

Abstract
Hyperexcitability of peripheral nociceptive pathways is often associated with inflammation and is an important mechanism underlying inflammatory pain. Here we describe a completely novel mechanism via which nociceptor G-protein-coupled receptor kinase 2 (GRK2) contributes to regulation of inflammatory hyperalgesia. We show that nociceptor GRK2 is downregulated during inflammation. In addition, we show for the first time that prostaglandin E2 (PGE2)-induced hyperalgesia is prolonged from <6 h in wild-type (WT) mice to 3 d in mice with low GRK2 in Nav1.8+ nociceptors (SNS–GRK2 +/− mice). This prolongation of PGE2 hyperalgesia in SNS–GRK2 +/− mice does not depend on changes in the sensitivity of the prostaglandin receptors because prolonged hyperalgesia also developed in response to 8-Br-cAMP. PGE2 or cAMP-induced hyperalgesia in WT mice is PKA dependent. However, PKA activity is not required for hyperalgesia in SNS–GRK2 +/− mice. SNS–GRK2 +/− mice developed prolonged hyperalgesia in response to the Exchange proteins directly activated by cAMP (Epac) activator 8-pCPT-2′-O-Me-cAMP (8-pCPT). Coimmunoprecipitation experiments showed that GRK2 binds to Epac1. In vitro, GRK2 deficiency increased 8-pCPT-induced activation of the downstream effector of Epac, Rap1, and extracellular signal-regulated kinase (ERK). In vivo, inhibition of MEK1 or PKCε prevented prolonged PGE2, 8-Br-cAMP, and 8-pCPT hyperalgesia in SNS–GRK2 +/− mice. In conclusion, we discovered GRK2 as a novel Epac1-interacting protein. A reduction in the cellular level of GRK2 enhances activation of the Epac–Rap1 pathway. In vivo, low nociceptor GRK2 leads to prolonged inflammatory hyperalgesia via biased cAMP signaling from PKA to Epac–Rap1, ERK/PKCε pathways.
Introduction
Inflammatory mediators sensitize primary sensory neurons, causing increased responsiveness to noxious stimuli (hyperalgesia). The well-known analgesic effect of inhibition of prostaglandin E2 (PGE2) synthesis by COX-2 inhibitors clearly points toward a major role for this eicosanoid in inflammatory pain (Aley and Levine, 1999; Southall and Vasko, 2001; Moriyama et al., 2005). The action of PGE2 on primary neuronal afferents can be mediated via various subtypes of PGE2 receptors (EP1–EP4) that all belong to the class of G-protein-coupled receptors (GPCRs). At the molecular level, it is known that local administration of PGE2 induces transient hyperalgesia and nociceptor sensitization via activation of the cAMP/PKA-dependent second-messenger system (Minami et al., 2001; Southall and Vasko, 2001; Lin et al., 2006). When animals are primed by a previous transient inflammation, PGE2 hyperalgesia is prolonged and the signaling pathway switches from a cAMP/PKA-dependent pathway to a PKA, PKCε, and extracellular signal-regulated kinase (ERK)-mediated route (Aley et al., 2000; Dina et al., 2003).
The kinase GRK2 regulates GPCR signaling via interference at various levels of the signal transduction cascade. GRK2 phosphorylates agonist-occupied GPCR, leading to receptor desensitization and internalization (Zhang et al., 1997; Ribas et al., 2007). GRK2 also limits signaling via direct interaction with specific downstream intracellular kinases such as Akt, MEK1/2, phosphoinositide-3 kinase, and p38, leading to inhibition of their activity (Reiter and Lefkowitz, 2006; Ribas et al., 2007). In addition, GRK2 binds and phosphorylates a variety of cytoskeletal proteins, including tubulin, ezrin, and synuclein (Haga et al., 1998; Pitcher et al., 1998; Pronin et al., 2000; Cant and Pitcher, 2005).
We and others have shown that the intracellular level of GRK2 is tightly regulated. In vivo, the level of GRK2 in leukocytes from patients with autoimmune diseases such as rheumatoid arthritis and multiple sclerosis is decreased, and similar changes in GRK2 expression occur in animals with experimental autoimmune encephalomyelitis and adjuvant arthritis (Lombardi et al., 1999; Vroon et al., 2005, 2006). Moreover, neuropathic pain in animals subjected to nerve ligation is associated with decreased spinal cord neuronal and microglial GRK2 expression, suggesting that GRK2 may contribute to regulation of pain signaling under inflammatory conditions (Kleibeuker et al., 2007, 2008; Eijkelkamp et al., 2010).
Recently, we described that a 50% reduction of GRK2 in nociceptors (SNS–GRK2 +/− mice) increased and prolonged carrageenan-induced inflammatory hyperalgesia in mice (Eijkelkamp et al., 2010). The aim of this study is to investigate the mechanisms as to how neuronal GRK2 biases signaling responsible for the increased duration of hyperalgesia induced by a single injection of the prototypic inflammatory mediator PGE2 in conditions of low nociceptor GRK2.
Materials and Methods
Animals.
Female C57BL/6 (12–14 weeks) SNS–GRK2 +/−, SNS–GRK2 −/−, and control SNS–GRK2 +/+ offspring was generated by breeding homozygous Nav1.8–CRE transgenic mice (Stirling et al., 2005; Abrahamsen et al., 2008) with heterozygous GRK2–LOX mice (Matkovich et al., 2006). Mice heterozygous for targeted deletion of the GRK2 gene (GRK2 +/−) and their wild-type (WT) littermates were used (Jaber et al., 1996). Because GRK2 −/− mice die in utero, only GRK2 +/− mice can be used for experiments. Inducible GRK2 +/− mice were generated by crossing transgenic mice overexpressing Cre-recombinase protein fused to two mutant estrogen receptor ligand-binding domains under the control of the α-myosin heavy chain (αMHC) promoter (αMHC–MerCreMer) (Sohal et al., 2001) (The Jackson Laboratory) with heterozygous mice with the floxed GRK2 alleles. To induce Cre expression, adult mice were treated with tamoxifen (2 mg/mouse, i.p., in saline containing 9% ethanol and 51% sunflower oil for 5 consecutive days) (Sigma-Aldrich). GRK2 expression in dorsal root ganglia (DRGs) homogenates was determined 2 weeks after the last injection of tamoxifen. Female C57BL/6 GRK6 −/− and WT littermates were generated by breeding heterozygous GRK6 +/− mice (Eijkelkamp et al., 2007).
Mice were genotyped by PCR analysis on genomic DNA. Mice were bred and maintained in the Animal facility of the University of Utrecht (Utrecht, The Netherlands). All experiments were performed in accordance with international guidelines and approved by the University Medical Center Utrecht Experimental Animal Committee.
Measurement of thermal hyperalgesia.
Heat-withdrawal latency times were determined using the Hargreaves (IITC Life Science) test as described previously (Hargreaves et al., 1988). Intensity of the light beam was chosen to induce a heat-withdrawal latency time at baseline of ∼8 s. Baseline withdrawal latencies were determined three times before intraplantar injection of the inflammatory agent.
Pharmacological agents.
All pharmacological agents were injected intraplantarly in a volume of 2.5 μl, except that H89 (N-[2-(p-bromo-cinnamylamino)-ethyl]-5-isoquinoline-sulfon-amide 2HCl) was injected in a volume of 5 μl. A solution of 40 μg/ml PGE2 (Sigma-Aldrich) was made in saline containing <1% ethanol. EP receptor agonists were applied at a dose equivalent to the effective dose of PGE2 for that particular receptor subtype. Dissociation constant of the compounds were obtained from manufactures and http://www.iuphar-db.org/index.jsp. The EP4 agonist L-902688 (5-[(1E, 3R)-4, 4-difluoro-3-hydroxy-4-phenyl-1-buten-1-yl]-1-[6-(2H-tetrazol-5R-yl)hexyl]-2-pyrrolidinone) (18.85 μg/ml; generous gift from Merck Frosst Canada Ltd.), the EP1/3 agonist sulprostone [N-(methylsulfonyl)-9-oxo-11α, 15R-dihydroxy-16-phenoxy-17,18,19,20-tetranor-prosta-5Z, 13E-dien-1-amide] (40 μg/ml; Sigma-Aldrich), the EP2 agonist ONO-AE1-259-01 [(z)-7-[(1R, 2R, 3R, 5R)-5-chloro-3-hydroxy-2-[(E, 4S)-4-hydroxy-4-(1-prop-2-enylcyclobutyl)but-1-enyl]cyclopentyl]hept-5-enoic acid] (12.23 μg/ml; generous gift from Ono Pharmaceutical Co. Ltd.), or the specific EP3 agonist ONO-AE-248 [(z)-7-[1R, 2R, 3R)-3-methoxy-2-[(E, 3S)-3-methoxyoct-1-enyl]-5-oxocyclopentyl]hept-5-enoic acid] (0.41 mg/ml; generous gift from Ono Pharmaceuticals) were dissolved in saline containing 10–20% DMSO. 8-Br-cAMP (4 μg/ml; Sigma-Aldrich), the PKA activator 6-Bnz-cAMP (4.46 μg/ml; Biolog LifeScience Institute), and the Epac activator 8-pCPT-2′-O-Me-cAMP (8-pCPT) (5.04 μg/ml; Biolog LifeScience Institute) were dissolved in saline.
Inhibitors were injected intraplantarly 15 min before the injection of the pharmacological agents that induced hyperalgesia. The MEK1 inhibitor PD98059 [2-(2-amino-3-methyoxyphenyl)-4H-1-benzopyran-4-one] (1 mg/ml Sigma-Aldrich) (Dina et al., 2003) was dissolved in saline containing 20% DMSO. The PKA inhibitor H89 (5.4 mg/ml; Sigma-Aldrich) (Cunha et al., 1999) or the PKCε translocation inhibitory peptide (TAT-PKCεV1–2; YGRKKRRQRRRCCEAVSLKPT-COOH) or scrambled control peptide [1 mg/ml (W. M. Keck Facility, Yale University, New Haven, CT); YGRKKRRQRRRCCLSETKPAV-COOH] were dissolved in saline.
Carrageenan paw inflammation and immunohistochemical staining of dorsal root ganglia.
Mice received an intraplantar injection of 20 μl of λ-carrageenan [2% (w/v); Sigma-Aldrich] in saline in both hindpaws. At day 6 after carrageenan, mice were deeply anesthetized with sodium pentobarbital (50 mg/kg, i.p.) and perfused intracardially with 0.9% saline containing, followed by 4% paraformaldehyde in PBS. DRGs of lumbar level 2–6 (L2–L6) were isolated, postfixed for 4 h at 4°C, and then kept overnight in 20% sucrose in PBS at 4°C, followed by 30% sucrose in PBS for 24–48 h. Dissected tissue was mounted in OCT compound and frozen at −20°C. DRG sections (10 μm) were cut using a cryostat. Sections were washed in PBS and blocked in 5% normal goat serum, 2% bovine serum albumin, and 0.3% Triton X-100. Sections were then incubated overnight at 4°C with rabbit anti-GRK2 (1:200; Santa Cruz Biotechnology). GRK2 staining was controlled by using primary anti-GRK2 antibody blocked with a GRK2 blocking peptide (Santa Cruz Biotechnology). Tissue samples were then washed and incubated with Alexa Fluor 594-conjugated goat anti-rabbit antibody (1:200; Invitrogen). Cells were photographed with an EVOS fl (AMG Inc.), and GRK2 levels in small-diameter DRG neurons were analyzed with NIH ImageJ software.
The average background fluorescence (primary GRK2 antibody plus GRK2 blocking peptide) of 15 small-diameter neurons of DRGs from vehicle- and carrageenan-treated animals was subtracted before calculation of the percentage change in GRK2 staining. Exposure times of photographs were identical for all slices. All stainings were done in parallel for DRGs from vehicle- and carrageenan-treated mice.
DRG cell culture.
Dorsal root ganglia were obtained from adult WT and GRK2 +/− mice. Ganglia were digested in collagenase (type XI, 0.6 mg/ml; Sigma-Aldrich), protease (Streptomycis griseus, 0.4 mg/ml; Sigma-Aldrich), and glucose (1.8 mg/ml; Sigma-Aldrich) in Ca2+ and Mg2+-free PBS for 40 min before mechanical trituration. Cells were then washed and resuspended in DMEM (Invitrogen) containing 10% fetal bovine serum (Invitrogen), 2 mm glutamine (Invitrogen), 10,000 IU/ml penicillin–streptomycin (Invitrogen), and 100 ng/ml nerve growth factor (NGF) (Sigma-Aldrich) and plated in a 24-well plate coated with poly-l-lysine and laminin. Cells were stimulated 15–25 h after plating and after 4 h of NGF starvation. Cells were homogenized in RAL lysis buffer [200 mm NaCl, 50 mm Tris-HCl, pH 7.5, 2 mm MgCl2, 10% glycerol, 1% NP-40 containing a mixture of protease inhibitors (Sigma) with the addition of 2 mm NaF, 10 mm β-glycerol-phosphate, and 2 mm phenylmethylsulfonyl fluoride].
Splenocyte isolation.
Single-cell suspensions of spleens from adult WT and GRK2 +/− mice were obtained by dispersing the tissue through a 70 μm filter in cold RPMI (Invitrogen). Erythrocytes were removed by lysis in hypotonic buffer for 2 min. Subsequently, splenocytes were washed twice in RPMI supplemented with 5% fetal bovine serum and 50 μm β-mercaptoethanol. Cells were stimulated for the indicated time periods with 8-pCPT (100 μm; Biolog LifeScience Institute).
Rap1 activation assay using RalGDS–RBD.
We used splenocytes as a source of primary cells from WT and GRK2 +/− mice because these cells are easily available in large amounts and do respond to 8-pCPT. After stimulation with 8-pCPT, isolated splenocytes were lysed by addition of 700 μl of cold RAL lysis buffer. Lysates were clarified by centrifugation at maximal speed in an Eppendorf centrifuge for 10 min at 4°C. Rap activation assays were performed as described previously (Franke et al., 1997). Briefly, equal amounts of cell lysates were incubated with glutathione–agarose beads that were precoupled with glutathione S-transferase fused to the minimal Ras-binding domain (RBD) of Ral-guanine nucleotide-dissociation stimulator (GDS) to specifically pulldown-activated GTP-bound forms of Rap1. The amount of Rap1 was quantified by Western blotting. Total Rap1 in cell lysates was determined to verify equal input into the Rap1–GTP pulldown assay.
Cell transfection and immunoprecipitation.
HEK293a cells were transfected with hemagglutinin (HA)-tagged Epac1 and as control HA–tuberous sclerosis 1 (TSC1) using Lipofectamine (Invitrogen) following the protocol of the manufacturer. Transfected HEK293a cells or spinal cord or dorsal root ganglion tissue were washed in PBS and lysed in ice-cold modified radioimmunoprecipitation assay buffer [20 mm Tris-HCl, pH 8, 1% Triton X-100, 10% glycerol, 137 mm NaCl, 2 mm EDTA, containing a mixture of protease inhibitors (Sigma) with the addition of 2 mm NaF and 10 mm β-glycerol-phosphate]. The lysate was centrifuged at 14,000 × g for 10 min at 4°C. The cell lysate (200 μg) was precleared by adding 50 μl of 50% Protein A Sepharose bead slurry per 200 μl of cell lysate. Mouse anti-GRK2/3 (Millipore Corporation) or mouse anti-HA (Invitrogen) were coupled to Sepharose A beads overnight at 4°C with gentle tumbling. These beads were added to the lysate for 3 h at 4°C with gentle tumbling. The Sepharose beads were washed three times with ice-cold wash buffer (5 mm Tris-HCl, pH 7.4, 20 mm NaCl, and 0.5% Triton X-100) and resuspended in 50 μl of 2× sample buffer, followed by elution through boiling.
Western blot analysis.
Proteins were separated by SDS-PAGE and transferred to polyvinylidene difluoride membranes (Millipore Corporation). Blots were stained with rabbit anti-GRK2 (Santa Cruz Biotechnology), mouse anti-p-ERK1/2 (Santa Cruz Biotechnology), rabbit anti-ERK1/2 (Cell Signaling Technology), rabbit anti-Rap1 (Santa Cruz Biotechnology), mouse anti-Epac1 (Price et al., 2004), mouse anti-Epac2 (generated at Department of Physiological Chemistry of Dr. J. L. Bos, University Medical Center Utrecht, Utrecht, The Netherlands), mouse anti-HA–HRP (Miltenyi), mouse anti-α-tubulin (Santa Cruz Biotechnology), and goat anti-β-actin (Santa Cruz Biotechnology). Subsequently, blots were incubated for 1 h with goat anti-mouse peroxidase IgG plus IgM (heavy and light chain) (Jackson ImmunoResearch) or donkey anti-rabbit peroxidase IgG (GE Healthcare) and developed by enhanced chemiluminescence (GE Healthcare). Band density was determined using a GS-700 Imaging Densitometer (Bio-Rad).
Data analysis.
Data are expressed as mean ± SEM. Measurements were compared using Student's t test, one-way ANOVA, or two-way ANOVA followed by Bonferroni's analysis. A p value <0.05 was considered to be statistically significant.
Results
GRK2 protein levels in sensory neurons during peripheral paw inflammation
To test whether ongoing inflammation changes GRK2 levels in sensory neurons, we injected a dose of carrageenan that induces long-lasting (>6 d) paw inflammation (Honore et al., 2006). We quantified GRK2 levels in small-diameter neurons in DRG from mice that had received an intraplantar injection of 20 μl of a 2% carrageenan solution. Intraplantar injection of this high dose of carrageenan induced hyperalgesia that was still present at day 6 (Fig. 1 A). At this time point, there was a reduction in GRK2 levels in small-diameter sensory neurons of ∼35% compared with vehicle-treated animals (Fig. 1 B). The average diameter of measured small-diameter neurons in which GRK2 was quantified was 14.30 ± 0.08 μm in saline-treated and 14.65 ± 0.08 μm in carrageenan-treated animals.
Figure 1.

Chronic carrageenan hyperalgesia is associated with reduced GRK2 in DRG neurons. A, Percentage decrease in heat-withdrawal latency time after intraplantar carrageenan injection (20 μl, 2%) in WT mice (n = 8–12 per genotype). B, GRK2 expression in dorsal root ganglia isolated 6 d after intraplantar carrageenan or saline administration was compared by immunofluorescence analysis. Representative pictures of GRK2 staining of dorsal root ganglia. C, Bar graph represents average GRK2 immunofluorescence intensity of ∼40 neurons on two to three different slides per animal (n = 4 animals per group). Data are expressed as mean ± SEM fluorescence intensity. *p < 0.05.
Contribution of GRK2 to PGE2-induced hyperalgesia
To examine the contribution of nociceptor GRK2 to PGE2 hyperalgesia, we used mice homozygous (SNS–GRK2 −/−) or heterozygous (SNS–GRK2 +/−) for deletion of the GRK2 gene in Nav1.8+ sensory neurons. Mice homozygous for GRK2 deletion in all cells die in utero. In SNS–GRK2 +/− mice, GRK2 protein levels are reduced by ∼50% in DRGs compared with SNS–GRK2 +/+ (WT) controls (Eijkelkamp et al., 2010). This reduction is similar to the reduction in GRK2 observed in small-diameter sensory neurons during chronic carrageenan-induced paw inflammation (Fig. 1).
SNS–GRK2 −/− mice, SNS–GRK2 +/− mice, and SNS–GRK2 +/+ (WT) littermates were injected intraplantarly with PGE2. PGE2 hyperalgesia was significantly increased in SNS–GRK2 −/− mice (Fig. 2 A) and SNS–GRK2 +/− mice (Fig. 2 B) compared with SNS–GRK2 +/+ (WT) littermates. Moreover, PGE2-induced hyperalgesia was significantly prolonged and lasted 3 d in SNS–GRK2 −/− mice and SNS–GRK2 +/− mice, whereas SNS–GRK2 +/+ (WT) littermates recovered within 4–6 h (Fig. 2 A,B). The severity and duration of PGE2 hyperalgesia was similar in SNS–GRK2 −/− and SNS–GRK2 +/− mice. Intraplantar saline injection did not induce a detectable change in thermal sensitivity in any genotype. Baseline latency times of SNS–GRK2 −/− mice were significantly longer than in SNS–GRK2 +/+ (WT) mice (8.1 ± 0.2 vs 10.7 ± 0.5 s; n = 14, p < 0.001). However, in SNS–GRK2 +/− mice, baseline heat-withdrawal latency times were comparable with WT littermates (8.3 ± 0.1 vs 8.3 ± 0.1 s; n = 40) (Eijkelkamp et al., 2010). In view of the fact that inflammation reduces GRK2 from 35 to 50% in various cell types including DRG, we decided to use mice heterozygous for deletion of the GRK2 gene in Nav1.8+ nociceptors (SNS–GRK2 +/− mice) and SNS–GRK2 +/+ (WT) littermate controls for additional experiments.
Figure 2.

Reduced nociceptor GRK2 prolongs PGE2 hyperalgesia. A, Percentage decrease in heat-withdrawal latency time after intraplantar PGE2 injection (100 ng/paw) in SNS–GRK2 +/+ control animals and in SNS–GRK2 −/− mice (n = 8 per genotype). B, Percentage decrease in heat-withdrawal latency times after intraplantar PGE2 injection in SNS–GRK2 +/+ control animals and SNS–GRK2 +/− mice (n = 8 per genotype). C, Percentage decrease in heat-withdrawal latency time after intraplantar PGE2 injection (100 ng/paw) in WT and GRK2 +/− mice (n = 8 per genotype). D, GRK2 levels in dorsal root ganglia of inducible GRK2 +/− and WT mice after tamoxifen treatment (n = 3). E, Percentage decrease in heat-withdrawal latency time after intraplantar PGE2 injection in WT mice and inducible GRK2 +/− (n = 7–8 per genotype). F, Percentage decrease in heat-withdrawal latency time after intraplantar PGE2 injection in WT and GRK6 −/− mice (n = 7–8 per genotype). Data are expressed as mean ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001.
To address the question whether the increased and prolonged PGE2 hyperalgesia in GRK2-deficient mice was caused by developmental adaptation to GRK2 deficiency, we also used tamoxifen-treated inducible GRK2 +/− mice compared with GRK2 +/− mice that have a 40–60% decrease in GRK2 protein in all cells throughout development (Kleibeuker et al., 2008; Eijkelkamp et al., 2010).
PGE2-induced thermal hyperalgesia was increased and prolonged in GRK2 +/− mice (Fig. 2 C). Baseline heat-withdrawal latencies in GRK2 +/− mice did not differ from WT mice as we described previously (7.8 ± 0.1 vs 8.0 ± 0.1 s; n = 30) (Eijkelkamp et al., 2010). Tamoxifen-treated inducible GRK2 +/− mice show an ∼50% reduction in GRK2 levels in DRG tissue extracts compared with WT mice (Fig. 2 D). Importantly, tamoxifen-inducible GRK2 +/− mice developed increased and prolonged PGE2 hyperalgesia similar to that observed in GRK2 +/− mice (Fig. 2 E). Tamoxifen-treated GRK2 +/+ mice responded like WT mice and only developed a transient PGE2 hyperalgesia (Fig. 2 E). Baseline heat-withdrawal latency times did not differ between tamoxifen-treated GRK2 +/− and GRK2 +/+ mice (8.5 ± 0.2 vs 8.4 ± 0.3 s; n = 8).
To test whether the effect on the regulation of the hyperalgesic response to PGE2 is specific for GRK2, we also determined the magnitude and duration of PGE2 hyperalgesia in GRK6 −/− mice. In Figure 2 F, it is shown that deletion of GRK6 does not affect the intensity and duration of PGE2 hyperalgesia.
Contribution of PKA in PGE2 hyperalgesia in WT and SNS–GRK2 +/− mice
In WT rats, PGE2 hyperalgesia is mediated via a cAMP/PKA-dependent pathway (Dina et al., 2001, 2003). To evaluate whether the increased and prolonged hyperalgesia in SNS–GRK2 +/− mice is mediated via (increased) activation of PKA, mice were pretreated with an intraplantar injection of the PKA inhibitor H89. In line with data in the literature, H89 completely prevented PGE2 hyperalgesia in SNS–GRK2 +/+ (WT) mice (Fig. 3 A). In contrast, H89 did not affect PGE2 hyperalgesia in mice with reduced nociceptor GRK2 (Fig. 3 A), indicating that PGE2 activates signaling cascades independent of PKA, leading to prolonged hyperalgesia in mice with reduced nociceptor GRK2. In GRK2 +/− mice with low GRK2 in all cells, PGE2 hyperalgesia was also independent of PKA activity (Fig. 3 B).
Figure 3.

PGE2 hyperalgesia is PKA independent in SNS–GRK2 +/− and GRK2 +/− mice. Percentage change in heat-withdrawal latencies after intraplantar injection of the PKA inhibitor H89 (27 μg/paw) before PGE2 injection in SNS–GRK2 +/+ (WT) animals and SNS–GRK2 +/− mice (n = 4 per group) (A) or WT and GRK2 +/− mice (n = 4 per group) (B). Data are expressed as mean ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001 SNS–GRK2 +/+ (WT) versus SNS–GRK2 +/+ (WT) treated with inhibitor.
Role of nociceptor GRK2 in EP receptor subtype agonist-induced hyperalgesia
All four cloned PGE2 receptor subtypes (EP receptors 1–4) are members of the G-protein-coupled receptor family and are expressed on nerve terminals of nociceptors (Oida et al., 1995). The EP receptors couple to different G proteins, leading to the activation of different signaling cascades. EP1 receptors couple to Gq, and activation of the receptor lead to an increase in inositol trisphosphate; EP2 and EP4 receptors couple to Gs and cause an increase in cAMP formation. EP3 receptors couple to Gi, leading to a decrease in cAMP formation (Coleman et al., 1994). In rodents, EP4 and to a lesser extent EP1 and EP3 have been shown to mediate PGE2 hyperalgesia (Minami et al., 2001; Lin et al., 2006). We used EP receptor subtype-specific agonists to determine whether prolonged hyperalgesia in SNS–GRK2 +/− mice was caused by a switch in the use of EP receptor subtype(s). The EP1/3 agonist sulprostone induced transient hyperalgesia that did not differ between genotypes (Fig. 4 A). Intraplantar injection of the specific EP2 agonist ONO-AE1-259-01 or the specific EP3 agonist ONO-AE-248 did not significantly affect thermal sensitivity in both genotypes (ONO-AE1-259-01: 9.7 ± 4.6 vs 8.0 ± 6.0% maximal decrease in latency, n = 8; ONO-AE-248: 6.5 ± 3.7 vs 12.0 ± 5.4% maximal decrease in latency, n = 8, p = 0.42). However, the EP4 agonist L-902688 completely mimicked the effect of PGE2; L-902688-induced transient hyperalgesia in WT mice and L-902688 hyperalgesia was increased and prolonged in SNS–GRK2 +/− mice (Fig. 4 B). Because the specific receptor antagonists were not available, we could not test the receptor specificity in more detail. Nonetheless, our current data indicate that the prolonged PGE2-induced hyperalgesia in SNS–GRK2 +/− mice is likely to rely on EP4 receptor signaling.
Figure 4.
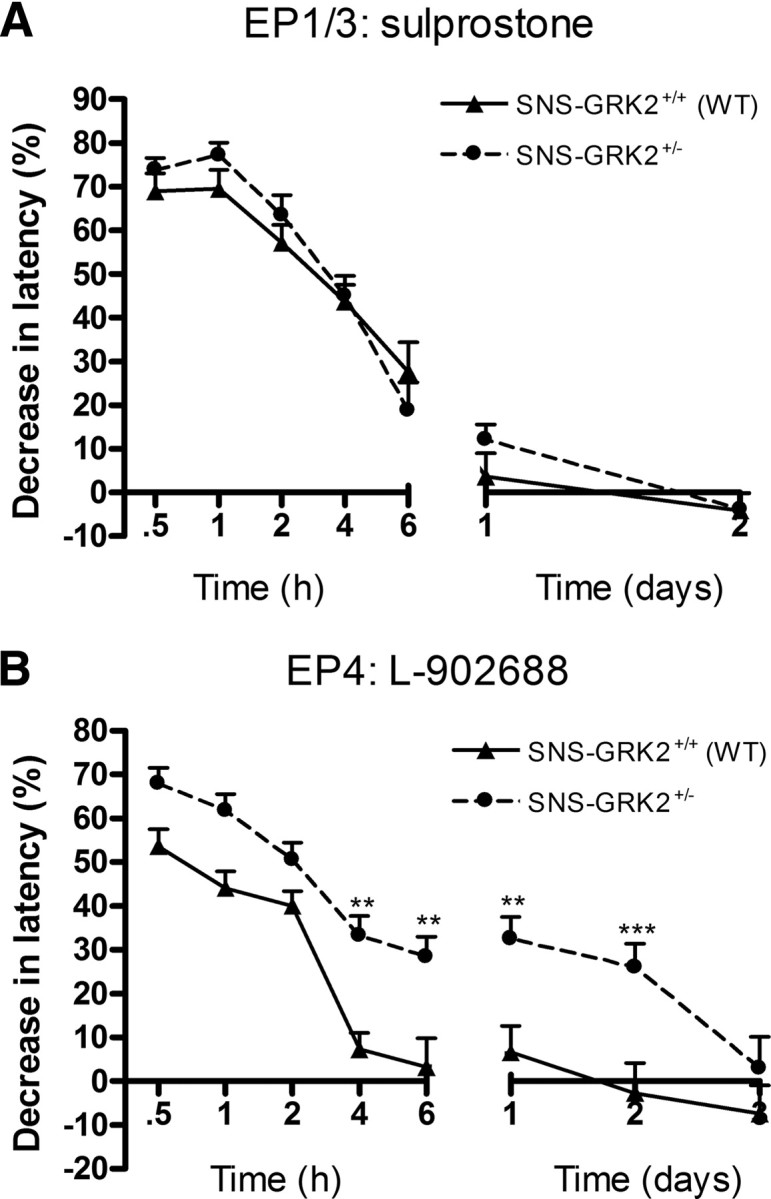
Reduced nociceptor GRK2 only affects hyperalgesia induced by an EP4 agonist. A, Percentage decrease in heat-withdrawal latency times after intraplantar sulprostrone (EP1/3 agonist) injection in SNS–GRK2 +/+ (WT) and SNS–GRK2 +/− mice (n = 8 per genotype). B, Percentage decrease in heat-withdrawal latency time after intraplantar L-902668 (EP4 agonist) injection in SNS–GRK2 +/+ control animals and in SNS–GRK2 +/− (n = 8 per genotype). Data are expressed as mean ± SEM. **p < 0.01, ***p < 0.001.
Reduced nociceptor GRK2 regulates hyperalgesia downstream of cAMP
To test whether the prolongation of hyperalgesia in mice with reduced nociceptor GRK2 is cAMP-mediated but PKA independent, we used the cAMP analog 8-Br-cAMP; H89 was applied to block PKA activation. 8-Br-cAMP-induced hyperalgesia was significantly prolonged in SNS–GRK2 +/− mice compared with control SNS–GRK2 +/+ littermates; 8-Br-cAMP-induced hyperalgesia lasted for 3 d in SNS–GRK2 +/− mice, whereas SNS–GRK2 +/+ (WT) littermates recovered within 6 h (Fig. 5 A). Acute 8-Br-cAMP-induced hyperalgesia was also significantly elevated in SNS–GRK2 +/− mice compared with control littermates (Fig. 4 A).
Figure 5.

Reduced nociceptor GRK2 specifically prolongs cAMP- and Epac-mediated hyperalgesia but not PKA-mediated hyperalgesia. A, Percentage change in heat-withdrawal latencies after intraplantar injection of 8-Br-cAMP (10 ng/paw) in SNS–GRK2 +/+ (WT) animals and SNS–GRK2 +/− mice (n = 8 per genotype). B, Percentage change in heat-withdrawal latencies after intraplantar injection of the PKA inhibitor H89 (27 μg/paw) before 8-Br-cAMP injection in SNS–GRK2 +/+ (WT) animals and SNS–GRK2 +/− mice (n = 4 per group). C, Percentage decrease in heat-withdrawal latency time after intraplantar injection of 8-pCPT; 12.6 ng/paw) in SNS–GRK2 +/+ control animals and in SNS–GRK2 +/− (n = 12 per genotype). D, Percentage decrease in heat-withdrawal latency times after intraplantar injection of 6-Bnz-cAMP (cAMP analog that specifically activates PKA; 11.2 ng/paw) in SNS–GRK2 +/+ control mice and SNS–GRK2 +/− mice (n = 8 per genotype). Data are expressed as mean ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001.
Pretreatment with H89 totally prevented 8-Br-cAMP hyperalgesia in SNS–GRK2 +/+ (WT) but failed to affect 8-Br-cAMP hyperalgesia in mice with reduced nociceptor GRK2 (Fig. 5 B). These findings indicate that GRK2 reduction in nociceptors causes biased signaling toward a PKA-independent pathway downstream of cAMP production, resulting in increased and prolonged hyperalgesia.
cAMP not only regulates cellular activity via activation of PKA but also of a family of guanine nucleotide exchange factors (GEFs) known as exchange proteins directly activated by cAMP (Epac). Specific inhibitors of Epac are not available. Therefore, we used the specific Epac activator 8-pCPT to determine the contribution of Epac-mediated pathways to the prolonged hyperalgesia in SNS–GRK2 +/− mice. Hyperalgesia induced by intraplantar injection of the Epac activator 8-pCPT lasted 3 d in SNS–GRK2 +/−, whereas SNS–GRK2 +/+ (WT) littermates recovered within 6 h (Fig. 5 C), similar to what was observed after PGE2 or 8-Br-cAMP. To verify the contribution of a PKA-mediated pathway in prolonged hyperalgesia in mice with low nociceptor GRK2, we used the specific PKA activator 6-Bnz-cAMP. Although acute hyperalgesia induced by the specific PKA-activator 6-Bnz-cAMP was slightly increased in SNS–GRK2 +/− mice compared with SNS–GRK2 +/+ (WT) mice, PKA activation was not sufficient to induce prolonged hyperalgesia in SNS–GRK2 +/− mice (Fig. 5 D).
GRK2 modulates Rap1 activation via Epac
To test whether reduced GRK2 leads to changes in the level of Epac expression that may affect the duration of PGE2 hyperalgesia, we determined Epac protein expression in isolated DRG neurons. Western blot analysis of isolated DRG neurons from WT and GRK2 +/− mice showed that reduced GRK2 did not affect the level of Epac1 or Epac2 protein (Fig. 6 A).
Figure 6.

Low primary sensory neuron GRK2 enhances 8-pCPT-induced Rap1 and ERK1/2 activation in vitro. A, Epac1 and Epac2 expression levels in isolated primary sensory neurons of WT and GRK2 +/− mice (n = 5 per genotype). B, Rap1 expression levels in isolated primary sensory neurons of WT and GRK2 +/− mice (n = 4 per genotype). C, 8-pCPT-induced Rap1 activation in splenocytes from WT and GRK2 +/− mice. Bar graphs show quantification of Rap1 activation after 8-pCPT (100 μm) treatment (n = 4–6 per genotype). D, HA–Epac1, HA–TSC1, or GRK2 was immunoprecipitated (IP) from lysates of HEK293 cells that were transfected with HA–Epac or with HA–TSC1 as a control. 8-pCPT (100 μm) was added during the precipitation to keep Epac in an activated conformation. E, Immunoprecipitates of GRK2 from lysates of either spinal cord or DRG immunoblotted for Epac1. F, 8-pCPT-induced ERK1/2 phosphorylation in splenocytes from WT and GRK2 +/− mice. Bar graphs show quantification of activated p-ERK1/2 after 8-pCPT treatment (100 μm; n = 3–6 per genotype). G, 8-pCPT-induced (100 μm) ERK1/2 phosphorylation in primary sensory neurons of WT and GRK2 +/− mice. H, cAMP-induced (1 mm) ERK1/2 phosphorylation in primary sensory neurons of WT and GRK2 +/− mice. Data are expressed as mean ± SEM. *p < 0.05, **p < 0.01.
The GEFs Epac1 and Epac 2 activate Rap1 and Rap2. Rap1 and Rap2 are small GTP binding proteins of the Ras family of GTPases. In its inactive state, Rap is bound to GDP. After binding of cAMP, the conformation of Epac changes so that Rap can bind and this induces the exchange of GDP for GTP, resulting in activation of Rap. Activated Rap induces downstream signaling, including the activation of PKCε (Wang et al., 2007; Oestreich et al., 2009). Finally, Rap1 activation has been implicated in the modulation of nociceptor functioning (Delcroix et al., 2003; Taniguchi et al., 2008). Western blot analysis of isolated DRG neurons of WT and GRK2 +/− neurons showed that Rap1 is expressed and that expression levels are similar between WT and GRK2 +/− neurons (Fig. 6 B).We investigated whether GRK2 deficiency affects Epac signaling to Rap1. The data in Figure 6 C demonstrate that GRK2 deficiency significantly enhances 8-pCPT-induced Rap1 activation (Fig. 6 C). To investigate whether the effect of low GRK2 on Epac-to-Rap1 signaling could be mediated via a direct interaction between GRK2 and Epac, we performed coimmunoprecipitation studies. Cells transfected with HA-tagged Epac1 or with control HA-tagged TSC1 were stimulated with 8-pCPT or medium to get Epac in an active or non-active conformation and coimmunoprecipitated using either anti-GRK2 or anti-HA antibodies. HA–Epac1 was coimmunoprecipitated with GRK2, especially in 8-pCPT-stimulated extracts, whereas the control, HA–TSC1 was not found in the GRK2 immunoprecipitate (Fig. 6 D). Conversely, only after stimulation with 8-pCPT, GRK2 was coprecipitated with HA–Epac1 (Fig. 6 D). Finally, coimmunoprecipitation studies using a specific GRK2 antibody on tissue extracts of DRG and spinal cord tissue revealed that the low-molecular-weight splice variant of Epac1 binds GRK2 in these tissues (Fig. 6 E).
Apart from Rap1 activation, we also show here that 8-pCPT moderately induced ERK1/2 phosphorylation in GRK2 +/− lymphocytes and sensory neurons but not in WT cells (Fig. 6 F,G). Similar results were obtained for 8-Br-cAMP-induced ERK1/2 phosphorylation in GRK2 +/− primary sensory neurons (Fig. 6 H).
In vivo contribution of the MEK/ERK and PKCε pathway in PGE2, 8-Br-cAMP, and 8-pCPT-induced hyperalgesia
Our in vitro results indicated that low sensory neuron GRK2 facilitates cAMP signaling toward Epac/Rap1 and ERK. To test whether in vivo low nociceptor GRK2 prolongs PGE2, 8-Br-cAMP, and 8-pCPT hyperalgesia via biased signaling to the mitogen-activated protein kinase kinase (MEK)/ERK1/2 pathway, we used the MEK1 inhibitor PD98059. In SNS–GRK2 +/+ (WT) mice, intraplantar pretreatment with PD98059 did not affect PGE2 or 8-Br-cAMP-induced hyperalgesia (Fig. 7 A,B). In contrast, the MEK1 inhibitor PD98059 inhibited the prolonged PGE2, 8-Br-cAMP, and 8-pCPT hyperalgesia in SNS–GRK2 +/− mice (Fig. 7 A–C). In addition, after PD98059 treatment, we no longer observed the increased acute hyperalgesia in SNS–GRK2 +/− mice (Fig. 7 A–C). PD98059 slightly reduced acute 8-pCPT hyperalgesia in SNS–GRK2 +/+ mice (Fig. 7 C). Vehicle treatment did not affect PGE2, 8-Br-cAMP, or 8-pCPT hyperalgesia (data not shown).
Figure 7.
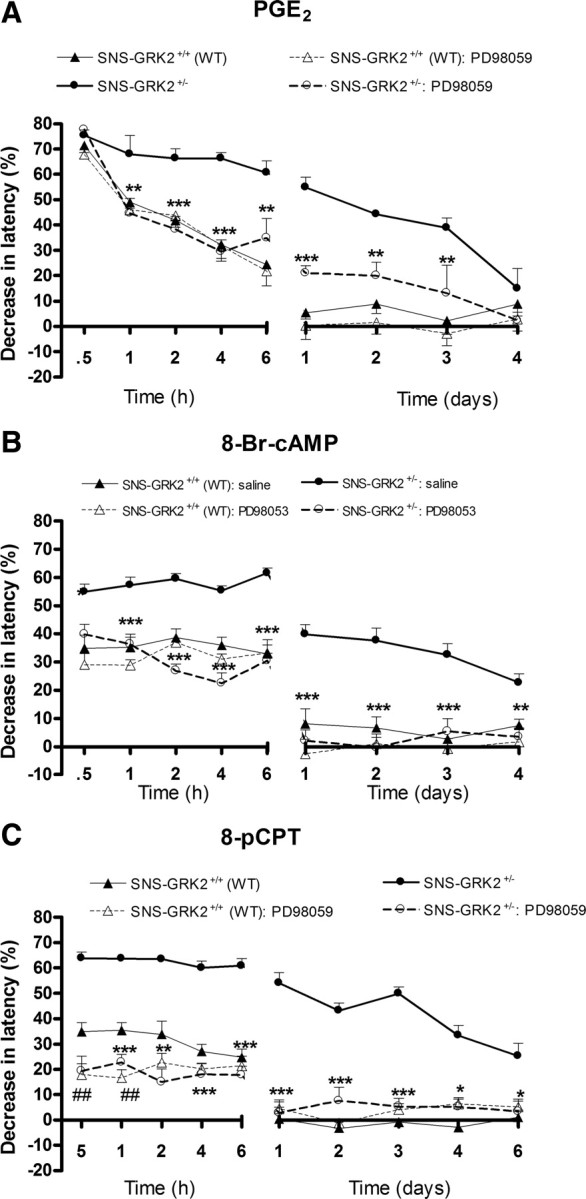
PGE2, 8-Br-cAMP, and 8-pCPT-induced hyperalgesia in SNS–GRK2 +/− mice is biased to MEK/ERK. Percentage change in heat-withdrawal latencies after intraplantar injection of the MEK inhibitor PD98059 (2.5 μg/paw) before PGE2 (A), 8-Br-cAMP (B), or 8-pCPT (C) injection in SNS–GRK2 +/+ (WT) animals and SNS–GRK2 +/− mice (n = 8 per group). Data are expressed as mean ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001 SNS–GRK2 +/− versus SNS–GRK2 +/− treated with inhibitor. ## p < 0.01 SNS–GRK2 +/+ (WT) versus SNS–GRK2 +/+ (WT) treated with inhibitor.
PKCε is a well-defined target of Epac activation (Hucho et al., 2005; Oestreich et al., 2009). We used the inhibitory peptide TAT–PKCεv1–2 that prevents translocation of PKCε to the membrane (Cesare et al., 1999). In control SNS–GRK2 +/+ mice, intraplantar TAT–PKCε v1–2 administration 20 min before intraplantar PGE2 did not have any effect on PGE2 hyperalgesia (Fig. 8 A). In contrast, intraplantar pretreatment with TAT–PKCε v1–2 completely prevented the prolonged phase of PGE2 hyperalgesia in SNS–GRK2 +/− mice (Fig. 8 A). In addition, intraplantar TAT–PKCε v1–2 attenuated acute PGE2 hyperalgesia in SNS–GRK2 +/− mice (Fig. 8 A). Similarly, in SNS–GRK2 +/− mice, the enhanced and prolonged 8-pCPT hyperalgesia was completely prevented by the PKCε inhibitory peptide (Fig. 8 B). In control SNS–GRK2 +/+ mice, 8-pCPT-induced hyperalgesia was also attenuated by intraplantar pretreatment with TAT-PKCεV1–2 (Fig. 8 B). Pretreatment with a TAT-coupled scrambled peptide that does not affect PKCε translocation did not affect PGE2 or 8-pCPT-induced hyperalgesia in SNS–GRK2 +/+ and SNS–GRK2 +/− mice at any of the time points tested (data not shown).
Figure 8.

PGE2, 8-pCPT-induced hyperalgesia in SNS–GRK2 +/− mice is biased to PKCε. Percentage change in heat-withdrawal latencies after intraplantar injection of the PKC inhibitor PKCεv1–2 (2.5 μg/paw) before PGE2 injection (A) or 8-pCPT (B) in SNS–GRK2 +/+ (WT) animals and SNS–GRK2 +/− mice (n = 4 per group). As a control, mice were treated with scrambled peptide before PGE2 injection (scr). Data are expressed as mean ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001 SNS–GRK2 +/− versus SNS–GRK2 +/− treated with inhibitor. # p < 0.05, ## p < 0.01 SNS–GRK2 +/+ (WT) versus SNS–GRK2 +/+ (WT) treated with inhibitor. C, Model of the role of reduced nociceptor GRK2 in prolonged PGE2 hyperalgesia. Low levels of nociceptor GRK2 promote cAMP-to-Epac signaling, whereas in nociceptor with “normal” GRK2 levels cAMP signals mainly to PKA. Additionally, low levels of GRK2 enhance ERK1/2 activation at the MEK/ERK1/2 interface. The effect of low nociceptor GRK2 levels ultimately lead to enhanced cAMP-to-Epac and ERK/PKCε signaling resulting in prolonged hyperalgesia.
Discussion
This study demonstrates that low nociceptor GRK2 protein levels such as those occurring during ongoing inflammatory hyperalgesia can induce biased PGE2/cAMP signaling from a PKA to an Epac/PKCε/ERK-dependent pathway, resulting in markedly prolonged PGE2/cAMP hyperalgesia. PGE2 hyperalgesia lasted ∼12 times longer in mice with a cell-specific reduction of GRK2 in Nav1.8-expressing sensory neurons than in littermate controls. The biased PGE2 signaling in SNS–GRK2 +/− mice occurred downstream of cAMP, because injection of a cAMP analog that activates both PKA and Epac, or a specific Epac activator, also induced PKA-independent prolonged hyperalgesia in these mice. Moreover, our in vitro studies showed that GRK2 physically binds to Epac1. This Epac1–GRK2 interaction may well underlie our finding that reduced GRK2 results in enhanced Rap1 activation in response to stimulation with a specific activator of Epac, leading to prolongation of hyperalgesia.
The maximal effect of low nociceptor GRK2 on PGE2 hyperalgesia was already obtained in heterozygous SNS–GRK2 f/+ mice with a 40–60% reduction in GRK2. Recently, we have observed a similar phenomenon when comparing the effect of partial or total GRK2 deletion in microglia/macrophages on IL-1β-induced hyperalgesia (Willemen et al., 2010). These findings indicate that there is a threshold level for the effect of reduction of GRK2 in both neurons and microglia. We have described previously a similar threshold level for the effect of GRK6 on colitis; experimental colitis was prolonged to the same extent in mice that were heterozygous or homozygous for deletion of GRK6 (Eijkelkamp et al., 2007).
Using specific EP receptors agonists, we showed that the prolonged hyperalgesia in mice with reduced nociceptor GRK2 does not result from a switch to an EP receptor subtype that signals to second messengers other than cAMP. Only activation of the Gs-coupled EP4 receptor mimicked the increased and prolonged PGE2 hyperalgesia in mice with reduced nociceptor GRK2.
GRK2 is known to desensitize signaling via a wide variety of GPCR. This classical model of GRK2 action predicts that GRK2 reduction would enhance EP4-mediated signaling, leading to a stronger cAMP response and increased PKA activation (Ribas et al., 2007). There is indeed evidence that human EP4 receptors can be phosphorylated by GRK2 (Neuschäfer-Rube et al., 1999). Conversely, PGE2-induced cAMP production in smooth muscle cells is unaffected in conditions of low GRK2 (Kong et al., 2008). We cannot exclude that low GRK2 in nociceptors has an additional effect upstream of cAMP, e.g., at the level of G-protein coupling to the receptor (Dina et al., 2009). However, we show that the cAMP analog 8-Br-cAMP also induced increased and prolonged hyperalgesia in SNS–GRK2 +/− mice. These findings indicate that the prolongation of hyperalgesia in SNS–GRK2 +/− mice is not dependent on GRK2 regulation of EP4 receptor desensitization but depends of GRK2 regulation at a level downstream of cAMP.
We recently showed that CCL3-induced hyperalgesia was increased but not prolonged in mice with reduced nociceptor GRK2 (Eijkelkamp et al., 2010). The chemokine CCL3 acts via GPCRs regulated by GRK2 (Oppermann et al., 1999; Vroon et al., 2004). However, CCL3 receptors do not signal to cAMP but to an IP3/calcium-dependent pathway. Apparently, regulation of the duration of inflammation-induced hyperalgesia by low nociceptor GRK2 levels occurs only if a cAMP-dependent but not an IP3/calcium-dependent signaling cascade is activated.
For many years, the downstream effects of an intracellular rise in cAMP have been attributed to PKA only. However, our present data clearly demonstrate that PGE2 or cAMP-induced hyperalgesia are no longer PKA dependent in mice with low GRK2 in nociceptors. Nowadays, the contribution of another cAMP target, Epac, has become widely accepted (de Rooij et al., 1998). We show here for the first time using coimmunoprecipitation studies that Epac1 is present in a protein complex with GRK2, especially after stimulation with the specific Epac activator 8-pCPT. Moreover, in vitro GRK2 reduction enhanced 8-pCPT-induced activation of the direct downstream target of Epac Rap1. The direct physical interaction between GRK2 and activated Epac may well contribute to this enhanced Epac/Rap1 activation in cells with low GRK2. In vivo PGE2-, cAMP-, and 8-pCPT-induced hyperalgesia in mice with reduced nociceptor GRK2 is prolonged and occurs via a PKA-independent pathway. Based on our in vivo and in vitro findings, we hypothesize that reduced GRK2 reorganizes the cAMP signaling cascade to favor Epac/Rap1 activation (Fig. 8 C). We also propose that this enhanced activation of the Epac/Rap1 pathway underlies the prolongation of PGE2 hyperalgesia that we observed in SNS–GRK2 +/− mice.
Studies on the functional role of Epac in the neuronal system have shown that Epac enhances neurotransmitter release in glutamatergic synapses (Sakaba and Neher, 2003; Zhong and Zucker, 2005; Gekel and Neher, 2008) and that Epac1 and Epac2 modulate neuronal excitability (Ster et al., 2007). Additionally, PGE2-induced sensitization of ATP currents in sensory neurons is enhanced in DRG neurons primed by inflammation in an Epac-dependent manner (Wang et al., 2007). Finally, Epac has been implicated in integrin signaling (Bos, 2006), which is important in the maintenance of inflammatory hyperalgesia (Dina et al., 2004). Thus, if low GRK2 biases cAMP signaling to Epac, as our present findings indicate, such activation of Epac may ultimately result in long-lasting changes in neurotransmitter release, neuronal excitability, and/or integrin signaling and thereby cause prolonged hyperalgesia.
Epac activation has been shown to directly stimulate PKCε activation (Wang et al., 2007; Oestreich et al., 2009). It has also been described that Epac-to-PKCε signaling is critical in P2X3 sensitization in primed neurons (Wang et al., 2007). Additionally, hyperalgesic priming that significantly prolongs hyperalgesia appears to be highly dependent on PKCε signaling (for review, see Reichling and Levine, 2009). Finally, PKCε activation can potentiate Nav1.8 and TRPV1 currents (Srinivasan et al., 2008; Cang et al., 2009). We show here that inhibition of PKCε completely blocked the prolongation of PGE2 or 8-pCPT-induced hyperalgesia in SNS–GRK2 +/− mice. Therefore, we propose that the switch to Epac/Rap1 activation in the nociceptors with low GRK2 leads to PKCε activation downstream of enhanced Epac/Rap1 activation, finally resulting in prolongation of inflammatory hyperalgesia (Fig. 8 C).
Our in vivo data showed that not only inhibition of PKCε but also inhibition of the MEK/ERK pathway completely prevented prolonged PGE2-, cAMP-, and 8-pCPT-induced hyperalgesia in mice with reduced nociceptor GRK2. In contrast, inhibition of the MEK/ERK pathway did not affect hyperalgesia in WT mice. These findings indicate that PKCε and MEK/ERK signaling act in parallel, and activation of both is required for the induction of prolonged hyperalgesia in SNS–GRK2 +/− mice. It is also possible, however, that PKCε and MEK are part of the same pathway and one is downstream of the other. At present, there is no evidence that activation of Epac by cAMP can lead to direct activation of MEK/ERK pathway in vitro (Enserink et al., 2002; Lin et al., 2003; Gelinas et al., 2008). Consistently, we observed a small 8-pCPT-induced increase in p-ERK in GRK2-deficient cells but not in WT cells. Nevertheless, in vivo, inhibition of the MEK–ERK pathway inhibits 8-pCPT-induced prolonged hyperalgesia in GRK2-deficient mice. These data indicate that, in vivo in SNS–GRK2 +/− mice, activation of Epac can lead to MEK–ERK activation. It remains to be determined whether this activation of the MEK–ERK pathway in SNS–GRK2 +/− mice is induced via indirect or direct pathways downstream of Epac and potentially via PKCε. In this respect, it is of interest that GRK2 binds to MEK1 and inhibits ERK activation at the MEK1–ERK interface (Jiménez-Sainz et al., 2006). Thus, the possibility exists that, in our model, low nociceptor GRK2 prolongs hyperalgesia via enhancing cAMP-induced Epac/Rap1 activation in combination with increased MEK1-to-ERK signaling that results from a direct interaction of GRK2 with MEK1 (Fig. 8 C).
Our finding that marked prolongation of PGE2-induced hyperalgesia occurs in the condition of a 50% reduction in nociceptor GRK2 levels raises the question whether similar changes in GRK2 levels occur in (patho)physiological conditions. Previously, we have shown that GRK2 protein levels in peripheral blood mononuclear cells from patients with rheumatoid arthritis or multiple sclerosis was reduced compared with healthy controls. Similarly, in animal models of rheumatoid arthritis or multiple sclerosis, splenocyte GRK2 protein levels were reduced (Lombardi et al., 1999; Vroon et al., 2005). Our present results show that GRK2 levels are reduced in small-diameter DRG neurons during carrageenan-induced hyperalgesia. In addition, we described that spinal cord neuronal and microglial GRK2 was reduced in rat or mouse models of neuropathic pain (Kleibeuker et al., 2007, 2008; Eijkelkamp et al., 2010). It is therefore tempting to speculate that an inflammation-induced decrease in GRK2 levels may contribute to the chronic pain that develops in conditions of chronic inflammation or nerve damage.
Overall, this study shows that low nociceptor GRK2 is a novel and important risk factor for the switch from acute to persistent pain by inducing biased intracellular cAMP signaling. Moreover, we propose that GRK2 restrains Epac/Rap1 signaling. Our data strongly indicate that low nociceptor GRK2 favors cAMP signaling to Epac/Rap1, leading to PKCε and MEK/ERK-dependent signaling and prolonged hyperalgesia.
Footnotes
We thank Dr. Gerald W. Dorn II at Washington University (St. Louis, MO) for providing GRK2–LOX mice.
References
- Abrahamsen B, Zhao J, Asante CO, Cendan CM, Marsh S, Martinez-Barbera JP, Nassar MA, Dickenson AH, Wood JN. The cell and molecular basis of mechanical, cold, and inflammatory pain. Science. 2008;321:702–705. doi: 10.1126/science.1156916. [DOI] [PubMed] [Google Scholar]
- Aley KO, Levine JD. Role of protein kinase A in the maintenance of inflammatory pain. J Neurosci. 1999;19:2181–2186. doi: 10.1523/JNEUROSCI.19-06-02181.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aley KO, Messing RO, Mochly-Rosen D, Levine JD. Chronic hypersensitivity for inflammatory nociceptor sensitization mediated by the epsilon isozyme of protein kinase C. J Neurosci. 2000;20:4680–4685. doi: 10.1523/JNEUROSCI.20-12-04680.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bos JL. Epac proteins: multi-purpose cAMP targets. Trends Biochem Sci. 2006;31:680–686. doi: 10.1016/j.tibs.2006.10.002. [DOI] [PubMed] [Google Scholar]
- Cang CL, Zhang H, Zhang YQ, Zhao ZQ. PKCepsilon-dependent potentiation of TTX-resistant Nav1.8 current by neurokinin-1 receptor activation in rat dorsal root ganglion neurons. Mol Pain. 2009;5:33. doi: 10.1186/1744-8069-5-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cant SH, Pitcher JA. G protein-coupled receptor kinase 2-mediated phosphorylation of ezrin is required for G protein-coupled receptor-dependent reorganization of the actin cytoskeleton. Mol Biol Cell. 2005;16:3088–3099. doi: 10.1091/mbc.E04-10-0877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cesare P, Dekker LV, Sardini A, Parker PJ, McNaughton PA. Specific involvement of PKC-epsilon in sensitization of the neuronal response to painful heat. Neuron. 1999;23:617–624. doi: 10.1016/s0896-6273(00)80813-2. [DOI] [PubMed] [Google Scholar]
- Coleman RA, Smith WL, Narumiya S. International Union of Pharmacology classification of prostanoid receptors: properties, distribution, and structure of the receptors and their subtypes. Pharmacol Rev. 1994;46:205–229. [PubMed] [Google Scholar]
- Cunha FQ, Teixeira MM, Ferreira SH. Pharmacological modulation of secondary mediator systems—cyclic AMP and cyclic GMP—on inflammatory hyperalgesia. Br J Pharmacol. 1999;127:671–678. doi: 10.1038/sj.bjp.0702601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delcroix JD, Valletta JS, Wu C, Hunt SJ, Kowal AS, Mobley WC. NGF signaling in sensory neurons: evidence that early endosomes carry NGF retrograde signals. Neuron. 2003;39:69–84. doi: 10.1016/s0896-6273(03)00397-0. [DOI] [PubMed] [Google Scholar]
- de Rooij J, Zwartkruis FJ, Verheijen MH, Cool RH, Nijman SM, Wittinghofer A, Bos JL. Epac is a Rap1 guanine-nucleotide-exchange factor directly activated by cyclic AMP. Nature. 1998;396:474–477. doi: 10.1038/24884. [DOI] [PubMed] [Google Scholar]
- Dina OA, Aley KO, Isenberg W, Messing RO, Levine JD. Sex hormones regulate the contribution of PKCepsilon and PKA signalling in inflammatory pain in the rat. Eur J Neurosci. 2001;13:2227–2233. doi: 10.1046/j.0953-816x.2001.01614.x. [DOI] [PubMed] [Google Scholar]
- Dina OA, McCarter GC, de Coupade C, Levine JD. Role of the sensory neuron cytoskeleton in second messenger signaling for inflammatory pain. Neuron. 2003;39:613–624. doi: 10.1016/s0896-6273(03)00473-2. [DOI] [PubMed] [Google Scholar]
- Dina OA, Parada CA, Yeh J, Chen X, McCarter GC, Levine JD. Integrin signaling in inflammatory and neuropathic pain in the rat. Eur J Neurosci. 2004;19:634–642. doi: 10.1111/j.1460-9568.2004.03169.x. [DOI] [PubMed] [Google Scholar]
- Dina OA, Khasar SG, Gear RW, Levine JD. Activation of Gi induces mechanical hyperalgesia poststress or inflammation. Neuroscience. 2009;160:501–507. doi: 10.1016/j.neuroscience.2009.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eijkelkamp N, Heijnen CJ, Lucas A, Premont RT, Elsenbruch S, Schedlowski M, Kavelaars A. G protein-coupled receptor kinase 6 controls chronicity and severity of dextran sodium sulphate-induced colitis in mice. Gut. 2007;56:847–854. doi: 10.1136/gut.2006.107094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eijkelkamp N, Heijnen CJ, Willemen HL, Deumens R, Joosten EA, Kleibeuker W, den Hartog IJ, van Velthoven CT, Nijboer C, Nassar MA, Dorn GW, 2nd, Wood JN, Kavelaars A. GRK2: a novel cell-specific regulator of severity and duration of inflammatory pain. J Neurosci. 2010;30:2138–2149. doi: 10.1523/JNEUROSCI.5752-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enserink JM, Christensen AE, de Rooij J, van Triest M, Schwede F, Genieser HG, Døskeland SO, Blank JL, Bos JL. A novel Epac-specific cAMP analogue demonstrates independent regulation of Rap1 and ERK. Nat Cell Biol. 2002;4:901–906. doi: 10.1038/ncb874. [DOI] [PubMed] [Google Scholar]
- Franke B, Akkerman JW, Bos JL. Rapid Ca2+-mediated activation of Rap1 in human platelets. EMBO J. 1997;16:252–259. doi: 10.1093/emboj/16.2.252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gekel I, Neher E. Application of an Epac activator enhances neurotransmitter release at excitatory central synapses. J Neurosci. 2008;28:7991–8002. doi: 10.1523/JNEUROSCI.0268-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gelinas JN, Banko JL, Peters MM, Klann E, Weeber EJ, Nguyen PV. Activation of exchange protein activated by cyclic-AMP enhances long-lasting synaptic potentiation in the hippocampus. Learn Mem. 2008;15:403–411. doi: 10.1101/lm.830008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haga K, Ogawa H, Haga T, Murofushi H. GTP-binding-protein-coupled receptor kinase 2 (GRK2) binds and phosphorylates tubulin. Eur J Biochem. 1998;255:363–368. doi: 10.1046/j.1432-1327.1998.2550363.x. [DOI] [PubMed] [Google Scholar]
- Hargreaves K, Dubner R, Brown F, Flores C, Joris J. A new and sensitive method for measuring thermal nociception in cutaneous hyperalgesia. Pain. 1988;32:77–88. doi: 10.1016/0304-3959(88)90026-7. [DOI] [PubMed] [Google Scholar]
- Honore P, Wade CL, Zhong C, Harris RR, Wu C, Ghayur T, Iwakura Y, Decker MW, Faltynek C, Sullivan J, Jarvis MF. Interleukin-1alphabeta gene-deficient mice show reduced nociceptive sensitivity in models of inflammatory and neuropathic pain but not post-operative pain. Behav Brain Res. 2006;167:355–364. doi: 10.1016/j.bbr.2005.09.024. [DOI] [PubMed] [Google Scholar]
- Hucho TB, Dina OA, Levine JD. Epac mediates a cAMP-to-PKC signaling in inflammatory pain: an isolectin B4+ neuron-specific mechanism. J Neurosci. 2005;25:6119–6126. doi: 10.1523/JNEUROSCI.0285-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaber M, Koch WJ, Rockman H, Smith B, Bond RA, Sulik KK, Ross J, Jr, Lefkowitz RJ, Caron MG, Giros B. Essential role of beta-adrenergic receptor kinase 1 in cardiac development and function. Proc Natl Acad Sci U S A. 1996;93:12974–12979. doi: 10.1073/pnas.93.23.12974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiménez-Sainz MC, Murga C, Kavelaars A, Jurado-Pueyo M, Krakstad BF, Heijnen CJ, Mayor F, Jr, Aragay AM. G protein-coupled receptor kinase 2 negatively regulates chemokine signaling at a level downstream from G protein subunits. Mol Biol Cell. 2006;17:25–31. doi: 10.1091/mbc.E05-05-0399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleibeuker W, Ledeboer A, Eijkelkamp N, Watkins LR, Maier SF, Zijlstra J, Heijnen CJ, Kavelaars A. A role for G protein-coupled receptor kinase 2 in mechanical allodynia. Eur J Neurosci. 2007;25:1696–1704. doi: 10.1111/j.1460-9568.2007.05423.x. [DOI] [PubMed] [Google Scholar]
- Kleibeuker W, Gabay E, Kavelaars A, Zijlstra J, Wolf G, Ziv N, Yirmiya R, Shavit Y, Tal M, Heijnen CJ. IL-1 beta signaling is required for mechanical allodynia induced by nerve injury and for the ensuing reduction in spinal cord neuronal GRK2. Brain Behav Immun. 2008;22:200–208. doi: 10.1016/j.bbi.2007.07.009. [DOI] [PubMed] [Google Scholar]
- Kong KC, Gandhi U, Martin TJ, Anz CB, Yan H, Misior AM, Pascual RM, Deshpande DA, Penn RB. Endogenous Gs-coupled receptors in smooth muscle exhibit differential susceptibility to GRK2/3-mediated desensitization. Biochemistry. 2008;47:9279–9288. doi: 10.1021/bi801056w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin CR, Amaya F, Barrett L, Wang H, Takada J, Samad TA, Woolf CJ. Prostaglandin E2 receptor EP4 contributes to inflammatory pain hypersensitivity. J Pharmacol Exp Ther. 2006;319:1096–1103. doi: 10.1124/jpet.106.105569. [DOI] [PubMed] [Google Scholar]
- Lin SL, Johnson-Farley NN, Lubinsky DR, Cowen DS. Coupling of neuronal 5-HT7 receptors to activation of extracellular-regulated kinase through a protein kinase A-independent pathway that can utilize Epac. J Neurochem. 2003;87:1076–1085. doi: 10.1046/j.1471-4159.2003.02076.x. [DOI] [PubMed] [Google Scholar]
- Lombardi MS, Kavelaars A, Schedlowski M, Bijlsma JW, Okihara KL, Van de Pol M, Ochsmann S, Pawlak C, Schmidt RE, Heijnen CJ. Decreased expression and activity of G-protein-coupled receptor kinases in peripheral blood mononuclear cells of patients with rheumatoid arthritis. FASEB J. 1999;13:715–725. doi: 10.1096/fasebj.13.6.715. [DOI] [PubMed] [Google Scholar]
- Matkovich SJ, Diwan A, Klanke JL, Hammer DJ, Marreez Y, Odley AM, Brunskill EW, Koch WJ, Schwartz RJ, Dorn GW., 2nd Cardiac-specific ablation of G-protein receptor kinase 2 redefines its roles in heart development and beta-adrenergic signaling. Circ Res. 2006;99:996–1003. doi: 10.1161/01.RES.0000247932.71270.2c. [DOI] [PubMed] [Google Scholar]
- Minami T, Nakano H, Kobayashi T, Sugimoto Y, Ushikubi F, Ichikawa A, Narumiya S, Ito S. Characterization of EP receptor subtypes responsible for prostaglandin E2-induced pain responses by use of EP1 and EP3 receptor knockout mice. Br J Pharmacol. 2001;133:438–444. doi: 10.1038/sj.bjp.0704092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moriyama T, Higashi T, Togashi K, Iida T, Segi E, Sugimoto Y, Tominaga T, Narumiya S, Tominaga M. Sensitization of TRPV1 by EP1 and IP reveals peripheral nociceptive mechanism of prostaglandins. Mol Pain. 2005;1:3–9. doi: 10.1186/1744-8069-1-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neuschäfer-Rube F, Oppermann M, Möller U, Böer U, Püschel GP. Agonist-induced phosphorylation by G protein-coupled receptor kinases of the EP4 receptor carboxyl-terminal domain in an EP3/EP4 prostaglandin E(2) receptor hybrid. Mol Pharmacol. 1999;56:419–428. doi: 10.1124/mol.56.2.419. [DOI] [PubMed] [Google Scholar]
- Oestreich EA, Malik S, Goonasekera SA, Blaxall BC, Kelley GG, Dirksen RT, Smrcka AV. Epac and phospholipase Cepsilon regulate Ca2+ release in the heart by activation of protein kinase Cepsilon and calcium-calmodulin kinase II. J Biol Chem. 2009;284:1514–1522. doi: 10.1074/jbc.M806994200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oida H, Namba T, Sugimoto Y, Ushikubi F, Ohishi H, Ichikawa A, Narumiya S. In situ hybridization studies of prostacyclin receptor mRNA expression in various mouse organs. Br J Pharmacol. 1995;116:2828–2837. doi: 10.1111/j.1476-5381.1995.tb15933.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oppermann M, Mack M, Proudfoot AE, Olbrich H. Differential effects of CC chemokines on CC chemokine receptor 5 (CCR5) phosphorylation and identification of phosphorylation sites on the CCR5 carboxyl terminus. J Biol Chem. 1999;274:8875–8885. doi: 10.1074/jbc.274.13.8875. [DOI] [PubMed] [Google Scholar]
- Pitcher JA, Hall RA, Daaka Y, Zhang J, Ferguson SS, Hester S, Miller S, Caron MG, Lefkowitz RJ, Barak LS. The G protein-coupled receptor kinase 2 is a microtubule-associated protein kinase that phosphorylates tubulin. J Biol Chem. 1998;273:12316–12324. doi: 10.1074/jbc.273.20.12316. [DOI] [PubMed] [Google Scholar]
- Price LS, Hajdo-Milasinovic A, Zhao J, Zwartkruis FJ, Collard JG, Bos JL. Rap1 regulates E-cadherin-mediated cell-cell adhesion. J Biol Chem. 2004;279:35127–35132. doi: 10.1074/jbc.M404917200. [DOI] [PubMed] [Google Scholar]
- Pronin AN, Morris AJ, Surguchov A, Benovic JL. Synucleins are a novel class of substrates for G protein-coupled receptor kinases. J Biol Chem. 2000;275:26515–26522. doi: 10.1074/jbc.M003542200. [DOI] [PubMed] [Google Scholar]
- Reichling DB, Levine JD. Critical role of nociceptor plasticity in chronic pain. Trends Neurosci. 2009;32:611–618. doi: 10.1016/j.tins.2009.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiter E, Lefkowitz RJ. GRKs and beta-arrestins: roles in receptor silencing, trafficking and signaling. Trends Endocrinol Metab. 2006;17:159–165. doi: 10.1016/j.tem.2006.03.008. [DOI] [PubMed] [Google Scholar]
- Ribas C, Penela P, Murga C, Salcedo A, García-Hoz C, Jurado-Pueyo M, Aymerich I, Mayor F., Jr The G protein-coupled receptor kinase (GRK) interactome: role of GRKs in GPCR regulation and signaling. Biochim Biophys Acta. 2007;1768:913–922. doi: 10.1016/j.bbamem.2006.09.019. [DOI] [PubMed] [Google Scholar]
- Sakaba T, Neher E. Direct modulation of synaptic vesicle priming by GABA(B) receptor activation at a glutamatergic synapse. Nature. 2003;424:775–778. doi: 10.1038/nature01859. [DOI] [PubMed] [Google Scholar]
- Sohal DS, Nghiem M, Crackower MA, Witt SA, Kimball TR, Tymitz KM, Penninger JM, Molkentin JD. Temporally regulated and tissue-specific gene manipulations in the adult and embryonic heart using a tamoxifen-inducible Cre protein. Circ Res. 2001;89:20–25. doi: 10.1161/hh1301.092687. [DOI] [PubMed] [Google Scholar]
- Southall MD, Vasko MR. Prostaglandin receptor subtypes, EP3C and EP4, mediate the prostaglandin E2-induced cAMP production and sensitization of sensory neurons. J Biol Chem. 2001;276:16083–16091. doi: 10.1074/jbc.M011408200. [DOI] [PubMed] [Google Scholar]
- Srinivasan R, Wolfe D, Goss J, Watkins S, de Groat WC, Sculptoreanu A, Glorioso JC. Protein kinase C epsilon contributes to basal and sensitizing responses of TRPV1 to capsaicin in rat dorsal root ganglion neurons. Eur J Neurosci. 2008;28:1241–1254. doi: 10.1111/j.1460-9568.2008.06438.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ster J, De Bock F, Guérineau NC, Janossy A, Barrère-Lemaire S, Bos JL, Bockaert J, Fagni L. Exchange protein activated by cAMP (Epac) mediates cAMP activation of p38 MAPK and modulation of Ca2+-dependent K+ channels in cerebellar neurons. Proc Natl Acad Sci U S A. 2007;104:2519–2524. doi: 10.1073/pnas.0611031104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stirling LC, Forlani G, Baker MD, Wood JN, Matthews EA, Dickenson AH, Nassar MA. Nociceptor-specific gene deletion using heterozygous NaV1.8-Cre recombinase mice. Pain. 2005;113:27–36. doi: 10.1016/j.pain.2004.08.015. [DOI] [PubMed] [Google Scholar]
- Taniguchi J, Fujitani M, Endo M, Kubo T, Fujitani M, Miller FD, Kaplan DR, Yamashita T. Rap1 is involved in the signal transduction of myelin-associated glycoprotein. Cell Death Differ. 2008;15:408–419. doi: 10.1038/sj.cdd.4402278. [DOI] [PubMed] [Google Scholar]
- Vroon A, Heijnen CJ, Lombardi MS, Cobelens PM, Mayor F, Jr, Caron MG, Kavelaars A. Reduced GRK2 level in T cells potentiates chemotaxis and signaling in response to CCL4. J Leukoc Biol. 2004;75:901–909. doi: 10.1189/jlb.0403136. [DOI] [PubMed] [Google Scholar]
- Vroon A, Kavelaars A, Limmroth V, Lombardi MS, Goebel MU, Van Dam AM, Caron MG, Schedlowski M, Heijnen CJ. G protein-coupled receptor kinase 2 in multiple sclerosis and experimental autoimmune encephalomyelitis. J Immunol. 2005;174:4400–4406. doi: 10.4049/jimmunol.174.7.4400. [DOI] [PubMed] [Google Scholar]
- Vroon A, Heijnen CJ, Kavelaars A. GRKs and arrestins: regulators of migration and inflammation. J Leukoc Biol. 2006;80:1214–1221. doi: 10.1189/jlb.0606373. [DOI] [PubMed] [Google Scholar]
- Wang C, Gu Y, Li GW, Huang LY. A critical role of the cAMP sensor Epac in switching protein kinase signalling in prostaglandin E2-induced potentiation of P2X3 receptor currents in inflamed rats. J Physiol. 2007;584:191–203. doi: 10.1113/jphysiol.2007.135616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willemen HL, Eijkelkamp N, Wang H, Dantzer R, Dorn GW, 2nd, Kelley KW, Heijnen CJ, Kavelaars A. Microglial/macrophage GRK2 determines duration of peripheral IL-1beta-induced hyperalgesia: contribution of spinal cord CX3CR1, p38 and IL-1 signaling. Pain. 2010;150:550–560. doi: 10.1016/j.pain.2010.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Ferguson SS, Barak LS, Aber MJ, Giros B, Lefkowitz RJ, Caron MG. Molecular mechanisms of G protein-coupled receptor signaling: role of G protein-coupled receptor kinases and arrestins in receptor desensitization and resensitization. Receptors Channels. 1997;5:193–199. [PubMed] [Google Scholar]
- Zhong N, Zucker RS. cAMP acts on exchange protein activated by cAMP/cAMP-regulated guanine nucleotide exchange protein to regulate transmitter release at the crayfish neuromuscular junction. J Neurosci. 2005;25:208–214. doi: 10.1523/JNEUROSCI.3703-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]