

Abstract
The continuous release of neurotransmitter could be seen to place a persistent burden on presynaptic proteins, one that could compromise nerve terminal function. This supposition and the molecular mechanisms that might protect highly active synapses merit investigation. In hippocampal cultures from knock-out mice lacking the presynaptic cochaperone cysteine string protein-α (CSP-α), we observe progressive degeneration of highly active synaptotagmin 2 (Syt2)-expressing GABAergic synapses, but surprisingly not of glutamatergic terminals. In CSP-α knock-out mice, synaptic degeneration of basket cell terminals occurs in vivo in the presence of normal glutamatergic synapses onto dentate gyrus granule cells. Consistent with this, in hippocampal cultures from these mice, the frequency of miniature IPSCs, caused by spontaneous GABA release, progressively declines, whereas the frequency of miniature excitatory AMPA receptor-mediated currents (mEPSCs), caused by spontaneous release of glutamate, is normal. However, the mEPSC amplitude progressively decreases. Remarkably, long-term block of glutamatergic transmission in cultures lacking CSP-α substantially rescues Syt2-expressing GABAergic synapses from neurodegeneration. These findings demonstrate that elevated neural activity increases synapse vulnerability and that CSP-α is essential to maintain presynaptic function under a physiologically high-activity regimen.
Introduction
Nerve terminals are known to achieve fast neuronal communication through special molecular machineries that release neurotransmitters and recycle synaptic vesicles (Südhof, 2004). This raises the question of whether the maintained synaptic activity poses a burden on nerve terminals that compromises synaptic integrity. Cysteine string protein-α (CSP-α) is a synaptic vesicle protein that contains a DNAJ domain typical of cochaperones from the Hsp40 family (Gundersen and Umbach, 1992; Schmitz and Fernández-Chacón, 2009). CSP-α was originally proposed to act as a mandatory element in synaptic vesicle fusion or the regulation of calcium influx through voltage-dependent channels (Gundersen and Umbach, 1992; Umbach et al., 1994; Zinsmaier et al., 1994). However, more recently, it has been proposed as a key element of the synaptic molecular machinery devoted to the rescue of synaptic proteins that have been unfolded by activity-dependent stress (Fernández-Chacón et al., 2004; Chandra et al., 2005). Mice lacking CSP-α survive <3 months because of neurological deficits (Fernández-Chacón et al., 2004). During early postnatal development, CSP-α is not essential for neurotransmitter release at the calyx of Held, but at later ages, the absence of CSP-α leads to deficits in neurotransmitter release because of neurodegeneration of presynaptic terminals (Fernández-Chacón et al., 2004). The fastest synaptic degeneration occurs in photoreceptor ribbon synapses, which become severely damaged by postnatal day 14 (P14) (Schmitz et al., 2006), whereas degeneration at the neuromuscular junction develops more slowly (Fernández-Chacón et al., 2004; Ruiz et al., 2008). These observations suggest that the requirement for CSP-α might be greater at synapses subject to heavy membrane trafficking because of intense synaptic vesicle cycling, such as occurs in the photoreceptors (Sterling and Matthews, 2005), or in synapses specialized in conveying fast reliable signals at high frequencies, as is the case in the calyx of Held (Joshi and Wang, 2002). However, experimental evidence that synaptic activity is the primary cause of the degenerative phenotype is still lacking. Interestingly, the soluble N-ethylmaleimide-sensitive factor attachment protein receptor (SNARE) complex (Fasshauer et al., 1998; Südhof, 2004; Rizo and Rosenmund, 2008) requires CSP-α to function correctly: the protein levels of SNAP25 are significantly decreased and the SNARE complex becomes unstable in the brain of CSP-α knock-out mice (Chandra et al., 2005). Remarkably, the overexpression of α-synuclein partially restores SNARE complex stability, indicating that CSP-α and α-synuclein cooperate to chaperone the SNARE complex (Chandra et al., 2005). However, although compelling biochemical data suggest a general role for CSP-α in the brain, its function at small central synapses has been poorly explored. Here, we used long-term hippocampal cultures from mice lacking CSP-α to reveal that a population of GABAergic synapses develops a progressive neurodegenerative phenotype. In contrast, glutamatergic synapses are less sensitive to the loss of CSP-α but indirectly undergo homeostatic plasticity changes that are likely to compensate for the loss of GABAergic input. Remarkably, we show that degeneration of GABAergic synapses is partially rescued by pharmacological reduction of network excitability with glutamatergic blockers. These observations demonstrate that high synaptic activity increases the vulnerability of nerve terminals and that CSP-α is a critical factor in preventing synapse rundown under these conditions.
Materials and Methods
Neuronal cultures.
Primary hippocampal neurons were prepared essentially as previously described (Fernández-Chacón et al., 2001) from P0 wild-type (WT) and CSP-α knock-out (KO) littermate mice (Fernández-Chacón et al., 2004) and grown for up to 40 d in a chemically defined medium (Neurobasal A medium supplemented with Glutamax and B-27; Invitrogen) containing antibiotics. The medium was partially replaced every 4 d. Neurons were plated on coverslips covered with continuous (no microdots) rat astrocyte feeder layers treated with the mitotic inhibitor FUdR (fluorodeoxyuridine), at a density of 35,000 cells per well. For deprivation of glutamatergic activity, neuronal cultures were incubated with 25 μm (R)-amino-5-phosphonovaleric acid (APV) and 10 μm 2,3-dihydroxy-6-nitro-7-sulfamoyl-benzo[f]quinoxaline-2,3-dione (NBQX) throughout a 3 week period from the 10th to the 30th day in vitro (DIV). Drugs were added every day, and the medium was partially replaced every 4 d.
Tissue preparation.
After deep anesthesia with 2% tribromoethanol (0.15 ml/10 mg), control and CSP-α mutant mice were transcardially perfused with 0.1 m PBS, pH 7.4, followed by 4% paraformaldehyde (PFA) in 0.1 m phosphate buffer (PB), pH 7.4. The brains were removed, postfixed for 6–8 h in 4% PFA at 4°C, cryoprotected in 30% sucrose, and then sectioned at 40 μm thickness in the coronal plane on a freezing microtome. All experiments with animals were performed in accordance with the National Institutes of Health (NIH) Guide for the Care and Use of Laboratory Animals and approved by the Committee of Animal Use for Research at the University of Seville.
Immunocytochemistry.
Hippocampal neuron cultures were fixed in 4% PFA for 30 min, rinsed three times with PBS, and then permeabilized with 0.3% Triton X-100 for 5 min, after which they were again rinsed three times with PBS. Cultures were blocked in 3% normal fetal bovine serum (FBS) for 2 h and subsequently incubated overnight with different primary antibodies at 4°C, followed by three washes in PBS. Alexa 488-conjugated goat anti-mouse (1:1000; Invitrogen) and Cy3-conjugated goat anti-rabbit (1:1000; Jackson ImmunoResearch) secondary antibodies were then applied for 2 h at room temperature, followed by three PBS washes. Coverslips were mounted with ProLong Gold antifade reagent (Invitrogen) and examined with a confocal microscope (Leica TCS-SP2). Hippocampal slices were permeabilized with 0.5% Triton X-100 for 10 min, rinsed three times with PBS, and blocked in 2% normal FBS, 0.1% Triton X-100 in PBS for 1 h. Slices were then incubated overnight with different primary antibodies at 4°C, followed by three washes in PBS. Alexa 488-conjugated goat anti-mouse (1:1000; Invitrogen) and Cy3-conjugated goat anti-rabbit (1:1000; Jackson ImmunoResearch) secondary antibodies were then applied for 1 h at room temperature, followed by three PBS washes. Sections were mounted onto slides and coverslipped with ProLong Gold antifade reagent (Invitrogen).
Primary antibodies.
The following mouse monoclonal primary antibodies and concentrations were used in this study: glutamic acid decarboxylase 65 (GAD65) (GAD-6; 1:1000; developed by Dr. D. I. Gottlieb (Washington University School of Medicine, St. Louis, MO) and obtained from the Developmental Studies Hybridoma Bank developed under the auspices of the National Institute of Child Health and Human Development and maintained by The University of Iowa), gephyrin (1:1000; Synaptic Systems), GluR1 (1:50; Santa Cruz Biotechnology), MAP2a-b (1:1000; Millipore Bioscience Research Reagents), synaptobrevin 2/VAMP2 (Cl69.1; 1:1000; Synaptic Systems), and synaptotagmin 2 (znp-1; 1:200; Zebrafish International Resource Center, which is supported by NIH–National Center for Research Resources Grant P40 RR012546). The following rabbit polyclonal primary antibodies and concentrations were used: calretinin (CR) (1:1000; Swant), CSP [R807; 1:1000; generous gift from Dr. Thomas Südhof (Stanford University School of Medicine, Palo Alto, CA) (Fernández-Chacón et al., 2004)], parvalbumin (PV) (1:1000; Swant), synapsins [E028; 1:1000; generous gift from Dr. Thomas Südhof (Südhof et al., 1989)], vesicular GABA transporter (VGAT) (1:1000; Synaptic Systems), and vesicular glutamate transporter 1 (VGLUT1) (1:1000; Synaptic Systems).
Quantitative analysis of immunocytochemical images.
A Leica TCS-SP2 confocal microscope (objective: HCX PL APO 40×/63×) was used to obtain the maximum projection from a series of z-axis sections, which were then analyzed with NIH ImageJ software. To restrict our analysis to synaptic puncta and to avoid contamination by overall background and diffuse synapsin staining, we first manually removed the synapsin-positive pixels that were not associated with microtubule-associated protein (MAP2) staining. Next, grayscale images from red (synapsin) and green (MAP2) channels were converted separately to binary images using the same threshold level for both channels. Synapse density was estimated using thresholded images based on the ratio provided by the number of synapsin-positive pixels divided by the number of MAP2-positive pixels. The proportion (percentage) of GABAergic synapses was calculated by subtracting the pixels from the thresholded red channel (VGAT fluorescence) from the thresholded green channel (synaptobrevin 2/VAMP2 fluorescence). The pixels left after the subtraction were considered to be non-GABAergic synapses. Similar experiments were performed using GAD65 and synapsin antibodies. The relationship between GABAergic and glutamatergic puncta was established by dividing the thresholded GAD65 and VGLUT1 signals. Computer-Assisted Stereological Toolbox (CAST) software associated with an Olympus BX61 microscope was used to quantify the different populations of neurons in culture. The MAP2 signal revealed the total number of neurons. PV-positive interneurons were doubly labeled using antibodies against MAP2 and PV. CR-positive interneurons were identified using MAP2 and CR antibodies. To calculate the percentage of synaptotagmin 2 (Syt2)-positive GABAergic synapses, the thresholded Syt2 fluorescence of the green channel was subtracted from the thresholded VGAT fluorescence of the red channel. The intensity of fluorescence signals detected with antibodies against synaptic proteins was weaker in cultures on activity deprivation. For those conditions, the laser power required to detect fluorescence with confocal microscopy was higher than the laser power used to analyze nontreated cultures. In any case, the laser intensity was always the same to compare treated WT and treated KO cultures.
Stereological analysis.
Peroxidase-labeled PV-immunopositive cells were quantified according to the optical dissector method, using an Olympus BX61 microscope (Olympus). The CAST-Grid software package (Olympus) generated sampling frames with a known frame area (aF) and directed the motorized X–Y stage (Prior Proscan; Prior Scientific Instruments). The number of neurons was quantified in every seventh section (a distance of 280 μm) through the entire anteroposterior axis of the hippocampus. Nine to 12 serial sections were measured in each animal. One side of the entire hippocampus was defined using a 4× objective, and the number of neurons was counted using a 40× objective. The numerical density (ND) (in cells per cubic millimeter) of immunopositive cells was estimated using the following expression:
where I is the number of dissector-counted somatic profiles, n is the number of analyzed sections, F is the number of frames for each slice, aF is the area of frame (15,686.7 μm2), and SF is the volumetric shrinkage factor of the sample, determined as previously described (Jinno et al., 1998). The average area shrinkage factor was 0.95, and the average thickness shrinkage factor was 0.80. The neuronal density was corrected by the average volumetric shrinkage factor of the sample (0.76), which was considered the same for control and mutant brain sections.
Quantification of immunofluorescence intensity.
The images were acquired using an Olympus Fluoview FV1000 confocal microscope (objective: PLAPON 60×; numerical aperture, 1.42). Two stacks with 1.4 μm intervals between optical slice were collected per region (CA1 and dentate gyrus of hippocampus) and per genotype. Illumination intensity determined by laser power was always the same for each litter. Images were analyzed using Analyze particles module of NIH ImageJ software. First, grayscale images from red (PV) and green (Syt2) channels were converted separately to binary images using the same threshold level for each genotype. Puncta density was calculated using the thresholded image of each stack (in total, ∼36 images for each stack) and the Analyze particles module. To avoid background contamination, the minimum size of puncta was set at 10 pixels. For analysis of parvalbumin staining, the maximum puncta size was set at 200 pixels to exclude somatodendritic staining. Then, the collected data were analyzed using SPSS statistical software.
Colocalization analysis.
The degree of colocalization between synaptic markers was studied by intensity correlation analysis using an ImageJ plug-in: http://www.uhnres.utoronto.ca/facilities/wcif/imagej/color_analysis.htm#_ftn1, based on the study by Li et al. (2004). Briefly, pixel intensity for each channel (12 bits grayscale, vertical axis) was plotted against the product of the differences from the mean (PDM), for each pixel: PDM = (red intensity − mean red intensity) × (green intensity − mean green intensity). From this plot, two parameters were obtained: the calculated intensity correlation quotient (ICQ) and the Mander's overlap coefficient (R). Positive ICQ values indicated a dependency relationship between the two signals. R values ranged between 1 and 0, with values close to 1 indicative of high colocalization.
Protein biochemistry.
For immunoblotting analyses, brains from CSP-α WT and KO mice were removed in ice-cold PBS, and the hippocampi were extracted and then homogenized in lysis solution [50 mm HEPES, pH 7.4, 1% Triton X-100, 150 mm NaCl, 5 mm EDTA, 10 mm Na2HPO4, and supplemented with protease inhibitors (Sigma-Aldrich): 1 mm NaF, 1 mm Na3VO4, 1 mm PMSF, leupeptin (2 μg/ml), aprotinin (2 μg/ml), and pepstatin A (1 μg/ml)] using a glass homogenizer. Protein concentrations were determined using BCA assay (Pierce). Equivalent amounts of hippocampal proteins from wild-type and CSP-α KO mice were analyzed by SDS-PAGE and immunoblotting using antibodies as follows: CSP-α (R807; 1:2000), Hsc70 (3C5; Synaptic Systems; 1:1500), synaptotagmin 2 (znp-1; ZIRC Antibodies; 1:1500), PV (Synaptic Systems; 1:1500), synaptobrevin 2 (Cl 69.1; Synaptic Systems; 1:2000), synaptotagmin 1 (Cl 41.1; Synaptic Systems; 1:2000), and α-actin (A 2066; Sigma-Aldrich; 1:5000) as loading control. For quantitations, peroxidase-labeled secondary antibodies (Promega; anti-mouse, 1:10,000; anti-rabbit, 1:15,000) were revealed using ECL Plus (GE Healthcare) and imaged with ChemiDoc XRS System (Bio-Rad).
Electrophysiology.
Whole-cell recordings were performed using an EPC10 amplifier (HEKA). Data were filtered at 1 kHz and acquired at 20 kHz using Pulse 8.74 software (HEKA). Cells were voltage clamped at −70 mV. Patch electrodes had a resistance of 2–3 MΩ. Series resistance (75%) and cell capacitance (5–25 pF) were compensated and only recordings with series resistance <10 MΩ were included in the analysis. The external solution for AMPA receptor (AMPAR) miniature evoked postsynaptic current (mEPSC) recordings contained 140 mm NaCl, 2.4 mm KCl, 10 mm HEPES, 10 mm glucose, 4 mm CaCl2, and 4 mm MgCl2, 305 mOsm and pH 7.4. The internal pipette solution consisted of 10 mm NaCl, 120 mm K-gluconate, 10 mm HEPES, 1 mm K-EGTA, 4.6 mm MgCl2, 4 mm Na2ATP, 15 mm creatine phosphate, and 20 U/ml phosphocreatine kinase, 290 mOsm and pH 7.4. Recordings were usually performed for at least 60 s in the presence of 1 μm tetrodotoxin, 25 μm APV, and 25 μm bicuculline. In the case of NMDA receptor (NMDAR) mEPSC recordings, the external solution contained 140 mm NaCl, 2.4 mm KCl, 10 mm HEPES, 10 mm glucose, 2.7 mm CaCl2, and 10 μm glycine, 305 mOsm and pH 7.4 (Rosenmund et al., 1993). The internal pipette solution was the same as that used for AMPAR mEPSC recordings. Recordings were usually performed for at least 60 s in the presence of 1 μm tetrodotoxin, 10 μm NBQX, and 25 μm bicuculline. When recording GABA receptor miniature IPSCs (mIPSCs), the external solution was the same as that used for AMPA mEPSC recordings. The internal pipette solution for mIPSC recordings consisted of 135 mm CsCl, 10 mm HEPES, 1 mm K-EGTA, 4.6 mm MgCl2, 4 mm Na2ATP, 0.3 mm Na2GTP, 15 mm creatine phosphate, and 20 U/ml phosphocreatine kinase, 305 mOsm and pH 7.4. Recordings (60 s) were performed with 1 μm tetrodotoxin, 10 μm NBQX, and 25 μm APV. All drugs were acquired from Ascent Scientific. All miniature postsynaptic currents were analyzed with the program Axograph X (Molecular Devices) using a very sensitive method based on automated search of putative synaptic events by comparison with a template selected by the user (Clements and Bekkers, 1997). Recordings were first digitally filtered at 0.5 kHz. Few (six) random events were handpicked from every recording (60 s duration) and averaged to get a representative template. The program used that template to adjust the values of a function that generated a new template with a linear basal region followed by an idealized synaptic time course. The program used the new template for the detection of synaptic events. Threshold for detection was set to 2.5 times the baseline SD for mEPSCs (AMPA and NMDA), and 3.2 times the baseline SD for mIPSCs. Captured miniature currents of individual cells were averaged to determine mean amplitude and waveform per cell. The relationship between control and CSP-α KO amplitude distribution was determined by random selection of 100 miniature postsynaptic current amplitudes per cell from 25 cells recorded from both genotypes, to produce an equal number of events. WT and KO events were sorted in ascending order, after which the KO amplitudes were plotted against the WT amplitudes, according to previously published methods (Turrigiano et al., 1998). The best fit to the data of additive, random additive, or multiplicative functions was defined in each case.
Electron microscopy.
Two types of samples were processed for electron microscopy: hippocampal neuron cultures and hippocampi dissected from mouse brains. Hippocampal neuron cultures were fixed in 2% PFA and 1.5% glutaraldehyde in 0.1 m PB, pH 7.4, for 4 h. To obtain hippocampi, mice were deeply anesthetized by intraperitoneal injection of tribromoethanol (300 mg/kg), and perfused with 2% PFA and 2% glutaraldehyde, in 0.1 m PB, pH 7.4, after which the brains were removed and the hippocampi were dissected. Both the cultures and tissue samples were rinsed three times with PBS and processed for standard electron microscopy. This included treatment with osmium tetroxide (1% in 0.1 m PB), block staining with uranyl acetate, dehydration in a graded series of ethanol, and flat-embedding on glass slides in Durcupan (Fluka) resin. Regions of interest were cut at 70–90 nm on an ultramicrotome (Reichert Ultracut E; Leica) and collected on 200 mesh nickel grids. Staining was performed on drops of 1% aqueous uranyl acetate followed by Reynolds' lead citrate. Ultrastructural analyses were performed in a JEOL-1010 electron microscope. To establish the relative abundance of GABAergic and glutamatergic synapses in the WT and KO cultures, quantification was performed in cultures from three different animals of each genotype. Randomly selected areas were captured at a final magnification of 35,000×. The number of GABAergic synapses on the soma and dendritic shafts of 15 cells/animal (totaling 45 cells) and 5 dendrites/cell (totaling 225 dendrites) were counted. We also counted the number of glutamatergic synapses on the dendritic shafts and spines in 10 samples of 5.25 μm2 each per animal (totaling an area of 157.5 μm2). As GABAergic synapses are mainly established on dendritic shafts, we calculated the density of GABAergic synapses as number per 100 μm length of dendrite. However, as glutamatergic synapses are mainly established on dendritic spines, as well as on shafts, we calculated the density of glutamatergic synapses as a function of area (in square micrometers). We did not study the relative abundance of GABAergic versus glutamatergic synapses within individual samples. Rather, we compared the number of both types of synapses between the WT and KO mice. Consequently, the different metric used for GABAergic and glutamatergic synapses was very efficient and quantitatively useful for our purpose. Analysis of brain sections was performed in three pairs of littermate mice (P30–P36) including three KO, one WT, and two heterozygous mice.
Immunohistochemistry for electron microscopy.
Electron microscopic examination of immunoreactivity for CSP-α in the hippocampus of mice was performed as described previously using the preembedding immunogold method (Lujan et al., 1996). Briefly, brains were fixed by perfusion with 4% paraformaldehyde, 0.05% glutaraldehyde in 0.1 m phosphate buffer, pH 7.4. Next, sections (60 μm) were cut on a Vibratome (Leica VT1000S) and then incubated in Tris-buffered saline containing 10% of normal goat serum (TBS-NGS) during 1 h. After blocking, sections were incubated during 48 h in anti-CSP-α at a final protein concentration of 1–2 μg/ml diluted in TBS containing 1% NGS. We successfully used two rabbit polyclonal antibodies: R807 (Fernández-Chacón et al., 2004) and 154-003 from Synaptic Systems. Both antibodies gave virtually the same presynaptic labeling pattern. After several washes in TBS, sections were incubated for 2 h in goat anti-rabbit IgG coupled to 1.4 nm gold (Nanoprobes) diluted 1:100 in TBS containing 1% NGS. After several washes in PBS, the sections were postfixed in 1% glutaraldehyde diluted in the same buffer for 10 min. They were washed in double-distilled water, followed by silver enhancement of the gold particles with an HQ Silver kit (Nanoprobes). The sections were then treated with osmium tetroxide (1% in 0.1 m PB), block stained with uranyl acetate, dehydrated in graded series of ethanol, and flat-embedded on glass slides in Durcupan (Fluka) resin. Regions of interest were cut at 70–90 nm on an ultramicrotome (Reichert Ultracut E; Leica) and collected on 200 mesh nickel grids. Staining was performed on drops of 1% aqueous uranyl acetate followed by Reynolds' lead citrate. Ultrastructural analyses were performed in a JEOL-1010 electron microscope. To test method specificity in the procedures for electron microscopy, the primary antibody was either omitted or replaced with 5% (v/v) normal serum of the species of the primary antibody. Under these conditions, no selective labeling was observed.
Statistics.
Values are expressed as mean ± SEM. In general, every culture corresponds to a different mouse. Error bars in figures indicate SEM.
Results
Progressive and specific decrease of GABAergic synapses in CSP-α KO cultures
Hippocampal neurons from newborn CSP-α KO mice (Fernández-Chacón et al., 2004) and their WT littermates were cultured on rat astrocytes, which, compared with mouse astrocytes, proved more reliable in terms of maintaining long-term neuronal cultures (up to 40 DIV). Synapse density was assessed from thresholded binary images as the ratio between the number of pixels labeled with a general presynaptic marker (synapsins) and the pixels labeled with a somatodendritic marker (MAP2) (see Materials and Methods). Mutant and control neurons developed equally well, with a progressive increase in the total number of synapses observed for up to ∼3 weeks in vitro (11 DIV WT: synapsin/MAP2 ratio, 0.49 ± 0.06, n = 2 cultures, 22 fields; KO: 0.48 ± 0.05, n = 2 cultures, 20 fields; 19 DIV WT: 1.12 ± 0.11, n = 2 cultures, 20 fields; KO: 0.99 ± 0.06, n = 2 cultures, 20 fields) (Fig. 1A,B). Nevertheless, examination of the synapses after 1 month (30 d) in culture revealed that neurons lacking CSP-α failed to maintain the typically high number of synapses observed in control littermate cultures (WT: synapsin/MAP2 ratio, 1.03 ± 0.06, n = 3 cultures, 30 fields; KO: 0.75 ± 0.04, n = 3 cultures, 30 fields) (Fig. 1A,B). Those observations were consistent with a role for CSP-α in maintaining synapse stability. We then investigated whether the phenotype impacted every synapse equally or whether its effect was restricted to specific synaptic populations. Specific markers of GABAergic (GAD65) and glutamatergic (VGLUT1) synapses were used to quantify the proportion of inhibitory and excitatory synapses within the same cultures (Fig. 1C). Unexpectedly, we found that the proportion of GABAergic synapses in 30 DIV cultures from CSP-α KO mice was significantly decreased compared with that in WT cultures [WT, VGAT/(VGAT + VGLUT1) ratio, 0.35 ± 0.02, n = 3 cultures, 30 fields; KO, 0.12 ± 0.025, n = 3 cultures, 28 fields]. This observation strongly suggested that the number of GABAergic synapses was specifically reduced in the CSP-α KO cultures. However, an alternative interpretation, albeit less likely, was that the number of glutamatergic synapses in the CSP-α KO was upregulated despite a general decrease in synapse number.
Figure 1.
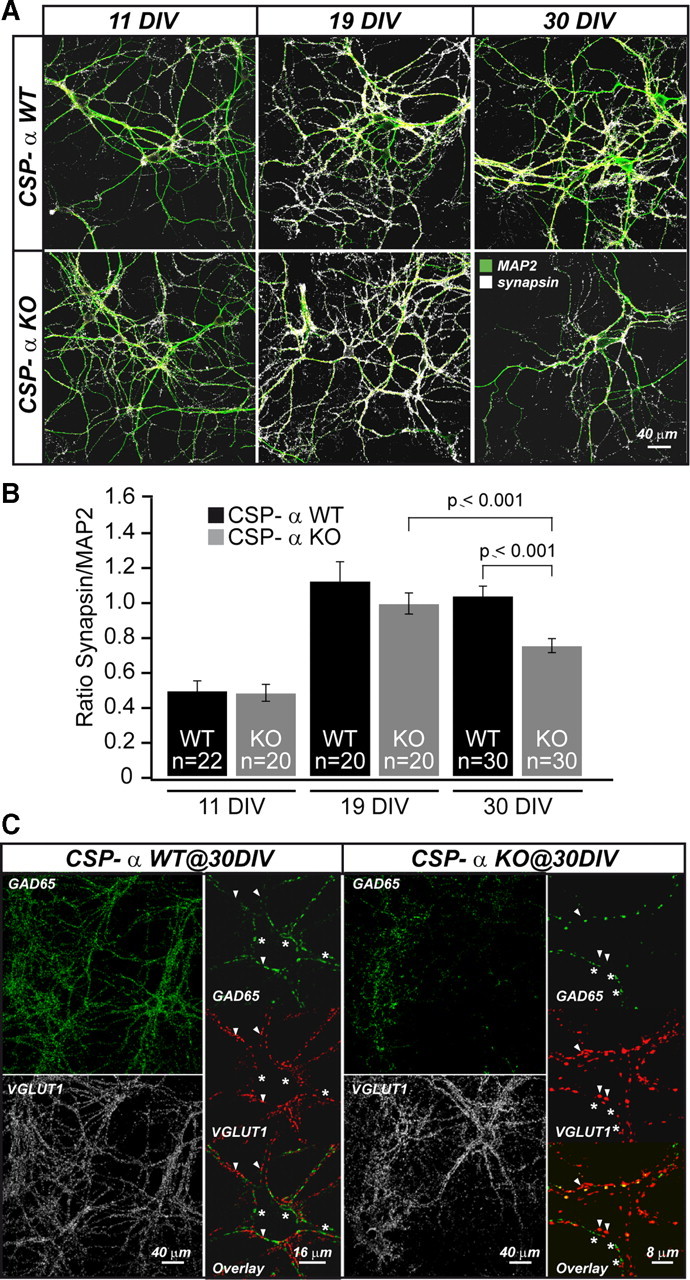
Progressive decrease in total synapse number in the absence of CSP-α. A, Confocal images of MAP2 (green) and synapsin (white) expression in WT and CSP-α KO neuronal cultures at three different time points. B, The synapsin/MAP2 ratio in CSP-α KO compared with WT cultures is only reduced at 30 DIV. Error bars indicate SEM. C, Confocal images of GABAergic (anti-GAD65; green; asterisks) and glutamatergic (anti-VGLUT1; white or red; arrowheads) synaptic puncta indicate a decreased proportion of GABAergic puncta in CSP-α KO cultures at 30 DIV.
To distinguish between these two possibilities, we directly quantified the number of GABAergic synapses, first using specific antibodies and then with electron microscopy. Antibodies against the general presynaptic marker synaptobrevin 2/VAMP2 were used to label all the synapses, after which we quantified the proportion that corresponded to GABAergic terminals by additional labeling with antibodies against the GABA transporter VGAT (Fig. 2). No significant differences in the proportion of GABAergic terminals existed between the mutant and control neurons at 11 DIV (31 ± 1.5%, n = 3 cultures, 30 fields, for WT; 25.5 ± 1.4%, n = 3 cultures, 30 fields, for KO). In contrast, older CSP-α KO cultures (30 DIV) suffered from a dramatic decrease in the number of GABAergic synapses compared with controls (42.5 ± 3.6%, n = 3 cultures, 31 fields, for WT; 21.6 ± 2.3%, n = 3 cultures, 30 fields, for KO; p ≤ 0.001, Mann–Whitney rank sum test). Similar results were obtained using GAD65 as a GABAergic marker (data not shown). These observations suggested that, in the absence of CSP-α, GABAergic synapses are initially established but later degenerate, perhaps because of a maintenance failure. CSP-α has been proposed to participate in the transport of GABA into synaptic vesicles by promoting the stability of protein complexes between GABA-synthesizing enzymes (GAD65 and GAD67) and VGAT (Hsu et al., 2000). Thus, it is possible that the decreased detection of GABAergic labeling with antibodies against GAD65 and VGAT was attributable to the specific degradation of these proteins within otherwise normal GABAergic presynaptic terminals.
Figure 2.

Progressive reduction in GABAergic synaptic puncta of CSP-α KO hippocampal neuron cultures. Confocal images at two different magnifications of WT and CSP-α KO hippocampal neuron cultures at 11 DIV labeled with anti-synaptobrevin 2/VAMP2 (green; asterisks) and anti-VGAT (white and red) antibodies identify GABAergic synaptic puncta at colocalization points (arrowheads) that coexist with non-GABAergic puncta (asterisks). A, At 11 DIV, the percentage of GABAergic puncta is similar in WT and CSP-α KO cultures. B, At 30 DIV, the percentage of GABAergic puncta is significantly reduced in CSP-α KO (21.6%) compared with WT cultures (42.5%).
To explore this possibility, we examined aged (30 DIV) hippocampal cultures with electron microscopy. Glutamatergic terminals were identified as small axon terminals filled with rounded synaptic vesicles, and forming asymmetric synapses with a prominent electron-dense specialization at the postsynaptic site (Fig. 3; supplemental Fig. 1, available at www.jneurosci.org as supplemental material). No changes were found in the number of glutamatergic terminals between control and mutant cultures (1.14 ± 0.09 synapses/μm2, n = 3 mice for WT; 0.99 ± 0.05 synapses/μm2, n = 3 mice for KO). GABAergic synapses were identified as medium to large terminals, filled with flattened synaptic vesicles, and with no prominent postsynaptic specialization at the postsynaptic site (Fig. 3; supplemental Fig. 1, available at www.jneurosci.org as supplemental material). WT cultures were rich in GABAergic terminals, unlike the CSP-α KO cultures in which GABAergic synapses were clearly diminished in size and number (7.1 ± 0.55 synapses/100 μm, n = 3 mice for WT, and 2.8 ± 0.33 synapses/100 μm, n = 3 mice for KO at 30 DIV; p = 0.002, Student's t test), equivalent to a 61.3% reduction in the CSP-α KO compared with the WT cultures. Furthermore, in the CSP-α KO neurons, otherwise normal dendrites often had no GABAergic inputs (Fig. 3H). These observations are consistent with the notion that, in CSP-α KO cultures, a subpopulation of GABAergic synapses is progressively lost, probably because of presynaptic degeneration, whereas the glutamatergic synapses are preserved. Next, we investigated whether this susceptibility to the absence of CSP-α was different for specific types of GABAergic synapses.
Figure 3.
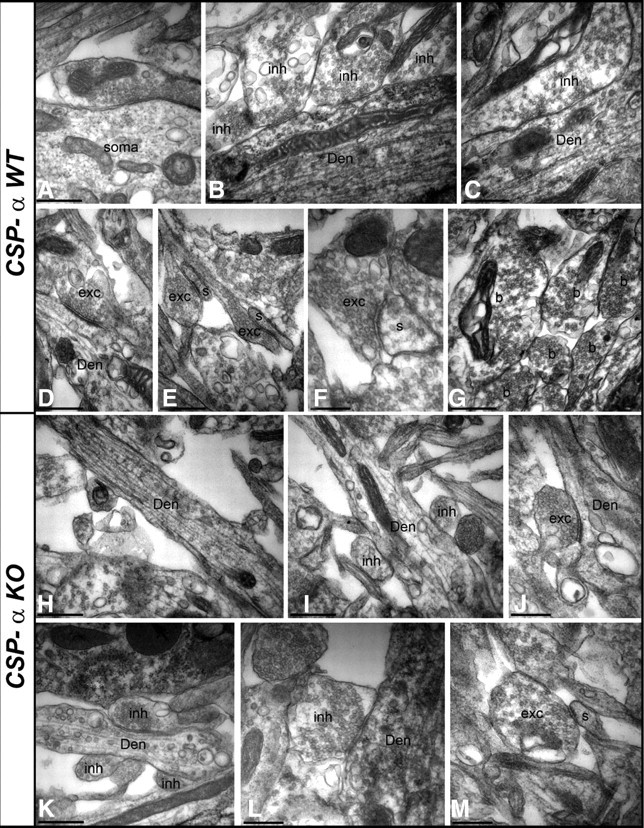
Ultrastructural analysis of GABAergic and glutamatergic synapses in culture. EM micrographs of glutamatergic and GABAergic synapses in WT (A–G) and KO (H–M) neuronal cultures. A–G, In the WT, GABAergic axon terminals (inh) establish inhibitory synapses with cell bodies (soma) and dendritic shafts (Den), and glutamatergic axon terminals (b) establish excitatory synapses with dendritic shafts (Den) and spines (s) of hippocampal neurons. H–M, In the KO, GABAergic (inh) and glutamatergic (exc) axon terminals were also detected, but the GABAergic axon terminals were generally smaller in size than those in the WT. Scale bars, 0.5 μm. The density of glutamatergic synapses did not change between WT and KO cultures, but there was a significant reduction in GABAergic synapses in the KO cultures (see Results).
Higher vulnerability of synapses from basket cells expressing Syt2 in CSP-α KO GABAergic neurons
CA1 pyramidal cells are supported by at least 16 distinct types of GABAergic neurons with different physiological, morphological, and neurochemical properties (Freund and Buzsáki, 1996; Somogyi and Klausberger, 2005; Klausberger and Somogyi, 2008). Among all the hippocampal interneurons, the PV-containing interneurons, the basket and axo-axonic cells, have the highest action potential firing rate (Pawelzik et al., 2002; Klausberger et al., 2003). Interestingly, cortical PV-positive interneurons in culture also develop fast-spiking properties (Kinney et al., 2006). The synaptic vesicle protein Syt2 is a presynaptic marker of a subpopulation of hippocampal basket cell terminals (Pang et al., 2006; Fox and Sanes, 2007; Kerr et al., 2008) (supplemental Fig. 2, available at www.jneurosci.org as supplemental material). In addition, Syt2 is also detected in smaller synaptic populations at stratum lacunosum moleculare (supplemental Fig. 2, available at www.jneurosci.org as supplemental material) and in the subiculum (Su et al., 2010). We therefore first stained 1-month-old cultures with antibodies against the GABA transporter VGAT and Syt2 (Fig. 4A). WT cultures were rich in synapses containing Syt2, which always colocalized with VGAT (Fig. 4A). In contrast, in CSP-α KO cultures, none of the GABAergic terminals that survived after 30 DIV expressed Syt2 (Fig. 4A,B). This observation suggested a higher vulnerability of Syt2-expressing GABAergic terminals in the absence of CSP-α. Almost all Syt2-positive puncta presented PV immunoreactivity in WT cultures (Fig. 4C), suggesting that the majority of Syt2-expressing synapses in culture were formed by PV-expressing basket cells. Next, we used antibodies against PV and CR to label interneurons with high- and low-activity levels, respectively (Gulyás et al., 2006). We also used antibodies against MAP2 to label the somata of all cultured neurons. The total number of WT neurons in aged cultures (36–39 DIV) was reduced by 24.7% compared with that in younger (18–20 DIV) cultures (Fig. 4C, left panel). During the same period, however, the decrease in the number of neurons in the CSP-α KO cultures was much greater (50.9% reduction), suggesting that CSP-α is important for neuronal survival. We then examined the number of CR-positive interneurons and found that, although the number of neurons was reduced in old compared with young cultures, the reduction was similar for both the WT (48.6%) and the CSP-α mutant (51.2%) neurons (Fig. 4C, middle panel). In contrast, we found a dramatic difference in the number of PV-containing neurons: in old WT cultures, the number of PV-containing neurons was reduced by 41.9% compared with the number found in young WT cultures. Surprisingly, however, we failed to detect any PV-positive neurons in the CSP-α KO cultures at the two in vitro ages examined (18–20 and 36–39 DIV) (Fig. 4C, right panel). To determine whether our culture results reflected a similar situation in vivo, we examined P31 hippocampal sections labeled with antibodies against PV (Fig. 5) and performed quantitative stereological measurements. Surprisingly, in contrast to our in vitro observations, we found no differences in the number of hippocampal PV-containing neurons between WT and littermate mutant brains (1610 ± 185 neurons/mm3, n = 7 mice in WT, compared with 1659 ± 182 neurons/mm3, n = 7 mice in CSP-α KO; p = 0.853, Student's t test). Strikingly, however, we found that the PV labeling of the pyramidal and granular layers, to which PV-positive axons project, was much weaker in the mutant than in the control brains (Fig. 5A,B,C′–F′; supplemental Fig. 3, available at www.jneurosci.org as supplemental material), suggesting a reduction in the number of PV-positive terminals, as would occur with synaptic degeneration. Consistent with this, we also found that Syt2 labeling of these layers was much weaker in the mutant than in the WT (Fig. 5A,B,C–F; supplemental Fig. 3, available at www.jneurosci.org as supplemental material).
Figure 4.
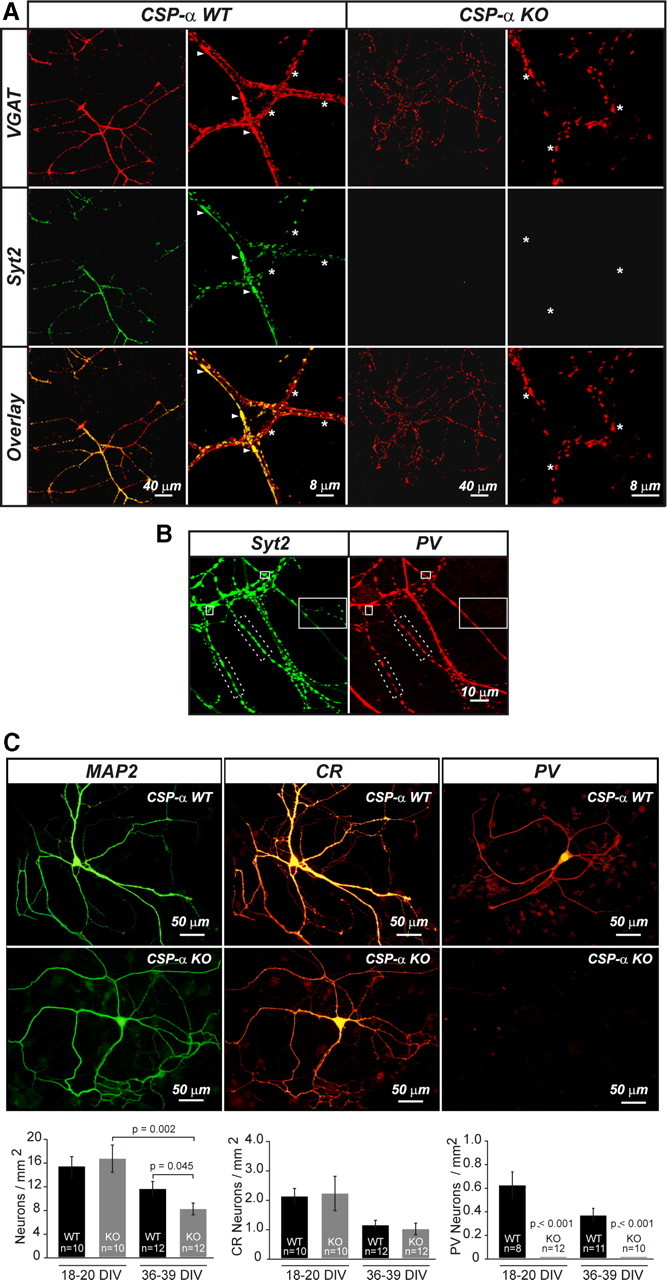
Syt2-positive GABAergic synapses are strongly reduced in cultures lacking CSP-α. A, Confocal images at two different magnifications of hippocampal neuron cultures labeled with anti-VGAT (red) and anti-Syt2 (green) antibodies identify GABAergic synaptic puncta colocalizing (arrowheads) and not colocalizing (asterisks) with Syt2 in WT cultures. In CSP-α KO cultures, GABAergic puncta (asterisks), but not Syt2-positive puncta, are present. In WT cultures, the percentage of colocalized VGAT/Syt2-positive puncta at 30 DIV is 55.2 ± 2.7% (n = 4 cultures; 60 fields). In CSP-α KO cultures, Syt2-positive puncta is not detected (n = 4 cultures; 60 fields). B, Maximum projection of confocal images from WT neuron cultures showing Syt2- (green) and PV-positive puncta (red). Most of Syt2-positive puncta come from PV-positive neurons (dotted border rectangle), and only a few spots show no Syt2/PV colocalization (continuous border rectangle). That image is not representative and it was selected to show infrequent Syt2 synapses that do not colocalize with PV. C, Left, Stronger progressive decrease in the number of CSP-α KO neurons labeled with anti-MAP2 antibodies compared with WT (average density: 18–20 DIV WT, 15.3 ± 1.8 neurons/mm2, n = 2 cultures, 10 coverslips; KO, 16.6 ± 2.3 neurons/mm2, n = 2 cultures, 10 coverslips, nonsignificant differences; 36–39 DIV WT, 11.6 ± 1.3 neurons/mm2, n = 2 cultures, 12 coverslips; KO, 8.1 ± 1.0 neurons/mm2, n = 2 cultures, 12 coverslips; p = 0.045, Student's t test) and between KO cultures at different time points (p = 0.002, Student's t test). Middle, Similar progressive decrease in the number of WT and CSP-α KO neurons labeled with anti-CR antibodies (average density: 18–20 DIV WT, 2.1 ± 0.3 neurons/mm2, n = 2 cultures, 10 coverslips; KO, 2.2 ± 0.6 neurons/mm2, n = 2 cultures, 10 coverslips; 36–39 DIV WT, 1.1 ± 0.2 neurons/mm2, n = 2 cultures, 12 coverslips; KO, 1.0 ± 0.2 neurons/mm2, n = 2 cultures, 12 coverslips). Right, Progressive decrease in the number PV-expressing neurons in WT contrasts with no detection of PV-positive neurons in CSP-α KO cultures (average density in WT at 18–20 DIV: 0.6 ± 0.1 neurons/mm2, n = 3 cultures, 8 coverslips; 36–39 DIV: 0.4 ± 0.1 neurons/mm2, n = 3 cultures, 11 coverslips; p ≤ 0.001, Mann–Whitney rank sum test). Error bars indicate SEM.
Figure 5.

Reduction in synaptic Syt2 and PV labeling and degeneration of basket cell terminals in CSP-α KO hippocampus. A, B, Confocal images from WT (A) and CSP-α KO (B) hippocampal slices labeled with anti-Syt2 (green) and anti-PV (red) antibodies. Stereological analysis shows no differences in the number of PV-positive somata (arrowheads) between the CSP-α WT and CSP-α KO hippocampus at P31–P40. However, note the different intensity in synaptic labeling (arrows) in the stratum pyramidale and granule cell layer. Syt2 staining also detected at the stratum lacunosum moleculare (dots). Labeling profiles through the z-axis demonstrate homogenous antibody penetration through the whole slice. C–D″, CA1 region in WT (C–C″) and KO (D–D″) hippocampal slices. Weaker Syt2 (green) and PV (red) labeling can be seen in the KO stratum pyramidale (asterisks). E–F″, Dentate gyrus in WT (E–E″) and KO (F–F″). There is a reduction in PV-positive puncta in the CSP-α KO (asterisks). Data from four WT and four KO littermates. G–L, EM analysis of excitatory (H, L) and inhibitory (G, I–K) synapses in the dentate gyrus. In the WT, GABAergic axon terminals (bt) (G) from basket cells establishing GABAergic synapses around the somata of granule cells (GC) are medium to large in size and are filled with synaptic vesicles. In the CSP-α KO mice (I–K), these terminals are smaller in size with less synaptic vesicles. In contrast, glutamatergic axon terminals (b) establishing excitatory synapses with dendritic shafts (Den) or spines (s) in the molecular layer of the dentate gyrus or close to the somatic inhibitory synapses are virtually similar in both WT (H) and CSP-α KO (L) mice. SO, Stratum oriens; SP, stratum pyramidale; SR, stratum radiatum; ML, molecular layer; GCL, granule cell layer.
These differences in staining intensity were less evident with antibodies against general GABAergic markers (VGAT and GAD65) (data not shown), suggesting that the phenotype did not impact as strongly on other types of GABAergic synapses as it did on the Syt2-positive synapses of basket cells. In accordance with this interpretation, electron microscopy examination of the dentate gyrus in brain sections from 1-month-old WT and CSP-α KO mice (Fig. 5G–L) revealed clear signs of degeneration at the basket cell terminals projecting onto granule cells in the mutant mice (Fig. 5I,J, bt). These changes included a reduction in the size of the inhibitory axon terminals. In contrast, glutamatergic synapses in the molecular layer of the dentate gyrus or close to GABAergic synapses in the granule cell layer were similar in the WT and KO brain sections (Fig. 5G–K, GC).
Next, we isolated hippocampi from mice at different ages to quantify protein levels (Fig. 6). No differences in the levels of key synaptic proteins such as synaptobrevin 2/VAMP2 and synaptotagmin1 were observed between WT and CSP-α KO mice at any age. In contrast, Syt2 and PV protein levels were significantly reduced in the CSP-α KO hippocampus by 3 weeks of age (Fig. 6). In summary, therefore, Syt2-expressing, hippocampal GABAergic synapses, from fast-spiking basket cells, underwent degeneration in the absence of CSP-α, whereas glutamatergic synapses appeared normal. However, it could be that, in the mutant, the apparently normal glutamatergic and young GABAergic synapses might have functional abnormalities undetectable by morphological analysis. Thus, to examine presynaptic function, we studied the spontaneous release of neurotransmitters at both inhibitory and excitatory synapses in cultured neurons.
Figure 6.

Postnatal decrease in Syt2 and PV hippocampal protein levels in CSP-α KO mice. A, Representative immunoblots of hippocampal proteins from WT and CSP-α KO mice at P16, P20, P28, and P32 labeled with antibodies against CSP-α, Hsc70, Syt2, PV, synaptobrevin 2/VAMP2 (Syb2), synaptotagmin 1 (Syt1), and α-actin. B, Normalized protein levels assessed by measurement of direct chemiluminescence and relative to α-actin levels. Data are plotted as mean ± SEM; n = 4 for each genotype and for each marker; statistical significance was determined by Student's t test.
Progressive decrease in mIPSC frequency in CSP-α KO cultures
In 1-month-old cultures (30–33 DIV), we recorded mIPSCs caused by the spontaneous release of GABA from single synaptic vesicles, in the presence of tetrodotoxin, to block action potentials, and NBQX and APV, to block glutamatergic inputs. In the WT cultures, all the recorded neurons presented inhibitory inputs, which were reversibly blocked with the GABAA receptor antagonist bicuculline. In just over one-half of the WT neurons (52%), the mIPSC frequency was low enough (<13 Hz) to reliably distinguish and isolate single events (supplemental Fig. 4, available at www.jneurosci.org as supplemental material). We classified those recordings as “type I spontaneous responses.” However, the remainder of the recordings (48%) revealed a remarkably high frequency of mIPSCs in which temporal overlapping of single events impeded reliable event detection and quantification of mIPSC frequency. We termed those recordings “type II spontaneous responses.” We reasoned that neurons with type I responses simply received fewer inhibitory inputs than neurons with type II responses. Both types of response were also detected in CSP-α KO cultures, although in different proportions: 75% of type I (low-frequency) and 10% of type II (high-frequency) responses. We interpreted this shift toward type I responses as a decrease in functional inhibitory input in the mutant, probably because of a reduction in the number of actual GABAergic contacts. Consistent with this view, we also determined that 15% of the recorded mutant neurons showed no mIPSCs (supplemental Fig. 4, available at www.jneurosci.org as supplemental material), presumably because they received no GABAergic contacts. In addition, we further analyzed type I responses and found a significant reduction in mIPSC frequency in the mutant cultures (9.1 ± 0.3 Hz, n = 4 cultures, 37 cells, for WT, and 5.0 ± 0.4 Hz, n = 4 cultures, 53 cells, for KO at 30 DIV; p ≤ 0.001, Mann–Whitney rank sum test) but no change in the size of individual mIPSCs (38.5 ± 2.9 pA, n = 4 cultures, 37 cells, for WT, and 36.0 ± 2.6 pA, n = 4 cultures, 53 cells, for KO at 30 DIV) (Fig. 7A,B). Once again, this probably reflected a functional decrease in the number of inhibitory inputs. In contrast to the reduced mIPSC frequency at 30 DIV, there was clearly no decrease in either frequency (4.9 ± 0.4 Hz, n = 3 cultures, 52 cells, for WT, and 7.1 ± 0.6 Hz, n = 3 cultures, 40 cells, for KO; p = 0.007, Mann–Whitney rank sum test) or mIPSC size (48.7 ± 3.6 pA, n = 3 cultures, 52 cells, for WT, and 58.8 ± 3.8 pA, n = 3 cultures, 40 cells, for KO; p = 0.056 Mann–Whitney rank sum test) in younger mutant cultures (11 DIV) compared with littermate controls (Fig. 7C,D). Intriguingly, the frequency at 11 DIV was even slightly higher in the CSP-α KO cultures. Although this observation reinforced the idea that there was no reduction in the number of inhibitory inputs at younger ages, it did suggest that the absence of CSP-α might produce a subtle early instability of presynaptic function. The above set of experiments, consistent with the decrease in GABAergic synapse number, strongly supported the notion that CSP-α is crucial to keep the integrity of inhibitory inputs for extended periods of time. We therefore next evaluated the ability of CSP-α to specifically support glutamatergic synaptic function over time.
Figure 7.

Decrease in mIPSC frequency, but not amplitude, in CSP-α KO hippocampal cultures at 30 DIV. A, C, mIPSCs in WT and KO neuron cultures at 30 and 11 DIV; bicuculline reversibly abolishes the mIPSCs. B, Histogram and cumulative distributions of mIPSC amplitudes from WT and KO hippocampal neuron cultures at 30 DIV. Inset, Average mIPSC traces for both genotypes. D, Histograms and cumulative distributions of mIPSC amplitudes from WT and KO hippocampal neuron cultures at 11 DIV. Inset, Average mIPSC traces for both genotypes.
Progressive decrease in AMPAR-mediated mEPSC size in CSP-α KO cultures
To investigate the spontaneous release of glutamate, we recorded AMPAR-type mEPSCs in the presence of tetrodotoxin in aged cultures (30–33 DIV). This analysis revealed no changes in the mEPSC frequency in mutant cultures compared with controls (14.8 ± 0.8 Hz, n = 5 cultures, 34 cells, for WT, and 13.0 ± 0.8 Hz, n = 5 cultures, 25 cells, for KO at 30 DIV; p = 0.136, Student's t test) (Fig. 8A,B). Nevertheless, we were surprised to find that the mEPSC amplitude was strongly reduced to approximately one-half of the control mEPSC size (22.3 ± 2.2 pA, n = 5 cultures, 34 cells, for WT, and 11.7 ± 1.1 pA, n = 5 cultures, 25 cells, for KO at 30 DIV; p ≤ 0.001, Mann–Whitney rank sum test). This observation was confirmed in all the cultures that we studied. No kinetic alterations were associated with the size reduction: Scaling up the average trace obtained from multiple mEPSCs yielded a trace coincident with the average WT mEPSC trace. This finding prompted the idea that, although we had not found any structural changes in cultured glutamatergic synapses, CSP-α might have some regulatory role in presynaptic function not related to the preservation of synaptic terminals, as for example the filling of synaptic vesicles with glutamate. Given that such a defect, instead of being progressive, should already be detectable in younger cultures, we determined the amplitude and frequency of AMPAR-type mEPSCs at 11 and at 21 DIV but found no differences between the mEPSCs recorded in mutant cultures and controls in terms of either amplitude (22.2 ± 3.3 pA, n = 4 cultures, 19 cells, for WT, and 19.5 ± 3.6 pA, n = 4 cultures, 16 cells, for KO at 11 DIV; 20.1 ± 3.4 pA, n = 4 cultures, 17 cells, for WT, and 18.9 ± 3.5 pA, n = 4 cultures, 15 cells, for KO at 21 DIV) or frequency (13.6 ± 0.5 Hz, n = 4 cultures, 19 cells, for WT, and 11.9 ± 0.9 Hz, n = 4 cultures, 16 cells, for KO at 11 DIV; 15 ± 1.0 Hz, n = 4 cultures, 17 cells, for WT, and 13.9 ± 0.9 Hz, n = 4 cultures, 15 cells, for KO at 21 DIV) (Fig. 8C,D) (data not shown). Thus, we concluded that, similar to what happened to GABAergic synapses, the functional changes in glutamatergic synapses were also progressive and depended on the culture period. However, in contrast to the changes in GABAergic terminals, we could not detect any structural defect in the glutamatergic terminals. To get additional insight into the functional changes in glutamatergic transmission, we therefore expanded our study to analyze mEPSCs mediated by NMDARs.
Figure 8.
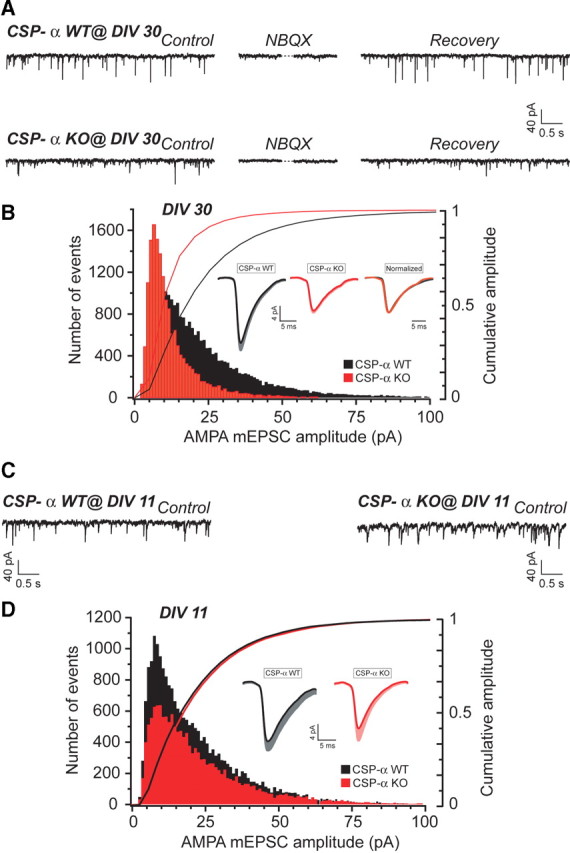
Downscaling of AMPAR-mediated mEPSCs in long-term CSP-α KO hippocampal cultures. A, Normal frequency of AMPAR-mediated mEPSCs in CSP-α KO cultures compared with WT at 30 DIV. NBQX reversibly blocks AMPAR mEPSCs. B, Strong reduction in the size of AMPAR-mediated mEPSCs in CSP-α KO cultures at 30 DIV. Histograms and cumulative distributions of amplitudes for both genotypes (error bars indicate SEM). Inset, Averaged AMPAR mEPSC waveforms for both genotypes and overlaid normalized KO response show no kinetic differences. C, Normal frequency of AMPAR-mediated mEPSCs for both genotypes at 11 DIV. D, Histograms and cumulative distributions of amplitudes for both genotypes show similar size of AMPAR-mediated mEPSCs in CSP-α KO and WT cultures at 11 DIV. Inset, Averaged AMPAR mEPSC waveforms for both genotypes (error bars indicate SEM).
Synaptic downscaling of AMPAR in the absence of CSP-α
In the presence of tetrodotoxin and NBQX in a bath solution with no added Mg2+, we measured the mEPSCs produced by the activation of NMDA receptors on glutamate release from single synaptic vesicles in aged hippocampal cultures (30 DIV). In contrast to the significant decrease in AMPAR-type mEPSC amplitude, we found no significant differences in amplitude (17.7 ± 2.2 pA, n = 3 cultures, 18 cells, for WT, and 20.8 ± 2.4 pA, n = 3 cultures, 19 cells, for KO at 30 DIV; p = 0.054, Mann–Whitney rank sum test) or kinetics in NMDAR-type mEPSCs between cultures from CSP-α KO and WT littermate mice (Fig. 9A,B). We did, however, find a small, but significant, increase in the frequency of NMDAR-mediated mEPSCs (3.2 ± 0.2 Hz, n = 3 cultures, 18 cells, for WT, and 4.1 ± 0.2 Hz, n = 3 cultures, 19 cells, for KO; p = 0.028, Student's t test). The possible involvement of silent synapses in this phenomenon (Gomperts et al., 1998) remains to be investigated in future studies. In the case of a presynaptic defect in glutamate release, we would have expected a reduction in the amplitude of both AMPAR-type and NMDAR-type mEPSCs. Thus, since we only observed a reduction in AMPAR-type mEPSCs, we reasoned that the functional changes in glutamatergic synapses emerged from a postsynaptic alteration in either the expression or the trafficking of glutamatergic AMPARs (Malinow and Malenka, 2002). This was a somewhat unexpected possibility, given that CSP-α had not previously been localized at the postsynaptic terminal. Nonetheless, we directly investigated postsynaptic expression of CSP-α. This was achieved by carrying out immunocytochemical labeling of hippocampal cultures with antibodies against CSP-α, first with the presynaptic marker synaptobrevin 2/VAMP2 (Baumert et al., 1989), and then with the postsynaptic markers GluR1 (Craig et al., 1993) and gephyrin (Kirsch and Betz, 1995). As expected, CSP-α and synaptobrevin 2/VAMP2 colocalized at presynaptic terminals. In contrast, spots of GluR1 and gephyrin labeling appeared in front of, but not overlapping with, the CSP-α labeling (supplemental Fig. 5, available at www.jneurosci.org as supplemental material). These observations suggested that CSP-α is a presynaptic protein. Nevertheless, since confocal fluorescence microscopy might still lack the high spatial resolution required to totally separate presynaptic and postsynaptic labeling, we performed immunoelectron microscopy techniques on mouse hippocampal sections using anti-CSP-α antibodies. This demonstrated, independently with two different antibodies (see Materials and Methods), a remarkable labeling of CSP-α in presynaptic terminals of excitatory and inhibitory neurons (supplemental Fig. 6, available at www.jneurosci.org as supplemental material). Although it was possible to detect some labeling at the postsynaptic terminal, the level was extremely low (only 18 of 1297 gold particles, representing 0.08% of all the gold particles detected, were in dendrites) and probably because of residual background staining. Thus, we concluded that CSP-α is only a presynaptic and not a postsynaptic protein.
Figure 9.
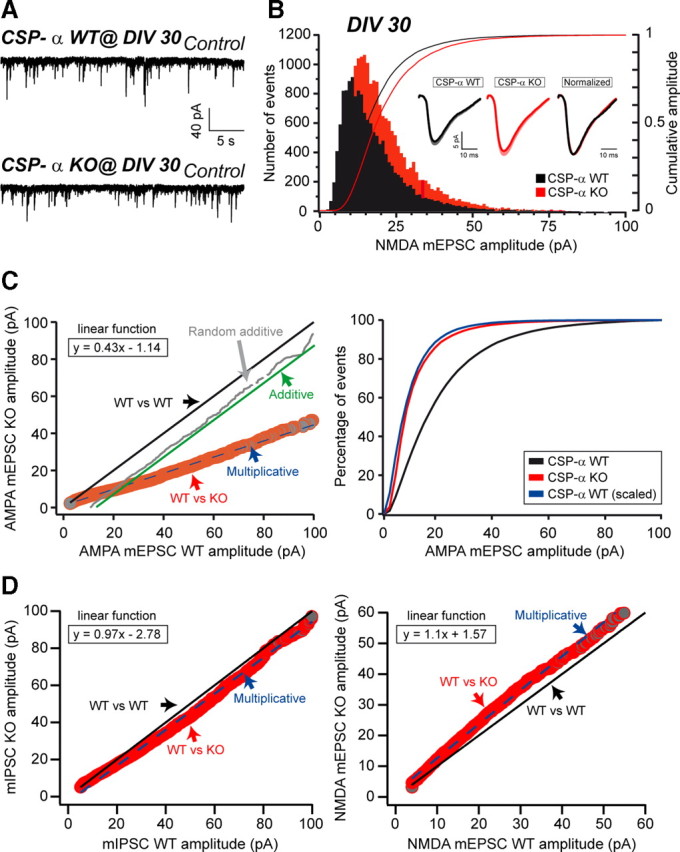
Multiplicative downscaling of AMPAR mEPSCs in the absence of CSP-α. A, NMDAR mEPSCs in WT and CSP-α KO neuronal cultures at 30 DIV. B, Histograms and cumulative distributions of NMDAR mEPSC amplitudes from WT and KO cultures at 30 DIV. Inset, Average NMDAR mEPSC traces for both genotypes and overlaid normalized response. C, Left, Ranked control AMPAR mEPSC amplitudes plotted against ranked CSP-α KO AMPAR mEPSC amplitudes. The best fit to the data of additive, random additive, or multiplicative functions was defined. Best fit: KO = WT × 0.43 − 1.14 (multiplicative function). Right, Cumulative amplitude histograms for WT and KO AMPAR mEPSCs. WT distribution was transformed according to the preceding multiplicative function and plotted together with the KO data. D, Left, Ranked control mIPSC amplitudes plotted against ranked KO mIPSC amplitudes. The best fit to the data was a multiplicative function with slope of 0.97 (KO = WT × 0.97 − 2.78). Right, Similar plot for comparison of WT and KO NMDAR mEPSC amplitudes. Best fit: KO = WT × 1.1 + 1.57 (multiplicative function). Slopes close to 1 indicate no size escalation of mIPSCs and NMDAR-mEPSCs in CSP-α KO cultures.
Having excluded a direct postsynaptic role of CSP-α, we queried whether the changes at excitatory synapses could be a consequence of the initial deficit of inhibitory inputs. Removal of synaptic inhibition increases the action potential firing of neuronal networks. Under these conditions, homeostatic plasticity mechanisms restore the neuronal firing to normal levels by weakening excitatory synapses through the removal of AMPARs (Turrigiano et al., 1998; Turrigiano and Nelson, 2004). Indeed, in cultured neurons, blocking of GABAergic synaptic transmission with bicuculline, an antagonist of GABAA receptors, leads to synaptic downscaling of glutamatergic synapses typically characterized by a reduction in the size of AMPAR-type mEPSCs (Turrigiano et al., 1998). Since such a feature was strikingly similar to the phenotype of the CSP-α KO glutamatergic synapses, we hypothesized that the observed downscaling of AMPAR-mediated mEPSCs was attributable to synaptic scaling through homeostatic plasticity mechanisms. To investigate this, we studied the relationship between the WT and CSP-α KO mEPSC amplitude distributions by ranking mEPSC amplitudes from all the cells recorded from both control and mutant cultures in ascending order. As has been previously described (Turrigiano et al., 1998), we plotted the control amplitudes against the CSP-α KO amplitudes, and the resulting relationship was fit to different models (additive, random additive, and multiplicative function) (Fig. 9C, left panel). The data were only well fit by a linear function with a slope of 0.43, indicating that the relationship between control and mutant amplitudes followed a multiplicative model. After scaling the events from the control cultures according to that linear function, the resulting cumulative distribution overlapped with the cumulative mutant distribution (Fig. 9C, right panel). When the same type of analysis was applied to the distributions of GABA-mediated mIPSCs and NMDAR-mediated mEPSCs, it revealed that, for both of these distributions, the relationship between WT and CSP-α KO events was linear, with a slope close to 1 (slope = 0.97 for GABAergic mIPSCs and slope = 1.1 for NMDA-mEPSCs) (Fig. 9D). Thus, our data suggested that, in the absence of CSP-α, hippocampal glutamatergic synapses underwent homeostatic plasticity changes by specific regulation of AMPA-type receptors on the postsynaptic surface. This is the expected response in the face of a functional decrease in inhibitory input and, therefore, is consistent with the observed reduction of GABAergic synapses in the absence of CSP-α. However, this raised a new question: Why is the CSP-α requirement for long-lasting synaptic maintenance higher in GABAergic than in glutamatergic terminals? One possible explanation is that GABAergic synaptic vesicles specifically require CSP-α (Hsu et al., 2000; Jin et al., 2003), but we did not see a general GABAergic phenotype or a reduction in mIPSC size. However, since certain GABAergic neurons show severalfold higher discharge rates than principal cells (Csicsvari et al., 1999; Jonas et al., 2004), the action potential firing rate could be a key factor in triggering the degeneration of inhibitory terminals earlier than that of other terminals. If this hypothesis were correct, then the phenotype should be ameliorated by reducing network excitability in the culture, a possibility we next investigated.
GABAergic synapses rescued from degeneration on blockade of excitatory transmission
To reduce the excitability of the neuronal network in culture, we used APV and NBQX to block glutamatergic synapses (Thiagarajan et al., 2005; Mateos et al., 2007) for 3 weeks (from 10 to 30 DIV) and compared the relative number of GABAergic synapses in control conditions for WT and CSP-α KO cultures. Figure 10A shows that WT and CSP-α KO cultures maintained a robust number of synapses labeled with synaptobrevin 2/VAMP2 antibodies. However, as previously observed (Fig. 2B), the staining of hippocampal cultures with the GABAergic marker VGAT was dramatically weaker in the CSP-α KO cultures compared with the WT. The level of synaptobrevin 2/VAMP2 labeling was also reduced in cultures of both genotypes treated with glutamatergic blockers, compared with nontreated cultures. In contrast, the labeling of GABAergic synapses was remarkably increased in the CSP-α KO cultures treated with glutamatergic blockers in comparison with the nontreated cultures (Fig. 10A).
Figure 10.
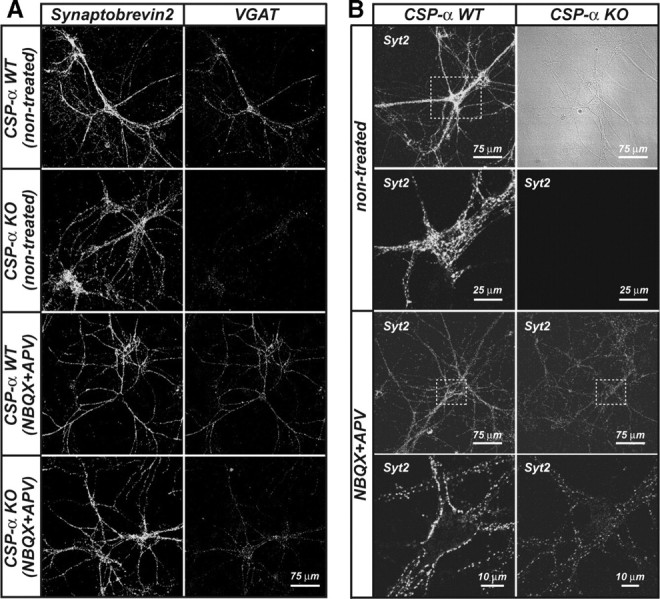
Recovery of GABAergic synaptic puncta in cultures lacking CSP-α after long-term blocking of glutamatergic synaptic transmission. A, Confocal images of synaptobrevin 2/VAMP2 and VGAT presynaptic staining of WT and KO neuronal cultures at 30 DIV after 3 weeks of glutamatergic transmission blockade. B, Recovery of Syt2 synaptic labeling at 30 DIV after glutamatergic blocking in KO cultures (n = 3 cultures). The right panels show magnification of selected areas from left panels, except for panel CSP-α (control), in which a bright-field image of the right panel demonstrates the presence of nonlabeled neurons.
The percentage of GABAergic synapses was calculated from the ratio between pixels doubly positive for VGAT and Syb2 versus the total number of Syb2-positive pixels for neuronal cultures at 30 DIV. Data from mutant cultures were normalized to the changes observed between treated and nontreated littermate WT cultures. Although the rescue in the number of GABAergic synapses in the mutant was only partial compared with the WT, the increase in the number of GABAergic synapses was dramatic: almost three times as many GABAergic synapses were labeled in the treated versus untreated mutant cultures (nontreated KO ratio: 0.19 ± 0.03, n = 3 cultures, 30 fields; treated KO ratio: 0.54 ± 0.043, n = 3 cultures, 40 fields; p ≤ 0.001, Mann–Whitney rank sum test).
Furthermore, the absence of Syt2 labeling in the mutant cultures at every stage examined (Fig. 4A) was reversed in the mutant cultures deprived of excitatory synaptic transmission (Fig. 10B). We also stained the cultures with antibodies against PV, but we did not see recovery of PV labeling in the treated CSP-α KO cultures. However, interpreting these results is difficult, given that PV expression is triggered by high levels of neural activity (Jiang and Swann, 2005) and blocked by NMDAR antagonists (Kinney et al., 2006), as evidenced by the fact that the level of PV labeling was also greatly reduced in treated WT cultures (data not shown). These observations are consistent with the notion that synaptic activity is a major cause of the synaptic phenotype produced by the absence of CSP-α, and support the concept of CSP-α as a chaperone particularly suited to protect synapses from activity-induced stress.
Discussion
Previous work on KO mice lacking the synaptic vesicle protein CSP-α led to two important hypotheses: (1) synaptic activity increases nerve terminal vulnerability and (2) CSP-α is part of the molecular machinery that prevents synapse rundown caused by the molecular stress induced by synaptic activity (Fernández-Chacón et al., 2004; Chandra et al., 2005; Schmitz et al., 2006). Here, we provide experimental evidence in support of these concepts in hippocampal synapses. More specifically, we have shown that the Syt2-expressing GABAergic synapses (Pang et al., 2006; Fox and Sanes, 2007; Kerr et al., 2008) established by neurons which typically fire action potentials at very high frequencies (∼200 Hz) (Ylinen et al., 1995; Csicsvari et al., 1999) are extremely dependent on CSP-α to maintain their functional and structural integrity over time, and therefore suffer from progressive degeneration on genetic removal of CSP-α. In contrast, neighboring glutamatergic synapses from neurons that normally trigger action potentials at a lower frequency (∼1 Hz) (Csicsvari et al., 1999; Frerking et al., 2005) do not show signs of presynaptic degeneration in the absence of CSP-α. These conclusions are based on the following findings: First, using immunofluorescence and electron microscopy, we have demonstrated progressive degeneration of GABAergic synapses both in vitro (Figs. 1–4) and in vivo (Fig. 5). Remarkably, basket cell terminals that form synapses onto dentate gyrus granule cells show clear signs of presynaptic neurodegeneration (Fig. 5I–K). Second, we have identified a significant in vivo reduction in Syt2 protein levels (Fig. 6) and Syt2-positive basket cell terminals in the hippocampus of mutant mice (Fig. 5; supplemental Fig. 3, available at www.jneurosci.org as supplemental material), and shown that this reduction is greater in neuronal cultures in which basically no Syt2-positive terminals were detected (Fig. 4). Syt2 is also detected in small synaptic populations, for example, at the stratum lacunosum moleculare, that do not express or express low levels of PV (Fig. 5A, dots). Likely, those neurons are poorly represented in our cultures because the number of Syt2-expressing synapses that do not colocalize with PV is extremely low (Fig. 4C). We cannot rule out that other interneurons coexpressing Syt2 and PV are mixed with basket cells in our cultures, but, in any case they would also be very sensitive to the lack of CSP-α. In addition, PV protein levels (Fig. 6) and PV-positive terminals were also decreased in vivo but no changes were detected in the number of PV-positive neurons as quantified with stereological methods (Fig. 5; supplemental Fig. 3, available at www.jneurosci.org as supplemental material). Intriguingly, although there was no in vitro change in CR-positive GABAergic neurons in the mutant compared with the WT, no PV-positive neurons were detected in cultures lacking CSP-α (Fig. 4). This might reflect the higher vulnerability of PV-positive neurons, given that the majority of hippocampal PV-expressing cells are fast-spiking (Pawelzik et al., 2002) and that this population includes the most active hippocampal interneurons: basket cells and axoaxonic chandelier cells (Howard et al., 2005; Freund and Katona, 2007). In contrast, CR-positive cells are less active and have lower energy requirements (Gulyás et al., 2006). It is therefore possible that initial presynaptic disturbance produced by the removal of CSP-α compromises the uptake and retrograde transport of the neurotrophic factors that are essential for survival of PV-expressing neurons (Tao and Poo, 2001). Likely, such a dying-back process is much faster in culture because neurons are removed from a more nurturing physiological hippocampal niche. Since CSP-α KO mice die rather early (6–8 weeks age), only significant changes in PV protein levels and in PV-synaptic staining are detected in vivo (Figs. 5, 6). Predictably, if the mice survived several months, then a decreased number of PV-positive somata will be detected. Future experiments based on neuron-specific knockdown of CSP-α in vivo, allowing prolonged longevity, will help to investigate that issue. Third, electrophysiological measurements of GABAergic synaptic transmission, consistent with our morphological observations, showed a progressive decrease in the frequency of mIPSCs in CSP-α KO synapses (Fig. 7) but no changes in the size or kinetics of individual mIPSCs. In contrast, the frequency of AMPAR-mediated mEPSCs was normal in the mutant compared with the WT (Fig. 8), although the amplitude of these postsynaptic currents was dramatically (50%) reduced. Two key findings support the idea that the reduction in mEPSC size is produced by homeostatic plasticity changes: (1) the mechanism is postsynaptic as no changes were detected in the size or kinetics of NMDAR-mediated mEPSCs, and (2) the comparison of ranked amplitudes between AMPAR-mediated EPSCs from the mutant and the WT followed a multiplicative relationship (slope = 0.43), as would be expected for the uniform reduction in postsynaptic receptors function that is a hallmark of homeostatic plasticity changes (Turrigiano et al., 1998). Such a plastic response of glutamatergic synapses is expected for increased network excitability and, therefore, independently supports the notion that GABAergic function is impaired in CSP-α neuronal cultures. A model outlining this interpretation is presented in supplemental Figure 7 (available at www.jneurosci.org as supplemental material).
Together, the above observations suggest that high synaptic activity is a major factor in increased nerve terminal vulnerability and that this can be prevented by a CSP-α-mediated mechanism. This conclusion is further reinforced by our results showing that deprivation of excitatory transmission in hippocampal cultures is enough to rescue Syt2-expressing GABAergic synapses devoid of CSP-α from neurodegeneration (Fig. 10). In control conditions, in which glutamatergic transmission is a major promoter of network excitability (Thiagarajan et al., 2005; Mateos et al., 2007), we failed to detect Syt2 staining in cultures lacking CSP-α (Figs. 4A,B, 10B). In contrast, on prolonged (3 week) AMPAR and NMDAR blockade, we detected, for the first time in CSP-α KO cultures, Syt2-positive puncta (Fig. 10B), together with a significant increase (three times) in the number of GABAergic synapses (Fig. 10A). In WT cultures, the synaptic expression of Syt2 was robustly immunodetected only after the second week in culture (data not shown). We believe that, under control culture conditions, Syt2-expressing synapses lacking CSP-α are labile, and quickly degenerate (Figs. 3, 4), making it difficult to detect them with immunotechniques. This situation is ameliorated in vivo, in which, although significantly reduced, Syt2 staining is still detectable (Fig. 5), probably because the physiological hippocampal niche provides better trophic support than the cultures or is better controlled in terms of network excitability. Irrespectively, the remarkable increase in Syt2-positive synapses on glutamatergic deprivation in culture might be attributable to the formation of new synapses and to the reactivation of less compromised terminals, which become more viable once they have been released from the burden of high synaptic activity. In WT cultures deprived of excitatory activity (Fig. 10A), we observed a 30.4% reduction in the number of GABAergic synapses, compared with nontreated cultures, consistent with previously reported results (Hartman et al., 2006). Given that there is no increase in the number of GABAergic synapses in WT cultures, we cannot explain the increase in GABAergic synapses in the mutant cultures in terms of secondary neurotrophic effects of the pharmacological treatment. We cannot rule out that Syt2 expression on glutamatergic blockade is attributable to a loss of activity-dependent suppression of Syt2 in some other cell type different to basket cell. However, that is unlikely because it would mean the unexpected activation of a GABAergic synapse on activity deprivation. Our most parsimonious explanation is that glutamatergic blockers (1) directly limit postsynaptic depolarization of GABAergic neurons, (2) indirectly reduce the number of action potentials reaching the presynaptic GABAergic terminals, and therefore (3) prevent presynaptic degeneration by reducing evoked exocytosis of GABA-loaded synaptic vesicles. In addition, glutamatergic synaptic inactivity reduces the Ca2+ influx through L-type Ca2+-channels in hippocampal cultures (Thiagarajan et al., 2005), thereby probably reducing Ca2+ transients triggered by spike depolarizations and lowering the resting [Ca2+] (Magee et al., 1996). Importantly, L-type Ca2+ channels are not involved in low-frequency synaptic transmission at GABAergic synapses (Jensen et al., 1999; Jensen and Mody, 2001). They are, however, essential for GABA release at fast-spiking basket cells during the high-frequency trains (40 Hz) typical of hippocampal gamma rhythms (Jensen and Mody, 2001). Therefore, the secondary reduction of L-type Ca2+ channels could be critical in protecting Syt2-positive presynaptic terminals by preventing Ca2+ overload and massive GABA release. Regardless, the present study reports, for the first time in cultured neurons, a CSP-α-dependent neurodegenerative and synaptic phenotype suitable for future investigations of molecular mechanisms linking CSP-α to the prevention of activity-dependent rundown of synapses.
In summary, our study shows that neurons with a high basal action potential firing rate are prone to presynaptic degeneration. The synaptic vesicle protein CSP-α is a key component of an essential physiological mechanism that maintains presynaptic function under potentially deleterious conditions, such as those that occur during high-frequency synaptic vesicle cycling. Our study has highlighted remarkable differences in the properties of two hippocampal synapses: (1) Syt2-positive GABAergic synapses, which are highly dependent on CSP-α, and (2) glutamatergic synapses, which apparently do not require CSP-α for their normal function. This scenario is in principle surprising for a protein that is evolutionarily conserved (Schmitz and Fernández-Chacón, 2009) and universally expressed in every synaptic type (Kohan et al., 1995). However, the precise role of CSP-α in many synapses might become evident only during physiologically extreme or pathophysiological circumstances, such as synapse aging or synapse overload during epilepsy. Our observations in cultured neurons pave the way for additional studies aimed at understanding the molecular mechanisms underlying the activity of synaptic chaperones in health and disease.
Footnotes
This work was supported by Spanish Ministry of Science and Innovation (MICINN) Grant BFU2007-66008, Junta de Andalucía Grant P07-CVI-02854, and Instituto de Salud Carlos III (ISCIII) (R.F.-C.); Junta de Comunidades de Castilla–La Mancha Grant PAI08-0174-6967 (R.L.); Fondo Europeo de Desarrollo Regional, Junta de Andalucía Grant P06-CVI-02392, and the Sixth Framework Program of the European Union (Network of European Neuroscience Institutes) Grant LSHM-CT-2005-019063. G.C. is supported by the Sara Borrell Program (ISCIII) and L.G.-S. by the Formación de Personal Investigador Program (MICINN). We are grateful to Dr. Thomas C. Südhof for advice and for comments on a previous version of this manuscript, to Dr. Christian Rosenmund and Dr. Mariluz Montesinos for advice, to Dr. José L. Rozas for comments on this manuscript, to Alejandro Arroyo, Maria C. Rivero, César de Cires, Itziar Benito, and Dr. Juan L. Ribas (Centro de Investigación, Tecnología e Innovación de la Universidad de Sevilla) for excellent technical assistance.
References
- Baumert M, Maycox PR, Navone F, De Camilli P, Jahn R. Synaptobrevin: an integral membrane protein of 18,000 daltons present in small synaptic vesicles of rat brain. EMBO J. 1989;8:379–384. doi: 10.1002/j.1460-2075.1989.tb03388.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandra S, Gallardo G, Fernández-Chacón R, Schlüter OM, Südhof TC. Alpha-synuclein cooperates with CSPalpha in preventing neurodegeneration. Cell. 2005;123:383–396. doi: 10.1016/j.cell.2005.09.028. [DOI] [PubMed] [Google Scholar]
- Clements JD, Bekkers JM. Detection of spontaneous synaptic events with an optimally scaled template. Biophys J. 1997;73:220–229. doi: 10.1016/S0006-3495(97)78062-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Craig AM, Blackstone CD, Huganir RL, Banker G. The distribution of glutamate receptors in cultured rat hippocampal neurons: postsynaptic clustering of AMPA-selective subunits. Neuron. 1993;10:1055–1068. doi: 10.1016/0896-6273(93)90054-u. [DOI] [PubMed] [Google Scholar]
- Csicsvari J, Hirase H, Czurkó A, Mamiya A, Buzsáki G. Oscillatory coupling of hippocampal pyramidal cells and interneurons in the behaving rat. J Neurosci. 1999;19:274–287. doi: 10.1523/JNEUROSCI.19-01-00274.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fasshauer D, Eliason WK, Brünger AT, Jahn R. Identification of a minimal core of the synaptic SNARE complex sufficient for reversible assembly and disassembly. Biochemistry. 1998;37:10354–10362. doi: 10.1021/bi980542h. [DOI] [PubMed] [Google Scholar]
- Fernández-Chacón R, Königstorfer A, Gerber SH, García J, Matos MF, Stevens CF, Brose N, Rizo J, Rosenmund C, Südhof TC. Synaptotagmin I functions as a calcium regulator of release probability. Nature. 2001;410:41–49. doi: 10.1038/35065004. [DOI] [PubMed] [Google Scholar]
- Fernández-Chacón R, Wölfel M, Nishimune H, Tabares L, Schmitz F, Castellano-Muñoz M, Rosenmund C, Montesinos ML, Sanes JR, Schneggenburger R, Südhof TC. The synaptic vesicle protein CSP alpha prevents presynaptic degeneration. Neuron. 2004;42:237–251. doi: 10.1016/s0896-6273(04)00190-4. [DOI] [PubMed] [Google Scholar]
- Fox MA, Sanes JR. Synaptotagmin I and II are present in distinct subsets of central synapses. J Comp Neurol. 2007;503:280–296. doi: 10.1002/cne.21381. [DOI] [PubMed] [Google Scholar]
- Frerking M, Schulte J, Wiebe SP, Stäubli U. Spike timing in CA3 pyramidal cells during behavior: implications for synaptic transmission. J Neurophysiol. 2005;94:1528–1540. doi: 10.1152/jn.00108.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freund TF, Buzsáki G. Interneurons of the hippocampus. Hippocampus. 1996;6:347–470. doi: 10.1002/(SICI)1098-1063(1996)6:4<347::AID-HIPO1>3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]
- Freund TF, Katona I. Perisomatic inhibition. Neuron. 2007;56:33–42. doi: 10.1016/j.neuron.2007.09.012. [DOI] [PubMed] [Google Scholar]
- Gomperts SN, Rao A, Craig AM, Malenka RC, Nicoll RA. Postsynaptically silent synapses in single neuron cultures. Neuron. 1998;21:1443–1451. doi: 10.1016/s0896-6273(00)80662-5. [DOI] [PubMed] [Google Scholar]
- Gulyás AI, Buzsáki G, Freund TF, Hirase H. Populations of hippocampal inhibitory neurons express different levels of cytochrome c. Eur J Neurosci. 2006;23:2581–2594. doi: 10.1111/j.1460-9568.2006.04814.x. [DOI] [PubMed] [Google Scholar]
- Gundersen CB, Umbach JA. Suppression cloning of the cDNA for a candidate subunit of a presynaptic calcium channel. Neuron. 1992;9:527–537. doi: 10.1016/0896-6273(92)90190-o. [DOI] [PubMed] [Google Scholar]
- Hartman KN, Pal SK, Burrone J, Murthy VN. Activity-dependent regulation of inhibitory synaptic transmission in hippocampal neurons. Nat Neurosci. 2006;9:642–649. doi: 10.1038/nn1677. [DOI] [PubMed] [Google Scholar]
- Howard A, Tamas G, Soltesz I. Lighting the chandelier: new vistas for axo-axonic cells. Trends Neurosci. 2005;28:310–316. doi: 10.1016/j.tins.2005.04.004. [DOI] [PubMed] [Google Scholar]
- Hsu CC, Davis KM, Jin H, Foos T, Floor E, Chen W, Tyburski JB, Yang CY, Schloss JV, Wu JY. Association of l-glutamic acid decarboxylase to the 70-kDa heat shock protein as a potential anchoring mechanism to synaptic vesicles. J Biol Chem. 2000;275:20822–20828. doi: 10.1074/jbc.M001403200. [DOI] [PubMed] [Google Scholar]
- Jensen K, Mody I. L-type Ca2+ channel-mediated short-term plasticity of GABAergic synapses. Nat Neurosci. 2001;4:975–976. doi: 10.1038/nn722. [DOI] [PubMed] [Google Scholar]
- Jensen K, Jensen MS, Lambert JD. Role of presynaptic L-type Ca2+ channels in GABAergic synaptic transmission in cultured hippocampal neurons. J Neurophysiol. 1999;81:1225–1230. doi: 10.1152/jn.1999.81.3.1225. [DOI] [PubMed] [Google Scholar]
- Jiang M, Swann JW. A role for L-type calcium channels in the maturation of parvalbumin-containing hippocampal interneurons. Neuroscience. 2005;135:839–850. doi: 10.1016/j.neuroscience.2005.06.073. [DOI] [PubMed] [Google Scholar]
- Jin H, Wu H, Osterhaus G, Wei J, Davis K, Sha D, Floor E, Hsu CC, Kopke RD, Wu JY. Demonstration of functional coupling between γ-aminobutyric acid (GABA) synthesis and vesicular GABA transport into synaptic vesicles. Proc Natl Acad Sci U S A. 2003;100:4293–4298. doi: 10.1073/pnas.0730698100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jinno S, Aika Y, Fukuda T, Kosaka T. Quantitative analysis of GABAergic neurons in the mouse hippocampus, with optical disector using confocal laser scanning microscope. Brain Res. 1998;814:55–70. doi: 10.1016/s0006-8993(98)01075-0. [DOI] [PubMed] [Google Scholar]
- Jonas P, Bischofberger J, Fricker D, Miles R. Interneuron diversity series: fast in, fast out—temporal and spatial signal processing in hippocampal interneurons. Trends Neurosci. 2004;27:30–40. doi: 10.1016/j.tins.2003.10.010. [DOI] [PubMed] [Google Scholar]
- Joshi I, Wang LY. Developmental profiles of glutamate receptors and synaptic transmission at a single synapse in the mouse auditory brainstem. J Physiol. 2002;540:861–873. doi: 10.1113/jphysiol.2001.013506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerr AM, Reisinger E, Jonas P. Differential dependence of phasic transmitter release on synaptotagmin 1 at GABAergic and glutamatergic hippocampal synapses. Proc Natl Acad Sci U S A. 2008;105:15581–15586. doi: 10.1073/pnas.0800621105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinney JW, Davis CN, Tabarean I, Conti B, Bartfai T, Behrens MM. A specific role for NR2A-containing NMDA receptors in the maintenance of parvalbumin and GAD67 immunoreactivity in cultured interneurons. J Neurosci. 2006;26:1604–1615. doi: 10.1523/JNEUROSCI.4722-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirsch J, Betz H. The postsynaptic localization of the glycine receptor-associated protein gephyrin is regulated by the cytoskeleton. J Neurosci. 1995;15:4148–4156. doi: 10.1523/JNEUROSCI.15-06-04148.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klausberger T, Somogyi P. Neuronal diversity and temporal dynamics: the unity of hippocampal circuit operations. Science. 2008;321:53–57. doi: 10.1126/science.1149381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klausberger T, Magill PJ, Márton LF, Roberts JD, Cobden PM, Buzsáki G, Somogyi P. Brain-state- and cell-type-specific firing of hippocampal interneurons in vivo. Nature. 2003;421:844–848. doi: 10.1038/nature01374. [DOI] [PubMed] [Google Scholar]
- Kohan SA, Pescatori M, Brecha NC, Mastrogiacomo A, Umbach JA, Gundersen CB. Cysteine string protein immunoreactivity in the nervous system and adrenal gland of rat. J Neurosci. 1995;15:6230–6238. doi: 10.1523/JNEUROSCI.15-09-06230.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Q, Lau A, Morris TJ, Guo L, Fordyce CB, Stanley EF. A syntaxin 1, Gαo, and N-type calcium channel complex at a presynaptic nerve terminal: analysis by quantitative immunocolocalization. J Neurosci. 2004;24:4070–4081. doi: 10.1523/JNEUROSCI.0346-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lujan R, Nusser Z, Roberts JD, Shigemoto R, Somogyi P. Perisynaptic location of metabotropic glutamate receptors mGluR1 and mGluR5 on dendrites and dendritic spines in the rat hippocampus. Eur J Neurosci. 1996;8:1488–1500. doi: 10.1111/j.1460-9568.1996.tb01611.x. [DOI] [PubMed] [Google Scholar]
- Magee JC, Avery RB, Christie BR, Johnston D. Dihydropyridine-sensitive, voltage-gated Ca2+ channels contribute to the resting intracellular Ca2+ concentration of hippocampal CA1 pyramidal neurons. J Neurophysiol. 1996;76:3460–3470. doi: 10.1152/jn.1996.76.5.3460. [DOI] [PubMed] [Google Scholar]
- Malinow R, Malenka RC. AMPA receptor trafficking and synaptic plasticity. Annu Rev Neurosci. 2002;25:103–126. doi: 10.1146/annurev.neuro.25.112701.142758. [DOI] [PubMed] [Google Scholar]
- Mateos JM, Lüthi A, Savic N, Stierli B, Streit P, Gähwiler BH, McKinney RA. Synaptic modifications at the CA3-CA1 synapse after chronic AMPA receptor blockade in rat hippocampal slices. J Physiol. 2007;581:129–138. doi: 10.1113/jphysiol.2006.120550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pang ZP, Melicoff E, Padgett D, Liu Y, Teich AF, Dickey BF, Lin W, Adachi R, Südhof TC. Synaptotagmin-2 is essential for survival and contributes to Ca2+ triggering of neurotransmitter release in central and neuromuscular synapses. J Neurosci. 2006;26:13493–13504. doi: 10.1523/JNEUROSCI.3519-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pawelzik H, Hughes DI, Thomson AM. Physiological and morphological diversity of immunocytochemically defined parvalbumin- and cholecystokinin-positive interneurones in CA1 of the adult rat hippocampus. J Comp Neurol. 2002;443:346–367. doi: 10.1002/cne.10118. [DOI] [PubMed] [Google Scholar]
- Rizo J, Rosenmund C. Synaptic vesicle fusion. Nat Struct Mol Biol. 2008;15:665–674. doi: 10.1038/nsmb.1450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenmund C, Clements JD, Westbrook GL. Nonuniform probability of glutamate release at a hippocampal synapse. Science. 1993;262:754–757. doi: 10.1126/science.7901909. [DOI] [PubMed] [Google Scholar]
- Ruiz R, Casañas JJ, Südhof TC, Tabares L. Cysteine string protein-alpha is essential for the high calcium sensitivity of exocytosis in a vertebrate synapse. Eur J Neurosci. 2008;27:3118–3131. doi: 10.1111/j.1460-9568.2008.06301.x. [DOI] [PubMed] [Google Scholar]
- Schmitz F, Fernández-Chacón R. Cysteine string proteins. In: Squire LR, editor. Encyclopedia of neuroscience. Oxford: Academic; 2009. pp. 285–292. [Google Scholar]
- Schmitz F, Tabares L, Khimich D, Strenzke N, de la Villa-Polo P, Castellano-Muñoz M, Bulankina A, Moser T, Fernández-Chacón R, Südhof TC. CSPα-deficiency causes massive and rapid photoreceptor degeneration. Proc Natl Acad Sci U S A. 2006;103:2926–2931. doi: 10.1073/pnas.0510060103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Somogyi P, Klausberger T. Defined types of cortical interneurone structure space and spike timing in the hippocampus. J Physiol. 2005;562:9–26. doi: 10.1113/jphysiol.2004.078915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sterling P, Matthews G. Structure and function of ribbon synapses. Trends Neurosci. 2005;28:20–29. doi: 10.1016/j.tins.2004.11.009. [DOI] [PubMed] [Google Scholar]
- Su J, Gorse K, Ramirez F, Fox MA. Collagen XIX is expressed by interneurons and contributes to the formation of hippocampal synapses. J Comp Neurol. 2010;518:229–253. doi: 10.1002/cne.22228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Südhof TC. The synaptic vesicle cycle. Annu Rev Neurosci. 2004;27:509–547. doi: 10.1146/annurev.neuro.26.041002.131412. [DOI] [PubMed] [Google Scholar]
- Südhof TC, Czernik AJ, Kao HT, Takei K, Johnston PA, Horiuchi A, Kanazir SD, Wagner MA, Perin MS, De Camilli P, Greengard P. Synapsins: mosaics of shared and individual domains in a family of synaptic vesicle phosphoproteins. Science. 1989;245:1474–1480. doi: 10.1126/science.2506642. [DOI] [PubMed] [Google Scholar]
- Tao HW, Poo M. Retrograde signaling at central synapses. Proc Natl Acad Sci U S A. 2001;98:11009–11015. doi: 10.1073/pnas.191351698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thiagarajan TC, Lindskog M, Tsien RW. Adaptation to synaptic inactivity in hippocampal neurons. Neuron. 2005;47:725–737. doi: 10.1016/j.neuron.2005.06.037. [DOI] [PubMed] [Google Scholar]
- Turrigiano GG, Nelson SB. Homeostatic plasticity in the developing nervous system. Nat Rev Neurosci. 2004;5:97–107. doi: 10.1038/nrn1327. [DOI] [PubMed] [Google Scholar]
- Turrigiano GG, Leslie KR, Desai NS, Rutherford LC, Nelson SB. Activity-dependent scaling of quantal amplitude in neocortical neurons. Nature. 1998;391:892–896. doi: 10.1038/36103. [DOI] [PubMed] [Google Scholar]
- Umbach JA, Zinsmaier KE, Eberle KK, Buchner E, Benzer S, Gundersen CB. Presynaptic dysfunction in Drosophila csp mutants. Neuron. 1994;13:899–907. doi: 10.1016/0896-6273(94)90255-0. [DOI] [PubMed] [Google Scholar]
- Ylinen A, Bragin A, Nádasdy Z, Jandó G, Szabó I, Sik A, Buzsáki G. Sharp wave-associated high-frequency oscillation (200 Hz) in the intact hippocampus: network and intracellular mechanisms. J Neurosci. 1995;15:30–46. doi: 10.1523/JNEUROSCI.15-01-00030.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zinsmaier KE, Eberle KK, Buchner E, Walter N, Benzer S. Paralysis and early death in cysteine string protein mutants of Drosophila. Science. 1994;263:977–980. doi: 10.1126/science.8310297. [DOI] [PubMed] [Google Scholar]