

Abstract
Phosphodiesterase 10A (PDE10A) is a striatum-enriched, dual-specific cyclic nucleotide phosphodiesterase that has gained considerable attention as a potential therapeutic target for psychiatric disorders such as schizophrenia. As such, a PDE10A-selective inhibitor compound, MP-10, has recently entered clinical testing. Since little is known about the cellular regulation of PDE10A, we sought to elucidate the mechanisms that govern its subcellular localization in striatal medium spiny neurons. Previous reports suggest that PDE10A is primarily membrane bound and is transported throughout medium spiny neuron axons and dendrites. Moreover, it has been shown in PC12 cells that the localization of the major splice form, PDE10A2, may be regulated by protein kinase A phosphorylation at threonine 16 (Thr-16). Using an antibody that specifically recognizes phosphorylated Thr-16 (pThr-16) of PDE10A2, we provide evidence that phosphorylation at Thr-16 is critical for the regulation of PDE10A subcellular localization in vivo. Furthermore, we demonstrate in primary mouse striatal neuron cultures that PDE10A membrane association and transport throughout dendritic processes requires palmitoylation of cysteine 11 (Cys-11) of PDE10A2, likely by the palmitoyl acyltransferases DHHC-7 and -19. Finally, we show that Thr-16 phosphorylation regulates PDE10A trafficking and localization by preventing palmitoylation of Cys-11 rather than by interfering with palmitate–lipid interactions. These data support a model whereby PDE10A trafficking and localization can be regulated in response to local fluctuations in cAMP levels. Given this, we propose that excessive striatal dopamine release, as occurs in schizophrenia, might exert differential effects on the regulation of PDE10A localization in the two striatal output pathways.
Introduction
Cyclic nucleotides are highly ubiquitous second messenger signaling molecules that play central roles in a wide range of intracellular signal transduction pathways involved in multiple cellular processes (Beavo and Brunton, 2002). That these ubiquitous, soluble molecules can simultaneously participate in multiple, distinct signaling pathways with an astonishingly high degree of specificity has been the subject of intense study. Such specificity is achieved in part through the tight spatial regulation of multiple signaling components, including the cyclic nucleotide phosphodiesterases (PDEs) (Dessauer, 2009). PDEs are a superfamily of structurally related enzymes that catalyze the hydrolysis of cAMP and/or cGMP. In this way, it is thought that PDEs act as cyclic nucleotide diffusion barriers through their spatially confined zones of enzymatic activity, contributing to the subcellular compartmentalization of distinct signaling cascades (Beavo and Brunton, 2002; Houslay, 2010). Moreover, PDEs are the focus of considerable attention as therapeutic targets for a range of human diseases, since pharmacological manipulation of these enzymes can have profound effects on specific signal transduction pathways (Menniti et al., 2006).
Cyclic nucleotide PDEs are encoded by 21 genes identified to date which are segregated into 11 gene families based on comparative functional and structural analysis (Conti and Beavo, 2007). Phosphodiesterase 10A (PDE10A) is a dual-specific PDE (Fujishige et al., 1999a; Soderling et al., 1999), hydrolyzing both cAMP and cGMP and encoded by a single gene mapped to human chromosome 6q26 (Fujishige et al., 1999a), which gives rise to multiple splice forms (Fujishige et al., 1999b; Kotera et al., 1999; Fujishige et al., 2000). PDE10A is highly enriched in the brain across multiple mammalian species (Fujishige et al., 1999a; Loughney et al., 1999; Soderling et al., 1999; Coskran et al., 2006). Furthermore, high expression levels of PDE10A mRNA and protein are confined to the medium spiny output neurons (MSNs) of the striatum (Seeger et al., 2003; Coskran et al., 2006; Xie et al., 2006). Within the MSNs, but not within other neuron types, PDE10A is primarily membrane -bound and associated with postsynaptic densities on dendritic spines (Kotera et al., 2004; Xie et al., 2006). This distribution pattern predicts that PDE10A inhibition might affect basal ganglia circuitry in a manner that is consistent with the potential for antipsychotic efficacy. Indeed, multiple studies using selective PDE10A inhibitors and PDE10A knock-out mutant mice suggest that PDE10A inhibition produces neurochemical, neurophysiological, and behavioral effects predictive of antipsychotic efficacy (Siuciak et al., 2006a,b; Nishi et al., 2008; Schmidt et al., 2008; Siuciak et al., 2008; Grauer et al., 2009; Threlfell et al., 2009).
Given the importance of PDE localization in the compartmentalization of cyclic nucleotide-mediated signal cascades, combined with the growing interest in PDE10A as a novel pharmaceutical target for psychiatric disease, we sought to further investigate the mechanisms underlying the regulation of PDE10A localization. Our results show that both membrane localization and long-distance trafficking of PDE10A in neurons are mediated by N-linked palmitoylation of PDE10A2, a major splice form in rodent and human, and that this process is regulated by protein kinase A (PKA)-mediated phosphorylation.
Materials and Methods
Antibodies.
An affinity-purified rabbit polyclonal phospho-specific anti-PDE10A2 antibody was custom made using an antigenic peptide corresponding to amino acids 1-23 of the PDE10A2 polypeptide (MEDGPSNNASCFRRLTECFLSPS), where threonine 16 (Thr-16) (underlined) was phosphorylated (Open Biosystems). An affinity-purified rabbit polyclonal antibody that recognizes all splice forms of PDE10A was custom made (Enzo Life Sciences) using an antigenic peptide corresponding to amino acids 303-321 of the PDE10A2 polypeptide (DHKNKELYSDLFDIGEEKE), within the GAF-A regulatory domain. A rabbit anti-HaloTag antibody (Promega) was used to detect PDE10A2 by immunofluorescence. A rabbit anti-actin antibody was from Sigma-Aldrich. A mouse anti-green fluorescent protein (GFP) antibody (Santa Cruz Biotechnology) was used to enhance enhanced GFP (EGFP) fluorescence. For all immunofluorescence experiments, secondary antibodies were donkey anti-mouse or donkey anti-rabbit Alexa Fluor 488 and donkey anti-rabbit Alexa Fluor 594 (Invitrogen).
Fractionation of striatal tissue or cultured HEK293 cells.
Fractionation of mouse striatal tissue or cultured HEK293 cells was performed as described by Kotera et al. (2004). Briefly, male C57BL/6J mice were killed by cervical dislocation, and brains were harvested and dissected. Striatal tissue was suspended (100 μg/ml) in ice-cold HB-A buffer (20 mm Tris-HCl, pH 7.5, 2 mm magnesium acetate, 0.3 mm calcium chloride, 1 mm dithiothreitol, 1.3 mm benzamidine, protease inhibitor cocktail (Roche Diagnostics), and phosphatase inhibitor cocktail (Sigma-Aldrich) and homogenized by brief sonication. Homogenates were centrifuged at 1000 × g for 10 min at 4°C. Supernatants were recovered and centrifuged at 100,000 × g for 60 min at 4°C, yielding a cytosolic fraction in the supernatant. Pellets were resuspended by brief sonication in an identical volume of ice-cold HB-A buffer containing 0.5% Triton X-100, followed by agitation at 4°C for 30 min. Samples were centrifuged at 100,000 × g for 60 min, yielding a membrane fraction in the supernatant. Total protein concentration in each fraction was measured using a Bio-Rad DC protein assay. Identical amounts of total protein from homogenate, membrane, and cytosolic fractions were subjected to SDS-PAGE and Western blot analysis using the indicated antibodies. The same procedure was performed for cultured HEK293 cell fractionation.
cDNA construction and site-directed mutagenesis.
For phospho-specific antibody validation, the cDNA corresponding to amino acids 2-91 of human PDE10A2 was amplified by PCR and subcloned to pGEX-4T1 for bacterial expression and purification. For full-length PDE10A2 used in all subcellular fractionation and immunofluorescence experiments, full-length human PDE10A2 was amplified by PCR and subcloned to pFc8A in-frame with cDNA encoding the HaloTag located at the 3′ end, yielding a fusion protein with HaloTag fused to the C terminus of PDE10A2. Mutagenesis was conducted with the above full-length PDE10A2 construct as the template, using the QuikChange II site directed mutagenesis kit according to the manufacturer's instructions (Stratagene).
Culture and transfection of primary striatal neurons and HEK293 cells.
HEK293 cells were cultured in 10 cm or 60 mm culture dishes (for subcellular fractionation) or on 12 mm glass coverslips (for immunofluorescence) in HEK293 culture medium [DMEM, 10% FBS, nonessential amino acids, and penicillin/streptomycin (Pen/Strep)] at 37°C/5% CO2. Cells were transfected with the indicated cDNA constructs using Lipofectamine 2000 (Invitrogen) according to the manufacturer's instructions. For primary embryonic striatal neuron cultures, striata were collected from embryonic day 17 rat embryos in dissection buffer [HBSS (without Ca/Mg) containing 10 mm HEPES]. Striata were triturated in dissection buffer and seeded on 60 mm, poly-d-lysine-treated,12 mm glass coverslips at a density of 1200 cells/mm2 in plating medium (MEM containing 10% horse serum, 10% fetal bovine serum, and pen/strep). After 24 h at 37°C/5% CO2, plating medium was removed and replaced with striatal maintenance medium (Neurobasal medium supplemented with B27 plus Pen/Strep plus 0.5 mm l-glutamine), and neurons were incubated at 37°C/5% CO2. Neurons were transfected with the indicated cDNA constructs at 12 days in vitro (d.i.v.) using the CalPhos transfection kit (Invitrogen) according to the manufacturer's instructions.
Immunocytochemistry of primary striatal neuron cultures and HEK293 cells.
After transfection, cells were left for 48 h, followed by fixation at room temperature (RT) with 4% paraformaldehyde/4% sucrose for 15 min at 14 d.i.v. Cells were washed with PBS, followed by incubation with blocking buffer (2% normal donkey serum and 0.1% Triton X-100 in PBS, pH 7.5) at RT for 1 h. Cells were incubated with the indicated primary antibodies diluted in blocking buffer for either 3 h at RT or overnight at 4°C, washed with PBS, and incubated with the appropriate Alexa Fluor-conjugated secondary antibodies for 1 h at RT. Cells were washed with PBS and coverslips were mounted using Fluoromount-G (Southern Biotech).
Confocal microscopy and image analysis.
Confocal images were acquired on a Leica SP5 confocal microscope using a 63× oil-immersion objective. Images were analyzed using MetaMorph Image Analysis software suite (Molecular Devices). For measurements of membrane versus cytosolic enrichment in transfected primary striatal neuron cultures, the outer borders of neuronal somata were traced and segmented regions of interest (ROIs; 5 μm × 3 m) were automatically generated on the inner face of the trace (capturing plasma membrane fluorescence). The average fluorescence intensity in all segmented ROIs was calculated (outer ring). A second set of segmented ROIs (5 × 3 μm) was generated internal to the first set (capturing cytosolic fluorescence), and the average fluorescence intensity of these segments was calculated (inner ring). The ratio of the fluorescence intensity of the outer ring to that of the inner ring was used to assess the degree of membrane versus cytosolic enrichment. For measurements of protein trafficking, images were acquired such that PDE10A signal was just below saturation. EGFP signal was binarized, and segmented regions (5 × 3 μm) were generated along the length of three dendrites per neuron from the soma boundary to the 100 μm mark from the soma boundary. In each segment, the Metamorph Integrated Morphometry Analysis tool was used to measure the total area occupied by the binarized EGFP signal, while the total fluorescence intensity was measured for the same segment in the PDE10A2 channel. For each segment, the PDE10A2 fluorescence normalized to dendritic area (to eliminate changes in PDE10A2 fluorescence intensity caused by variability of dendritic area) was plotted against the distance of that segment from the soma boundary.
Palmitate labeling.
Transfected HEK293 cells were grown to 90% confluence in 10 cm tissue culture dishes, followed by preincubation with HEK293 media containing 10 mg/ml fatty acid-free BSA without serum for 30 min at 37°C/5% CO2. Cells were labeled with 0.5 mCi/ml [3H]palmitate in preincubation media for 4 h, followed by 1 wash with PBS. Cells were scraped into 1 ml of ice-cold immunoprecipitation buffer (10 mm Tris-HCl, pH 7.5, 150 mm NaCl, 1 mm EDTA, 1% Triton X-100, protease inhibitor cocktail, phosphatase inhibitor cocktail) and needle lysed, followed by agitation at 4°C for 1 h. Insoluble material was removed by centrifugation at 20,000 × g for 15 min at 4°C, and supernatants were precleared with 50 μl of protein A-Sepharose. Precleared lysates were subjected to immunoprecipitation with a rabbit anti-PDE10A antibody. Immunoprecipitates were eluted by boiling samples in SDS-PAGE sample buffer without reducing agent and subjected to SDS-PAGE. [3H]Palmitate incorporation was visualized using Amplify fluorography reagent according to the manufacturer's instructions (GE Healthcare).
Drug treatment of cultured primary striatal neurons and HEK293 cells.
For 2-bromopalmitate treatments, transfected HEK293 or primary striatal neuron cultures were treated with 100 μm 2-bromohexadecanoic acid (Sigma-Aldrich) or with the identical volume of DMSO for 24 h, followed by fractionation or immunofluorescence analysis. For acute treatments, transfected HEK293 cells were incubated with either 100 μm Sp-6-Phe-cAMPS (BioLog), 100 μm Sp-8-pCPT-cGMPS (BioLog), 100 nm phorbol 12-myristate 13-acetate (Calbiochem), or the identical volume of DMSO for 1 or 8 h (see Results) in the presence of 10 nm okadaic acid (Calbiochem) at 37°C/5% CO2, followed by subcellular fractionation and Western blot analysis.
Data analysis and statistical methods.
For all quantitative Western blot analysis, films were scanned using a Bio-Rad GS-800 densitometer, and the area under the curve for each immunoreactive protein band was calculated using ImageJ image analysis software (Abramoff et al., 2004). Densitometric measurements of immunoreactive bands of the indicated antibodies were normalized to that of rabbit anti-actin. For all quantitative immunofluorescence analysis, confocal images were acquired on a Leica SP5 confocal microscope using a 63× oil-immersion objective. Images were analyzed using MetaMorph Image Analysis software suite (Molecular Devices). For statistical analysis, Student's t tests were performed for all experiments, comparing one treatment condition to control condition. For comparison of more than one experimental condition to the control condition, one-way ANOVA followed by Dunnet's post hoc analysis was performed. When each experimental condition involved multiple measurements (see Fig. 4), we performed two-way repeated-measures ANOVA with post hoc analysis by least square difference. Significance was determined at p < 0.05, and all data are expressed as mean ± SEM.
Figure 4.
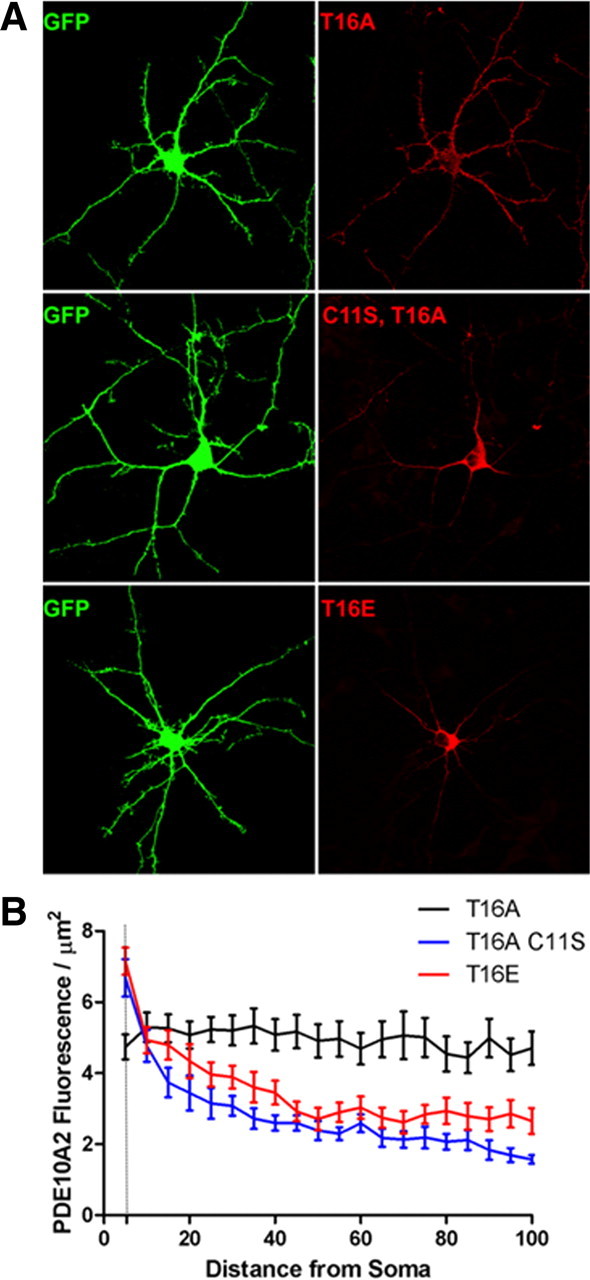
Distal dendritic trafficking of PDE10A2 requires Cys-11 in cultured striatal neurons. A, Representative primary cultures of striatal neurons transfected with cDNA encoding EGFP and either the phospho-resistant PDE10A2T16A, the phosphomimetic PDE10A2T16E, or the membrane binding-deficient PDE10A2C11S mutant. B, Confocal images were analyzed by generating 5 × 3 μm bins at identical positions in both the EGFP channel and the PDE10A2 immunofluorescence channel that extended sequentially for 100 μm along dendritic arbors beginning at the soma boundaries. For each bin, total area occupied by EGFP and the average PDE10A2 fluorescence intensity was measured. The PDE10A2 fluorescence normalized to dendritic area was plotted against the distance of that segment from the soma boundary. Fluorescence/μm2 for the phospho-resistant PDE10A2T16A mutant, which contained an intact cysteine at position 11, did not differ as function of distance from the soma. In contrast, fluorescence/μm2 of the phosphomimetic PDE10A2T16E mutant or the membrane binding-deficient PDE10A2C11S mutant was greatest near the soma boundary and sharply decayed with increasing distance from the soma boundary. Two-way repeated-measures ANOVA, effect of distance × treatment, p < 0.0001. Post hoc analysis by least significant difference: PDE10A2T16A, 0 μm versus any other distance, NS; PDE10A2C11S, 0 μm versus 5–100 μm, p < 0.0001 for each comparison; PDE10A2T16E, 0 μm versus 5–100 μm, p < 0.0001 for each comparison.
Results
A pool of endogenous PDE10A2 is phosphorylated and enriched in a cytosolic fraction of the mouse striatum
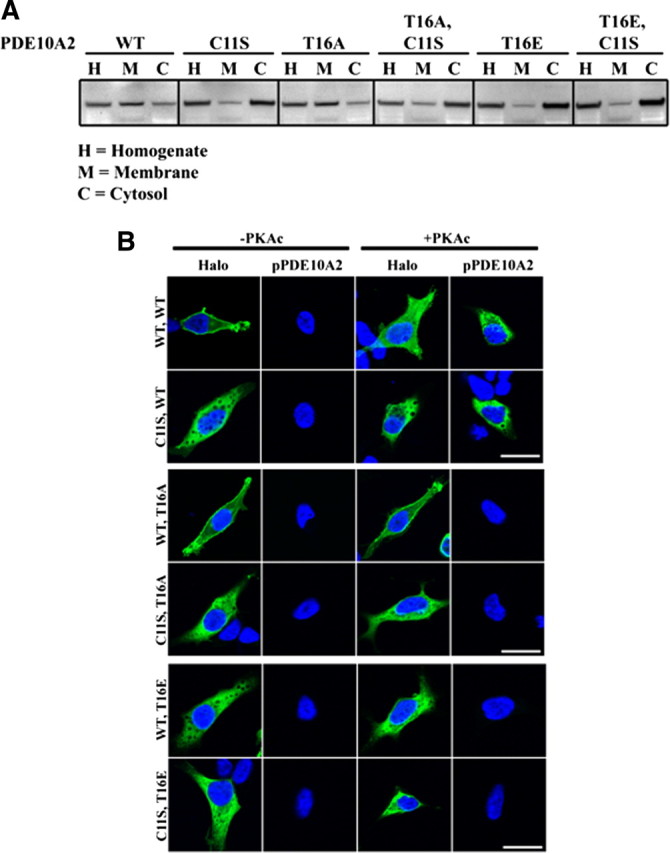
It was reported previously that recombinant forms of the two major PDE10A splice variants in rodent and human, PDE10A1 and PDE10A2, differ in their localization patterns such that PDE10A1 is enriched in a cytosolic fraction, whereas PDE10A2 is enriched in a membrane fraction of transfected PC12 cells (Kotera et al., 2004). The difference in localization pattern between these two isoforms was further shown to be attributable to alternative splicing in the regions encoding the extreme N terminus of PDE10A (Fig. 1 A). Moreover, it was shown that PDE10A2, but not PDE10A1, can be phosphorylated by PKA on Thr-16 and that this event resulted in a redistribution of membrane-bound PDE10A2 to a cytosolic fraction of these cells (Kotera et al., 2004). To further examine the regulation of PDE10A2 localization, we generated a rabbit polyclonal antibody that specifically recognizes the PDE10A2-specific N-terminal peptide containing a phosphothreonine at position 16 (Fig. 1 A,B). This antibody reacted strongly with a bacterially expressed N-terminal fragment of PDE10A2 only when this fragment was phosphorylated in vitro by PKA and contained a threonine, but not an alanine, at position 16 (Fig. 1 B). In addition, the immunoreactive band was abolished when this antibody was preincubated with the phosphorylated, but not the unphosphorylated, immunogenic peptide, further confirming that this antibody is specific for PDE10A2 only when it is phosphorylated at Thr-16 (Fig. 1 B). When coexpressed with the catalytic subunit of PKA in transfected HEK293 cells, wild-type (WT) PDE10A2 (PDE10A2WT) was phosphorylated at Thr-16, as determined with the phospho-specific PDE10A2 antibody (anti-pPDE10A2), and was enriched in the cytosolic fraction compared with the membrane fraction of these cells (Fig. 1 C, left). In contrast, a phospho-resistant T16A mutant (PDE10A2T16A) was enriched in the membrane fraction compared with the cytosolic fraction and was not phosphorylated by PKA (Fig. 1 C, middle). A phosphomimetic T16E mutant (PDE10A2T16E) was enriched in the cytosolic fraction compared with the membrane fraction, but was not phosphorylated by PKA (Fig. 1 C, right). These data confirm the previously reported findings that PKA phosphorylation of PDE10A2 on Thr-16 results in a cytosolic localization (Kotera et al., 2004).
Figure 1.

Phosphorylation at Thr-16 leads to a cytosolic localization of endogenous PDE10A2 in mouse striatum. A, The difference in localization pattern between the two major PDE10A splice variants in rodent and human, PDE10A1 and PDE10A2, is caused by alternative splicing in the region encoding the extreme N terminus of PDE10A. B, Anti-pPDE10A2 strongly reacted with a bacterially expressed N-terminal fragment of PDE10A2 only when this fragment was phosphorylated in vitro by PKA and contained a threonine, but not an alanine, at position 16. The immunoreactive band was displaced by a phosphorylated, but not an unphosphorylated, immunogenic peptide. C, When coexpressed with PKAc in transfected HEK293 cells, both PDE10A2WT and PDE10A2T16E, the latter carrying the phosphomimetic mutation at amino acid position 16, were enriched in the cytosolic (Cyt) fraction over the membrane (Mem) fraction (top), but only PDE10A2WT was phosphorylated at Thr-16 (bottom). In contrast, PDE10A2T16A, which is predicted to be resistant to phosphorylation at Thr-16 by PKAc, is enriched in the membrane fraction over the cytosolic fraction (top) and is not phosphorylated by PKAc (bottom). D, Phospho-PDE10A2 was detected in immunoprecipitates (IP) of total PDE10A in mouse striatal extracts. The anti-pPDE10A2 immunoreactive band was displaced by the phosphorylated, but not the nonphosphorylated, immunogenic peptide. The signal strength of the anti-pPDE10A2 band was further elevated by in vitro phosphorylation by PKAc. E, Crude membrane and cytosolic fractions of mouse striatum were immunoblotted with anti-pPDE10A2 or anti-PDE10A. The ratio of immunoreactive band intensities in the cytosolic versus membrane fractions are shown for both anti-pPDE10A2 and anti-PDE10A. Phosphorylated PDE10A2 is highly enriched in the cytosolic fraction over the membrane fraction of the mouse striatum. Conversely, total PDE10A is enriched in the membrane fraction over the cytosolic fraction of mouse striatum. ***p < 0.001 by Student's t test.
To further extend these findings, we used anti-pPDE10A2 to test the notion that PDE10A2 is phosphorylated in vivo. Immunoblot analysis using anti-pPDE10A2 could not detect phosphorylated PDE10A2 in total detergent extracts of mouse striatum. However, when total PDE10A was enriched by immunoprecipitation with an antibody to the GAF-B domain (anti-PDE10A) present in all isoforms of PDE10A (Fig. 1), an immunoreactive band was detected with anti-pPDE10A2 in these immunoprecipitates, and this band comigrated with that detected by anti-PDE10A (Fig. 1 D). This immunoreactive band was specific for PDE10A2 phosphorylated at Thr-16, since it was displaced by the phospho-immunogenic peptide but not the nonphospho-immunogenic peptide (Fig. 1 D). Furthermore, the signal strength of the anti-pPDE10A2 band was elevated by in vitro phosphorylation of immunoprecipitated PDE10A by PKA, suggesting that a portion of PDE10A2 also exists in an unphosphorylated state in the mouse striatum (Fig. 1 D). To test the notion that phosphorylation of PDE10A2 at Thr-16 results in cytosolic localization in vivo as suggested previously (Kotera et al., 2004), we prepared crude membrane and cytosolic fractions of mouse striatum and immunoblotted these fractions with anti-pPDE10A2 (Fig. 1 E). Results indicated that while phosphorylated PDE10A2 cannot be detected in total striatal homogenates or membrane fractions, it is highly enriched in the cytosolic fraction of the mouse striatum (Fig. 1 E). This is in contrast to total PDE10A, which is enriched in the membrane fraction of the striatum (Fig. 1 E). These results support the notion that phosphorylation at Thr-16 results in a cytosolic localization of endogenous PDE10A2 in mouse medium spiny neurons.
Plasma membrane binding of PDE10A2 requires cysteine 11
To elucidate the mechanism by which PDE10A2 localization is regulated by phosphorylation at Thr-16, we sought to identify those amino acids required for the association of PDE10A2 with cell membranes. Since only PDE10A2 can associate with membranes in vitro and in vivo (Kotera et al., 2004), we confined this analysis to the N-terminal peptide that distinguishes PDE10A2 from other isoforms, such as PDE10A1 (Fig. 1 A). Mutagenic analysis identified a single amino acid, cysteine 11 (Cys-11), as necessary for enrichment in the membrane fraction of transfected HEK293 cells, since a mutant form of PDE10A2 containing a serine in place of Cys-11 (PDE10A2C11S) was enriched in the cytosolic fraction of these cells (Fig. 2 A). This localization pattern was independent of the amino acid identity at position 16, since PDE10A2C11S/T16A and PDE10A2C11S/T16E mutants were also enriched in the cytosolic fraction (Fig. 2 A). This was confirmed by immunofluorescence confocal microscopy using antibodies to total and phosphorylated PDE10A2 (Fig. 2 B). As shown, the C11S mutation abolished membrane binding independent of the amino acid at position 16 and also independent of the phosphorylation state of PDE10A2, as determined by pPDE10A2 immunofluoresence labeling (Fig. 2 B). Given these data, we reasoned that Cys-11 was necessary for PDE10A2 membrane binding but not sufficient since Cys-11-mediated membrane binding required that Thr-16 remains unphosphorylated. Therefore, we hypothesized that phosphorylation of Thr-16 might interfere with Cys-11-mediated membrane binding.
Figure 2.
Plasma membrane binding of PDE10A2 requires Cys-11 in HEK293 cells. A, A mutant form of PDE10A2 containing a serine in place of Cys-11 (PDE10A2C11S) was enriched in the cytosol as determined by Western blot analysis of crude membrane and cytosolic fractions of HEK293 cells transfected with the indicated PDE10A2 mutants. B, Immunofluorescence confocal microscopy using antibodies to total and phosphorylated PDE10A2 showed that the C11S mutation abolished plasma membrane binding independent of the amino acid at position 16 and of the phosphorylation state of PDE10A2, as determined by pPDE10A2 immunofluorescence labeling. Halo, HaloTag antibody.
Curiously, in all HEK293 cells examined by confocal immunofluorescence microscopy, we noted that PDE10A2WT and PDE10A2T16A immunofluorescence labeling was distinctly enriched at the plasma membrane of these cells. This is in contrast to that reported previously using undifferentiated PC12 cells, where PDE10A2WT and PDE10A2T16A were only associated with membranes of the trans-Golgi network (TGN), as shown by colocalization with TGN38 in a perinuclear domain (Kotera et al., 2004). To determine whether this apparent discrepancy was arbitrarily related to the choice of cell line in our study, we transfected rat primary striatal neuron cultures with the same cDNA constructs used in HEK293 cells (Fig. 3). These cultures are expected to be more biologically relevant to the study of PDE10A localization, since medium spiny neurons, the only cell type in which native PDE10A is highly expressed and localized to membranes, comprise >95% of neurons in the striatum (Gerfen, 1992). Consistent with the distribution in HEK293 cells, both PDE10A2WT and PDE10A2T16A immunofluorescence was strongly enriched in the plasma membrane of both the somata and the dendrites (Fig. 3 A, insets) of transfected primary striatal neurons. In contrast, PDE10A2T16E, PDE10A2C11S, PDE10A2C11S/T16A, and PDE10A2C11S/T16E immunofluorescence was strongly accumulated in the cytosol, both in the soma and in dendrites (Fig. 3 A, insets) of these cells. We devised a method for quantifying the degree of membrane enrichment of immunofluorescent labeling over the cytosol (see Materials and Methods for details) and applied it to these transfected striatal neurons (Fig. 3 B). As expected, the outer ring to inner ring ratio was significantly greater for both PDE10A2WT and PDE10A2T16A than either PDE10A2T16E, PDE10A2C11S, PDE10A2C11S/T16A, or PDE10A2C11S/T16E, consistent with our qualitative observations. Thus, Cys-11 is required for association of PDE10A with the cytoplasmic face of the plasma membrane, and this association is likely disrupted by phosphorylation of Thr-16.
Figure 3.
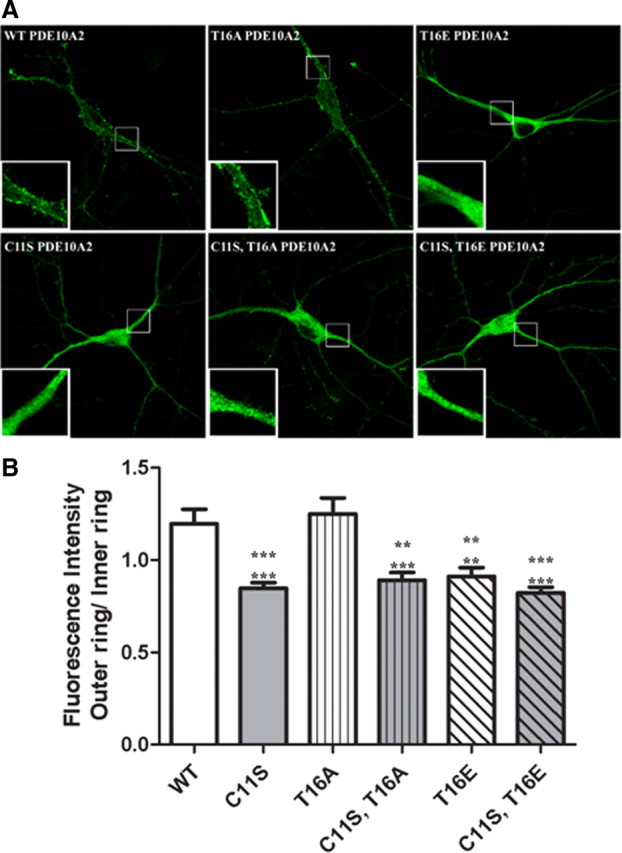
Cys-11 is required for association of PDE10A2 to the cytoplasmic face of the plasma membrane in cultured striatal neurons. A, Both PDE10A2WT and PDE10A2T16A immunofluorescence was strongly accumulated in the plasma membrane in the somata and dendrites (insets) of transfected primary striatal neurons. In contrast, PDE10A2T16E, PDE10A2C11S, PDE10A2C11S/T16A, and PDE10A2C11S/T16E immunofluorescence was strongly accumulated in the cytosol, both in the soma and in dendrites (insets) of these cells. B, Quantification of the degree of membrane enrichment of immunofluorescent labeling over the cytosol (see Materials and Methods for details) in transfected striatal neuron cultures. The outer ring (membrane immunofluorescence) to inner ring (cytosolic immunofluorescence) ratio was significantly greater for both PDE10A2WT and PDE10A2T16A compared with PDE10A2T16E, PDE10A2C11S, PDE10A2C11S/T16A, or PDE10A2C11S/T16E. PDE10A2T16D and PDE10A2C11S/T16D (data not shown) also showed significant reductions in outer ring to inner ring ratio compared with both PDE10A2WT and PDE10A2T16A. ***p < 0.001; **p < 0.01 by one-way ANOVA followed by Dunnett's multiple comparison test. Upper asterisks denote comparison with PDE10A2WT; lower asterisks denote comparison with PDE10A2T16A. No significant difference between PDE10A2WT and PDE10A2T16A was observed.
Distal dendritic trafficking of PDE10A2 requires cysteine 11 in cultured striatal neurons
It has been reported that PDE10A mRNA and protein are present at high levels in MSN cell bodies, whereas only PDE10A protein is present in the dendrites of these neurons, suggesting that PDE10A protein is transported throughout the dendritic tree (Seeger et al., 2003). We therefore hypothesized that Cys-11 was involved in the efficient trafficking of PDE10A2 over long dendritic distances (Fig. 4). To test this hypothesis, we cotransfected primary cultures of striatal neurons with cDNA encoding EGFP and either the phospho-resistant PDE10A2T16A, the phosphomimetic PDE10A2T16E, or the membrane binding-deficient PDE10A2C11S mutant (Fig. 4). Confocal images were analyzed by generating 5 × 3 μm bins at identical positions in both the EGFP channel and the PDE10A2 immunofluorescence channel that extended sequentially for 100 μm along dendritic arbors beginning at the soma boundaries. For each bin, we simultaneously measured the total area occupied by EGFP and the average PDE10A2 fluorescence intensity. In this way, we analyzed the relationship between PDE10A2 expression and distance from the soma boundary, eliminating reductions in PDE10A2 fluorescence intensity caused by restriction of the dendritic shaft in more distal portions of the neuron. Thus, we found that the phospho-resistant PDE10A2T16A mutant, which contained an intact cysteine at position 11, was uniformly distributed along the length of the dendritic arbors, whereas the expression of the phosphomimetic PDE10A2T16E mutant or the membrane binding-deficient PDE10A2C11S mutant was greatest near the soma boundary but sharply decayed with increasing distance from the soma boundary to levels that were significantly lower than that of PDE10A2T16A (Fig. 4 B). These results suggest that Cys-11 is necessary to facilitate the efficient transport of PDE10A2 over long dendritic distances but is not sufficient, since this function also requires that Thr-16 not be phosphorylated, consistent with the observation that the phosphomimetic PDE10A2T16E mutant cannot be efficiently transported despite the presence of Cys-11. Thus, we hypothesize that phosphorylation of PDE10A2 Thr-16 interferes with the ability of PDE10A2 Cys-11 to facilitate efficient distal trafficking of PDE10A2.
Phosphorylation of PDE10A2 interferes with N-terminal palmitoylation
Numerous studies have shown that the association of soluble cellular proteins with the cytoplasmic leaf of lipid bilayers, including the plasma membrane and transport vesicles, is promoted by protein palmitoylation, a posttranslational modification that results from the addition of a palmitate to a cysteine residue (Huang and El-Husseini, 2005; Linder and Deschenes, 2007). Since Cys-11 of PDE10A2 is required for plasma membrane association and trafficking to distal dendrites, we asked whether these effects could be mediated by palmitoylation of Cys-11. We first tested this notion by treating transfected HEK293 cells (Fig. 5 A) or primary striatal neurons (Fig. 5 B) with 2-bromopalmitate, a palmitate analog that nonspecifically interferes with the ability of protein acyltransferases to catalyze the transfer of a palmitate moiety to cysteine residues. Crude fractionation of HEK293 cells expressing the phospho-resistant PDE10A2T16A mutant showed that the PDE10A2 membrane to cytosol ratio was significantly reduced after treatment with 100 μm 2-bromopalmitate compared with vehicle control (Fig. 5 A). Specifically, this reduction in membrane to cytosol ratio was caused by a reduction of PDE10A2 levels in the membrane fraction with a concurrent increase in PDE10A2 levels in the cytosolic fraction (Fig. 5 A). Likewise, the ratio of outer ring to inner ring fluorescence intensity was significantly reduced after primary striatal neurons, expressing the phospho-resistant PDE10A2T16A mutant, were treated with 100 μm 2-bromopalmitate (Fig. 5 B). These results suggested that the association of PDE10A2 with the plasma membrane required not only Cys-11, but also protein palmitoylation.
Figure 5.

PDE10A2 membrane binding is disrupted by inhibiting palmitoyltransferase activity. A, Crude fractionation of HEK293 cells expressing the phospho-resistant PDE10A2T16A mutant showed that the PDE10A2 membrane to cytosol ratio was significantly reduced after treatment with 100 μm 2-bromopalmitate compared with vehicle control. B, The ratio of outer ring to inner ring fluorescence intensity was significantly reduced after primary striatal neurons, transfected with the phospho-resistant PDE10A2T16A mutant, were treated with 100 μm 2-bromopalmitate. **p < 0.01; ***p < 0.001 by Student's t test.
To directly confirm that PDE10A2 is palmitoylated on Cys-11, we performed in vitro palmitoylation assays to test the incorporation of [3H]palmitate by PDE10A2 (Fig. 6). As shown, PDE10A2WT and PDE10A2T16A incorporated [3H]palmitate, whereas PDE10A2C11S and PDE10A2T16E did not (Fig. 6 A). Thus, Cys-11 is necessary for palmitoylation of PDE10A2 but not sufficient, since the phosphomimetic mutant cannot be palmitoylated on Cys-11, suggesting that Thr-16 must also be unphosphorylated. Thus, these results parallel our fractionation and immunolocalization data (Figs. 2, 3) demonstrating that both Cys-11 and unphosphorylated Thr-16 are required for membrane binding. Together, these results suggest that phosphorylation of Thr-16 prevents membrane association of PDE10A2 by interfering with protein palmitoylation, rather than directly interfering with phospholipid interactions.
Figure 6.

PDE10A2 is palmitoylated on Cys-11. A, Palmitoylation assays show that PDE10A2WT and PDE10A2T16A incorporated [3H]palmitate, whereas PDE10A2C11S and PDE10A2T16E did not, suggesting that Cys-11 is necessary for palmitoylation of PDE10A2, but Thr-16 must also be unphosphorylated. B, To identify the PATs likely to mediate the palmitoylation of native PDE10A2, HEK293 cells were transfected with the 23 cloned mouse ZDHHC proteins and PDE10A2 (WT or C11S) using the [3H]palmitate incorporation assay. As shown, ZDHHC-7 and -19 elicited robust [3H]palmitate signals over basal levels.
To identify the protein acyltransferases (PATs) likely to mediate the palmitoylation of native PDE10A2, we screened the 23 cloned mouse zinc finger domain-containing, DHHC domain-containing (ZDHHC) proteins (Fukata et al., 2004; Fukata and Fukata, 2010) against PDE10A2 using the [3H]palmitate incorporation assay (Fig. 6 B). As shown, ZDHHC-7 and -19 elicited robust [3H]Palmitate signals over basal levels in this assay.
Acute phosphorylation of Thr-16 does not cause translocation of PDE10A2
If PDE10A2 phosphorylation at Thr-16 prevents palmitoylation, as suggested above, then the notion that phosphorylation causes a translocation from the membrane to the cytosol, as proposed previously (Kotera et al., 2004), might not be correct. Instead, phosphorylation of Thr-16 might prevent membrane binding by interfering with palmitoylation at Cys-11. To distinguish between these two possibilities, we acutely treated HEK293 cells expressing PDE10A2 with a membrane-permeant, PDE-resistant cAMP analog (Sp-6-Phe-cAMPS). We also used a membrane-permeant, PDE-resistant cGMP analog (Sp-8-pCPT-cGMPS) and phorbol 12-myristate 13-acetate (PMA) to test whether PDE10A2 phosphorylation can also be facilitated by PKG or PKC, respectively. If phosphorylation caused a true translocation from the membrane to the cytosol, then cyclic nucleotide-mediated phosphorylation of PDE10A2 at Thr-16, as detected by anti-pPDE10A2, should be accompanied by a shift in total PDE10A2 from the membrane to the cytosolic fraction. As shown in Figure 7, 1 h treatment with the cAMP analog led to a robust increase in phosphorylation of PDE10A2 at Thr-16 in total HEK293 cell homogenates, as well as in the cytosolic fraction of these cells. We also noted that the cGMP analog elicited significant increases in phosphorylation of PDE10A2 at Thr-16 in total homogenates, but the effect of the cGMP analog was too small to detect significant increases of phosphorylation in the cytosolic fraction (Fig. 7). We also noted that PMA did not alter the phosphorylation state of PDE10A2 at Thr-16, suggesting that PKC is not involved in PDE10A2 phosphorylation at Thr-16. Furthermore, while PDE10A2 was robustly phosphorylated at Thr-16 by the cAMP analog, there was no detectable shift in localization of total PDE10A2, as detected by anti-PDE10A, from the membrane to the cytosolic fraction (Fig. 7). These results suggest that phosphorylation of PDE10A2 at Thr-16 does not cause a true translocation of membrane-localized PDE10A2 to the cytosol, since acute phosphorylation is not accompanied by a shift in localization of total PDE10A2 from the membrane to the cytosolic fraction. This result is in contrast to that observed when the catalytic subunit of PKA (PKAc) is coexpressed with PDE10A2 in HEK293 cells or when PDE10A2 is expressed with a phosphomimetic mutation in place of Thr-16, which yielded an apparent shift in the localization of PDE10A2 from the membrane to the cytosolic compartments (Figs. 1 C, 2 B). Together, our results suggest that phosphorylation of nascent PDE10A2, as might occur in the presence of coexpressed PKAc, prevents membrane binding by interfering with posttranslational palmitoylation. Conversely, these results argue against the notion that phosphorylation of PDE10A2 at Thr-16 causes the release of PDE10A2 from the membrane.
Figure 7.

Acute phosphorylation of Thr-16 does not cause translocation of PDE10A2. A, HEK293 cells expressing PDE10A2 were treated for 1 h with a membrane-permeant, PDE-resistant cAMP analog (Sp-6-Phe-cAMPS), a membrane-permeant, PDE-resistant cGMP analog (Sp-8-pCPT-cGMPS), or PMA, and crude fractions were immunoblotted with anti-pPDE10A2 and anti-PDE10A to test whether acute phosphorylation of Thr-16 leads to a disengagement of PDE10A2 with cellular membranes. H, Homogenate, M, membrane, C, cytosol. B, Treatment with Sp-6-Phe-cAMPS lead to a robust increase in phosphorylation of PDE10A2 at Thr-16 in total homogenates, as well as in the cytosolic fraction. Treatment with Sp-8-pCPT-cGMPS elicited a significant increase in phosphorylation of PDE10A2 at Thr-16 in total homogenates only. PMA did not alter the phosphorylation state of PDE10A2 at Thr-16, suggesting that PKC is not involved in PDE10A2 phosphorylation at Thr-16. Although PDE10A2 was robustly phosphorylated at Thr-16 by Sp-6-Phe-cAMPS, there was no detectable shift in localization of total PDE10A2. ***p < 0.001; *p < 0.05 by one-way ANOVA followed by Dunnett's multiple comparisons test against vehicle control.
If acute phosphorylation of PDE10A2 at Thr-16 prevented membrane binding but could not cause the release of PDE10A2 from the membrane, then we might expect to find phosphorylated PDE10A2 still present in the membrane fraction after acute treatment with the cAMP analog. However, we only found phosphorylated PDE10A2 in the cytosolic fraction after treatment for 1 h with the cAMP analog. We hypothesized that this observation might be caused by the greater availability of cytosolic PDE10A2 to endogenous PKA phosphorylation, compared with that associated with the plasma membrane. To test this hypothesis, we increased the period of treatment with the cAMP analog to 8 h and assessed the extent to which PDE10A2 was phosphorylated at Thr-16 in both the membrane and cytosolic compartments (Fig. 8). Our results show that the degree of PDE10A2 phosphorylation at Thr-16 was significantly elevated in the membrane fraction as well as the cytosolic fraction compared with the vehicle control. Moreover, this was not accompanied by a shift in the total pool of PDE10A2 from the membrane to the cytosolic fraction (Fig. 8). These results suggest the following: (1) membrane-associated PDE10A2 can be phosphorylated at Thr-16; and (2) this event does not cause the disengagement of PDE10A2 from cellular membranes. These results are in agreement with the notion that phosphorylation of newly synthesized PDE10A2 at Thr-16 prevents palmitoylation, but phosphorylation of membrane-localized PDE10A2 at Thr-16 does not cause a redistribution of PDE10A2 to cytosolic compartments.
Figure 8.

Phosphorylation of membrane-localized PDE10A2 does not lead to cytosolic translocation. A, The period of treatment with Sp-6-Phe-cAMPS was increased to 8 h, and crude fractions were immunoblotted with anti-pPDE10A2 and anti-PDE10A. H, Homogenate, M, membrane, C, cytosol. B, The degree of PDE10A2 phosphorylation at Thr-16 was significantly elevated in the membrane fraction as well as in the cytosolic fraction compared with the vehicle control. The increase in Thr-16 phosphorylation was not accompanied by a shift in the total pool of PDE10A2 from the membrane to the cytosolic fraction. *p < 0.05; **p < 0.01; ***p < 0.001 by Student's test.
Discussion
It is now widely accepted that the high degree of specificity of cyclic nucleotide-mediated signaling pathways is achieved in part through the specific spatial localization of phosphodiesterases. Since multiple lines of evidence point to the striatum-enriched PDE10A as a potential therapeutic target for schizophrenia and related disorders (Siuciak et al., 2006a,b; Nishi et al., 2008; Schmidt et al., 2008; Siuciak et al., 2008; Grauer et al., 2009; Threlfell et al., 2009), we sought to further characterize the mechanism by which PDE10A localization is regulated. Previous reports suggest that PDE10A protein, but not mRNA, is specifically transported from the cell bodies to the axons and dendrites of medium spiny neurons (Seeger et al., 2003; Coskran et al., 2006; Xie et al., 2006). Moreover, fractionation studies indicate that striatal PDE10A is primarily membrane-bound and enriched in synaptosomal fractions, while immunogold electron microscopy studies have demonstrated that PDE10A is localized to the boundaries of postsynaptic densities on the dendritic spines of medium spiny neurons (Xie et al., 2006). While the above cited fractionation and histological studies were performed using antibodies or hybridization probes that did not distinguish between specific PDE10A splice forms, it can be inferred that the membrane-bound form of PDE10A localized to axons and neuropils of the medium spiny neurons corresponds to PDE10A2 because of the following: (1) this isoform represents a major PDE10A transcript in both rodent and human brain (Kotera et al., 1999); (2) using antisera to PDE10A1, PDE10A2, or PDE10A3, only PDE10A2 protein was adequately detected in rodent striata (Kotera et al., 2004); and (3) PDE10A2 is the only isoform that possesses the ability to bind membranes in striatum and recombinant systems (Kotera et al., 1999; Kotera et al., 2004; Dlaboga et al., 2008). Thus, we focused on the unique 23 amino acid N-terminal peptide of PDE10A2 to elucidate the mechanism by which PDE10A associates with membranes and is transported into dendrites of medium spiny neurons.
Our results indicate that PDE10A2 associates with the plasma membrane in both HEK293 cells and primary striatal neurons and that this association requires cysteine 11 of PDE10A2. We further show that membrane binding is achieved through palmitoylation of Cys-11, likely through ZDHHC-7 and -19. This finding is supported by the recent identification of PDE10A as a member of the neuronal palmitoyl proteome (Kang et al., 2008). Interestingly, ZDHHC-7 mRNA was shown to be actively translated in MSNs of the striatonigral but not the striatopallidal pathway, suggesting that PDE10A localization may be differentially regulated in these two output pathways (Doyle et al., 2008). We further show that palmitoylation of PDE10A2 can be regulated by phosphorylation of threonine 16 of PDE10A2, such that Thr-16 phosphorylation interferes with palmitoylation of Cys-11 and thus prevents membrane binding. Experiments using an antibody to phosphorylated PDE10A2 at Thr-16 suggested that native PDE10A2 localization is regulated by phosphorylation, such that phosphorylation at Thr-16 of PDE10A2 leads to a cytosolic localization, consistent with our in vitro findings as well as that reported by others (Kotera et al., 2004). Moreover, our results suggest that Cys-11 palmitoylation and Thr-16 phosphorylation regulate not only membrane binding but also the efficient trafficking of PDE10A2 to distal dendrites of primary striatal neurons. This latter observation may also be related to the ability of palmitoylated Cys-11 to facilitate association of PDE10A2 with intracellular transport vesicles. These findings are consistent with and provide a mechanistic account of the previously reported observations noted above, namely that striatal PDE10A is primarily membrane bound (Kotera et al., 2004; Xie et al., 2006; Dlaboga et al., 2008) and that PDE10A protein, but not mRNA, is localized to the axons and neuropils of striatal MSNs, suggesting that the PDE10A is transported along axons and throughout the dendritic tree of these neurons (Seeger et al., 2003; Coskran et al., 2006; Xie et al., 2006).
Our results also suggest that rather than interfering with the interaction of the palmitate moiety with phospholipids of the plasma membrane, phosphorylation of PDE10A2 at Thr-16 prevents palmitoylation at Cys-11 (Fig. 6). This alternate mechanism has been demonstrated for large-conductance calcium- and voltage-gated potassium (BK) channels, where acute treatment with a membrane-permeant cAMP analog lead to the phosphorylation-induced dissociation of palmitoylated BK channels from the plasma membrane (Tian et al., 2008). This is in direct contrast to our own experiments, where membrane-bound PDE10A2 became phosphorylated under similar treatment conditions but did not become dissociated from the membrane fraction of HEK293 cells (Figs. 7, 8). Our acute treatment results also contrasts that from cotransfection studies, where the coexpression of the catalytic subunit of PKA, PKAc, resulted in the phosphorylation and cytoplasmic localization of PDE10A2 (Figs. 1, 2 B) (Kotera et al., 2004). This suggests that palmitoylation at Cys-11 might occur shortly after protein synthesis and, thus, phosphorylation of PDE10A2 at Thr-16 must occur during this time period to interfere with palmitoylation. This proposed mechanism assumes that the palmitoylation of PDE10A2 is not readily reversible, otherwise Thr-16 phosphorylation would prevent repalmitoylation of PDE10A2 and the net effect on PDE10A2 localization might be a membrane to cytosolic translocation, contrary to our results in Figures 7 and 8. Indeed, for most palmitoylated proteins a palmitic acid moiety is linked to cysteine residues through a reversible thioester bond, a process called S-palmitoylation (Linder and Deschenes, 2007). However, when a palmitic acid is linked to an N-terminal cysteine, as is the case for PDE10A2, a thioester intermediate is thought to be immediately converted to a stable amide linkage, a process call N-palmitoylation (Linder and Deschenes, 2007). This is best exemplified by the stable N-linked palmitoylation of sonic hedgehog (Shh), where palmitoylation of Cys-24 first results in a thioester intermediate, followed by transfer of the palmitate moiety to the N terminus through the formation of a cyclic intermediate (Pepinsky et al., 1998). This event was initially inferred from the observation that an N-terminal HIS-tag prevented the incorporation of [3H]palmitate by Cys-24 of Shh, suggesting the requirement of a free NH2 terminus (Pepinsky et al., 1998). Interestingly, a recombinant form of PDE10A2 with an N-terminal FLAG tag was primarily localized to the cytosolic fraction of COS-7 cells, consistent with the notion that a free NH2 terminus is required for N-linked palmitoylation and thus for membrane association (Kotera et al., 1999). Thus, the membrane targeting and neuronal transport of PDE10A is likely mediated by irreversible N-linked palmitoylation of PDE10A2, a process that can be inhibited by PKA-mediated phosphorylation.
Taking these results together, we propose a model whereby PDE10A localization can be regulated in response to local fluctuations in cAMP levels at the site of PDE10A2 synthesis (Fig. 9). Thus, under conditions of high local cAMP levels near the site of synthesis, PDE10A2 becomes phosphorylated at Thr-16 because of increased cAMP-mediated activation of PKA interfering with palmitoylation at Cys-11, resulting in the local cytosolic accumulation of PDE10A2 where it can normalize cAMP levels through its catalytic activity (Fig. 9, right). Under conditions of low local cAMP at the site of PDE10A2 synthesis, PDE10A2 can become palmitoylated, facilitating the association with intracellular transport vesicles and permitting distal transport and plasma membrane targeting where it may serve to regulate intracellular signaling cascades associated with dopaminergic and glutamatergic synapses (Fig. 9, left).
Figure 9.

Proposed model for the regulation of PDE10A localization in response to local fluctuations in cAMP levels. Under conditions of high local cAMP levels near the site of synthesis, PDE10A2 becomes phosphorylated at Thr-16 because of increased cAMP-mediated activation of PKA. Thr-16 phosphorylation interferes with palmitoylation at Cys-11, resulting in the local cytosolic accumulation of PDE10A2 where it can normalize cAMP levels through its catalytic activity. Under conditions of low local cAMP at the site of PDE10A2 synthesis, PDE10A2 can become palmitoylated, facilitating the association with intracellular transport vesicles and permitting distal transport and plasma membrane targeting where it may serve to regulate intracellular signaling cascades associated with dopaminergic and glutamatergic synapses.
The above model has profound implications for the treatment of schizophrenia and related pathological conditions involving excessive striatal dopamine release. Under these conditions, dopamine D1 receptor activation in striatonigral MSNs is expected to lead to an elevation in cAMP production, whereas dopamine D2 receptor activation in striatopallidal MSNs is expected to lead to a reduction in cAMP production (Nishi et al., 1997, 2008). Incorporating the above model, excessive striatal dopamine release would exert differential effects on the regulation of PDE10A localization in the two striatal output pathways. In this way, PDE10A would be expected to shift toward a somatic, cytosolic localization in striatonigral MSNs caused by elevations in pPDE10A2, whereas it would be expected to shift toward an axonal and dendritic localization in striatopallidal MSNs caused by disinhibition of PDE10A2 palmitoylation. Moreover, it is likely that palmitoylation of PDE10A2 also facilitates targeting to specific signal transduction microdomains, since it was previously shown that the majority of PDE activity in brain-derived lipid rafts was caused by the presence of PDE10A and PDE2A in these microdomains, the latter of which is myristoylated and also likely to be palmitoylated (Noyama and Maekawa, 2003; Russwurm et al., 2009). Multiple studies have underscored the importance of palmitoylation in the localization of cellular proteins to lipid rafts, as well as the function of lipid rafts as key structures involved in signal transduction and subcellular trafficking (Brown, 2006). Thus, activation of the striatopallidal pathway might also facilitate the localization of PDE10A to specific signal transduction microdomains associated with dopaminergic and glutamatergic synapses. Although the differential regulation of PDE10A localization in the two striatal output pathways has not been directly tested, it is interesting to note that PDE10A inhibition was shown to have a preferential effect on cAMP/PKA-mediated signaling in striatopallidal neurons (Nishi et al., 2008) and also on the response of striatopallidal neurons to cortically evoked activity (Threlfell et al., 2009). Results of these studies might reflect a subtle bias of PDE10A localization to dendritic signal transduction microdomains in striatopallidal neurons compared with striatonigral neurons, consistent with our model of spatial regulation of PDE10A by palmitoylation and cAMP-mediated phosphorylation.
Footnotes
We thank Prof. Masaki Fukata (National Institute for Physiological Sciences, National Institute of Natural Sciences, Myodaiji, Japan) for generously providing the 23 mouse PAT cDNA clones. We also thank Drs. Thorsten Hage, Ute Egerland, and Tom Kronbach (Biotie Therapies, Radebeul, Germany) for their constructive comments toward the preparation of this manuscript. Finally, we thank Dr. Michele Kelly (Pfizer Global Research and Development, Groton, CT) for her guidance in statistical analysis.
References
- Abramoff MD, Magelhaes PJ, Ram SJ. Image processing with ImageJ. Biophotonics Int. 2004;11:36–42. [Google Scholar]
- Beavo JA, Brunton LL. Cyclic nucleotide research – still expanding after half a century. Nat Rev Mol Cell Biol. 2002;3:710–718. doi: 10.1038/nrm911. [DOI] [PubMed] [Google Scholar]
- Brown DA. Lipid rafts, detergent-resistant membranes, and raft targeting signals. Physiology (Bethesda) 2006;21:430–439. doi: 10.1152/physiol.00032.2006. [DOI] [PubMed] [Google Scholar]
- Conti M, Beavo J. Biochemistry and physiology of cyclic nucleotide phosphodiesterases: essential components in cyclic nucleotide signaling. Annu Rev Biochem. 2007;76:481–511. doi: 10.1146/annurev.biochem.76.060305.150444. [DOI] [PubMed] [Google Scholar]
- Coskran TM, Morton D, Menniti FS, Adamowicz WO, Kleiman RJ, Ryan AM, Strick CA, Schmidt CJ, Stephenson DT. Immunohistochemical localization of phosphodiesterase 10A in multiple mammalian species. J Histochem Cytochem. 2006;54:1205–1213. doi: 10.1369/jhc.6A6930.2006. [DOI] [PubMed] [Google Scholar]
- Dessauer CW. Adenylyl cyclase–A-kinase anchoring protein complexes: the next dimension in cAMP signaling. Mol Pharmacol. 2009;76:935–941. doi: 10.1124/mol.109.059345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dlaboga D, Hajjhussein H, O'Donnell JM. Chronic haloperidol and clozapine produce different patterns of effects on phosphodiesterase-1B, -4B, and -10A expression in rat striatum. Neuropharmacology. 2008;54:745–754. doi: 10.1016/j.neuropharm.2007.12.002. [DOI] [PubMed] [Google Scholar]
- Doyle JP, Dougherty JD, Heiman M, Schmidt EF, Stevens TR, Ma G, Bupp S, Shrestha P, Shah RD, Doughty ML, Gong S, Greengard P, Heintz N. Application of a translational profiling approach for the comparative analysis of CNS cell types. Cell. 2008;135:749–762. doi: 10.1016/j.cell.2008.10.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujishige K, Kotera J, Michibata H, Yuasa K, Takebayashi S, Okumura K, Omori K. Cloning and characterization of a novel human phosphodiesterase that hydrolyzes both cAMP and cGMP (PDE10A) J Biol Chem. 1999a;274:18438–18445. doi: 10.1074/jbc.274.26.18438. [DOI] [PubMed] [Google Scholar]
- Fujishige K, Kotera J, Omori K. Striatum- and testis-specific phosphodiesterase PDE10A isolation and characterization of a rat PDE10A. Eur J Biochem. 1999b;266:1118–1127. doi: 10.1046/j.1432-1327.1999.00963.x. [DOI] [PubMed] [Google Scholar]
- Fujishige K, Kotera J, Yuasa K, Omori K. The human phosphodiesterase PDE10A gene genomic organization and evolutionary relatedness with other PDEs containing GAF domains. Eur J Biochem. 2000;267:5943–5951. doi: 10.1046/j.1432-1327.2000.01661.x. [DOI] [PubMed] [Google Scholar]
- Fukata M, Fukata Y, Adesnik H, Nicoll RA, Bredt DS. Identification of PSD-95 palmitoylating enzymes. Neuron. 2004;44:987–996. doi: 10.1016/j.neuron.2004.12.005. [DOI] [PubMed] [Google Scholar]
- Fukata Y, Fukata M. Protein palmitoylation in neuronal development and synaptic plasticity. Nat Rev Neurosci. 2010;11:161–175. doi: 10.1038/nrn2788. [DOI] [PubMed] [Google Scholar]
- Gerfen CR. The neostriatal mosaic: multiple levels of compartmental organization. Trends Neurosci. 1992;15:133–139. doi: 10.1016/0166-2236(92)90355-c. [DOI] [PubMed] [Google Scholar]
- Grauer SM, Pulito VL, Navarra RL, Kelly MP, Kelley C, Graf R, Langen B, Logue S, Brennan J, Jiang L, Charych E, Egerland U, Liu F, Marquis KL, Malamas M, Hage T, Comery TA, Brandon NJ. Phosphodiesterase 10A inhibitor activity in preclinical models of the positive, cognitive, and negative symptoms of schizophrenia. J Pharmacol Exp Ther. 2009;331:574–590. doi: 10.1124/jpet.109.155994. [DOI] [PubMed] [Google Scholar]
- Houslay MD. Underpinning compartmentalised cAMP signalling through targeted cAMP breakdown. Trends Biochem Sci. 2010;35:91–100. doi: 10.1016/j.tibs.2009.09.007. [DOI] [PubMed] [Google Scholar]
- Huang K, El-Husseini A. Modulation of neuronal protein trafficking and function by palmitoylation. Curr Opin Neurobiol. 2005;15:527–535. doi: 10.1016/j.conb.2005.08.001. [DOI] [PubMed] [Google Scholar]
- Kang R, Wan J, Arstikaitis P, Takahashi H, Huang K, Bailey AO, Thompson JX, Roth AF, Drisdel RC, Mastro R, Green WN, Yates JR, 3rd, Davis NG, El-Husseini A. Neural palmitoyl-proteomics reveals dynamic synaptic palmitoylation. Nature. 2008;456:904–909. doi: 10.1038/nature07605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kotera J, Fujishige K, Yuasa K, Omori K. Characterization and phosphorylation of PDE10A2, a novel alternative splice variant of human phosphodiesterase that hydrolyzes cAMP and cGMP. Biochem Biophys Res Commun. 1999;261:551–557. doi: 10.1006/bbrc.1999.1013. [DOI] [PubMed] [Google Scholar]
- Kotera J, Sasaki T, Kobayashi T, Fujishige K, Yamashita Y, Omori K. Subcellular localization of cyclic nucleotide phosphodiesterase type 10A variants, and alteration of the localization by cAMP-dependent protein kinase-dependent phosphorylation. J Biol Chem. 2004;279:4366–4375. doi: 10.1074/jbc.M308471200. [DOI] [PubMed] [Google Scholar]
- Linder ME, Deschenes RJ. Palmitoylation: policing protein stability and traffic. Nat Rev Mol Cell Biol. 2007;8:74–84. doi: 10.1038/nrm2084. [DOI] [PubMed] [Google Scholar]
- Loughney K, Snyder PB, Uher L, Rosman GJ, Ferguson K, Florio VA. Isolation and characterization of PDE10A, a novel human 3′,5′-cyclic nucleotide phosphodiesterase. Gene. 1999;234:109–117. doi: 10.1016/s0378-1119(99)00171-7. [DOI] [PubMed] [Google Scholar]
- Menniti FS, Faraci WS, Schmidt CJ. Phosphodiesterases in the CNS: targets for drug development. Nat Rev Drug Discov. 2006;5:660–670. doi: 10.1038/nrd2058. [DOI] [PubMed] [Google Scholar]
- Nishi A, Snyder GL, Greengard P. Bidirectional regulation of DARPP-32 phosphorylation by dopamine. J Neurosci. 1997;17:8147–8155. doi: 10.1523/JNEUROSCI.17-21-08147.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishi A, Kuroiwa M, Miller DB, O'Callaghan JP, Bateup HS, Shuto T, Sotogaku N, Fukuda T, Heintz N, Greengard P, Snyder GL. Distinct roles of PDE4 and PDE10A in the regulation of cAMP/PKA signaling in the striatum. J Neurosci. 2008;28:10460–10471. doi: 10.1523/JNEUROSCI.2518-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noyama K, Maekawa S. Localization of cyclic nucleotide phosphodiesterase 2 in the brain-derived Triton-insoluble low-density fraction (raft) Neurosci Res. 2003;45:141–148. doi: 10.1016/s0168-0102(02)00208-0. [DOI] [PubMed] [Google Scholar]
- Pepinsky RB, Zeng C, Wen D, Rayhorn P, Baker DP, Williams KP, Bixler SA, Ambrose CM, Garber EA, Miatkowski K, Taylor FR, Wang EA, Galdes A. Identification of a palmitic acid-modified form of human Sonic hedgehog. J Biol Chem. 1998;273:14037–14045. doi: 10.1074/jbc.273.22.14037. [DOI] [PubMed] [Google Scholar]
- Russwurm C, Zoidl G, Koesling D, Russwurm M. Dual acylation of PDE2A splice variant 3: targeting to synaptic membranes. J Biol Chem. 2009;284:25782–25790. doi: 10.1074/jbc.M109.017194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt CJ, Chapin DS, Cianfrogna J, Corman ML, Hajos M, Harms JF, Hoffman WE, Lebel LA, McCarthy SA, Nelson FR, Proulx-LaFrance C, Majchrzak MJ, Ramirez AD, Schmidt K, Seymour PA, Siuciak JA, Tingley FD, 3rd, Williams RD, Verhoest PR, Menniti FS. Preclinical characterization of selective phosphodiesterase 10A inhibitors: a new therapeutic approach to the treatment of schizophrenia. J Pharmacol Exp Ther. 2008;325:681–690. doi: 10.1124/jpet.107.132910. [DOI] [PubMed] [Google Scholar]
- Seeger TF, Bartlett B, Coskran TM, Culp JS, James LC, Krull DL, Lanfear J, Ryan AM, Schmidt CJ, Strick CA, Varghese AH, Williams RD, Wylie PG, Menniti FS. Immunohistochemical localization of PDE10A in the rat brain. Brain Res. 2003;985:113–126. doi: 10.1016/s0006-8993(03)02754-9. [DOI] [PubMed] [Google Scholar]
- Siuciak JA, McCarthy SA, Chapin DS, Fujiwara RA, James LC, Williams RD, Stock JL, McNeish JD, Strick CA, Menniti FS, Schmidt CJ. Genetic deletion of the striatum-enriched phosphodiesterase PDE10A: evidence for altered striatal function. Neuropharmacology. 2006a;51:374–385. doi: 10.1016/j.neuropharm.2006.01.012. [DOI] [PubMed] [Google Scholar]
- Siuciak JA, Chapin DS, Harms JF, Lebel LA, McCarthy SA, Chambers L, Shrikhande A, Wong S, Menniti FS, Schmidt CJ. Inhibition of the striatum-enriched phosphodiesterase PDE10A: a novel approach to the treatment of psychosis. Neuropharmacology. 2006b;51:386–396. doi: 10.1016/j.neuropharm.2006.04.013. [DOI] [PubMed] [Google Scholar]
- Siuciak JA, McCarthy SA, Chapin DS, Martin AN, Harms JF, Schmidt CJ. Behavioral characterization of mice deficient in the phosphodiesterase-10A (PDE10A) enzyme on a C57BL/6N congenic background. Neuropharmacology. 2008;54:417–427. doi: 10.1016/j.neuropharm.2007.10.009. [DOI] [PubMed] [Google Scholar]
- Soderling SH, Bayuga SJ, Beavo JA. Isolation and characterization of a dual-substrate phosphodiesterase gene family: PDE10A. Proc Natl Acad Sci U S A. 1999;96:7071–7076. doi: 10.1073/pnas.96.12.7071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Threlfell S, Sammut S, Menniti FS, Schmidt CJ, West AR. Inhibition of phosphodiesterase 10A increases the responsiveness of striatal projection neurons to cortical stimulation. J Pharmacol Exp Ther. 2009;328:785–795. doi: 10.1124/jpet.108.146332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian L, Jeffries O, McClafferty H, Molyvdas A, Rowe IC, Saleem F, Chen L, Greaves J, Chamberlain LH, Knaus HG, Ruth P, Shipston MJ. Palmitoylation gates phosphorylation-dependent regulation of BK potassium channels. Proc Natl Acad Sci U S A. 2008;105:21006–21011. doi: 10.1073/pnas.0806700106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie Z, Adamowicz WO, Eldred WD, Jakowski AB, Kleiman RJ, Morton DG, Stephenson DT, Strick CA, Williams RD, Menniti FS. Cellular and subcellular localization of PDE10A, a striatum-enriched phosphodiesterase. Neuroscience. 2006;139:597–607. doi: 10.1016/j.neuroscience.2005.12.042. [DOI] [PMC free article] [PubMed] [Google Scholar]