

Abstract
Selective degeneration of neuronal projections and neurite pruning are critical for establishment and maintenance of functional neural circuits in both insects and mammals. However, the molecular mechanisms that govern developmental neurite pruning versus injury-induced neurite degeneration are still mostly unclear. Here, we show that the effector caspases 6 and 3 are both expressed within axons and that, on trophic deprivation, they exhibit distinct modes of activation. Surprisingly, inhibition of caspases is not sufficient for axonal protection and a parallel modulation of a NAD+-sensitive pathway is required. The proapoptotic protein BAX is a key element in both pathways as its genetic ablation protected sensory axons against developmental degeneration both in vitro and in vivo. Last, we demonstrate that both pathways are also involved in developmental dendritic pruning in Drosophila. More specifically, the mouse WldS (Wallerian degeneration slow) protein, which is mainly composed of the full-length sequence of the NAD+ biosynthetic Nmnat1 enzyme, can suppress dendritic pruning in C4da (class IV dendritic arborization) sensory neurons in parallel to the fly effector caspases. These findings indicate that two distinct autodestruction pathways act separately or in concert to regulate developmental neurite pruning.
Introduction
Autodestruction of neurites (axons or dendrites) can occur during development (i.e., neurite pruning) or after injury (i.e., Wallerian degeneration) (Raff et al., 2002; Luo and O'Leary, 2005). These two processes share anatomical and morphological similarities, including breakdown of axonal microtubules (MTs) and neurofilaments (NFs), and axonal fragmentation (Waller, 1850; Griffin et al., 1996). The discovery of the Wallerian degeneration slow (WldS) mutant mouse, in which transected axons exhibit prolonged survival, suggested that Wallerian degeneration is an active process (Lunn et al., 1989; Glass et al., 1993). The WldS mutation is a translocation resulting in a fusion of two genes: one encoding the N-terminal 70 aa of the ubiquitination factor E4b, and the other, the full-length NAD+ biosynthetic molecule nicotinamide mononucleotide adenylyl transferase 1 (Nmnat1) (Mack et al., 2001). The mechanisms underlying Wallerian degeneration are also conserved in flies, as transgenic expression of mouse WldS fusion protein in Drosophila protected neurons against injury-induced axon degeneration (Hoopfer et al., 2006; MacDonald et al., 2006). The mechanisms of axon protection by the WldS mutation are under intense investigations. Recent studies strongly argue that the enzymatic activity of Nmnat1 within the cytoplasm can account for the neuroprotective effects of WldS (Avery et al., 2009; Conforti et al., 2009; Sasaki et al., 2009; Yahata et al., 2009). Moreover, bath-applied NAD+ was shown to significantly delay axonal degeneration after nerve injury in vitro (Araki et al., 2004; Wang et al., 2005; Sasaki et al., 2006). Therefore, regulation of a NAD+-sensitive pathway emerges as the hallmark of the WldS protection, which is conserved from insects to mammals.
Despite the morphological resemblance between Wallerian degeneration and developmental neurite pruning, it was suggested that these processes may be governed by distinct mechanisms (Hoopfer et al., 2006). This concept is based on the findings that no effect on developmental axon pruning was detected in WldS mice nor by transgenic expression of WldS in Drosophila mushroom body neurons (Hoopfer et al., 2006). However, these observations do not rule out the possibility that additional destruction mechanisms may act redundantly. One such mechanism may involve the apoptotic caspases. Although previous studies have not assigned a role for the apoptotic machinery in axon pruning (Sagot et al., 1995; Finn et al., 2000; Watts et al., 2003; Whitmore et al., 2003; Luo and O'Leary, 2005; Awasaki et al., 2006; Saxena and Caroni, 2007), recent findings in flies and mice challenge this paradigm (Kuo et al., 2006; Williams et al., 2006; Nikolaev et al., 2009). Nevertheless, the interaction between caspases and the NAD+-sensitive pathway remains unclear.
Here, we examined the roles of the apoptotic machinery and the NAD+-sensitive pathway during developmental neurite pruning in mice and Drosophila sensory neurons. We found that genetic ablation of the proapoptotic protein BAX was sufficient for axonal protection in vitro, and we also demonstrate, for the first time, a protective role in vivo. We next show that the effector caspases 6 and 3 are expressed and activated within degenerating axons through distinct mechanisms. Furthermore, we found that a NAD+-sensitive pathway is operating in parallel to caspases and that efficient axonal protection is achieved only on inhibition of both pathways. Significantly, transgenic expression in Drosophila of the mouse WldS, but not an “enzyme-dead” WldS, produced a marked suppression of dendritic pruning, which was further enhanced after inhibition of the effector caspases. Notably, mutations in Dronc, the Drosophila ortholog of initiator caspase-9, attenuated dendritic pruning more efficiently than inhibition of the effector caspases, transgenic expression of WldS, or both, indicating additional roles of Dronc in this process. These results demonstrate an evolutionarily conserved role for the parallel actions of the effector caspases and the NAD+-sensitive pathway in developmental neurite destruction.
Materials and Methods
Antibodies.
Antibodies and dilutions used for immunofluorescent staining were as follows: 2H3 anti-NF (Developmental Studies Hybridoma Bank; 1:100), neuronal class III β-tubulin (Tuj1; Covance; MRB-435P; 1:500–1:2000), caspase-3 antibody (Cell Signaling; 9662; 1:50), cleaved caspase-3 (Asp175) antibody (Cell Signaling; 9664; 1:100), mouse anti-green fluorescent protein (GFP) (Roche), rat anti-mouse CD8α (Southern Biotechnology Associates; 1:100), and rabbit anti-cleaved poly(ADP-ribose) polymerase (PARP) (Abcam; 1:500). FITC or Rhodamine Red X-conjugated secondary antibodies were used at 1:200 (Jackson ImmunoResearch Laboratories). For immunoblotting, the following antibodies and dilutions were used: caspase-3 antibody (1:1000), cleaved caspase-3 (Asp175) antibody (1:1000), caspase-6 antibody (Cell Signaling; 9762; 1:1000), cleaved caspase-6 (Asp162) antibody (Cell Signaling; 9761; 1:1000), X-linked inhibitor of apoptosis protein (XIAP) (R&D Systems; MAB822; 1:1000), Tuj1 (1:15,000), and 2H3 (1:10).
Mouse strains. Plexin A3/A4 double knock-out mouse strain was previously described (Yaron et al., 2005). Bax knock-out (KO) mouse strain B6.129X1-Baxtm1Sjk/J (stock number 002994) was purchased from The Jackson Laboratory. Transgenic Brn3a-LacZ mouse strain was generated using standard procedures with a construct that was previously described (Eng et al., 2001).
Fly strains and genetics.
The fly strains used in this study were as follows: ppk-eGFP and ppk-GAL4 (Grueber et al., 2003), UAS- mCD8::GFP (Lee and Luo, 1999), UAS-CD8::PARP::Venus (Williams et al., 2006), UAS-WldS (MacDonald et al., 2006), UAS-WldS-dead (Avery et al., 2009), UAS-p35 (Hay et al., 1994), UAS-drice RNAi (E. A. and H. Steller, unpublished data), droncI24 (a premature stop codon at positions 28) (Xu et al., 2005), drice17 (a conserved Asn116 to Tyr change) (Xu et al., 2006), driceΔ1 (lacks the entire Drice-coding region) (Muro et al., 2006), dcp-1Prev (a 40 bp insertion generating a frameshift mutation) (Laundrie et al., 2003), GMR-GAL4 (Takahashi et al., 1999), engrailed (en)-GAL4 (Bloomington Stock Center), and UAS-hid (Grether et al., 1995). For live imaging experiments, the mCD8::GFP construct was expressed in the ddaC neurons using the ppk-GAL4 driver or by using the direct fusion line ppk-eGFP. For visualization of caspase activity within the ddaC dendrites, the UAS-CD8::PARP::Venus line was crossed to the ppk-GAL4 line. To monitor neuroprotective effects, mutant or transgenic lines were crossed to flies of the genotype ppk-GAL4, UAS-CD8::GFP, and the progeny were maintained as stable lines. For expression in the posterior compartment of the wing imaginal disc, the UAS-CD8::PARP::Venus was crossed to the en-GAL4 line. Hid expression in the eye was driven by GMR-GAL4.
Whole-mount embryo X-gal staining.
For whole-mount staining for β-gal (β-galactosidase) activity, embryos were fixed in 4% formaldehyde in PBS for 30 min [up to 2 h for embryonic day 16.5 (E16.5) embryos] and then rinsed in PBS. Staining is performed in 100 mm potassium phosphate, pH 7.4, containing 5 mm EGTA, 2 mm MgCl2, 0.02% Nonidet P-40, and 0.01% sodium deoxycholate, to which 0.5 mg/ml X-gal, 5 mm potassium ferricyanide, and 5 mm potassium ferrocyanide were added. Embryos are stained over one to two nights at 37°C, and then rinsed in PBS, dehydrated in graded ethanols, and then cleared in 1:2 benzyl alcohol/benzyl benzoate.
Explant culture.
Dorsal root ganglion explants of E13.5 mice were cultured on poly-d-lysine (PDL)-laminin-coated plates. The explants were grown in Neurobasal medium supplemented with 2% B-27, 1% glutamine, 1% penicillin–streptomycin, and 12.5 ng/ml mNGF 2.5S (Alomone Labs; N-100) for 2 d before treatments. For NGF deprivation, the medium was exchanged to medium lacking NGF with addition of rabbit anti-NGF neutralizing antibodies (Alomone Labs; AN-240). Other conditions required the addition of 20 mm NAD+ (nicotinamide adenine dinucleotide hydrate) (Sigma-Aldrich; N3014) and/or caspase inhibitors: 25 or 50 μm pancaspase inhibitor [Z-VAD(OMe)-FMK; Calbiochem; 627610], 25 or 50 μm caspase-6 inhibitor (Z-VEID-FMK; R&D Systems; FMK006), 25 or 50 μm caspase-3 inhibitor (Z-DEVD-FMK; R&D Systems; FMK004).
Axonal preparations.
Cell culture inserts adequate to a six-well plate with 1 μm pore size were coated on both sides with PDL for 1 h at room temperature, rinsed three times with double-distilled water, and coated with laminin for additional 2 h at 37°C/5% CO2. The bottom side was then coated with 200 μl of collagen-MEM to form an even thin layer. After the collagen has consolidated, the inserts were placed upright in a six-well plate, each well containing 2 ml of medium with 50 ng/ml NGF. The inner side of the insert was filled with 1 ml of medium without NGF. DRG explants were placed within the inserts and grown as described above, allowing the axons, but not cell bodies, to pass through the pores and to grow on the bottom surface of the insert. For NGF deprivation experiments, the medium in the lower compartment was exchanged to an NGF-free medium with 0.1 μg/ml rabbit anti-NGF neutralizing antibodies (Alomone Labs; AN-240).
Caspase activity assay.
For the caspase activity assay, the axon-containing collagen layer was scraped off the bottom of the insert and transferred to a 1.5 ml standard microtube and kept on ice. A 50 μm final concentration of Z-VAD(OMe)-FMK was added to the negative control samples. Each sample was transferred to a 96-well assay white plate (Costar 3610; Corning), and caspase-Glo 3/7 reagent (Promega; G8090) was added to a final volume of 200 μl. Caspase-3-like (DEVDase) activity was assessed by the amount of luminescence released from the cleaved Ac-DEVD-pNA substrate every 2 min for a 1 h period using multiwell plate reader. For Western blot analysis, the collagen layer was scraped off and transferred to an Eppendorf and lysed with sample buffer by boiling at 100°C for 10 min. Samples were then loaded onto a SDS-PAGE gel.
Immunohistochemistry of paraffin sections.
For the in vivo visualization of cleaved caspase-3 within axons, E13.5 embryos were fixed for 24 h in 4% formaldehyde. The following day, the embryos were embedded in paraffin, and 4 μm transversal sections were taken. The slides then underwent deparaffinization with xylene and ethanol. Antigen retrieval was performed in a pressure cocker in a sodium citrate buffer at 125°C. Staining of the slides with anti-cleaved caspase-3 antibody (Cell Signaling) and anti Tuj-1 antibody (Covance) was done as described above.
Detection of apoptotic cells.
DRGs from E13 mice embryos were dissociated as described previously and plated on a PDL/laminin-coated plate. The cells were grown for 1 d and then were either maintained in the presence of NGF for additional 24 h or deprived of NGF in the presence or absence of the caspases inhibitors Z-VAD, Z-VEID, or Z-DEVD (50 μm) with or without 20 mm NAD+. Terminal deoxynucleotidyl transferase-mediated biotinylated UTP nick end labeling (TUNEL)-positive cells were detected with In Situ Cell Death Detection kit, TMR red (Roche; 11767291910) and counted manually using the ImageJ program.
Fly staining procedures and imaging.
Pupae of the desired fly lines were aged to the appropriate time points, dissected along the dorsal longitudinal axis, anchored to a wax plate with four pins, and fixed and stained using standard procedures. For live imaging 18 h after puparium formation (APF), the interior body of the pupa was removed from its case, placed on double-sided tape slides, and imaged under the microscope. Wing imaginal discs were dissected from third-instar larvae 3–4 h after irradiation and subjected to fixation and staining procedures essentially as described by Tomlinson and Ready (1987). All fluorescent images were obtained using Zeiss confocal microscopy LSM 510 or 710. Pictures of adult eyes were taken under visible light with a Leica MZ 16F stereomicroscope using a Nikon DS-L2 digital camera.
Results
Establishment of an in vitro system for DRG axonal degeneration
To gain insights into the mechanisms that execute axonal degeneration, we set up an in vitro system based on embryonic DRGs and the Twiss filter system (Zheng et al., 2001). In this system, growing axons, but not cell bodies, pass through the pores of the filter, enabling efficient purification of axonal material for biochemical analysis (for details, see Materials and Methods) (Fig. 1). Cellular analysis of this filter culture using 4′,6′-diamidino-2-phenylindole (DAPI) labeling of the nuclei revealed that the cell bodies are mainly retained on top of the filter, whereas the growing axons, visualized by neurofilament staining, are present in both compartments (top and bottom) of the filter (Fig. 1C–F). We next used antibodies against molecular protein markers of cell bodies and axons and examined the purification level of protein extracts from each compartment by Western blot (Fig. 1B). Nuclear histones were only detected in extracts from the top compartment, whereas extracellular signal-regulated kinase (ERK), a known resident of DRG axons, was present in preparations from both compartments. Therefore, this filter system can be used to effectively produce highly purified axon preparations by harvesting the projections in the bottom compartment.
Figure 1.
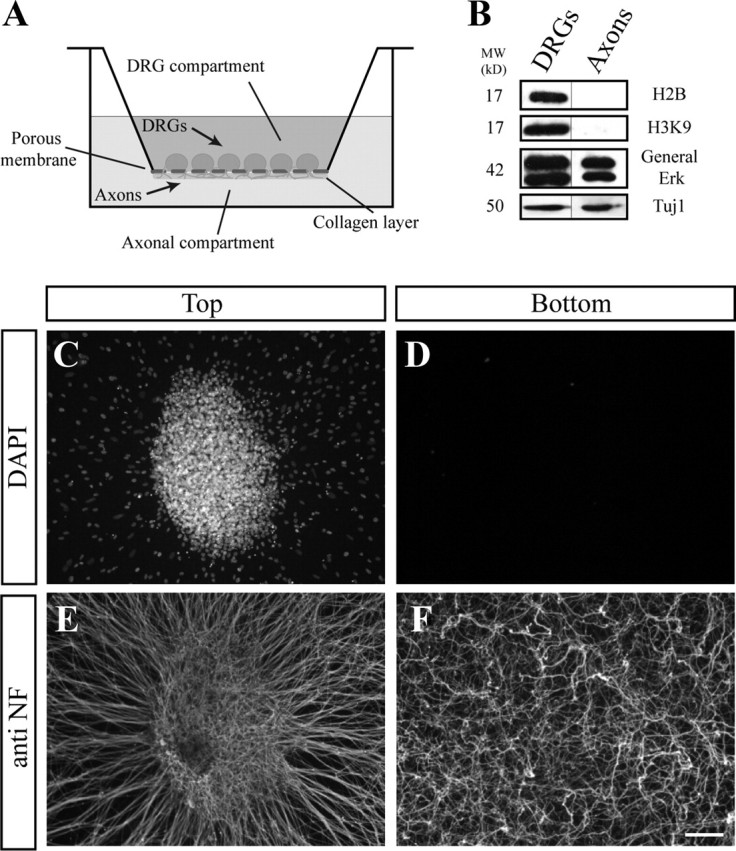
Axonal preparations. A, Schematic illustration of the modified Twiss filter system. C–F, DRG explants were grown on the filter for 48 h, allowing the axons to cross through the 1 μm pores to the bottom compartment and extend within the collagen layer on the bottom side of the membrane. C, E, The upper compartment contains DRG neurons and migrating cells (DAPI) (C) and axons stained with anti-neurofilament (E). In contrast, in the bottom compartment, although numerous axons have crossed the filter (F), no cell bodies are seen (D). Scale bar, 200 μm. B, Biochemical analysis of DRG extracts (left) and axonal preparation (right) using Western blot. The nuclear proteins histones (H2B and H3K9) are present solely in the DRG preparation collected from the upper compartment. Importantly, ERK, which was previously shown to reside in axons, can be detected in our axonal extract.
The apoptotic machinery is activated within axons on trophic withdrawal
Recent work has demonstrated the involvement of the apoptotic machinery in the process of axonal degeneration after NGF deprivation (Nikolaev et al., 2009). In this system, the proapoptotic protein BAX is shown to be required for axonal degeneration. Since we are using a different setting of DRG cultured explants, we first asked whether we can detect similar effects of BAX on axonal degeneration in our system. Expression levels of BAX were examined by Western blot in both healthy and degenerating DRG axons (i.e., axonal preparations were made before and after NGF deprivation, respectively). As can be seen in Figure 2A, BAX is present in the cell bodies and axons of the neurons.
Figure 2.

The active forms of caspase-3 and -6 are present within axons. Axonal preparations were produced as described in Materials and Methods and in Figure 1. A, Immunoblotting with anti-BAX antibody confirmed that BAX protein is missing in Bax KO DRGs and present within WT axons (bottom band; the top bands are unspecific, n.s.). B, Immunoblotting with anti-caspase-6 antibody shows that, whereas the full-length caspase is present in axons regardless of the presence of NGF, the cleaved form of caspase-6 was only present after NGF deprivation, first seen 5 h after deprivation and peaking at 10 h after NGF removal. C, Immunoblotting with anti-caspase-3 or with anti-cleaved caspase-3 revealed the existence of both the full-length and the cleaved forms of caspase-3 within axons, regardless of NGF deprivation. D, Neurons were maintained in NGF-containing medium (+NGF) or deprived of NGF for 16–24 h (−NGF). Caspase-3-like (DEVDase) activity was determined as the amount of luminescence released from the cleaved Ac-DEVD-pNA substrate. Measurements were obtained every 2 min over a time period of 1 h. Caspase activity was completely blocked with 50 μm caspase inhibitor Z-VAD-FMK (Z-VAD). Shown is the average of three repeats (±SEM). E, To control for the protein levels, Tuj1 immunolabeling of the examined samples shows similar protein content. F, Immunoblotting with anti-XIAP shows a reduction in the protein levels after NGF deprivation.
To test whether BAX is required for axonal degeneration in this system, we next induced axonal degeneration either by complete deprivation of NGF or after axotomy (supplemental Fig. S1A–F, available at www.jneurosci.org as supplemental material). In accordance with the recent report (Nikolaev et al., 2009) on NGF deprivation, axonal degeneration was completely blocked in DRG explants from Bax KO mice. In contrast, no significant effect on axonal degeneration was detected after axotomy, which is consistent with previous observations using transected axons from the adult optic nerve and the sciatic nerve (Whitmore et al., 2003). These findings suggest that the apoptotic machinery is activated after NGF deprivation and that BAX is a key element in mediating axonal degeneration induced by trophic withdrawal.
The effectors caspase-6 and caspase-3 are both activated within degenerating axons
Caspases are the main downstream executioners of BAX in the apoptotic pathway (Yuan et al., 1993; Nicholson and Thornberry, 1997; Bergmann et al., 1998; Budihardjo et al., 1999; Wolf and Green, 1999; Lamkanfi et al., 2007; Salvesen and Riedl, 2008). Recently, it was suggested that the effector caspase-6 is required for axonal degeneration, whereas the main apoptotic effector caspase-3 is not involved in this process, and these functional differences were attributed to the differential expression of the two caspases in axons and cell bodies, respectively (Nikolaev et al., 2009). We, therefore, went on to examine the expression levels of the effector caspases 3 and 6 in DRG axons. Unexpectedly, the zymogen forms of both caspase-6 and -3 were detected in axons grown in the presence of NGF (Fig. 2B,C). Moreover, after NGF withdrawal, these two caspases were activated in axons, exhibiting distinct modes of activation. Caspase-6 was activated in a “classical manner,” as its cleaved (active) form started accumulating 5 h after NGF withdrawal (Fig. 2B). However, most of the cleaved (active) form of caspase-3 was already present in cultures supplied with NGF, and even after 20 h of trophic withdrawal, no significant change in the level of the cleaved form was noticed (Fig. 2C).
Since caspase-3 is the most potent apoptotic effector and its activity can normally lead to cell death within a few hours (Bursch et al., 1990; Maeno et al., 2000), we reasoned that caspase-3 activity must be restrained by other mechanisms in healthy axonal cultures. Indeed, when we measured effector caspase-3 activity by monitoring DEVD cleavage in axonal lysates from neurons maintained in NGF or after trophic deprivation, a relatively high level of DEVDase activity was measured in lysates from axons deprived of NGF, whereas only a minor activity was detected in the NGF-maintained axons (Fig. 2D). We conclude that both caspase-6 and caspase-3 are activated in degenerating DRG axons and that, in healthy axons maintained with NGF, caspase-3 activity is kept below the threshold level required for axonal degeneration.
Caspase-3 is activated in axons in vitro and in vivo in a BAX-dependent fashion
To corroborate our biochemical findings that caspase-3 is expressed and activated in degenerating axons, we examined whether caspase-3 can be directly visualized within axons. To this end, we first stained cultured DRG sensory neurons with antibodies that detect either the full-length (zymogen; procaspase) or cleaved (active) forms of caspase-3. Whereas the full-length form was readily detected within axons both before and after NGF withdrawal, the active form was detected only after NGF deprivation (Fig. 3A–L). Furthermore, whereas procaspase-3 was distributed throughout the entire axon, the active form was mainly localized with tubulin aggregates. Because the presence of the cleaved caspase-3 in healthy, NGF-maintained, axons was demonstrated biochemically (Fig. 2C), we attribute the lack of active caspase-3 staining in healthy axons to possible epitope masking caused by binding of the caspase inhibitor protein XIAP to active caspase-3 (see below) (Huesmann and Clayton, 2006).
Figure 3.
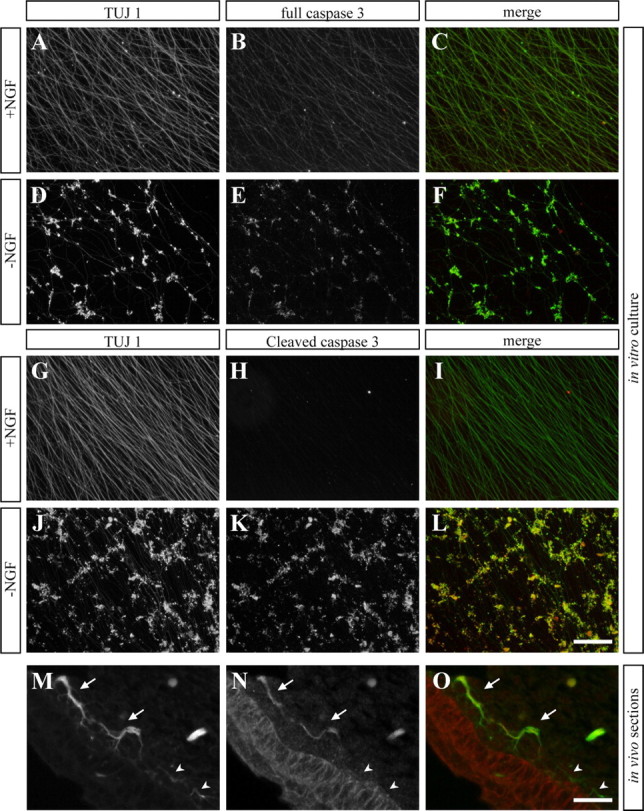
Caspase-3 is present within axons both in vitro and in vivo. A–L, In vitro cultures of DRG explants. A–C, G–I, Explants grown in NGF-containing media for 72 h. D–F, J, K, Cultures were deprived of NGF for 24 h after 2 d of growth. Left row, Anti-Tuj1 antibody staining. Middle row, Anti-full-length caspase-3 antibody (B, E) or anti-cleaved caspase-3 antibody (H, K, N). Right row, Merge. Whereas the full-length form of caspase-3 is visualized already within the intact axons of neurons grown in the presence of NGF (B), a very low signal of the cleaved (active) form is detected under these conditions (H). However, after NGF deprivation, high levels of both the full-length and the cleaved forms of caspase-3 are detected at the sites of MT aggregations (E, F; K, L). Scale bar, 200 μm. M–O, Paraffin sections of E13.5 embryos. Shown are peripheral axons adjacent to the skin labeled with anti-Tuj1 antibody (M). N, Cleaved caspase-3 labeling is evident in some, but not all of the axons (marked by arrows and arrowheads, respectively). Scale bar, 100 μm.
We next tested for the presence of active caspases 3 in axons, in vivo. During embryonic development, elimination of sensory neurons and axons reaches a peak at E13 (White et al., 1996). Immunostaining of E13 embryonic sections revealed that some of sensory axons that innervate the developing skin were positively labeled for active caspase-3 (Fig. 3M–O). Finally, in all the assays examined (i.e., biochemical assay and in vitro and in vivo staining), active caspase-3 expression was absent in axons of the Bax KO mice (supplemental Fig. S2, available at www.jneurosci.org as supplemental material). We conclude that the activated form of caspase-3 is present in healthy axons, albeit in an inactive state, and that it is activated in degenerating axons in a BAX-dependent fashion.
XIAP undergoes degeneration after NGF deprivation
The idea that the active form of caspase-3 is present in healthy axons, but that it is normally inhibited, raised the possibility of the involvement of caspase inhibitory proteins. One important family of caspase inhibitors is the inhibitor of apoptosis proteins (IAPs) (Deveraux et al., 1998; Goyal, 2001; Salvesen and Duckett, 2002). The XIAP is the best studied mammalian IAP and is considered the most potent caspase inhibitor in vitro (Eckelman et al., 2006). XIAP can specifically bind to and inhibit the activity of many caspases including caspase-3. During apoptosis, XIAP can undergo autoubiquitination and degradation, which facilitates caspase activation (Deveraux et al., 1998; Yang et al., 2000; Riedl et al., 2001; Scott et al., 2005; Schile et al., 2008). Prompted by this bulk of data, we examined the presence of XIAP in healthy and degenerating DRG axons by Western blot analysis. Using a specific antibody for XIAP, this protein was readily detected in extracts from healthy axons (Fig. 2F). Significantly, a major reduction in XIAP level was observed at 20 h after NGF withdrawal (Fig. 2F), the time when caspase-3 activity reaches a peak. Therefore, XIAP may function to modulate caspase-3 activity in healthy axons.
Caspase-6 and caspase-3 operate in parallel to the NAD+-sensitive pathway
Specific inhibition of caspase-6, but not caspase-3, using peptide inhibitors or short interfering RNA was previously shown to block axonal degeneration after NGF deprivation as visualized by MT fragmentation in vitro (Nikolaev et al., 2009). To examine the significance of effector caspases activation for axonal degeneration in our DRG cultures, we used peptide inhibitors specific for caspase-3, caspase-6, or a pancaspase inhibitor, and monitored both MT fragmentation and NF degradation on trophic withdrawal or axotomy. In this system, the caspase-6 inhibitor, Z-VEID-FMK, or caspase-3 inhibitor, Z-DEVD-FMK, did not show any effect (Fig. 4D,E,J; supplemental Fig. S5, available at www.jneurosci.org as supplemental material). Treatment with the pancaspase inhibitor Z-VAD-FMK did produce some level of axonal protection, albeit not to the level observed in the Bax KO neurons (Fig. 4F,I,J; supplemental Figs. S1, S5, available at www.jneurosci.org as supplemental material), indicating that additional pathway is operating in parallel to caspases and downstream of BAX. Since the NAD+-sensitive pathway has been shown to protect axon degeneration after injury, we investigated the role of this pathway. In accordance with previously published data, supplement of 20 mm NAD+ to the DRG cultures significantly delayed (up to 8 h) axonal degeneration induced by axotomy (Araki et al., 2004; Wang et al., 2005) (supplemental Fig. S4, available at www.jneurosci.org as supplemental material). However, it had only a minor effect on trophic deprivation-induced axonal degeneration (Fig. 4C,J; supplemental Fig. S5, available at www.jneurosci.org as supplemental material). Surprisingly, the combined addition of NAD+ and caspase-6 or caspase-3 peptide inhibitors completely protected NGF-deprived axons against degeneration (Fig. 4G–J), inhibiting both MT fragmentation as well as NF degradation (Fig. 4J; supplemental Fig. S5, available at www.jneurosci.org as supplemental material). Unexpectedly, we also detected a role for NAD+ in cell death of the NGF-deprived neurons (supplemental Fig. S3, available at www.jneurosci.org as supplemental material). Although the physiological relevance of this protection is still unclear, a recent study has demonstrated that NAD+ depletion is necessary for neuronal cell death (Alano et al., 2010). In summary, these results indicate that two parallel pathways act in the execution of axonal degeneration, the NAD+-sensitive pathway and the caspase-dependent pathway.
Figure 4.
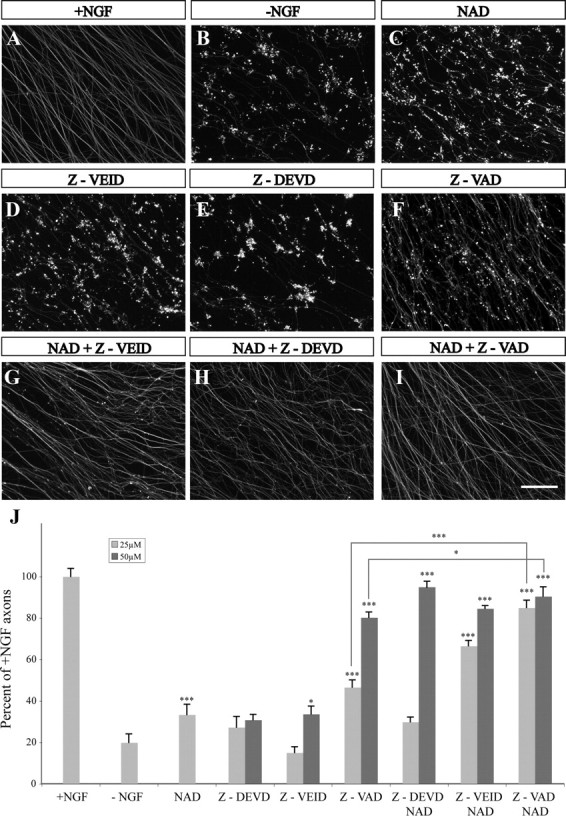
Combined supplement of caspase inhibitors and NAD+ protects axons against NGF deprivation-induced degeneration. DRG explants were cultured for 48 h in NGF-containing medium, and then deprived of NGF for an additional 24 h. Axons were stained with anti-Tuj1 antibody. A, Control. B–I, NGF deprivation. Addition of 20 mm NAD+ (C), 50 μm caspase-6 inhibitor (Z-VEID) (D), or 50 μm caspase-3 inhibitor (Z-DEVD) (E), did not block axon degeneration, whereas addition of 50 μm pancaspase inhibitor Z-VAD (F) produced a neuroprotective effect. However, addition of 20 mm NAD+ in combination with either 50 μm Z-VEID (G) or 50 μm Z-DEVD (H) successfully protected DRG axons from MT fragmentation. I, The combined addition of NAD+ and Z-VAD significantly increased the protective effect obtained with Z-VAD or NAD+ alone. Scale bar, 100 μm. J, Quantification of Tuj1-labeled axons after exposure to the different treatments as described above. Caspase inhibitors were supplemented in two concentrations: 25 and 50 μm. Presented are the percentages of positively labeled axons in each treatment compared with the number of labeled axons in NGF maintained DRGs + SEM. Student's t test was conducted between each treatment and NGF deprivation (−NGF). *p < 0.05; ***p < 0.001.
BAX is required for degeneration of misprojecting sensory axons in vivo
The findings that the NAD+-sensitive pathway and the caspase-dependent pathway are jointly required for axonal protection after NGF deprivation and that these two pathways are downstream of BAX, prompted us to examine the role of BAX in axonal degeneration in vivo. Since it is virtually impossible to capture the elimination of DRG sensory axons during development, we decided to use a sensitized background in which elimination of sensory axons is more obvious. A previous study demonstrated that, in the absence of the axonal repulsive cue Sema3A, many DRG sensory axons that misproject early during development are eventually eliminated (White and Behar, 2000). To avoid any potential compensatory effect by other Semaphorins and since Sema3A KO mice are homozygous lethal, we first examined whether this axonal elimination also occurs in mice mutant for the Sema3A signaling receptors Plexin-A3 and -A4. DRG axons from the Plexin-A3/A4 double knock-out mouse were visualized by crossing this viable mutant mouse line to a reporter line that carries the LacZ gene under control of the neuronal specific promoter Brn3a (Eng et al., 2001). Using this approach, we could effectively visualize DRG axons up to E16.5 simply by X-gal staining of whole-mount preparations (supplemental Fig. S6, available at www.jneurosci.org as supplemental material). Early in development, mutant DRG axons displayed severe defasciculation and guidance defects, phenotypes that are typical of lack in Semaphorin signaling (Fig. 5A,B). By embryonic day E15.5, many of the misprojecting axons underwent degeneration defined by fragmented morphology and appearance of bulblike structures along the axons, a phenomenon that was never observed in wild-type embryos (Fig. 5C–F). These structures are not retraction bulbs as they appear along the axons and not at their tips (Fig. 5D,E). Eventually, by day E16.5, the vast majority of misprojecting axons were already eliminated (Fig. 6D). We conclude that elimination of misguided DRG axons involves mechanisms of axonal degeneration and that this system enables additional investigation of the role of the apoptotic machinery in this process.
Figure 5.
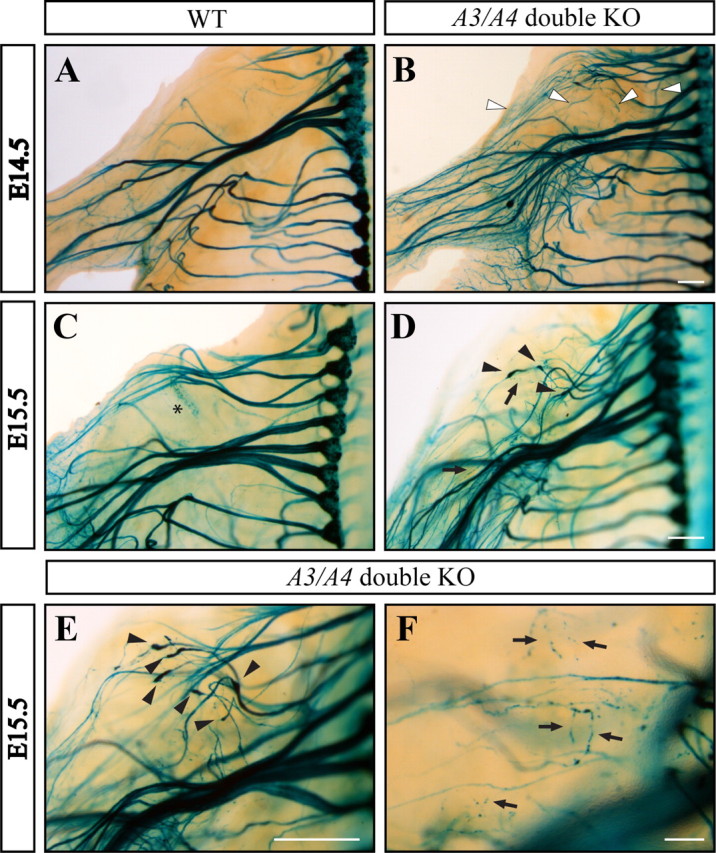
The Plexin-A3/A4 double knock-out mice exhibit axonal defasciculation and multiple misprojections that degenerate at subsequent developmental stages. An “open-book embryo” carrying the LacZ gene was fixed and stained with X-gal at different developmental stages (see also supplemental Fig. S5, available at www.jneurosci.org as supplemental material). A, B, At E14.5, neurons from the Plexin-A3/A4 double KO mouse (B) exhibit severe defasciculation of axon bundles compared with the wild type (A). In addition, many misprojecting axons can be detected (B, white arrowheads). D, F, By E15.5, most of the misprojecting axons in the double KO mouse are undergoing degeneration (D, F, black arrows), which are associated with bulblike structures (D, E, black arrowheads). The asterisk in C indicates a staining artifact. Scale bars, 300 μm.
Figure 6.
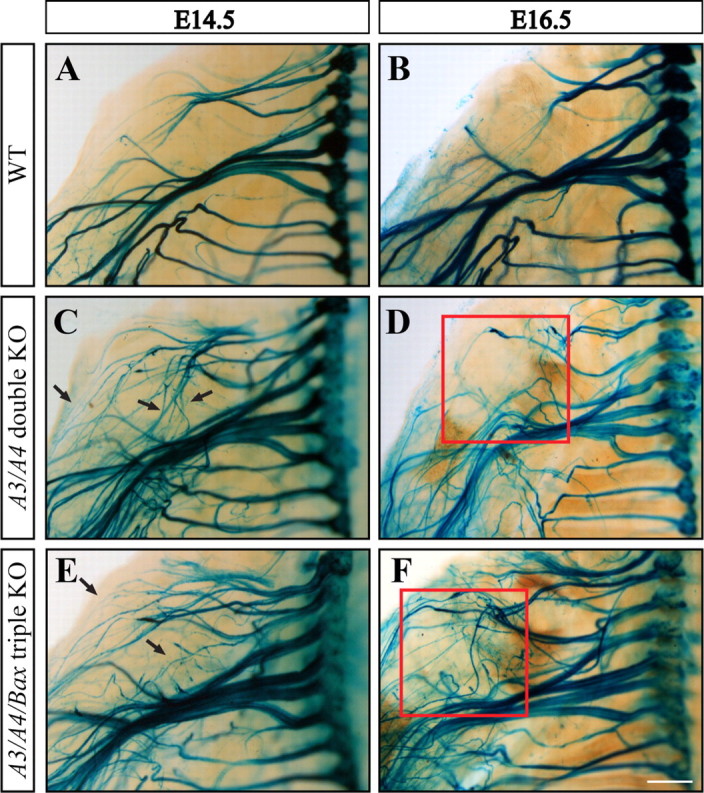
BAX is required for developmental elimination of misprojecting axons. A, B, In wild-type mice, no major changes in axonal architecture are observed between developmental days E14.5 and E16.5. C, D, In contrast, great ultrastructural modification occurs in the Plexin A3/A4 double KO mouse. Most of the misprojecting axons seen at E14.5 (C, black arrows) no longer exist at E16.5, resulting in axonal architecture more similar to that seen in the wild type (compare B, D). In contrast, whereas at E14.5 the excess of misprojecting axons seen in the Plexin-A3/A4/Bax triple KO mouse are similar to those seen in the double KO mouse (compare C, E), the typical axonal elimination seen in the double KO at E16.5 is lacking in the triple KO embryos (compare D, F). Scale bar, 500 μm. Quantification of axonal degeneration was done by counting the number of axonal bulbs in the shoulder apparent at E14.5 and E15.5 in the double KO embryos (n = 21) versus the triple KO embryos (n = 16). Quantification of misprojecting axons was done by counting axonal bundles present in upper limb (D, F, red square) at E16.5 in the double KO embryos (n = 9) versus the triple KO embryos (n = 6).
We then set out to test whether axonal degeneration in the Plexin-A3/A4 double KO mice is BAX dependent. Using the in vitro setting, we readily detected axonal degeneration in DRG explants from the Plexin-A3/A4 double KO mice, which was completely blocked by coablation of Bax (supplemental Fig. S1, available at www.jneurosci.org as supplemental material). Next, to examine the role of BAX in vivo, we generated a Plexin-A3/A4/Bax triple KO mice. We then compared the extent of elimination of DRG sensory axons in the Plexin-A3/A4 double KO with that in the Plexin-A3/A4/Bax triple KO mouse in vivo. The axonal projections of the double and triple KOs were visualized using the Brn3a-LacZ transgene (Fig. 6). A marked decrease in the average number of degenerating axons was observed in the triple KO compared with the double KO mice already at E15.5 (4 vs 6.63; p < 0.05). This reduction was accompanied by a pronounced increase in the number of misprojecting axons by day E16.5 (Fig. 6, compare D, F) (9.5 vs 4; p < 0.0001). These results suggest that BAX can mediate elimination of misprojecting axons in vivo and that the apoptotic machinery may be active within these axons during embryonic development.
Caspases are required during dendritic pruning in Drosophila
Another system that has been used to investigate the mechanisms of neurite degeneration in a physiological setting is the dendritic pruning in class IV dendritic arborization (C4da) sensory neurons, which occurs during larval-to-adult transformation in Drosophila (Grueber and Jan, 2004; Luo and O'Leary, 2005; Williams and Truman, 2005). Two studies have suggested a role of the apoptotic machinery during pruning in C4da neurons (Kuo et al., 2006; Williams et al., 2006). However, our understanding of the involvement of different key apoptotic components in this process is still incomplete (for review, see Feinstein-Rotkopf and Arama, 2009). Furthermore, a role for the NAD+-sensitive pathway in this system has never been reported. We, therefore, went on to test whether the mechanisms identified in our mouse culture system may be also conserved in flies. To start addressing these questions, we focused on dendritic pruning in ddaC, a specific C4da sensory neuron of the dorsal cluster (Grueber et al., 2002). C4da neurons were labeled using the pickpocket (ppk)-eGFP marker and/or expression of mCD8::GFP using the ppk-GAL4 driver, and the ddaC neuron within this cluster was identified during late larval and metamorphosis stages based on its morphology and position (Grueber et al., 2003; Williams et al., 2006; Lee et al., 2009). In wild-type larvae before puparium formation, dendritic branches of ddaC neurons are intact (Fig. 7A). At 7.5 h APF, these branches appeared patchy and many of them were already severed from their parental neurons, whereas at 18 h APF all branches were completely eliminated (Fig. 7B,C) (Williams et al., 2006; Lee et al., 2009). Similar to wild type, dendrites of flies with a strong mutation in the initiator caspase-9 ortholog Dronc also exhibited an intact morphology before puparium formation, as expected. However, unlike the wild-type dendrites, at 7.5 h APF the dronc mutant dendrites continued to possess an overall regular morphology and were mostly attached to the parental neurons (Fig. 7D,E). These branches persisted even at 18 h APF, albeit with less homogenous morphology (Fig. 7F). Similar findings using different dronc alleles were also reported (Kuo et al., 2006; Williams et al., 2006).
Figure 7.

Suppression of caspases causes defects in dendritic pruning in ddaC neurons during metamorphosis. A–I, Live projected confocal z-stack images of ddaC neurons during metamorphosis visualized by ppk-eGFP and/or mCD8::GFP expression under the control of the ppk-GAL4 driver. This driver was also used to drive expression of the indicated transgenes to ddaC neurons. Wild-type (A–C) ddaC neurons are shown at 0, 7.5, and 18 h APF. The arrowheads indicate soma; the asterisks mark the axon. D–F, ddaC neurons at similar stages as in wild type were documented in flies homozygous for the droncI24 allele. Note that, whereas dendrites are completely eliminated in wild type (C), they persist in dronc mutant flies (F). G–I, Fragmented ddaC dendritic branches are persisting at 18 h APF in the following indicated fly strains: transheterozygotes for the two drice alleles, driceΔ1 and drice17 (G), drice RNAi (H), and homozygotes for the dcp-1Prev allele (I). Scale bar, 20 μm.
Drosophila possesses four effector-like caspases defined by their amino acid sequence, of which Drice and Dcp-1 are the main functional effector caspases (Song et al., 1997; Muro et al., 2006; Xu et al., 2006). Whereas one report demonstrated an inhibitory effect on ddaC dendritic pruning on expression of the viral effector caspases inhibitor p35 (Williams et al., 2006), a study by another group failed to detect any effect with p35 using a similar setting (Kuo et al., 2006). To directly investigate the involvement of the effector caspases in ddaC pruning, we examined flies with mutations in either drice or dcp-1. Transheterozygosity between two loss-of-function alleles of drice or gene silencing of drice by RNA interference (RNAi) resulted in a mild suppression of branch removal at 18 h APF (Fig. 7G,H). However, only a very weak effect was detected in flies carrying a dcp-1-null allele (Fig. 7I). These findings are consistent with a role of Drice as the major effector caspase acting downstream of Dronc (Muro et al., 2006; Xu et al., 2006). Expression of p35 in the C4da neurons (using the ppk-Gal4 line) provided much greater protection of the ddaC dendrites than in drice mutants alone (compare Fig. 8A with Fig. 7G,H). Therefore, in addition to Drice or Dcp-1, other effector caspases may also function in ddaC dendritic pruning. Nonetheless, the overall inhibition of effector caspases by p35 was appreciably weaker than that observed in dronc mutants, suggesting that Dronc may exert more functions in addition to activation of effector caspases in this system (compare Fig. 7F with Fig. 8A). We conclude that key apoptotic caspases are required during the non-cell death process of dendritic pruning in Drosophila.
Figure 8.

Cooperation of caspases and the NAD+ pathway in ddaC neuron dendrite pruning. A, B, In flies overexpressing p35 (A) and WldS (B), fragmented ddaC dendritic branches are persisting at 18 h APF. C, However, in the fly strain overexpressing an enzyme dead Wlds (WldS-dead), no ddaC dendritic branches are detected. D–F, Combined expression of both p35 and WldS results in a greater protection than each of them alone. Scale bar, 20 μm. Branch length of ddaC neurons was measured manually within regions of interest using the ImageJ program. Statistical analysis was performed using nonparametrical ANOVA (Kruskal–Wallis). Error bars represent the confidence interval of the median. E, Median dendritic length. F, Median of total dendritic length. *p < 0.05, **p < 0.01.
The NAD+-sensitive pathway operates during dendritic pruning in Drosophila
Our studies in mice demonstrated a cooperative function between effector caspases and the NAD+-sensitive pathway during neurite degeneration. We asked whether the NAD+-sensitive pathway might also be involved in ddaC dendritic pruning. Previous studies in Drosophila using transgenic expression of the mouse WldS fusion protein, which is considered to mainly act in the NAD+-sensitive pathway, demonstrated that this transgene could protect axons in the olfactory receptor neurons from injury-induced degeneration, but failed to inhibit naturally occurring axon pruning in the mushroom body γ neurons (Hoopfer et al., 2006; MacDonald et al., 2006). To test whether the NAD+-sensitive pathway is involved in ddaC dendritic pruning, we expressed the mouse WldS gene in ddaC neurons (using the ppk-Gal4 driver). Significantly, WldS expression delayed removal of ddaC dendritic braches to a greater extent than that seen in transgenic expression of p35 (Fig. 8B,E,F). Moreover, this effect was completely absent in flies expressing an “enzyme-dead” version of WldS (WldS-dead) (Fig. 8C) (Avery et al., 2009), suggesting that the activity of the Nmnat1 portion of the molecule is required for its neuroprotective function in ddaC neurons. Most importantly, transgenic flies expressing both WldS and p35 in their ddaC neurons exhibited significantly better protection than flies with p35 or WldS alone (Fig. 8D–F), indicating that, similar to the mammalian system, these two pathways operate in parallel.
If indeed the apoptotic and NAD+-sensitive pathways act independently of each other, expression of the WldS protein should not interfere with activation of the effector caspases. To address this, we used the previously described genetically encoded detector of caspase activity CD8::PARP::Venus (Williams et al., 2006). This hybrid protein contains a 40-aa-long peptide from human PARP, which includes the DEVD consensus sequence for cleavage by effector caspases. We have found that cleavage of this artificial caspase substrate, detected by an anti-cleaved PARP antibody, can reflect the activity of Drice and Dcp-1, in vivo (A. Florentin and E. Arama, unpublished results). As was previously shown, cleaved PARP was readily detected within degenerating ddaC dendrites at 7.5–10 h APF, but not in the cell bodies or axons, demonstrating the compartmentalized nature of caspase activity in these cells (Fig. 9A) (Williams et al., 2006). Surviving ddaC dendrites of WldS transgenic flies also displayed effector caspase activity at 7.5 h APF (Fig. 9B). Similarly, the WldS protein could not suppress caspase activity during irradiation-induced apoptosis in the Drosophila wing-imaginal disc or the apoptosis in the Drosophila compound eye induced by ectopic expression of the proapoptotic protein Hid (supplemental Fig. S7, available at www.jneurosci.org as supplemental material). We conclude that the neuroprotective function of WldS and suppression of pruning by the effector caspase inhibitor p35 affect distinct destruction pathways and may act in concert to provide efficient dendritic pruning during development.
Figure 9.

Effector caspase activity is not altered in ddaC dendrites expressing WldS. A, B, The genetically encoded caspase activity reporter CD8::PARP::Venus reveals local caspase activity in dendrites of pruning da neurons. Caspase activity indicated by the presence of cleaved PARP (cPARP) (magenta; arrows) is localized to degenerating dendritic branches in both wild-type (A) and WldS-expressing ddaC (B) neurons at the indicated hours APF. The entire ddaC neuron is visualized in green using anti-CD8 antibody (CD8; green). The cell bodies and axons are marked by arrowheads and asterisks, respectively.
Discussion
The role of the NAD+-sensitive pathway in neurite degeneration
In the present study, we uncovered an evolutionarily conserved role of two cellular destruction systems during developmental neurite pruning: the apoptotic caspases and a NAD+-sensitive pathway (Fig. 10). Until recently, however, it had been widely accepted that neurite degeneration during developmental pruning involves a novel destruction system, distinct from the apoptotic cell death machinery and the injury-induced NAD+-sensitive pathway of Wallerian degeneration. This notion stems from the observations that inhibition of one of these pathways did not block developmental pruning and that caspases do not appear to play a role in the model of distal axon degeneration after transection (Finn et al., 2000; Watts et al., 2003; Whitmore et al., 2003; Awasaki et al., 2006). Although we cannot rule out the possibility that a third destruction system may also exist, the fact that to obtain a pronounced neuroprotective effect the simultaneous inhibition of both the apoptotic machinery and the NAD+-sensitive pathway is required in mice, strongly suggests that other types of neurons may also use similar destruction systems. Moreover, the conserved involvement of these pathways in neurite degeneration from flies to mammals supports the notion that the molecular mechanisms of neurite destruction may be less diverse than has been previously appreciated. Additional cellular and genetic analyses of these destruction systems in different neuronal systems may provide additional insights into the mechanisms underlying neurite elimination during development.
Figure 10.

Conserved pathways control neurite degeneration. A, In mammalian sensory axons, BAX, likely through its action in the mitochondria, activates caspases and a parallel NAD+-sensitive pathway to execute axonal degeneration in response to trophic withdrawal. The two effector caspases 3 and 6 are activated by different mechanisms, caspase-6 by cleavage of the procaspase and caspase-3 through regulation by XIAP. In contrast, axonal destruction after severing occurs only through the NAD+-sensitive pathway. B, In the fly, developmental pruning of sensory neuron dendrites is initiated by the initiator caspase Dronc, which induces the severing of the dendrites. The effector caspases and the NAD+-sensitive pathway are then operating in parallel to execute the degeneration process.
Using the Drosophila system, our studies demonstrate, for the first time, a role for a NAD+-sensitive pathway during naturally occurring neurite pruning. Interestingly, unlike DRG neurons in which supplement of NAD+ alone produced only a minor protection, ectopic expression of WldS by itself can cause a marked attenuation of dendritic pruning in Drosophila ddaC neurons, an effect that depends on the enzymatic activity of the Nmnat1 portion of WldS (i.e., production of NAD+). This neuroprotctive effect of WldS is not a consequence of indirect inhibition of caspases for two reasons. First, caspase activity can be readily detected within WldS-expressing neuritis, and WldS cannot block caspase-dependent cell death. Second, the combination of WldS and p35 provides better protection from degeneration than either one of them alone. A similar phenomenon of delayed onset of neurite disintegration was also observed with neurons derived from postnatal sympathetic superior ganglia (SCG) of WldS mutant mice (Deckwerth and Johnson, 1994) or wild-type SCG neurons deprived of NGF and supplemented with NAD+ (Z. Schoenmann and A. Yaron, unpublished results). This inhibition was detected despite the fact that inhibition of caspase activity using Z-VAD-FMK (Zhai et al., 2003) or ablation of BAX (Z. Schoenmann and A. Yaron, unpublished results) could also effectively suppress axon degeneration in these neurons. Thus, it appears that the level of requirement for the combined actions of the apoptotic machinery and the NAD+-sensitive pathway may vary in different types of neurons.
The role of the apoptotic machinery in neurite degeneration
Our studies using a primary culture of murine DRG sensory neurons indicate roles for BAX and the effector caspase-3 and -6 during axonal degeneration. We found that these two effector caspases are activated by distinct mechanisms within degenerating axons. Whereas caspase-6 is cleaved and activated only after trophic withdrawal, caspase-3 is already present in its active form before NGF deprivation, albeit its activity is kept below the threshold level required to induce axonal degeneration. The corresponding reduction in the level of the endogenous caspase inhibitor XIAP after trophic deprivation suggests that caspase-3 activity may be regulated by this inhibitor. Other nonapoptotic roles of caspase-3 in neurons were also reported. For example, using a peptide inhibitor, a role for caspase-3 in modulating synaptic activity and synaptic loss was suggested (D'Amelio et al., 2009). In another study, caspase-3 was shown to be activated in dendritic spines of the zebra finch auditory forebrain after memory-associated stimuli (Huesmann and Clayton, 2006). Significantly, similar to our system, the active form of caspase-3 was present in the unstimulated brain, yet it exhibited very low activity, which was also attributed to inhibition by XIAP. Although the significance of this mode of caspase regulation is still unclear, these results raise the attractive hypothesis that, during apoptosis, caspase-3 may be activated preferentially by cleavage, whereas activation of caspase-3 during nonapoptotic processes may rely mostly on degradation of the endogenous inhibitors.
A recent report demonstrated a requirement for BAX and caspase-6 during axonal degeneration in cultured dissociated DRG neurons (Nikolaev et al., 2009). However, several apparent discrepancies between the two studies are noted. More specifically, whereas Nikolaev et al. reported that inhibition of caspase-6 alone is sufficient to block axonal degeneration and showed no effect of caspase-3 inhibition on this process, we demonstrate that an efficient block of axonal degeneration is obtained by the combination of either caspase-6 or caspase-3 inhibition and NAD+ supplement. We believe that these discrepancies may reflect the differences between the two experimental systems. In particular, in our system, the DRG neurons are not dispersed but rather maintained as intact ganglia. Furthermore, to induce axonal degeneration, the entire filter culture is deprived of NGF and not just the axonal compartment; hence axonal degeneration is accompanied by cell death as often occurs during neuronal development. More studies are needed to pinpoint the exact cause for these differences and, more importantly, to determine which of these molecular mechanisms, if not both, operates in vivo.
Our results provide the first evidence for a requirement of BAX during axonal degeneration in vivo, demonstrating that BAX is required for elimination of misprojecting sensory axons during PNS development. Although it is possible that the elevated number of misprojecting axons may be in part attributable to the already increased neuronal survival in Bax KO mice, we consider this explanation very unlikely for the following reasons. First, no differences between the double and triple KO mice in the number and pattern of misprojecting axons were observed before the elimination phase early in development. Second, the elevation in the number of misprojecting axons in the Plexin A3/A4/Bax triple knock-out mice at E16.5 is inversely associated with a reduction in the number of degenerating axons during earlier developmental stages, supporting the idea that this elevation is a consequence of suppression of degeneration of misprojecting axons. These results imply that, similar to BAX, other factors that were characterized in culture, such as the effector caspases, may have also a role during axonal degeneration in vivo.
Emerging data now indicate that apoptosis and autophagy may be linked (Debnath et al., 2005; Kroemer and Jäättelä, 2005; Bialik and Kimchi, 2010). Furthermore, several key factors and regulators of apoptosis, such as Bcl-2, DAP (death-associated protein)-kinase, and some of the Drosophila effector caspases, were shown to also control some aspects of the autophagic process (Liang et al., 1999; Gozuacik et al., 2008; Denton et al., 2009). Our finding that the proapoptotic Bcl-2 family member BAX regulates axonal degeneration raised the possibility that some components of the autophagic system may also play a role in axonal degeneration. However, we could not detect any effect on axonal degeneration after trophic deprivation by supplementing these cultures with 3-methyladenine, a potent inhibitor of autophagy (data not shown). Therefore, although we cannot completely exclude some roles for the autophagic system in axonal elimination in vivo or in other neuronal systems, it is unlikely that autophagy plays a major role in axonal degeneration in our system.
The first indications for a role of the apoptotic machinery in neurite destruction came from two genetic studies in Drosophila demonstrating that caspases are involved in developmental dendritic pruning in C4da sensory neurons (Kuo et al., 2006; Williams et al., 2006). Our study both corroborates and extends the idea that dendritic pruning in these neurons uses key components of the apoptotic machinery. We show that, in addition to the initiator caspase Dronc, the main effector caspase Drice is also required for efficient elimination of dendritic branches. Unexpectedly, however, we found that dronc mutant flies exhibit a more pronounced attenuation of dendritic pruning than drice mutants or transgenic flies expressing the pan effector caspase inhibitor p35. Although persisting at 18 h APF, dendritic branches were always fragmented and detached from their parental neurons in drice mutants or flies expressing p35, whereas in dronc mutants, the morphology of these branches was highly preserved and they often remained attached to the parental neurons even at 18 h APF. Likewise, strong suppression was also observed with a gain-of-function allele of the inhibitor of apoptosis protein Diap1, a major negative regulator of Dronc (data not shown) (Kuo et al., 2006). Furthermore, recent work from the Jan Laboratory indicated a strong suppression of dendritic pruning in mutants for Ik2, the Drosophila IKK (IκB kinase)-related kinase, which was previously described as a negative regulator of Diap1 in another caspase-dependent nonapoptotic process (Kuranaga et al., 2006; Lee et al., 2009). We conclude that Dronc exerts other roles during this process in addition to its role in the activation of the effector caspases (Fig. 10). Significantly, these data strongly suggest that the effector caspase activity peaks only after detachment of dendritic branches from the parental neuron, thus providing an explanation for how dendrite-confined effector caspase activity does not affect the soma.
In conclusion, our study sheds light on the mechanisms of developmental axonal and dendritic pruning (Fig. 10). Surprisingly, we find high evolutionary conservation in these mechanisms, suggesting that they may operate in other neuronal systems as well. Axonal degeneration is an early pathological event in various neurodegenerative diseases responsible for the clinical progression and death in patients (Raff et al., 2002; Saxena and Caroni, 2007; Hilliard, 2009). Additional studies of models of neurodegenerative conditions may uncover the extent of involvement of these cellular pathways in the etiology of such diseases.
Footnotes
This work was supported by Human Frontier Science Program Career Development Award CDA0024/2006-C, The Joseph D. Shane Fund for Neurosciences, and The Estelle Funk Foundation for Biomedical Research (laboratory of A.Y.). E.A.-K. is supported by a postdoctoral fellowship from the Israel Cancer Research Fund. E.A. is an Alon Fellow with the Council for Higher Education of the Israeli Academy of Sciences and is also supported by grants from the Israel Science Foundation, Lord Mitchell, The Henry S. and Anne S. Reich Research Fund for Mental Health, The Samuel M. Soref and Helene K. Soref Foundation, and The Chais Family Fellows Program for New Scientists. E.A. is the Incumbent of the Corinne S. Koshland Career Development Chair. We are grateful to A. Bergmann, M. R. Freeman, W. B. Grueber, K. McCall, D. W. Williams, and the Bloomington Stock Center for providing fly stocks and reagents. We also thank E. Truner for the Brn3a-LacZ plasmid and The Weizmann Transgenic Facility for generation of the Brn3a-LacZ transgenic mouse. We are indebted to Marc Tessier-Lavigne for helpful discussions and for sharing results before publication. We thank the Yaron and Arama laboratory members for advice and criticism, Michael Fainzilber and Orly Reiner for critically reading this manuscript, Yossef Kaplan for help with photographing fly eyes, and Anat Florentin for help with the caspase activity assays.
References
- Alano CC, Garnier P, Ying W, Higashi Y, Kauppinen TM, Swanson RA. NAD+ depletion is necessary and sufficient for poly(ADP-ribose) polymerase-1-mediated neuronal death. J Neurosci. 2010;30:2967–2978. doi: 10.1523/JNEUROSCI.5552-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Araki T, Sasaki Y, Milbrandt J. Increased nuclear NAD biosynthesis and SIRT1 activation prevent axonal degeneration. Science. 2004;305:1010–1013. doi: 10.1126/science.1098014. [DOI] [PubMed] [Google Scholar]
- Avery MA, Sheehan AE, Kerr KS, Wang J, Freeman MR. Wld S requires Nmnat1 enzymatic activity and N16-VCP interactions to suppress Wallerian degeneration. J Cell Biol. 2009;184:501–513. doi: 10.1083/jcb.200808042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Awasaki T, Tatsumi R, Takahashi K, Arai K, Nakanishi Y, Ueda R, Ito K. Essential role of the apoptotic cell engulfment genes draper and ced-6 in programmed axon pruning during Drosophila metamorphosis. Neuron. 2006;50:855–867. doi: 10.1016/j.neuron.2006.04.027. [DOI] [PubMed] [Google Scholar]
- Bergmann A, Agapite J, Steller H. Mechanisms and control of programmed cell death in invertebrates. Oncogene. 1998;17:3215–3223. doi: 10.1038/sj.onc.1202586. [DOI] [PubMed] [Google Scholar]
- Bialik S, Kimchi A. Lethal weapons: DAP-kinase, autophagy and cell death DAP-kinase regulates autophagy. Curr Opin Cell Biol. 2010;22:199–205. doi: 10.1016/j.ceb.2009.11.004. [DOI] [PubMed] [Google Scholar]
- Budihardjo I, Oliver H, Lutter M, Luo X, Wang X. Biochemical pathways of caspase activation during apoptosis. Annu Rev Cell Dev Biol. 1999;15:269–290. doi: 10.1146/annurev.cellbio.15.1.269. [DOI] [PubMed] [Google Scholar]
- Bursch W, Paffe S, Putz B, Barthel G, Schulte-Hermann R. Determination of the length of the histological stages of apoptosis in normal liver and in altered hepatic foci of rats. Carcinogenesis. 1990;11:847–853. doi: 10.1093/carcin/11.5.847. [DOI] [PubMed] [Google Scholar]
- Conforti L, Wilbrey A, Morreale G, Janeckova L, Beirowski B, Adalbert R, Mazzola F, Di Stefano M, Hartley R, Babetto E, Smith T, Gilley J, Billington RA, Genazzani AA, Ribchester RR, Magni G, Coleman M. Wld S protein requires Nmnat activity and a short N-terminal sequence to protect axons in mice. J Cell Biol. 2009;184:491–500. doi: 10.1083/jcb.200807175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D'Amelio M, Cavallucci V, Cecconi F. Neuronal caspase-3 signaling: not only cell death. Cell Death Differ. 2009 doi: 10.1038/cdd.2009.180. [DOI] [PubMed] [Google Scholar]
- Debnath J, Baehrecke EH, Kroemer G. Does autophagy contribute to cell death? Autophagy. 2005;1:66–74. doi: 10.4161/auto.1.2.1738. [DOI] [PubMed] [Google Scholar]
- Deckwerth TL, Johnson EM., Jr Neurites can remain viable after destruction of the neuronal soma by programmed cell death (apoptosis) Dev Biol. 1994;165:63–72. doi: 10.1006/dbio.1994.1234. [DOI] [PubMed] [Google Scholar]
- Denton D, Shravage B, Simin R, Mills K, Berry DL, Baehrecke EH, Kumar S. Autophagy, not apoptosis, is essential for midgut cell death in Drosophila. Curr Biol. 2009;19:1741–1746. doi: 10.1016/j.cub.2009.08.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deveraux QL, Roy N, Stennicke HR, Van Arsdale T, Zhou Q, Srinivasula SM, Alnemri ES, Salvesen GS, Reed JC. IAPs block apoptotic events induced by caspase-8 and cytochrome c by direct inhibition of distinct caspases. EMBO J. 1998;17:2215–2223. doi: 10.1093/emboj/17.8.2215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eckelman BP, Salvesen GS, Scott FL. Human inhibitor of apoptosis proteins: why XIAP is the black sheep of the family. EMBO Rep. 2006;7:988–994. doi: 10.1038/sj.embor.7400795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eng SR, Gratwick K, Rhee JM, Fedtsova N, Gan L, Turner EE. Defects in sensory axon growth precede neuronal death in Brn3a-deficient mice. J Neurosci. 2001;21:541–549. doi: 10.1523/JNEUROSCI.21-02-00541.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feinstein-Rotkopf Y, Arama E. Can't live without them, can live with them: roles of caspases during vital cellular processes. Apoptosis. 2009;14:980–995. doi: 10.1007/s10495-009-0346-6. [DOI] [PubMed] [Google Scholar]
- Finn JT, Weil M, Archer F, Siman R, Srinivasan A, Raff MC. Evidence that Wallerian degeneration and localized axon degeneration induced by local neurotrophin deprivation do not involve caspases. J Neurosci. 2000;20:1333–1341. doi: 10.1523/JNEUROSCI.20-04-01333.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glass JD, Brushart TM, George EB, Griffin JW. Prolonged survival of transected nerve fibres in C57BL/Ola mice is an intrinsic characteristic of the axon. J Neurocytol. 1993;22:311–321. doi: 10.1007/BF01195555. [DOI] [PubMed] [Google Scholar]
- Goyal L. Cell death inhibition: keeping caspases in check. Cell. 2001;104:805–808. doi: 10.1016/s0092-8674(01)00276-8. [DOI] [PubMed] [Google Scholar]
- Gozuacik D, Bialik S, Raveh T, Mitou G, Shohat G, Sabanay H, Mizushima N, Yoshimori T, Kimchi A. DAP-kinase is a mediator of endoplasmic reticulum stress-induced caspase activation and autophagic cell death. Cell Death Differ. 2008;15:1875–1886. doi: 10.1038/cdd.2008.121. [DOI] [PubMed] [Google Scholar]
- Grether ME, Abrams JM, Agapite J, White K, Steller H. The head involution defective gene of Drosophila melanogaster functions in programmed cell death. Genes Dev. 1995;9:1694–1708. doi: 10.1101/gad.9.14.1694. [DOI] [PubMed] [Google Scholar]
- Griffin JW, George EB, Chaudhry V. Wallerian degeneration in peripheral nerve disease. Baillieres Clin Neurol. 1996;5:65–75. [PubMed] [Google Scholar]
- Grueber WB, Jan YN. Dendritic development: lessons from Drosophila and related branches. Curr Opin Neurobiol. 2004;14:74–82. doi: 10.1016/j.conb.2004.01.001. [DOI] [PubMed] [Google Scholar]
- Grueber WB, Jan LY, Jan YN. Tiling of the Drosophila epidermis by multidendritic sensory neurons. Development. 2002;129:2867–2878. doi: 10.1242/dev.129.12.2867. [DOI] [PubMed] [Google Scholar]
- Grueber WB, Ye B, Moore AW, Jan LY, Jan YN. Dendrites of distinct classes of Drosophila sensory neurons show different capacities for homotypic repulsion. Curr Biol. 2003;13:618–626. doi: 10.1016/s0960-9822(03)00207-0. [DOI] [PubMed] [Google Scholar]
- Hay BA, Wolff T, Rubin GM. Expression of baculovirus P35 prevents cell death in Drosophila. Development. 1994;120:2121–2129. doi: 10.1242/dev.120.8.2121. [DOI] [PubMed] [Google Scholar]
- Hilliard MA. Axonal degeneration and regeneration: a mechanistic tug-of-war. J Neurochem. 2009;108:23–32. doi: 10.1111/j.1471-4159.2008.05754.x. [DOI] [PubMed] [Google Scholar]
- Hoopfer ED, McLaughlin T, Watts RJ, Schuldiner O, O'Leary DD, Luo L. Wlds protection distinguishes axon degeneration following injury from naturally occurring developmental pruning. Neuron. 2006;50:883–895. doi: 10.1016/j.neuron.2006.05.013. [DOI] [PubMed] [Google Scholar]
- Huesmann GR, Clayton DF. Dynamic role of postsynaptic caspase-3 and BIRC4 in zebra finch song-response habituation. Neuron. 2006;52:1061–1072. doi: 10.1016/j.neuron.2006.10.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kroemer G, Jäättelä M. Lysosomes and autophagy in cell death control. Nat Rev Cancer. 2005;5:886–897. doi: 10.1038/nrc1738. [DOI] [PubMed] [Google Scholar]
- Kuo CT, Zhu S, Younger S, Jan LY, Jan YN. Identification of E2/E3 ubiquitinating enzymes and caspase activity regulating Drosophila sensory neuron dendrite pruning. Neuron. 2006;51:283–290. doi: 10.1016/j.neuron.2006.07.014. [DOI] [PubMed] [Google Scholar]
- Kuranaga E, Kanuka H, Tonoki A, Takemoto K, Tomioka T, Kobayashi M, Hayashi S, Miura M. Drosophila IKK-related kinase regulates nonapoptotic function of caspases via degradation of IAPs. Cell. 2006;126:583–596. doi: 10.1016/j.cell.2006.05.048. [DOI] [PubMed] [Google Scholar]
- Lamkanfi M, Festjens N, Declercq W, Vanden Berghe T, Vandenabeele P. Caspases in cell survival, proliferation and differentiation. Cell Death Differ. 2007;14:44–55. doi: 10.1038/sj.cdd.4402047. [DOI] [PubMed] [Google Scholar]
- Laundrie B, Peterson JS, Baum JS, Chang JC, Fileppo D, Thompson SR, McCall K. Germline cell death is inhibited by P-element insertions disrupting the dcp-1/pita nested gene pair in Drosophila. Genetics. 2003;165:1881–1888. doi: 10.1093/genetics/165.4.1881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee HH, Jan LY, Jan YN. Drosophila IKK-related kinase Ik2 and Katanin p60-like 1 regulate dendrite pruning of sensory neuron during metamorphosis. Proc Natl Acad Sci U S A. 2009;106:6363–6368. doi: 10.1073/pnas.0902051106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee T, Luo L. Mosaic analysis with a repressible cell marker for studies of gene function in neuronal morphogenesis. Neuron. 1999;22:451–461. doi: 10.1016/s0896-6273(00)80701-1. [DOI] [PubMed] [Google Scholar]
- Liang XH, Jackson S, Seaman M, Brown K, Kempkes B, Hibshoosh H, Levine B. Induction of autophagy and inhibition of tumorigenesis by beclin 1. Nature. 1999;402:672–676. doi: 10.1038/45257. [DOI] [PubMed] [Google Scholar]
- Lunn ER, Perry VH, Brown MC, Rosen H, Gordon S. Absence of wallerian degeneration does not hinder regeneration in peripheral nerve. Eur J Neurosci. 1989;1:27–33. doi: 10.1111/j.1460-9568.1989.tb00771.x. [DOI] [PubMed] [Google Scholar]
- Luo L, O'Leary DD. Axon retraction and degeneration in development and disease. Annu Rev Neurosci. 2005;28:127–156. doi: 10.1146/annurev.neuro.28.061604.135632. [DOI] [PubMed] [Google Scholar]
- MacDonald JM, Beach MG, Porpiglia E, Sheehan AE, Watts RJ, Freeman MR. The Drosophila cell corpse engulfment receptor Draper mediates glial clearance of severed axons. Neuron. 2006;50:869–881. doi: 10.1016/j.neuron.2006.04.028. [DOI] [PubMed] [Google Scholar]
- Mack TG, Reiner M, Beirowski B, Mi W, Emanuelli M, Wagner D, Thomson D, Gillingwater T, Court F, Conforti L, Fernando FS, Tarlton A, Andressen C, Addicks K, Magni G, Ribchester RR, Perry VH, Coleman MP. Wallerian degeneration of injured axons and synapses is delayed by a Ube4b/Nmnat chimeric gene. Nat Neurosci. 2001;4:1199–1206. doi: 10.1038/nn770. [DOI] [PubMed] [Google Scholar]
- Maeno E, Ishizaki Y, Kanaseki T, Hazama A, Okada Y. Normotonic cell shrinkage because of disordered volume regulation is an early prerequisite to apoptosis. Proc Natl Acad Sci U S A. 2000;97:9487–9492. doi: 10.1073/pnas.140216197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muro I, Berry DL, Huh JR, Chen CH, Huang H, Yoo SJ, Guo M, Baehrecke EH, Hay BA. The Drosophila caspase Ice is important for many apoptotic cell deaths and for spermatid individualization, a nonapoptotic process. Development. 2006;133:3305–3315. doi: 10.1242/dev.02495. [DOI] [PubMed] [Google Scholar]
- Nicholson DW, Thornberry NA. Caspases: killer proteases. Trends Biochem Sci. 1997;22:299–306. doi: 10.1016/s0968-0004(97)01085-2. [DOI] [PubMed] [Google Scholar]
- Nikolaev A, McLaughlin T, O'Leary DD, Tessier-Lavigne M. APP binds DR6 to trigger axon pruning and neuron death via distinct caspases. Nature. 2009;457:981–989. doi: 10.1038/nature07767. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Raff MC, Whitmore AV, Finn JT. Axonal self-destruction and neurodegeneration. Science. 2002;296:868–871. doi: 10.1126/science.1068613. [DOI] [PubMed] [Google Scholar]
- Riedl SJ, Renatus M, Schwarzenbacher R, Zhou Q, Sun C, Fesik SW, Liddington RC, Salvesen GS. Structural basis for the inhibition of caspase-3 by XIAP. Cell. 2001;104:791–800. doi: 10.1016/s0092-8674(01)00274-4. [DOI] [PubMed] [Google Scholar]
- Sagot Y, Dubois-Dauphin M, Tan SA, de Bilbao F, Aebischer P, Martinou JC, Kato AC. Bcl-2 overexpression prevents motoneuron cell body loss but not axonal degeneration in a mouse model of a neurodegenerative disease. J Neurosci. 1995;15:7727–7733. doi: 10.1523/JNEUROSCI.15-11-07727.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salvesen GS, Duckett CS. IAP proteins: blocking the road to death's door. Nat Rev Mol Cell Biol. 2002;3:401–410. doi: 10.1038/nrm830. [DOI] [PubMed] [Google Scholar]
- Salvesen GS, Riedl SJ. Caspase mechanisms. Adv Exp Med Biol. 2008;615:13–23. doi: 10.1007/978-1-4020-6554-5_2. [DOI] [PubMed] [Google Scholar]
- Sasaki Y, Araki T, Milbrandt J. Stimulation of nicotinamide adenine dinucleotide biosynthetic pathways delays axonal degeneration after axotomy. J Neurosci. 2006;26:8484–8491. doi: 10.1523/JNEUROSCI.2320-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasaki Y, Vohra BP, Baloh RH, Milbrandt J. Transgenic mice expressing the Nmnat1 protein manifest robust delay in axonal degeneration in vivo. J Neurosci. 2009;29:6526–6534. doi: 10.1523/JNEUROSCI.1429-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saxena S, Caroni P. Mechanisms of axon degeneration: from development to disease. Prog Neurobiol. 2007;83:174–191. doi: 10.1016/j.pneurobio.2007.07.007. [DOI] [PubMed] [Google Scholar]
- Schile AJ, García-Fernández M, Steller H. Regulation of apoptosis by XIAP ubiquitin-ligase activity. Genes Dev. 2008;22:2256–2266. doi: 10.1101/gad.1663108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott FL, Denault JB, Riedl SJ, Shin H, Renatus M, Salvesen GS. XIAP inhibits caspase-3 and -7 using two binding sites: evolutionarily conserved mechanism of IAPs. EMBO J. 2005;24:645–655. doi: 10.1038/sj.emboj.7600544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song Z, McCall K, Steller H. DCP-1, a Drosophila cell death protease essential for development. Science. 1997;275:536–540. doi: 10.1126/science.275.5299.536. [DOI] [PubMed] [Google Scholar]
- Takahashi Y, Hirose F, Matsukage A, Yamaguchi M. Identification of three conserved regions in the DREF transcription factors from Drosophila melanogaster and Drosophila virilis. Nucleic Acids Res. 1999;27:510–516. doi: 10.1093/nar/27.2.510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomlinson A, Ready DF. Neuronal differentiation in Drosophila ommatidium. Dev Biol. 1987;120:366–376. doi: 10.1016/0012-1606(87)90239-9. [DOI] [PubMed] [Google Scholar]
- Waller A. Experiments on the section of glossopharyngeal and hypoglossal nerves of the frog and observations of the alternatives produced thereby in the structure of their primitive fibers. Philos Trans R Soc Lond B Biol Sci. 1850;140:423. [Google Scholar]
- Wang J, Zhai Q, Chen Y, Lin E, Gu W, McBurney MW, He Z. A local mechanism mediates NAD-dependent protection of axon degeneration. J Cell Biol. 2005;170:349–355. doi: 10.1083/jcb.200504028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watts RJ, Hoopfer ED, Luo L. Axon pruning during Drosophila metamorphosis: evidence for local degeneration and requirement of the ubiquitin-proteasome system. Neuron. 2003;38:871–885. doi: 10.1016/s0896-6273(03)00295-2. [DOI] [PubMed] [Google Scholar]
- White FA, Behar O. The development and subsequent elimination of aberrant peripheral axon projections in Semaphorin3A null mutant mice. Dev Biol. 2000;225:79–86. doi: 10.1006/dbio.2000.9822. [DOI] [PubMed] [Google Scholar]
- White FA, Silos-Santiago I, Molliver DC, Nishimura M, Phillips H, Barbacid M, Snider WD. Synchronous onset of NGF and TrkA survival dependence in developing dorsal root ganglia. J Neurosci. 1996;16:4662–4672. doi: 10.1523/JNEUROSCI.16-15-04662.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitmore AV, Lindsten T, Raff MC, Thompson CB. The proapoptotic proteins Bax and Bak are not involved in Wallerian degeneration. Cell Death Differ. 2003;10:260–261. doi: 10.1038/sj.cdd.4401147. [DOI] [PubMed] [Google Scholar]
- Williams DW, Truman JW. Remodeling dendrites during insect metamorphosis. J Neurobiol. 2005;64:24–33. doi: 10.1002/neu.20151. [DOI] [PubMed] [Google Scholar]
- Williams DW, Kondo S, Krzyzanowska A, Hiromi Y, Truman JW. Local caspase activity directs engulfment of dendrites during pruning. Nat Neurosci. 2006;9:1234–1236. doi: 10.1038/nn1774. [DOI] [PubMed] [Google Scholar]
- Wolf BB, Green DR. Suicidal tendencies: apoptotic cell death by caspase family proteinases. J Biol Chem. 1999;274:20049–20052. doi: 10.1074/jbc.274.29.20049. [DOI] [PubMed] [Google Scholar]
- Xu D, Li Y, Arcaro M, Lackey M, Bergmann A. The CARD-carrying caspase Dronc is essential for most, but not all, developmental cell death in Drosophila. Development. 2005;132:2125–2134. doi: 10.1242/dev.01790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu D, Wang Y, Willecke R, Chen Z, Ding T, Bergmann A. The effector caspases drICE and dcp-1 have partially overlapping functions in the apoptotic pathway in Drosophila. Cell Death Differ. 2006;13:1697–1706. doi: 10.1038/sj.cdd.4401920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yahata N, Yuasa S, Araki T. Nicotinamide mononucleotide adenylyltransferase expression in mitochondrial matrix delays Wallerian degeneration. J Neurosci. 2009;29:6276–6284. doi: 10.1523/JNEUROSCI.4304-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Fang S, Jensen JP, Weissman AM, Ashwell JD. Ubiquitin protein ligase activity of IAPs and their degradation in proteasomes in response to apoptotic stimuli. Science. 2000;288:874–877. doi: 10.1126/science.288.5467.874. [DOI] [PubMed] [Google Scholar]
- Yaron A, Huang PH, Cheng HJ, Tessier-Lavigne M. Differential requirement for Plexin-A3 and -A4 in mediating responses of sensory and sympathetic neurons to distinct class 3 Semaphorins. Neuron. 2005;45:513–523. doi: 10.1016/j.neuron.2005.01.013. [DOI] [PubMed] [Google Scholar]
- Yuan J, Shaham S, Ledoux S, Ellis HM, Horvitz HR. The C. elegans cell death gene ced-3 encodes a protein similar to mammalian interleukin-1 beta-converting enzyme. Cell. 1993;75:641–652. doi: 10.1016/0092-8674(93)90485-9. [DOI] [PubMed] [Google Scholar]
- Zhai Q, Wang J, Kim A, Liu Q, Watts R, Hoopfer E, Mitchison T, Luo L, He Z. Involvement of the ubiquitin-proteasome system in the early stages of wallerian degeneration. Neuron. 2003;39:217–225. doi: 10.1016/s0896-6273(03)00429-x. [DOI] [PubMed] [Google Scholar]
- Zheng JQ, Kelly TK, Chang B, Ryazantsev S, Rajasekaran AK, Martin KC, Twiss JL. A functional role for intra-axonal protein synthesis during axonal regeneration from adult sensory neurons. J Neurosci. 2001;21:9291–9303. doi: 10.1523/JNEUROSCI.21-23-09291.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]