

Abstract
CollagenQ (ColQ) plays an important structural role at vertebrate neuromuscular junctions (NMJs) by anchoring and accumulating acetylcholinesterase (AChE) in the extracellular matrix (ECM). Moreover, ColQ interacts with perlecan/dystroglycan and the muscle-specific receptor tyrosine kinase (MuSK), key molecules in the NMJ formation. MuSK promotes acetylcholine receptor (AChR) clustering in a process mediated by rapsyn, a cytoplasmic protein that stimulates AChR packing in clusters and regulates synaptic gene transcription. Here, we investigated a regulatory role for ColQ by comparing the clustering and expression of synaptic proteins in wild type and ColQ-deficient muscle cells in culture and at NMJ. We show first that AChR clusters are smaller and more densely packed in the absence of ColQ both in vitro and in vivo. Second, we find that like AChRs and rapsyn, MuSK mRNA levels are increased in cultured cells but not in muscles lacking ColQ. However, membrane-bound MuSK is decreased both in vitro and in vivo suggesting that ColQ controls MuSK sorting or stabilization in the muscle membrane. In line with this, our data show that activation of the MuSK signaling pathway is altered in the absence of ColQ leading to (1) perturbation of AChR clustering and/or β-AChR subunit phosphorylation and (2) modifications of AChR mRNA level due to the lack of ColQ-MuSK interaction. Together, our results demonstrate that ColQ, in addition to its structural role, has important regulatory functions at the synapse by controlling AChR clustering and synaptic gene expression through its interaction with MuSK.
Introduction
Depolarization of the motoneuron at vertebrate neuromuscular junctions (NMJs) evokes release of acetylcholine (ACh) into the synaptic cleft, which activates acetylcholine receptors (AChRs) on the postsynaptic membrane. Once released in the synaptic space, ACh level is largely controlled by acetylcholinesterase (AChE; EC 3.1.1.7) (Legay, 2000). The most abundant AChE form accumulated at the NMJ is the AChET variant bound to a specific collagen, CollagenQ (ColQ) (Krejci et al., 1991, 1997). The physiological importance of the AChE-ColQ complex has been highlighted by identification of >30 COLQ mutations in the human gene (Donger et al., 1998; Ohno et al., 1998; Schreiner et al., 2007; Mihaylova et al., 2008). Patients bearing COLQ mutations and mice deficient for COLQ present a congenital myasthenic syndrome with AChE deficiency (CMS-1c) (Ohno et al., 1998; Feng et al., 1999; Mihaylova et al., 2008). To date, most observed defects have been assigned to the complete absence or reduced level of AChE at NMJs (Feng et al., 1999; Hantaï et al., 2004).
ColQ is a nonfibrillar collagen composed of a central collagenous domain flanked by noncollagenous N terminus and C terminus regions (Krejci et al., 1997). Through its three domains, ColQ is able to interact with elements of the extracellular matrix (ECM) and postsynaptic membrane of vertebrate NMJs. ColQ N terminus is responsible for AChE association (Bon et al., 1997; Dvir et al., 2004). The collagen domain interacts with perlecan, which itself binds dystroglycan (Peng et al., 1999; Jacobson et al., 2001; Arikawa-Hirasawa et al., 2002). The ColQ C terminus binds the muscle-specific receptor tyrosine kinase (MuSK), an interaction that is thought to be largely responsible for the synaptic localization of AChE-ColQ complex (Cartaud et al., 2004).
MuSK is indispensable to NMJ formation and triggers AChR clustering even before the nerve contacts the muscle (DeChiara et al., 1996; Lin et al., 2001; Yang et al., 2001). Innervation provides a factor called agrin that binds the low density lipoprotein receptor-related protein 4 (LRP4) in the MuSK-LRP4 complex and stimulates MuSK phosphorylation (Kim et al., 2008; Zhang et al., 2008). Once phosphorylated, MuSK recruits rapsyn, a membrane associated cytoplasmic protein via complex pathways. Rapsyn then interacts with AChR cytoplasmic loops and β-AChR subunits are phosphorylated leading to AChR clustering (Sanes and Lichtman, 2001). MuSK is also a signaling platform that either stimulates or represses synaptic gene transcription (Moore et al., 2001; Lacazette et al., 2003; Strochlic et al., 2004). Given the essential role of MuSK in AChR clustering and in the regulation of synaptic gene transcription, we hypothesized that in addition to its structural role, ColQ could control postsynaptic differentiation through this partner. This hypothesis is consistent with the fact that collagens have been previously shown to participate in signal transduction mechanisms, mostly through interaction with integrins (Barczyk et al., 2010) and tyrosine kinase receptors (Schlessinger, 1997).
To explore the function of ColQ in postsynaptic differentiation, we compared the phenotypes of AChR clusters in wild type and ColQ-deficient muscle cell lines and mouse muscles. We demonstrate that ColQ, in addition to its structural role in anchoring AChE, exerts regulatory functions in AChR clustering and expression involving its interaction with MuSK.
Materials and Methods
Antibodies and reagents.
α-Bungarotoxin (α-BTX) Alexa Fluor 488 or 594 conjugate, rabbit monoclonal or polyclonal anti-GFP, Alexa Fluor 488- or 594-conjugated goat anti-rabbit or anti-mouse and mouse monoclonal anti-transferrin receptor (TfR) were purchased from Invitrogen. Cy3-conjugated goat anti-rabbit were from Interchim. Mouse monoclonal anti-α-AChR subunit was purchased from Transduction Laboratories (BD Biosciences). Antibody to the β-AChR subunit (mAb124) was a gift from J. Lindstrom, University of Pennsylvania, Philadelphia, PA (Gullick and Lindstrom, 1983). Rabbit anti-pAChRβ1 (Y390) antiserum (sc-17087R) was obtained from Santa Cruz Biotechnology. Mouse monoclonal anti-GAPDH antibody (clone 6C5) and rabbit anti-MuSK antiserum for Western blot were purchased from Abcam. Rabbit antiserum to MuSK (194T) for immunofluorescence was a gift from M. Ruegg, University of Basel, Basel, Switzerland (Scotton et al., 2006). The mouse monoclonal anti-rapsyn (clone 1234) and anti-Flag M2 were obtained from Sigma-Aldrich. Rabbit polyclonal anti-AChE antibody was a gift from P. Taylor, University of California, San Diego, San Diego, CA (Jennings et al., 2003). Horseradish peroxidase-conjugated donkey anti-rabbit IgG and sheep anti-mouse IgG antiserum were obtained from GE Healthcare.
Constructs.
The rat AChE and ColQ1a cDNA clones have been previously described (Krejci et al., 1999). The GFP-ColQ construct previously used in Cartaud et al. (2004) was generated by introduction of a GFP tag sequence after the triplet coding for amino acid 20 (numbering from the first methionine) in the ColQ1a rat cDNA that contains the sequence coding for the peptide signal. The GFP-ColQ-Y431S cDNA clone has been previously described by Cartaud et al. (2004). The GFP-ColQ-ΔCt and GFP-ColQ-ΔCol cDNA clones were generated from ΔCt and ΔCol constructs described by Deprez et al. (2003) in which the GFP tag sequence was introduced at the same position as for GFP-ColQ construct. The GFP-ColQ-ΔColΔCt cDNA clone was generated by inserting four stop codons in the GFP-ColQ-ΔCol construct after the ColQ trimerization domain (amino acid 370). The Flag-ColQ cDNA clone was generated by the introduction of a sequence encoding the Flag epitope (GAC TAC AAG GAT GAC GAT GAC AAG) after the triplet coding for amino acid 26 in the ColQ1a coding sequence.
Animals.
Generation of ColQ knock-out mice was described by Feng et al. (1999). The use of animals is in compliance with the Society for Neuroscience Policies on the Use of Animals and Humans in Neuroscience Research. Standard breeding methods were used to generate double mutants.
Muscle cell culture and transfection.
The two muscle cell lines, wild type (wt) and ColQ-deficient (ColQ−/−) were generated as described by Cartaud et al. (2004). The MuSK-deficient (MK−/−) muscle cell line has been described previously by Herbst and Burden (2000). Myoblasts were cultured on plates coated with collagen type I (Iwaki) maintained in DMEM supplemented with 10% fetal bovine serum, 20% horse serum, 2 mm glutamine, 2% penicillin/streptomycin (5000 U) and 20 U/ml γ-interferon (Roche Diagnostics) at 33°C in 8% CO2. All reagents for the culture medium were purchased from Invitrogen. Cells were differentiated into myotubes in the same medium with 5% horse serum and without γ-interferon (differentiation medium). Treatment of myotubes with agrin was made with 0.4 μg/ml Recombinant Rat Sf21-derived C-terminal agrin (R&D Systems).
Myoblasts were transfected using ExGen 500 (Euromedex) 1 d after fusion was induced by addition of differentiation medium. Transfections were performed according to the manufacturer's instructions and resulted in 50–70% of cells expressing the transfected cDNAs.
COS-7 cell culture, transfection, and immunoprecipitation.
COS-7 cells (purchased from American Type Culture Collection) were cultured in DMEM supplemented with 10% fetal bovine serum, 2 mm glutamine, and 2% penicillin/streptomycin (5000 U) at 37°C in 5% CO2. Cells were grown to 70% confluence on coverslips and transfected using ExGen 500. Protein extraction and immunoprecipitation analyses were performed 48 h later. GFP and Flag immunoprecipitations were performed on total cell lysates incubated with 4 μg of anti-GFP or anti-Flag antibodies. GFP-ColQ and Flag-ColQ proteins were precipitated with protein G Sepharose (3 h at 4°C), according to the manufacturer's instructions (GE Healthcare). Immunoprecipitates were then washed in lysis buffer (TrisHCl 50 mm, NaCl 150 mm, EDTA 2 mm, Sodium orthovanadate 2 mm and TritonX100 1%), suspended in 1× SDS sample buffer, resolved by 7% NuPAGE Novex Tris-Acetate Gels and analyzed by Western blot using anti-GFP or anti-Flag antibodies.
Immunofluorescence.
Immunostaining was performed on myotubes after fixation in 2% paraformaldehyde (PFA)-phosphate buffer (PBS). Before incubation with antibodies, nonspecific binding was blocked using DakoCytomation Protein block for 5 min. Cells were first incubated with the primary antibody for 1 h at room temperature, then washed in Glycine-BSA (0.15%:0.5%) buffer and incubated with a fluorochrome-conjugated secondary antibody for 30 min. Slides were mounted in Vectashield Hard Set Mounting Medium for fluorescence (Vector Laboratories).
Immunohistochemistry was performed on two different muscles: the sternomastoid (STM), a fast-type muscle and the soleus (Sol), a slow-type muscle from postnatal day 7 (P7) wt and ColQ−/− mice. Muscles were fixed in 3% PFA-PBS buffer, followed by successive incubations in 5%, 10% and 25% sucrose-PBS buffer at 4°C and frozen in isopentane cooled with liquid nitrogen. 14 μm cryostat cross-sections were incubated with DakoCytomation Protein block, then with the primary antibody for 1 h at room temperature, washed in Glycine-BSA (0.15%:0.5%) buffer and incubated with a fluorochrome-conjugated secondary antibody for 1 h.
Images were collected using a microscope (Olympus BX61) equipped with a Fast 1394 Digital CCD FireWire camera (model Retiga 2000R; Qimaging) and a 40× oil objective (numerical aperture: 1.0; Olympus). Images were captured using ImageProPlus software (version 5.1). For quantitative analysis, images were collected on a confocal microscope (Zeiss) equipped with 40× (in vitro) or 63× (in vivo) oil objectives (numerical aperture: 1.3 and 1.4, respectively; Zeiss). Collected confocal image stacks and image capture were made using an LSM Image Browser, with exposure time kept constant and below grayscale pixel saturation. All confocal images presented are single-projected images derived from overlaying each set of stacks taken at 0.47 μm (in vitro) or 0.9 μm (in vivo) intervals.
For quantification of area and fluorescence intensity, the perimeters of individual clusters were delimited. The LSM Image Browser software calculated the area in μm2 and the fluorescence intensity within the cluster, giving a measurement of pixel intensity. The cluster outline was then placed in an adjacent area devoid of clusters to record the background fluorescence intensity. This reading was subtracted from the cluster reading, yielding the background-corrected intensity.
Western blot analysis.
Proteins were extracted from cells using lysis buffer (TrisHCl 50 mm, NaCl 150 mm, EDTA 2 mm, Sodium orthovanadate 2 mm and TritonX100 1%) and lysates were precleared by centrifugation. Equal amounts of proteins (10–30 μg/sample) were first separated by 7% NuPAGE Novex Tris-Acetate Gels according to Invitrogen protocols, then electrotransferred to nitrocellulose membranes (TransBlot Transfer Medium, Bio-Rad) (Towbin et al., 1979). Membranes were blocked in PBS supplemented with Tween 0.1% (PBST) and 5% nonfat dry milk or 3% BSA for phosphorylated protein analysis and incubated with various primary antibodies in PBST-milk 5% or BSA 1.5%. After washing in PBST, membranes were incubated with Horseradish Peroxidase-conjugated secondary antibodies in PBST-milk 5% or BSA 1.5%. Bound antibodies were revealed using ECL plus detection reagents (GE Healthcare) and visualized with a Storm fluorescence scanner (Storm 860, Molecular Dynamics; GE Healthcare) or with hyperfilm ECL. Protein loading was assessed by probing with anti-GAPDH or anti-TfR antibodies. Results obtained with the Storm scanner were quantified with ImageQuant (version 5.2, Molecular Dynamics), and results obtained on hyperfilms were scanned and quantified using UnScanIt gel Automated Digitizing System (version 4.1).
Biotinylation of surface proteins.
Myotubes were treated or not with agrin for 1 h, then washed with PBS supplemented with 0.1 mm CaCl2, 1 mm MgCl2 (PBS-Ca-Mg) and incubated with 0.5 mg/ml EZ-link sulfo-NHS-LC-biotin (Pierce) in PBS-Ca-Mg at room temperature for 30 min, according to Herbst and Burden (2000). Cells were rinsed twice with PBS-Ca-Mg and incubated with DMEM at 33°C and 8% CO2. After 30 min, the cells were washed with ice-cold PBS and proteins were extracted from cells using lysis buffer. Lysates were precleared by centrifugation, and biotin-labeled proteins were recovered by incubating with streptavidin-agarose for 4 h at 4°C, followed by four washes with lysis buffer. Bound and unbound (2% of supernatant) proteins were resolved by a 7% NuPAGE Novex Tris-Acetate Gel and detected by Western blot.
RNA preparation.
Total RNAs from wt and ColQ−/− muscle cells were extracted by using the RNeasy Protect Mini-Kit (Qiagen) and by performing DNA digestion according to the manufacturer's instructions. RNA quality was assessed by polyacrylamide-gel microelectrophoresis (Bioanalyser 2100, Agilent Technologies) and by RT-PCR. Lack of genomic DNA contamination was verified by PCR. Total gastrocnemius fast- and slow-muscle RNAs from wt and ColQ−/− P7 mice were extracted by using Lysing Matrix D (MP Biomedicals), proteinase K treatment, and RNeasy Fibrous Tissue Mini-Kit (Qiagen) according to the manufacturer's instructions.
TaqMan real-time RT-PCR.
The TaqMan Low Density Array (TLDA, Applied Biosystems) was used in a two-step RT-PCR process (Marionneau et al., 2005). First-strand cDNA was synthesized from 220 ng of total RNA using the High-Capacity cDNA Reverse Transcription Kit (Applied Biosystems). PCRs were then performed on TLDA with the ABI Prism 7900HT Sequence Detection System (Applied Biosystems). The 384 wells of each card were preloaded with predesigned fluorogenic TaqMan probes and primers corresponding mainly to synaptic genes, and five of these genes were selected for normalization (HPRT1, MTA1, REXO1, RPL39, and SEC63). Data were collected with Applied Biosystems SDS 2.1 software, and then analyzed with the threshold cycle (Ct) relative-quantification method (Livak and Schmittgen, 2001). Since SEC63 is the most uniformly distributed gene between the different conditions, we selected it for data normalization. The relative expression of each gene versus SEC63 (2−ΔCt, ΔCt indicating normalized data: ΔCt = target gene Ct − SEC63 Ct) was calculated and then averaged for each condition. TLDA technique was used in a large screen experiment performed in vitro. From the results obtained by TLDA, we focused on a few genes of interest that have subsequently been analyzed in vivo and in in vitro transfection experiments using SyBR Green technique.
SyBR Green RT-PCR.
The QuantiTect SyBR Green RT-PCR Kit (Qiagen) was used in a one-step RT-PCR protocol for mouse mRNA analysis, for β-AChR subunit analysis in vitro and for transfection experiments. α-ACHR, β-ACHR, ε-ACHR, MUSK, RAPSYN, GAPDH, and SEC63 QuantiTect primers were also purchased from Qiagen. Data were normalized to values for SEC63 (RNAs from cell culture) or GAPDH (RNAs from mice muscles). RT-PCRs were then performed in triplicates on a 384-well plate with the ABI Prism 7900HT Sequence Detection System (Applied Biosystems). The wells were loaded with RT-PCR Master Mix, QuantiTect RT Mix, QuantiTect Primer Assay and 9 ng of total RNAs for each sample. The relative expression of each gene versus SEC63 or GAPDH (2−ΔCt) was calculated and then averaged for each condition.
Statistical analysis.
Data are expressed as means ± SEM or were normalized against the control groups to allow for comparisons between experiments and are presented as percentage of control ± SEM. Statistical analyses were done by pairwise comparisons between two conditions using Student's unpaired t tests for n ≥ 30 and Mann–Whitney U test for n < 30 (p < 0.05 considered significant).
Results
ColQ regulates AChR clustering and packing in vitro
ColQ expression in myoblasts is first upregulated during their fusion into myotubes and before AChR cluster formation (Guerra et al., 2005). A second dramatic increase in ColQ level is observed concomitantly with AChE upregulation as muscle cells further differentiate and leads to AChE cluster formation (Guerra et al., 2005). Given that ColQ is expressed early during postsynaptic differentiation, we investigated its role in AChR clustering by comparing wt and ColQ−/− muscle cell lines 2 d after myotube formation. At this time-point, AChR clusters were observed in both cell lines (Fig. 1) and were in greater number in cells lacking ColQ (+125%, p < 0.001, n ≥ 80 myotubes: wt, 4.1 ± 0.3 clusters per myotube; ColQ−/−, 8.7 ± 0.7 clusters per myotube) (Fig. 1). Similar to the C2C12 muscle cell line, only a subset of wt and ColQ−/− myotubes bore AChR clusters and the percentage of myotubes presenting AChR clusters was similar in wt and ColQ-deficient cell lines (data not shown). However, quantitative fluorescence analysis of α-BTX labeling (see Materials and Methods) revealed that AChR clusters were smaller in the absence of ColQ (−49%, p < 0.001, n ≥160 AChR clusters: wt clusters, 132 ± 9.06 μm2; ColQ−/− clusters, 67 ± 4.35 μm2) (Fig. 1). These results prompted us to examine whether ColQ deficiency modifies the number of AChR molecules per cluster. The fluorescence intensity per AChR cluster and per μm2 that reflects AChR density within a cluster were both higher in ColQ−/− myotubes than in wt myotubes (+49% and +125% respectively, p < 0.001, n ≥ 160 AChR clusters) (Fig. 1). These data indicate that AChR clusters in ColQ-deficient myotubes are smaller but more densely packed with AChRs than wt myotubes.
Figure 1.

AChR clustering in wt and ColQ-deficient muscle cells. AChR clusters in wt (a) and ColQ−/− (b) myotubes (2 d old) labeled with Alexa 488 α-BTX. Projections of confocal stacks. Scale bar, 20 μm.
Restoring ColQ reverses the effects of ColQ deficiency on AChR clustering
We next tested whether restoring ColQ levels would reverse the effects of ColQ deficiency on AChR clustering. To assess the effects of ColQ independently of AChE, ColQ−/− cells were transfected with a ColQ construct in which the GFP tag position did not modify ColQ interaction with MuSK and perlecan both required for ColQ-AChE anchoring in the ECM (Cartaud et al., 2004), but severely limits its association with AChE. This last result was obtained by comparing AChE binding to GFP-ColQ and Flag-ColQ, a construct in which the Flag was inserted in the ColQ cDNA at about the same position as the GFP tag (see Materials and Methods). Coimmunoprecipitations were performed after cotransfection of COS-7 cells with GFP-ColQ or Flag-ColQ together with AChE and analyzed by Western blot (supplemental Fig. 1, available at www.jneurosci.org as supplemental material). Western blot quantification showed that GFP-ColQ is less efficient in AChE binding than Flag-ColQ (−88%, p < 0.05, n = 3). Moreover, no collagen-tailed form of AChE were detected by velocity sedimentation analysis when GFP-ColQ and AChE were cotransfected in Xenopus oocytes (E. Krejci, personal communication), suggesting that the GFP sterically hinders the binding of AChE to GFP-ColQ.
The effects of GFP-ColQ on AChR clustering were analyzed by fluorescence analysis of α-BTX labeling and revealed that ColQ−/− cells overexpressing GFP-ColQ had fewer AChR clusters than untransfected myotubes (Fig. 2Aa,b). Indeed, 75% of cells transfected had zero to two AChR clusters (1.5 ± 0.3) (Fig. 2B, black bars), whereas 53% of untransfected myotubes had three to five clusters (4.4 ± 0.5) (Fig. 2B, white bars). This decrease of AChR clustering was not specific to the ColQ−/− cell line since wt and C2C12 myotubes transfected with GFP-ColQ were also nearly devoid of AChR clusters (Fig. 2Ac–f). In addition, the areas of the remaining clusters were higher in transfected cells than in untransfected ColQ−/− cells (+84%, p < 0.01, n ≥30 AChR clusters). The fluorescence intensity per AChR cluster was decreased in ColQ-deficient transfected myotubes (−26%, p < 0.01, n ≥ 30 AChR clusters) as well as AChR density in clusters (−60%, p < 0.001, n ≥ 30 AChR clusters). In contrast to the full-length construct, a similar number of AChR clusters was observed in ColQ−/− myotubes and ColQ−/− myotubes transfected with a GFP-ColQ construct lacking the collagenous and C terminus domains (GFP-ColQ-ΔColΔCt) (Fig. 2Ag,h). Thus, restoring the full-length ColQ in ColQ-deficient cells reversed the loss of function with regard to AChR cluster number, size variations and packing of AChRs in clusters. Moreover, these data revealed that GFP-ColQ effects on AChR clustering was not due to GFP per se and suggest that the collagenous domain and/or the C terminus domain of ColQ are implicated in the control of AChR clustering, whereas interaction of ColQ with AChE is not necessary to regulate clustering.
Figure 2.

Restoring ColQ reverses the effects of ColQ deficiency on AChR clustering. A, AChR clusters labeled with Alexa 594 α-BTX in ColQ−/−, wt, and C2C12 myotubes overexpressing GFP-ColQ or GFP-ColQ-ΔColΔCt. ColQ−/− cells (a, b) as well as wt (c, d) and C2C12 cells (e, f) overexpressing GFP-ColQ (white arrows) were nearly devoid of AChR clusters, whereas clusters were observed in adjacent untransfected myotubes (white stars). ColQ−/− myotubes overexpressing GFP-ColQ-ΔColΔCt (g, h; white arrows) had similar number of AChR clusters compared to ColQ−/− myotubes. B, Relative frequency distribution for the number of AChR clusters per myotube (expressed as bins) observed in ColQ−/− (white bars) and ColQ−/− cells transfected with GFP-ColQ (black bars). A significant decrease in AChR cluster number was observed in cells transfected with GFP-ColQ. C, Non permeabilized ColQ−/− cells overexpressing GFP-ColQ (a) and stained with anti-GFP antibody (b), indicating that GFP-ColQ was secreted. D, Western blot of cell surface GFP from ColQ−/− cells transfected with GFP-ColQ showing that the GFP tagged ColQ was secreted. Cell surface TfR was used as a loading control. E, AChR clusters labeled with Alexa 594 α-BTX in ColQ−/− myotubes transfected with GFP-ColQ. In transfected ColQ−/− cells overexpressing GFP-ColQ, remaining AChR clusters colocalized with GFP-ColQ (a, b and white arrows). In ColQ−/− myotubes overexpressing GFP-ColQ (white arrows), agrin treatment was able to stimulate AChR clustering (c, d). Data are presented as means ± SEM from three independent experiments (n ≥36 myotubes). **p < 0.01; NS, not significant; unpaired Mann–Whitney's U test. Scale bars, 20 μm.
A series of control experiments were performed on ColQ−/− cells transfected with GFP-ColQ. First, we showed that the chimeric GFP-ColQ protein, like endogenous ColQ, was secreted. Thus, the GFP tag could be readily visualized by immunofluorescence with anti-GFP antibodies on nonpermeabilized cells (Fig. 2Ca,b). Moreover, when cell surface proteins were labeled with a membrane-impermeable biotinylated N-hydroxysuccimide ester (NHS-biotin) and isolated with streptavidin after protein extraction (Herbst and Burden, 2000), GFP-ColQ was precipitated by streptavidin (Fig. 2D). Second, surface expressed GFP-ColQ colocalized with AChR clusters consistent with its effect on clustering (Fig. 2Ea,b). To address the possibility that a steric bulk caused by the GFP tag inserted in ColQ could perturb the process of AChR cluster formation, myotubes overexpressing GFP-ColQ were treated with the C terminus of agrin, known to stimulate AChR cluster formation and stabilization through a cascade of intracellular events (Sanes and Lichtman, 2001). Agrin was able to stimulate AChR clustering in myotubes overexpressing GFP-ColQ, showing that GFP inserted in ColQ did not block the process of AChR clustering (Fig. 2Ec,d).
Decreased AChR cluster size and increased AChR packing in ColQ−/− mice
We then wondered whether the modifications of AChR clustering detected in ColQ−/− myotubes were also observed in muscle from ColQ−/− mice. To address this issue, AChR clusters were visualized in sternomastoid (STM) and soleus (Sol) muscle cross-sections from P7 wt and ColQ−/− mice (Fig. 3A). Quantitative analysis of α-BTX labeling performed on confocal stacks revealed that AChR cluster areas were smaller in both muscle types lacking ColQ (−35% in both muscles) (Fig. 3B). However, the muscle fibers have smaller diameters in the ColQ-deficient mice and NMJ size is correlated with fiber diameter (Kuno et al., 1971). When AChR cluster areas were normalized to fiber diameters they still revealed a decrease in the absence of ColQ (−30% in both muscles) (Fig. 3C). Consistent with these observations, smaller AChR cluster area and fiber diameter were previously observed in the adult ColQ−/− mice and there is a tendency for this to worsen with age (Nguyen-Huu et al., 2005). On the basis of these results, we examined whether ColQ deficiency modifies the number of AChRs per synapse. The fluorescence intensity per AChR cluster was increased in ColQ−/− STM muscle (+16%) and remained unchanged in ColQ−/− Sol muscle (Fig. 3D). The AChR density in clusters was higher in ColQ−/− STM and Sol muscles than in wt muscles (+88% and +73%, respectively) (Fig. 3E). Therefore, both in vitro and in vivo, ColQ deficiency leads to smaller AChR clusters in which AChRs are more densely packed.
Figure 3.
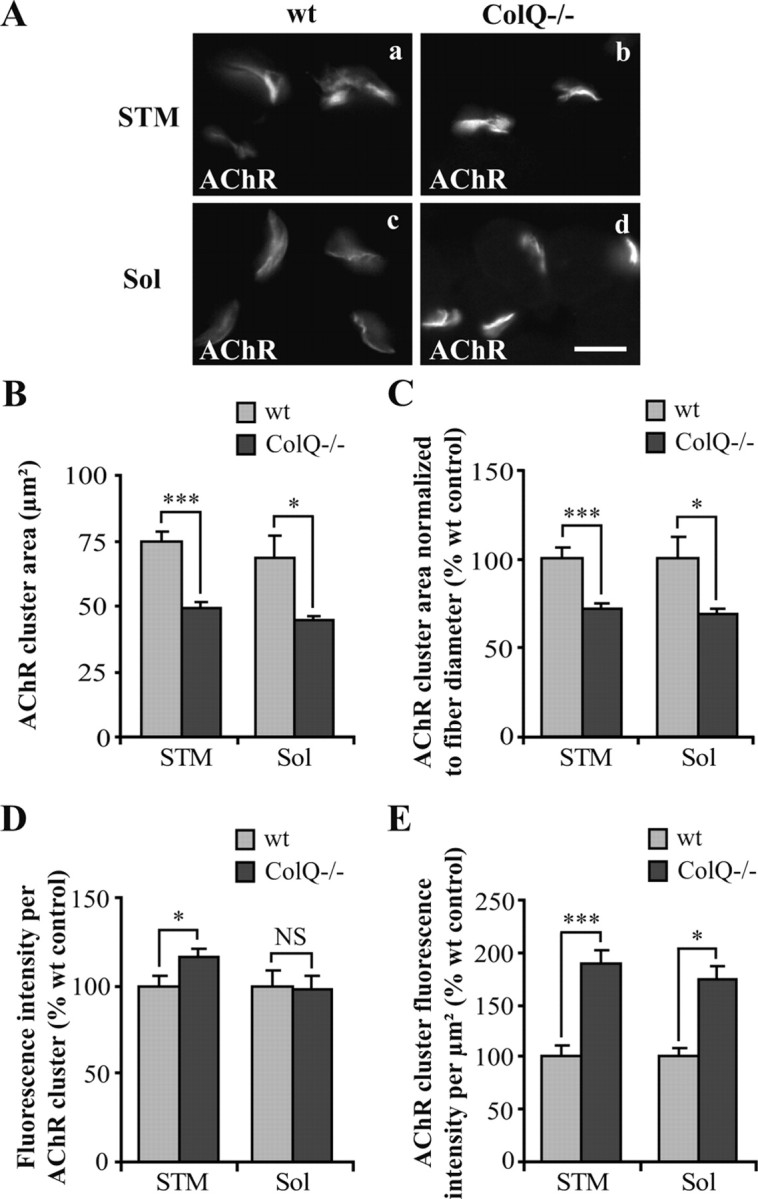
ColQ deficiency decreases cluster size and increases AChR packing in vivo. A, AChR clusters labeled with Alexa 488 α-BTX on cross-sections of STM (a, b) and Sol (c, d) muscles from wt (a, c) and ColQ−/− (b, d) P7 mice. B–E, Area and fluorescence intensity of individual NMJs measured from projection of confocal stacks. AChR cluster area (B) and AChR cluster area normalized to fiber diameter (C) were decreased in ColQ-deficient STM and Sol muscles. AChR cluster fluorescence intensity was increased in STM muscle but not in Sol muscle from ColQ−/− mice (D). The fluorescence intensity per μm2 was significantly increased both in STM and Sol muscles from ColQ−/− mice (E). Data are presented as means ± SEM or percentage of wt control ± SEM (n ≥ 32 NMJs from 3 mice per group). *p < 0.05; ***p < 0.001; NS, not significant; unpaired Student's t test. Scale bar, 20 μm.
Upregulation of synaptic gene expression in the absence of ColQ both in vitro and in vivo
To get insights into the mechanisms leading to ColQ−/− AChR cluster phenotypes, we wondered whether ColQ might control AChR expression as well as MuSK and rapsyn expression, both critical components for AChR clustering and packing. In cultures of myotubes, quantification of the mRNAs coding for these genes relative to Sec63 mRNAs revealed that the lack of ColQ induced an increase in mRNA levels for the five AChR subunits, MuSK and rapsyn (α-AChR, +57%; β-AChR, +26%; δ-AChR, +39%; γ-AChR, +111%; ε-AChR, +211%; MuSK, +40% and rapsyn, +69%) (Fig. 4A). In addition, quantitative Western blot analysis revealed that ColQ deficiency resulted in an increase in total protein amounts for α-AChR subunit (+25%, p < 0.05, n = 4), MuSK (+107%, p < 0.05, n = 4) and rapsyn (+40%, p < 0.05, n = 4) (Fig. 4B). Therefore, increased α-AChR subunit, MuSK and rapsyn protein levels in ColQ-deficient myotubes reflected the increased levels of corresponding mRNAs.
Figure 4.

AChR, MuSK and rapsyn mRNA and protein levels are upregulated in the absence of ColQ in vitro. A, Quantification of AChR subunits, MuSK and rapsyn mRNAs in wt and ColQ−/− myotubes by real-time RT-PCR. AChR subunits, MuSK and rapsyn mRNA levels relative to Sec63 mRNAs were higher in ColQ−/− cells compared to wt cells. B, Western blot of total α-AChR subunit, MuSK and rapsyn proteins in ColQ−/− cells compared to wt cells. GAPDH was used as a loading control. Loss of ColQ led to significant increases in α-AChR subunit, MuSK, and rapsyn protein levels relative to GAPDH. C, Quantification of α- and β-AChR subunits, MuSK and rapsyn mRNAs in ColQ−/− cells transfected with GFP-ColQ by real-time RT-PCR. A decrease of α- and β-AChR subunits and MuSK mRNAs was observed in ColQ−/− cells overexpressing GFP-ColQ, whereas mRNA levels of rapsyn remained unchanged. Levels of mRNAs are represented as relative expression (2−ΔCt vs reference gene × 100). Data are presented as percentage of control ± SEM from three to five independent experiments. *p < 0.05; **p < 0.01; NS, not significant; unpaired Mann–Whitney's U test.
We next tested whether ColQ restoration in ColQ−/− myotubes would reverse the mRNA level modifications observed for synaptic gene. GFP-ColQ overexpression in ColQ−/− myotubes led to a downregulation of mRNAs coding for α-AChR subunit (−36%), β-AChR subunit (−22%) and MuSK (−26%), but unexpectedly did not significantly change rapsyn relative expression (Fig. 4C). This result might be explained by the high rapsyn mRNA level detected before fusion of myoblasts into myotubes, at the time of GFP-ColQ transfection (data not shown). The same results were obtained in ColQ−/− cells overexpressing a nontagged ColQ construct allowing ColQ-AChE association (supplemental Fig. 2, available at www.jneurosci.org as supplemental material). Together, these results confirmed the regulatory role of ColQ on synaptic gene expression in vitro.
We then turned to in vivo experiments to see if the absence of ColQ would lead to similar regulation of synaptic genes in muscles from P7 mice. Higher levels of mRNAs coding for α-, β-, ε-AChR subunits and rapsyn relative to GAPDH mRNAs were observed in gastrocnemius muscle from ColQ−/− mice although this upregulation was only significant for β-AChR subunit and rapsyn (+63% and +106%, respectively) (Fig. 5A). In contrast to our in vitro result (Fig. 4A), MuSK mRNA level remained unchanged in ColQ−/− muscle compared to wt muscle (Fig. 5A).
Figure 5.
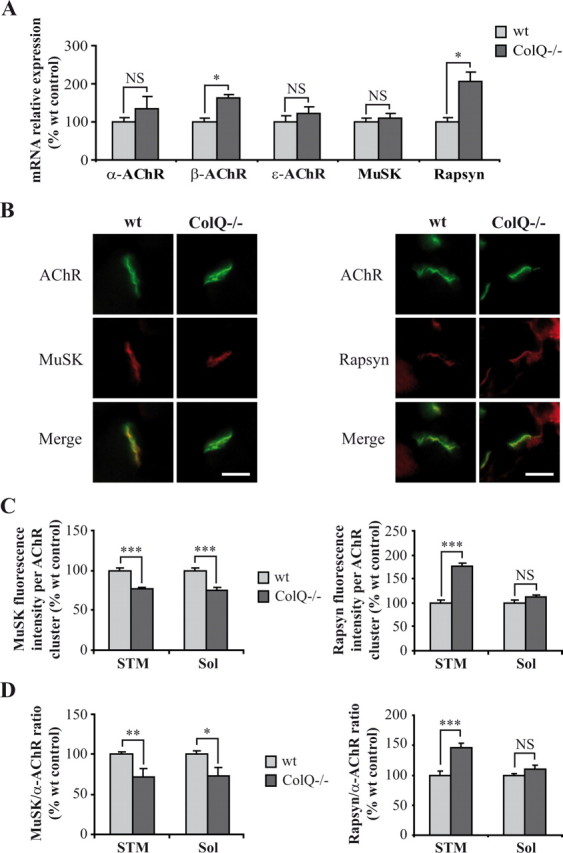
ColQ regulates AChR and rapsyn mRNA levels and MuSK membrane insertion at neonatal NMJs. A, Quantification of AChR subunits, MuSK, and rapsyn mRNAs on wt and ColQ−/− P7 gastrocnemius muscles by real-time RT-PCR. Levels of mRNAs are represented as relative expression (2−ΔCt versus reference gene × 100). β-AChR subunit and rapsyn mRNA levels relative to GAPDH mRNAs were higher in ColQ−/− mice compared to wt mice whereas other AChR subunits and MuSK mRNAs were not significantly increased. B, AChR, MuSK and rapsyn clusters labeled respectively with Alexa 488 α-BTX, MuSK and rapsyn antibodies on cross-sections of STM muscle from wt and ColQ−/− P7 mice. C, D, Quantification of MuSK and rapsyn fluorescence intensities from projection of confocal stacks. MuSK fluorescence intensity per AChR cluster was decreased in the absence of ColQ whereas rapsyn was increased (C). STM and Sol synapses from ColQ−/− mice displayed lower MuSK-to-α-AChR fluorescence ratio but higher rapsyn-to-α-AChR ratio compared to wt synapses (D). Data are presented as percentage of wt control ± SEM from five mice for mRNA analysis (A) and from three mice for immunofluorescence analysis (n ≥32 NMJs for C, D). *p < 0.05; **p < 0.01; ***p < 0.001; NS, not significant; unpaired Mann–Whitney's U test for mRNA analyses and unpaired Student's t test for immunofluorescence analyses. Scale bars, 20 μm.
In addition, to determine MuSK and rapsyn protein levels at the NMJ, fluorescence intensities were analyzed in STM (Fig. 5B) and Sol (data not shown) muscles from wt and ColQ−/− mice. MuSK fluorescence intensity per AChR cluster was decreased in STM and Sol NMJs from ColQ-deficient mice (−23% in both muscles) (Fig. 5C) whereas rapsyn fluorescence intensity per AChR cluster was increased (STM: +75% and Sol: +12% not significant) (Fig. 5C). Moreover, STM and Sol ColQ-deficient synapses displayed lower MuSK-to-α-AChR fluorescence intensity ratio (−28% in both muscles) (Fig. 5D) but higher rapsyn-to-α-AChR fluorescence intensity ratio than wt synapses (STM: +46% and Sol: +11% not significant) (Fig. 5D). Together, our data show that ColQ regulates synaptic gene mRNA and protein levels both in vitro and in vivo and suggest that ColQ may control MuSK incorporation or stabilization in the muscle membrane.
ColQ deficiency leads to a decrease in membrane-bound MuSK level and alters the activity of the MuSK signaling pathway
Given that our in vivo results showed a decrease in membrane-bound MuSK in ColQ-deficient NMJ (Fig. 5C), we decided to further quantify the amount of MuSK at the membrane of myotubes. For this purpose, wt and ColQ−/− cell surface proteins were labeled with a membrane-impermeable biotinylated N-hydroxysuccimide ester (NHS-biotin) and isolated with streptavidin after protein extraction (Herbst and Burden, 2000). The membrane protein transferrin receptor (TfR) was used as a loading control as the total level of this protein was not affected by ColQ deficiency (p = 0.62 not significant, n = 3) (Fig. 6A). Quantitative analysis of the amount of MuSK associated to the membrane of ColQ−/− myotubes revealed a 35% decrease compared to cell surface MuSK in wt myotubes (p < 0.05, n = 4) (Fig. 6B). Consistent with our in vivo result (Fig. 3D,E), we found in these same samples higher levels of α-AChR subunit on the membrane of ColQ-deficient cells compared to wt cells (+178%, p < 0.05, n = 4) (Fig. 6B), indicating that MuSK is not efficiently localized at the membrane unlike α-AChR subunit.
Figure 6.
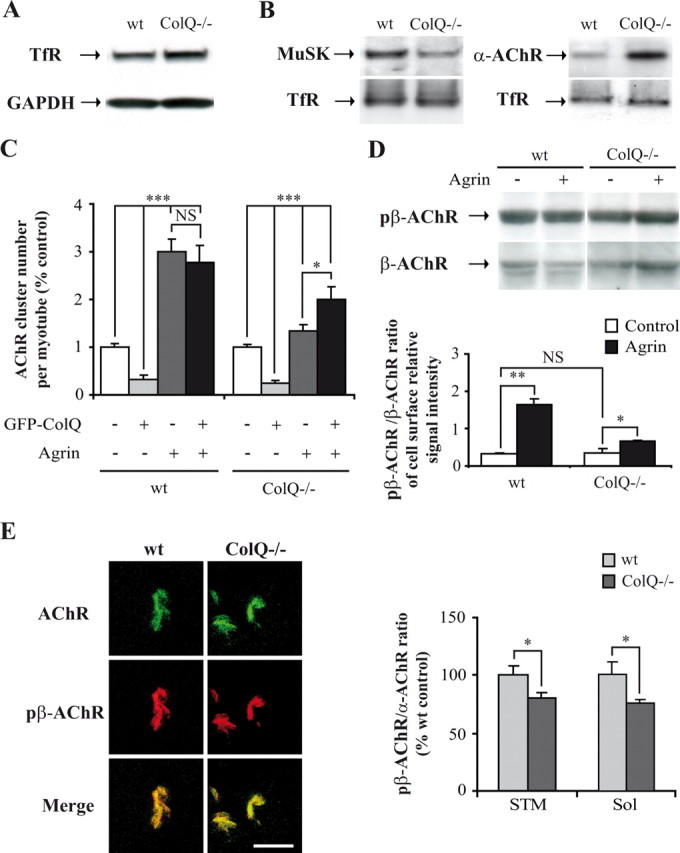
ColQ deficiency decreases membrane-bound MuSK in vitro and alters the activity of MuSK signaling pathway. A, Western blot of total transferrin receptor proteins (TfR) in wt and ColQ−/− myotubes. GAPDH was used as a loading control. Total TfR protein level relative to GAPDH was not significantly different between wt and ColQ−/− myotubes. B, Western blot of membrane-bound MuSK and α-AChR subunit in wt and ColQ−/− myotubes. Membrane-bound proteins were labeled with NHS-biotin and isolated with streptavidin. TfR was used as a loading control. MuSK surface protein level relative to TfR was decreased by 35% in the absence of ColQ. Conversely, loss of ColQ resulted in a 178% increase of α-AChR subunit surface protein level relative to TfR. C, Fluorescence analyses of AChR cluster number in wt and ColQ−/− myotubes treated with agrin and/or transfected with GFP-ColQ. Compared to untransfected cells (white bars), AChR cluster number was drastically decreased both in wt and ColQ−/− cells overexpressing GFP-ColQ (light gray bars). Agrin treatment had less effect on the number of AChR clusters in ColQ−/− cells compared to wt cells (dark gray bars), whereas GFP-ColQ transfection restored agrin capacity to induce normal AChR clustering (black bars). D, Western blot (up panel) and quantification (bottom panel) of membrane-bound β-AChR subunit and β-AChR subunit phosphorylated on Y390 residue (pβ-AChR). Agrin treatment increased β-AChR subunit phosphorylation in both cell lines but was threefold less efficient in ColQ−/− cells as shown by the pβ-AChR-to-β-AChR ratio. E, AChR clusters labeled with Alexa 488 α-BTX and pAChRβ1 antibody on cross-sections of STM muscle from wt and ColQ−/− P7 mice (left panel). Quantification of pβ-AChR fluorescence intensity from projection of confocal stacks showed that STM and Sol synapses from ColQ−/− mice displayed lower pβ-AChR-to-α-AChR fluorescence ratio than muscles from wt mice (right panel). Data are presented as means ± SEM or percentage of wt or ColQ−/− control ± SEM from three to five independent experiments (n ≥ 35 NMJs for E). *p < 0.05; **p < 0.01; ***p < 0.001; NS, not significant; unpaired Mann–Whitney's U test for Western blot analysis, unpaired Student's t test for immunofluorescence analyses. Scale bar, 20 μm.
Could the observed decrease in membrane-bound MuSK affect agrin/MuSK signaling? AChR cluster number was analyzed in wt and ColQ−/− myotubes treated with the C terminus of agrin. Agrin treatment was fourfold less efficient on AChR clustering in ColQ−/− cells compared to wt cells (+54% and +222%, respectively) (Fig. 6C, white and dark gray bars), a defect that was compensated by GFP-ColQ restoration (Fig. 6C, black bars). We also analyzed cell surface β-AChR subunit phosphorylation on Y390 residue, a process known to increase AChR-rapsyn interactions and AChR clustering in presence of agrin (Borges et al., 2008). The pβ-AChR-to-β-AChR ratio was two to threefold less important in ColQ−/− cells treated with agrin compared to wt cells (Fig. 6D).
We then wondered whether reduced levels of MuSK in vivo in ColQ-deficient AChR clusters would also affect the phosphorylation of β-AChR subunit by neural agrin. Immunofluorescence analysis performed on STM and Sol muscles revealed that the pβ-AChR-to-α-AChR ratio was decreased in ColQ−/− AChR clusters compared to wt clusters (STM: −19% and Sol: −24%) (Fig. 6E). Altogether, our results suggest that the activity of the agrin/MuSK signaling pathway is altered in the absence of ColQ, probably due to the decrease in membrane-bound MuSK.
ColQ-MuSK interaction regulates AChR mRNA levels
Since ColQ binds MuSK, which is involved in the control of gene expression at the synapse, we asked whether the effects of ColQ on synaptic mRNA levels are mediated by ColQ-MuSK interaction. For this purpose, we compared the mRNA levels of α- and β-AChR subunits in untransfected ColQ−/− cells and cells transfected with GFP-ColQ or a GFP-ColQ construct lacking the C terminus domain (GFP-ColQΔCt) or point mutated in the C terminus (GFP-ColQ-Y431S), both of which impair ColQ-MuSK association (Cartaud et al., 2004). As previously shown in Figure 4C, GFP-ColQ restoration in ColQ−/− cells led to a significant decrease in α- and β-AChR mRNA levels compared to untransfected cells (Fig. 7A, white and black bars). Importantly, impairing ColQ interaction with MuSK abolished the effect of ColQ on α- and β-AChR mRNA levels (Fig. 7A, light and dark gray bars). In addition, when GFP-ColQ was overexpressed in MuSK-deficient (MK−/−) myotubes, the levels of α- and β-AChR mRNAs remained unchanged compared to untransfected MK−/− cells (Fig. 7B). We conclude that at least part of the ColQ-dependent regulation of AChR mRNA levels is mediated by ColQ-MuSK interaction.
Figure 7.

ColQ-dependent regulation of AChR mRNA levels is mediated by ColQ-MuSK interaction. A, B, Quantification of α- and β-AChR mRNA levels by real-time RT-PCR. Levels of mRNAs are represented as relative expression (2−ΔCt versus reference gene × 100). A, α- and β-AChR mRNA levels were decreased in ColQ−/− transfected with GFP-ColQ but not in cells transfected with GFP-ColQ-ΔCt or GFP-ColQ-Y431S. B, α- and β-AChR mRNA levels were not modified by GFP-ColQ overexpression in MuSK-deficient myotubes (MK−/−). Data are presented as percentage of control ± SEM from three to five independent experiments. *p < 0.05; **p < 0.01; NS, not significant; unpaired Mann–Whitney's U test.
Discussion
Here, we demonstrate that ColQ has regulatory functions on AChR clustering both in muscle cell culture and in neonatal mouse muscles. In the absence of ColQ, AChR clusters are smaller and AChRs are more densely packed in these clusters both in vitro and in vivo. These complex effects generated by ColQ on AChR clustering result from regulation of both synaptic gene expression and MuSK membrane trafficking: (1) ColQ downregulates the expression of AChR subunits, a process that seems to be mediated by ColQ-MuSK interaction; (2) ColQ controls AChR packing, presumably through rapsyn; and (3) ColQ increases membrane-bound MuSK and thus could regulate the size of AChR clusters.
ColQ regulates the expression of AChRs and synaptically associated proteins
Our results show that the absence of ColQ leads to an upregulation of the mRNA levels encoding most of AChR subunits in vitro and in vivo while, in contrast, overexpression of ColQ downregulates AChR mRNA levels. Regulation of mRNA levels could result from transcriptional or posttranscriptional mechanisms. At NMJs, MuSK is essential for synaptic transcription and activates downstream pathways either directly through a JNK pathway (Moore et al., 2001; Lacazette et al., 2003) or indirectly via neuregulins and their receptors ErB (Sanes and Lichtman, 2001). According to our data, ColQ inhibitory effect on AChR mRNA levels is mediated by MuSK. So far, two molecules have been reported to negatively regulate transcription at the NMJ, 14-3-3 γ and erbin. The adaptor protein 14-3-3 γ binds MuSK and is a transcriptional repressor of synaptic genes (Strochlic et al., 2004). Erbin has been shown to interact with ErB2 and suppress neuregulin-induced Erk activation mediated by ErB (Huang et al., 2001, 2003). Thus, in the transcriptional hypothesis, it will remain to investigate the possibility that ColQ by interacting with MuSK recruits one of these molecules.
Alternatively, ColQ could regulate the stability of synapse specific mRNAs. In line with this hypothesis, members of the Hu family of RNA-binding proteins have been found to stabilize AChE mRNAs in muscle cells (Deschênes-Furry et al., 2005). Moreover, the canonical binding sequence for Hu proteins is present in the 3′UTR of most of AChR subunit coding transcripts. Together, these observations suggest an alternative mechanism to ColQ-induced transcriptional regulation in which ColQ would control AChR subunit expression through a posttranscriptional mechanism mediated by MuSK.
ColQ controls AChR packing in postsynaptic aggregates
Our results demonstrate that the density of AChR molecules is increased in the absence of ColQ, an observation that can be correlated to higher expression of rapsyn and an increased ratio of rapsyn-to-AChR. Indeed, data suggest that rapsyn is involved in AChR packing. Rapsyn interacts with AChR cytoplasmic loops and is indispensable for the formation of AChR clusters at the synapse (Froehner et al., 1990; Gautam et al., 1995). This AChR-associated protein not only triggers AChR aggregation but also stimulates cytoskeletal anchoring of AChRs (Moransard et al., 2003). Rapsyn was originally found to occur in a 1:1 stoichiometry with AChRs (LaRochelle and Froehner, 1986). However, several studies suggest that this ratio is modulated and is correlated with AChR degree of packing. First, developmental regulation induces changes in rapsyn-to-AChR ratio linked to higher packing and stability of AChRs in the postsynaptic membrane (Frail et al., 1989; Schaeffer et al., 2001; Gervásio et al., 2007). Second, agrin stimulates rapsyn-AChR interactions in vitro and in vivo, a process that is also correlated with tighter AChR packing in clusters (Moransard et al., 2003; Gervásio et al., 2007; Brockhausen et al., 2008). Our observation that clusters of AChRs are more densely packed in ColQ-deficient clusters could thus be related to higher expression of rapsyn with an increased ratio of rapsyn-to-AChR in vivo.
Interestingly, the regulatory effect of ColQ is more pronounced in STM muscle compared to Sol muscle regarding AChR density in clusters and rapsyn-to-AChR ratio. At neonatal stage, two ColQ transcripts, ColQ1 and ColQ1A are expressed in muscles whereas only ColQ1A remains expressed in adult STM and is restricted to synaptic domains in this muscle (Krejci et al., 1999). It is therefore difficult to correlate ColQ effects with a specific variant in our studies. One explanation could be that ColQ transcripts are expressed at higher levels in NMJs of STM muscle compared to Sol muscle at P7 and therefore drive stronger effects.
Increased packing of AChRs and rapsyn-AChR binding have been reported to correlate with higher phosphorylation of tyrosine Y390 in the β-AChR subunit loop in response to agrin (Borges et al., 2008). In our in vitro experiments and in the absence of agrin treatment, AChR packing at cell surface is increased but the ratio of the basal level of phosphorylated β-AChR subunits over total β-AChR subunits is unchanged which seems at odds with the above data. However, Borges et al. (2008) recently suggested that AChR and rapsyn could interact in a regulated manner dependent on β-AChR subunit tyrosine phosphorylation and in a constitutive manner independent of phosphorylation. It is therefore possible that the absence of ColQ stimulates interactions between rapsyn and AChR mostly through increased phosphorylation-independent binding of rapsyn to AChR.
Reduced levels of MuSK at the muscle cell surface in the absence of ColQ
Although the level of MuSK mRNAs and proteins are increased in ColQ-deficient cells, level of MuSK at the plasma membrane is severely reduced in culture and in vivo. Since this is observed in both systems and that ColQ has been found to interact with MuSK, reduced membrane-bound MuSK could be directly related to the absence of ColQ in the MuSK complex. A possibility is that ColQ stimulates MuSK sorting at the membrane. We previously showed that ColQ and MuSK codistribute in the same vesicles in cotransfected COS cells (Cartaud et al., 2004). These two molecules could be already associated in these vesicles and the lack of one of them would reduce the delivery of the complex to the membrane. In this context and conversely, the absence of MuSK leads to reduced levels of ColQ at the cell surface (Cartaud et al., 2004). Another possibility would be that ColQ promotes MuSK stability at the membrane through interactions of MuSK extracellular domain with the ECM.
In the absence of ColQ, we observed a reduction in AChR cluster area in vitro and in neonatal mice even when the area was normalized to the muscle fiber diameter, a phenotype that has been observed in adult mice (Feng et al., 1999; Nguyen-Huu et al., 2005). AChR cluster size has previously been correlated to the level of MuSK expression as the overexpression of MuSK leads to an increase in AChR cluster size (Fu et al., 2001). Therefore, the decrease in AChR cluster area observed in the absence of ColQ could be correlated to the decrease in membrane-bound MuSK. In addition, reduced levels of membrane MuSK probably explain the lower effect of agrin on AChR clustering and the reduced levels of agrin-induced β-AChR subunit phosphorylation in vitro. The levels of β-AChR subunit phosphorylation were also found to be reduced in STM and Sol muscles of neonatal mice, which can also be correlated to a weaker activity of the agrin/MuSK pathway.
CMS-1c patients share some common syndromes with CMS patients bearing mutations in MUSK and DOK7, a gene coding for a cytoplasmic adaptor of MuSK (Chevessier et al., 2004, 2008; Mihaylova et al., 2008). Dok-7 and MuSK are both essential to NMJ formation (Okada et al., 2006). Animal models for these myasthenic syndromes associated to mutations in COLQ, MUSK or DOK7 have been generated (Feng et al., 1999; Okada et al., 2006; Chevessier et al., 2008). For the three mutants, the size of AChR clusters, the levels of membrane-bound MuSK and MuSK activation are reduced (Chevessier et al., 2004, 2008; Anderson et al., 2008; Hamuro et al., 2008). Similar to our results in ColQ-deficient P7 mice, an increased AChR density is also detected in P5 MuSK mutant mice (Chevessier et al., 2008). It is therefore tempting to speculate that some of the mutations in COLQ and DOK7 result in reduced activity of the Dok-7/MuSK pathway that would explain similar phenotypes at least in young patients. The fact that a majority of the CMS-1c patients present an onset of the disease at birth argues in favor of a role of ColQ in initial synapse formation, in agreement with our results.
Comparison of NMJ phenotypes between cholinesterase mutants
Specificity of ColQ effects on postsynaptic differentiation is reinforced by the comparison with other mutants in which cholinesterase activity is affected. In the absence of ColQ as in the absence of AChE, AChR clusters are smaller and fragmented, but in contrast to ColQ mutant mice, the density of AChRs is decreased in adult AChE mutant mice (Xie et al., 2000; Adler et al., 2004). This last phenotype was only evident in mature synapses as neonatal AChE mutant NMJs were normal. Thus, the phenotypes of ColQ- and AChE-deficient NMJs are clearly different. In addition to AChE, butyrylcholinesterase (BChE) is expressed during early postnatal development and can also associate with ColQ (Krejci et al., 1997; Feng et al., 1999). However, BChE-deficient mice have normal NMJs (Li et al., 2006, 2008). More interesting will be to compare the double AChE-BChE mutant with ColQ mutant.
In summary, our results reveal that ColQ is not only a structural molecule but also fulfills a specific regulatory function in postsynaptic differentiation or maintenance. They bring new insights into the comprehension of myasthenic syndromes.
Footnotes
This work was funded by the Institut National de la Santé et de la Recherche Médicale, the Association Française contre les Myopathies (grants to C.L.), the Agence Nationale de la Recherche, and Université Paris Descartes. S.M.S. is the recipient of a PhD grant from the French Ministry of Research. We thank E. Girard for providing wt and ColQ-deficient mice, O. Bignolais for help with TLDA experiment, and M. Ruegg, J. Lindstrom, and P. Taylor for the gifts of antibodies.
The authors declare no competing financial interests.
References
- Adler M, Manley HA, Purcell AL, Deshpande SS, Hamilton TA, Kan RK, Oyler G, Lockridge O, Duysen EG, Sheridan RE. Reduced acetylcholine receptor density, morphological remodeling, and butyrylcholinesterase activity can sustain muscle function in acetylcholinesterase knockout mice. Muscle Nerve. 2004;30:317–327. doi: 10.1002/mus.20099. [DOI] [PubMed] [Google Scholar]
- Anderson JA, Ng JJ, Bowe C, McDonald C, Richman DP, Wollmann RL, Maselli RA. Variable phenotypes associated with mutations in DOK7. Muscle Nerve. 2008;37:448–456. doi: 10.1002/mus.20944. [DOI] [PubMed] [Google Scholar]
- Arikawa-Hirasawa E, Rossi SG, Rotundo RL, Yamada Y. Absence of acetylcholinesterase at the neuromuscular junctions of perlecan-null mice. Nat Neurosci. 2002;5:119–123. doi: 10.1038/nn801. [DOI] [PubMed] [Google Scholar]
- Barczyk M, Carracedo S, Gullberg D. Integrins. Cell Tissue Res. 2010;339:269–280. doi: 10.1007/s00441-009-0834-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bon S, Coussen F, Massoulié J. Quaternary associations of acetylcholinesterase. II. The polyproline attachment domain of the collagen tail. J Biol Chem. 1997;272:3016–3021. doi: 10.1074/jbc.272.5.3016. [DOI] [PubMed] [Google Scholar]
- Borges LS, Yechikhov S, Lee YI, Rudell JB, Friese MB, Burden SJ, Ferns MJ. Identification of a motif in the acetylcholine receptor beta subunit whose phosphorylation regulates rapsyn association and postsynaptic receptor localization. J Neurosci. 2008;28:11468–11476. doi: 10.1523/JNEUROSCI.2508-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brockhausen J, Cole RN, Gervásio OL, Ngo ST, Noakes PG, Phillips WD. Neural agrin increases postsynaptic ACh receptor packing by elevating rapsyn protein at the mouse neuromuscular synapse. Dev Neurobiol. 2008;68:1153–1169. doi: 10.1002/dneu.20654. [DOI] [PubMed] [Google Scholar]
- Cartaud A, Strochlic L, Guerra M, Blanchard B, Lambergeon M, Krejci E, Cartaud J, Legay C. MuSK is required for anchoring acetylcholinesterase at the neuromuscular junction. J Cell Biol. 2004;165:505–515. doi: 10.1083/jcb.200307164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chevessier F, Faraut B, Ravel-Chapuis A, Richard P, Gaudon K, Bauché S, Prioleau C, Herbst R, Goillot E, Ioos C, Azulay JP, Attarian S, Leroy JP, Fournier E, Legay C, Schaeffer L, Koenig J, Fardeau M, Eymard B, Pouget J, Hantaï D. MUSK, a new target for mutations causing congenital myasthenic syndrome. Hum Mol Genet. 2004;13:3229–3240. doi: 10.1093/hmg/ddh333. [DOI] [PubMed] [Google Scholar]
- Chevessier F, Girard E, Molgó J, Bartling S, Koenig J, Hantaï D, Witzemann V. A mouse model for congenital myasthenic syndrome due to MuSK mutations reveals defects in structure and function of neuromuscular junctions. Hum Mol Genet. 2008;17:3577–3595. doi: 10.1093/hmg/ddn251. [DOI] [PubMed] [Google Scholar]
- DeChiara TM, Bowen DC, Valenzuela DM, Simmons MV, Poueymirou WT, Thomas S, Kinetz E, Compton DL, Rojas E, Park JS, Smith C, DiStefano PS, Glass DJ, Burden SJ, Yancopoulos GD. The receptor tyrosine kinase MuSK is required for neuromuscular junction formation in vivo. Cell. 1996;85:501–512. doi: 10.1016/s0092-8674(00)81251-9. [DOI] [PubMed] [Google Scholar]
- Deprez P, Inestrosa NC, Krejci E. Two different heparin-binding domains in the triple-helical domain of ColQ, the collagen tail subunit of synaptic acetylcholinesterase. J Biol Chem. 2003;278:23233–23242. doi: 10.1074/jbc.M301384200. [DOI] [PubMed] [Google Scholar]
- Deschênes-Furry J, Bélanger G, Mwanjewe J, Lunde JA, Parks RJ, Perrone-Bizzozero N, Jasmin BJ. The RNA-binding protein HuR binds to acetylcholinesterase transcripts and regulates their expression in differentiating skeletal muscle cells. J Biol Chem. 2005;280:25361–25368. doi: 10.1074/jbc.M410929200. [DOI] [PubMed] [Google Scholar]
- Donger C, Krejci E, Serradell AP, Eymard B, Bon S, Nicole S, Chateau D, Gary F, Fardeau M, Massoulié J, Guicheney P. Mutation in the human acetylcholinesterase-associated collagen gene, COLQ, is responsible for congenital myasthenic syndrome with end-plate acetylcholinesterase deficiency (Type Ic) Am J Hum Genet. 1998;63:967–975. doi: 10.1086/302059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dvir H, Harel M, Bon S, Liu WQ, Vidal M, Garbay C, Sussman JL, Massoulié J, Silman I. The synaptic acetylcholinesterase tetramer assembles around a polyproline II helix. EMBO J. 2004;23:4394–4405. doi: 10.1038/sj.emboj.7600425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng G, Krejci E, Molgó J, Cunningham JM, Massoulié J, Sanes JR. Genetic analysis of collagen Q: roles in acetylcholinesterase and butyrylcholinesterase assembly and in synaptic structure and function. J Cell Biol. 1999;144:1349–1360. doi: 10.1083/jcb.144.6.1349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frail DE, Musil LS, Buonanno A, Merlie JP. Expression of RAPsyn (43K protein) and nicotinic acetylcholine receptor genes is not coordinately regulated in mouse muscle. Neuron. 1989;2:1077–1086. doi: 10.1016/0896-6273(89)90232-8. [DOI] [PubMed] [Google Scholar]
- Froehner SC, Luetje CW, Scotland PB, Patrick J. The postsynaptic 43K protein clusters muscle nicotinic acetylcholine receptors in Xenopus oocytes. Neuron. 1990;5:403–410. doi: 10.1016/0896-6273(90)90079-u. [DOI] [PubMed] [Google Scholar]
- Fu AK, Cheung J, Smith FD, Ip FC, Ip NY. Overexpression of muscle specific kinase increases the transcription and aggregation of acetylcholine receptors in Xenopus embryos. Brain Res Mol Brain Res. 2001;96:21–29. doi: 10.1016/s0169-328x(01)00253-4. [DOI] [PubMed] [Google Scholar]
- Gautam M, Noakes PG, Mudd J, Nichol M, Chu GC, Sanes JR, Merlie JP. Failure of postsynaptic specialization to develop at neuromuscular junctions of rapsyn-deficient mice. Nature. 1995;377:232–236. doi: 10.1038/377232a0. [DOI] [PubMed] [Google Scholar]
- Gervásio OL, Armson PF, Phillips WD. Developmental increase in the amount of rapsyn per acetylcholine receptor promotes postsynaptic receptor packing and stability. Dev Biol. 2007;305:262–275. doi: 10.1016/j.ydbio.2007.02.008. [DOI] [PubMed] [Google Scholar]
- Guerra M, Cartaud A, Cartaud J, Legay C. Acetylcholinesterase and molecular interactions at the neuromuscular junction. Chem Biol Interact. 2005;157–158:57–61. doi: 10.1016/j.cbi.2005.10.110. [DOI] [PubMed] [Google Scholar]
- Gullick WJ, Lindstrom JM. Comparison of the subunit structure of acetylcholine receptors from muscle and electric organ of Electrophorus electricus. Biochemistry. 1983;22:3801–3807. doi: 10.1021/bi00285a014. [DOI] [PubMed] [Google Scholar]
- Hamuro J, Higuchi O, Okada K, Ueno M, Iemura S, Natsume T, Spearman H, Beeson D, Yamanashi Y. Mutations causing DOK7 congenital myasthenia ablate functional motifs in Dok-7. J Biol Chem. 2008;283:5518–5524. doi: 10.1074/jbc.M708607200. [DOI] [PubMed] [Google Scholar]
- Hantaï D, Richard P, Koenig J, Eymard B. Congenital myasthenic syndromes. Curr Opin Neurol. 2004;17:539–551. doi: 10.1097/00019052-200410000-00004. [DOI] [PubMed] [Google Scholar]
- Herbst R, Burden SJ. The juxtamembrane region of MuSK has a critical role in agrin-mediated signaling. EMBO J. 2000;19:67–77. doi: 10.1093/emboj/19.1.67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang YZ, Wang Q, Xiong WC, Mei L. Erbin is a protein concentrated at postsynaptic membranes that interacts with PSD-95. J Biol Chem. 2001;276:19318–19326. doi: 10.1074/jbc.M100494200. [DOI] [PubMed] [Google Scholar]
- Huang YZ, Zang MZ, Xiong WC, Luo Z, Mei L. Erbin suppresses the MAP kinase pathway. J Biol Chem. 2003;278:1108–1114. doi: 10.1074/jbc.M205413200. [DOI] [PubMed] [Google Scholar]
- Jacobson C, Côté PD, Rossi SG, Rotundo RL, Carbonetto S. The dystroglycan complex is necessary for stabilization of acetylcholine receptor clusters at neuromuscular junctions and formation of the synaptic basement membrane. J Cell Biol. 2001;152:435–450. doi: 10.1083/jcb.152.3.435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jennings LL, Malecki M, Komives EA, Taylor P. Direct analysis of the kinetic profiles of organophosphate-acetylcholinesterase adducts by MALDI-TOF mass spectrometry. Biochemistry. 2003;42:11083–11091. doi: 10.1021/bi034756x. [DOI] [PubMed] [Google Scholar]
- Kim N, Stiegler AL, Cameron TO, Hallock PT, Gomez AM, Huang JH, Hubbard SR, Dustin ML, Burden SJ. Lrp4 is a receptor for Agrin and forms a complex with MuSK. Cell. 2008;135:334–342. doi: 10.1016/j.cell.2008.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krejci E, Coussen F, Duval N, Chatel JM, Legay C, Puype M, Vandekerckhove J, Cartaud J, Bon S, Massoulié J. Primary structure of a collagenic tail peptide of Torpedo acetylcholinesterase: co-expression with catalytic subunit induces the production of collagen-tailed forms in transfected cells. EMBO J. 1991;10:1285–1293. doi: 10.1002/j.1460-2075.1991.tb08070.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krejci E, Thomine S, Boschetti N, Legay C, Sketelj J, Massoulié J. The mammalian gene of acetylcholinesterase-associated collagen. J Biol Chem. 1997;272:22840–22847. doi: 10.1074/jbc.272.36.22840. [DOI] [PubMed] [Google Scholar]
- Krejci E, Legay C, Thomine S, Sketelj J, Massoulié J. Differences in expression of acetylcholinesterase and collagen Q control the distribution and oligomerization of the collagen-tailed forms in fast and slow muscles. J Neurosci. 1999;19:10672–10679. doi: 10.1523/JNEUROSCI.19-24-10672.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuno M, Turkanis SA, Weakly JN. Correlation between nerve terminal size and transmitter release at the neuromuscular junction of the frog. J Physiol. 1971;213:545–556. doi: 10.1113/jphysiol.1971.sp009399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lacazette E, Le Calvez S, Gajendran N, Brenner HR. A novel pathway for MuSK to induce key genes in neuromuscular synapse formation. J Cell Biol. 2003;161:727–736. doi: 10.1083/jcb.200210156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaRochelle WJ, Froehner SC. Determination of the tissue distributions and relative concentrations of the postsynaptic 43-kDa protein and the acetylcholine receptor in Torpedo. J Biol Chem. 1986;261:5270–5274. [PubMed] [Google Scholar]
- Legay C. Why so many forms of acetylcholinesterase? Microsc Res Tech. 2000;49:56–72. doi: 10.1002/(SICI)1097-0029(20000401)49:1<56::AID-JEMT7>3.0.CO;2-R. [DOI] [PubMed] [Google Scholar]
- Li B, Duysen EG, Saunders TL, Lockridge O. Production of the butyrylcholinesterase knockout mouse. J Mol Neurosci. 2006;30:193–195. doi: 10.1385/JMN:30:1:193. [DOI] [PubMed] [Google Scholar]
- Li B, Duysen EG, Lockridge O. The butyrylcholinesterase knockout mouse is obese on a high-fat diet. Chem Biol Interact. 2008;175:88–91. doi: 10.1016/j.cbi.2008.03.009. [DOI] [PubMed] [Google Scholar]
- Lin W, Burgess RW, Dominguez B, Pfaff SL, Sanes JR, Lee KF. Distinct role of nerve and muscle in postsynaptic differentiation of the neuromuscular synapse. Nature. 2001;410:1057–1064. doi: 10.1038/35074025. [DOI] [PubMed] [Google Scholar]
- Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(−Delta Delta C(T)) Method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- Marionneau C, Couette B, Liu J, Li H, Mangoni ME, Nargeot J, Lei M, Escande D, Demolombe S. Specific pattern of ionic channel gene expression associated with pacemaker activity in the mouse heart. J Physiol. 2005;562:223–234. doi: 10.1113/jphysiol.2004.074047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mihaylova V, Müller JS, Vilchez JJ, Salih MA, Kabiraj MM, D'Amico A, Bertini E, Wölfle J, Schreiner F, Kurlemann G, Rasic VM, Siskova D, Colomer J, Herczegfalvi A, Fabriciova K, Weschke B, Scola R, Hoellen F, Schara U, Abicht A, Lochmüller H. Clinical and molecular genetic findings in COLQ-mutant congenital myasthenic syndromes. Brain. 2008;131:747–759. doi: 10.1093/brain/awm325. [DOI] [PubMed] [Google Scholar]
- Moore C, Leu M, Müller U, Brenner HR. Induction of multiple signaling loops by MuSK during neuromuscular synapse formation. Proc Natl Acad Sci U S A. 2001;98:14655–14660. doi: 10.1073/pnas.251291598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moransard M, Borges LS, Willmann R, Marangi PA, Brenner HR, Ferns MJ, Fuhrer C. Agrin regulates rapsyn interaction with surface acetylcholine receptors, and this underlies cytoskeletal anchoring and clustering. J Biol Chem. 2003;278:7350–7359. doi: 10.1074/jbc.M210865200. [DOI] [PubMed] [Google Scholar]
- Nguyen-Huu T, Dobbertin A, Barbier J, Minic J, Krejci E, Duvaldestin P, Molgó J. Cholinesterases and the resistance of the mouse diaphragm to the effect of tubocurarine. Anesthesiology. 2005;103:788–795. doi: 10.1097/00000542-200510000-00017. [DOI] [PubMed] [Google Scholar]
- Ohno K, Brengman J, Tsujino A, Engel AG. Human endplate acetylcholinesterase deficiency caused by mutations in the collagen-like tail subunit (ColQ) of the asymmetric enzyme. Proc Natl Acad Sci U S A. 1998;95:9654–9659. doi: 10.1073/pnas.95.16.9654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okada K, Inoue A, Okada M, Murata Y, Kakuta S, Jigami T, Kubo S, Shiraishi H, Eguchi K, Motomura M, Akiyama T, Iwakura Y, Higuchi O, Yamanashi Y. The muscle protein Dok-7 is essential for neuromuscular synaptogenesis. Science. 2006;312:1802–1805. doi: 10.1126/science.1127142. [DOI] [PubMed] [Google Scholar]
- Peng HB, Xie H, Rossi SG, Rotundo RL. Acetylcholinesterase clustering at the neuromuscular junction involves perlecan and dystroglycan. J Cell Biol. 1999;145:911–921. doi: 10.1083/jcb.145.4.911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanes JR, Lichtman JW. Induction, assembly, maturation and maintenance of a postsynaptic apparatus. Nat Rev Neurosci. 2001;2:791–805. doi: 10.1038/35097557. [DOI] [PubMed] [Google Scholar]
- Schaeffer L, de Kerchove d'Exaerde A, Changeux JP. Targeting transcription to the neuromuscular synapse. Neuron. 2001;31:15–22. doi: 10.1016/s0896-6273(01)00353-1. [DOI] [PubMed] [Google Scholar]
- Schlessinger J. Direct binding and activation of receptor tyrosine kinases by collagen. Cell. 1997;91:869–872. doi: 10.1016/s0092-8674(00)80477-8. [DOI] [PubMed] [Google Scholar]
- Schreiner F, Hoppenz M, Klaeren R, Reimann J, Woelfle J. Novel COLQ mutation 950delC in synaptic congenital myasthenic syndrome and symptomatic heterozygous relatives. Neuromuscul Disord. 2007;17:262–265. doi: 10.1016/j.nmd.2006.11.010. [DOI] [PubMed] [Google Scholar]
- Scotton P, Bleckmann D, Stebler M, Sciandra F, Brancaccio A, Meier T, Stetefeld J, Ruegg MA. Activation of muscle-specific receptor tyrosine kinase and binding to dystroglycan are regulated by alternative mRNA splicing of agrin. J Biol Chem. 2006;281:36835–36845. doi: 10.1074/jbc.M607887200. [DOI] [PubMed] [Google Scholar]
- Strochlic L, Cartaud A, Mejat A, Grailhe R, Schaeffer L, Changeux JP, Cartaud J. 14-3-3 gamma associates with muscle specific kinase and regulates synaptic gene transcription at vertebrate neuromuscular synapse. Proc Natl Acad Sci U S A. 2004;101:18189–18194. doi: 10.1073/pnas.0406905102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Towbin H, Staehelin T, Gordon J. Electrophoretic transfer of proteins from polyacrylamide gels to nitrocellulose sheets: procedure and some applications. Proc Natl Acad Sci U S A. 1979;76:4350–4354. doi: 10.1073/pnas.76.9.4350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie W, Stribley JA, Chatonnet A, Wilder PJ, Rizzino A, McComb RD, Taylor P, Hinrichs SH, Lockridge O. Postnatal developmental delay and supersensitivity to organophosphate in gene-targeted mice lacking acetylcholinesterase. J Pharmacol Exp Ther. 2000;293:896–902. [PubMed] [Google Scholar]
- Yang X, Arber S, William C, Li L, Tanabe Y, Jessell TM, Birchmeier C, Burden SJ. Patterning of muscle acetylcholine receptor gene expression in the absence of motor innervation. Neuron. 2001;30:399–410. doi: 10.1016/s0896-6273(01)00287-2. [DOI] [PubMed] [Google Scholar]
- Zhang B, Luo S, Wang Q, Suzuki T, Xiong WC, Mei L. LRP4 serves as a coreceptor of agrin. Neuron. 2008;60:285–297. doi: 10.1016/j.neuron.2008.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]