.61, N 5.40.
4,5,6,7-Tetrafluoro-2-isopropylisoindoline-1,3-dione (2b)[22] was prepared from 1 and isopropylamine (89 mg, 0.13 mL); yield 51% (0.20 g); mp: 148–151°C; 1H NMR ([D6]DMSO): δ=1.38 (d, J=7.0 Hz, 6H, CH3), 4.34 (sept, J=6.9 Hz, 1H, CH);13C NMR ([D6]DMSO): δ=19.61 (CH3), 43.38 (CH), 114.09 (m, 3J(C,F)=9.9 Hz, C3a, C7a), 142.46 (m, 1J(C,F)=264.1 Hz, C4, C7), 144.02 (m, 1J(C,F)= 258.7 Hz, C5, C6), 162.62 (CO). Elemental analysis calcd (%) for C11H7F4NO2: C 50.59, H 2.70, N 5.36, found: C 50.59, H 2.90, N 5.36.
4,5,6,7-Tetrafluoro-2-isobutylisoindoline-1,3-dione (2c) was prepared from 1 and isobutylamine (110 mg, 0.15 mL); yield 99%(0.41 g); mp: 136–138°C; 1H NMR ([D6]DMSO): δ=0.87 (d, J=6.7 Hz, 6H, CH3), 1.91–2.02 (m, 1H, CH), 3.35 (d, J=7.3 Hz, 2H, CH2); 13C NMR ([D6]DMSO): δ=20.02 (CH3), 27.40 (CH), 45.57 (CH2), 114.11 (m, 3J(C,F)=7.4 Hz, C3a, C7a), 142.55 (m, 1J(C,F)=263.8 Hz, C4, C7), 144.16 (m, 1J(C,F)=262.5 Hz, C5, C6), 163.06 (CO). Elemental analysis calcd (%) for C12H9F4NO2: C 52.37, H 3.30, N 5.09, found: C 52.63, H 3.21, N 5.09.
4,5,6,7-Tetrafluoro-2-cyclohexylisoindoline-1,3-dione (2d)[22] was prepared from 1 and cyclohexylamine (150 mg, 0.17 mL); yield 80% (0.36 g); mp: 155–158°C; 1H NMR ([D6]DMSO): δ=1.10–2.04 (m, 10H, CH2), 3.93 (tt, J=12.5, 3.9 Hz, 1H, CH); 13C NMR ([D6]DMSO): δ=24.94 (C4′), 25.48 (C3′, C5′), 29.22 (C2′, C6′), 51.12 (C1′), 113.98 (m, 3J(C,F)=7.7 Hz, C3a, C7a), 142.49 (m, 1J(C,F)= 264.4 Hz, C4, C7), 144.06 (m, 1J(C,F)=262.5 Hz, C5, C6), 162.64 (CO). Elemental analysis calcd (%) for C14H11F4NO2: C 55.82, H 3.68, N 4.65, found: C 55.58, H 3.76, N 4.72.
4,5,6,7-Tetrafluoro-2-benzylisoindoline-1,3-dione (2e) was prepared from 1 and benzylamine (161 mg, 0.16 mL); yield 93%(0.43 g); mp: 177–179°C; lit; mp: 179–181°C;[3] 1H NMR ([D6]DMSO): δ=4.74 (s, 2H, CH2), 7.26–7.35 (m, 5H, Ar-H); 13C NMR ([D6]DMSO): δ=41.65 (CH2), 114.19 (m, 3J(C,F)=8.3 Hz, C3a, C7a), 127.73, 128.75 (C2′, C6′, C3′, C5′), 127.81 (C4′), 135.92 (C1′), 142.70 (m, 1J(C,F)=267.0 Hz, C4, C7), 144.27 (m, 1J(C,F)=263.6 Hz, C5, C6), 162.69 (CO). Elemental analysis calcd (%) for C15H7F4NO2: C 58.26, H 2.28, N 4.53, found: C 58.19, H 2.32, N 4.56.
4,5,6,7-Tetrafluoro-2-tert-butylisoindoline-1,3-dione (2 f) was prepared from 1 and tert-butylamine (110 mg, 0.16 mL); yield 11% (45 mg); mp: 174–176°C; 1H NMR ([D6]DMSO): δ=1.59 (s, 9H, CH3); 13C NMR ([D6]DMSO): δ=28.41 (CH3), 59.47 (C(CH3)3), 114.01 (m,3J(C,F)=7.2 Hz, C3a, C7a), 142.36 (m, 1J(C,F)=264.9 Hz, C4, C7), 144.06 (m, 1J(C,F)=262.5 Hz, C5, C6), 163.49 (CO). Elemental analysis calcd (%) for C12H9F4NO2: C 52.37, H 3.30, N 5.09, found: C 52.36, H 3.12, N 5.04.
Synthesis of ammonium tetrafluorophthalamates 3a–f: General Procedure. A mixture of the primary amine (15 mmol) and tetrafluorophthalic anhydride (1, 1.65 g, 7.5 mmol) in dry toluene (500 mL) was stirred under reflux for 3 h. The mixture was allowed to cool down to room temperature, the resulting solid was filtered off, washed with n-hexanes (2×20 mL) and dried under reduced pressure.
Propylammonium 2,3,4,5-tetrafluoro-6-(propylcarbamoyl)benzoate (3a) was prepared from 1 and propylamine (0.89 g, 1.24 mL); yield 65% (1.65 g); mp: 160–164°C; 1H NMR ([D6]DMSO): δ=0.87 (t, J=7.6 Hz, 3H, CH3), 0.87 (t, 7.3 Hz, 3H, CH3), 1.46 (sext, J=7.3 Hz, 2H, CH2CH3), 1.53 (sext, J=7.6 Hz, 2H, CH2CH3), 2.69 (t, J=7.6 Hz, 2H, NCH2), 3.08–3.12 (m, 2H, NCH2), 8.05 (brs, 3H, NH3),8.36 (t, J=5.7 Hz, 1H, NH); 13C NMR ([D6]DMSO): δ=11.01 (CH3), 11.48 (CH3), 20.62 (CH2CH3), 22.19 (CH2CH3), 40.53 (NCH2), 41.02 (NCH2), 119.69 (m, 2J(C,F)=15.4 Hz, C6), 127.96 (m, 2J(C,F)=21.6 Hz,C1), 137.75 (m 1J(C,F)=239.6 Hz), 139.67 (m, 1J(C,F)=241.6 Hz), 142.57 (m, 1J(C,F)=241.1 Hz), 143.60 (m, 1J(C,F)=245.6 Hz, C2, C3, C4, C5), 161.51 (CO), 163.11 (CO). Elemental analysis calcd (%) for C14H18F4N2O3·0.25H2O: C 49.05, H 5.44, N 8.17, found: C 48.77, H 5.89, N 8.20.
Isopropylammonium 2,3,4,5-tetrafluoro-6-(isopropylcarbamoyl)-benzoate (3b) was prepared from 1 and isopropylamine (0.89 g, 1.29 mL); yield 88% (2.24 g); mp: 178–180°C; 1H NMR ([D6]DMSO): δ=0.87 (d, J=6.7 Hz, 6H, CH3), 1.16 (d, J=6.6 Hz, 6H, CH3), 3.25 (sept, J=6.6 Hz, 1H, CH), 3.87–3.97 (m, 1H, CH); 8.03 (s, 3H, NH3), 8.24 (d, J=7.6 Hz, 1H, NH); 13C NMR ([D6]DMSO): δ=20.56 (CH3),22.16 (CH3), 41.21 (CH), 42.96 (CH), 119.77 (m, 2J(C,F)=15.9 Hz, C6), 127.43 (m, 2J(C,F)=21.3 Hz, C1), 137.87 (m, 1J(C,F)=229.2 Hz), 139.73 (m, 1J(C,F)=233.6 Hz), 142.68 (m, 1J(C,F)=242.9 Hz), 143.66 (m, 1J(C,F)=244.5 Hz, C2, C3, C4, C5), 160.59 (CO), 163.06 (CO). Elemental analysis calcd (%) for C14H18F4N2O3·0.1H2O: C 49.44, H 5.39, N 8.24, found: C 49.20, H 5.78, N 8.27.
Isobutylammonium 2,3,4,5-tetrafluoro-6-(isobutylcarbamoyl)-benzoate (3c) was prepared from 1 and isobutylamine (1.10 g, 1.50 mL); yield 33% (0.92 g); mp: 168–170°C; 1H NMR ([D6]DMSO): δ=0.87 (d, J=6.6 Hz, 6H, CH3), 0.89 (d, J=6.7 Hz, 6H, CH3), 1.71–1.87 (m, 2H, 2×CH), 2.58 (d, J=7.0 Hz, 2H, CH2), 2.98 (t, J=6.0 Hz, 2H, CH2), 8.00 (brs, 3H, NH3), 8.37 (t, J=6.0 Hz, 1H, NH); 13C NMR ([D6]DMSO): δ=19.87 (CH3), 20.27 (CH3), 26.59 (CH), 28.06 (CH),45.87 (CH2), 46.78 (CH2), 119.53 (m, 2J(C,F)=15.1 Hz, C6), 128.12 (m,2J(C,F)=22.1 Hz, C1), 137.69 (m 1J(C,F)=242.5 Hz), 139.64 (m,1J(C,F)=244.1 Hz), 142.48 (m, 1J(C,F)=240.6 Hz), 143.69 (m,1J(C,F)=247.4 Hz, C2, C3, C4, C5), 161.53 (CO), 163.00 (CO). Elemental analysis calcd (%) for C16H22F4N2O3·H2O: C 50.00, H 6.29, N 7.29, found: C 49.82, H 5.98, N 7.30.
Cyclohexylammonium 2,3,4,5-tetrafluoro-6-(cyclohexylcarbamoyl)benzoate (3d) was prepared from 1 and cyclohexylamine (1.49 g, 1.72 mL); yield 91% (2.86 g); mp: 212–214°C; 1H NMR ([D6]DMSO): δ=1.07–1.30 (m, 10H, CH2), 1.51–189 (m, 10H, CH2),2.89–2.93 (m, 1H, CH), 3.59–3.66 (m, 1H, CH), 8.01 (3H, NH3), 8.25 (d, J=7.9 Hz, 1H, NH); 13C NMR ([D6]DMSO): δ=23.96 (CH2), 24.44 (CH2), 24.72 (CH2), 25.37 (CH2), 30.55 (CH2), 32.05 (CH2), 48.15 (CH),49.36 (CH), 119.30 (m, 2J(C,F)=13.9 Hz, C6), 128.31 (m, 2J(C,F)=22.8 Hz, C1), 137.66 (m 1J(C,F)=241.3 Hz), 139.62 (m, 1J(C,F)= 249.1 Hz), 142.41 (m, 1J(C,F)=240.4 Hz), 143.78 (m, 1J(C,F)= 245.5 Hz, C2, C3, C4, C5), 160.65 (CO), 162.88 (CO). Elemental analysis calcd (%) for C20H26F4N2O3: C 57.41, H 6.26, N 6.69, found: C57.58, H 6.41, N 6.71.
Benzylammonium 2,3,4,5-tetrafluoro-6-(benzylcarbamoyl)benzoate (3e) was prepared from 1 and benzylamine (1.61 g, 1.64 mL); yield 61% (1.98 g); mp: 181–183°C; 1H NMR ([D6]DMSO): δ=3.98 (s, 2H, CH2), 4.40 (d, J=5.9 Hz, 2H, CH2), 7.20–7.45 (m, 10H, 2×Ar-H), 8.34 (brs, 3H, NH3), 8.94 (t, J=5.9 Hz, 1H, NH); 13C NMR ([D6]DMSO): δ=42.49 (CH2), 42.71 (CH2), 119.49 (m,2J(C,F)=15.4 Hz, C6), 126.77 (C-Ar), 127.26 (C-Ar), 128.26 (C-Ar), 128.31 (C-Ar), 128.63 (C-Ar), 128.87 (C-Ar), 134.91 (C-Ar), 139.00 (C-Ar), 137.76 (m 1J(C,F)=251.4 Hz), 139.75 (m, 1J(C,F)=245.4 Hz), 142.70 (m, 1J(C,F)=241.1 Hz), 143.68 (m, 1J(C,F)=245.5 Hz, C2, C3, C4, C5), 161.91 (CO), 163.00 (CO). One carbon signal (C1) was not observed. Elemental analysis calcd (%) for C22H18F4N2O3: C 58.41, H 4.46, N 6.19, found: C 58.47, H 4.60, N 6.19.
tert-Butylammonium 2,3,4,5-tetrafluoro-6-(tert-butylcarbamoyl)-benzoate (3 f) was prepared from 1 and tertbutylamine (1.10 g, 1.58 mL); yield 81% (2.22 g); mp: 178–180°C; 1H NMR ([D6]DMSO): δ=1.24 (s, 9H, CH3), 1.29 (s, 9H, CH3), 7.96 (s, 1H, NH), 8.10 (brs, 3H, NH3); 13C NMR ([D6]DMSO): δ=27.34 (CH3), 28.42 (CH3), 50.80 (C(CH3)3), 51.00 (C(CH3)3), 119.91 (m, 2J(C,F)=14.6 Hz, C6), 127.97 (m, 2J(C,F)=22.1 Hz, C1), 137.67 (m 1J(C,F)=236.5 Hz), 139.57 (m,1J(C,F)=239.3 Hz), 142.39 (m, 1J(C,F)=241.4 Hz), 143.82 (m,1J(C,F)=247.0 Hz, C2, C3, C4, C5), 160.94 (CO), 163.18 (CO). Elemental analysis calcd (%) for C16H22F4N2O3: C 52.46, H 6.05, N 7.65, found C 52.03, H 6.80, N 7.17.
Synthesis of tetrafluorophthalamic acids 4a–f: General Procedure. A solution of the ammonium tetrafluorophthalamate (5 mmol) in H2O was acidified with HCl (2M) to pH 1–2 and kept overnight at 4°C. The colorless precipitate was filtered off, washed with H2O and dried under reduced pressure.
2,3,4,5-Tetrafluoro-6-(propylcarbamoyl)benzoic acid (4a) was prepared from 3a (1.69 g) in H2O (30 mL); yield 73% (1.02 g); mp: 132–134°C; 1H NMR ([D6]DMSO): δ=0.89 (t, J=7.3 Hz, 3H, CH3), 1.48 (sext, J=7.3 Hz, 2H, CH2CH3), 3.14–3.18 (m, 2H, NCH2), 8.66 (t, J=5.5 Hz, 1H, NH), 14.02 (brs, 1H, CO H); 132 C NMR ([D6]DMSO): δ= 11.42 (CH3), 22.08 (CH2CH3), 41.19 (NCH2), 117.86 (m,2J(C,F)=13.6 Hz, C1), 122.27 (m, 2J(C,F)=15.4 Hz, C6), 140.24 (m 1J(C,F)= 252.0 Hz), 140.78 (m, 1J(C,F)=254.4 Hz), 143.63 (m, 1J(C,F)= 245.0 Hz), 144.88 (m, 1J(C,F)=250.7 Hz, C2, C3, C4, C5), 160.10 (CO), 162.63 (CO). MS (EI) (m/z, %) 279 (M+, 13), 235 (M+–CO2, 25), 221 (M+–C3H8N, 61), 177 (M+–C3H8N–CO2, 100), 149 (M+–C3H8N CO2 CO, 25). Elemental analysis calcd (%) for C11H9F4NO3: C 47.32, H 3.25, N 5.02, found: C 47.18, H 3.38, N 5.13.
2,3,4,5-Tetrafluoro-6-(isopropylcarbamoyl)benzoic acid (4b) was prepared from 3b (1.69 g) in H2O (36 mL); yield 81% (1.13 g); mp: 144–146°C; 1H NMR ([D6]DMSO): δ=1.11 (d, J=6.6 Hz, 6H, CH3),3.92–4.02 (m, 1H, CH), 8.55 (d, J=7.9 Hz, 1H, NH), 14.00 (brs, 1H, CO H); 132 C NMR ([D6]DMSO): δ=22.04 (CH3), 41.53 (CH), 117.68 (m,2J(C,F)=14.6 Hz, C1), 122.50 (m, 2J(C,F)=15.9 Hz, C6), 140.18 (m,1J(C,F)=251.4 Hz), 140.80 (m, 1J(C,F)=255.2 Hz), 143.59 (m,1J(C,F)=244.6 Hz), 144.93 (m, 1J(C,F)=251.4 Hz, C2, C3, C4, C5), 159.19 (CO), 162.60 (CO). MS (EI) (m/z, %) 279 (M+, 6), 235 (M+–CO2, 19), 221 (M+–C3H8N, 38), 177 (M+–C3H8N–CO2, 100), 149 (M+–C3H8N–CO2–CO, 25). Elemental analysis calcd (%) for C11H9F4NO3·0.1H2O: C 47.02, H 3.30, N 4.98, found: C 46.72, H 3.70, N 5.19.
2,3,4,5-Tetrafluoro-6-(isobutylcarbamoyl)benzoic acid (4c) was prepared from 3c (1.83 g) in H2O (100 mL); yield 58% (0.85 g); mp: 148–150°8C; 1H NMR ([D6]DMSO): δ=0.88 (d, J=6.7 Hz, 6H, CH3),1.74–1.82 (m, 1H, CH), 3.02–3.04 (m, 2H, CH2), 8.67 (t, J=5.8 Hz, 1H, NH), 14.04 (brs, 1H, CO2 H); 13 C NMR ([D6]DMSO): δ=20.18 (CH3), 28.00 (CH), 46.94 (CH2), 117.90 (m,2J(C,F)=14.6 Hz, C1), 122.27 (m, 2J(C,F)=19.1 Hz, C6), 140.21 (m 1J(C,F)=249.7 Hz), 140.74 (m, 1J(C,F)=253.4 Hz), 143.66 (m, 1J(C,F)=244.2 Hz), 144.82 (m, 1J(C,F)=250.6 Hz, C2, C3, C4, C5), 160.15 (CO), 162.65 (CO). MS (EI) (m/z, %) 293 (M+, 3), 249 (M+–CO2, 8), 221 (M+–C4H10N, 36),177 (M+–C4H10N–CO2, 100), 149 (M+–C4H10N–CO2 – CO, 18). Elemental analysis calcd (%) for C12H11F4NO3 ·0.1H2O: C 48.86, H 3.83, N 4.75, found: C 48.62, H 4.11, N 4.96.
2,3,4,5-Tetrafluoro-6-(cyclohexylcarbamoyl)benzoic acid (4d)[23]was prepared from 3d (2.09 g) in H2O (700 mL); yield 85% (1.36 g); mp: 174–178°C; 1H NMR ([D6]DMSO): δ=1.11–1.81 (m, 10H, CH2),3.64–3.71 (m, 1H, CH), 8.54 (d, J=7.9 Hz, 1H, NH), 14.00 (brs, 1H, CO2H);13C NMR ([D6]DMSO): δ=24.43 (CH2), 25.27 (CH2), 32.00 (CH2), 48.51 (CH), 117.61 (m,2J(C,F)=13.6 Hz, C1), 122.54 (m,2J(C,F)=18.6 Hz, C6), 140.15 (m 1J(C,F)=252.7 Hz), 140.79 (m,1J(C,F)=255.3 Hz), 143.58 (m, 1J(C,F)=245.2 Hz), 144.92 (m,1J(C,F)=251.3 Hz, C2, C3, C4, C5), 159.19 (CO), 162.54 (CO). MS (EI) (m/z, %) 319 (M+, 5), 275 (M+–CO2, 18), 221 (M+–C6H12N, 23), 177 (M+–C6H12N–CO2, 100), 149 (M+–C6H12N–CO2–CO, 21). Elemental analysis calcd (%) for C14H13F4 NO3:C 52.67, H 4.10, N 4.39, found: C52.56, H 4.15, N 4.43.
2,3,4,5-Tetrafluoro-6-(benzylcarbamoyl)benzoic acid (4e) was prepared from 3e (2.17 g) in H2O (450 mL); yield 68% (1.11 g); mp: 153–156°C; 1H NMR ([D6]DMSO): δ=4.45 (d, J=5.9 Hz, 2H, CH2),7.24–7.35 (m, 5H, Ar-H), 9.21 (t, J=5.9 Hz, 1H, NH), 14.14 (brs, 1H, CO2H); 13C NMR ([D6]DMSO): δ=42.94 (CH2), 117.94 (m, 2J(C,F)=14.4 Hz, C1), 121.89 (m, 2J(C,F)=17.4 Hz, C6), 127.08 (C-Ar), 127.39 (C-Ar), 128.43 (C-Ar), 138.58 (C-Ar), 140.40 (m 1J(C,F)=255.3 Hz), 140.83 (m, 1J(C,F)=253.5 Hz), 143.82 (m, 1J(C,F)=245.7 Hz), 144.94 (m, 1J(C,F)=249.6 Hz, C2, C3, C4, C5), 160.40 (CO), 162.71 (CO). MS (EI) (m/z, %) 328 (M+H+, 9), 283 (M+–CO2, 100), 177 (M+–C7H8N–CO2–,95), 149 (M+–C7H8N–CO2–CO, 27). Elemental analysis (%) for C15H9F4NO3·0.7H2O: C 53.01, H 3.08, N 4.12, found: C 53.02, H 2.94, N 4.27.
2,3,4,5-Tetrafluoro-6-(tert-butylcarbamoyl)benzoic acid (4 f) was prepared from 3 f (1.83 g) in H2O (115 mL); yield 52% (0.76 g); mp: 148–151°C; 1H NMR ([D6]DMSO): δ=1.31 (s, 9H, CH3), 8.28 (s, 1H,NH), 14.02 (brs, 1H, CO2H); 13C NMR ([D6]DMSO): δ=28.35 (CH3),51.43 (C(CH3)3), 117.56 (m, 2J(C,F)=12.9 Hz, C1), 123.17 (m, 2J(C,F)=17.1 Hz, C6), 139.98 (m 1J(C,F)=251.8 Hz), 140.71 (m, 1J(C,F)= 255.2 Hz), 143.61 (m, 1J(C,F)=245.7 Hz), 144.83 (m, 1J(C,F)= 251.1 Hz, C2, C3, C4, C5), 159.44 (CO), 162.69 (CO). MS (EI) (m/z, %) 293 (M+, 10), 249 (M+–CO2, 8), 221 (M+–C4H10H, 44), 177(M+–C4H10N–CO2, 100), 149 (M+–C4H10N–CO2–CO, 21). Elemental analysis calcd (%) for C12H11F4NO3: C 49.15, H 3.78, N 4.78, found: C 48.83, H 4.06, N 4.98.
Synthesis of tetrafluorobenzamides 5a–f: General Procedure. A mixture of the tetrafluorophthalamic acid (1.5 mmol) and dry DMSO (1 mL) was heated at 153°C for 4 h. After cooling to room temperature, H2O was added (10 mL). The oil formed in the reaction mixture was removed and the aqueous solution was kept for 3 days at room temperature. The precipitate was filtered off, dried under reduced pressure and further purified as indicated.
2,3,4,5-Tetrafluoro-N-propylbenzamide (5a) was prepared from 4a (0.42 g) and purified by column chromatography using petroleum ether/EtOAc (4:1); yield 26% (92 mg); mp: 46–50°C; 1H NMR ([D6]DMSO): δ=0.89 (t, J=7.4 Hz, 3H, CH3), 1.48–1.55 (m, 2H,CH2CH3), 3.20 (q, J=6.5 Hz, 2H, NCH2), 7.53–7.59 (m, 1H, 6-H), 8.49 (brs, 1H, NH); 13C NMR ([D6]DMSO): δ=11.43 (CH3), 22.19 (CH2CH3), 41.25 (NCH2), 111.70 (m,2J(C,F)=20.3 Hz, C6), 121.20 (m, C1), 140.00 (m 1J(C,F)=247.6 Hz), 140.77 (m, 1J(C,F)=252.2 Hz), 144.69 (m, 1J(C,F)=246.7 Hz), 146.18 (m, 1J(C,F)=245.5 Hz, C2, C3, C4, C5), 160.89 (CO). MS (EI) (m/z, %) 235 (M+, 19), 220 (M+–CH3, 7), 206(M+–C2H5, 11), 177 (M+–C3H8N, 100), 149 (M+–C3H8N–CO, 14). Elemental analysis calcd (%) for C10H9F4NO: C 51.07, H 3.86, N 5.96, found: C 51.32, H 4.44, N 5.69.
2,3,4,5-Tetrafluoro-N-isopropylbenzamide (5b) was prepared from 4b (0.42 g) and recrystallized from n-hexanes; yield 43%(0.15 g); mp: 111–113°C; 1H NMR ([D6]DMSO): δ=1.14 (d, J=6.7 Hz, 6H, CH3), 3.97–4.06 (m, 1H, CH), 7.52–7.57 (m, 1H, 6-H),8.36 (d, J=6.3 Hz, 1H, NH); 13C NMR ([D6]DMSO): δ=22.18 (CH3),41.62 (CH), 111.68 (m, 2J(C,F)=20.1 Hz, C6), 121.51 (m, C1), 139.98 (m, 1J(C,F)=250.9 Hz), 140.68 (m, 1J(C,F)=252.0 Hz), 144.63 (m,1J(C,F)=247.1 Hz), 146.16 (m, 1J(C,F)=245.0 Hz, C2, C3, C4, C5), 160.12 (CO). MS (EI) (m/z, %) 235 (M+, 19), 220 (M+–CH3, 21), 177 (M+–C3H8N, 100). Elemental analysis calcd (%) for C10H9F4NO: C 51.07, H 3.86, N 5.96, found: C 50.81, H 4.23, N 5.97.
2,3,4,5-Tetrafluoro-N-isobutylbenzamide (5c) was prepared from 4c (0.44 g); yield 80% (0.30 g); mp: 88–90°C; 1H NMR ([D6]DMSO): δ=0.89 (d, J=6.6 Hz, 6H, CH3), 1.81 (non, J=6.7 Hz, 1H, CH), 3.07 (t, J=6.5 Hz, 2H, CH2), 7.53–7.58 (m, 1H, 6-H), 8.50 (brs 1H, NH);13C NMR ([D6]DMSO): δ=20.16 (CH3), 28.06 (CH), 46.89 (CH2), 111.67 (m, 2J(C,F)=20.3 Hz, C6), 121.36 (m, C1), 139.99 (m 1J(C,F)= 248.3 Hz), 140.74 (m, 1J(C,F)=252.2 Hz), 144.64 (m, 1J(C,F)= 247.8 Hz), 146.21 (m, 1J(C,F)=244.5 Hz, C2, C3, C4, C5), 161.02 (CO). MS (EI) (m/z, %) 249 (M+, 7), 234 (M+–CH3, 9), 177 (M+–C4H10N,100). Elemental analysis calcd (%) for C11H11F4 NO: C 53.02, H 4.45, N5.62, found: C 52.27, H 4.75, N 5.68.
2,3,4,5-Tetrafluoro-N-cyclohexylbenzamide (5d) was prepared from 4d (0.48 g) and recrystallized from n-hexanes; yield 56%(0.23 g); mp: 124–126°C; 1H NMR ([D6]DMSO): δ=1.09–1.82 (m, 10H, CH2), 3.67–3.74 (m, 1H, CH), 7.51–7.56 (m, 1H, 6-H), 8.35 (d,J=7.6 Hz, 1H, NH); 13C NMR ([D6]DMSO): δ=24.61 (CH2), 25.25 (CH2), 32.16 (CH2), 48.70 (CH), 111.68 (m,2J(C,F)=20.3 Hz, C6), 121.59 (m, C1), 139.98 (m 1J(C,F)=251.6 Hz), 140.66 (m, 1J(C,F)= 250.6 Hz), 144.62 (m, 1J(C,F)=248.6 Hz), 146.15 (m, 1J(C,F)= 244.0 Hz, C2, C3, C4, C5), 160.13 (CO). MS (EI) (m/z, %) 275 (M+,19), 232 (M+–C3H7, 18), 194 (M+–C6H9, 64), 177 (M+–C6H12N, 100). Elemental analysis calcd (%) for C13H13F4NO: C 56.73, H 4.76, N 5.09, found: C 57.10, H 4.96, N 5.21.
2,3,4,5-Tetrafluoro-N-benzylbenzamide (5e) was prepared from 4e (0.49 g) and recrystallized from n-hexanes; yield 47% (0.20 g); mp: 96–99°C; 1H NMR ([D6]DMSO): δ=4.46 (d, J=6.0 Hz, 2H, CH2),7.23–7.27 (m, 1H, 4′-H), 7.32–7.36 (m, 4H, 2′-H, 3′-H, 5′-H, 6′-H),7.60–7.66 (m, 1H, 6-H), 9.06 (t, J=5.1 Hz, 1H, NH); 13C NMR ([D6]DMSO): δ=43.00 (CH2), 111.84 (m, 2J(C,F)=20.6 Hz, C6), 120.76 (m, C1), 127.08 (C4′), 127.37, 128.48 (C2′, C6′, C3′, C5′), 138.78 (C1′), 140.08 (m 1J(C,F)=249.4 Hz), 140.96 (m, 1J(C,F)=252.5 Hz), 144.88 (m, 1J(C,F)=247.7 Hz), 146.23 (m, 1J(C,F)=244.4 Hz, C2, C3, C4, C5), 161.08 (CO). MS (EI) (m/z, %) 283 (M+, 100), 177 (M+–C7H8N, 80). Elemental analysis calcd (%) for C14H9F4NO: C 59.37, H 3.20, N 4.95, found: C 58.99, H 3.80, N 5.25.
2,3,4,5-Tetrafluoro-N-tert-butylbenzamide (5 f) was prepared from 4 f (0.44 g); yield 56% (0.21 g); mp: 62–65–151°C; 1H NMR ([D6]DMSO): δ=1.34 (s, 9H, CH3), 7.49–7.54 (m, 1H, 6-H), 8.06 (brs, 1H, NH); 13C NMR ([D6]DMSO): δ=28.45 (CH3), 51.51 (C(CH3)3), 111.62 (m, 2J(C,F)=20.1 Hz, C6), 122.44 (m, C1), 139.84 (m 1J(C,F)= 246.3 Hz), 140.46 (m, 1J(C,F)=251.7 Hz), 144.48 (m, 1J(C,F)= 246.5 Hz), 146.12 (m, 1J(C,F)=244.0 Hz, C2, C3, C4, C5), 160.10 (CO). MS (EI) (m/z, %) 234 (M+–CH3, 42), 177 (M+–C4H10N, 100). Elemental analysis calcd (%) for C11H11F4NO: C 53.02, H 4.45, N 5.62, found: C 52.61, H 4.73, N 5.51.
Synthesis of 4-amino-2,3,5-trifluorobenzamides 6–7. General Procedure A: A mixture of 1 mmol of the tetrafluorobenzamide 5b (0.24 g) or 5d (0.28 g) and the corresponding primary amine was heated for 4 h at an indicated oil bath temperature. Upon cooling and stirring, 2N HCl was added until a weak acidic pH was reached. A saturated aqueous solution of NaHCO3 was then added to adjust a pH value of 7. The isolation and purification of the product was carried out as indicated.
Synthesis of 4-amino-2,3,5-trifluorobenzamides 7c and 7d. General Procedure B: A mixture of 0.5 mmol of 2,3,4,5-tetrafluoro-N-cyclohexylbenzamide 5d (0.14 g) and the corresponding primary amine (1 mmol) in DMSO (3 mL) was reacted in a sealed microwave vessel for 10 min at 110°C in a CEM Discovery reactor (max.100 W). After addition of half-saturated brine (50 mL), it was extracted with ethyl acetate (2×25 mL), washed with 10% KHSO4 (50 mL) and brine (50 mL), dried (Na2SO4) and the filtrate was evaporated to dryness. The crude product was purified by column chromatography using a gradient of petroleum ether/ethyl acetate (19:1 to 10:1).
2,3,5-Trifluoro-N-isopropyl-4-(propylamino)benzamide (6a) was prepared from 5b and propylamine (7 mL) following General Procedure A at oil bath temperature of 55°C. The aqueous mixture was kept overnight at 4°C and the supernatant was decanted. The residue was dried in a desiccator and recrystallized from n-hexanes; yield 51% (0.14 g); mp: 41–42°C; 1H NMR ([D6]DMSO): δ=0.86 (t, J=7.3 Hz, 3H, CH2CH3), 1.12 (d, J=6.7 Hz, 6H, CHCH3),1.49–1.54 (m, 2H, CH2CH3), 3.23–3.27 (m, 2H, NHCH2), 3.98–4.02 (m, 1H, CH), 5.95–5.97 (m, 1H, NHCH2), 7.11–7.16 (m, 1H, 6-H), 7.77 (d,J=6.3 Hz, 1H, CONH); 13C NMR ([D6]DMSO): δ=11.11 (CH2CH3),22.31 (CHCH3), 23.54 (CH2CH3), 41.26 (CH), 46.14 (NHCH2), 110.08 (m, 2,3J(C,F)=12.8, 6.9 Hz, C1), 110.55 (m, 2J(C,F)=23.3 Hz, C6), 129.68 (m, C4), 139.33 (m, 1,2,3J(C,F)=238.4, 17.2, 8.6 Hz, C3), 145.97 (m, 1,2J(C,F)=242.9, 11.6 Hz, C2), 146.53 (m, 1,3J(C,F)=235.1, 6.6 Hz, C5), 161.14 (CO). MS (EI) (m/z, %) 274 (M+, 52), 245 (M+–C2H5, 100), 216 (M+–C3H8 N, 64). Elemental analysis calcd (%) for C13H17F3N2O: C 56.93, H 6.25, N 10.21, found: C 56.93, H 6.38, N10.38.
2,3,5-Trifluoro-N-isopropyl-4-(isopropylamino)benzamide (6b) was prepared from 5b and isopropylamine (5 mL) following General Procedure A at oil bath temperature of 55°C. The aqueous mixture was extracted with ethyl acetate (3×15 mL), the combined organic layers were dried (Na2SO4) and the filtrate was evaporated to dryness. The residue was recrystallized from n-hexanes; yield 33% (91 mg); mp: 56–59°C; 1H NMR ([D6]DMSO): δ=1.12 (d, J=6.3 Hz, 6H, CH3), 1.16 (d, J=7.3 Hz, 6H, CH3), 3.84–3.90 (m, 1H, CH), 3.97–4.04 (m, 1H, CH), 5.36 (d, J=9.2 Hz, 1H, 4-NH), 7.13–7.17 (m, 1H, 6-H), 7.82 (d, J=7.0 Hz, 1H, CONH); 13C NMR ([D6]DMSO): δ=22.29 (CH3), 23.39 (CH3), 41.26 (CH), 46.19 (CH), 110.58 (m,2J(C,F)=23.6 Hz, C6), 110.99 (m, 2,3J(C,F)=12.5, 7.2 Hz, C1), 128.98 (m, C4), 139.60 (m, 1,2,3J(C,F)=239.2, 17.1, 8.3 Hz, C3), 145.88 (m, 1,2J(C,F)= 241.9, 14.0 Hz, C2), 146.79 (m, 1,3J(C,F)=235.6, 6.1 Hz, C5), 161.09 (CO). MS (EI) (m/z, %) 274 (M+, 45), 259 (M+–CH3, 100), 216 (M+–C3H8N, 20). Elemental analysis calcd (%) for C13H17F3N2O: C 56.93, H 6.25, N 10.21, found: C 56.94, H 6.48, N 9.80.
2,3,5-Trifluoro-4-(isobutylamino)-N-isopropylbenzamide (6c) was prepared from 5b and isobutylamine (8 mL) following General Procedure A at oil bath temperature of 70°C. The aqueous mixture was extracted with ethyl acetate (3×15 mL), the combined organic layers were dried (Na2SO4) and the filtrate was evaporated to dryness. The residue was purified by column chromatography using a gradient of petroleum ether/CH2Cl2 (1:1) to CH2Cl2; yield 26%(75 mg); mp: 42–43°C; 1H NMR ([D6]DMSO): δ=0.86 (d, J=6.6 Hz, 6H, CH2CHCH3), 1.12 (d, J=6.6 Hz, 6H, NHCHCH3), 1.73–1.81 (m, 1H, CH2CH), 3.07–3.10 (m, 2H, CH2), 3.97–4.03 (m, 1H, NHCH), 6.02 (t, J=6.5 Hz, 1H, 4-NH), 7.11–7.16 (m, 1H, 6-H), 7.77 (d, J=6.7 Hz, 1H, CONH); 13C NMR ([D6]DMSO): δ=19.90 (CH2CHCH3), 22.30 (NHCHCH3), 28.95 (CH2CH), 41.26 (NHCH), 51.90 (CH2), 110.08 (m,2,3J(C,F)=12.5, 7.1 Hz, C1), 110.56 (m, 2J(C,F)=23.3 Hz, C6), 129.74 (m, C4), 139.33 (m, 1,2,3J(C,F)=238.5, 17.4, 8.4 Hz, C3), 145.98 (m,1,2J(C,F)=243.9, 12.6 Hz, C2), 146.54 (m, 1,3J(C,F)=235.1, 7.3 Hz, C5), 161.14 (CO). MS (EI) (m/z, %) 274 (M+, 30), 245 (M+–C3H7, 100). Elemental analysis calcd (%) for C14H19F3N2O: C 58.32,H 6.64, N 9.72, found: C 57.81, H 6.69, N 9.37.
4-(Cyclohexylamino)-2,3,5-trifluoro-N-isopropylbenzamide (6d) was prepared from 5b and cyclohexylamine (8 mL) following General Procedure A at oil bath temperature of 130°C. The aqueous mixture was extracted with ethyl acetate (3×20 mL), the combined organic layers were dried (Na2SO4) and the filtrate was evaporated to dryness. The residue was recrystallized from n-hexanes; yield 17% (53 mg); mp: 83–85°C; 1H NMR ([D6]DMSO): δ=1.12 (d, J=6.7 Hz, 6H, CH3), 1.30–2.00 (m, 10H, CH2), 3.41–3.52 (m, 1H, CHCH2), 3.96–4.05 (m, 1H, CHCH3), 5.35 (d, J=9.2 Hz, 1H, 4-NH),7.12–7.17 (m, 1H, 6-H), 7.80 (d, J=6.3 Hz, 1H, CONH); 13C NMR([D6]DMSO): δ=22.30 (CH3), 24.86, 25.31 (CHCH2CH2CH2), 33.68 (CHCH2), 41.29 (CHCH3), 53.55 (CHCH2), 110.59 (m, 2J(C,F)=23.8 Hz, C6), 110.90 (m, 2,3J(C,F)=12.6, 7.2 Hz, C1), 128.80 (m, C4), 139.61 (m, 1,2,3J(C,F)=239.0, 17.1, 8.4 Hz, C3), 145.90 (m, 1,2,4J(C,F)=244.1,12.8, 2.2 Hz, C2), 146.81 (m, 1,3J(C,F)=235.8, 6.3 Hz, C5), 161.11 (CO). MS (EI) (m/z, %) 314 (M+, 76), 271 (M+–C3H7, 100), 174 (M+–C3H8N–C6H11, 42). Elemental analysis calcd (%) for C16H21F3N2O: C61.13, H 6.73, N 8.91, found: C 60.72, H 6.91, N 8.83.
N-Cyclohexyl-2,3,5-trifluoro-4-(propylamino)benzamide (7a) was prepared from 5d and propylamine (9 mL) following General Procedure A at oil bath temperature of 55°C. The aqueous mixture was extracted with ethyl acetate (3×15 mL), the combined organic layers were dried (Na2SO4) and the filtrate was evaporated to dryness. The residue was recrystallized from n-hexanes; yield 38% (0.12 g); mp: 86–88°C; 1H NMR ([D6]DMSO): δ=0.86 (t, J=7.4 Hz, 3H, CH3), 1.11–1.79 (m, 10H, CHCH2CH2CH2), 1.51 (sext, J=7.3 Hz, 2H, CH2CH3), 3.24 (q, J=6.9 Hz, 2H, NHCH2), 3.67–3.69 (m, 1H,CHCH2), 5.95 (brs, 1H, 4-NH), 7.10–7.14 (m, 1H, 6-H), 7.76 (d, J=6.6 Hz, 1H, CONH); 13C NMR ([D6]DMSO): δ=11.10 (CH3), 23.52 (CH2CH3), 24.74, 25.30 (CHCH2CH2CH2), 32.29 (CHCH2), 46.14 (NHCH2), 48.41 (CH), 110.15 (m, 2,3J(C,F)=12.4, 6.9 Hz, C1), 110.54 (m, 2J(C,F)=23.3 Hz, C6), 129.65 (m, C4), 139.32 (m, 1,2,3J(C,F)= 238.4, 17.2, 8.6 Hz, C3), 145.93 (m, 1,2J(C,F)=242.9, 11.6 Hz, C2), 146.53 (m, 1,3J(C,F)=235.1, 6.6 Hz, C5), 161.09 (CO). MS (EI) (m/z, %) 314 (M+, 80), 285 (M+–C2H5, 34), 232 (M+–C6H10, 100). Elemental analysis calcd (%) for C16 H21F3N2O: C 61.13, H 6.73, N 8.91, found: C61.00, H 6.96, N 8.80.
N-Cyclohexyl-2,3,5-trifluoro-4-(isopropylamino)benzamide (7b) was prepared from 5d and isopropylamine (6 mL) following General Procedure A at oil bath temperature of 35°C. The aqueous mixture was extracted with ethyl acetate (3×15 mL), the combined organic layers were dried (Na2SO4) and the filtrate was evaporated to dryness. The residue was purified by column chromatography using a gradient of petroleum ether/CH2Cl2 (1:1) to CH2Cl2; yield 30% (94 mg); mp: 83–85°C; 1H NMR ([D6]DMSO): d=1.16 (d, J=6.7 Hz, 6H, CH3), 1.24–1.82 (m, 10H, CH2), 3.66–3.68 (m, 1H, CHCH2), 3.87 (oct, J=6.8 Hz, CHCH3), 5.35 (d, 1H, J=9.1 Hz, 4-NH),7.12–7.16 (m, 1H, 6-H), 7.80 (d, J=7.0 Hz, 1H, CONH); 13C NMR([D6]DMSO): δ=23.39 (CH3), 24.75, 25.32 (CHCH2CH2CH2), 32.30 (CHCH2), 46.21 (CHCH3), 48.44 (CHCH2), 110.39 (m, 2,3J(C,F)=12.5,7.2 Hz, C1), 110.60 (m, 2J(C,F)=23.6 Hz, C6), 128.95 (m, C4), 139.64 (m, 1,2,3J(C,F)=239.2, 17.1, 8.3 Hz, C3), 145.37 (m, 1,2J(C,F)=241.9,14.0 Hz, C2), 146.81 (m, 1,3J(C,F)=235.6, 6.1 Hz, C5), 160.14 (CO). MS (EI) (m/z, %) 314 (M+, 100), 299 (M+–CH3, 75), 232 (M+–C6H10 Elemental analysis calcd (%)for C16H21F3N2O: C 61.13, H 6.73, N 8.91, found: C 61.19, H 6.94, N 8.61.
N-Cyclohexyl-2,3,5-trifluoro-4-(isobutylamino)benzamide (7c) was prepared from 5d and isobutylamine (9 mL) following General Procedure A at oil bath temperature of 80°C. The aqueous mixture was extracted with ethyl acetate (3×15 mL), the combined organic layers were dried (Na2SO4) and the filtrate was evaporated to dryness. The residue was recrystallized from n-hexanes; yield 37% (0.12 g); mp: 102–105°C;1H NMR ([D6]DMSO): δ=0.86 (d, J=7.0 Hz, 6H, CH3), 1.11–1.81 (m, 11H, CHCH2CH2CH2, CHCH3), 3.08 (t, J=6.8 Hz, 2H, NHCH2), 3.65–3.72 (m, 1H, NHCH), 6.02 (t, J=6.5 Hz, 1H, 4-NH), 7.10–7.14 (m, 1H, 6-H), 7.76 (d, J=6.6 Hz, 1H, CONH); 13C NMR ([D6]DMSO): δ=19.89 (CH3), 24.75, 25.32 (CHCH2CH2CH2), 28.94 (CHCH3), 32.29 (CHCH2), 48.41 (NHCH), 51.90 (NHCH2), 110.16 (m, 2,3J(C,F)=12.5, 7.1 Hz, C1), 110.56 (m, 2J(C,F)=23.3 Hz, C6), 129.74 (m, C4), 139.34 (m, 1,2,3J(C,F)=238.5, 17.4, 8.4 Hz, C3), 145.95 (m, 1,2J(C,F)=243.9, 12.6 Hz, C2), 146.54 (m, 1,3J(C,F)=235.1, 7.3 Hz, C5), 161.12 (CO). MS (EI) (m/z, %) 328 (M+, 56), 285 (M+–C3H7,100), 246 (M+–C6H10, 55). Elemental analysis calcd (%) forC17H23F3N2O: C 62.18, H 7.06, N 8.53, found: C 62.01, H 7.32, N 8.24. In a separate experiment, compound 5d was reacted with isobutylamine under microwave conditions (General Procedure B). After column chromatography, the product 7c was obtained in 45% yield.
N-Cyclohexyl-2,3,5-trifluoro-4-(cyclohexylamino)benzamide (7d) was prepared from 5d and cyclohexylamine (6 mL) following General Procedure A at oil bath temperature of 130°C. The aqueous mixture was extracted with ethyl acetate (3×15 mL), the combined organic layers were dried (Na2SO4) and the filtrate was evaporated to dryness. The residue was recrystallized from n-hexanes; yield 27% (96 mg); mp: 98–102°C; 1H NMR ([D6]DMSO): δ=1.09–1.87 (m, 20H, CH2), 3.40–3.51 (m, 1H, CH), 3.62–3.70 (m, 1H, CH), 5.34 (d, J=9.2 Hz, 1H, 4-NH), 7.11–7.15 (m, 1H, 6-H), 7.79 (d, J=6.3 Hz, 1H, CONH); 13C NMR ([D6]DMSO): δ=24.74, 24.85, 25.30, 25.30, (CHCH2CH2CH2), 32.29, 33.67 (CHCH2), 48.43 (CONHCH), 53.55 (4-NHCH), 110.58 (m, 2J(C,F)=23.8 Hz, C6), 111.00 (m, 2,3J(C,F)=12.6,7.2 Hz, C1), 128.77 (m, C4), 139.59 (m, 1,2,3J(C,F)=239.0, 17.1, 8.4 Hz, C3), 145.85 (m, 1,2, 4J(C,F)=241.1, 12.8, 2.2 Hz, C2), 146.81 (m,1,3J(C,F)=235.8, 6.3 Hz, C5), 161.07 (CO). MS (EI) (m/z, %) 354 (M+,80), 272 (M+–C6H10, 100). Elemental analysis calcd (%) for C19H25F3N2O: C 64.39, H 7.11, N 7.90, found: C 64.56, H 7.32, N 7.90. In a separate experiment, compound 5d was reacted with cyclohexylamine under microwave conditions (General Procedure B). After column chromatography, the product 7d was obtained in 43% yield.
4-(Benzylamino)-N-cyclohexyl-2,3,5-trifluorobenzamide (7e) was prepared from 5d and benzylamine (9 mL) following General Procedure A at oil bath temperature of 130°C. The aqueous mixture was extracted with ethyl acetate (3×15 mL), the combined organic layers were dried (Na2SO4) and the filtrate was evaporated to dryness. The residue was recrystallized from n-hexanes; yield 45% (0.16 g); mp: 133–136°C; 1H NMR ([D6]DMSO): δ=1.09–1.80 (m, 10H, CH2), 3.62–3.70 (m, 1H, CH), 4.47 (d, J=6.9 Hz, 2H, NHCH2),6.66–6.69 (m, 1H, 4-NH), 7.18–7.21 (m, 1H, 6-H), 7.27–7.31 (m, 5H, Ar-H), 7.79 (d, J=6.6 Hz, 1H, CONH); 13C NMR ([D6]DMSO): δ=24.71, 25.31 (CHCH2CH2CH2), 32.32 (CHCH2), 47.63 (NHCH2), 48.41 (NHCH), 110.48 (m, 2J(C,F)=22.8 Hz, C6), 111.25 (m, 2J(C,F)=12.7 Hz, C1), 126.84, 127.46, 128.34 (Ar-CH), 128.46 (m, C4), 139.60 (m,1,2,3J(C,F)=239.1, 16.9, 8.7 Hz, C3), 140.43 (Ar-C), 145.70 (m,1,2,4J(C,F)=244.1, 12.8, 1.8 Hz, C2), 146.81 (m, 1,3J(C,F)=236.4,6.8 Hz, C5), 161.01 (CO). MS (EI) (m/z, %) 362 (M+, 46), 280 (M+–C6H10, 48), 91 (C7H7). Elemental analysis calcd (%) for C20H21F3N2O: C 66.29, H 5.84, N 7.73, found: C 66.55, H 5.91, N 7.44.
4-Fluoroisoindoline (8). 13C NMR ([D6]DMSO): δ=47.32 (C3), 50.38 (C1), 114.99 (m, 2J(C,F)=18.9 Hz, C5), 119.45 (m, 4J(C,F)=3.0 Hz, C7), 121.85 (m, 2J(C,F)=18.9 Hz, C3a), 131.23 (m, 3J(C,F)=6.9 Hz, C6), 139.04 (m, 3J(C,F)=4.7 Hz, C7a), 157.29 (m, 1J(C,F)=247.6 Hz, C4).
2-Fluorobenzamide (9). 13C NMR ([D6]DMSO): δ=116.23 (m,2J(C,F)=22.7 Hz, C3), 123.99 (m, 1J(C,F)=14.2 Hz, C1), 124.54 (m,4J(C,F)=3.4 Hz, C5), 130.38 (m, 3J(C,F)=2.9 Hz, C6), 132.61 (m,3J(C,F)=8.6 Hz, C4), 159.46 (m, 1J(C,F)=249.5 Hz, C2), 165.38 (CO).
3-Fluorobenzamide (10). 13C NMR ([D6]DMSO): δ=114.39 (m,2J(C,F)=22.6 Hz, C2), 118.24 (m, 2J(C,F)=21.2 Hz, C4), 123.78 (m,4J(C,F)=2.9 Hz, C6), 130.51 (m, 3J(C,F)=7.9 Hz, C5), 136.89 (m,3J(C,F)=6.7 Hz, C1), 162.13 (m, 1J(C,F)=244.3 Hz, C3), 166.68 (m,4J(C,F)=2.5 Hz, CO).
Biological investigations
Rat aortic ring assay.
To determine the extent of the antiangiogenic effects of the test compounds, the rat aortic ring assay was carried out as described elsewhere.[8] Briefly, 24-well tissue culture plates were covered with 250 μL of Matrigel (Becton-Dickinson) and allowed to gel for 30 to 45 min at 37°C and 5% CO2. Sections of thoracic aorta were removed from 8- to 10-week-old male Sprague Dawley rats. Following excision of fibroadipose tissue, the aortic sections were cut into 1-mm-long cross-sections, placed on Matrigel-coated wells, and layered with additional Matrigel (250 mL). These were then allowed to set, after which the cross-sectional rings were covered with endothelial cell growth media (EGM-II) and incubated under 5% CO2 at 37°C overnight. EGM-II consists of endothelial cell basal medium (EBM-II) and endothelial cell growth factors. After 24 h, the medium was removed and replaced with EBM-II (1 mL) with the vehicle (0.5% DMSO), positive control TNP-470 (50 mm), or test compound (50 μM). This was replicated three times. The area of angiogenic sprouting was quantified using Adobe Photoshop.
Cytotoxicity assay.
This assay was determined as described elsewhere.[8] Briefly, HUVECs were treated with compounds at 10 μM. After 24 hours of incubation with compounds, 10 μL of CCK-8 solution (Dojindo, MD) was added to each well. The plates were left to incubate for 2 hours, and then read at a wavelength of 450 nm using a microplate reader.
Disclaimer
The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organization imply endorsement by the U.S. Government.
Acknowledgement
G.S. thanks Prof. A. C. Filippou for support. The authors thank Ioana Silly and Andreas Bock for synthetic support.
Footnotes
Conflict of interest
The authors declare no conflict of interest.
References
- [1].a) Weidner N, Carroll PR, Flax J, Blumenfeld W, Folkman J, Am. J. Pathol 1993, 143, 401–409; [PMC free article] [PubMed] [Google Scholar]; b) Panigrahy D, Singer S, Shen LQ, Butterfield CE, Freedman DA, Chen EJ, Moses MA, Kilroy S, Duensing S, Fletcher C, Fletcher JA, Hlatky L, Hahnfeldt P, Folkman J, Kai-painen A, J. Clin. Invest 2002, 110, 923–932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].a) Folkman J, Nat. Rev. Drug Discovery 2007, 6, 273–286; [DOI] [PubMed] [Google Scholar]; b) Aragon-Ching JB, Li H, Gardner ER, Figg WD, Recent Pat. Anti-Cancer Drug Discovery 2007, 2, 167–174; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Belotti D, Foglieni C, Resovi A, Giavazzi R, Taraboletti G, Int. J. Biochem. Cell. Biol 2011, 43, 1674–1685; [DOI] [PubMed] [Google Scholar]; d) Papa A, Zaccarelli E, Caruso D, Vici P, Benedetti P, Tomao F, Expert Opin. Invest. Drugs 2016, 25, 31–49; [DOI] [PubMed] [Google Scholar]; e) Foglieni C, Pagano K, Lessi M,Bugatti A, Moroni E, Pinessi D, Resovi A, Ribatti D, Bertini S, Ragona L, Bellina F, Rusnati M, Colombo G, Taraboletti G, Sci. Rep 2016, 6, 23432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Capitosti SM, Hansen TP, Brown ML, Bioorg. Med. Chem 2004, 12, 327–336. [DOI] [PubMed] [Google Scholar]
- [4].a) Ng SSW, Gütschow M, Weiss M, Hauschildt S, Teubert U, Hecker TK, Luzzio FA, Kruger EA, Eger K, Figg WD, Cancer Res. 2003, 63, 3189–3194; [PubMed] [Google Scholar]; b) Lepper ER, Ng SS, Gütschow M, Weiss M, Hauschildt S, Hecker TK, Luzzio FA, Eger K, Figg WD, J. Med. Chem 2004, 47, 2219–2227; [DOI] [PubMed] [Google Scholar]; c) Ng SS, MacPherson GR, Gütschow M, Eger K,Figg WD, Clin. Cancer Res. 2004, 10, 4192–4197. [DOI] [PubMed] [Google Scholar]
- [5].Gütschow M, Hecker T, Thiele A, Hauschildt S, Eger K, Bioorg. Med. Chem 2001, 9, 1059–1065. [DOI] [PubMed] [Google Scholar]
- [6].a) Kumar S, Raje N, Hideshima T, Ishitsuka K, Roccaro A, Shiraishi N,Hamasaki M, Yasui H, Munshi NC, Richardson P, Figg WD, Anderson KC, Leukemia 2005, 19, 1253–1261; [DOI] [PubMed] [Google Scholar]; b) Warfel NA, Lepper ER, Zhang C, Figg WD, Dennis PA, Clin. Cancer Res. 2006, 12, 3502–3509; [DOI] [PubMed] [Google Scholar]; c) Therapontos C, Erskine L, Gardner ER, Figg WD, Vargesson N, Proc. Natl. Acad. Sci. USA 2009, 106, 8573–8578; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Beedie SL, Mahony C, Walker HM, Chau CH, Figg WD, Vargesson N, Sci. Rep 2016, 6, 30038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Zalazar F, De Luca P, Gardner K, Figg WD, Meiss R, Spallanzani RG,Vallecorsa P, Elguero B, Cotignola J, Vazquez E, De Siervi A, Curr. Pharm. Biotechnol 2015, 16, 553–563. [DOI] [PubMed] [Google Scholar]
- [8].Beedie SL, Peer CJ, Pisle S, Gardner ER, Mahony C, Barnett S, Ambrożak A, Gütschow M, Chau CH, Vargesson N, Figg WD, Mol. Cancer Ther. 2015, 14, 2228–2237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Ambrożak A, Steinebach C, Gardner ER, Beedie SL, Schnakenburg G, Figg WD, Gütschow M, ChemMedChem 2016, 11, 2621–2629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].a) Hagmann WK, J. Med. Chem 2008, 51, 4359–4369; [DOI] [PubMed] [Google Scholar]; b) Wang J, Sánchez-Roselló M, Aceña JL, del Pozo C, Sorochinsky AE, Fustero S,Soloshonok VA, Liu H, Chem. Rev 2014, 114, 2432–2506; [DOI] [PubMed] [Google Scholar]; c) Zhou Y, Wang J, Gu Z, Wang S, Zhu W, AceÇa JL, Soloshonok VA, Izawa K, Liu H, Chem. Rev 2016, 116, 422–518. [DOI] [PubMed] [Google Scholar]
- [11].Champagne PA, Desroches J, Paquin JF, Synthesis 2015, 47, 306–322. [Google Scholar]
- [12].a) Olsen JA, Banner DW, Seiler P, Wagner B, Tschopp T, ObstSander U, Kansy M, Müller K, Diederich F, ChemBioChem 2004, 5, 666–675; [DOI] [PubMed] [Google Scholar]; b) Behrends M, Wagner S, Kopka K, Schober O, Schäfers M, Kumbhar S, Waller M, Haufe G, Bioorg. Med. Chem 2015, 23, 3809–3818. [DOI] [PubMed] [Google Scholar]
- [13].a) Kim D, Wang L, Beconi M, Eiermann GJ, Fisher MH, He H, Hickey GJ, Kowalchick JE, Leiting B, Lyons K, Marsilio F, McCann ME,Patel RA, Petrov A, Scapin G, Patel SB, Roy RS, Wu JK, Wyvratt MJ, Zhang BB, Zhu L, Thornberry NA, Weber AE, J. Med. Chem 2005, 48, 141–151; [DOI] [PubMed] [Google Scholar]; b) Podichetty AK, Wagner S, Schrçer S, Faust A, Schäfers M, Schober O, Kopka K, Haufe G, J. Med. Chem 2009, 52, 3484–3495; [DOI] [PubMed] [Google Scholar]; c) Fustero S, Rodrigo V, Sánchez-Roselló M, del Pozo C, Timoneda J, Frizler M, Sisay MT, Bajorath J, Calle LP, Cañada FJ, Jiménez-Barbero J, Gütschow M, Chem. Eur. J 2011, 17, 5256–5260. [DOI] [PubMed] [Google Scholar]
- [14].a) Xia G, Chen W, Zhang J, Shao J, Zhang Y, Huang W, Zhang L, Qi W, Sun X, Li B, Xiang Z, Ma C, Xu J, Deng H, Li Y, Li P, Miao H, Han J, Liu Y, Shen J, Yu Y, J. Med. Chem 2014, 57, 9889–9900; [DOI] [PubMed] [Google Scholar]; b) Lou Y,Sweeney ZK, Kuglstatter A, Davis D, Goldstein DM, Han X, Hong J,Kocer B, Kondru RK, Litman R, McIntosh J, Sarma K, Suh J, Taygerly J, Owens TD, Bioorg. Med. Chem. Lett 2015, 25, 367–371. [DOI] [PubMed] [Google Scholar]
- [15].Puskas L, Feher L, Molnar E (Avidin Kft.), Int. PCT Pub. No. WO2008155594 A2, 2008.
- [16].a) Lawrence CD, Friedlos F, Hedley D, Martin J, Ogilvie LM, Scanlon IJ, Springer CJ, J. Med. Chem 2005, 48, 5321–5328; [DOI] [PubMed] [Google Scholar]; b) Nosova EV, Lipunova GN, Laeva AA, Charushin VN, Russ. J. Org. Chem 2005, 41, 1671–1677; [Google Scholar]; c) Layeva AA, Nosova EV, Lipunova GN, Trashakhova TV, Charushin VN, J. Chem 2007, 128, 748–754; [Google Scholar]; d) Lipunova GN, Nosova EV, Laeva AA, Trashakhova TV, Slepukhin PA, Charushin VN, Russ. J. Org. Chem 2008, 44, 741–749. [Google Scholar]
- [17].Chuikov IP, Fadeev DS, Mamatyuk VI, Vaganova TA, Malykhin EV, J. Fluorine Chem 2011, 132, 512–515. [Google Scholar]
- [18].Schwarzer A, Weber E, Cryst. Growth Des. 2008, 8, 2862–2874. [Google Scholar]
- [19].A HSQC spectrum of 9 was recorded and applied for the assignment of the 13C NMR signals of 9.
- [20].Kato Y, Sakurai K, Bull. Chem. Soc. Jpn 1982, 55, 1643–1644. [Google Scholar]
- [21].The X-ray crystallographic data collection of 4a and 7e were performed on a Bruker D8 Venture diffractometer at 100(2)K. The diffractometer was equipped with a low-temperature device (Cryostream 800er series, Oxford Cryosystems) and used Cu-Kα radiation (λ= 1.54178 Å). Intensities were measured by fine-slicing φ- and ω-scans and corrected for background, polarization and Lorentz effects. Semi-empirical absorption corrections were applied for all data sets by using Bruker’s SADABS program. The structures were solved by direct methods and refined aniso-tropically by the least-squares procedure implemented in the ShelX-2014/7 program system. The hydrogen atoms were included isotropically using the riding model on the bound carbon atoms. CCDC 1838436 (4a) and CCDC 1838437 (7e) contain the supplementary crystallographic data for this paper. These data are provided free of charge by The Cambridge Crystallographic Data Centre.
- [22].Ito H, Matsushita U, Shimizu T, Ishikawa N, Shimizu M (SDS Biotech), Eur. Pat. No. EP259663 A1, 1986.
- [23].Mobashery S, Hesek D, Chang M (University of Notre Dame), Int. PCT Pub. No. WO2011026107 A1, 2011.