

Abstract
Cerebral amyloid angiopathy (CAA) is a common feature of Alzheimer's disease (AD). More advanced stages are accompanied by microhemorrhages and vasculitis. Peripheral blood-borne macrophages are intimately linked to cerebrovascular pathology coincident with AD. Magnetic resonance imaging (MRI) was used to noninvasively study microvascular lesions in amyloid precursor protein transgenic mouse AD models. Foci of signal attenuation were detected in cortical and thalamic brain regions of aged APP23 mice. Their strength and number was considerably enhanced by intravenous administration of iron oxide nanoparticles, which are taken up by macrophages through absorptive endocytosis, 24 h before image acquisition. The number of cortical sites displaying signal attenuation increased with age. Histology at these sites demonstrated the presence of iron-containing macrophages in the vicinity of CAA-affected blood vessels. A fraction of the sites additionally showed thickened vessel walls and vasculitis. Consistent with the visualization of CAA-associated lesions, MRI detected a much smaller number of attenuated signal sites in APP23xPS45 mice, for which a strong presenilin mutation caused a shift toward amyloid β42, thus reducing vascular amyloid. Similar results were obtained with APP24 and APP51 mice, which develop significantly less CAA and microvascular pathology than APP23. In a longitudinal study, we noninvasively demonstrated the reinforced formation of microvascular pathology during passive amyloid β immunotherapy of APP23 mice. Histology confirmed that foci of signal attenuation reflected an increase in CAA-related lesions. Our data demonstrate that MRI has the sensitivity to noninvasively monitor the development of vascular pathology and its possible enhancement by amyloid β immunotherapy in transgenic mice modeling AD.
Introduction
The pathology of Alzheimer's disease (AD) is characterized by amyloid β (Aβ)-peptide-containing plaques, neurofibrillary tangles consisting of hyperphosphorylated tau, extensive neuritic and synaptic degeneration, and distinct neuron loss. Vascular abnormalities coexist with these AD features. At autopsy, ∼80% of the AD patients exhibit cerebral amyloid angiopathy (CAA) (Jellinger, 2002; Greenberg et al., 2004), characterized by deposition of Aβ in the walls of cerebral vessels. The contribution of CAA to the onset of dementia remains unknown. Advanced CAA leads to degeneration of the smooth muscle cell layer, fibrinoid necrosis, and microaneurisms, while it may also occlude capillaries (Thal et al., 2008). Although relatively rare, CAA is associated with hemorrhages ranging from microbleeds to stroke, and with vasculitis presenting a variable degree of lymphocyte infiltration and vascular thickening (Vinters et al., 1998; Kinnecom et al., 2007). Both conditions are accompanied by the presence of macrophages/microglia, which surround affected vessels (Maat-Schieman et al., 1997; Vinters et al., 1998). Microbleeds, also referred to as microhemorrhages, have long been considered clinically silent. More recently, their potential contribution to the development of cognitive dysfunction or as a risk factor of hemorrhagic stroke has been extensively investigated (Cordonnier et al., 2007).
Detecting such events in vivo can help to better understand the role of CAA in the development of dementia as well as in complications potentially related to Aβ immunization, a therapeutic approach that has been the subject of intense investigation [see Brody and Holtzman (2008) for a recent review]. In animal models, Aβ immunotherapy has been shown to reduce parenchymal amyloid, while it may increase cerebral microhemorrhages associated with amyloid-laden vessels (Pfeifer et al., 2002; Wilcock et al., 2007). A recent histological follow-up study of the first active Aβ immunotherapy in humans also suggested an increase in CAA and associated microhemorrhages (Boche et al., 2008). These results hint at a link between an increase in CAA and possible complications of Aβ immunization.
Following intravenous administration, superparamagnetic iron oxide (SPIO) particles are taken up by macrophages through absorptive endocytosis (Weissleder et al., 1990). Macrophages loaded with iron particles present in damaged areas can lead to local signal changes in magnetic resonance (MR) images, because of magnetic susceptibility effects of the iron. This property has been explored to follow with MR imaging (MRI) the infiltration of macrophages into inflamed areas in several disease models in rodents and in patients [for a recent review, see (Beckmann et al., 2009)].
In this work, we have used APP23 transgenic mice (Sturchler-Pierrat et al., 1997), which develop a considerable amount of vascular (CAA) and parenchymal (plaque) amyloid, to demonstrate the feasibility of detecting CAA-related microvascular lesions by MRI in combination with SPIO administration. The specificity of the method was further shown by comparison with three additional amyloid precursor protein (APP) transgenic lines differing in the relative amount of CAA and compact or diffuse parenchymal amyloid plaques. Moreover, we demonstrated the suitability of this imaging approach to detect and longitudinally follow vascular alterations induced by passive Aβ immunotherapy of APP23 mice.
Materials and Methods
Animals
All transgenic mice used expressed the transgene under control of a murine Thy-1 promoter element. The generation of APP23 [human APP with the K670N/M671L mutation (Sturchler-Pierrat et al., 1997)], APP24 [human APP with the K670N/M671L and V717L mutations (Abramowski et al., 2008)], APP51 [human wild-type APP (Bodendorf et al., 2002)], and PS45 [human presenilin-1 with the G384A mutation (Herzig et al., 2004)] mice has been described earlier. APP23xPS45 were obtained by crossbreeding the respective lines (Busche et al., 2008). All mice were hemizygous for the transgene(s) of interest and were bred in-house. Male and female APP23, male APP24, female APP51, male APP23xPS45, and age- and gender-matched wild-type mice have been used throughout the study as indicated in the figure legends.
Animals were housed under standard conditions (temperature 20–24°C, relative humidity minimum 40%, light/dark cycle 12 h) and fed a standard diet (Kliba Nr. 3893.025, Frantschach, Rothrist) and water supply ad libitum. Mice of different ages were measured by MRI. Age-matched nontransgenic littermate mice served as controls. Animal handling, care, and experimental use were in line with the Swiss federal law for animal protection. The study has been approved by the Veterinary Department of the City of Basel (animal licenses BS-1094 and -1283).
Passive immunization
Male mice were passively immunized weekly by intraperitoneal injections of 0.5 mg of β1 mouse monoclonal IgG2a antibody that recognizes amino acids 3–6 of human Aβ (Paganetti et al., 1996). Age-matched control mice received weekly intraperitoneal injections of 0.5 mg of a control antibody (monoclonal mouse anti-wheat auxin IgG2a antibody; AMS Biotechnology). Four groups of mice were formed: (1) APP23 mice immunized with the β1 antibody (n = 9 at the beginning of the immunization); (2) APP23 mice immunized with the control antibody (n = 10 at the beginning of the immunization); (3) wild-type mice immunized with the β1 antibody (n = 10 at the beginning of the immunization); (4) wild-type mice immunized with the control antibody (n = 10 at the beginning of the immunization). All animals were 18 months old at the beginning of the immunization.
MRI
For the MRI investigations, animals were anesthetized with 1.3% isoflurane (Abbott) in a mixture of oxygen/N2O (1:2) administered via a face mask. The body temperature of the mice was kept at 37°C. No stereotactic holding was used.
Measurements were performed with a Biospec 47/40 spectrometer (Bruker Medical Systems) operating at 4.7 T, equipped with an actively shielded gradient system. The operational software of the scanner was Paravision (Bruker). Images were obtained using a three-dimensional (3D) gradient-echo sequence with the following imaging parameters: repetition time 40 ms; echo time 8 ms; matrix 256 × 192 × 48; field-of-view 2.8 × 1.44 × 1.44 cm3, 2 averages. Total acquisition time for an image having a pixel size of 109 × 133 × 300 μm3 was of 12.3 min. The datasets were reconstructed to 2563.
MR image analysis
Analysis was performed by a blinded investigator. Sites in the cortex presenting signal attenuation with a minimum diameter of 150 μm were counted throughout the whole brain. To ensure that the same site was not counted multiple times, its presence was carefully controlled over several consecutive slices from the 3D dataset. In an initial phase, two investigators who were unaware of the genotype and of the age of the mice analyzed fifteen 3D datasets, with 10–16 sites of signal attenuation registered for each dataset. The agreement between the observers was 90%.
MRI contrast agent
Colloid-based SPIO (Endorem, 11.2 mg of Fe/ml; Guerbet) particles, so-called ferumoxides, were used. Ferumoxides represent Fe2O3 and Fe3O4 particles with a mean particle diameter of 150 nm, consisting of nonstoichiometric magnetic crystalline cores covered with a 3-nm-thick dextran T-10 layer (Jung, 1995). Animals received a bolus of Endorem (0.2 ml) intravenously 24 h before an imaging session. To exclude that the injection of SPIO itself could not have elicited vascular damage, 28- to 30-month-old APP23 mice (n = 6) were imaged by MRI before and 1 h after intravenous administration of physiological saline (0.2 ml) as a bolus.
Postmortem analyses
Mice (n = 40 male APP23, n = 7 female APP51, n = 6 male APP23xPS45, n = 18 male APP24) were overdosed with pentobarbital. The brains were removed from the skull and fixed for histology, or both hemispheres were separated. One brain hemisphere was then fixed for histology, while the forebrain from the second hemisphere (excluding olfactory bulb, brainstem and cerebellum) was used for assessments of Aβ levels. Analyses were performed blind to the genotype and treatment of the mice. Brains from n = 20 APP23 mice were used solely for histology, and from n = 13 APP23 and n = 13 APP24 animals for determination of Aβ levels only. Brains from the remaining animals were submitted to both analyses.
Histology.
Removed whole or half brains were fixed by immersion in 4% formaldehyde in 0.1 m PBS at 4°C for 1 week at minimum. After dehydration through increasing graded series of ethylic alcohol (50%, 70%, 80%, and 100%), brains were trimmed to get horizontal (coronal) sections and embedded in paraffin. Acquisition of 3D MRI datasets facilitated the detection of lesions, as these could be searched for through the whole brain. For the validation, the horizontal (coronal) coordinate of the slice showing a lesion in the MRI served as basis to localize the corresponding lesion by histology. Eight consecutive 5-μm-thick sections were cut with a microtome at the horizontal level provided by MRI. Sections were mounted onto Superfrost slides, dried at 37°C, deparaffinized in xylene, hydrated through graded ethylic alcohol, and washed in deionized water. Hematoxylin and eosin staining according to standard protocols was used to assess the general morphology. The Perls/Prussian blue reaction and the Congo red staining were used respectively to visualize ferric iron and to demonstrate amyloid. Grading of iron-containing microglia was done according to Pfeifer et al. (2002). Severity grades 1 and 2 (≤10 cells) were summarized as moderate, in contrast to grade 3 (>10 cells, abundant).
Immunohistochemistry was performed on paraffin sections according to previously published protocols (Calhoun et al., 1999) by using the avidin-biotin-peroxidase complex method (Vector Laboratories) with diaminobenzidine as chromogen. NT12 antiserum raised against Aβ40 (Paganetti et al., 1996) was diluted 1:2000 in PBS with 3% goat serum and incubated overnight at 4°C. After rinsing, sections were incubated for 1 h with biotinylated anti-rabbit IgG secondary antibody (BA1000, Vector Laboratories) diluted 1:200 in PBS. Following another rinsing, sections were further processed with the avidin-biotin-peroxidase technique (ABC-Elite Kit PK6100; Vector Laboratories). Finally, sections were reacted with metal-enhanced diaminobenzidine substrate (Code 1718096, Boehringer), counterstained with hemalum, dehydrated, cleared in xylene, and coverslipped.
Quantification of total Aβ in brain homogenates.
Forebrain hemispheres, excluding olfactory bulb, brainstem, and cerebellum, were weighed, homogenized by sonication in 9 volumes of Tris-buffered saline, 20 mm Tris-HCl, pH 7.6, 137 mm sodium chloride, and protease inhibitor cocktail Complete (Roche Molecular Biochemicals), and extracted with 70% formic acid. The extracts were neutralized by addition of 19 volumes of 1 m Tris-base and centrifuged for 15 min at 20,000 × g. The supernatants were analyzed by electrochemoluminescence-linked immunoassays (Human Aβ40 and Aβ42 Ultrasensitive Kits, Meso Scale Discovery) (Abramowski et al., 2008).
Data analysis
Mean values (±SEM) from n individual animals are presented. Mann–Whitney analyses were performed on the counts at the different time points with SYSTAT 11 (Systat Software). In addition, for the longitudinal assessment of the effects of immunization, the log10 of the counts were analyzed using an extension of ANOVA, called “Mixed model analysis” or “ANOVA with random effects” in SYSTAT 12. For multiple comparisons, a Bonferroni correction followed the ANOVA. Significance was assumed at the 5% probability level.
Results
Microvascular lesions in APP23 mice revealed by MRI
The feasibility of detecting CAA-related changes in mice was tested using MRI in the absence and in combination with iron oxide nanoparticles. Optimization of the method with APP23 mice, which develop significant CAA (Calhoun et al., 1999), led to a sensitive T2*-weighted gradient-echo imaging sequence used throughout the present study. Similar to previous experience with MRI detection of macrophage infiltration into inflamed peripheral tissues (Beckmann et al., 2009), an interval of 24 h between SPIO administration and image acquisition turned out to be well suited. Figure 1A shows coronal MR images extracted from a 3D dataset acquired from a 28-month-old mouse. Focal signal attenuations were observed throughout the cerebral cortex (indicated by arrows) as well as in thalamic regions. Instead, diffuse contrast changes over an extended region are detected in peripheral tissues with macrophage infiltration or in the brain as shown for stroke and multiple sclerosis [see Beckmann et al., 2009 for a review]. No signal change was detected in the brains of age-matched wild-type controls (Fig. 1A). Although rare, sites of attenuated signal could be detected in cortical areas of APP23 mice without contrast agent injection (Fig. 1B). After SPIO administration, these sites were more prominent and additional foci of signal attenuation became clearly visible as demonstrated in Figure 1B. For comparison, in 28-month-old APP23 mice, the mean number of sites showing MRI signal attenuation in the cortex was 0.5 ± 0.2 (n = 10 animals) and 10.1 ± 1.5 (n = 8 animals) without and with SPIO administration, respectively.
Figure 1.
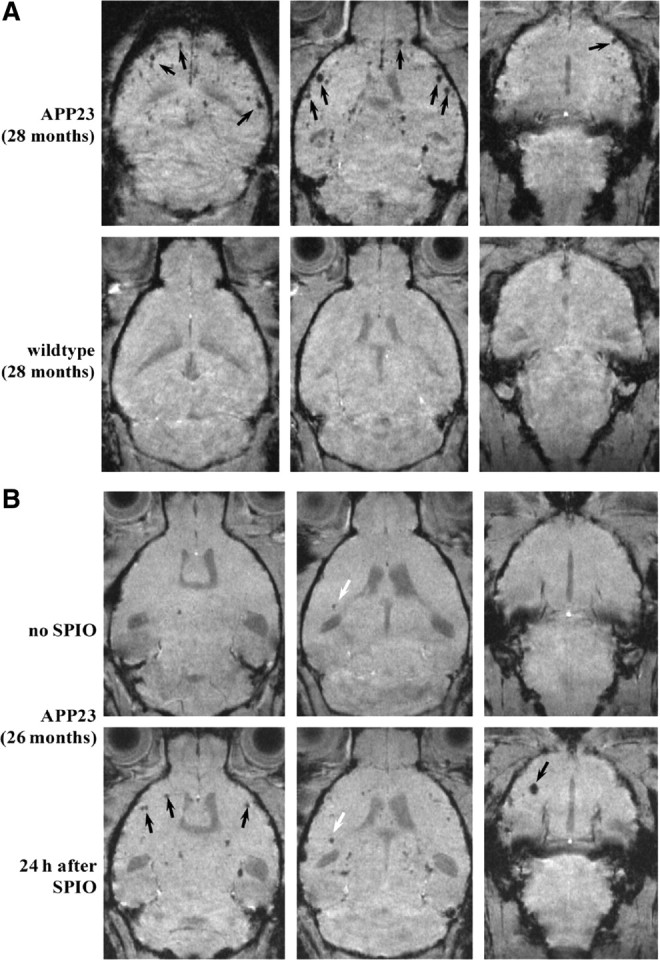
MRI from old male mice. A, Representative images extracted from 3D datasets acquired from 28-month-old mice 24 h after SPIO administration. Sites of signal attenuation (arrows) were detected throughout the cerebral cortex of the APP23, but not in the wild-type mouse. B, Images from a 26-month-old APP23 mouse, extracted from a 3D dataset acquired before (upper row) and 24 h after SPIO administration (lower row). The interval between both measurements was 2 days. Before SPIO, one site of signal attenuation was encountered in the cortex (white arrow). This site became more prominent following SPIO. In addition, many other foci of signal attenuation became apparent (black arrows).
Histology revealed iron-containing macrophages at the sites of MRI signal attenuation in all SPIO-injected APP23 mice. Figure 2 shows the histological analysis of regions in cerebral cortex matching three foci of attenuated MRI signal. At two sites, iron-containing macrophages were found in the wall of thickened vessels, which also contained amyloid deposits. One of the vessels (site β) in addition presented vasculitis with typical infiltration of lymphocytes. At the third site, isolated iron-loaded macrophages (severity grade 1) were present close to amyloid-laden vessels. Detailed histological analyses of 60 regions of MRI signal attenuation (five APP23 animals; 26–28 months of age) revealed clusters of iron-containing macrophages of all severity grades (grades 1, 2: moderate: ≤10 cells; grade 3: abundant: >10 cells), mostly in close vicinity to CAA-affected blood vessels. Grade 1 and 2 lesions had sizes up to 250 μm in the MR images, whereas grade 3 lesions appeared typically at sizes of 300–400 μm in the MRI. No blood vessel association was apparent for a fraction of the moderate sites (17 ± 11% of sites), but it may have been missed on the thin 5 μm sections. In contrast, CAA-affected blood vessels were present at the majority of the sites with moderate (65 ± 17%) and at all sites with abundant (18 ± 16%) iron-containing macrophages. The majority of these cells were located outside the blood vessels. In the walls of CAA-affected vessels, they were relatively rare (8 ± 1% of CAA vessels). Additional cerebrovascular alterations such as a thickened vessel wall and vasculitis were found in ∼30% of the sites of MRI signal attenuation. None of the sites showed an acute hemorrhage. Histological analyses of wild-type mice after SPIO administration did not identify any iron or other pathological alteration in the brain consistent with the absence of MRI signal foci. Together these data indicated that the MRI protocol detected a range of microvascular lesions from minor to more severe pathologies.
Figure 2.
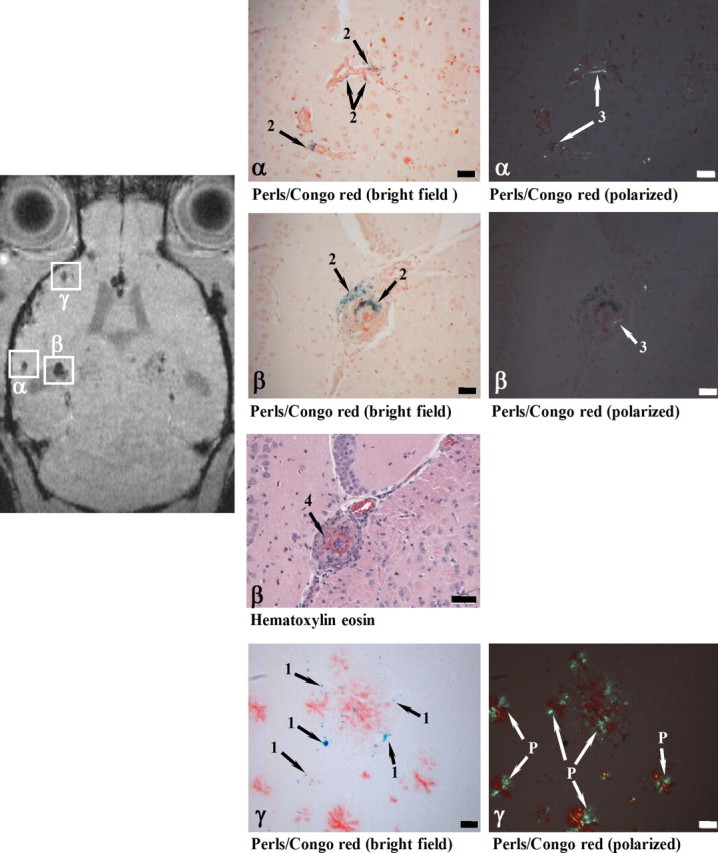
Histological examination of cerebral cortex sites with foci of attenuated MRI signal (α, β, γ). At 24 h following SPIO administration, a male 28-month-old APP23 mouse was analyzed in vivo by MRI and processed for histology immediately thereafter. Perls/Prussian blue staining showed iron-loaded macrophages in CAA-laden vessels (Congo red positive) at both sites (α and β). While the vessel walls were thickened at both α and β locations, only site β in addition showed vasculitis characterized by lymphocyte infiltration (Hematoxylin eosin). At site γ, isolated iron-loaded macrophages (severity grade 1) were present close to amyloid vessels. 1, Iron in isolated macrophages; 2, iron in macrophages at the vessel wall; 3, amyloid deposit in vessel wall; 4, vasculitis; P, amyloid plaque. Scale bars, 50 μm. Congo red-stained sections were observed under bright field or polarized light.
Brains of three APP23 animals (age 26–28 months) that had not received SPIO were also examined by histology. At sites of MRI signal attenuation, iron-containing macrophages (moderate number) associated with CAA-affected vessels were detected. In addition, a thickened vessel wall and vasculitis were detected at these sites. Iron-containing macrophages were only rarely found in the vessel wall (1 ± 0.8% of analyzed CAA vessels).
To investigate whether the injection of SPIO itself might elicit vascular damage, APP23 male mice (n = 6) 28–30 months of age were measured by MRI before and 1 h after intravenous injection of saline as a bolus. No additional sites of signal attenuation were observed following saline administration (data not shown). Moreover, a careful histological analysis of 28- to 30-month-old APP23 mice did not reveal increased signs of inflammation or CAA pathology in animals that received SPIO. These observations demonstrate that the injection of SPIO per se did not cause vascular damage.
Age-dependent increase in microvascular lesions in APP23 mice
The number of pathological blood vessels increases with age and amyloid deposition in APP23 transgenic mice (Winkler et al., 2001). Consistently, we found an age-dependent elevation of attenuated signal foci in the cerebral cortex of male APP23 mice that had received SPIO before MRI (Fig. 3). The number of sites displaying signal attenuation increased from 16 months of age onwards. In wild-type controls, only a very small increase in number of foci was observed after an age of 18 months. The number of sites displaying signal loss in the cortex of female transgenic mice developed in a similar way and was not significantly different from that in male APP23 mice.
Figure 3.
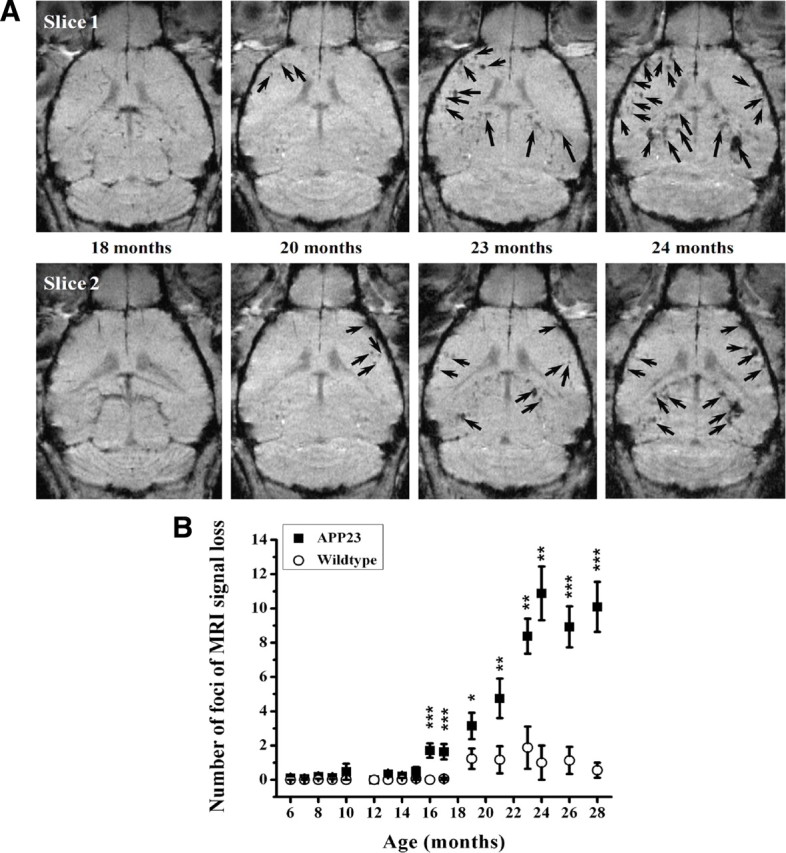
Age-related effects in male APP23 mice. A, Foci of attenuated signal are shown in two slices extracted from 3D MRI datasets acquired at different ages from a representative animal. SPIO was administered 24 h before each image acquisition. B, Age-dependent increase in the number of sites presenting signal attenuation in the brain cortex of male APP23 mice and age-matched littermate controls as determined from 3D MRI datasets. Data are given as mean ± SEM (n = 8–16 mice at each time point). The levels of significance *0.01 < p < 0.05, **0.001 < p < 0.01, and ***0.0001 < p < 0.001 refer to Mann–Whitney statistical tests performed between APP23 and wild-type mice at each specified age. The contrast agent, SPIO, was administered 24 h before each image acquisition.
We next examined the long-term stability of the signal attenuation following SPIO injection. Animals labeled with SPIO at 16 months completely lost the signal when reanalyzed 1 month later. In contrast, for APP23 mice older than 23 months, sites presenting signal attenuation were visible even 1 or 2 months after SPIO administration (Fig. 4). Therefore, we verified whether repeated administration of SPIO might have led to different results compared to a single dose. Similar numbers of foci were encountered in 28-month-old APP23 mice that had received one or multiple injections of SPIO [10.0 ± 2.7 foci (means ± SEM) for a single injection of SPIO to n = 5 mice; 10.2 ± 1.7 foci for seven administrations of SPIO to 6 mice, starting at an age of 19 months, with a minimum interval of 1 month between consecutive contrast agent administrations], demonstrating that any residual label did not affect the total number of sites detected after acute SPIO administration.
Figure 4.

Effect of repeated SPIO administration on number of sites presenting signal attenuation in male APP23 mice. The same animals were repeatedly measured by MRI at the specified ages, just before and 24 h following SPIO administration. For APP23 mice older than 21 months, some foci of signal loss were detected even 1 or 2 months after the last SPIO administration. Data are given as mean ± SEM. The significance levels p refer to Mann–Whitney comparisons.
MRI analyses of APP transgenic mouse lines differing in cerebral amyloid angiopathy
To further evaluate the specificity of the method for microvascular lesions, these changes were reduced by crossbreeding APP23 with mutated presenilin transgenic mice (APP23xPS45, analyzed at 15–17 months). We also used transgenic lines forming less CAA as they express human wild-type APP (APP51 mice, 28–30 months) or APP with the Swedish and London mutations (APP24 mice, 24 months). Histological analysis of the neocortex (Fig. 5A) confirmed that vascular Aβ was most pronounced in 24- to 28-month-old APP23 mice. Most parenchymal amyloid plaques were compact, but a considerable amount of diffuse deposits were present as well at this advanced age. In contrast, only scattered vascular amyloid was seen in APP23xPS45 mice, although they contained as much compact and more diffuse Aβ deposits at this younger age (Fig. 5A). Biochemically this histological change was associated with a shift of Aβ isoforms from Aβ40 to Aβ42 (Fig. 5B) as expected (Herzig et al., 2004; Busche et al., 2008). APP51 mice showed a similar distribution of the different forms of Aβ deposits as APP23, but the deposits were generally less abundant (Fig. 5A). This was reflected in the lower Aβ level but an unchanged Aβ42/40 ratio (Fig. 5B). For APP24 mice, Aβ deposits in the cerebral cortex were predominantly diffuse. To a lesser degree, compact amyloid plaques could also be found. The amount of vascular Aβ was lower than that in APP23 animals (Fig. 5A). In agreement, the Aβ level was generally lower than in APP23 mice and the shift from Aβ40 to Aβ42 was moderate compared to APP23xPS45 (Fig. 5B).
Figure 5.
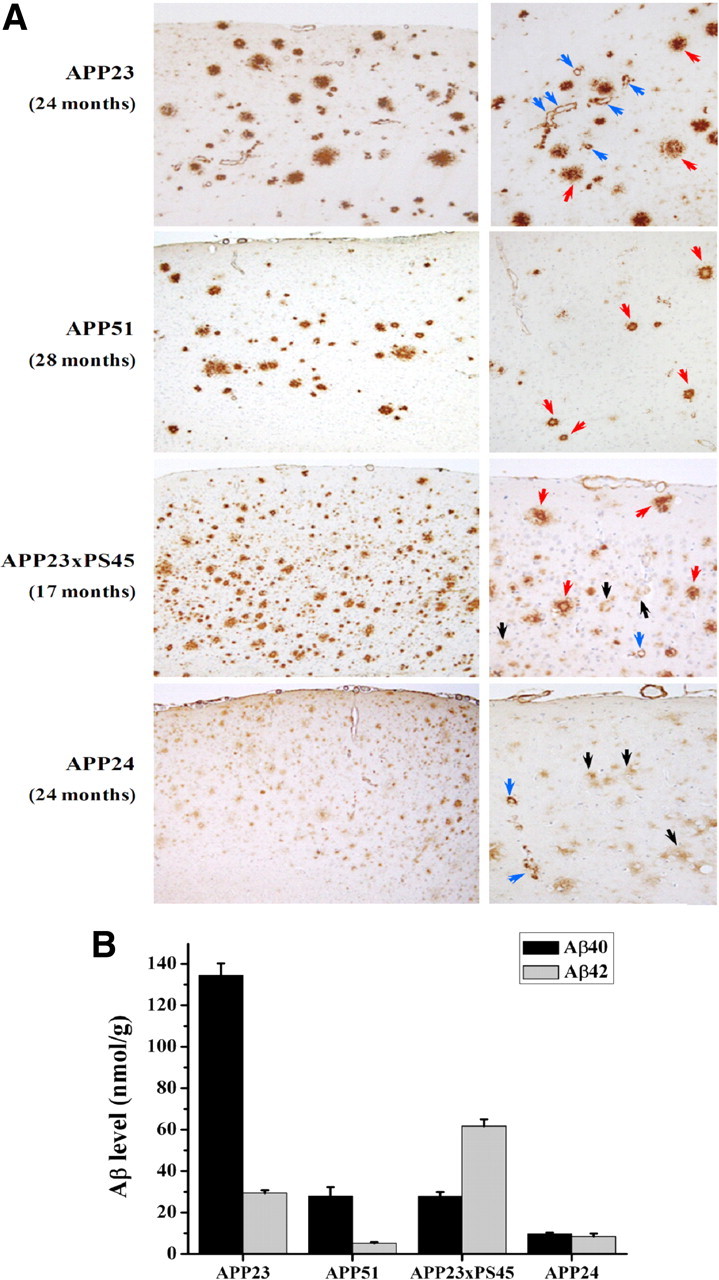
Characterization of various transgenic mouse lines differing by the relative amount of CAA and parenchymal amyloid plaques. A, Histological sections were stained with a rabbit polyclonal antibody to Aβ (NT12) indicating the presence of amyloid plaques (red arrows), diffuse plaques (black arrows), and vascular amyloid (blue arrows). Note the difference in CAA frequency as well as severity between the lines. B, Forebrain concentrations (means ± SEM) of Aβ40 and Aβ42 for the different APP transgenic mouse lines at the ages used: APP23 (24- to 28-month-old male mice, n = 20), APP51 (28-month-old female mice, n = 7), APP23xPS45 (13- to 17-month-old male mice, n = 6), and APP24 (26-month-old male mice, n = 18) animals.
In contrast to the MRI results obtained with APP23 animals, no significant increases in number of sites displaying MRI signal attenuation were found in the cortex or other brain regions of APP23xPS45, APP24, and APP51 transgenic mice (Fig. 6). A minor, nonsignificant increase may have occurred in APP51 mice at 30 months of age. These observations are consistent with the histological results presented above and summarized in Table 1, indicating a more pronounced presence of vascular Aβ in APP23 than in the other three lines.
Figure 6.

Comparison of the different APP transgenic mouse lines for foci of MRI signal attenuation in the cerebral cortex. Representative images from each line are presented with corresponding signal attenuation foci indicated by arrows. The number of sites is given as means ± SEM. Five to twelve animals were analyzed per group and time point. The level of significance p refers to Mann–Whitney statistical comparisons. Images were acquired 24 h after SPIO injection.
Table 1.
Comparison of the different forms of Aβ deposition in the cerebral cortex of the four transgenic lines (5–7 brains per line, 3 slices per brain) used in the present study
| Transgenic line/age | Compact amyloid plaques | Diffuse plaques | Vascular amyloid (CAA) |
|---|---|---|---|
| APP23 (24 months) | ++++ | ++ | +++ |
| APP51 (28–30 months) | +++ | ++ | ++ |
| APP23xPS45 (15–17 months) | ++++ | +++ | + |
| APP24 (24 months) | ++ | +++ | ++ |
The total number of amyloid plaques and vascular amyloid profiles per slice was estimated and classified with the following grading system: +, 5–10 deposits; ++, 11–50 deposits; +++, 51–100 deposits; ++++, more than 100 deposits. All animals were male except for the APP51 mice.
MRI detection of increased microvascular lesions following passive Aβ immunotherapy of APP23 mice
Treatment of plaque-bearing APP23 mice with Aβ antibody β1 was demonstrated to increase microhemorrhages (Pfeifer et al., 2002). We have used a similar passive immunization protocol to determine whether the MRI paradigm developed was able to detect these microvascular lesions in vivo. Starting at 18 months of age, APP23 mice received either 0.5 mg of β1 antibody (recognizing amino acids 3–6 of Aβ) or unrelated control antibody and were imaged at baseline and at different time points during the treatment. The number of foci presenting signal attenuation in the cerebral cortex increased during β1 immunization (Fig. 7). In contrast, there was only a small and nonsignificant trend toward an increase in the control antibody-treated animals. No sites of signal attenuation were detected in the brains of age-matched wild-type mice that received either the β1 or the control antibody. Histology performed at the end of the study showed a significant increase of blood vessels associated with CAA and of iron-containing macrophages in the cerebral cortex of β1 antibody-treated APP23 mice (Table 2). This confirms the described increase in CAA-related microvascular lesions in the cerebral cortex of APP23 mice following β1 antibody treatment. Our data demonstrate that MRI imaging is suited to detect such treatment-related alterations in mice. This includes minor lesions as these were primarily increased during treatment.
Figure 7.
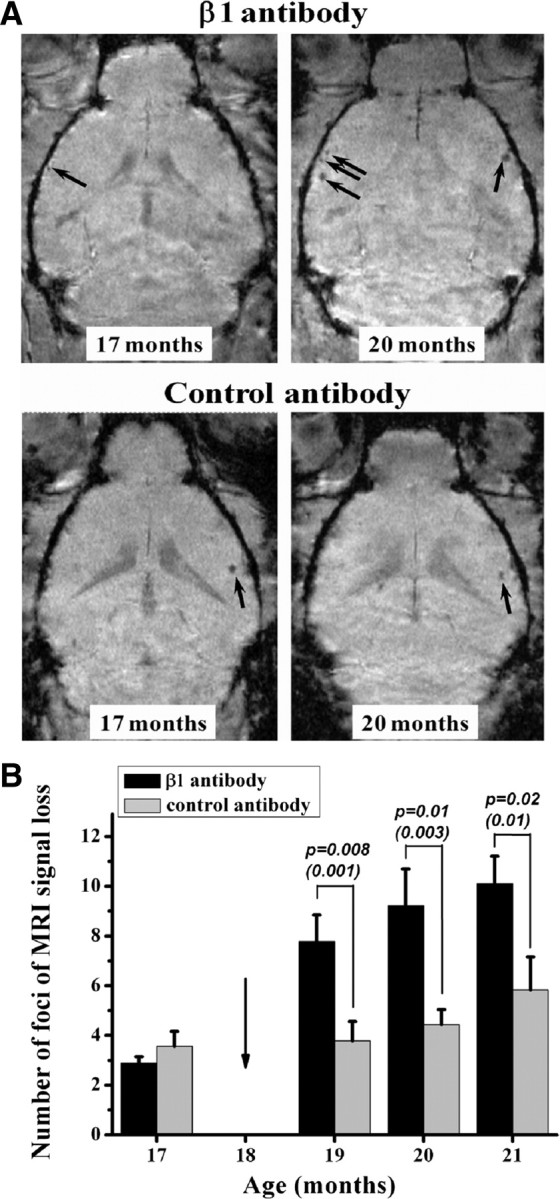
Immunization study with male APP23 mice. A, MR images from two animals at baseline (17 months of age) and at 2 months after beginning of immunization (20 months of age). Additional sites of signal attenuation were detected only in the brain cortex of the mouse receiving the β1 antibody (arrows). Both animals had received SPIO 24 h before being imaged. B, Number of signal attenuation foci (means ± SEM, n = 8–10 mice in each group and time point) in cerebral cortex, 24 h after SPIO administration to immunized mice. Treatment with the β1 antibody increased the number of foci with MRI signal attenuation. A baseline measurement was done at 17 months. The arrow indicates the beginning of the immunization, at 18 months of age. The significance levels p correspond to Mann–Whitney comparisons at the specified time points and, in parentheses, to the longitudinal comparisons of the log10 of the individual counts using ANOVA with random effects.
Table 2.
Summary of histological analysis performed on the cerebral cortex from 6 APP23 male mice treated with the β1 antibody and on 5 male APP23 mice that had received the control antibody
| Parameter | Control antibody | β1 antibody | Statistics |
|---|---|---|---|
| Blood vessels with CAA | 25.6 ± 3.8 | 33.9 ± 3.3 | p = 0.04 |
| Areas with iron containing macrophages (total) | 3.6 ± 1.2 | 15.2 ± 3.0 | p = 0.03 |
| By severity grade | |||
| Low: 1–3 macrophages | 0.9 ± 0.6 | 7.1 ± 1.6 | p = 0.006 |
| Moderate: 4–10 macrophages | 2.5 ± 0.9 | 6.7 ± 1.3 | p = 0.01 |
| Abundant: >10 macrophages | 0.3 ± 0.2 | 1.4 ± 0.5 | n.s. |
| Amyloid vessels with iron-containing macrophages | 2.6 ± 0.7 | 3.0 ± 0.5 | n.s. |
Numbers (means ± SEM) refer to counts of areas in the vicinity of CAA vessels presenting iron-containing macrophages, averaged over 11 sections per animal.
Discussion
In vivo detection of microvascular pathologies such as microhemorrhages and vasculitis in humans gains increasing importance as these lesions potentially contribute to cognitive dysfunction or indicate a risk of hemorrhagic stroke. Moreover, microhemorrhages may be elevated by Aβ immunotherapy, various approaches of which are currently developed for AD. In patients, focal hypointensities have been detected using T2*-weighted gradient-echo protocols (Fazekas et al., 1999; Tanaka et al., 1999; Imaizumi et al., 2004; Pettersen et al., 2008; Ayaz et al., 2010). While the signal attenuation is expected to result from the paramagnetic properties of the hemoglobin degradation product hemosiderin, the relationship to histologically detected microhemorrhages could not be studied satisfactorily in humans. In the present study, we have demonstrated that MRI can detect microvascular pathologies in a CAA-containing mouse model of AD and that an increase during Aβ immunotherapy can be followed in vivo.
T2*-weighted gradient-echo images acquired from the brain of aged APP23 mice without contrast agent showed only a small number of foci of signal loss in cortical and thalamic areas, which resulted from hemosiderin in macrophages as confirmed by Berlin blue histological staining. After administration of SPIO, 24 h before the measurements, the hypointense foci identified in precontrast images could be enhanced considerably and many more sites of signal loss became clearly apparent. Histology at the sites of signal loss demonstrated the presence of iron entrapped in macrophages in the vicinity of pathologic, CAA-affected blood vessels, or more rarely in the vessel wall. A fraction of the sites showed additional pathology such as thickened vessel walls and vasculitis. The same spectrum of lesions was found with or without SPIO treatment. Hence, sites of signal attenuation in the absence of labeling could pathologically not be distinguished from sites detected after SPIO labeling. This assertion is based on the analysis of histological slices obtained from the sites of attenuated signals as detected in vivo, rather than on a systematic slicing of whole brains. The number of cortical sites displaying signal attenuation increased with age. These observations suggest that administration of iron oxide nanoparticles improved the detection of microhemorrhages in the brains of APP23 mice and also led to the labeling of additional sites of microvascular alterations. Our findings match previous observations in the same transgenic line showing a very similar location of iron-containing macrophages (siderophages) at areas displaying cerebrovascular amyloid, additional vascular pathology at a subset of sites, and an age-related increase in these microhemorrhages (Calhoun et al., 1999; Winkler et al., 2001). Interestingly, at the sites analyzed in the present study, we did not detect any acute hemorrhages in agreement with their very rare occurrence in previous investigations (Calhoun et al., 1999; Winkler et al., 2001). The vast majority of the sites of signal attenuation, therefore, represent older lesions.
To further confirm that the sites of attenuated MRI signal in the APP23 brain reflected CAA-associated microvascular pathology, we introduced a strong presenilin mutation (APP23xPS45 mice), which shifted Aβ generation toward the 42 isoform, thus increasing parenchymal and reducing vascular amyloid (Herzig et al., 2004). Consistent with this change, MRI detected a much smaller number of attenuated signal sites than in APP23 mice. Very similar results were obtained with APP24 and APP51 mice, both of which develop significantly less CAA and microvascular pathology than APP23 mice. Overall, these observations strongly indicate that following administration of SPIO, MRI detects CAA-related pathology, and not changes reflecting deposition of parenchymal amyloid.
Different mechanisms may have contributed to the foci of signal attenuation detected by MRI after administration of contrast agent. (1) Since iron was found entrapped in macrophages, it is conceivable that SPIO nanoparticles were endocytosed in the blood circulation by macrophages, which then infiltrated the brain at sites of pathologic vessels. Although these sites coincided to a certain extent with the location of previous microhemorrhages, they also represented additional CAA-related microvasculopathy, characterized by the presence of macrophages (Vinters et al., 1998; Thal et al., 2008). Circulating macrophages have been shown to migrate from the lumen into the vessel wall (Vinters et al., 1998) and may be recruited into amyloid-containing AD or APP transgenic mouse brain (Fiala et al., 2002; Stalder et al., 2005; Simard et al., 2006). Macrophages are able to transmigrate the blood–brain barrier (BBB) when attracted by chemokines produced by amyloid β-stimulated cells (Fiala et al., 1998; El Khoury et al., 2007; Hickman and El Khoury, 2010). (2) A partial, local BBB breakdown could also have caused leakage of contrast agent, which would then be taken up by microglia in brain. However, studies in old APP23 mice with the extracellular contrast agent, gadolinium-tetraazacyclododecanetetraacetic acid (Gd-DOTA), administered intravenously did not show significant hyperintense signals, indicating that the agent did not leak through the BBB (data not shown). With a mean molecular diameter of 0.9 nm, Gd-DOTA is considerably smaller than the SPIO nanoparticles (mean diameter of 150 nm) used in this study (Corot et al., 2003). These results are in agreement with histological analyses performed by Winkler et al. (2001), demonstrating an absence of significant BBB disruption in old APP23 animals.
The present investigation also demonstrates that SPIO-enhanced MRI is well suited to detect the treatment-induced elevation of microhemorrhage in mice as observed in some studies of Aβ immunotherapy (Pfeifer et al., 2002; Wilcock et al., 2007). While chronically treated with the Aβ antibody β1, APP23 mice showed an increase in the number of sites with attenuated MRI signals, which could be followed with time. Lack of a change in control antibody-treated APP23 mice and β1-treated wild-type animals demonstrated the specificity of the measurements. Postmortem histological analysis confirmed a corresponding increase in the number of CAA-associated iron-containing macrophages in brain. These results are in agreement with previous studies of β1-treated aged APP23 mice demonstrating a twofold increase in the frequency and severity of microhemorrhages (Pfeifer et al., 2002; Burbach et al., 2007). Overall, our in vivo and postmortem observations show an increase in CAA-related microvascular pathology in the cerebral cortex of APP23 mice as a consequence of passive Aβ immunotherapy. They demonstrate the sensitivity of the MRI method used to detect this increase in living animals. As SPIO is approved for clinical use, the approach has translational value and can be used to screen or to monitor patients for microvascular lesions.
In conclusion, the present results demonstrate that MRI in combination with the administration of SPIO can be used noninvasively to study CAA-related microvascular lesions in transgenic mouse models of AD. The technique is complementary to MR angiography, which has detected flow disturbances at the level of the circle of Willis in old APP23 mice (Beckmann et al., 2003; Thal et al., 2009). Overall, these approaches visualize the loss of vascular integrity, which provides the basis for the age-related impairment of the cerebral blood volume response to pharmacological or electrical stimulation shown in brain functional MRI studies in APP23 mice (Mueggler et al., 2003). These observations support the idea that cerebral microcirculatory abnormalities developing progressively contribute to AD pathogenesis and cognitive impairment (Meyer et al., 2008). Although not well understood, certain forms of Aβ immunotherapy may have the potential to enhance such pathologies, which could be monitored noninvasively by an approach as described here.
References
- Abramowski D, Wiederhold KH, Furrer U, Jaton AL, Neuenschwander A, Runser MJ, Danner S, Reichwald J, Ammaturo D, Staab D, Stoeckli M, Rueeger H, Neumann U, Staufenbiel M. Dynamics of Aβ turnover and deposition in different APP transgenic mouse models following gamma-secretase inhibition. J Pharmacol Exp Ther. 2008;327:411–424. doi: 10.1124/jpet.108.140327. [DOI] [PubMed] [Google Scholar]
- Ayaz M, Boikov AS, Haacke EM, Kido DK, Kirsch WM. Imaging cerebral microbleeds using susceptibility weighted imaging: one step toward detecting vascular dementia. J Magn Reson Imaging. 2010;31:142–148. doi: 10.1002/jmri.22001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beckmann N, Schuler A, Mueggler T, Meyer EP, Wiederhold KH, Staufenbiel M, Krucker T. Age-dependent cerebrovascular abnormalities and blood flow disturbances in APP23 mice modeling Alzheimer's disease. J Neurosci. 2003;23:8453–8459. doi: 10.1523/JNEUROSCI.23-24-08453.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beckmann N, Cannet C, Babin AL, Blé FX, Zurbruegg S, Kneuer R, Dousset V. In vivo visualization of macrophage infiltration and activity in inflammation using MRI. Wiley Interdiscip Rev Nanomed Nanobiotechnol. 2009;1:272–298. doi: 10.1002/wnan.16. [DOI] [PubMed] [Google Scholar]
- Boche D, Zotova E, Weller RO, Love S, Neal JW, Pickering RM, Wilkinson D, Holmes C, Nicoll JA. Consequence of A{beta} immunization on the vasculature of human Alzheimer's disease brain. Brain. 2008;131:3299–3310. doi: 10.1093/brain/awn261. [DOI] [PubMed] [Google Scholar]
- Bodendorf U, Danner S, Fischer F, Stefani M, Sturchler-Pierrat C, Wiederhold KH, Staufenbiel M, Paganetti P. Expression of human beta-secretase in the mouse brain increases the steady-state level of beta-amyloid. J Neurochem. 2002;80:799–806. doi: 10.1046/j.0022-3042.2002.00770.x. [DOI] [PubMed] [Google Scholar]
- Brody DL, Holtzman DM. Active and passive immunotherapy for neurodegenerative disorders. Annu Rev Neurosci. 2008;31:175–193. doi: 10.1146/annurev.neuro.31.060407.125529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burbach GJ, Vlachos A, Ghebremedhin E, Del Turco D, Coomaraswamy J, Staufenbiel M, Jucker M, Deller T. Vessel ultrastructure in APP23 transgenic mice after passive anti-Abeta immunotherapy and subsequent intracerebral hemorrhage. Neurobiol Aging. 2007;28:202–212. doi: 10.1016/j.neurobiolaging.2005.12.003. [DOI] [PubMed] [Google Scholar]
- Busche MA, Eichhoff G, Adelsberger H, Abramowski D, Wiederhold KH, Haass C, Staufenbiel M, Konnerth A, Garaschuk O. Clusters of hyperactive neurons near amyloid plaques in a mouse model of Alzheimer's disease. Science. 2008;321:1686–1689. doi: 10.1126/science.1162844. [DOI] [PubMed] [Google Scholar]
- Calhoun ME, Burgermeister P, Phinney AL, Stalder M, Tolnay M, Wiederhold KH, Abramowski D, Sturchler-Pierrat C, Sommer B, Staufenbiel M, Jucker M. Neuronal overexpression of mutant amyloid precursor protein results in prominent deposition of cerebrovascular amyloid. Proc Natl Acad Sci U S A. 1999;96:14088–14093. doi: 10.1073/pnas.96.24.14088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cordonnier C, Al-Shahi Salman R, Wardlaw J. Spontaneous brain microbleeds: systematic review, subgroup analyses and standards for study design and reporting. Brain. 2007;130:1988–2003. doi: 10.1093/brain/awl387. [DOI] [PubMed] [Google Scholar]
- Corot C, Violas X, Robert P, Gagneur G, Port M. Comparison of different types of blood pool agents (P792, MS325, USPIO) in a rabbit MR angiography-like protocol. Invest Radiol. 2003;38:311–319. [PubMed] [Google Scholar]
- El Khoury J, Toft M, Hickman SE, Means TK, Terada K, Geula C, Luster AD. Ccr2 deficiency impairs microglial accumulation and accelerates progression of Alzheimer-like disease. Nat Med. 2007;13:432–438. doi: 10.1038/nm1555. [DOI] [PubMed] [Google Scholar]
- Fazekas F, Kleinert R, Roob G, Kleinert G, Kapeller P, Schmidt R, Hartung HP. Histopathologic analysis of foci of signal loss on gradient-echo T2*-weighted MR images in patients with spontaneous intracerebral hemorrhage: evidence of microangiopathy-related microbleeds. AJNR Am J Neuroradiol. 1999;20:637–642. [PMC free article] [PubMed] [Google Scholar]
- Fiala M, Zhang L, Gan X, Sherry B, Taub D, Graves MC, Hama S, Way D, Weinand M, Witte M, Lorton D, Kuo YM, Roher AE. Amyloid-beta induces chemokine secretion and monocyte migration across a human blood–brain barrier model. Mol Med. 1998;4:480–489. [PMC free article] [PubMed] [Google Scholar]
- Fiala M, Liu QN, Sayre J, Pop V, Brahmandam V, Graves MC, Vinters HV. Cyclooxygenase-2-positive macrophages infiltrate the Alzheimer's disease brain and damage the blood-brain barrier. Eur J Clin Invest. 2002;32:360–371. doi: 10.1046/j.1365-2362.2002.00994.x. [DOI] [PubMed] [Google Scholar]
- Greenberg SM, Gurol ME, Rosand J, Smith EE. Amyloid angiopathy-related vascular cognitive impairment. Stroke. 2004;35:2616–2619. doi: 10.1161/01.STR.0000143224.36527.44. [DOI] [PubMed] [Google Scholar]
- Herzig MC, Winkler DT, Burgermeister P, Pfeifer M, Kohler E, Schmidt SD, Danner S, Abramowski D, Stürchler-Pierrat C, Bürki K, van Duinen SG, Maat-Schieman ML, Staufenbiel M, Mathews PM, Jucker M. Abeta is targeted to the vasculature in a mouse model of hereditary cerebral hemorrhage with amyloidosis. Nat Neurosci. 2004;7:954–960. doi: 10.1038/nn1302. [DOI] [PubMed] [Google Scholar]
- Hickman SE, El Khoury J. Mechanisms of mononuclear phagocyte recruitment in Alzheimer's disease. CNS Neurol Disord Drug Targets. 2010;9:168–173. doi: 10.2174/187152710791011982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imaizumi T, Horita Y, Chiba M, Hashimoto Y, Honma T, Niwa J. Dot-like hemosiderin spots on gradient echo T2*-weighted magnetic resonance imaging are associated with past history of small vessel disease in patients with intracerebral hemorrhage. J Neuroimaging. 2004;14:251–257. doi: 10.1177/1051228404265714. [DOI] [PubMed] [Google Scholar]
- Jellinger KA. Alzheimer disease and cerebrovascular pathology: an update. J Neural Transm. 2002;109:813–836. doi: 10.1007/s007020200068. [DOI] [PubMed] [Google Scholar]
- Jung CW. Surface properties of superparamagnetic iron oxide MR contrast agents: ferumoxides, ferumoxtran, ferumoxsil. Magn Reson Imaging. 1995;13:675–691. doi: 10.1016/0730-725x(95)00023-a. [DOI] [PubMed] [Google Scholar]
- Kinnecom C, Lev MH, Wendell L, Smith EE, Rosand J, Frosch MP, Greenberg SM. Course of cerebral amyloid angiopathy-related inflammation. Neurology. 2007;68:1411–1416. doi: 10.1212/01.wnl.0000260066.98681.2e. [DOI] [PubMed] [Google Scholar]
- Maat-Schieman ML, van Duinen SG, Rozemuller AJ, Haan J, Roos RA. Association of vascular amyloid beta and cells of the mononuclear phagocyte system in hereditary cerebral hemorrhage with amyloidosis (Dutch) and Alzheimer disease. J Neuropathol Exp Neurol. 1997;56:273–284. doi: 10.1097/00005072-199703000-00006. [DOI] [PubMed] [Google Scholar]
- Meyer EP, Ulmann-Schuler A, Staufenbiel M, Krucker T. Altered morphology and 3D architecture of brain vasculature in a mouse model for Alzheimer's disease. Proc Natl Acad Sci U S A. 2008;105:3587–3592. doi: 10.1073/pnas.0709788105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mueggler T, Baumann D, Rausch M, Staufenbiel M, Rudin M. Age-dependent impairment of somatosensory response in the amyloid precursor protein 23 transgenic mouse model of Alzheimer's disease. J Neurosci. 2003;23:8231–8236. doi: 10.1523/JNEUROSCI.23-23-08231.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paganetti PA, Lis M, Klafki HW, Staufenbiel M. Amyloid precursor protein truncated at any of the gamma-secretase sites is not cleaved to beta-amyloid. J Neurosci Res. 1996;46:283–293. doi: 10.1002/(SICI)1097-4547(19961101)46:3<283::AID-JNR1>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]
- Pettersen JA, Sathiyamoorthy G, Gao FQ, Szilagyi G, Nadkarni NK, St George-Hyslop P, Rogaeva E, Black SE. Microbleed topography, leukoaraiosis, and cognition in probable Alzheimer disease from the Sunnybrook dementia study. Arch Neurol. 2008;65:790–795. doi: 10.1001/archneur.65.6.790. [DOI] [PubMed] [Google Scholar]
- Pfeifer M, Boncristiano S, Bondolfi L, Stalder A, Deller T, Staufenbiel M, Mathews PM, Jucker M. Cerebral hemorrhage after passive anti-Abeta immunotherapy. Science. 2002;298:1379. doi: 10.1126/science.1078259. [DOI] [PubMed] [Google Scholar]
- Simard AR, Soulet D, Gowing G, Julien JP, Rivest S. Bone marrow-derived microglia play a critical role in restricting senile plaque formation in Alzheimer's disease. Neuron. 2006;49:489–502. doi: 10.1016/j.neuron.2006.01.022. [DOI] [PubMed] [Google Scholar]
- Stalder AK, Ermini F, Bondolfi L, Krenger W, Burbach GJ, Deller T, Coomaraswamy J, Staufenbiel M, Landmann R, Jucker M. Invasion of hematopoietic cells into the brain of amyloid precursor protein transgenic mice. J Neurosci. 2005;25:11125–11132. doi: 10.1523/JNEUROSCI.2545-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sturchler-Pierrat C, Abramowski D, Duke M, Wiederhold KH, Mistl C, Rothacher S, Ledermann B, Bürki K, Frey P, Paganetti PA, Waridel C, Calhoun ME, Jucker M, Probst A, Staufenbiel M, Sommer B. Two amyloid precursor protein transgenic mouse models with Alzheimer disease-like pathology. Proc Natl Acad Sci U S A. 1997;94:13287–13292. doi: 10.1073/pnas.94.24.13287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka A, Ueno Y, Nakayama Y, Takano K, Takebayashi S. Small chronic hemorrhages and ischemic lesions in association with spontaneous intracerebral hematomas. Stroke. 1999;30:1637–1642. doi: 10.1161/01.str.30.8.1637. [DOI] [PubMed] [Google Scholar]
- Thal DR, Griffin WS, de Vos RA, Ghebremedhin E. Cerebral amyloid angiopathy and its relationship to Alzheimer's disease. Acta Neuropathol. 2008;115:599–609. doi: 10.1007/s00401-008-0366-2. [DOI] [PubMed] [Google Scholar]
- Thal DR, Capetillo-Zarate E, Larionov S, Staufenbiel M, Zurbruegg S, Beckmann N. Capillary cerebral amyloid angiopathy is associated with vessel occlusion and cerebral blood flow disturbances. Neurobiol Aging. 2009;30:1936–1948. doi: 10.1016/j.neurobiolaging.2008.01.017. [DOI] [PubMed] [Google Scholar]
- Vinters HV, Natté R, Maat-Schieman ML, van Duinen SG, Hegeman-Kleinn I, Welling-Graafland C, Haan J, Roos RA. Secondary microvascular degeneration in amyloid angiopathy of patients with hereditary cerebral hemorrhage with amyloidosis, Dutch type (HCHWA-D) Acta Neuropathol. 1998;95:235–244. doi: 10.1007/s004010050793. [DOI] [PubMed] [Google Scholar]
- Weissleder R, Elizondo G, Wittenberg J, Rabito CA, Bengele HH, Josephson L. Ultrasmall superparamagnetic iron oxide: characterization of a new class of contrast agents for MR imaging. Radiology. 1990;175:489–493. doi: 10.1148/radiology.175.2.2326474. [DOI] [PubMed] [Google Scholar]
- Wilcock DM, Jantzen PT, Li Q, Morgan D, Gordon MN. Amyloid-beta vaccination, but not nitro-nonsteroidal anti-inflammatory drug treatment, increases vascular amyloid and microhemorrhage while both reduce parenchymal amyloid. Neuroscience. 2007;144:950–960. doi: 10.1016/j.neuroscience.2006.10.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winkler DT, Bondolfi L, Herzig MC, Jann L, Calhoun ME, Wiederhold KH, Tolnay M, Staufenbiel M, Jucker M. Spontaneous hemorrhagic stroke in a mouse model of cerebral amyloid angiopathy. J Neurosci. 2001;21:1619–1627. doi: 10.1523/JNEUROSCI.21-05-01619.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]