

Abstract
Associative learning depends on multiple cortical and subcortical structures, including striatum, hippocampus, and amygdala. Both glutamatergic and dopaminergic neurotransmitter systems have been implicated in learning and memory consolidation. While the role of glutamate is well established, the role of dopamine and its receptors in these processes is less clear. In this study, we used two models of dopamine D1 receptor (D1R, Drd1a) loss, D1R knock-out mice (Drd1a−/−) and mice with intrahippocampal injections of Drd1a-siRNA (small interfering RNA), to study the role of D1R in different models of learning, hippocampal long-term potentiation (LTP) and associated gene expression. D1R loss markedly reduced spatial learning, fear learning, and classical conditioning of the eyelid response, as well as the associated activity-dependent synaptic plasticity in the hippocampal CA1–CA3 synapse. These results provide the first experimental demonstration that D1R is required for trace eyeblink conditioning and associated changes in synaptic strength in hippocampus of behaving mice. Drd1a-siRNA mice were indistinguishable from Drd1a−/− mice in all experiments, indicating that hippocampal knockdown was as effective as global inactivation and that the observed effects are caused by loss of D1R and not by indirect developmental effects of Drd1a−/−. Finally, in vivo LTP and LTP-induced expression of Egr1 in the hippocampus were significantly reduced in Drd1a−/− and Drd1a-siRNA, indicating an important role for D1R in these processes. Our data reveal a functional relationship between acquisition of associative learning, increase in synaptic strength at the CA3–CA1 synapse, and Egr1 induction in the hippocampus by demonstrating that all three are dramatically impaired when D1R is eliminated or reduced.
Introduction
Recent studies demonstrate that dopamine plays an important role in learning and memory. Moreover, integration of glutamate- and dopamine-mediated signals at the cellular level is required for persistent long-term potentiation (LTP) (O'Carroll and Morris, 2004), learning (Smith-Roe and Kelley, 2000; Baldwin et al., 2002), and long-term memory (O'Carroll et al., 2006). Exposure to a novel environment facilitates LTP (Li et al., 2003), linking dopamine signaling with enhanced LTP and with new information acquisition and storage (Lisman and Grace, 2005). Conversely, dopaminergic dysfunction significantly alters spatial learning and short- and long-term memory in rodents and in nonhuman primates (Whishaw and Dunnett, 1985; Williams and Goldman-Rakic, 1995). Dopamine depletion causes cognitive deficits in Parkinson's disease patients (Dubois and Pillon, 1997; Levin and Katzen, 2005), in agreement with studies in dopamine-deficient mice (Palmiter, 2008; Darvas and Palmiter, 2009), stressing the importance of dopamine in learning and associated synaptic plasticity.
The dopamine D1 receptor (D1R), in particular, has been implicated in mediating dopamine's effects in learning and synaptic plasticity. Pharmacological blockade of D1/D5 receptors significantly diminishes early and late phases of LTP in rat hippocampal slices (Otmakhova and Lisman, 1996) and blocks long-term memory storage (O'Carroll et al., 2006; Rossato et al., 2009) in vivo. Selective genetic inactivation of the dopamine D1R subtype (Drd1a) differentiated between the roles of D1 and D5 receptor subtypes in LTP (Granado et al., 2008) and spatial learning (El-Ghundi et al., 1999; Granado et al., 2008). However, the role of the D1R in associative learning and classical conditioning is less clear, as is its role in the synaptic changes that occur in hippocampal networks in vivo during the acquisition of new information. Most, if not all, of the electrophysiological studies involving D1R have been performed in vitro.
Trace eyeblink conditioning, a form of associative learning, was recently shown to induce a progressive increase in strength at the hippocampal CA3–CA1 synapse in awake mice (Gruart et al., 2006; Madroñal et al., 2009) that correlates with the progressive increase in conditioned responses. To directly demonstrate the relationship between LTP and associative learning, we studied the role of D1R in associative learning and synaptic plasticity in adult behaving mice. LTP is well established as a form of synaptic memory but is usually studied under nonphysiological conditions. Our approach here is unique in that we simultaneously assess trace eyeblink conditioning and synaptic efficiency by measuring changes in evoked extracellular field EPSPs (fEPSPs) at the CA3–CA1 synapse in behaving animals during conditioning. We compared wild-type (WT) mice to genetically engineered mice lacking D1R (Drd1a−/−). In addition, we used small interfering RNA (siRNA) technology to silence Drd1a in adult mice in vivo. Our data reveal a functional relationship between acquisition of associative learning, increase in synaptic strength at the CA3–CA1 synapse, and Egr1 expression in the hippocampus by revealing that all three are dramatically impaired when D1R is eliminated or reduced. These results indicate an important role for hippocampal D1R in associative learning and its physiological and molecular correlates.
Materials and Methods
Animals.
All experiments were performed on 3- to 6-month-old (25–30 g) male mice. Drd1a−/− mice (Xu et al., 1994; Moratalla et al., 1996) were backcrossed to C57BL/6 for >10 generations. WT and Drd1a−/− mice used in this study were derived from the mating of heterozygous mice. Animal genotypes were determined by Southern blot analysis (Xu et al., 1994). RNA interference procedures were performed on WT C57BL/6 mice. Before surgery, animals were housed in separate cages (n = 10 per cage) on a 12 h light/dark cycle with constant ambient temperature (21 ± 1°C) and humidity (55 ± 9%). Food and water were available ad libitum. Electrophysiological and behavioral studies were performed in accordance with the guidelines of the European Union (2003/65/CE) and Spanish regulations (BOE 252/34367-91, 2005) for the use of laboratory animals in chronic experiments. Experiments were also approved by the local ethical committee.
Spatial learning: Barnes maze.
In the Barnes maze, animals receive reinforcement to escape from the open platform surface to a small dark recessed chamber located under the platform called an “escape box.” The paradigm consists of a circular platform (90 cm in diameter) with 20 holes (hole diameter, 5 cm) along the perimeter. Spatial cues were placed in the walls of the room during the experiment. The experiment was divided in three different phases. During the first 11 d, mice were trained to enter into the escape box; during the second part, mice rested for 3 d and were tested for long-term spatial memory. In the last phase, animals were trained again in the Barnes maze for 3 d, but the escape box was placed in a new position 180° from the original position. The task conditions during all phases were identical; the mouse was placed in the middle of the maze in a black cylindrical start. After 10 s elapsed, the cylinder was lifted, and the mouse was free to explore the maze. The trial ended when the mouse entered the escape box or after 2 min had elapsed; in this case, the mouse was guided to the escape box. In all cases, mice were allowed to stay 30 s in the escape box. All animals were given four training trials per day, and trials were separated by 20 min. After each trial, the maze was cleaned with 70% alcohol to eliminate the use of intramaze cues. Trials were recorded using a computerized tracking analyzer system (SMART, Panlab).
Active avoidance.
For this test, we used a two-way shuttle box (Ugo Basile) with acrylic walls and stainless steel bars in the floor controlled by a programming/recording unit with a shock generator (Ugo Basile). Animals were given one training session each day for 7 consecutive days. Each training session consisted of an adaptation period of 3 min, in which animals were allowed to move freely from one compartment to the other, followed by 20 trials separated by an intertrial interval (ITI) of 20 s (±5 to counteract any time associations). In each trial, a white light and a tone (100 GHz, 100 dB) were presented simultaneously for 10 s in the compartment where the animal stayed and were used as the conditioned stimulus (CS). After 5 s of the CS, mice received a 0.2 mA electric footshock as the unconditioned stimulus (US) for a maximal duration of 10 s. An avoidance response was defined as when the animal crossed to the opposite compartment of the box after the CS started but before the US was delivered. Crossings while the shock was being delivered were considered escape responses. Response latencies were counted as the time (in seconds) from the onset of the CS until the animal crossed into the opposite compartment. The number of crosses during the ITI was determined as a measure of general activity. The test session was performed 3 d after the end of the training phase, on day 10 of the experiment. The apparatus was cleaned with water after each animal.
Passive avoidance.
This test was performed as described previously (Pittenger et al., 2006). Mice were placed into the passive avoidance box (Ugo Basile) with two different compartments, one dark and black and the other illuminated and white. On the first test day, we measured how long the mice spent in the lighted compartment. As soon as the animal crossed to the dark compartment, the automatic door closed, and mice received an electrical footshock (0.4 or 0.8 mA, 1 s). At 1 and 24 h after this first trial with footshock, animals were tested in the box using the same conditions without the electrical shock.
Fear conditioning and extinction.
This behavioral task was performed as described previously (Alarcón et al., 2004). On training day, mice were placed in the conditioning chamber for 2 min before onset of the CS, a 30 s tone. During the last 2 s of the tone, the US, an electrical shock of 0.7 mA, was presented. Mice were maintained in the chamber for an additional 30 s and returned to the home cage. Conditioning was tested 24 h later by measuring freezing behavior with a tracking video system (Panlab). Mice were re-placed into the conditioning chamber, and the freezing time was measured for 5 min without the tone to assess contextual conditioning. Mice were returned to home cages for 3 h and placed into a novel chamber to test cued fear conditioning. After 1 min in the novel context, the tone was presented for 30 s, and freezing time was measured for 2 min.
To study fear extinction, the US was modified to achieve equal freezing times in the two genotypes. The new US consisted of three consecutive electrical shocks (0.7 mA for 2 s, with 2 min intershock intervals) delivered in the conditioning chamber followed by a 2 min measurement of freezing time before returning animals to home cages. Extinction was assessed by measuring freezing for 5 min after the animals were re-placed in the same conditioning chamber, every day for 6 consecutive days.
Sensitivity to electric shock.
This test was performed as described by El-Ghundi et al. (2001). Briefly, mice were subjected to a series of mild footshocks with gradually increasing amperage (0.02, 0.04, 0.06, 0.08, 0.1, 0.15, 0.2, 0.25, 0.3, 0.4, 0.5, 0.6 mA). Duration of footshock was 1 s with 20 s intershock intervals. For each mouse, we determined the shock intensity that produced each of the following initial sensation responses: sniffing and staring at the floor bars, licking and biting the floor bars, alternately lifting the paws off the floor, startle response, jumping, and vocalization.
Elevated plus maze.
We used an apparatus with four 30 × 5 cm arms, elevated 50 cm above the floor (Cibertec). Two arms were enclosed by 15 cm walls, and the other two had a 3 mm edge to prevent slipping. All arms were illuminated equally. A 5 × 5 platform at the center was considered a neutral area. Animals were habituated to the experimenter, and 1 h before the experiment, animals were placed in the testing room. At the beginning of the test, animals were placed in the center of the maze facing the open arm and allowed to explore for 5 min. The percentage of time spent in the open arms compared with the total time minus time in the center as well as the number of entries to the open arms was used as the primary measure of anxiety-like behavior.
Nociceptive thresholds.
We used three different nociceptive tests: hot plate, plantar, and tail immersion tests. We used the plantar test apparatus (Ugo Basile) to measure paw withdrawal latencies in response to radiant heat (55°C). A cutoff time of 20 s was used to prevent tissue damage in the absence of response. Mean paw withdrawal latencies were determined from the average of three separate trials, taken at 5 min intervals in each group of mice. The hot plate test was performed with a hot plate apparatus (Ugo Basile) at 52°C. We measured the time (in seconds) to paw licking or paw withdrawal in response to heat. For the tail immersion test, about 3 cm of the distal part of the tail was immersed into a temperature-controlled water bath (52 ± 0.5°C). Latency was the time from tail immersion until it was removed or vigorously pulled away. The cutoff time was 20 s to prevent tissue damage.
Construction of Lenti-Drd1a-siRNAs.
To silence dopamine D1R expression in vitro and in vivo, three sequences were designed, targeted to different regions of the Drd1a mRNA sequence: (1) bp 82–89; (2) bp 1322–1329; (3) bp 720–729. These targets were selected based on Hannon's design criterion as indicated previously (Bahi et al., 2004a; Dreyer, 2010; Ramiro-Fuentes et al., 2010). An XhoI restriction site was added at the 3′ end of each oligo, and a U6–3′-specific 10mer was added at the 5′ end. Using the pSilencer 1.0-U6 (Ambion) as a template and a U6 promoter-specific forward primer containing the BamHI restriction site (GCGGATCCCGCTCTAGAACTAGTGC), each siRNA target was added to the mouse U6 promoter by PCR. Initial denaturation was 120 s at 94°C, followed by 35 cycles of the following program: 45 s at 94°C, 45 s at 64°C, and 45 s at 72°C. The PCR contained 4% dimethyl sulfoxide (Sigma). PCR products were digested with BamHI and XhoI, cloned into similar sites in pTK431, and sequenced to verify the integrity of each construct.
Lentivirus production.
The vector plasmids pTK-Drd1a-siRNA and pTK433-GFP together with the packaging construct plasmid pΔNRF and the envelope plasmid pMDG-VSV-G were cotransfected into HEK293T cells to produce the viral particles, Lv-GFP or Lv-Drd1a-siRNA (Bahi et al., 2004a,b). Viral titers were determined by p24 antigen measurements (KPL). For in vivo experiments, viral stocks were matched for viral particle content and used at 2 × 109 particles/μl.
Determination of lentivirus silencing efficiency in vitro.
The efficiency of the lentiviruses at silencing was tested in vitro in HEK293T cells. A total of 1 × 105 HEK293T cells were plated per well in six-well plates. The next day, lentivirus (Lv) stocks were mixed with 10 μg/ml Polybrene (Sigma), incubated for 30 min at room temperature, added to the cells, and incubated at 37°C. After 48 h, the medium was replaced with normal growth medium, and cells were left for an additional 48 h. Cells were then collected, and total RNA was isolated for real-time PCR. For in vitro silencing of Drd1a, cells were infected with 4 μl of Lv preparation, either 4 μl of Lenti-GFP used as control or 2 μl of Lenti-GFP plus 2 μl of one of the three Lenti-Drd1a-siRNA or plus 2 μl of all three Lenti-Drd1a-siRNAs together.
Quantitative real-time PCR.
Primer sets for rat and mouse Drd1a, Drd2, and Gapdh were designed to amplify 100 to 200 bp products. The following specific primer pairs were used: Drd1a, 5′-AGGATTGCCCAGAAGCAAAT-3′ and 5′-GGGCACCATACAGTTCGAGA-3′; Drd2, 5′-CATTGTCTGGGTCCTGTTCCT-3′ and 5′-GACCAGCAGAGTGACGATGA-3′; Gapdh, 5′-ATGACTCTACCCACGGCAAG-3′ and 5′-CATACTCAGCACCAGCATCAC-3′. Gapdh was used as an endogenous control for normalization. Total RNA was extracted from the HEK293T cells (for in vitro quantification) or from the brains of treated animals (for in vivo quantification) using TRIzol reagent (Invitrogen) including an RNase-free DNase step. RNA was quantified by spectrophotometry, and its integrity was verified by agarose gel electrophoresis visualized with ethidium bromide. First-strand cDNA was generated from 2 μg of total RNA and Oligo (dT12–18) primer with the Moloney murine leukemia virus reverse transcription kit (Invitrogen) in a total volume of 20 μl. Quantitative real-time PCR was performed in a real-time PCR iCycler (Bio-Rad). Five microliters of cDNA, 0.5 μm of forward and reverse primers, and 10 μl of IQ SYBR Green Supermix (Bio-Rad) were combined in a total volume of 20 μl. PCR was performed as follows: 3 min at 95°C (initial denaturation); 20°C/s temperature transition up to 95°C for 45 s, 45 s at 62°C, repeated for 40 cycles (amplification). The PCR was evaluated by melting-curve analysis and by checking the PCR products on a 2% agarose gel.
The PCR cycle number at which each assay target reached the threshold detection line was determined (“threshold cycles,” Ct value). The Ct of each gene was normalized against that of Gapdh or B-actin. To determine the linearity and detection limit of the assay, successive 10-fold dilutions of each cDNA sample were amplified in a series of real-time PCRs, using duplicate assays for each dilution, so that the correlation coefficient could be calculated from the standard curve of Ct values. Comparisons were made between the different animal groups, and significance was calculated using two-tailed Student's t test. The level of statistical significance was set at p < 0.05. Data were expressed as mean ± SEM. The ΔCt for each candidate was calculated as ΔCt = [Ct (candidate) − Ct (Gapdh or B-actin)]. The relative abundance of each target in each protocol was calculated as the ratio between treated and untreated samples (Bahi and Dreyer, 2004; Bahi et al., 2004b; Mühlbauer et al., 2004).
Western blotting.
The hippocampal region surrounding the siRNA injection site was dissected, homogenized in buffer (50 mm Tris, 300 mm NaCl, 1% Triton X-100, 25 mm NaF, 1 mm sodium orthovanadate, 4 mm sodium pyrophosphate, 1 mm EDTA, and 1 tablet of Complete protease inhibitor; Roche), and incubated at 4°C with shaking for 20 min. The samples were centrifuged at 10,000 × g at 4°C for 15 min. The protein concentration of the supernatants was quantified using the Bradford reaction. Equal amounts of total protein were subjected to SDS-PAGE, transferred to nitrocellulose membranes, and Western blotted following standard protocols. Membranes were blocked with 5% BSA in Tris-buffered saline–Tween 20, incubated with a primary antibody against D1R (1:1000; Santa Cruz Biotechnology) overnight at 4°C, washed again, incubated for 1 h at room temperature with a peroxidase-conjugated secondary antibody, and visualized using ECL (GE Healthcare) and exposed to film for 1 min. Membranes were stripped, washed, and reprobed with glyceraldehyde-3-phosphate dehydrogenase (1:1000; Abcam). Exposed films were digitized and quantified with Quantity One software.
Surgery.
As illustrated in Figure 7, we performed input–output curves, paired-pulse facilitation, and the LTP study in one set of four groups of animals: wild type, Drd1a−/−, WT-GFP, and Drd1a-siRNA (n = 10 animals per group). We performed classical trace eyeblink conditioning in a separate set of animals including the same four genotypes/conditions (also n = 10 animals per group) (see Fig. 8).
Figure 7.

Input–output curves, paired-pulse facilitation, and LTP induction in the CA1 area in wild type, Drd1a−/−, WT-GFP, and Drd1a-siRNA. A, Representative example of an input–output curve collected from a Drd1a−/− mouse. Stimulus consisted of paired (S1, S2) pulses (40 ms interpulse interval) presented at increasing intensities in 20 μA steps. Note the sigmoid shape of S1 + S2 value. B, The four experimental groups presented similar sigmoid curves for S1 + S2 values (n = 10 animals per group). Representative sample records collected from the four experimental groups are illustrated on the right. C, Paired-pulse facilitation of fEPSPs recorded in the CA1 area after stimulation of Schaffer collaterals. The data shown are mean ± SEM. slopes of the second fEPSP expressed as a percentage of the first for the six interpulse intervals. No significant differences were observed between the four experimental groups. Extracellular fEPSP paired traces were collected from a representative Drd1a-siRNA animal at the indicated interstimulus intervals. D, Top, Representative fEPSPs recorded from WT and Drd1a−/− animals before (baseline), and 15–30 min (1) and 24 h (2) after HFS. Graphs illustrate the time course of changes in fEPSPs (mean ± SEM) after HFS stimulation of the Schaffer collaterals. The HFS train was presented after 15 min of baseline recordings, at the time indicated by the dashed line. fEPSP slopes are expressed as a percentage of the baseline (100%) slope. WT mice exhibited significantly greater LTP than Drd1a−/− mice (*p < 0.001). E, Same analysis as in D for WT-GFP and Drd1a-siRNA groups. Here again, the control group (WT-GFP) presented significantly larger LTP than Drd1a-siRNA mice (*p < 0.01). St., Stimulating electrode.
Figure 8.
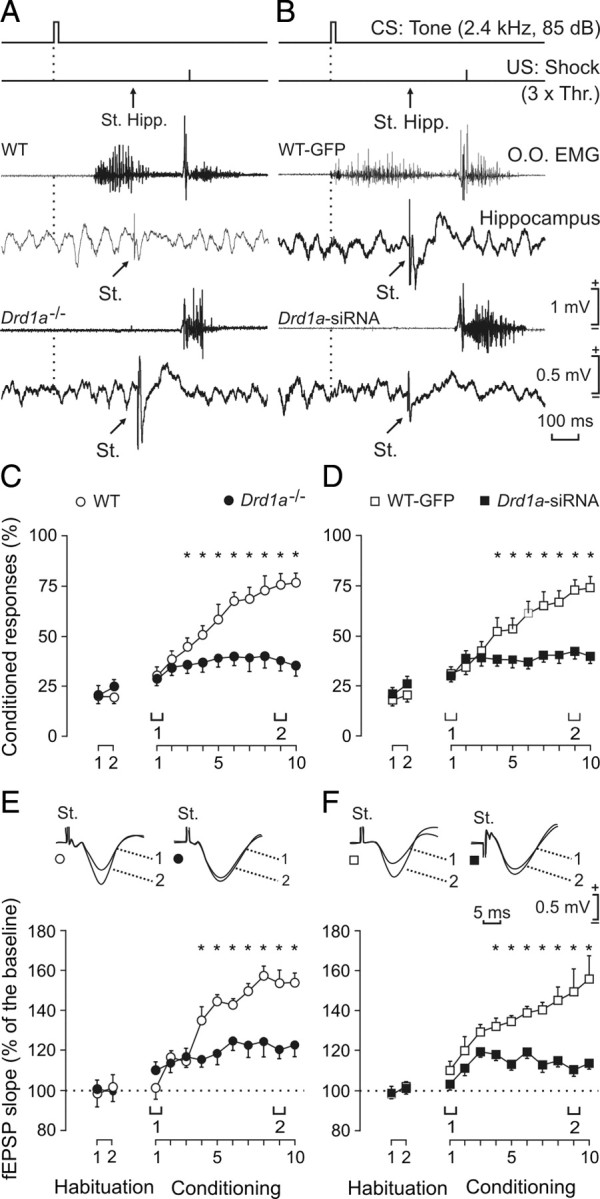
Evolution of CA3–CA1 synaptic field potentials and learning curves for WT, Drd1a−/−, WT-GFP, and Drd1a-siRNA. A, B, Top to bottom, the conditioning paradigm, representative EMG, and representative hippocampal recordings during paired CS–US presentations for WT and Drd1a−/− mice (A) and for WT-GFP and Drd1a-siRNA mice (B). The time of stimulus presentation at Schaffer collaterals (St. Hipp.) is indicated, as are the times of delivery of CS (dashed line) and US. Data shown were collected during the ninth conditioning session. 3× Thr., Three times threshold. C, D, Percentage of eyelid CRs reached by the four experimental groups. The acquisition curve presented by the WT group (open circles) was significantly larger than values reached by the Drd1a−/− group (C, filled circles; *p < 0.001). Similarly, the acquisition curve of the WT-GFP group was also significantly larger than that presented by Drd1a-siRNA animals (D; *p < 0.001). E, F, Evolution of fEPSPs evoked at the CA3–CA1 synapse across conditioning for WT and Drd1a−/− mice (E) and for WT-GFP and Drd1a-siRNA animals (F). fEPSP slope is expressed as a percentage of fEPSP slope during habituation for each group. Differences in fEPSP slopes between WT and Drd1a−/− groups were statistically significant from the 4th to the 10th conditioning sessions (E; *p < 0.006), as well as between WT-GFP and Drd1a-siRNA animals (F; *p < 0.001), indicating that activity-dependent synaptic plasticity was severely impaired in both Drd1a−/− and Drd1a-siRNA mice. St., Stimulating electrode.
Animals were anesthetized with 0.8–3% halothane (AstraZeneca). The gas mixture was delivered using a small anesthesia mask (David Kopf Instruments) connected to a calibrated Fluotec 5 (Fluotec-Ohmeda) vaporizer at a flow rate of 1–4 L/min oxygen. In the first surgical step, animals in groups WT-GFP and Drd1a-siRNA received a sterotaxic injection of 2 μl of Lv-GFP (WT-GFP) or a mix of Lv-Drd1a-siRNAs of concentrated lentiviral stocks (2 × 109 particles/μl) into the hippocampus. The injection was performed with a Hamilton syringe and performed unilaterally at the following coordinates, calculated from bregma and skull surface: anterior, −2.4; lateral, +1.5 (right side); ventral, −2.0 (Paxinos and Franklin, 2001).
As illustrated in Figure 1A, all animals included in the eight groups mentioned above were implanted with bipolar stimulating electrodes in the right Schaffer collateral–commissural pathway of the dorsal hippocampus (2 mm lateral and 1.5 mm posterior to bregma; depth from the brain surface, 1.0–1.5 mm) (Paxinos and Franklin, 2001) and with a recording electrode in the ipsilateral stratum radiatum underneath the CA1 area (1.2 mm lateral and 2.2 mm posterior to bregma; depth from the brain surface, 1.0–1.5 mm). These electrodes were made of 50 μm Teflon-coated tungsten wire (Advent Research Materials). The final position of hippocampal electrodes was determined as described previously (Gruart et al., 2006). The recording electrode was implanted in the CA1 area using as a guide the field potential depth profile evoked by paired (40 ms interval) pulses presented to the ipsilateral Schaffer collateral pathway. The recording electrode was fixed at the site where a reliable monosynaptic (≤5 ms) fEPSP was recorded.
Figure 1.

Experimental design for classical conditioning and LTP. Classical eyelid conditioning was achieved with a trace paradigm, using a tone as a CS. The loudspeaker was located 30 cm from the animal's head. Animals were implanted with bipolar stimulating electrodes on the left supraorbital nerve for US presentations. Eyelid conditioned responses were recorded with EMG electrodes implanted in the ipsilateral orbicularis oculi (O.O.) muscle. The top diagram illustrates that animals were also implanted with stimulating (St.) and recording (Rec.) electrodes to activate Schaffer collaterals and to record fEPSPs evoked at the pyramidal CA1 area of the right hippocampus and indicates the injection point for Drd1a-siRNA. A, Photomicrographs illustrating the location of stimulating and recording electrodes and lentivirus injection site. Scale bars, 200 μm. DG, Dentate gyrus; Sub., subiculum; D, dorsal; L, lateral; M, medial; V, ventral. B, The two sets of traces on the left illustrate the following: 1, a fEPSP evoked at the CA3–CA1 synapse; 2, an EMG recording evoked at the O.O. muscle by a single suprathreshold pulse presented to the supraorbital nerve. Both traces were collected during the ninth conditioning session of a control animal. Calibrations are as indicated.
Animals selected for the classical conditioning of eyelid responses were also implanted with stimulating electrodes on the left supraorbital nerve and with recording electrodes in the ipsilateral orbicularis oculi muscle (Fig. 1A). Electrodes were made of 50 μm Teflon-coated, annealed stainless steel wire (A-M Systems) bared at the tips for ∼0.5 mm. The tips were bent into a hook to facilitate stable insertion in the upper eyelid.
A 0.1 mm bare silver wire was affixed to the skull as a ground. All the wires were connected to two four-pin sockets (RS-Amidata). The sockets were fixed to the skull with the help of two small screws and dental cement. The implantation procedures used in this chronic preparation have been described in detail (Gruart et al., 2006). Experimental sessions started 1 week after surgery. To verify the location of stimulating and recording electrodes after completion of experiments, mice were deeply reanesthetized (50 mg/kg sodium pentobarbital) and perfused/fixed transcardially with saline and 4% phosphate-buffered paraformaldehyde. Selected brain sections (50 μm thick) including the dorsal hippocampus were obtained in a microtome (Leica), mounted on gelatinized glass slides, and Nissl stained with 0.1% toluidine blue.
Electrophysiology.
Recordings were made using six differential amplifiers with a bandwidth of 0.1 Hz to 10 kHz (P511; Grass-Telefactor) (Fig. 1B). Hippocampal recordings were made with a high-impedance probe (2 × 1012 Ω, 10 pF) (Fig. 1B).
For input–output curves, the stimulus intensity was raised to 0.3 mA in steps of 20 μA. The selected interstimulus interval was 40 ms, because it results in maximum facilitation of the CA3–CA1 synapse (Madroñal et al., 2007). For paired-pulse facilitation, pulse intensity (50–400 μA) was set at 30–40% of the amount necessary to evoke a maximum fEPSP response, and the following interstimulus intervals were used: 10, 20, 40, 100, 200, and 500 ms (Gureviciene et al., 2004). To avoid unwanted interactions between successive pairs of stimuli, the interpulse delay was always ≥20 s.
For evoking LTP, we used a high-frequency stimulation (HFS) train consisting of five 200 Hz, 100 ms trains of pulses at a rate of one per second. This protocol was presented six times, at intervals of 1 min. As indicated above for paired-pulse facilitation, pulse intensity was set at 30–40% of the amount necessary to evoke a maximum fEPSP response for baseline recordings and after the HFS train. To avoid evoking a population spike and/or unwanted EEG seizures, the stimulus intensity during the HFS train was set at the same intensity used for generating baseline records. Before presenting the animals with the HFS train, we collected baseline records for 15 min, by presenting single pulses (a 100 μs, square, negative–positive pulse) at a rate of one per 20 s. After the HFS train, we presented the same set of pulses for 30 min. An additional recording session lasting for 15 min was performed 24 h after the HFS session.
Classical eyeblink conditioning.
For classical conditioning, using a trace paradigm, three animals at a time were placed in separate small (5 × 5 × 10 cm) plastic chambers located inside a larger (30 × 30 × 20 cm) Faraday box. Classical conditioning was achieved using a trace paradigm consisting of a tone (20 ms, 2.4 kHz, 85 dB) presented as a CS. The US consisted of a cathodal, square pulse applied to the supraorbital nerve (500 μs, three times the threshold) 500 ms after the end of the CS. A total of two habituation and 10 conditioning sessions were performed for each animal. A conditioning session consisted of 60 CS–US presentations and lasted ∼30 min. For proper observation of conditioned response (CR) profiles, the CS was presented alone in 10% of the cases. CS–US presentations were separated at random by 30 ± 5 s. For habituation sessions, only the CS was presented, at the same frequency of 30 ± 5 s. Our criteria for CR were the presence, during the CS–US interval, of EMG activity lasting >10 ms and initiated > 50 ms after CS onset. In addition, the integrated EMG activity recorded during the CS–US interval had to be at least 2.5 times greater than the averaged activity recorded immediately before CS presentation (Porras-García et al., 2005). The total number of CRs per session was computed and expressed as a percentage of the maximum (60 CRs per session equal 100%).
Synaptic field potentials in the CA1 area were evoked during habituation and conditioning sessions by a single 100 μs square, biphasic (negative–positive) pulse applied to Schaffer collaterals 300 ms after CS presentation. Stimulus intensities ranged from 50 to 250 μA. For each animal, the stimulus intensity was selected according to data collected from the input–output curves, usually at ∼30% of the intensity necessary for evoking a maximum fEPSP response (Gureviciene et al., 2004). An additional criterion for selecting stimulus intensity was that a second stimulus, presented 40 ms after a conditioning pulse, evoked a larger (>20%) synaptic field potential (Bliss and Gardner-Medwin, 1973).
Immnohistochemistry for Egr1.
Mice used in the electrophysiology studies were fixed in 4% paraformaldehyde overnight, and brains were cut into 30 μm sections. Immunohistochemistry was done in free-floating sections with standard avidin–biotin immunocytochemical protocols (Rivera et al., 2002; Grande et al., 2004; Pavón et al., 2006; Granado et al., 2008) with specific polyclonal rabbit antisera raised against Egr1, formerly Zif268 (diluted 1:400; Santa Cruz Biotechnology). To enhance the staining, after incubation with the primary (one night) and secondary (2 h) antisera, sections were incubated for 1 h in a streptavidin–peroxidase complex (diluted 1:2000 in PBS-Triton X-100; Sigma). Peroxidase reactions were developed in 0.05% 3,3′-diaminobenzidine (Sigma) and 0.002% H2O2. Sections were then mounted on gelatin-coated slides, air dried, dehydrated in graded series of ethanol, cleared in xylene, and coverslipped with Permount mounting medium. Quantification of Egr1-positive nuclei in hippocampal sections was performed using an image analysis system (AIS; Imaging Research). Before counting, images were thresholded at a standardized gray-scale level, empirically determined by two different observers to allow detection of stained nuclei from low to high intensity, with suppression of the very lightly stained nuclei. The number of nuclei positive for Egr1 was determined and expressed as the number of positive nuclei per square millimeter (Granado et al., 2008). Counts were obtained from four hippocampal slices per animal (n = 10–12), for each group.
Statistical analysis.
EMG and hippocampal activity, and 1 V rectangular pulses corresponding to CS and US presentations, were stored digitally on a computer through an analog/digital converter (1401 Plus; CED), at a sampling frequency of 11–22 kHz and an amplitude resolution of 12 bits. Commercial computer programs (Spike 2 and SIGAVG; CED) were modified to represent EMG and fEPSP recordings. Data were analyzed off-line for quantification of CRs and fEPSP slope using custom representation programs (Porras-García et al., 2005; Gruart et al., 2006). Computed data were processed for statistical analysis using the SPSS for Windows package. Unless otherwise indicated, data are represented as the mean ± SEM. Acquired data were analyzed using a two-way ANOVA, with group, session, or time as the repeated measure. Contrast analysis was added to further study significant differences. Regression analysis was used to study the relationship between the fEPSP slopes and the percentage of CRs.
Statistics on behavioral values to assess genotype and trial differences in the Barnes maze, active and passive avoidance, and the fear test were performed using repeated-measures, two-way ANOVA where genotype (wild type and Drd1a−/−) and time (day of trials for passive avoidance or freezing test) were entered as independent variables. Relevant differences were analyzed pairwise by post hoc comparisons with Tukey's test. Immunohistochemical and Western blotting studies were analyzed using the Student's t test. For all statistical studies, SigmaStat 2.03 software was used, and the threshold for statistical significance was set at p < 0.05.
Results
Barnes maze
To confirm the role of D1R in spatial learning, we used the Barnes maze because it is less aversive and stressful than the water maze (Barnes, 1979; Harrison et al., 2009), and because previous studies have shown that the same mouse strain can perform differently in different spatial learning tasks (Patil et al., 2009; Zheng et al., 2009). In the Barnes maze, WT mice quickly learn to escape the open field and reach the black escape box, as shown by the rapid decline in escape latency (Fig. 2A). By day 7 of training, escape latency has reached a minimum that was maintained throughout the training phase (11 d) and during the probe trial, 3 d later. In contrast, there was no reduction in escape latency for the Drd1a−/− mice, even after an 11 d training period (Fig. 2A).
Figure 2.

Hippocampus-dependent learning is impaired in dopamine Drd1a−/− mice. Data show the mean values ± SEM. A, Progression of escape latency during the training phase in the Barnes maze. Drd1a−/− mice did not reduce escape latency at any time during the experiment (*p < 0.005). B, Immobility during the first day of training. WT and Drd1a−/− mice showed similar levels of immobility. C, Probe trial performed 3 d after the training phase. Histograms represent the time spent searching for the escape hole. Drd1a−/− mice did not reduce searching time during the probe trial (*p < 0.005). D, Escape latency during the relearning phase. For this test, the escape hole was located opposite to its position in the training phase (*p < 0.001). Statistics were determined with repeated-measures two-way ANOVA followed by Tukey's test for post hoc analysis (A, D) and with Student's t test (C).
To rule out the possibility that inactivation of Drd1a increases anxiety levels in these mice, masking their capacity to respond in the Barnes maze, we evaluated the immobility time during the first day of training in the Barnes maze, as an indirect measure of anxiety. We chose the first day of training because on this day, the two groups showed similar latency times for crossing to the black escape box. WT and Drd1a−/− mice spend similar amounts of time immobile during the first day of training (Fig. 2B). Moreover, in the elevated-plus maze, which is commonly used as a direct test of anxiety, Drd1a−/− mice do not show higher anxiety levels than their WT littermates, as demonstrated by the number of entries and the time spent in the open arms (Fig. 3A).
Figure 3.

Anxiety levels are similar in both genotypes, but Drd1a−/− mice are more sensitive to pain than WT mice. A, Anxiety-like behavior of Drd1a−/− and WT mice illustrated by the number of entries and percentage of total time (mean ± SEM) spent in the open arms of the elevated plus maze test. Drd1a−/− mice make more entries and spend more time in the open arms, indicating lower anxiety levels than the WT mice. *p < 0.05 versus WT mice. B, Pain sensitivity thresholds (in seconds, mean ± SEM) of mice in tail flick, hot plate, and plantar tests. Drd1a−/− mice exhibit lower pain thresholds than WT mice in all three tests, indicating higher pain sensitivity. *p < 0.05 versus WT mice; n = 8–10 animals.
In addition, Drd1a−/− mice showed no reduction in escape latency in a probe trial performed 3 d after training, to evaluate memory consolidation (Fig. 2C), or during the relearning trials, when the escape hole was moved to the opposite side of the training arena (Fig. 2D). With the previous study in the Morris maze (Granado et al., 2008), these data indicate that the D1R is required for spatial learning in more than one paradigm. Our demonstration that loss of D1R does not increase indicators of anxiety supports the notion that our results are attributable to an important role of D1R in spatial learning.
Associative learning is impaired in Drd1a−/− mice
Active avoidance
Dopamine depletion impairs the acquisition and maintenance of conditioned avoidance responses (Shannon et al., 1999), suggesting that dopamine receptors are involved in this behavior. To determine whether the dopamine D1R plays a role in this associative learning task, we used the two-way active avoidance paradigm. In this paradigm, WT mice learned the avoidance response within the first 2 d of training, while Drd1a−/− mice were unable to learn it, even with an extensive period of training (Fig. 4). Differences between the two groups were first evident on the second day of training (p < 0.001) and persisted throughout the experiment, demonstrating complete impairment of avoidance learning in Drd1a−/− mice (Fig. 4A).
Figure 4.

Active avoidance performance is impaired in dopamine Drd1a−/− mice. Data shown are mean values ± SEM. A, Progression of active avoidance responses during the training phase. Drd1a−/− mice did not increase the number of avoidance responses during the training phase (*p < 0.001.). B, Time course of crossing latencies for WT and Drd1a−/− mice during the training phase. Drd1a−/− mice did not decrease escape latency at any point during training (*p < 0.001). C, Number of intertrial crosses. From day 3 on, there was no significant difference between WT and Drd1a−/− mice in the number of intertrial crosses. Statistics were performed with repeated-measures two-way ANOVA, followed by post hoc analysis with Tukey's test.
The crossing latency reflects how rapidly the animal crosses to the safe compartment after the onset of the CS to avoid the footshock (Smith et al., 2002). WT animals progressively reduced their crossing latency, whereas Drd1a−/− mice did not, again indicating that Drd1a−/− mice were unable to learn that the footshock would follow the CS (Fig. 4B). Differences in latency between WT and Drd1a−/− mice were first observed on the second day of the training phase (p < 0.001).
Except for the first 2 d, we found no difference between wild type and Drd1a−/− in baseline crossing behavior, determined by counting crossings during the ITIs in the training phase or on the test day (Fig. 4C). Thus, the poor performance of the Drd1a−/− mice in this paradigm suggests impaired associative learning rather than changes in locomotor behavior.
Passive avoidance
Passive avoidance learning depends on multiple cortical and subcortical structures, including both dorsal and ventral striatum as well as hippocampus and amygdala (Pittenger et al., 2006). In this test, avoidance response or entry latency increases with footshock intensity (Crawley, 2007). First, we determined the sensitivity to footshock for both genotypes by gradually increasing footshock intensity (0.01–0.6 mA) and monitoring the onset of behavioral indicators of sensation or pain. Wild type and Drd1a−/− showed similar sensitivity thresholds to footshock (Fig. 5A), responding with a sudden stare at floor bars, startle response, and jumping and vocalization at the same footshock intensities in both genotypes. Jumping and vocalization responses, indicative of pain threshold, were elicited with 0.12 mA in both groups of mice. Passive avoidance experiments were performed with a moderate (0.4 mA) and a strong (0.8 mA) electric stimulus (Viosca et al., 2009), both well above the pain threshold (Fig. 5A). Baseline entry latency times in the passive avoidance test were similar in all experimental groups. However, after training with either 0.4 or 0.8 mA footshock, Drd1a−/− mice exhibited a shorter latency than WT mice (Fig. 5B), indicative of reduced memory strength. When animals were tested 24 h after 0.4 mA footshock, WT mice showed a latency time of 250 s compared with 123 s in Drd1a−/− mice (p < 0.001). With an 0.8 mA shock, this difference was smaller but still statistically significant (p < 0.05) (Fig. 5B). To exclude the possibility that Drd1a−/− are simply less sensitive to electric shock/pain than WT animals, we determined pain thresholds for both genotypes using tail flick, hot plate, and plantar tests. In all three assays, the pain threshold was actually lower for Drd1a−/− than WT mice (Fig. 3B), indicating that Drd1a−/− mice are actually more sensitive to footshock than WT mice and supporting the conclusion that our results in the passive avoidance test reflect an important role of Drd1a−/− in this type of associative learning.
Figure 5.

Performance in the passive avoidance test and fear conditioning are impaired in Drd1a−/− mice. Data show mean ± SEM. A, Thresholds for footshock responses. Increasing intensity footshocks were delivered to WT and Drd1a−/− mice, and the threshold for each listed behavior was determined. Thresholds for all three response behaviors were similar in the two genotypes. B, Avoidance response. Latency refers to the time spent in the light compartment before mice enter the dark compartment, which was paired with footshock in a single training trial. Drd1a−/− mice show partial impairment of passive avoidance at both 0.4 and 0.8 mA. C, Cued and contextual fear conditioning are impaired in Drd1a−/− mice. Freezing time was measured in contextual and cued fear conditioning trials 24 h after training. *p < 0.01 and **p < 0.001 versus wild type; #p < 0.01 and ##p < 0.001 versus Pre-shock (0 h). Statistics were determined by repeated-measures two-way ANOVA followed by post hoc analysis with Tukey's test.
Fear conditioning learning
We studied the role of dopamine D1Rs in fear memory using both contextual fear conditioning, which is dependent on both the hippocampus and the amygdala, and cued fear conditioning, which is dependent only on the amygdala. Our protocol allowed us to test both contextual and cued fear conditioning in one experiment. Baseline levels of freezing were equal in wild type and Drd1a−/− during training, as were levels of freezing after footshock. Then, 24 h after training, animals were reexposed to the training context. As expected, freezing levels in WT mice were significantly elevated to 60% of the total time. Although freezing was also elevated in Drd1a−/− mice, it was much lower than in WT mice. Drd1a−/− mice spent only 22% of the time freezing (Fig. 5C). When animals were exposed to a new context, both genotypes decreased their freezing levels compared with context testing levels, but freezing levels in WT mice remained significantly higher than in Drd1a−/−. When the CS (tone) was presented in the new context, both genotypes increased their freezing levels significantly, but again, WT mice spent significantly more time freezing than Drd1a−/− (Fig. 5C). Altogether, these results indicate that the D1R is necessary for cued and contextual associative learning.
To study extinction, we wanted to start with equal freezing times in the two genotypes because 24 h after a single footshock, freezing times were significantly different in wild type and Drd1a−/−. Therefore, 48 h after the first footshock, we delivered three consecutive footshocks separated by 30 s, which resulted in equal freezing times in both genotypes. After this, mice were tested daily in the same context for 6 consecutive days, revealing similar extinction curves for both genotypes: by 1 week after the footshocks, both groups of mice had reduced freezing time by 50% (data not shown). These results suggest that D1R is not necessary for extinction.
In vitro and in vivo siRNA-mediated knockdown of dopamine D1R
To rule out developmental effects of the absence of the D1R in Drd1a−/− mice, we used Lv-based RNA interference to knock down dopamine D1R expression in adult animals. We designed three siRNAs targeted against different regions of the Drd1a mRNA. These were inserted into the transfer plasmid of the Lv system, and their efficiency at silencing Drd1a was assessed in HEK293T cells (Fig. 6A). Then, 4 d after infection, Drd1a transcripts were measured by quantitative reverse transcription (RT)-PCR. Infection with Lv-Drd1a-siRNA 1, 2, or 3 individually resulted in dopamine Drd1a downregulation, yielding residual Drd1a−/− expression levels of 40, 15, and 60%, respectively. However, coinfection with all three Lv-Drd1a-siRNAs decreased Drd1a mRNA expression by >93% (Fig. 6A). Control infection with Lv-GFP did not alter Drd1a expression in these cells (Fig. 6A).
Figure 6.
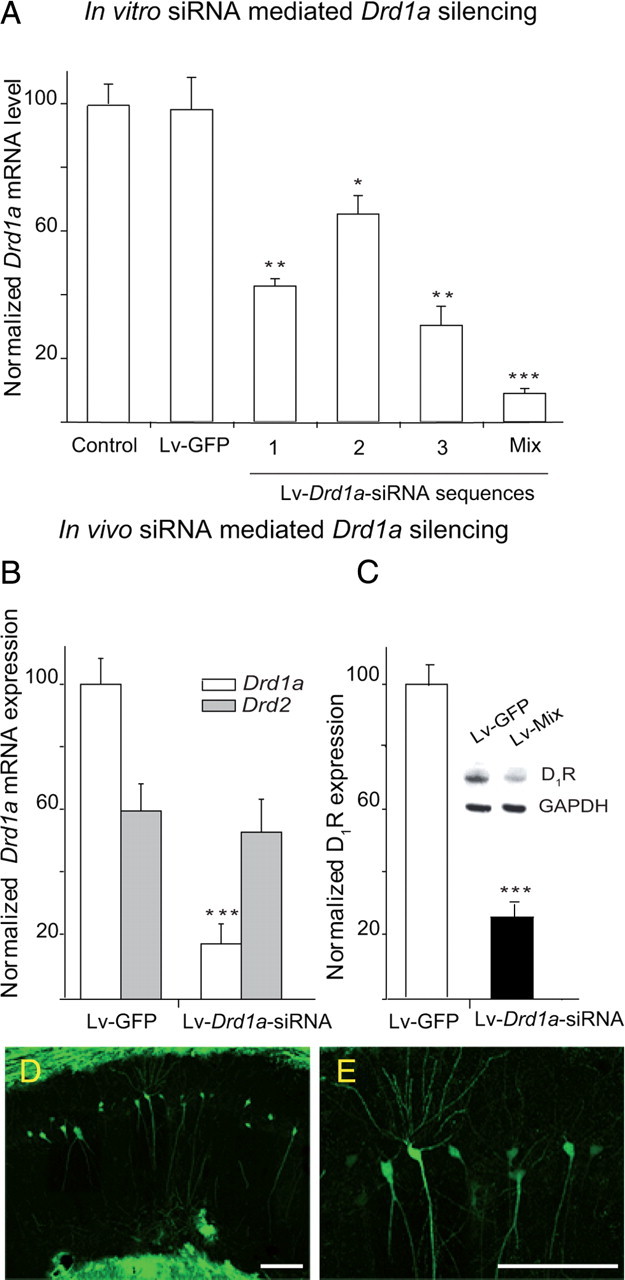
siRNA-mediated Drd1a silencing in vitro and in vivo. A, Drastic reduction of Drd1a mRNA expression in HEK cells in vitro after infection with Drd1a-siRNA constructs. mRNA levels were determined by quantitative RT-PCR 48 h after infection and normalized to Gapdh mRNA levels. *p < 0.05; **p < 0.01; ***p < 0.005; one-way ANOVA. B, Drd1a and Drd2 mRNA levels in NAc 48 h after intra-accumbal injection of a mixture of all three Drd1a-siRNAs. mRNA levels were determined by RT-PCR, normalized to Gapdh, and expressed as a percentage of Drd1a mRNA expression in Lv-GFP-injected animals. Injection of Drd1a-siRNAs specifically decreased Drd1a mRNA expression. C, D1R protein expression in hippocampus 48 h after injection of Drd1a-siRNAs. D1R protein levels were decreased ∼75%. ***p < 0.001, Student's t test. D, Photomicrograph of a coronal brain section illustrating the spread of lentiviral infection in the CA1 layer of the hippocampus of WT mice injected with 2 μl of Lenti-GFP particles. E, High-magnification image of infected pyramidal cells illustrated in D. Particles infect most of the dorsal hippocampus spreading through pyramidal CA1 cell layer. Scale bar, 100 μm.
To assess whether Lv-Drd1a-siRNAs can deplete D1R expression in vivo, the nucleus accumbens (NAc) and the hippocampus were injected stereotaxically with the mix of Lv-Drd1a-siRNAs (2 μl) or with Lv-GFP as a control. Two weeks after the injection, there were dramatic decreases in D1R protein or gene (Drd1a−/−) expression in these regions in Drd1a-siRNA-injected mice compared with animals given injections of Lv-GFP (WT-GFP). We include the NAc in this experiment because the expression of D1R in this nucleus is higher than that in the hippocampus, and this allows us to better assess the capacity of our vectors to silence D1R. Quantitative RT-PCR revealed an 80% decrease in Drd1a mRNA expression in the NAc (Fig. 6B), and Western blotting revealed a 73% decline in D1R protein in the hippocampus (Fig. 6C). Both decreases were statistically significant compared to WT-GFP. Drd2 receptor mRNA expression was not affected, indicating that the Drd1a siRNA mix was highly specific for dopamine D1R (Fig. 6B). We determined the spread of the virus within the hippocampus in the WT-GFP-injected mice using immunohistochemistry and found that particles infected ∼2 mm2 along the rostrocaudal axis, infecting most of the dorsal hippocampus, including the pyramidal cell layer and dentate gyrus (Fig. 6D,E).
In vivo basal synaptic transmission is normal in Drd1a−/− and in Drd1a-siRNA mice
We measured fEPSPs evoked at the CA3–CA1 synapse by in vivo electrical stimulation of Schaffer collaterals in the following groups of mice: wild type, Drd1a−/−, wild type injected with Lv-Drd1a−/−-siRNA (Drd1a-siRNA), and WT-GFP. Input–output curves using a wide range of stimulus intensities (0.02–0.4 mA) and paired (S1, S2) pulses at a fixed interval of 40 ms revealed no significant differences in basal synaptic transmission between the four groups (Fig. 7A,B). Interestingly, the combined value of fEPSPs evoked by both stimuli (S1 plus S2) presented an exponential relationship (r ≤ 0.98; p < 0.0001) with stimulus intensity, with no significant differences (p ≥ 0.1925) between the four groups. The stimulus intensities used in the remainder of this study were selected from within a range of 30–40% of the saturating intensity, intensities able to evoke facilitation of the second pulse.
Paired-pulse facilitation in vivo is normal in Drd1a−/−and in Drd1a-siRNA mice
Using the double-pulse test with interpulse intervals ranging from 10 to 500 ms, we found a significant (p < 0.001) increase in slope of fEPSPs evoked by the second pulse at short time intervals (20, 40, and 100 ms). There were no significant differences between WT, Drd1a−/−, WT-GFP, and Drd1a-siRNA mice (p = 0.959) (Fig. 7C). These results are similar to those obtained in Drd1a−/− in vitro (Granado et al., 2008) and confirm that D1Rs are not involved in this form of very short-term plasticity.
Hippocampal LTP in vivo is significantly reduced in Drd1a−/− and in Drd1a-siRNA mice
To explore the role of the D1R in hippocampal LTP, we compared CA3–CA1 fEPSPs after HFS of Schaffer collaterals in WT, Drd1a−/−, WT-GFP, and Drd1a-siRNA mice (Fig. 7D,E). To determine baseline responses, Schaffer collaterals were stimulated every 20 s for 15 min. The stimulus consisted of a 100 μs square, negative–positive pulse. After HFS, the same single stimulus was presented every 20 s for 30 min and repeated 24 h later, for 15 min. We found significant LTP in WT and WT-GFP mice. As expected, 15 min after HFS, the response to the single stimulus in both WT and WT-GFP groups was >200% of baseline values (p < 0.001) (Fig. 7D,E, open circles). Significant LTP persisted at 24 h post-HFS (p < 0.001) (Fig. 7D,E). In contrast, Drd1a−/− showed no LTP at the CA3–CA1 synapse after HFS. Indeed, 15–30 min after HFS, fEPSP slopes in Drd1a−/− mice were not significantly different (∼140%; p = 0.138) from baseline (Fig. 7D, filled circles). In Drd1a-siRNA mice, the fEPSP slope was not significantly different from baseline at any time after HFS (p = 0.146) (Fig. 7E, filled squares). These data suggest that D1R plays a crucial role in induction of LTP at the CA3–CA1 synapse after HFS of Schaffer collaterals in vivo.
Classical trace eyeblink conditioning is significantly reduced in Drd1a−/− and Drd1a-siRNA mice
First, we verified that the neural premotor circuits involved in the generation of eyelid responses function normally in the four groups of animals used in this study (wild type, Drd1a−/−, WT-GFP, and Drd1a-siRNA). As illustrated in Figure 1B2, the blink reflex is easily characterized by measuring the latency of its early (R1) and late (R2) components (Kugelberg, 1952) and the corresponding integrated EMG areas (Gruart et al., 1995). Eyeblinks evoked by electrical stimulation of the ipsilateral supraorbital nerve in the WT group presented values (R1 latency, 4.9 ± 1.3 ms; R2 latency, 11.2 ± 3.3 ms; R1+R2 integrated area, 79.2 ± 15.6 μV × s) in the range of previous descriptions in mice (Gruart et al., 2006). There were no significant differences between our four groups of mice in the baseline blink reflex (p ≤ 0.713).
To investigate the possible behavioral consequences of the deficit in synaptic plasticity at the CA3–CA1 synapse observed in Drd1a−/− and Drd1a-siRNA mice, we evaluated classical conditioning of eyeblink responses in the four groups of experimental animals using a trace paradigm (CS, tone; US, shock) with a 500 ms interval between the end of the CS and the beginning of the US (Fig. 8A,B). In WT mice, the percentage of CRs increased significantly across conditioning sessions (p < 0.001) (Fig. 8C), with a profile similar to that reported previously (Takatsuki et al., 2003; Gruart et al., 2006). In contrast, the percentage of CRs in the Drd1a−/− group was not significantly different from habituation values (p = 0.431) at any point over the course of conditioning. The percentage of CRs presented by the WT group was significantly different from that of the Drd1a−/− group from the 4th to the 10th conditioning session (p < 0.001) (Fig. 8C). Similarly, WT-GFP animals presented learning curves similar to those seen in WT mice, whereas CRs in Drd1a-siRNA mice were not significantly above baseline (p < 0.001) (Fig. 8D).
Evolution of CA3–CA1 fEPSP across classical conditioning: inactivation of D1R impairs CA3–CA1 synaptic efficiency induced during classical conditioning
As shown recently in behaving mice, trace eyeblink conditioning is associated with increases in synaptic efficiency at CA3–CA1 synapses (Gruart et al., 2006). We therefore evaluated the effect of D1R loss on CA3–CA1 fEPSPs. Electrical stimulation of Schaffer collaterals 300 ms after CS presentation evoked a fEPSP in the CA1 area in all four experimental groups (Fig. 8A,B). Although the stimuli presented to Schaffer collaterals disrupted the regular theta rhythm recorded in the CA1 area, the rhythm reappeared in phase ∼200 ms afterward. The slope of the evoked fEPSPs increased in the four experimental groups over the course of conditioning (Fig. 8E,F). These increases in fEPSP slopes across conditioning sessions were only observed for recordings collected during the CS–US interval with the animal placed in the recording box (i.e., fEPSPs were not modified by the mere repetitive stimulation of Schaffer collaterals).
There were clear differences between the two control (WT and WT-GFP) groups and the two experimental (Drd1a−/− and Drd1a-siRNA) groups. By the fourth conditioning session, fEPSP slopes recorded in WT (143%) and WT-GFP (134%) mice were significantly elevated over baseline values (p < 0.001) (Fig. 8E,F), consistent with previous studies (Gruart et al., 2006). In contrast, from the fourth conditioning session on, although the slopes of evoked fEPSPs in Drd1a−/− (114%) and Drd1a-siRNA (116%) mice were slightly (although nonsignificantly) elevated over baseline, they were significantly smaller than in corresponding control animals (Fig. 8E,F). Thus, the decreased performance in associative learning tasks seen in Drd1a−/− and Drd1a-siRNA mice compared with their respective controls is paralleled by a dramatic decline in activity-dependent increases in synaptic efficiency at the CA3–CA1 synapse during classical conditioning.
In vivo HFS-induced expression of Egr1 in the hippocampus is reduced in Drd1a−/−and Drd1a-siRNA mice
Hippocampal Egr1/zif268 expression is required for late LTP and memory formation (Guzowski et al., 2000; Hall et al., 2001; Jones et al., 2001; Kelly and Deadwyler, 2003; McIntyre et al., 2005; Granado et al., 2008). Our results reveal that the D1R is required for CA3–CA1 LTP induced by HFS of the Schaffer collaterals, as well as for classical eyeblink conditioning and the associated increase in CA3–CA1 synaptic efficiency. To explore whether Egr1 induction by HFS in vivo requires dopamine D1R, we examined Egr1 protein expression in the hippocampus of WT, Drd1a−/−, Drd1a-siRNA, and WT-GFP mice after HFS of the Schaffer collaterals. Basal expression of Egr1 in the hippocampus was similar in all groups (Fig. 9A–D, Table 1), with a moderate number of neurons expressing Egr1 constitutively. Then, 24 h after in vivo tetanic stimulation of the CA3 cell layer, Egr1 expression increased significantly in the hippocampus of WT and WT-GFP animals (Fig. 9, E and G, respectively) compared with nonstimulated animals (Fig. 9A,C). Staining was particularly intense in cells along the dorsal CA1 region. Genetic inactivation or siRNA silencing of Drd1a significantly inhibited induction of Egr1 expression after in vivo tetanic stimulation (Fig. 9F and H, respectively). Quantification revealed that HFS-induced Egr1 expression decreased by ∼50% in Drd1a−/− mice and by 60% in Drd1a-siRNA mice (Table 1). These results indicate that expression of D1R is important for induction of Egr1 by tetanization in vivo in the rodent hippocampus.
Figure 9.

D1R is required for activity-induced Egr1 in CA1 pyramidal cells after HFS of Schaffer collaterals. Photomicrographs of the CA1 pyramidal cell layer of the hippocampus from coronal brain slices showing immunohistochemistry for the Egr1 transcription factor are presented. A–D, Egr1 expression under basal conditions for WT, Drd1a−/−, WT-GFP, and Drd1a-siRNA mice, respectively. E–H, Expression 24 h after in vivo HFS of the Schaffer collaterals in the hippocampus of WT, Drd1a−/−, WT-GFP, and Drd1a-siRNA mice, respectively. Scale bar, 50 μm.
Table 1.
HFS-induced Egr1 expression in the hippocampus of alive mice
| Egr1-positive nuclei/mm2 |
||||
|---|---|---|---|---|
| WT | Drd1a−/− | WT-GFP | Drd1a-siRNA | |
| Basal | 103 ± 39 | 143 ± 78 | 341 ± 101 | 134 ± 103 |
| HFS | 1418 ± 106** | 734 ± 124*# | 1637 ± 112** | 643 ± 163*#Δ |
Quantification of HFS-induced Egr1 expression in hippocampal slices is shown. Immunostained cells were counted in hippocampal sections (examples in Fig. 9) obtained from different animals (n = 12 for each group). Numbers indicate immunostained nuclei per square millimeter (mean ± SEM).
*p < 0.05;
**p < 0.0005 compared with the same group of animals in basal conditions.
#p < 0.01 compared with HFS WT mice.
Δp < 0.01 compared with HFS WT-GFP mice. Statistics were determined by ANOVA followed by post hoc analysis with Tukey's test.
Discussion
Our principal finding is that associative learning is abolished or dramatically reduced in Drd1a−/− mice and mice given intrahippocampal injections of Lv-Drd1a-siRNA. In addition, both LTP induced at the CA3–CA1 synapse by in vivo HFS of the Schaffer collaterals and the physiological potentiation that occurs at CA3–CA1 synapses in parallel with acquisition of classical conditioning of eyelid responses are significantly impaired in Drd1a−/− and Drd1a-siRNA mice. These data extend our previous finding that the D1R is necessary for evoking early and late LTP in vitro (Granado et al., 2008). More importantly, our finding that acquisition of associative learning and the physiological increase in synaptic strength at the CA3–CA1 synapse in alert behaving mice are drastically reduced in D1R-depleted mice provides evidence for a functional relationship between associative learning and synaptic plasticity in this region.
It has been suggested that the Barnes maze discriminates spatial learning more clearly than the Morris water maze (Barnes, 1979; Harrison et al., 2009; Patil et al., 2009; Zheng et al., 2009) and is useful when the Morris water maze shows partial impairment. Drd1a−/− mice were tested in the Barnes maze to consolidate the finding that D1R is necessary for spatial learning in the Morris maze. Our results are in agreement with previous results from our laboratory (Granado et al., 2008) and others (Smith et al., 1998). Unlike a previous water maze study in which Drd1a−/− mice did learn after extended training (El-Ghundi et al., 1999), we saw no decrease in the escape latency for Drd1a−/− mice in the Barnes maze, even after extended training. Two indicators of anxiety level, immobility during the first day of training in the Barnes maze and performance in the elevated plus maze, revealed no increase in anxiety in the knock-out animals, so it is unlikely that the impaired performance of Drd1a−/− mice in the Barnes maze is attributable to an effect of the knockout on anxiety. Our results confirm the crucial role of D1R in spatial learning.
While spatial memory is hippocampus-dependent, other forms of associative learning also depend on the amygdala. To further assess the role of D1R in associative learning, we used three paradigms that evaluate fear memory. In the active avoidance test, Drd1a−/− mice did not decrease their escape latency at any point during the training trials. Similar random crossing scores for WT and Drd1a−/− mice indicate that this finding is not caused by decreased locomotor activity. The results of our passive avoidance trials also confirm that this learning impairment was not caused by locomotor deficits or freezing. In passive avoidance, Drd1a−/− showed some learning at 0.4 or 0.8 mA but were significantly impaired compared with WT mice 1 and 24 h after training. Sensitivity to footshock was similar in WT and Drd1a−/−, so altered sensitivity or pain threshold was not a factor. Thus, the avoidance impairment associated with D1R inactivation is likely attributable to abnormal acquisition of learning or to abnormal short-term memory retrieval, but not to deficits in memory consolidation. These mice showed similar avoidance responses at 1 and 24 h after the shock, indicating that they are able to retrieve what they have learned.
We used the fear conditioning test to discriminate between hippocampus- and amygdala-dependent associative learning. Drd1a−/− animals were impaired in both contextual (hippocampus-dependent) and cued (hippocampus- and amygdala-dependent) fear conditioning, with relatively less impairment in amygdala-dependent learning. These data are consistent with our passive avoidance test results and complement our other findings. However, they contradict a previous study that found increased fear-induced freezing in Drd1a−/− mice (El-Ghundi et al., 2001). This discrepancy is likely caused by differences in methodology.
To study extinction, we increased the footshock intensity to achieve similar retention in both groups of mice. Under these conditions, we found no difference between WT and Drd1a−/− mice in extinction of fear conditioning, consistent with a previous report (El-Ghundi et al., 2001). The same report showed that using higher-intensity footshocks (two consecutive 0.9 mA, 3 s shocks; four times at 5 min intervals) significantly increased retention (El-Ghundi et al., 2001) in Drd1a−/− mice. Together, these studies suggest that with moderate intensity stimuli, D1R are important for fear conditioning, but at higher intensities, D1R-independent mechanisms predominate. Similar results were obtained with Egr1 mutant mice, which exhibit deficits in spatial memory that can be rescued by extensive training (Jones et al., 2001).
It is widely accepted that hippocampal circuits are involved in the acquisition of classically conditioned eyelid responses (Berger et al., 1983; Thompson, 1988; Moyer et al., 1990; Gruart et al., 2006). Using unitary in vivo recordings, hippocampal pyramidal cell firing in response to CS presentations increases across conditioning sessions (McEchron and Disterhoft, 1997; Múnera et al., 2001; McEchron et al., 2003). Recently, it has been shown that trace eyeblink conditioning evokes a concomitant change in fEPSP strength at the hippocampal CA3–CA1 synapse in behaving mice (Gruart et al., 2006). We found that both acquisition of classically conditioned eyelid responses and the increase in CA3–CA1 synaptic strength across training are severely impaired in Drd1a−/− and Drd1a-siRNA mice, suggesting the D1R plays an important role in hippocampal mechanisms related to learning and memory. These data support our previous finding that the D1R is critical for certain forms of hippocampal synaptic plasticity in vitro (Granado et al., 2008). In addition, they also support the notion that learning and activity-dependent synaptic plasticity at the CA3–CA1 synapse are functionally related.
Both the generation of eyelid CRs and related changes in CA3–CA1 synaptic strength require a large number (>300) of paired CS-US presentations (Woody, 1986). In contrast, LTP is a fundamental property of most excitatory synapses in the mammalian brain that can be evoked by HFS of selected afferent pathways. Recent studies showed that trace eyeblink conditioning and the associated change in CA3–CA1 synaptic strength are prevented by HFS of the Schaffer collateral pathway, suggesting that the evoked LTP maximizes synaptic efficiency, occluding subsequent learning (Gruart et al., 2006). Moreover, learning-evoked changes in synaptic strength interfere with subsequent induction of LTP by HFS (Whitlock et al., 2006). Thus, these three processes (associative learning, activity-dependent synaptic plasticity, and LTP) seem to be functionally related (Gruart et al., 2006; Madroñal et al., 2007). In support of this, we found that D1R are also necessary for evoking LTP of the CA3–CA1 synapse after HFS of afferent Schaffer collaterals. Since input–output curves and paired-pulse potentiation were normal in both Drd1a−/− and Drd1a-siRNA mice, we conclude that the D1R does not affect normal transmission but contributes to synaptic plasticity during the acquisition and storage of new information.
We observed a marked reduction in Egr1 protein expression 24 h after in vivo tetanic stimulation of hippocampal CA3–CA1 in Drd1a−/− and Drd1a-siRNA mice. This inhibition is in agreement with the decrease in LTP in D1R-depleted mice and indicates that hippocampal D1R expression is necessary for these processes, consistent with results obtained in hippocampal slices from classical Drd1a−/− mice (Granado et al., 2008). Our results are consistent with previous data showing that inactivation of Egr1 blocks long-term memory and LTP maintenance (Davis et al., 2000; Guzowski et al., 2000; Jones et al., 2001). In addition, the decrease in Egr1 expression after tetanic stimulation in Drd1a−/− indicates that the signaling mechanisms linking synaptic activation in dendrites with nuclear gene expression require D1R activation in the hippocampus (Granado et al., 2008) and other brain areas including the striatum (Darmopil et al., 2009).
The mechanism by which D1R mediates associative learning is not known. One possibility is that D1R-triggered cAMP signaling phosphorylates CREB (cAMP response element-binding protein), which activates the histone acetyl transferase enzyme CREB-binding protein (Mayr et al., 2001; Alarcón et al., 2004), inducing expression of immediate-early genes, including Egr1 and arc, which play a crucial role in reference memory (Guzowski et al., 2000; Jones et al., 2001). Alternatively, D1Rs could directly phosphorylate NMDA receptors or the protein kinase C ζ isoform in hippocampus and amygdala, potentiating and maintaining synaptic strength (Gardner et al., 2001; Impey et al., 2002; Kelleher et al., 2004; Sacktor, 2008; Yao et al., 2008) by increasing the influx of calcium induced by NMDA receptor activation, as occurs in prefrontal cortex neurons (Kruse et al., 2009). Direct interaction between D1 and NMDA receptors within the plasma membrane of pyramidal neurons may also occur, since D1R can physically interact with NR1 and NR2A subunits, modulating receptor trafficking (Lee et al., 2002; Pei et al., 2004; Fiorentini et al., 2006). Coactivation of both receptors increases the presence of GluR1 receptors and facilitates their incorporation into synapses in hippocampal neurons (Smith et al., 2005; Gao et al., 2006; Sacktor, 2008). It is also possible that D1Rs are selectively activated by burst firing of dopamine neurons, mainly during the presentation of salient events (Zweifel et al., 2009).
This work contributes to our emerging understanding of dopamine system function in learning and memory by firmly establishing the critical role of the D1R in associative learning and the endogenous synaptic potentiation that accompanies it. In addition, it strengthens the proposed functional link between learning, synaptic potentiation, and hippocampal Egr1 expression.
Footnotes
This work was supported by Grant PI071073 from Plan Nacional Sobre Drogas from the Spanish Ministerio de Sanidad y Política Social and Spanish Ministerio de Ciencia e Innovación Grants BFU2010-20664 (R.M.) and BFU2005-01024 and BFU2005-02512 (J.M.D.-G. and A.G.). O.O. was supported by a Basque Government Ph.D. fellowship. We thank M. Esteban, E. Rubio, and M. de Mesa for technical assistance and Dr. Angel Barco for help and advice with the fear-conditioning test.
References
- Alarcón JM, Malleret G, Touzani K, Vronskaya S, Ishii S, Kandel ER, Barco A. Chromatin acetylation, memory, and LTP are impaired in CBP+/− mice: a model for the cognitive deficit in Rubinstein-Taybi syndrome and its amelioration. Neuron. 2004;6:947–959. doi: 10.1016/j.neuron.2004.05.021. [DOI] [PubMed] [Google Scholar]
- Bahi A, Dreyer JL. Cocaine-induced expression changes of axon guidance molecules in the adult rat brain. Mol Cell Neurosci. 2004;28:275–291. doi: 10.1016/j.mcn.2004.09.011. [DOI] [PubMed] [Google Scholar]
- Bahi A, Boyer F, Kafri T, Dreyer JL. CD81-induced behavioural changes during chronic cocaine administration: in vivo gene delivery with regulatable lentivirus. Eur J Neurosci. 2004a;19:1621–1633. doi: 10.1111/j.1460-9568.2004.03260.x. [DOI] [PubMed] [Google Scholar]
- Bahi A, Boyer F, Gumy C, Kafri T, Dreyer JL. In vivo gene delivery of urokinase-type plasminogen activator with regulatable lentivirus induces behavioural changes in chronic cocaine administration. Eur J Neurosci. 2004b;20:3473–3488. doi: 10.1111/j.1460-9568.2004.03771.x. [DOI] [PubMed] [Google Scholar]
- Baldwin AE, Sadeghian K, Kelley AE. Appetitive instrumental learning requires coincident activation of NMDA and dopamine D1 receptors within the medial prefrontal cortex. J Neurosci. 2002;22:1063–1071. doi: 10.1523/JNEUROSCI.22-03-01063.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnes CA. Memory deficits associated with senescence: a neurophysiological and behavioral study in the rat. J Comp Physiol Psychol. 1979;93:74–104. doi: 10.1037/h0077579. [DOI] [PubMed] [Google Scholar]
- Berger TW, Rinaldi P, Weisz DJ, Thompson RF. Single-unit analysis of different hippocampal cell types during classical conditioning of rabbit nictitating membrane response. J Neurophysiol. 1983;50:1197–1219. doi: 10.1152/jn.1983.50.5.1197. [DOI] [PubMed] [Google Scholar]
- Bliss TVP, Gardner-Medwin AR. Long-lasting potentiation of synaptic transmission in the dentate area of the unanaesthetized rabbit following stimulation of the perforant path. J Physiol. 1973;232:357–374. doi: 10.1113/jphysiol.1973.sp010274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crawley JN. Hoboken, NJ: Wiley; 2007. What's wrong with my mouse? Behavioral phenotyping of transgenic and knock out mice. [Google Scholar]
- Darmopil S, Martín AB, De Diego IR, Ares S, Moratalla R. Genetic inactivation of dopamine D1 but not D2 receptors inhibits L-DOPA-induced dyskinesia and histone activation. Biol Psychiatry. 2009;6:603–613. doi: 10.1016/j.biopsych.2009.04.025. [DOI] [PubMed] [Google Scholar]
- Darvas M, Palmiter RD. Restriction of dopamine signaling to the dorsolateral striatum is sufficient for many cognitive behaviors. Proc Natl Acad Sci U S A. 2009;106:14664–14669. doi: 10.1073/pnas.0907299106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis S, Vanhoutte P, Pages C, Caboche J, Laroche S. The MAPK/ERK cascade targets both Elk-1 and cAMP response element-binding protein to control long-term potentiation-dependent gene expression in the dentate gyrus in vivo. J Neurosci. 2000;20:4563–4572. doi: 10.1523/JNEUROSCI.20-12-04563.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dreyer JL. Lentiviral vector-mediated gene transfer and RNA silencing technology in neuronal dysfunctions. Methods Mol Biol. 2010;614:3–35. doi: 10.1007/978-1-60761-533-0_1. [DOI] [PubMed] [Google Scholar]
- Dubois B, Pillon B. Cognitive deficits in Parkinson's disease. J Neurol. 1997;244:2–8. doi: 10.1007/pl00007725. [DOI] [PubMed] [Google Scholar]
- El-Ghundi M, Fletcher PJ, Drago J, Sibley DR, O'Down BF, George SR. Spatial learning deficit in dopamine D1 receptor knockout mice. Eur J Pharmacol. 1999;383:95–106. doi: 10.1016/s0014-2999(99)00573-7. [DOI] [PubMed] [Google Scholar]
- El-Ghundi M, O'Dowd BF, George SR. Prolonged fear responses in mice lacking dopamine D1 receptor. Brain Res. 2001;892:86–93. doi: 10.1016/s0006-8993(00)03234-0. [DOI] [PubMed] [Google Scholar]
- Fiorentini C, Rizzetti MC, Busi C, Bontempi S, Collo G, Spano P, Missale C. Loss of synaptic D1 dopamine/N-methyl-D-aspartate glutamate receptor complexes in L-DOPA-induced dyskinesia in the rat. Mol Pharmacol. 2006;69:805–812. doi: 10.1124/mol.105.016667. [DOI] [PubMed] [Google Scholar]
- Gao C, Sun X, Wolf ME. Activation of D1 dopamine receptors increases surface expression of AMPA receptors and facilitates their synaptic incorporation in cultured hippocampal neurons. J Neurochem. 2006;5:1664–1677. doi: 10.1111/j.1471-4159.2006.03999.x. [DOI] [PubMed] [Google Scholar]
- Gardner B, Liu ZF, Jiang D, Sibley DR. The role of phosphorylation/ dephosphorylation in agonist-induced desensitization of D1 dopamine receptor function: evidence for a novel pathway for receptor dephosphorylation. Mol Pharmacol. 2001;59:310–321. doi: 10.1124/mol.59.2.310. [DOI] [PubMed] [Google Scholar]
- Granado N, Ortiz O, Suárez LM, Martín ED, Ceña V, Solís JM, Moratalla R. D1 but not D5 dopamine receptors are critical for LTP, spatial learning, and LTP-Induced arc and zif268 expression in the hippocampus. Cereb Cortex. 2008;18:1–12. doi: 10.1093/cercor/bhm026. [DOI] [PubMed] [Google Scholar]
- Grande C, Zhu H, Martín AB, Lee M, Ortíz O, Hiroi N, Moratalla R. Chronic treatment with atypical neuroleptics induces striosomal FosB/DeltaFosB expression in rats. Biol Psychiatry. 2004;55:457–463. doi: 10.1016/j.biopsych.2003.08.008. [DOI] [PubMed] [Google Scholar]
- Gruart A, Blázquez P, Delgado-García JM. Kinematics of unconditioned and conditioned eyelid movements in the alert cat. J Neurophysiol. 1995;74:226–248. doi: 10.1152/jn.1995.74.1.226. [DOI] [PubMed] [Google Scholar]
- Gruart A, Muñoz MD, Delgado-García JM. Involvement of the CA3–CA1 synapse in the acquisition of associative learning in behaving mice. J Neurosci. 2006;26:1077–1087. doi: 10.1523/JNEUROSCI.2834-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gureviciene I, Ikonen S, Gurevicius K, Sarkaki A, van Groen T, Pussinen R, Ylinen A, Tanila H. Normal induction but accelerated decay of LTP in APP + PS1 transgenic mice. Neurobiol Dis. 2004;15:188–195. doi: 10.1016/j.nbd.2003.11.011. [DOI] [PubMed] [Google Scholar]
- Guzowski JF, Lyford GL, Stevenson GD, Houston FP, McGaugh JL, Worley PF, Barnes CA. Inhibition of activity-dependent arc protein expression in the rat hippocampus impairs the maintenance of long-term potentiation and the consolidation of long-term memory. J Neurosci. 2000;20:3993–4001. doi: 10.1523/JNEUROSCI.20-11-03993.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall J, Thomas KL, Everitt BJ. Cellular imaging of Egr1 expression in the hippocampus and amygdala during contextual and cued fear memory retrieval: selective activation of hippocampal CA1 neurons during the recall of contextual memories. J Neurosci. 2001;21:2186–2193. doi: 10.1523/JNEUROSCI.21-06-02186.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrison FE, Hosseini AH, McDonald MP. Endogenous anxiety and stress responses in water maze and Barnes maze spatial memory tasks. Behav Brain Res. 2009:198247–198251. doi: 10.1016/j.bbr.2008.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Impey S, Fong AL, Wang Y, Cardinaux JR, Fass DM, Obrietan K, Wayman GA, Storm DR, Soderling TR, Goodman RH. Phosphorylation of CBP mediates transcriptional activation by neural activity and CaM kinase IV. Neuron. 2002;34:235–244. doi: 10.1016/s0896-6273(02)00654-2. [DOI] [PubMed] [Google Scholar]
- Jones MW, Errington ML, French PJ, Fine A, Bliss TV, Garel S, Charnay P, Bozon B, Laroche S, Davis S. A requirement for the immediate early gene Egr1 in the expression of late LTP and long-term memories. Nat Neurosci. 2001;4:289–296. doi: 10.1038/85138. [DOI] [PubMed] [Google Scholar]
- Kelleher RJ, 3rd, Govindarajan A, Jung HY, Kang H, Tonegawa S. Translational control by MAPK signaling in long-term synaptic plasticity and memory. Cell. 2004;116:467–479. doi: 10.1016/s0092-8674(04)00115-1. [DOI] [PubMed] [Google Scholar]
- Kelly MP, Deadwyler SA. Experience-dependent regulation of the immediate-early gene arc differs across brain regions. J Neurosci. 2003;23:6443–6451. doi: 10.1523/JNEUROSCI.23-16-06443.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kruse MS, Prémont J, Krebs MO, Jay TM. Interaction of dopamine D1 with NMDA NR1 receptors in rat prefrontal cortex. Eur Neuropsychopharmacol. 2009;19:296–304. doi: 10.1016/j.euroneuro.2008.12.006. [DOI] [PubMed] [Google Scholar]
- Kugelberg E. Facial reflexes. Brain. 1952;75:385–396. doi: 10.1093/brain/75.3.385. [DOI] [PubMed] [Google Scholar]
- Lee FJ, Xue S, Pei L, Vukusic B, Chery N, Wang Y, Wang YT, Niznik HB, Yu XM, Liu F. Dual regulation of NMDA receptor functions by direct protein-protein interactions with the dopamine D1 receptor. Cell. 2002;18:219–230. doi: 10.1016/s0092-8674(02)00962-5. [DOI] [PubMed] [Google Scholar]
- Levin BE, Katzen HL. Early cognitive changes and nondementing behavioral abnormalities in Parkinson's disease. Adv Neurol. 2005;98:84–94. [PubMed] [Google Scholar]
- Li S, Cullen WK, Anwyl R, Rowan MJ. Dopamine-dependent facilitation of LTP induction in hippocampal CA1 by exposure to spatial novelty. Nat Neurosci. 2003;6:526–531. doi: 10.1038/nn1049. [DOI] [PubMed] [Google Scholar]
- Lisman JE, Grace AA. The hippocampal-VTA loop: controlling the entry of information into long-term memory. Neuron. 2005;46:703–713. doi: 10.1016/j.neuron.2005.05.002. [DOI] [PubMed] [Google Scholar]
- Madroñal N, Delgado-García JM, Gruart A. Differential effects of long-term potentiation evoked at the CA3–CA1 synapse before, during, and after the acquisition of classical eyeblink conditioning in behaving mice. J Neurosci. 2007;27:12139–12146. doi: 10.1523/JNEUROSCI.3397-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madroñal N, Gruart A, Delgado-García JM. Differing presynaptic contributions to LTP and associative learning in behaving mice. Front Behav Neurosci. 2009;3:7. doi: 10.3389/neuro.08.007.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayr BM, Canettieri G, Montminy MR. Distinct effects of cAMP and mitogenic signals on CREB-binding protein recruitment impart specificity to target gene activation via CREB. Proc Natl Acad Sci U S A. 2001;98:10936–10941. doi: 10.1073/pnas.191152098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McEchron MD, Disterhoft JF. Sequence of single neuron changes in CA1 hippocampus of rabbits during acquisition of trace eyeblink conditioned responses. J Neurophysiol. 1997;78:1030–1044. doi: 10.1152/jn.1997.78.2.1030. [DOI] [PubMed] [Google Scholar]
- McEchron MD, Tseng W, Disterhoft JF. Single neurons in CA1 hippocampus encode trace interval duration during trace heart rate (fear) conditioning in rabbit. J Neurosci. 2003;23:1535–1547. doi: 10.1523/JNEUROSCI.23-04-01535.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McIntyre CK, Miyashita T, Setlow B, Marjon KD, Steward O, Guzowski JF, McGaugh JL. Memory-influencing intra-basolateral amygdala drug infusions modulate expression of Arc protein in the hippocampus. Proc Natl Acad Sci U S A. 2005;102:10718–10723. doi: 10.1073/pnas.0504436102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moratalla R, Xu M, Tonegawa S, Graybiel AM. Cellular responses to psychomotor stimulant and neuroleptic drugs are abnormal in mice lacking the D1 dopamine receptor. Proc Natl Acad Sci U S A. 1996;93:14928–14933. doi: 10.1073/pnas.93.25.14928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moyer JRJ, Deyo RA, Disterhoft JF. Hippocampectomy disrupts trace eye-blink conditioning in rabbits. Behav Neurosci. 1990;104:243–252. doi: 10.1037//0735-7044.104.2.243. [DOI] [PubMed] [Google Scholar]
- Mühlbauer M, Allard B, Bosserhoff AK, Kiessling S, Herfarth H, Rogler G, Schömerich J, Jobin C, Hellerbrand C. Differential effects of deoxycholic acid and taurodeoxycholic acid on NF-κB signal transduction and IL-8 gene expression in colonicepithelial cells. Am J Physiol Gastrointest Liver Physiol. 2004;286:G1000–G1008. doi: 10.1152/ajpgi.00338.2003. [DOI] [PubMed] [Google Scholar]
- Múnera A, Gruart A, Muñoz MD, Fernández-Más R, Delgado-García JM. Discharge properties of identified CA1 and CA3 hippocampus neurons during unconditioned and conditioned eyelid responses in cats. J Neurophysiol. 2001;86:2571–2582. doi: 10.1152/jn.2001.86.5.2571. [DOI] [PubMed] [Google Scholar]
- O'Carroll CM, Morris RG. Heterosynaptic co-activation of glutamatergic and dopaminergic afferents is required to induce persistent long-term potentiation. Neuropharmacology. 2004;47:324–332. doi: 10.1016/j.neuropharm.2004.04.005. [DOI] [PubMed] [Google Scholar]
- O'Carroll CM, Martin SJ, Sandin J, Frenguelli B, Morris RG. Dopaminergic modulation of the persistence of one-trial hippocampus-dependent memory. Learn Mem. 2006;13:760–769. doi: 10.1101/lm.321006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otmakhova NA, Lisman JE. D1/D5 dopamine receptor activation increases the magnitude of early long-term potentiation at CA1 hippocampal synapses. J Neurosci. 1996;16:7478–7486. doi: 10.1523/JNEUROSCI.16-23-07478.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmiter RD. Dopamine signaling in the dorsal striatum is essential for motivated behaviors: lessons from dopamine-deficient mice. Ann N Y Acad Sci. 2008;1129:35–46. doi: 10.1196/annals.1417.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patil SS, Sunyer B, Höger H, Lubec G. Evaluation of spatial memory of C57BL/6J and CD1 mice in the Barnes maze, the Multiple T-maze and in the Morris water maze. Behav Brain Res. 2009;198:58–68. doi: 10.1016/j.bbr.2008.10.029. [DOI] [PubMed] [Google Scholar]
- Pavón N, Martín AB, Mendialdua A, Moratalla R. ERK phosphorylation and FosB expression are associated with L-DOPA-induced dyskinesia in hemiparkinsonian mice. Biol Psychiatry. 2006;59:64–74. doi: 10.1016/j.biopsych.2005.05.044. [DOI] [PubMed] [Google Scholar]
- Paxinos G, Franklin KBJ. The mouse brain in stereotaxic coordinates. London: Academic; 2001. [Google Scholar]
- Pei L, Lee FJ, Moszczynska A, Vukusic B, Liu F. Regulation of dopamine D1 receptor function by physical interaction with the NMDA receptors. J Neurosci. 2004;4:1149–1158. doi: 10.1523/JNEUROSCI.3922-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pittenger C, Fasano S, Mazzocchi-Jones D, Dunnett SB, Kandel ER, Brambilla R. Impaired bidirectional synaptic plasticity and procedural memory formation in striatum-specific cAMP response element-binding protein-deficient mice. J Neurosci. 2006;10:2808–2813. doi: 10.1523/JNEUROSCI.5406-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porras-García E, Cendelin J, Domínguez-del-Toro E, Vožeh F, Delgado-García JM. Purkinje cell loss affects differentially the execution, acquisition and prepulse inhibition of skeletal and facial motor responses in Lurcher mice. Eur J Neurosci. 2005;21:979–988. doi: 10.1111/j.1460-9568.2005.03940.x. [DOI] [PubMed] [Google Scholar]
- Ramiro-Fuentes S, Ortiz O, Moratalla R, Fernandez-Espejo E. Intra-accumbal rimonabant is rewarding but induces aversion to cocaine in cocaine-treated rats, as does in vivo CB1R silencing: critical role for glutamate receptors. Neuroscience. 2010;167:205–215. doi: 10.1016/j.neuroscience.2010.02.019. [DOI] [PubMed] [Google Scholar]
- Rivera A, Alberti I, Martin AB, Narvaez JA, de la Calle A, Moratalla R. Molecular phenotype of rat striatal neurons expressing the dopamine D5 receptor subtype. Eur J Neurosci. 2002;16:2049–2058. doi: 10.1046/j.1460-9568.2002.02280.x. [DOI] [PubMed] [Google Scholar]
- Rossato JI, Bevilaqua LR, Izquierdo I, Medina JH, Cammarota M. Dopamine controls persistence of long-term memory storage. Science. 2009;325:1017–1020. doi: 10.1126/science.1172545. [DOI] [PubMed] [Google Scholar]
- Sacktor TC. PKMzeta, LTP maintenance, and the dynamic molecular biology of memory storage. Prog Brain Res. 2008;169:27–40. doi: 10.1016/S0079-6123(07)00002-7. [DOI] [PubMed] [Google Scholar]
- Shannon HE, Hart JC, Bymaster FP, Calligaro DO, DeLapp NW, Mitch CH, Ward JS, Fink-Jensen A, Sauerberg P, Jeppesen L, Sheardown MJ, Swedberg MD. Muscarinic receptor agonists, like dopamine receptor antagonist antipsychotics, inhibit conditioned avoidance response in rats. J Pharmacol Exp Ther. 1999;290:901–907. [PubMed] [Google Scholar]
- Smith DR, Striplin CD, Geller AM, Mailman RB, Drago J, Lawler CP, Gallagher M. Behavioural assessment of mice lacking D1A dopamine receptors. Neuroscience. 1998;86:135–146. doi: 10.1016/s0306-4522(97)00608-8. [DOI] [PubMed] [Google Scholar]
- Smith JW, Fetsko LA, Xu R, Wang Y. Dopamine D2L receptor knockout mice display deficits in positive and negative reinforcing properties of morphine and in avoidance learning. Neuroscience. 2002;113:755–765. doi: 10.1016/s0306-4522(02)00257-9. [DOI] [PubMed] [Google Scholar]
- Smith WB, Starck SR, Roberts RW, Schuman EM. Dopaminergic stimulation of local protein synthesis enhances surface expression of GluR1 and synaptic transmission in hippocampal neurons. Neuron. 2005;3:765–779. doi: 10.1016/j.neuron.2005.01.015. [DOI] [PubMed] [Google Scholar]
- Smith-Roe SL, Kelley AE. Coincident activation of NMDA and dopamine D1 receptors within the nucleus accumbens core is required for appetitive instrumental learning. J Neurosci. 2000;20:7737–7742. doi: 10.1523/JNEUROSCI.20-20-07737.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takatsuki K, Kawahara S, Kotani S, Fukunaga S, Mori H, Mishina M, Kirino Y. The hippocampus plays an important role in eyeblink conditioning with a short trace interval in glutamate receptor subunit delta 2 mutant mice. J Neurosci. 2003;23:17–22. doi: 10.1523/JNEUROSCI.23-01-00017.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson RF. The neural basis of basic associative learning of discrete behavioral responses. Trends Neurosci. 1988;11:152–155. doi: 10.1016/0166-2236(88)90141-5. [DOI] [PubMed] [Google Scholar]
- Viosca J, Lopez de Armentia M, Jancic D, Barco A. Enhanced CREB-dependent gene expression increases the excitability of neurons in the basal amygdala and primes the consolidation of contextual and cued fear memory. Learm Mem. 2009;16:193–197. doi: 10.1101/lm.1254209. [DOI] [PubMed] [Google Scholar]
- Whishaw IQ, Dunnett SB. Dopamine depletion, stimulation or blockade in the rat disrupts spatial navigation and locomotion dependent upon beacon or distal cues. Behav Brain Res. 1985;18:11–29. doi: 10.1016/0166-4328(85)90165-2. [DOI] [PubMed] [Google Scholar]
- Whitlock JR, Heynen AJ, Shuler MG, Bear MF. Learning induces long-term potentiation in the hippocampus. Science. 2006;313:1093–1097. doi: 10.1126/science.1128134. [DOI] [PubMed] [Google Scholar]
- Williams GV, Goldman-Rakic PS. Modulation of memory fields by dopamine D1 receptors in prefrontal cortex. Nature. 1995;376:572–575. doi: 10.1038/376572a0. [DOI] [PubMed] [Google Scholar]
- Woody CD. Understanding the cellular basis of memory and learning. Annu Rev Psychol. 1986;37:433–493. doi: 10.1146/annurev.ps.37.020186.002245. [DOI] [PubMed] [Google Scholar]
- Xu M, Moratalla R, Gold LH, Hiroi N, Koob GF, Graybiel AM, Tonegawa S. Dopamine D1 receptor mutant mice are deficient in striatal expression of dynorphin and in dopamine-mediated behavioral responses. Cell. 1994;79:729–742. doi: 10.1016/0092-8674(94)90557-6. [DOI] [PubMed] [Google Scholar]
- Yao Y, Kelly MT, Sajikumar S, Serrano P, Tian D, Bergold PJ, Frey JU, Sacktor TC. PKM zeta maintains late long-term potentiation by N-ethylmaleimide-sensitive factor/GluR2-dependent trafficking of postsynaptic AMPA receptors. J Neurosci. 2008;28:7820–7827. doi: 10.1523/JNEUROSCI.0223-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng JF, Patil SS, Chen WQ, An W, He JQ, Höger H, Lubec G. Hippocampal protein levels related to spatial memory are different in the Barnes maze and in the multiple T-maze. J Proteome Res. 2009;8:4479–4486. doi: 10.1021/pr9002596. [DOI] [PubMed] [Google Scholar]
- Zweifel LS, Parker JG, Lobb CJ, Rainwater A, Wall VZ, Fadok JP, Darvas M, Kim MJ, Mizumori SJ, Paladini CA, Phillips PE, Palmiter RD. Disruption of NMDAR-dependent burst firing by dopamine neurons provides selective assessment of phasic dopamine-dependent behavior. Proc Natl Acad Sci U S A. 2009;106:7281–7288. doi: 10.1073/pnas.0813415106. [DOI] [PMC free article] [PubMed] [Google Scholar]