

Abstract
The membrane protein Nogo-A, which is predominantly expressed by oligodendrocytes in the adult CNS and by neurons mainly during development, is well known for limiting neurite outgrowth and regeneration in the injured mammalian CNS. In addition, it has recently been proposed that abnormal Nogo-A expression or Nogo receptor (NgR) mutations may confer genetic risks for neuropsychiatric disorders of presumed neurodevelopmental origin, such as schizophrenia. We therefore evaluated whether Nogo-A deletion may lead to schizophrenia-like abnormalities in a mouse model of genetic Nogo-A deficiency. Here, we show that systemic, lifelong knock-out of the Nogo-A gene can lead to specific behavioral abnormalities resembling schizophrenia-related endophenotypes: deficient sensorimotor gating, disrupted latent inhibition, perseverative behavior, and increased sensitivity to the locomotor stimulating effects of amphetamine. These behavioral phenotypes were accompanied by altered monoaminergic transmitter levels in specific striatal and limbic structures, as well as changes in dopamine D2 receptor expression in the same brain regions. Nogo-A deletion was further associated with elevated expression of growth-related markers. In contrast, acute antibody-mediated Nogo-A neutralization in adult wild-type mice failed to produce such phenotypes, suggesting that the phenotypes observed in the knock-out mice might be of developmental origin, and that Nogo-A normally subserves critical functions in neurodevelopment. This study provides the first experimental demonstration that Nogo-A bears neuropsychiatric relevance, and alterations in its expression may be one etiological factor in schizophrenia and related disorders.
Introduction
Nogo-A is well known for its crucial role as a myelin-associated inhibitor of regenerative fiber growth and structural plasticity in the injured adult CNS (Schwab, 2004; Yiu and He, 2006). In the intact adult CNS, Nogo-A acts as a suppressor of growth and sprouting, thus stabilizing the wiring of the adult CNS (Buffo et al., 2000; Bareyre et al., 2002). Nogo-A is not only expressed in oligodendrocytes, but it is also present in subsets of neurons (Huber et al., 2002; Wang et al., 2002). Neuronal Nogo-A is especially highly expressed in the fetal and early postnatal brain and is downregulated in most anatomical structures in adulthood, except in some regions of high plasticity (e.g., hippocampus), in which neuronal Nogo-A expression remains high (Huber et al., 2002). Nogo-A might therefore assume critical control over neurodevelopmental processes as well as neural plasticity events. A recent report has emphasized Nogo's possible involvement in cortical development and neuronal maturation (Mingorance-Le Meur et al., 2007). Although controversial, human postmortem and genetic linkage studies have implicated Nogo-A and its chromosomal location in several neuropsychiatric disorders with a presumed neurodevelopmental origin, namely schizophrenia and bipolar disorder (Coon et al., 1998; Shaw et al., 1998; Novak et al., 2002; Tan et al., 2005). Moreover, Nogo receptor 1 (RTN4R, NgR1), a putative receptor subunit for Nogo-A, is encoded by a gene located on chromosome 22q11, which is a hotspot for genetic predisposition in schizophrenia (Liu et al., 2002). Recently, several NgR1 mutations have also been identified in schizophrenic patients (Sinibaldi et al., 2004; Hsu et al., 2007; Budel et al., 2008).
Schizophrenia is a severe and chronic neuropsychiatric disorder with a presumed etiology in abnormal neurodevelopment attributable to combinatorial effects of environmental and genetic risk factors (Harrison, 1997; Lewis and Levitt, 2002; Rapoport et al., 2005). Indeed, various genetic linkage and association studies have identified genomic regions that may harbor schizophrenia susceptibility genes (Harrison and Weinberger, 2005; Ross et al., 2006). Mouse deletion models of such genes are indispensable in characterizing their function at the system level and their potential contribution to disease pathophysiology in terms of the scope of cellular pathways, neural circuits, and behavior (Arguello and Gogos, 2006; Chen et al., 2006). Here, a mouse model of constitutive genetic Nogo-A deficiency was evaluated in a comprehensive series of behavioral tests with specific relevance to schizophrenia pathopsychology. Following the demonstration of multiple schizophrenia-like behavioral abnormalities in Nogo-A-deficient mice, we further characterized the concomitant alterations in brain neurotransmitters and specific markers by neurochemical and immunohistochemical analyses, focusing in particular on brain regions implicated in the pathophysiology of schizophrenia. Finally, we investigated whether the brain and behavioral aberrations identified in Nogo-A-deficient mice could also be induced by antibody-mediated Nogo-A suppression in adult wild-type mice, as the first test of whether lifelong Nogo-A deficiency since early life was necessary for the emergence of the Nogo-A knock-out phenotypes.
Materials and Methods
Animals
Subjects were all 2- to 4-month-old male mice on a C57BL/6J genetic background. They comprised Nogo-A knock-out (Nogo-A−/−) and wild-type (Nogo-A+/+) littermates. Full details of the generation of the Nogo-A−/− mouse line have been described before (Simonen et al., 2003; Dimou et al., 2006). Nogo-A was absent in the knock-outs, as expected (Fig. S1, available at www.jneurosci.org as supplemental material) (see also Simonen et al., 2003; Dimou et al., 2006). The animals were housed on a reversed 12/12 h light/dark cycle (lights on at 8:00 P.M.) under temperature-controlled (21°C) and humidity-controlled (55%) conditions and maintained under ad libitum food and water. Animals derived from multiple litters were used for each experiment. All animals were experimentally naive at the time of testing. Testing was performed during the dark phase of the light/dark cycle. All procedures described in the present study had been approved by the Cantonal Veterinary Office in Zurich and are in agreement with the Principles of Laboratory Animal Care (National Institutes of Health publication no. 86-23, revised 1985).
Behavioral analysis
Elevated plus maze.
The expression of spontaneous anxiety-like behavior was assessed in the elevated plus maze test. Two maze designs were used, differing in whether the maze walls were opaque or transparent. Such variation in the transparency of the closed arms has been suggested to be more congenial for the detection of anxiolytic or anxiogenic traits, respectively (Hagenbuch et al., 2006). The two mazes were identical in dimensions, consisting of two open and two enclosed arms, radiating from a central platform. Their construction has been fully described before (Willi et al., 2009). The test began by placing the animal on the central platform with its head facing one of the open arms. The mouse was then left to freely explore the maze for 5 min. A digital camera was mounted above the maze. Images were captured at a rate of 5 Hz and transmitted to a personal computer running the Ethovision tracking system (Noldus Technology). The following two indices of anxiety-related behavior were computed: the percentage open-arm entries [open-arm entries/(open-arm entries + closed-arm entries) × 100%], and the percentage time spent in the open arms [time in open arms/(time in open arms + time in closed arms) × 100%]. In addition, total distance traveled in the entire maze (in centimeters) was taken as a measure of activity.
Open-field exploration.
Spatial exploration was evaluated in four identical white open-field arenas (40 × 40 cm) as described previously (Meyer et al., 2006). Animals were allowed to freely explore the open field for 1 h. For the purpose of data collection, the arenas were conceptually partitioned into two zones: a center zone in the middle of the arena (measuring 13.5 × 13.5 cm) and a peripheral zone occupying the remaining area. Data collection was performed using the Ethovision tracking system. The dependent measures were as follows: distance traveled in the entire open-field arena and number of entries into the central area of the open field, expressed as a function of 5 min bins.
Prepulse inhibition.
Prepulse inhibition (PPI) refers to the reduction in startle reaction to a startle-eliciting pulse stimulus when it is shortly preceded by a subthreshold prepulse stimulus (Hoffman and Searle, 1965). A test of PPI was conducted using four acoustic startle chambers (SR-LAB, San Diego Instruments), in which the startle reflex was triggered by a 40 ms 120 dBA white noise pulse stimulus. The test apparatus and procedures have been fully described before (Meyer et al., 2005). Inhibition of the pulse-elicited startle reflex was achieved by using 20 ms white noise prepulse stimuli of various intensities (69, 73, 77, 81, and 85 dBA) that preceded the pulse stimulus by 100 ms. PPI was indexed by the percentage inhibition of the startle response at each level of prepulse intensity by the following formula: % PPI = [1 − (mean reactivity on prepulse-plus-pulse trials/mean reactivity on pulse-alone trials)] × 100%.
Latent inhibition.
Latent inhibition (LI) refers to the retardation in learning about the significance of a stimulus as a result of its prior repeated pre-exposures without consequence (Lubow and Moore, 1959). LI was assessed in a conditioned freezing paradigm using two sets of four chambers as previously described (Meyer et al., 2005). The test procedures consisted of four phases: pre-exposure, conditioning, context test, and tone test. Animals were randomly allocated to either the pre-exposed or non-pre-exposed condition. Pre-exposed animals received 40 presentations of a 30 s tone stimulus at a variable interstimulus interval of 40 ± 30 s, while non-pre-exposed animals were confined to the chamber for an equivalent time. Conditioning followed immediately at the end of pre-exposure without removing the animals from the chambers. Conditioning contained three discrete trials of tone–shock pairings. Each trial began with the 30 s tone stimulus followed immediately by the delivery of a 1 s footshock (0.25 mA). Each trial was preceded and followed by a 180 s interval. The context test took place 24 h later, when the subjects were returned to the same chambers for a period of 480 s in the absence of any discrete stimulus. The tone test was conducted another 24 h later in the same test chamber to assess the conditioned response to the tone. Following a 180 s acclimatization period, the tone was continuously presented for 180 s, and freezing (immobility) in the presence of the tone stimulus was evaluated. Freezing was expressed as the percentage time freezing for all test sessions.
Active avoidance learning.
The active avoidance learning task captures elements of both classical and instrumental conditioning, in which the animals learned to perform a specific operant act in response to a tone stimulus to avoid the delivery of an aversive footshock. The apparatus consisted of four identical two-way shuttle boxes as fully described before (Yee et al., 2006). Electric shocks (0.3 mA) could be delivered through the grid floor. The conditioned stimulus (CS) was an 86 dBA tone. Animals were placed in the shuttle chambers and received a total of 100 conditioned avoidance trials presented with an intertrial interval of 40 ± 15 s. A trial began with the onset of the tone. If the animal shuttled within 5 s of tone onset, the tone was terminated and the animal avoided the electric shock on that trial. Avoidance failure led immediately to an electric footshock presented in coincidence to the tone. This lasted for a maximum of 2 s but could be terminated earlier by a shuttle response during this period (i.e., an escape response). To index conditioned avoidance learning, the mean number of avoidance shuttles performed across successive 20-trial blocks was calculated. To account for general locomotor activity, the number of spontaneous shuttles performed during the intertrial intervals was also analyzed.
Discrimination reversal learning.
Discrimination reversal learning was examined using a water T-maze. The apparatus and procedures have been fully described before (Meyer et al., 2006). In acquisition training, the animals were required to learn to discriminate the left and right goal arms, with only one of them leading to an escape platform hidden just beneath the water surface at the far end. There were six trials per daily session, conducted at an intertrial interval of 10 min. Acquisition training continued until an animal had reached the criterion performance of 11 correct responses across 2 consecutive days. To assess reversal learning, the platform location was then moved to the other, previously incorrect, arm from the following day onwards. Reversal training continued until an animal had achieved criterion performance again. The percentage of correct arm choices per day was recorded for each animal during acquisition and reversal training.
Social interaction and social novelty.
Social behavior was assessed using methods adapted from previous studies (Smith et al., 2007; Moy et al., 2009). The testing apparatus comprised a rectangular, three-chambered Plexiglas box (61 × 40.5 cm). The chambers were separated by dividing walls, which had openings (6 × 4 cm) that allowed the animals to move between them. The doorways could be blocked by removable partitions. Cylindrical wire grid cages (10 cm height, 11 cm bottom diameter, bars horizontally and vertically spaced 9 mm apart) carrying a weighted cup on top of the cage to prevent the test mice from climbing and remaining on top of the wire cage were placed in each of the two side chambers. The test procedures consisted of three 10 min phases: habituation, social interaction, and social novelty. For habituation, each mouse was placed into the center compartment and allowed to freely explore the entire box. After this initial habituation period, the animal was enclosed in the center compartment, and an unfamiliar, adult C57BL/6J male mouse (stranger 1) was placed in one of the wire cages, whereas a dummy mouse was put into the other one. The doors were then reopened, and the animal was allowed to explore the three chambers. The location of the stranger and the dummy mouse alternated between the left and the right chambers across subjects. At the end of this social interaction test, the preference of each mouse for social novelty was assessed. The test animal was enclosed in the center chamber, and the dummy mouse was replaced by a novel unfamiliar mouse (stranger 2). The test animal was then again allowed to explore the entire box. For all test sessions, the time spent in each chamber and the time spent sniffing each cage (i.e., nose detection within a circular area of 10 mm in diameter surrounding the cage) were recorded using the Ethovision XT tracking system.
Amphetamine-induced locomotor activity.
The test for amphetamine-induced hyperlocomotor activity was conducted in the open-field apparatus described above. Animals were first allowed to explore the arena for 30 min, were then injected with vehicle solution (isotonic 0.9% NaCl solution, intraperitoneally, injection volume of 5 ml/kg), and were immediately returned to the same arena for 30 min. Subsequently, the animals were administered with d-amphetamine sulfate (2.5 mg/kg, intraperitoneally, injection volume of 5 ml/kg) and the locomotor response to the acute drug challenge was observed over a period of 120 min. Data collection was performed using the Ethovision tracking system. The distance traveled in the entire open-field arena was recorded and expressed as a function of 5 min bins.
High-performance liquid chromatography
Postmortem neurochemical analyses using high-performance liquid chromatography (HPLC) were performed as previously described (Enard et al., 2009; Winter et al., 2009). The levels of monoamines dopamine (DA) and serotonin (5-HT) and their metabolites dihydroxyphenylacetic acid (DOPAC), homovanillic acid (HVA), and 5-hydroxyindoleacetic acid (5-HIAA) in various brain areas were measured by HPLC with electrochemical detection. Glutamate, GABA, glutamine, and taurine levels in the same brain regions were determined by HPLC with fluorescence detection.
Immunohistochemistry
Tissue preparation.
Animals were deeply anesthetized with pentobarbital (Nembutal, 40 mg/kg body weight, intraperitoneally; Abbott Laboratories) and transcardially perfused with PBS (pH 7.4, room temperature), followed by ice-cold fixative [4% paraformaldehyde and 15% saturated picric acid in 0.15 m phosphate buffer (PB), pH 7.4] as described previously (Fritschy and Mohler, 1995). Brains were removed immediately after perfusion, postfixed in the same fixative for 4 h at 4°C, and immersed in 10%, 20%, and then 30% sucrose diluted in PBS for cryoprotection before freezing. Coronal sections (40 μm) were cut with a cryostat-microtome (model MGW Lauda 1720, Leitz) and collected in PBS. Sections were then transferred into antifreeze solution (15% sucrose and 30% ethylene glycol in 50 mm PB, pH 7.4) and stored at −20°C until used. Before immunostaining, sections for some antigen stainings were then processed with modified antigen retrieval protocols to optimize the signal-to-noise ratio (Table S1, available at www.jneurosci.org as supplemental material).
Immunostaining.
Free-floating, bregma-matched sections from Nogo-A−/− and Nogo-A+/+ mice were coded and batch processed under identical conditions to minimize staining variability. Sections were incubated overnight at 4°C with primary antibodies (Table S1, available at www.jneurosci.org as supplemental material) diluted in PBS containing 4% normal goat serum and 0.05% Triton X-100. For GAP-43, S100, and GFAP stainings, sections were then rinsed with 0.1 m PB and probed with fluorescence-conjugated secondary antibodies coupled to Cy2 or Cy3 (Jackson ImmunoResearch Laboratories). For DA D2 receptor (D2R) staining, primary antibodies were detected with biotinylated antibody, washed with PB, and probed with streptavidin Cy3 (Jackson ImmunoResearch Laboratories). For cytoarchitectonic reference, some sections were counterstained with Hoechst or NeuroTrace 640/660 deep-red fluorescent Nissl stain (Invitrogen). Sections were mounted on gelatin-coated slides, air dried, and finally coverslipped with Mowiol (Calbiochem).
Densitometry.
Microscopic analyses were conducted under strictly blind conditions. Images were acquired using a cooled CCD camera (CoolSnap HQ, Photometrics) attached to a Zeiss Axiophot microscope (Zeiss) and collected using computer-assisted image analysis software (MCID, Elite Software version 7.0, Imaging Research). For each animal, the marker immunoreactivity (IR) in a given region was obtained from bilateral densitometric measurements per section and evaluated using the software ImageJ (NIH, Bethesda, MD). The background-corrected optical densities were averaged per brain region and animal. The mean relative optical densities of the wild-type animals were set as 1.0 for each marker and brain region. For confirmation of the observed findings in some markers at higher magnification and their representative illustration, images were also acquired with a spectral confocal microscope (TCS SP2 AOBS, Leica Microsystems); only identical minimal contrast adjustments were applied.
D2R IR was evaluated in the medial prefrontal cortex (including layers II/III and V/VI of cingulate cortices 1/2), caudate–putamen, and nucleus accumbens core and shell. Digital images were acquired using a 20× objective (NA 0.5) and pixel brightness was measured in each brain area of interest by placing three or four rectangular boxes of 160 × 200 μm in the respective areas in both hemispheres. IRs for GAP-43, S100, and GFAP were investigated in the medial prefrontal cortex, caudate–putamen, nucleus accumbens core and shell, CA1 (including all layers), CA3 (including all layers), and dentate gyrus (including granular and molecular layers). Digital images were acquired using a 10× objective (NA 0.3) and pixel brightness was measured in each brain area of interest by placing three or four rectangular boxes of 325 × 400 μm in the respective areas in both hemispheres.
Analysis after acute Nogo-A neutralization in adult mice
Experimental groups. Two cohorts of adult Nogo-A+/+ mice treated with either anti-Nogo-A or control antibodies were used to assess whether acute blockade of Nogo-A function could induce changes similar to those observed after lifelong absence of Nogo-A function in the Nogo-A−/− mice. One cohort of mice was tested in the amphetamine-induced locomotor activity task. The second cohort was examined for D2R and GAP-43 expression. As antibody availability was limited, we focused on these particular analyses only. Behavioral and immunohistochemical analyses were conducted after 2 weeks of antibody treatment.
Pump implantation and application of antibodies.
Mice were deeply anesthetized with Hypnorm (30 μl/20 g of body weight; 0.315 mg of fentanyl citrate and 10 mg of fluanisone per ml; VetaPharma) and Dormicum (60 μl/20 g of body weight; 5 mg of Midazolamum per ml; Roche Pharmaceuticals). A hole was drilled into the dorsal lamina of the second lumbar vertebra, through which a fine catheter (32 G; Recathco) was inserted into the intrathecal space and pushed up to T10. The catheter was connected to the outlet of an osmotic minipump (Alzet Model 1002, Charles River Laboratories), which was placed under the skin on the back and filled with 600 μg of monoclonal anti-Nogo-A antibody (11C7) (for details, see the study by Weinmann et al., 2006) or control monoclonal IgG antibody (directed against the plant protein wheat auxin) (see also Weinmann et al., 2006) in PBS, to deliver a constant amount of antibodies into the CSF for at least up to 2 weeks. The anti-Nogo-A antibody 11C7 is monospecific for Nogo-A on Western blots and does not cross-react with other Nogo splice variants (Dodd et al., 2005). Pumps and antibody delivery to spinal cord and brain were verified at the end of the experiments. Infused antibodies were detected by immunoperoxidase staining and the use of anti-IgG antibodies (see also Weinmann et al., 2006).
Statistical analysis
Behavioral data were subjected to parametric ANOVA of the appropriate design. Data interpretation was further assisted by Fisher's least significant difference post hoc comparisons and the use of restricted ANOVAs whenever appropriate. HPLC and immunohistochemical data were analyzed using independent Student's t tests (two tailed). All statistical analyses were conducted using the statistical software SPSS for Windows (release 14.0).
Results
Schizophrenia-like behavioral phenotype in Nogo-A knock-out mice
In 2- to 4-month-old adult mice, we first examined the effect of genetic Nogo-A deletion on behavioral functions that are considered to be relevant to schizophrenia endophenotypes or overt psychopathology.
Anxiety-related behavior and spontaneous locomotor activity were examined in the elevated plus maze and the open-field exploration tests. For the interpretation of the data derived from the aversive conditioning paradigms, it is essential to ascertain whether the Nogo-A deletion had any effects on unconditioned anxiety-like behavior and spontaneous locomotor activity. Additionally, heightened anxiety has been found to be associated with schizophrenia (Lewis and Lieberman, 2000). In the two designs of the elevated plus maze test used here, no differences in anxiety-related behavior were detected between genotypes, as measured by the percentage of open-arm entries or the percentage of time spent in the open arms. Nogo-A−/− (n = 9/maze) and Nogo-A+/+ (n = 10/maze) mice displayed a mean percentage of open-arm entries of 32.5 ± 3.2% versus 28.0 ± 4.1% (opaque maze) and 46.5 ± 3.2% versus 43.5 ± 2.7% (transparent maze), respectively. The mean percentage of time spent in the open arms was 14.0 ± 3.2% versus 9.7 ± 1.9% (opaque maze) and 29.5 ± 5.6% versus 32.5 ± 4.9% (transparent maze), respectively. Examination of locomotion as measured by total distance traveled in the maze further indicated that the groups behaved similarly to each other (opaque maze, 1097.0 ± 69.7 cm versus 1151.7 ± 38.7 cm; transparent maze, 1108.9 ± 56.1 cm versus 1074.2 ± 37.4 cm). Spatial exploration in the open field was analyzed by focusing on the number of entries the animals ventured into the central area of the arena. Nogo-A−/− (n = 10) and Nogo-A+/+ (n = 14) mice showed similar values on this measure (11.4 ± 1.3 versus 8.9 ± 1.1 entries per 5 min bin, respectively). Overall locomotor activity as indicated by the distance traveled in the entire open field also did not differ between the groups (1341.8 ± 79.8 cm versus 1232.0 ± 67.0 cm per 5 min bin, respectively).
Sensorimotor gating was assessed by testing PPI. PPI deficiency has been linked to several neuropsychiatric disorders with a presumed neurodevelopmental origin, including schizophrenia and autism (Braff et al., 2001; Perry et al., 2007). As expected, the level of prepulse inhibition increased with increasing prepulse intensity (prepulse intensity: F(4,88) = 27.75, p < 0.001). Nogo-A−/− mice displayed clearly reduced PPI (percentage inhibition) independent of prepulse intensity compared with Nogo-A+/+ mice (genotype: F(1,22) = 5.24, p < 0.05) (Fig. 1A). Consistent with previous findings (Willi et al., 2009), neither startle reactivity responses recorded on pulse-alone (Fig. 1B) nor prepulse-alone trials (data not shown) were significantly altered in Nogo-A−/− mice, thus confirming that Nogo-A deletion did not result in any general hearing or startle reflex impairment.
Figure 1.

Decreased prepulse inhibition in Nogo-A−/− mice. A, PPI is indexed by the percentage PPI at each prepulse intensity. The histogram on the right illustrates the overall means across all prepulse intensities. Magnitude of the percentage PPI increased with increasing prepulse intensity. Nogo-A−/− mice displayed reduced PPI compared with Nogo-A+/+ mice (*p < 0.05). B, Startle reactivity to pulse-alone trials in arbitrary units. There was no significant difference between the genotypes. All values are mean ± SEM. KO, Nogo-A−/− (n = 12); WT, Nogo-A+/+ (n = 12).
Latent inhibition is a form of selective associative learning reported to be impaired in schizophrenic patients (Baruch et al., 1988; Gray et al., 1992). In the conditioned freezing paradigm used to assess LI here, the development of conditioned freezing was evaluated across the three tone–footshock pairings (Fig. 2A). LI was evident by elevated freezing levels in the non-pre-exposed animals relative to the pre-exposed animals (pre-exposure: F(1,44) = 21.48, p < 0.001). Although the LI effect appeared weaker in Nogo-A−/− relative to Nogo-A+/+ mice, the interaction between pre-exposure and genotype did not attain statistical significance (F(1,44) = 2.20, p = 0.145). The expression of conditioned freezing to the context was evaluated 24 h after conditioning (Fig. 2B). Contextual freezing was generally low in all groups and was not affected by the Nogo-A deletion. In the test of the conditioned response to the tone 48 h after conditioning in the same chambers (Fig. 2C), the presence of LI in Nogo-A+/+ mice was demonstrated by a clear reduction in the amount of freezing to the tone in the pre-exposed animals relative to the non-pre-exposed ones. In contrast, LI was substantially attenuated in Nogo-A−/− mice. This impression was supported by the presence of the significant interaction between pre-exposure and genotype (F(1,44) = 5.99, p < 0.05). Subsequent post hoc comparisons confirmed that a significant presence of LI was only statistically evident in Nogo-A+/+ mice (p < 0.001). It is apparent that the disruption of the LI effect seen in the Nogo-A−/− mice was mainly attributed to increased tone-freezing in the pre-exposed condition, compared with Nogo-A+/+ mice. Additional analyses restricted to either pre-exposure condition revealed that Nogo-A−/− mice displayed significantly higher freezing levels than Nogo-A+/+ mice in the pre-exposed condition (genotype: F(1,22) = 6.31, p < 0.05), whereas both non-pre-exposed groups were comparable. In the 3 min before tone onset, freezing was generally low, with mutant and control mice not differing in their overall level of freezing regardless of pre-exposure condition.
Figure 2.
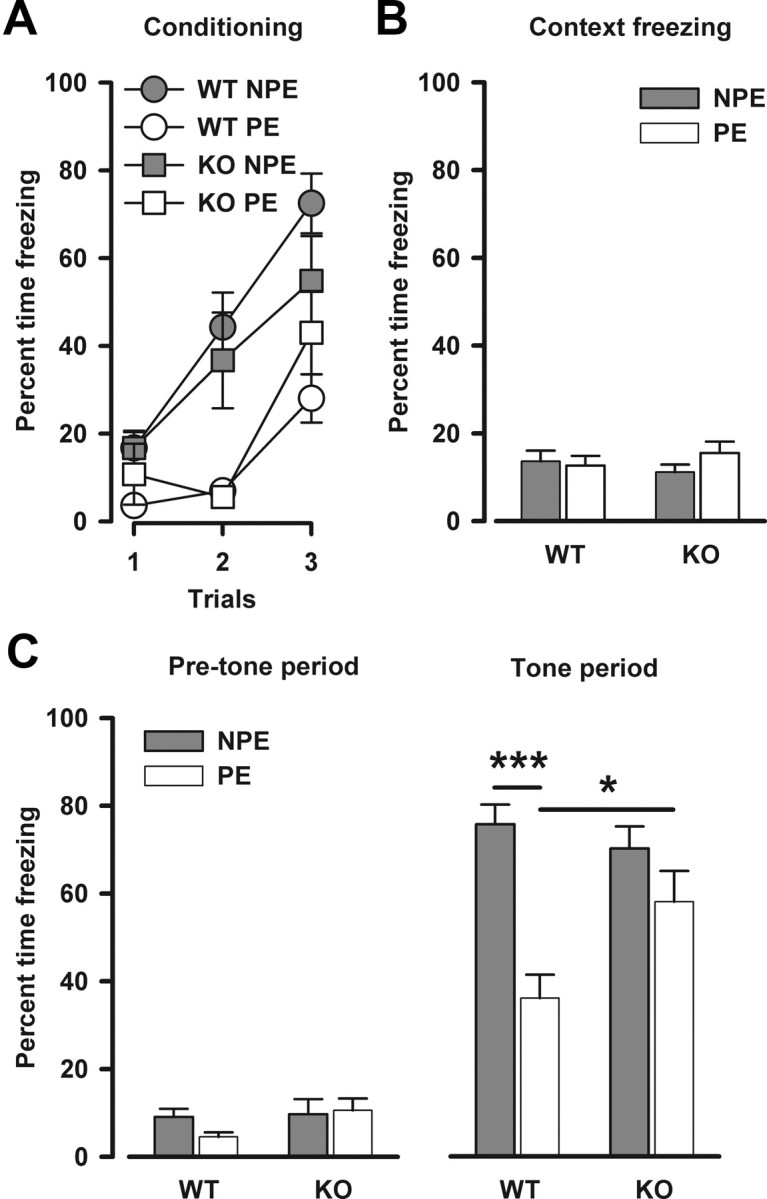
Impaired latent inhibition in Nogo-A−/− mice. A, Expression of freezing behavior toward the tone CS across the three conditioning trials immediately after pre-exposure in the conditioned freezing paradigm. Freezing is expressed as the percentage time freezing per trial. Freezing in the pre-exposed (PE) subjects was lower than in the non-pre-exposed (NPE) subjects, constituting the LI effect. B, Freezing to the context 24 h after conditioning. This is expressed as the overall mean percentage time freezing. There was no effect of genotype or pre-exposure. C, Freezing to the tone CS 48 h after conditioning. Freezing is expressed as the mean percentage time freezing before tone onset on the left and as the mean percentage time freezing across the tone period on the right. In the 3 min before the onset of the tone, there was no difference between any of the groups for the levels of freezing. Reduced freezing levels to the tone in the PE group compared with the NPE group in Nogo-A+/+ animals indicates the presence of LI (***p < 0.001 based on a priori contrast). In contrast, Nogo-A−/− mice failed to show significant LI. This difference between the genotypes was supported by the presence of a significant two-way interaction between pre-exposure and genotype (p < 0.05). Moreover, there was a genotype effect in the PE condition (*p < 0.05), whereas both NPE groups were comparable. All values are mean ± SEM. KO, Nogo-A−/− (n = 9 PE, 9 NPE); WT, Nogo-A+/+ (n = 15 PE, 15 NPE).
To further verify that the disrupted LI seen in Nogo-A−/− mice is unlikely to be due to a general impairment in the acquisition of a conditioned response, another associative learning paradigm (i.e., active avoidance) was conducted. Acquisition of the conditioned avoidance response was evident in both groups as indicated by the increase in the number of avoidance responses over successive 20-trial blocks (blocks: F(4,88) = 83.50, p < 0.001). Consistent with the outcome of the conditioned freezing experiment, no significant group difference was detected (Fig. S2A, available at www.jneurosci.org as supplemental material). In agreement with the results obtained in the elevated plus maze and open-field tests, spontaneous locomotor activity (as measured by the number of spontaneous shuttles performed during the intertrial interval periods) did not differ between genotypes (Fig. S2B, available at www.jneurosci.org as supplemental material).
Behavioral flexibility was then evaluated in a discrimination reversal learning task using a water T-maze. A learning deficit specific to the reversal phase, in the absence of an initial acquisition deficit, is indicative of perseverative behavior, as implicated in schizophrenia and autism (Ridley, 1994). Performance during the initial acquisition phase did not differ between genotypes, indicating that there is no general learning deficit after Nogo-A deletion (Fig. 3). In the reversal phase, Nogo-A−/− mice exhibited behavioral perseveration as evidenced by impaired choice accuracy compared with control mice (genotype × days: F(7,210) = 2.56, p < 0.05). As shown in Figure 3, the initial impact of reversal was comparable between the groups, but its influence on choice behavior had a higher impact on Nogo-A−/− animals, as they were more slowly adapting to the reversed contingency.
Figure 3.

Emergence of perseverative behavior in Nogo-A−/− mice. Discrimination reversal learning in the water T-maze. Performance is expressed as the percentage correct responses per day. Acquisition of the left–right discrimination learning was similar between the genotypes. A reversal effect was evident in both groups as suggested by the below-chance performance (represented by the dashed line) on the first day of the subsequent reversal training. However, Nogo-A−/− mice displayed a clear deficit in reversal learning as evidenced by a significant genotype × day interaction (p < 0.05). All values are mean ± SEM. **p < 0.01, ***p < 0.001 based on post hoc pairwise comparisons. KO, Nogo-A−/− (n = 16); WT, Nogo-A+/+ (n = 16).
Next, social behavior was examined. Social withdrawal is a characteristic that can be found in both schizophrenia and autism (American Psychiatric Association, 2000). In the social interaction assay, the animals generally spent more time in the chamber containing the stranger mouse (stranger 1) versus the chamber containing the dummy mouse (chambers: F(1,20) = 20.70, p < 0.001), and this effect was not dependent on the genotype (Fig. 4A). In the social novelty preference assay, a significant preference for spending time in the chamber with the novel stranger (stranger 2) versus the chamber with the familiar stranger (stranger 1) was observed (chambers: F(1,20) = 10.84, p < 0.01). Again, this effect did not depend on the genotype (Fig. 4B). Analysis of time spent sniffing each cage during both test phases (data not shown) yielded consistent results compared with the data on the time spent in each respective chamber, supporting the view that social behavior is similar in Nogo-A−/− and Nogo-A+/+ mice.
Figure 4.

Social interaction and social novelty preference levels are similar in Nogo-A−/− and Nogo-A+/+ mice. A, Social interaction is indexed by the time spent in the compartment containing a stranger mouse (stranger 1) versus the time spent in the compartment containing a dummy mouse. There was a general preference for the stranger mouse. No significant difference between the genotypes was detected. B, Time spent in the compartment containing a novel stranger mouse (stranger 2) versus the first stranger mouse (stranger 1) during the social novelty task. There was a general preference for the novel stranger mouse. No significant difference between the genotypes was detected. All values are mean ± SEM. KO, Nogo-A−/− (n = 11); WT, Nogo-A+/+ (n = 11).
To clarify whether Nogo-A−/− mice possess a psychotic-like phenotypic profile, we tested the locomotor stimulant effect of the indirect DA-receptor agonist amphetamine – a drug known to exacerbate and induce psychotic symptoms in humans (Laruelle et al., 1996). In a novel open field, locomotor activity for the baseline period and after vehicle (saline) injection was similar between genotypes (Fig. 5). Systemic administration of amphetamine resulted in markedly increased locomotor activity in both Nogo-A−/− and Nogo-A+/+ mice (bins: F(23,368) = 33.37, p < 0.001). However, the drug-induced hyperactivity was significantly more pronounced in Nogo-A−/− animals, particularly in the first half of the 120 min test session (genotype: F(1,16) = 5.71, p < 0.05, genotype × bins: F(23,368) = 2.09, p < 0.01) (Fig. 5).
Figure 5.

Increased sensitivity to the motor stimulant effects of systemic amphetamine in Nogo-A−/− mice. Locomotor activity in the open field is expressed as distance traveled per 5 min bin. The inset on the right illustrates the overall means across the amphetamine period. Locomotor activity for the baseline period and after vehicle (SAL, saline) injection was similar between both groups. Systemic administration of amphetamine (AMPH) resulted in a general increase in locomotor activity. The locomotor effect to the drug was potentiated in Nogo-A−/− mice compared with Nogo-A+/+ mice (*p < 0.05). All values are mean ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001 in the line plot based on post hoc pairwise comparisons. KO, Nogo-A−/− (n = 9); WT, Nogo-A+/+ (n = 9).
Alterations in brain dopamine and serotonin in Nogo-A knock-out mice
To investigate putative changes after Nogo-A deletion at the neurotransmitter level, specific anatomical regions were microdissected from fresh frozen brain slices and analyzed by HPLC. Levels of DA and its metabolites DOPAC and HVA were significantly reduced (DA: −62%, p < 0.001; DOPAC: −55%, p < 0.01; HVA: −55%, p < 0.01) in the dorsal striatum (i.e., caudate–putamen) of Nogo-A−/− mice. In the medial prefrontal cortex, the ventral striatum (i.e., nucleus accumbens), and the hippocampus, basal contents of DA and its metabolites were not affected by Nogo-A deletion (Table 1).
Table 1.
Alterations in brain monoamine levels and turnover in Nogo-A−/− mice
| DA | DOPAC | HVA | DOPAC/DA | HVA/DA | 5-HT | 5-HIAA | 5-HIAA/5-HT | |
|---|---|---|---|---|---|---|---|---|
| mPFC | ||||||||
| WT | 9.26 ± 1.58 | 1.01 ± 0.43 | 3.65 ± 1.53 | 0.11 ± 0.05 | 0.31 ± 0.12 | 73.95 ± 9.26 | 41.52 ± 6.77 | 0.59 ± 0.10 |
| KO | 6.50 ± 0.70 | 1.50 ± 0.29 | 4.69 ± 0.77 | 0.25 ± 0.05* | 0.75 ± 0.12* | 64.43 ± 6.28 | 56.48 ± 14.61 | 0.85 ± 0.18 |
| CPu | ||||||||
| WT | 1001.63 ± 124.89 | 63.29 ± 8.52 | 87.57 ± 13.93 | 0.06 ± 0.00 | 0.09 ± 0.00 | 64.89 ± 9.18 | 63.80 ± 9.57 | 1.03 ± 0.13 |
| KO | 378.72 ± 38.04*** | 28.76 ± 3.88** | 39.77 ± 4.81** | 0.08 ± 0.01 | 0.11 ± 0.02 | 16.55 ± 1.76*** | 6.29 ± 2.34*** | 0.42 ± 0.14** |
| NAc | ||||||||
| WT | 373.93 ± 50.33 | 45.58 ± 7.55 | 46.46 ± 7.55 | 0.12 ± 0.00 | 0.12 ± 0.01 | 174.83 ± 19.65 | 94.56 ± 9.53 | 0.60 ± 0.10 |
| KO | 322.99 ± 26.33 | 48.92 ± 5.56 | 47.22 ± 4.80 | 0.16 ± 0.02 | 0.15 ± 0.02 | 129.63 ± 9.40 | 64.80 ± 9.04 | 0.50 ± 0.11 |
| HPC | ||||||||
| WT | 6.43 ± 0.66 | ND | 2.55 ± 0.93 | ND | 0.41 ± 0.16 | 90.78 ± 7.94 | 102.09 ± 18.44 | 1.18 ± 0.23 |
| KO | 5.93 ± 0.69 | ND | 3.05 ± 1.11 | ND | 0.62 ± 0.31 | 66.15 ± 6.77* | 84.52 ± 15.12 | 1.90 ± 0.85 |
Levels are expressed as mean concentrations in nmol/mg protein. All values are mean ± SEM.
*p < 0.05, **p < 0.01, ***p < 0.001. mPFC, Medial prefrontal cortex; CPu, caudate-putamen; NAc, nucleus accumbens; HPC, hippocampus; ND, not detectable; KO, Nogo-A−/− (n = 6–9); WT, Nogo-A+/+ (n = 8–10).
Contents of 5-HT and its metabolite 5-HIAA were significantly reduced in the dorsal striatum (5-HT: −75%, p < 0.001; 5-HIAA: −90%, p < 0.001) of Nogo-A−/− mice. In addition, Nogo-A−/− mice displayed lower 5-HT levels in the hippocampus (−27%, p < 0.05). In contrast, Nogo-A deletion did not affect basal 5-HT and 5-HIAA contents in the medial prefrontal cortex (Table 1).
To further assess the impact of Nogo-A deletion on monoamine turnover in the adult brain, the ratios of DOPAC/DA, HVA/DA, and 5-HT/5-HIAA were analyzed (Table 1). DA turnover was significantly increased in the medial prefrontal cortex of Nogo-A−/− mice (DOPAC/DA: +136%, p < 0.05; HVA/DA: +144%, p < 0.05). 5-HT turnover was significantly reduced in the dorsal striatum (−60%, p < 0.01) of Nogo-A−/− mice. No other changes in DA or 5-HT turnover were detected.
Glutamate and GABA levels were not significantly altered in Nogo-A−/− mice in any of the brain regions examined. Similarly, contents of glutamine and the inhibitory amino sulfonic acid taurine were not altered in the brain of Nogo-A−/− mice (Table 2).
Table 2.
Brain glutamate, glutamine, taurine, and GABA levels in Nogo-A−/− and Nogo-A+/+ mice
| GLU | GLN | TAU | GABA | |
|---|---|---|---|---|
| mPFC | ||||
| WT | 108.46 ± 5.28 | 40.73 ± 7.20 | 87.06 ± 3.77 | 15.55 ± 1.03 |
| KO | 101.05 ± 1.62 | 31.86 ± 4.31 | 84.34 ± 4.26 | 14.05 ± 0.36 |
| CPu | ||||
| WT | 84.84 ± 6.24 | 45.76 ± 7.81 | 113.05 ± 6.42 | 15.07 ± 1.23 |
| KO | 76.10 ± 2.58 | 35.53 ± 3.62 | 103.61 ± 5.63 | 13.75 ± 0.50 |
| NAc | ||||
| WT | 74.26 ± 6.06 | 53.11 ± 6.96 | 70.97 ± 6.33 | 54.48 ± 1.89 |
| KO | 65.58 ± 2.14 | 42.74 ± 5.36 | 66.44 ± 3.37 | 45.40 ± 3.64 |
| HPC | ||||
| WT | 87.68 ± 5.22 | 41.58 ± 6.64 | 83.52 ± 4.52 | 23.90 ± 1.37 |
| KO | 81.68 ± 3.32 | 34.13 ± 4.36 | 77.44 ± 4.41 | 21.04 ± 0.98 |
Levels are expressed as mean concentrations in nmol/mg protein. All values are mean ± SEM. GLU, Glutamate; GLN, glutamine; TAU, taurine; mPFC, medial prefrontal cortex; CPu, caudate-putamen; NAc, nucleus accumbens; HPC, hippocampus; KO, Nogo-A−/− (n = 8–10); WT, Nogo-A+/+ (n = 8–10).
Changes of dopamine D2 receptor expression in Nogo-A knock-out brain
Next, we evaluated whether the observed alterations in DA levels and turnover in the striatum and the medial prefrontal cortex of Nogo-A−/− mice were paralleled by changes in the expression of the D2R, a receptor suggested to play an important role in the pathophysiology of schizophrenia, as most of the commonly used neuroleptics block this receptor (Seeman, 1987). Nogo-A deletion decreased D2R-IR in the medial prefrontal cortex (−34%, p < 0.05), whereas it led to an elevation of D2R-IR in the dorsal striatum (i.e., caudate–putamen; +27%, p < 0.01). In the ventral striatum (i.e., nucleus accumbens), D2R-IR was similar between Nogo-A−/− and Nogo-A+/+ mice (Fig. 6).
Figure 6.
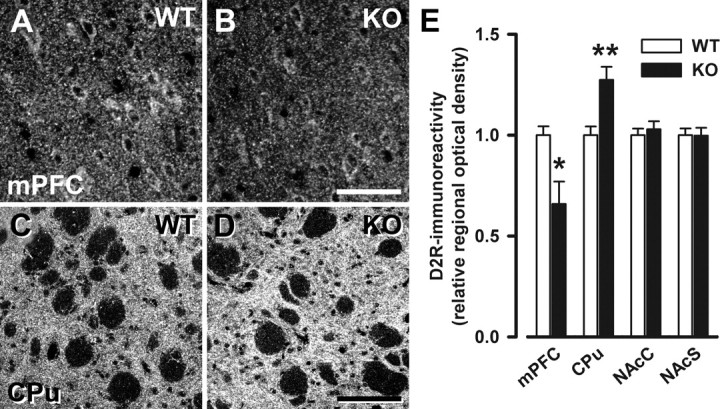
Selective changes in D2R immunoreactivity in Nogo-A−/− mice. A–D, Representative images of immunostaining with D2R in the medial prefrontal cortex (A, B) and the dorsal striatum (C, D) of Nogo-A+/+ and Nogo-A−/− mice. E, Quantitative analysis of the D2R staining. The relative optical density of D2R was reduced in the medial prefrontal cortex (mPFC; *p < 0.05) of Nogo-A−/− mice, whereas it was elevated in the dorsal striatum (CPu; **p < 0.01). No changes were observed in the nucleus accumbens core (NAcC) and shell (NAcS) regions. Note the differences in staining intensity between the striatum and the medial prefrontal cortex with the typically high levels in the striatum. All values are mean ± SEM. Scale bars: A, B, 50 μm; C, D, 100 μm. KO, Nogo-A−/− (n = 6–7); WT, Nogo-A+/+ (n = 6–7).
Increased expression of GAP-43 and S100B in Nogo-A knock-out brain
To seek further concomitant structural modifications in Nogo-A−/− brain, we examined putative changes in growth-related markers. We focused on the growth-associated protein GAP-43 and S100B, because elevations of both markers have been repeatedly reported in brains of schizophrenic patients (Perrone-Bizzozero et al., 1996; Blennow et al., 1999; Rothermundt et al., 2004). Moreover, GAP-43 expression was also found to be transiently upregulated after acute antibody-mediated Nogo-A neutralization in the intact adult rat CNS (Bareyre et al., 2002), and S100B is thought to exert some of its effects by inhibition of phosphorylation of synaptic and growth proteins such as GAP-43 (Sheu et al., 1994). IR for GAP-43 was significantly elevated in the medial prefrontal cortex (+24%, p < 0.05), the dentate gyrus (+31%, p < 0.05), and the hippocampal CA1 region (+10%, p < 0.05) of Nogo-A−/− mice, but remained unaltered in the hippocampal CA3 region and both dorsal and ventral striatum (Fig. 7A,B,E). Similar to GAP-43, S100-IR was elevated in the medial prefrontal cortex (+31%, p < 0.01) and the dentate gyrus (+22%, p < 0.05) of Nogo-A−/− mice, whereas its levels were comparable in the hippocampal subfields CA1 and CA3, and both dorsal and ventral striatum between Nogo-A−/− and Nogo-A+/+ mice (Fig. 7C,D,F).
Figure 7.

Increased immunoreactivity of GAP-43 and S100 in Nogo-A−/− mice. A–D, Representative images of immunostaining with GAP-43 (A, B) and S100 (C, D) in the dentate gyrus of Nogo-A+/+ and Nogo-A−/− mice. E, F, Quantitative analysis of the respective stainings. The relative optical density of GAP-43 was enhanced in the medial prefrontal cortex (mPFC; *p < 0.05), the dentate gyrus (DG; *p < 0.05), and the CA1 region (*p < 0.05) of Nogo-A−/− mice, whereas it was not altered in the CA3 region, the dorsal striatum (CPu), and the nucleus accumbens core (NAcC) and shell (NAcS) regions (E). Similarly, S100 immunoreactivity was elevated in the mPFC (**p < 0.01) and DG (*p < 0.05) of Nogo-A−/− mice, but it was not altered in the other brain areas investigated (F). All values are mean ± SEM. Scale bar, 200 μm. KO, Nogo-A−/− (n = 6–8); WT, Nogo-A+/+ (n = 5–8).
Increased S100-IR identified in specific brain regions in Nogo-A−/− mice was not associated with higher levels of the astrocyte marker GFAP, thus suggesting that Nogo-A deletion did not induce reactive gliosis (Fig. 8).
Figure 8.

No indication of reactive gliosis in Nogo-A−/− mice. A, B, Representative images of immunostaining with the astrocyte marker GFAP in the dentate gyrus of Nogo-A+/+ and Nogo-A−/− mice. C, Quantitative analysis of the GFAP staining. No difference between Nogo-A−/− and Nogo-A+/+ mice was detected in the relative optical density of GFAP. All values are mean ± SEM. Scale bar, 200 μm. DG, Dentate gyrus; mPFC, medial prefrontal cortex; CPu, dorsal striatum; NAcC, nucleus accumbens core; NAcS, nucleus accumbens shell; KO, Nogo-A−/− (n = 4–6); WT, Nogo-A+/+ (n = 4–6).
Absence of abnormal phenotype in mice treated with anti-Nogo-A antibodies in adulthood
Finally, we performed some of the major analyses described above in anti-Nogo-A versus control antibody-treated adult Nogo-A+/+ mice. Lack of effects after such acute blockade of Nogo-A function would support the hypothesis that neurodevelopmental aberrations due to Nogo-A deficiency in early life underlie the Nogo-A knock-out phenotypes, as would be anticipated based on the neurodevelopmental hypothesis of schizophrenia (Weinberger, 1987; Harrison, 1997; Lewis and Levitt, 2002; Rapoport et al., 2005). Two weeks of continuous intrathecal delivery of the function-blocking monoclonal mouse antibody 11C7 led to high levels of antibody throughout the brain and spinal cord (Fig. 9A–D), very similar to earlier studies in rats and macaque monkeys (Weinmann et al., 2006). Following amphetamine treatment, control and anti-Nogo-A antibody-treated groups displayed a similar hyperlocomotor response (Fig. 9E). Histologically, IRs for D2R were highly comparable in all regions of interest between the two groups (Fig. 9F–H). In contrast, expression of GAP-43 was found to be slightly increased after anti-Nogo-A antibody treatment in the dentate gyrus (+12%, p < 0.05), but it was not altered in hippocampal subfields CA1/CA3, the medial prefrontal cortex, and both dorsal and ventral striatum (Fig. S3, available at www.jneurosci.org as supplemental material).
Figure 9.

Absence of schizophrenia-like abnormalities in adult mice acutely treated with anti-Nogo-A antibodies for 2 weeks. A–C, Comparative regional antibody distribution in cervical spinal cord after intrathecal lumbar delivery of anti-Nogo-A or control IgG antibody via osmotic minipumps. Color coding of sections processed for immunoperoxidase staining ranges from black/dark blue, for background, to violet, red, orange, yellow, and white, for maximal staining intensity. Note the clear difference in antibody levels between the treatment groups. D, Representative image of antibody detection in the cortex of an anti-Nogo-A antibody-treated animal. Stained cell bodies reflect uptake of Nogo-A–anti-Nogo-A antibody complexes as shown earlier (Weinmann et al., 2006). E, Sensitivity of mice to the motor stimulant effects of systemic amphetamine. Locomotor activating effect of amphetamine is expressed as overall mean distance traveled during the 120 min observation period. Anti-Nogo-A antibody treatment did not significantly affect the locomotor reactivity to the drug (n = 9/group). F, G, Representative images of D2R immunostaining in the dorsal striatum of mice treated with either anti-Nogo-A or control antibodies. H, Quantitative analysis of the D2R staining. The relative optical density of D2R in the medial prefrontal cortex (mPFC), the dorsal striatum (CPu), and the nucleus accumbens core (NAcC) and shell (NAcS) regions, was not affected by the anti-Nogo-A antibody treatment (n = 5/group). All values are mean ± SEM. Scale bars: A–C, 0.5 mm; D, 20 μm; F, G, 100 μm. 11C7, Anti-Nogo-A antibody treated; IgG, control IgG treated; no pump, no pump control.
Discussion
This study represents the first systematic attempt to assess the impact of genetic Nogo-A deficiency on schizophrenia-associated traits and has identified multiple relevant phenotypes in Nogo-A−/− mice in terms of behavior, pharmacology, and neurochemistry.
Across a battery of translational behavioral paradigms relevant for assessing face validity of animal models of schizophrenia (Lipska and Weinberger, 2000; Arguello and Gogos, 2006; Powell and Miyakawa, 2006), Nogo-A−/− mice displayed deficits in both PPI and LI, indicating disruptions in sensorimotor gating and selective attention that are related to the symptoms (especially positive) of schizophrenia (Baruch et al., 1988; Braff et al., 2001). Nogo-A−/− mice also exhibited perseverative behavior in discrimination reversal, resembling another neuropsychological sign in schizophrenia (Crider, 1997). Furthermore, the motor stimulant response to systemic amphetamine in the open field was substantially potentiated in these mice, consolidating a similar finding obtained under familiar home cage conditions (Willi et al., 2009). Notably, enhanced susceptibility to acute amphetamine treatment has been directly linked to the genesis of positive symptoms in schizophrenia (Laruelle et al., 1996). On the other hand, spontaneous anxiety-like behavior as well as social behavior were not affected in the Nogo-A−/− mice. The latter is commonly used to model negative symptoms of schizophrenia in rodents. Constitutive knock-out of Nogo-A therefore produces a spectrum of selective behavioral and psychopharmacological aberrations in adulthood that are related to schizophrenia traits. Domains related primarily to the positive and cognitive symptoms of schizophrenia appear affected. The list of used tasks is not exhaustive, of course, as there are other tests that could be performed.
In light of the range of behavioral changes in Nogo-A−/− mice, it is not surprising that major neurochemical changes were identified in different striatal and limbic structures, and the observed neurochemical consequences of this constitutive Nogo-A absence may readily contribute to some of the behavioral and psychopharmacological dysfunctions present in Nogo-A−/− mice. For example, increased D2R expression in the dorsal striatum constitutes a condition that could well lead to the enhanced sensitivity to amphetamine seen in Nogo-A−/− mice (Wolinsky et al., 2007). Decreased basal DA levels in the dorsal striatum per se might rather predict an attenuation of amphetamine activation, but upregulation of D2R in this brain area may represent a compensatory mechanism and shift the balance to higher amphetamine reactivity. It has been postulated that an overactive DA transporter system and alterations in other regulatory mechanisms may lead to a reduction in basal striatal DA and ultimately to upregulation of DA receptors (Grace, 1993; Salahpour et al., 2008). Similar mechanisms may be responsible for the enhanced amphetamine-induced locomotor activation in Nogo-A−/− mice. In addition, D2R downregulation in the medial prefrontal cortex of Nogo-A−/− mice could also contribute to this phenotype, given that infusions of DA receptor agonists into the medial prefrontal cortex can block amphetamine-induced hyperactivity (Broersen et al., 1999). Furthermore, D2R downregulation seen in Nogo-A−/− mice is consistent with D2R antagonist infusion into the medial prefrontal cortex attenuating PPI (Ellenbroek et al., 1996). One should notice that the deficiency in 5-HT and its metabolite 5-HIAA in Nogo-A−/− mice may also contribute to the precipitation of the PPI deficit (Prinssen et al., 2002) and the increased sensitivity to systemic amphetamine (Marek, 2007). Additional studies are clearly warranted to further dissect the relative contributions of distinct neurochemical abnormalities to behavioral and pharmacological dysfunctions in mice lacking Nogo-A. One possibility would be to test whether the PPI and amphetamine phenotypes may be preferentially normalized by DA antagonists versus selective 5-HT reuptake inhibitors.
Given the substantial alterations revealed in specific DA circuits after constitutive Nogo-A deletion, it is crucial to assess the extent to which these DA changes are consistent with the DA pathophysiology identified in schizophrenia. Elevations of D2R densities in the striatum (Wong et al., 1986; Seeman, 1987; Seeman and Kapur, 2000; Hirvonen et al., 2005) and decreased levels of D2R in extrastriatal regions (Takahashi et al., 2006) have been reported in schizophrenic patients and correlate very well with our results in Nogo-A−/− mice. The dorsal striatal reduction in DA content in Nogo-A−/− mice may seem surprising. This appears to be inconsistent with the dopaminergic hypothesis of schizophrenia in terms of directional and regional selectivity, as increased dopaminergic activity localized to the ventral striatum is thought to be critical for schizophrenia (Seeman and Kapur, 2000). However, DA transmission is orchestrated by a broad network of brain areas, and disturbed mental functioning in schizophrenia is more likely attributable to complex dysregulations in multiple interconnected brain regions rather than a singular unidirectional change in a specific neurotransmitter (Grace, 1993; O'Donnell and Grace, 1998; Carlsson et al., 2001; Carlsson, 2006). One also has to keep in mind that Nogo-A−/− mice showed 5-HT deficits and a potential modulatory role for 5-HT in schizophrenia pathophysiology has been suggested (Abi-Dargham, 2007). However, it is important to emphasize that our knowledge of neuropathological disease markers in humans, and thus their availability in rodents, is limited. The claim of definite links between neurochemical markers and symptoms or behavioral phenotypes, respectively, is therefore complicated.
To date, it is not known whether Nogo-A is directly involved in dopaminergic transmission or related mechanisms. The fact that DA systems are affected after constitutive Nogo-A deletion is nonetheless not surprising. Interestingly, maturation of the different dopaminergic systems follows distinct developmental progression with differential temporal windows (Sturrock, 1980; Voorn et al., 1988), and wiring of dopaminergic projection systems appears to be extremely susceptible to neurodevelopmental insults. Various genetic as well as environmental manipulations, even when they are not directly targeting the dopaminergic systems, can lead to profound and specific long-term disturbances within distinct dopaminergic circuits (Roffler-Tarlov and Graybiel, 1984; Lipska et al., 2003; Fone and Porkess, 2008; Meyer et al., 2008; Winter et al., 2009). Thus, the defects in DA and its metabolites in Nogo-A−/− mice could well originate from selective, yet unknown, dysfunctions occurring during brain development. This would be in line with a role for Nogo-A in brain wiring processes during development (McGee et al., 2005; Mingorance-Le Meur et al., 2007).
During development, Nogo-A is present in early migrating neurons and during neurite outgrowth; Nogo-deficient mice show changes in cortical development and neuronal maturation (Mingorance-Le Meur et al., 2007). Aberrant neuronal development in cortical and subcortical areas due to the lack of neuronal Nogo-A might therefore contribute to the development of the behavioral and neurochemical changes described here, as seen in schizophrenia (Weinberger, 1987; Harrison, 1999; Ross et al., 2006) and several neurodevelopmental models of this disease in animals (e.g., Shen et al., 2008). In addition, increased expression levels of the growth-related proteins GAP-43 and S100B mainly in the medial prefrontal cortex and dentate gyrus of Nogo-A−/− mice suggest that lifelong absence of Nogo-A may lead to abnormal growth, entailing perturbed neurodevelopment (McGee et al., 2005). Growth dysregulations are likely due to the lack of Nogo-A in myelin (Schwab, 2004; McGee et al., 2005), which is in keeping with the notion that myelin-related abnormalities, such as lack of myelin-based restriction of axonal plasticity particularly during development, contributes to the risk of schizophrenia (Davis et al., 2003; Karoutzou et al., 2007). Although transitory suppression of Nogo-A function by antibodies applied to adult mice slightly increased GAP-43 expression in the dentate gyrus, this elevation was less pronounced than in Nogo-A−/− mice and did not affect behavior. It is also important to highlight that there was no indication of neurodegeneration in Nogo-A−/− mice as evidenced by the absence of reactive gliosis. Together, our data are consistent with the suggestion of developmental changes and dysregulated fiber growth and plasticity in the animals chronically lacking Nogo-A as one generic mechanism for the development of schizophrenia-like symptoms. This hypothesized association is highly interesting in view of a recent report linking schizophrenia to deficient Nogo/NgR1 signaling associated with a failure in restricting anatomical plasticity (Budel et al., 2008).
While the absence of major abnormalities in animals treated with anti-Nogo-A antibodies in adulthood may indicate that neurodevelopmental factors could play a role in the genesis of the Nogo-A knock-out phenotypes, one cannot exclude the possibility that the loss of Nogo-A in the adult brain as such could not contribute to their emergence. A rescue experiment by reintroducing Nogo-A into the knock-out mice would help to answer this question. Alternatively, examining the impact of Nogo-A blockade during early neurodevelopmental stages on brain function in the adult would also be highly instructive. Several critical time windows in fetal and early postnatal brain development would have to be taken into account in this respect, however (see Keshavan and Hogarty, 1999; Meyer et al., 2007). Because the feasibility of antibody pump implantations in mice is limited and its application in developing animals not possible, the generation of temporally controlled Nogo-A knock-out mice using specific gene induction systems (Wang et al., 1994; Kistner et al., 1996) would be necessary. Nevertheless, the fact that a transient loss of Nogo-A function in the adult CNS was not associated with schizophrenia-like effects is of utmost importance with regard to the ongoing clinical trials using function-blocking agents for Nogo-A in spinal cord injured patients.
In conclusion, the present study shows that Nogo-A is relevant for a wide spectrum of brain and behavioral functions. This implies an important role for Nogo-A in development in general, regardless of whether such behavioral, anatomical, and neurochemical differences faithfully mimic schizophrenic symptoms. Our findings thus bear broader significance for other neurodevelopmental mental disorders such as autism. This genetic mouse model holds additional heuristic value in elucidating the relevant neurodevelopmental, epigenetic, and physiological mechanisms that might be sensitive to Nogo-A polymorphisms. Characterization of relevant molecular pathways would also be instrumental in the identification of novel drug targets for therapeutic and protective interventions.
Footnotes
This work was supported by grants of the Swiss National Science Foundation (3100AO-122527/1), the National Center for Competence in Research “Neural Plasticity & Repair” of the Swiss National Science Foundation, the Spinal Cord Consortium of the Christopher and Dana Reeve Foundation (Springfield, NJ), and the EU-Network of Excellence NeuroNE. We are extremely grateful to Anissa Kempf and Peter Schmid for technical assistance and to Franziska Christ for animal breeding.
References
- Abi-Dargham A. Alterations of serotonin transmission in schizophrenia. Int Rev Neurobiol. 2007;78:133–164. doi: 10.1016/S0074-7742(06)78005-9. [DOI] [PubMed] [Google Scholar]
- American Psychiatric Association. Diagnostic and statistical manual of mental disorders (DSM-IV) Washington, DC: APA; 2000. [Google Scholar]
- Arguello PA, Gogos JA. Modeling madness in mice: one piece at a time. Neuron. 2006;52:179–196. doi: 10.1016/j.neuron.2006.09.023. [DOI] [PubMed] [Google Scholar]
- Bareyre FM, Haudenschild B, Schwab ME. Long-lasting sprouting and gene expression changes induced by the monoclonal antibody IN-1 in the adult spinal cord. J Neurosci. 2002;22:7097–7110. doi: 10.1523/JNEUROSCI.22-16-07097.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baruch I, Hemsley DR, Gray JA. Differential performance of acute and chronic schizophrenics in a latent inhibition task. J Nerv Ment Dis. 1988;176:598–606. doi: 10.1097/00005053-198810000-00004. [DOI] [PubMed] [Google Scholar]
- Blennow K, Bogdanovic N, Gottfries CG, Davidsson P. The growth-associated protein GAP-43 is increased in the hippocampus and in the gyrus cinguli in schizophrenia. J Mol Neurosci. 1999;13:101–109. doi: 10.1385/JMN:13:1-2:101. [DOI] [PubMed] [Google Scholar]
- Braff DL, Geyer MA, Swerdlow NR. Human studies of prepulse inhibition of startle: normal subjects, patient groups, and pharmacological studies. Psychopharmacology (Berl) 2001;156:234–258. doi: 10.1007/s002130100810. [DOI] [PubMed] [Google Scholar]
- Broersen LM, Feldon J, Weiner I. Dissociative effects of apomorphine infusions into the medial prefrontal cortex of rats on latent inhibition, prepulse inhibition and amphetamine-induced locomotion. Neuroscience. 1999;94:39–46. doi: 10.1016/s0306-4522(99)00287-0. [DOI] [PubMed] [Google Scholar]
- Budel S, Padukkavidana T, Liu BP, Feng Z, Hu F, Johnson S, Lauren J, Park JH, McGee AW, Liao J, Stillman A, Kim JE, Yang BZ, Sodi S, Gelernter J, Zhao H, Hisama F, Arnsten AF, Strittmatter SM. Genetic variants of Nogo-66 receptor with possible association to schizophrenia block myelin inhibition of axon growth. J Neurosci. 2008;28:13161–13172. doi: 10.1523/JNEUROSCI.3828-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buffo A, Zagrebelsky M, Huber AB, Skerra A, Schwab ME, Strata P, Rossi F. Application of neutralizing antibodies against NI-35/250 myelin-associated neurite growth inhibitory proteins to the adult rat cerebellum induces sprouting of uninjured Purkinje cell axons. J Neurosci. 2000;20:2275–2286. doi: 10.1523/JNEUROSCI.20-06-02275.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlsson A. The neurochemical circuitry of schizophrenia. Pharmacopsychiatry. 2006;39(Suppl 1):S10–S14. doi: 10.1055/s-2006-931483. [DOI] [PubMed] [Google Scholar]
- Carlsson A, Waters N, Holm-Waters S, Tedroff J, Nilsson M, Carlsson ML. Interactions between monoamines, glutamate, and GABA in schizophrenia: new evidence. Annu Rev Pharmacol Toxicol. 2001;41:237–260. doi: 10.1146/annurev.pharmtox.41.1.237. [DOI] [PubMed] [Google Scholar]
- Chen J, Lipska BK, Weinberger DR. Genetic mouse models of schizophrenia: from hypothesis-based to susceptibility gene-based models. Biol Psychiatry. 2006;59:1180–1188. doi: 10.1016/j.biopsych.2006.02.024. [DOI] [PubMed] [Google Scholar]
- Coon H, Myles-Worsley M, Tiobech J, Hoff M, Rosenthal J, Bennett P, Reimherr F, Wender P, Dale P, Polloi A, Byerley W. Evidence for a chromosome 2p13-14 schizophrenia susceptibility locus in families from Palau, Micronesia. Mol Psychiatry. 1998;3:521–527. doi: 10.1038/sj.mp.4000453. [DOI] [PubMed] [Google Scholar]
- Crider A. Perseveration in schizophrenia. Schizophr Bull. 1997;23:63–74. doi: 10.1093/schbul/23.1.63. [DOI] [PubMed] [Google Scholar]
- Davis KL, Stewart DG, Friedman JI, Buchsbaum M, Harvey PD, Hof PR, Buxbaum J, Haroutunian V. White matter changes in schizophrenia: evidence for myelin-related dysfunction. Arch Gen Psychiatry. 2003;60:443–456. doi: 10.1001/archpsyc.60.5.443. [DOI] [PubMed] [Google Scholar]
- Dimou L, Schnell L, Montani L, Duncan C, Simonen M, Schneider R, Liebscher T, Gullo M, Schwab ME. Nogo-A-deficient mice reveal strain-dependent differences in axonal regeneration. J Neurosci. 2006;26:5591–5603. doi: 10.1523/JNEUROSCI.1103-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dodd DA, Niederoest B, Bloechlinger S, Dupuis L, Loeffler JP, Schwab ME. Nogo-A, -B, and -C are found on the cell surface and interact together in many different cell types. J Biol Chem. 2005;280:12494–12502. doi: 10.1074/jbc.M411827200. [DOI] [PubMed] [Google Scholar]
- Ellenbroek BA, Budde S, Cools AR. Prepulse inhibition and latent inhibition: the role of dopamine in the medial prefrontal cortex. Neuroscience. 1996;75:535–542. doi: 10.1016/0306-4522(96)00307-7. [DOI] [PubMed] [Google Scholar]
- Enard W, Gehre S, Hammerschmidt K, Hölter SM, Blass T, Somel M, Brückner MK, Schreiweis C, Winter C, Sohr R, Becker L, Wiebe V, Nickel B, Giger T, Müller U, Groszer M, Adler T, Aguilar A, Bolle I, Calzada-Wack J, et al. A humanized version of Foxp2 affects cortico-basal ganglia circuits in mice. Cell. 2009;137:961–971. doi: 10.1016/j.cell.2009.03.041. [DOI] [PubMed] [Google Scholar]
- Fone KC, Porkess MV. Behavioural and neurochemical effects of post-weaning social isolation in rodents-relevance to developmental neuropsychiatric disorders. Neurosci Biobehav Rev. 2008;32:1087–1102. doi: 10.1016/j.neubiorev.2008.03.003. [DOI] [PubMed] [Google Scholar]
- Fritschy JM, Mohler H. GABAA-receptor heterogeneity in the adult rat brain: differential regional and cellular distribution of seven major subunits. J Comp Neurol. 1995;359:154–194. doi: 10.1002/cne.903590111. [DOI] [PubMed] [Google Scholar]
- Grace AA. Cortical regulation of subcortical dopamine systems and its possible relevance to schizophrenia. J Neural Transm. 1993;91:111–134. doi: 10.1007/BF01245228. [DOI] [PubMed] [Google Scholar]
- Gray NS, Hemsley DR, Gray JA. Abolition of latent inhibition in acute, but not chronic, schizophrenics. Neurol Psychiatry Brain Res. 1992;1:83–89. [Google Scholar]
- Hagenbuch N, Feldon J, Yee BK. Use of the elevated plus-maze test with opaque or transparent walls in the detection of mouse strain differences and the anxiolytic effects of diazepam. Behav Pharmacol. 2006;17:31–41. doi: 10.1097/01.fbp.0000189811.77049.3e. [DOI] [PubMed] [Google Scholar]
- Harrison PJ. Schizophrenia: a disorder of neurodevelopment? Curr Opin Neurobiol. 1997;7:285–289. doi: 10.1016/s0959-4388(97)80018-9. [DOI] [PubMed] [Google Scholar]
- Harrison PJ. The neuropathology of schizophrenia. A critical review of the data and their interpretation. Brain. 1999;122:593–624. doi: 10.1093/brain/122.4.593. [DOI] [PubMed] [Google Scholar]
- Harrison PJ, Weinberger DR. Schizophrenia genes, gene expression, and neuropathology: on the matter of their convergence. Mol Psychiatry. 2005;10:40–68. doi: 10.1038/sj.mp.4001558. [DOI] [PubMed] [Google Scholar]
- Hirvonen J, van Erp TG, Huttunen J, Aalto S, Någren K, Huttunen M, Lönnqvist J, Kaprio J, Hietala J, Cannon TD. Increased caudate dopamine D2 receptor availability as a genetic marker for schizophrenia. Arch Gen Psychiatry. 2005;62:371–378. doi: 10.1001/archpsyc.62.4.371. [DOI] [PubMed] [Google Scholar]
- Hoffman HS, Searle JL. Acoustic variables in the modification of startle reaction in the rat. J Comp Physiol Psychol. 1965;60:53–58. doi: 10.1037/h0022325. [DOI] [PubMed] [Google Scholar]
- Hsu R, Woodroffe A, Lai WS, Cook MN, Mukai J, Dunning JP, Swanson DJ, Roos JL, Abecasis GR, Karayiorgou M, Gogos JA. Nogo Receptor 1 (RTN4R) as a candidate gene for schizophrenia: analysis using human and mouse genetic approaches. PLoS One. 2007;2:e1234. doi: 10.1371/journal.pone.0001234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huber AB, Weinmann O, Brösamle C, Oertle T, Schwab ME. Patterns of Nogo mRNA and protein expression in the developing and adult rat and after CNS lesions. J Neurosci. 2002;22:3553–3567. doi: 10.1523/JNEUROSCI.22-09-03553.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karoutzou G, Emrich HM, Dietrich DE. The myelin-pathogenesis puzzle in schizophrenia: a literature review. Mol Psychiatry. 2008;13:245–260. doi: 10.1038/sj.mp.4002096. [DOI] [PubMed] [Google Scholar]
- Keshavan MS, Hogarty GE. Brain maturational processes and delayed onset in schizophrenia. Dev Psychopathol. 1999;11:525–543. doi: 10.1017/s0954579499002199. [DOI] [PubMed] [Google Scholar]
- Kistner A, Gossen M, Zimmermann F, Jerecic J, Ullmer C, Lübbert H, Bujard H. Doxycycline-mediated quantitative and tissue-specific control of gene expression in transgenic mice. Proc Natl Acad Sci U S A. 1996;93:10933–10938. doi: 10.1073/pnas.93.20.10933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laruelle M, Abi-Dargham A, van Dyck CH, Gil R, D'Souza CD, Erdos J, McCance E, Rosenblatt W, Fingado C, Zoghbi SS, Baldwin RM, Seibyl JP, Krystal JH, Charney DS, Innis RB. Single photon emission computerized tomography imaging of amphetamine-induced dopamine release in drug-free schizophrenic subjects. Proc Natl Acad Sci U S A. 1996;93:9235–9240. doi: 10.1073/pnas.93.17.9235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis DA, Levitt P. Schizophrenia as a disorder of neurodevelopment. Annu Rev Neurosci. 2002;25:409–432. doi: 10.1146/annurev.neuro.25.112701.142754. [DOI] [PubMed] [Google Scholar]
- Lewis DA, Lieberman JA. Catching up on schizophrenia: natural history and neurobiology. Neuron. 2000;28:325–334. doi: 10.1016/s0896-6273(00)00111-2. [DOI] [PubMed] [Google Scholar]
- Lipska BK, Weinberger DR. To model a psychiatric disorder in animals: schizophrenia as a reality test. Neuropsychopharmacology. 2000;23:223–239. doi: 10.1016/S0893-133X(00)00137-8. [DOI] [PubMed] [Google Scholar]
- Lipska BK, Lerman DN, Khaing ZZ, Weinberger DR. The neonatal ventral hippocampal lesion model of schizophrenia: effects on dopamine and GABA mRNA markers in the rat midbrain. Eur J Neurosci. 2003;18:3097–3104. doi: 10.1111/j.1460-9568.2003.03047.x. [DOI] [PubMed] [Google Scholar]
- Liu H, Abecasis GR, Heath SC, Knowles A, Demars S, Chen YJ, Roos JL, Rapoport JL, Gogos JA, Karayiorgou M. Genetic variation in the 22q11 locus and susceptibility to schizophrenia. Proc Natl Acad Sci U S A. 2002;99:16859–16864. doi: 10.1073/pnas.232186099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lubow RE, Moore AU. Latent inhibition: the effect of nonreinforced pre-exposure to the conditional stimulus. J Comp Physiol Psychol. 1959;52:415–419. doi: 10.1037/h0046700. [DOI] [PubMed] [Google Scholar]
- Marek GJ. Serotonin and dopamine interactions in rodents and primates: implications for psychosis and antipsychotic drug development. Int Rev Neurobiol. 2007;78:165–192. doi: 10.1016/S0074-7742(06)78006-0. [DOI] [PubMed] [Google Scholar]
- McGee AW, Yang Y, Fischer QS, Daw NW, Strittmatter SM. Experience-driven plasticity of visual cortex limited by myelin and Nogo receptor. Science. 2005;309:2222–2226. doi: 10.1126/science.1114362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer U, Feldon J, Schedlowski M, Yee BK. Towards an immuno-precipitated neurodevelopmental animal model of schizophrenia. Neurosci Biobehav Rev. 2005;29:913–947. doi: 10.1016/j.neubiorev.2004.10.012. [DOI] [PubMed] [Google Scholar]
- Meyer U, Nyffeler M, Engler A, Urwyler A, Schedlowski M, Knuesel I, Yee BK, Feldon J. The time of prenatal immune challenge determines the specificity of inflammation-mediated brain and behavioral pathology. J Neurosci. 2006;26:4752–4762. doi: 10.1523/JNEUROSCI.0099-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer U, Yee BK, Feldon J. The neurodevelopmental impact of prenatal infections at different times of pregnancy: the earlier the worse? Neuroscientist. 2007;13:241–256. doi: 10.1177/1073858406296401. [DOI] [PubMed] [Google Scholar]
- Meyer U, Nyffeler M, Schwendener S, Knuesel I, Yee BK, Feldon J. Relative prenatal and postnatal maternal contributions to schizophrenia-related neurochemical dysfunction after in utero immune challenge. Neuropsychopharmacology. 2008;33:441–456. doi: 10.1038/sj.npp.1301413. [DOI] [PubMed] [Google Scholar]
- Mingorance-Le Meur A, Zheng B, Soriano E, del Río JA. Involvement of the myelin-associated inhibitor Nogo-A in early cortical development and neuronal maturation. Cereb Cortex. 2007;17:2375–2386. doi: 10.1093/cercor/bhl146. [DOI] [PubMed] [Google Scholar]
- Moy SS, Nadler JJ, Young NB, Nonneman RJ, Grossman AW, Murphy DL, D'Ercole AJ, Crawley JN, Magnuson TR, Lauder JM. Social approach in genetically engineered mouse lines relevant to autism. Genes Brain Behav. 2009;8:129–142. doi: 10.1111/j.1601-183X.2008.00452.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novak G, Kim D, Seeman P, Tallerico T. Schizophrenia and Nogo: elevated mRNA in cortex, and high prevalence of a homozygous CAA insert. Mol Brain Res. 2002;107:183–189. doi: 10.1016/s0169-328x(02)00492-8. [DOI] [PubMed] [Google Scholar]
- O'Donnell P, Grace AA. Dysfunctions in multiple interrelated systems as the neurobiological bases of schizophrenic symptom clusters. Schizophr Bull. 1998;24:267–283. doi: 10.1093/oxfordjournals.schbul.a033325. [DOI] [PubMed] [Google Scholar]
- Perrone-Bizzozero NI, Sower AC, Bird ED, Benowitz LI, Ivins KJ, Neve RL. Levels of the growth-associated protein GAP-43 are selectively increased in association cortices in schizophrenia. Proc Natl Acad Sci U S A. 1996;93:14182–14187. doi: 10.1073/pnas.93.24.14182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perry W, Minassian A, Lopez B, Maron L, Lincoln A. Sensorimotor gating deficits in adults with autism. Biol Psychiatry. 2007;61:482–486. doi: 10.1016/j.biopsych.2005.09.025. [DOI] [PubMed] [Google Scholar]
- Powell CM, Miyakawa T. Schizophrenia-relevant behavioral testing in rodent models: a uniquely human disorder? Biol Psychiatry. 2006;59:1198–1207. doi: 10.1016/j.biopsych.2006.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prinssen EP, Assié MB, Koek W, Kleven MS. Depletion of 5-HT disrupts prepulse inhibition in rats: dependence on the magnitude of depletion, and reversal by a 5-HT precursor. Neuropsychopharmacology. 2002;26:340–347. doi: 10.1016/S0893-133X(01)00348-7. [DOI] [PubMed] [Google Scholar]
- Rapoport JL, Addington AM, Frangou S, Psych MR. The neurodevelopmental model of schizophrenia: update 2005. Mol Psychiatry. 2005;10:434–449. doi: 10.1038/sj.mp.4001642. [DOI] [PubMed] [Google Scholar]
- Ridley RM. The psychology of perserverative and stereotyped behaviour. Prog Neurobiol. 1994;44:221–231. doi: 10.1016/0301-0082(94)90039-6. [DOI] [PubMed] [Google Scholar]
- Roffler-Tarlov S, Graybiel AM. Weaver mutation has differential effects on the dopamine-containing innervation of the limbic and nonlimbic striatum. Nature. 1984;307:62–66. doi: 10.1038/307062a0. [DOI] [PubMed] [Google Scholar]
- Ross CA, Margolis RL, Reading SA, Pletnikov M, Coyle JT. Neurobiology of schizophrenia. Neuron. 2006;52:139–153. doi: 10.1016/j.neuron.2006.09.015. [DOI] [PubMed] [Google Scholar]
- Rothermundt M, Falkai P, Ponath G, Abel S, Bürkle H, Diedrich M, Hetzel G, Peters M, Siegmund A, Pedersen A, Maier W, Schramm J, Suslow T, Ohrmann P, Arolt V. Glial cell dysfunction in schizophrenia indicated by increased S100B in the CSF. Mol Psychiatry. 2004;9:897–899. doi: 10.1038/sj.mp.4001548. [DOI] [PubMed] [Google Scholar]
- Salahpour A, Ramsey AJ, Medvedev IO, Kile B, Sotnikova TD, Holmstrand E, Ghisi V, Nicholls PJ, Wong L, Murphy K, Sesack SR, Wightman RM, Gainetdinov RR, Caron MG. Increased amphetamine-induced hyperactivity and reward in mice overexpressing the dopamine transporter. Proc Natl Acad Sci U S A. 2008;105:4405–4410. doi: 10.1073/pnas.0707646105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwab ME. Nogo and axon regeneration. Curr Opin Neurobiol. 2004;14:118–124. doi: 10.1016/j.conb.2004.01.004. [DOI] [PubMed] [Google Scholar]
- Seeman P. Dopamine receptors and the dopamine hypothesis of schizophrenia. Synapse. 1987;1:133–152. doi: 10.1002/syn.890010203. [DOI] [PubMed] [Google Scholar]
- Seeman P, Kapur S. Schizophrenia: more dopamine, more D2 receptors. Proc Natl Acad Sci U S A. 2000;97:7673–7675. doi: 10.1073/pnas.97.14.7673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw SH, Kelly M, Smith AB, Shields G, Hopkins PJ, Loftus J, Laval SH, Vita A, De Hert M, Cardon LR, Crow TJ, Sherrington R, DeLisi LE. A genome-wide search for schizophrenia susceptibility genes. Am J Med Genet. 1998;81:364–376. doi: 10.1002/(sici)1096-8628(19980907)81:5<364::aid-ajmg4>3.0.co;2-t. [DOI] [PubMed] [Google Scholar]
- Shen S, Lang B, Nakamoto C, Zhang F, Pu J, Kuan SL, Chatzi C, He S, Mackie I, Brandon NJ, Marquis KL, Day M, Hurko O, McCaig CD, Riedel G, St Clair D. Schizophrenia-related neural and behavioral phenotypes in transgenic mice expressing truncated Disc1. J Neurosci. 2008;28:10893–10904. doi: 10.1523/JNEUROSCI.3299-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheu FS, Azmitia EC, Marshak DR, Parker PJ, Routtenberg A. Glial-derived S100b protein selectively inhibits recombinant beta protein kinase C (PKC) phosphorylation of neuron-specific protein F1/GAP43. Mol Brain Res. 1994;21:62–66. doi: 10.1016/0169-328x(94)90378-6. [DOI] [PubMed] [Google Scholar]
- Simonen M, Pedersen V, Weinmann O, Schnell L, Buss A, Ledermann B, Christ F, Sansig G, van der Putten H, Schwab ME. Systemic deletion of the myelin-associated outgrowth inhibitor Nogo-A improves regenerative and plastic responses after spinal cord injury. Neuron. 2003;38:201–211. doi: 10.1016/s0896-6273(03)00226-5. [DOI] [PubMed] [Google Scholar]
- Sinibaldi L, De Luca A, Bellacchio E, Conti E, Pasini A, Paloscia C, Spalletta G, Caltagirone C, Pizzuti A, Dallapiccola B. Mutations of the Nogo-66 receptor (RTN4R) gene in schizophrenia. Hum Mutat. 2004;24:534–535. doi: 10.1002/humu.9292. [DOI] [PubMed] [Google Scholar]
- Smith SE, Li J, Garbett K, Mirnics K, Patterson PH. Maternal immune activation alters fetal brain development through interleukin-6. J Neurosci. 2007;27:10695–10702. doi: 10.1523/JNEUROSCI.2178-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sturrock RR. A developmental study of the mouse neostriatum. J Anat. 1980;130:243–261. [PMC free article] [PubMed] [Google Scholar]
- Takahashi H, Higuchi M, Suhara T. The role of extrastriatal dopamine D2 receptors in schizophrenia. Biol Psychiatry. 2006;59:919–928. doi: 10.1016/j.biopsych.2006.01.022. [DOI] [PubMed] [Google Scholar]
- Tan EC, Chong SA, Wang H, Chew-Ping Lim E, Teo YY. Gender-specific association of insertion/deletion polymorphisms in the nogo gene and chronic schizophrenia. Mol Brain Res. 2005;139:212–216. doi: 10.1016/j.molbrainres.2005.05.010. [DOI] [PubMed] [Google Scholar]
- Voorn P, Kalsbeek A, Jorritsma-Byham B, Groenewegen HJ. The pre- and postnatal development of the dopaminergic cell groups in the ventral mesencephalon and the dopaminergic innervation of the striatum of the rat. Neuroscience. 1988;25:857–887. doi: 10.1016/0306-4522(88)90041-3. [DOI] [PubMed] [Google Scholar]
- Wang X, Chun SJ, Treloar H, Vartanian T, Greer CA, Strittmatter SM. Localization of Nogo-A and Nogo-66 receptor proteins at sites of axon-myelin and synaptic contact. J Neurosci. 2002;22:5505–5515. doi: 10.1523/JNEUROSCI.22-13-05505.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, O'Malley BW, Jr, Tsai SY, O'Malley BW. A regulatory system for use in gene transfer. Proc Natl Acad Sci U S A. 1994;91:8180–8184. doi: 10.1073/pnas.91.17.8180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinberger DR. Implications of normal brain development for the pathogenesis of schizophrenia. Arch Gen Psychiatry. 1987;44:660–669. doi: 10.1001/archpsyc.1987.01800190080012. [DOI] [PubMed] [Google Scholar]
- Weinmann O, Schnell L, Ghosh A, Montani L, Wiessner C, Wannier T, Rouiller E, Mir A, Schwab ME. Intrathecally infused antibodies against Nogo-A penetrate the CNS and downregulate the endogenous neurite growth inhibitor Nogo-A. Mol Cell Neurosci. 2006;32:161–173. doi: 10.1016/j.mcn.2006.03.007. [DOI] [PubMed] [Google Scholar]
- Willi R, Aloy EM, Yee BK, Feldon J, Schwab ME. Behavioral characterization of mice lacking the neurite outgrowth inhibitor Nogo-A. Genes Brain Behav. 2009;8:181–192. doi: 10.1111/j.1601-183X.2008.00460.x. [DOI] [PubMed] [Google Scholar]
- Winter C, Djodari-Irani A, Sohr R, Morgenstern R, Feldon J, Juckel G, Meyer U. Prenatal immune activation leads to multiple changes in basal neurotransmitter levels in the adult brain: implications for brain disorders of neurodevelopmental origin such as schizophrenia. Int J Neuropsychopharmacol. 2009;12:513–524. doi: 10.1017/S1461145708009206. [DOI] [PubMed] [Google Scholar]
- Wolinsky TD, Swanson CJ, Smith KE, Zhong H, Borowsky B, Seeman P, Branchek T, Gerald CP. The Trace Amine 1 receptor knockout mouse: an animal model with relevance to schizophrenia. Genes Brain Behav. 2007;6:628–639. doi: 10.1111/j.1601-183X.2006.00292.x. [DOI] [PubMed] [Google Scholar]
- Wong DF, Wagner HN, Jr, Tune LE, Dannals RF, Pearlson GD, Links JM, Tamminga CA, Broussolle EP, Ravert HT, Wilson AA, Toung JK, Malat J, Williams JA, O'Tuama LA, Snyder SH, Kuhar MJ, Gjedde A. Positron emission tomography reveals elevated D2 dopamine receptors in drug-naive schizophrenics. Science. 1986;234:1558–1563. doi: 10.1126/science.2878495. [DOI] [PubMed] [Google Scholar]
- Yee BK, Balic E, Singer P, Schwerdel C, Grampp T, Gabernet L, Knuesel I, Benke D, Feldon J, Mohler H, Boison D. Disruption of glycine transporter 1 restricted to forebrain neurons is associated with a procognitive and antipsychotic phenotypic profile. J Neurosci. 2006;26:3169–3181. doi: 10.1523/JNEUROSCI.5120-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yiu G, He Z. Glial inhibition of CNS axon regeneration. Nat Rev Neurosci. 2006;7:617–627. doi: 10.1038/nrn1956. [DOI] [PMC free article] [PubMed] [Google Scholar]