

Abstract
Inflammation is increasingly recognized as an important contributor to a host of CNS disorders; however, its regulation in the brain is not well delineated. Nucleotide-binding domain, leucine-rich repeat, pyrin domain containing 3 (NLRP3) is a key component of the inflammasome complex, which also includes ASC (apoptotic speck-containing protein with a card) and procaspase-1. Inflammasome formation can be triggered by membrane P2X7R engagement leading to cleavage-induced maturation of caspase-1 and interleukin-1β (IL-1β)/IL-18. This work shows that expression of the Nlrp3 gene was increased >100-fold in a cuprizone-induced demyelination and neuroinflammation model. Mice lacking the Nlrp3 gene (Nlrp3−/−) exhibited delayed neuroinflammation, demyelination, and oligodendrocyte loss in this model. These mice also showed reduced demyelination in the experimental autoimmune encephalomyelitis model of neuroinflammation. This outcome is also observed for casp1−/− and IL-18−/− mice, whereas IL-1β−/− mice were indistinguishable from wild-type controls, indicating that Nlrp3-mediated function is through caspase-1 and IL-18. Additional analyses revealed that, unlike the IL-1β−/− mice, which have been previously shown to show delayed remyelination, Nlrp3−/− mice did not exhibit delayed remyelination. Interestingly, IL-18−/− mice showed enhanced remyelination, thus providing a possible compensatory mechanism for the lack of a remyelination defect in Nlrp3−/− mice. These results suggest that NLRP3 plays an important role in a model of multiple sclerosis by exacerbating CNS inflammation, and this is partly mediated by caspase-1 and IL-18. Additionally, the therapeutic inhibition of IL-18 might decrease demyelination but enhance remyelination, which has broad implications for demyelinating diseases.
Introduction
CNS inflammation (neuroinflammation) is a key component of many neurological diseases including multiple sclerosis (MS), Parkinson's disease, and Alzheimer's disease. MS results from neuroinflammation characterized by lymphocyte/macrophage infiltration, microglial and astrocytic activation, enhanced cytokine/chemokine production, demyelination, and axonal loss (Sospedra and Martin, 2005; Pittock and Lucchinetti, 2007). Understanding the mechanisms by which neuroinflammation affects demyelination is important for therapeutic development.
Several families of innate immune receptors or sensors have been identified, with the nucleotide-binding domain, leucine-rich repeat containing (NLR) family receiving significant attention because of its genetic linkage to human immunologic diseases and its role in immune regulation (Harton et al., 2002; Ting et al., 2006). NLR family, Pyrin-domain containing 3 (NLRP3) (also Cryopyrin, NALP3, PYPAF1, CIAS1) represents a core component of a caspase-1-activating inflammasome complex, composed of an NLR protein, the adaptor molecule apoptotic speck-containing protein with a card (ASC), and procaspase-1 (Sutterwala et al., 2006a,b). Activated caspase-1 in turn cleaves >70 substrates including the proinflammatory cytokines interleukin-1β (IL-1β) and IL-18 (Shao et al., 2007; Keller et al., 2008). NLRP3 forms an inflammasome in response to bacterial RNA and toxins (Kanneganti et al., 2006a,b), viruses (Allen et al., 2009; Thomas et al., 2009), ATP (Mariathasan et al., 2006), uric acid (Martinon et al., 2006), hyaluronan (Yamasaki et al., 2009), amyloid-β (Halle et al., 2008), asbestos, silica (Dostert et al., 2008; Hornung et al., 2008), and alum (Hornung et al., 2008; Li et al., 2008). NLRP3 gene mutations have been identified in dominantly inherited autoinflammatory syndromes collectively referred to as Cryopyrin-associated periodic syndromes (CAPS) (Neven et al., 2004; Ting and Davis, 2005; Jha and Ting, 2009) characterized by hyperactivation of the inflammasome complex and increased IL-1β (Neven et al., 2004; Goldbach-Mansky et al., 2006). CAPS symptoms are treatable with the IL-1 receptor antagonist (Rigante et al., 2006). Recently, two studies reported MS-like lesions including periventricular infiltrates and corpus callosum demyelination in CAPS patients, suggesting a possible link between NLRP3 and neuroinflammation and demyelination (Lequerre et al., 2007; Compeyrot-Lacassagne et al., 2009). However, the role of NLRP3 in CNS inflammation and demyelination is not understood.
In this study, we used the cuprizone-induced demyelination model to evaluate the role of NLRP3 in CNS inflammation. This model has several advantages: (1) it exhibits type III and IV MS neuropathology characterized by microglial infiltration and astrogliosis without T-cell infiltrates (Lucchinetti et al., 2000; Liu et al., 2010), (2) it is easily induced by administering cuprizone through the chow, (3) the disease course follows a predictable time course and pathology, and (4) remyelination can be initiated by withdrawing the cuprizone (Matsushima and Morell, 2001). Using the cuprizone model, we studied mice deficient in genes encoding NLRP3 (Nlrp3−/−), IL-1β (IL-1β−/−), IL-18 (IL-18−/−), and caspase-1 (casp1−/−) as a way to examine the role of the NLRP3 inflammasome complex in CNS inflammation and demyelination in vivo. We found a significant role for the NLRP3 inflammasome pathway in the activation of neuroinflammation. In addition, we also describe a role for IL-18 in demyelination and remyelination.
Materials and Methods
Mice. Nlrp3
−/− mice were provided by Millennium Pharmaceuticals through Drs. Fayaz Sutterwala and Richard Flavell (Yale University, New Haven, CT) and were further backcrossed to C57BL/6 mice for a total of nine generations. Casp1−/− and IL-1β−/− were kindly provided by Dr. Richard Flavell (Yale University, New Haven, CT) and Dr. David Chaplin (University of Alabama, Birmingham, AL) (Shornick et al., 1996), respectively. C57BL/6 mice [wild type (WT)] were purchased from the National Cancer Institute (Bethesda, MD) and The Jackson Laboratory. IL-18−/− breeder mice were purchased from The Jackson Laboratory and bred in-house to generate mice for the cuprizone studies. All mice were 8–10 weeks of age before the start of treatment. All animal procedures conducted were approved by the Institutional Animal Care and Use Committee of University of North Carolina at Chapel Hill.
Cuprizone treatment.
Eight- to 10-week-old male mice were fed 0.2% cuprizone [oxalic bis(cyclohexylidenehydrazide)] (Sigma-Aldrich) mixed into ground chow ad libitum for 6 weeks to induce progressive demyelination. Untreated control mice were maintained on a diet of normal pellet chow. During cuprizone treatment, mice showed lethargic movement, weight loss (supplemental Fig. 1, available at www.jneurosci.org as supplemental material), ruffled hair, and altered gait as described previously (Arnett et al., 2001; Franco-Pons et al., 2007). For remyelination studies, mice were fed cuprizone mixed into ground chow ad libitum for 6 weeks followed by regular chow to allow for remyelination (Arnett et al., 2001).
Induction of experimental autoimmune encephalomyelitis.
Myelin oligodendrocyte glycoprotein (MOG)-induced experimental autoimmune encephalomyelitis (EAE) using the MOG35-55 peptide was performed as previously described (Zhang et al., 2010). Animals were scored daily by two independent investigators.
Tissue preparation.
Mice were deeply anesthetized and intracardially perfused with PBS followed by 4% paraformaldehyde (PFA). Brains were removed, postfixed in PFA, and embedded in paraffin. Coronal sections (5 μm) were cut at the fornix region of the corpus callosum. All analyses were restricted to the midline corpus callosum as described previously (Arnett et al., 2001). Some analyses were also performed lateral to the midline to confirm results at the midline.
Staining.
To examine demyelination, paraffin sections were rehydrated through a graded series of alcohol washes and stained with Luxol fast blue–periodic acid/Schiff base (LFB-PAS) (Sigma-Aldrich) as described previously (Arnett et al., 2002). Sections were read by three blinded readers and graded on a scale from 0 (complete myelination) to 3 (complete demyelination). Higher scores indicate greater pathology. For the detection of microglia/macrophages, tissue sections were rehydrated and permeabilized with 0.1% Triton/PBS for 20 min at room temperature (RT) and then incubated with Ricinus communis agglutinin-1 (RCA-1) lectin (1:500; Vector) at 37°C for 1 h. Only RCA-1+ cells with observable 4′,6′-diamidino-2-phenylindole (DAPI)-stained nuclei were included in the quantification.
Immunohistochemistry.
Immunohistochemistry was performed on 5 μm paraffin embedded sections that were deparaffinized and rehydrated through alcohols as described previously (Arnett et al., 2002). For the detection of mature oligodendrocytes, the sections were processed by boiling in antigen unmasking solution (1:100; Vector) for 13 min in a microwave. These sections were permeabilized with 0.1% Triton/PBS for 20 min and incubated with 2% normal goat serum (NGS) in 0.1% Triton/PBS for 20 min at RT. Subsequently, the sections were incubated with rabbit anti-mouse polyclonal antibody, anti-glutathione S-transferase π subunit (GSTπ) (1:500; Assay Designs), at 4°C overnight. Sections were then washed in PBS and incubated with the appropriate biotinylated antibody against primary antibody (1:100; Vector) and Texas Red-conjugated avidin (1:500; Vector). To detect astrocytes, sections were incubated with 5% NGS in 0.1% Triton/PBS for 20 min at RT. Subsequently, the sections were washed and incubated with rabbit anti-cow monoclonal antibody to glial fibrillary acidic protein (GFAP) (1:100; Dako) and goat anti-rabbit fluorescein-conjugated secondary antibody (1:100; Vector). To detect microglia, sections were processed by boiling in antigen unmasking solution (1:100; Vector) for 13 min in a microwave. These sections were permeabilized with 0.1% Triton/PBS for 10 min at RT. Subsequently, the sections were washed and incubated with goat anti-mouse polyclonal antibody to ionized calcium binding adaptor molecule-1 (Iba-1) (1:100; Abcam) and rabbit anti-goat biotin-conjugated secondary antibody (1:500; Vector). After PBS washes, the sections were incubated with Alexa 594-conjugated avidin (1:500; Invitrogen) for 1 h at RT. For the detection of 2′,3′-cyclic nucleotide phosphodiesterase (CNPase), the sections were processed by boiling in antigen unmasking solution (1:100; Vector) for 13 min in a microwave. These sections were permeabilized with 0.1% Triton/PBS for 20 min and incubated with 5% NGS in 0.1% Triton/PBS for 20 min at RT. Subsequently, the sections were incubated with chicken polyclonal antibody to CNPase (1:500; Millipore Bioscience Research Reagents) at RT for 1 h. Sections were then washed in PBS and incubated with rabbit polyclonal to chicken IgY biotin (1:500; Abcam) for 1 h at RT. After PBS washes, the sections were incubated with Alexa 594-conjugated avidin (1:500; Invitrogen) for 1 h at RT. Immunopositive cells with an observable DAPI-stained nucleus were counted blindly twice. Cell counts are averages of at least 9 and up to 14 mice per time point.
Terminal deoxynucleotidyl transferase-mediated biotinylated UTP nick end labeling.
To identify apoptotic cells in the brain, the terminal deoxynucleotidyl transferase-mediated biotinylated UTP nick end labeling (TUNEL) assay was used. Briefly, 5 μm paraffin embedded sections were rehydrated and permeabilized as described previously. TUNEL was performed using the in situ cell death detection kit (Roche). For the colocalization of TUNEL with GSTπ+ or RCA+ cells, sections were stained for GSTπ or RCA before TUNEL staining.
Imaging.
All cell counts were taken from the midline of the corpus callosum, confined to an area of 0.033 mm2 taken with a 50× oil-immersion objective. An Olympus BX-40 microscope with camera (Optronics Engineering) and Scion Image acquisition software was used for taking images.
Reverse transcription-PCR and quantitative real-time reverse transcription-PCR.
Total RNA was isolated from a dissected region of the brain containing the corpus callosum of wild-type and Nlrp3−/− mice at several points during and after cuprizone treatment. RNA isolation was performed using Trizol reagent (Invitrogen) under RNase-free conditions.
Protein analysis.
Total protein was extracted from the forebrains of cuprizone-treated and untreated control C57BL/6 mice. Briefly, corpus callosi were homogenized on ice in 600 μl of radioimmunoprecipitation assay buffer containing protease inhibitors. The homogenate was centrifuged at 10,000 × g for 15 min at 4°C. Supernatants were used for additional analyses. Protein concentrations were determined using a Coomassie (Bradford) protein assay kit (Pierce). IL-1β levels were determined by ELISA (optiEIA ELISA; BD Biosciences). Tumor necrosis factor-α (TNF-α) levels were determined by ELISA (optiEIA ELISA; BD Biosciences).
Statistical analysis.
Data are expressed as mean ± SEM. Unpaired Student's t tests were used to statistically evaluate significant differences. Differences were considered statistically significant if p < 0.05.
Results
Nlrp3 expression is increased in the cuprizone model of demyelination
Nlrp3 transcript expression in the CNS of cuprizone-treated C57BL/6 mice was examined by real-time quantitative PCR amplification. Nlrp3 expression was found to increase progressively to >120-fold by the 4 week time point of a course of cuprizone-induced demyelination (Fig. 1a). This increase coincided with the peak of inflammatory cell infiltration, demyelination, and mature oligodendrocyte death. Protein lysates from corpus callosi of cuprizone-treated Nlrp3−/− and control mouse brains analyzed by IL-1β ELISA showed a similar increase in IL-1β protein as Nlrp3 mRNA, supporting a functional increase in NLRP3 activity (Fig. 1b). This corroborates results from a previous study that quantified the number of IL-1β+ cells during cuprizone-induced demyelination and remyelination (Mason et al., 2001). Baseline cytokine levels were similar in Nlrp3−/− and control mouse brains as measured by IL-1β and TNF-α ELISA (supplemental Fig. 2, available at www.jneurosci.org as supplemental material). The lack of a difference in IL-1β levels between Nlrp3−/− and control mice is not surprising as this has been reported previously by others (Mayer-Barber et al., 2010). Many proteases can cleave pro-IL-1β, such as by Fas ligand (Miwa et al., 1998), human mast cell chymase (Omoto et al., 2006), trypsin and plasmin (Matsushima et al., 1986), proteinase 3 (Coeshott et al., 1999), neutrophil elastase (Black et al., 1988), cathepsin G (Hazuda et al., 1990), and matrix metalloproteinases (Schönbeck et al., 1998). However, the use of IL-1β−/− mice indicates that this cytokine is not a determining factor in the demyelination phase of the cuprizone model, which supports a previous publication (Mason et al., 2001). A lack of a difference in TNF-α is typical of NLRP3-deficient mice (Mariathasan et al., 2006; Sutterwala et al., 2006a).
Figure 1.
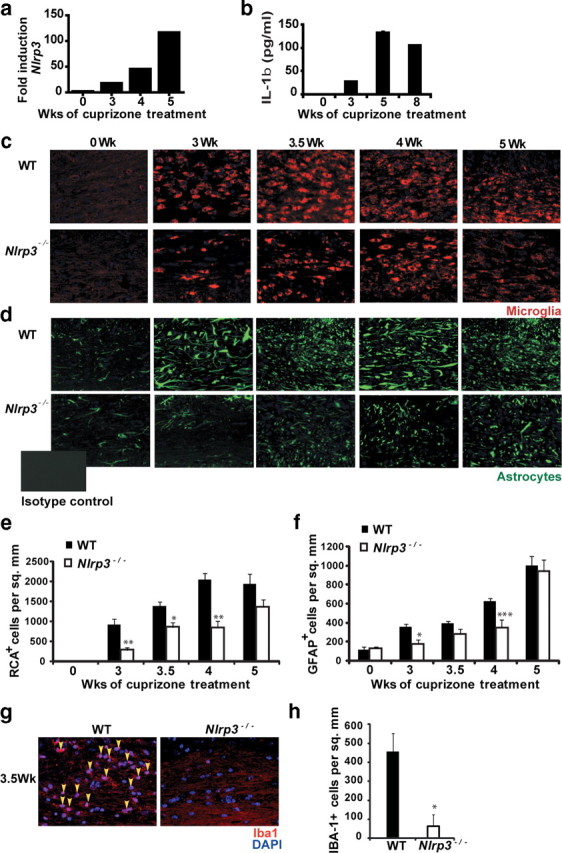
Increases in NLRP3 expression coincide with peak disease symptoms in the cuprizone model. a, Nlrp3 mRNA increases during cuprizone-induced demyelination. Nlrp3 transcript was detected by real-time quantitative PCR and the quantity at 0 week was set to 1. Nlrp3 mRNA increased progressively reaching a maximum of ∼120-fold after 5 weeks of cuprizone treatment concurrent with microglia/macrophage recruitment. b, Increase in IL-1β release is concurrent with the increase in Nlrp3 expression during demyelination. IL-1β release was measured by ELISA. c, Microglial recruitment to the corpus callosum was reduced in Nlrp3−/− mice. Microglia were detected by RCA lectin staining (red). DAPI was used to label nuclei (blue). d, Astrogliosis was reduced in Nlrp3−/− mice. GFAP (green) was used to detect astrocyte accumulation in the corpus callosum. The inset shows isotype control for GFAP antibody. RCA+ or GFAP+ cells with an observable DAPI-stained nucleus were counted blindly twice. Cell counts are averages of between 9 and 14 mice per time point. e, Microglial infiltration was quantitated and found to be significantly reduced in Nlrp3−/− mice (white bars) during demyelination (p = 0.02 at 3 weeks, p = 0.05 at 3.5 weeks, p = 0.01 at 4 weeks). f, Astrocyte accumulation was quantitated and found to be significantly reduced in Nlrp3−/− mice during demyelination (p = 0.03 at 3 weeks, p = 0.001 at 4 weeks). g, Microglial recruitment to the corpus callosum was reduced in Nlrp3−/− mice. Microglia were detected by Iba1 staining (red). DAPI was used to label nuclei (blue). h, Microglial infiltration in Figure 1g was quantitated and found to be significantly reduced in Nlrp3−/− mice (white bars) (p = 0.014 at 3.5 weeks). In e, f, and h, *p < 0.05, **p < 0.01, ***p < 0.005; n = 9–14. Error bars indicate SEM. Representative images for 9–14 mice per time point are shown.
Recruitment of microglia and astrocytes is delayed in Nlrp3−/− mice
To explore whether NLRP3 has a role during cuprizone-induced inflammation, we used mice lacking the Nlrp3 gene. WT and Nlrp3−/− mice showed similar reduction in weight during the course of cuprizone treatment (supplemental Fig. 1, available at www.jneurosci.org as supplemental material). We first examined whether deletion of this gene in mice had an effect on microglial accumulation and astrogliosis (Fig. 1c–f). Microglia are resident immune cells of the CNS (Hanisch and Kettenmann, 2007). Activated microglia can phagocytose cellular debris, present antigens to T-cells, and release cytokines and chemokines (Hanisch and Kettenmann, 2007; Napoli and Neumann, 2009). Activated microglia and astrocytes perform several overlapping roles during neuroinflammation (Dong and Benveniste, 2001). Microglial populations at the corpus callosum were identified by RCA-1 lectin (Fig. 1c) and Iba-1 staining (Fig. 1g). Astroglial populations at the corpus callosum were identified by GFAP staining (Fig. 1d). Untreated, age-matched (0 week) Nlrp3−/− mice and C57BL/6 (WT) controls showed no difference in the quantitation of microglia and astrocytes at the corpus callosum (Fig. 1c–h). A histological representation of these data is shown in Figure 1, c, d, and g. Quantitation is shown in Figure 1, e, f, and h. At 3, 3.5, and 4 weeks of cuprizone treatment, there was a progressive and significant (p = 0.02 at 3 weeks, p = 0.05 at 3.5 weeks, p = 0.01 at 4 weeks) reduction in microglial infiltration in Nlrp3−/− mice relative to WT controls as measured by RCA staining (Fig. 1e). Similarly, there was a progressive and statistically significant reduction in astrogliosis in the Nlrp3−/− mice at weeks 3 and 4 of cuprizone treatment (Fig. 1f) (p = 0.03 at 3 weeks, p = 0.001 at 4 weeks). These results indicate that NLRP3 enhances microglia accumulation and astrogliosis in the affected tissues. After 5 weeks of cuprizone treatment, microglial accumulation and astrogliosis between Nlrp3−/− and C57BL/6 control animals were similar (Fig. 1e,f). This is consistent with other studies of the cuprizone model wherein the removal of inflammatory genes has not affected neuropathology after this time point (Arnett et al., 2001; Mason et al., 2001). As an additional assessment of microglial cells, Iba-1 staining at 3.5 weeks of cuprizone treatment showed a similar reduction in microglial infiltration as RCA in Nlrp3−/− mice relative to WT controls (Fig. 1h) (p = 0.014 at 3.5 weeks). In addition to the midline corpus callosum, microglial infiltration and astrogliosis were also studied in two separate fields lateral to the midline corpus callosum as outlined by gray boxes in the schematic in Figure 2a. Both microglial infiltration and astrogliosis were significantly reduced in Nlrp3−/− mice compared with C57BL/6 controls, similar to the reduction observed at the midline corpus callosum (Fig. 2b–i).
Figure 2.
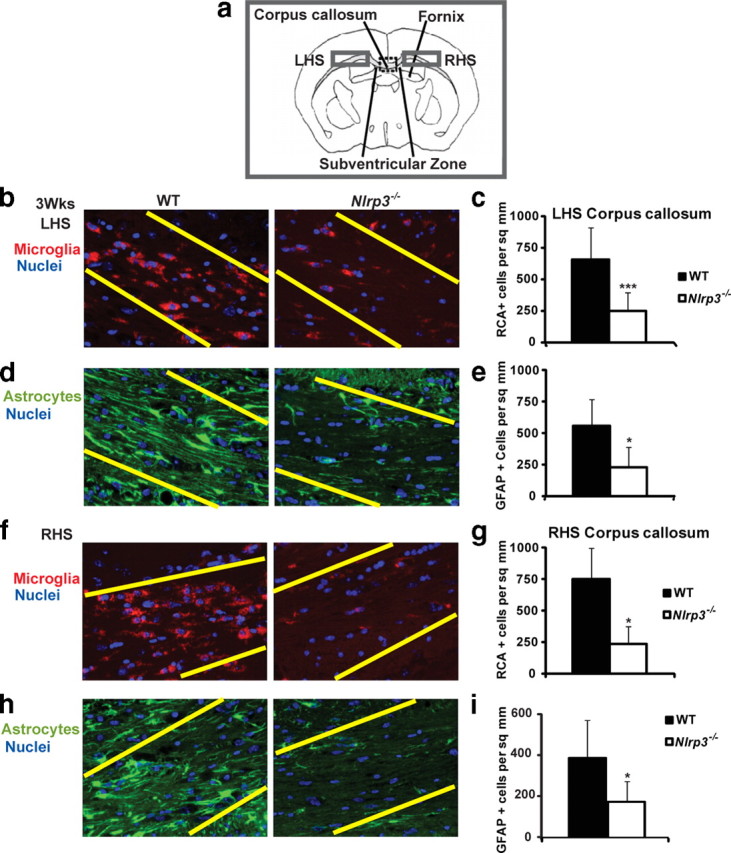
Microglial infiltration and astrogliosis in Nlrp3−/− are delayed in regions lateral to the midline corpus callosum relative to control WT mice after 3 weeks of cuprizone-induced demyelination. a, Microglial infiltration and astrogliosis were also studied in two separate fields on the left-hand side (LHS) and right-hand side (RHS), respectively, lateral to the midline corpus callosum as outlined by gray boxes in the schematic. b, Microglial recruitment on the LHS to the midline corpus callosum was reduced in Nlrp3−/− mice. c, Microglial infiltration on the LHS was quantitated and found to be significantly reduced in Nlrp3−/− mice (white bars) during demyelination (p = 0.0008 at 3 weeks). d, Astrogliosis on the LHS to the midline corpus callosum was reduced in Nlrp3−/− mice. e, Astrogliosis was quantified on the LHS and was reduced in Nlrp3−/− mice (p = 0.028 at 3 weeks). f, Microglial recruitment on the RHS to the midline corpus callosum was reduced in Nlrp3−/− mice. g, Microglial infiltration on the RHS was quantitated and found to be significantly reduced in Nlrp3−/− mice (white bars) during demyelination (p = 0.036 at 3 weeks). h, Astrogliosis on the LHS to the midline corpus callosum was reduced in Nlrp3−/− mice. i, Astrogliosis was quantified on the LHS and was reduced in Nlrp3−/− mice (p = 0.05 at 3 weeks). Microglia were detected by RCA lectin staining (red). DAPI was used to label nuclei (blue). GFAP (green) was used to detect astrocyte accumulation in the corpus callosum. RCA+ or GFAP+ cells with an observable DAPI stained nucleus were counted blindly twice. There was no statistically significant difference within genotypes on the LHS or RHS that would suggest any laterality. Cell counts are averages of between four and six mice per time point. *p < 0.05, ***p < 0.005. Error bars indicate SEM.
Demyelination is delayed in cuprizone-treated Nlrp3−/− mice
A second component of the cuprizone model that is relevant to human diseases is the demyelination process. To assess whether NLRP3 plays a role in demyelination and in the loss of mature oligodendrocytes, Nlrp3−/− mice along with age-matched WT control mice were treated with cuprizone for 3, 3.5, 4, and 5 weeks. Representative scoring of the extent of demyelination as measured by LFB-PAS staining is shown in Figure 3a. Slides were read by three blinded readers on a scale of 0 (no demyelination) to 3 (complete demyelination). WT mice showed significant demyelination initiating at the 3 week time point and continuing to the end of the study at the 5 week time point (Fig. 3b). Nlrp3−/− mice showed a significant delay in demyelination at the 3, 3.5, and 4 week time points when compared with WT controls (Fig. 3b) (p = 0.003 at 3 weeks, p = 0.012 at 3.5 weeks, p = 0.025 at 4 weeks). To assess whether the role of NLRP3 in exacerbating demyelination disease severity and demyelination is found in another disease model, we used EAE, a model of T-cell-mediated demyelination for multiple sclerosis. At the 3 week time point, Nlrp3−/− mice showed significantly reduced disease scores (Fig. 3c) and retained more myelin as determined by the CNPase stain for myelin (Fig. 3d) when compared with wild-type controls. A more comprehensive study of this gene in EAE is beyond the scope of this report and will be reported elsewhere.
Figure 3.

NLRP3 exacerbates demyelination in the corpus callosum during cuprizone-induced demyelination and experimental autoimmune encephalomyelitis. a, The schematic depicts the scale for scoring of demyelination. Each slide was scored by three independent blinded readers on a score of 0 (no demyelination) to 3 (complete demyelination). All scores are restricted to the midline corpus callosum (boxed area). b, Nlrp3−/− mice (open circles) show delayed demyelination compared with WT controls (filled circles). Demyelination was quantitated by LFB-PAS staining. Each circle represents the averaged observed LFB score from three readers for one mouse. The mean value of each data set is depicted by a red line (p = 0.003 at 3 weeks, p = 0.012 at 3.5 weeks, p = 0.025 at 4 weeks). c, Clinical scores for EAE showed significantly reduced disease in Nlrp3−/− mice relative to WT controls. d, CNPase staining of paraffin-embedded 5 μm spinal cord sections from Nlrp3−/− and WT control mice during EAE showed delayed loss of myelin in Nlrp3−/− mice. *p < 0.05, ***p < 0.005. Error bars indicate SEM.
LFB is a screening assay for myelin that requires additional verification with more specific stains such as GSTπ, a marker of the mature oligodendrocyte population. Before cuprizone treatment (0 week), Nlrp3−/− mice and WT controls showed no difference in mature oligodendrocyte populations at the corpus callosum as shown in Figure 4a. After 3, 3.5, and 4 weeks of cuprizone treatment, the reduction of mature oligodendrocytes was attenuated in Nlrp3−/− mice relative to C57BL/6 controls. A quantitation of the composite data is shown in Figure 4b (p = 0.004 at 3 weeks, p = 0.02 at 3.5 weeks, and p = 0.002 at 4 weeks). After 5 weeks of cuprizone treatment, Nlrp3−/− and WT control mice showed no difference in demyelination and mature oligodendrocyte depletion (Figs. 3b, 4a,b). Together, these data indicate that NLRP3 delays demyelination but does not obviate this process, suggesting the existence of other compensatory mechanisms. This is consistent with studies of other inflammatory genes in the cuprizone model (Arnett et al., 2001, 2002; Mason et al., 2001; Plant et al., 2007). A TUNEL assay was used to assess differences in cell death after 3 weeks of cuprizone-induced demyelination. Nlrp3−/− mice showed relatively less apoptotic cells compared with C57BL/6 controls at the midline corpus callosum (Fig. 4c, inset). These apoptotic cells were identified as mature oligodendrocytes by colocalization with the oligodendrocyte marker GSTπ (Fig. 4c). Double staining for apoptotic TUNEL+ cells with the microglial marker RCA showed that microglia were not undergoing apoptosis (Fig. 4d) as described previously (Arnett et al., 2001). As an additional analysis of oligodendrocytes and myelination, a CNPase stain was performed and showed similar results as the LFB and GSTπ data (Fig. 4e).
Figure 4.
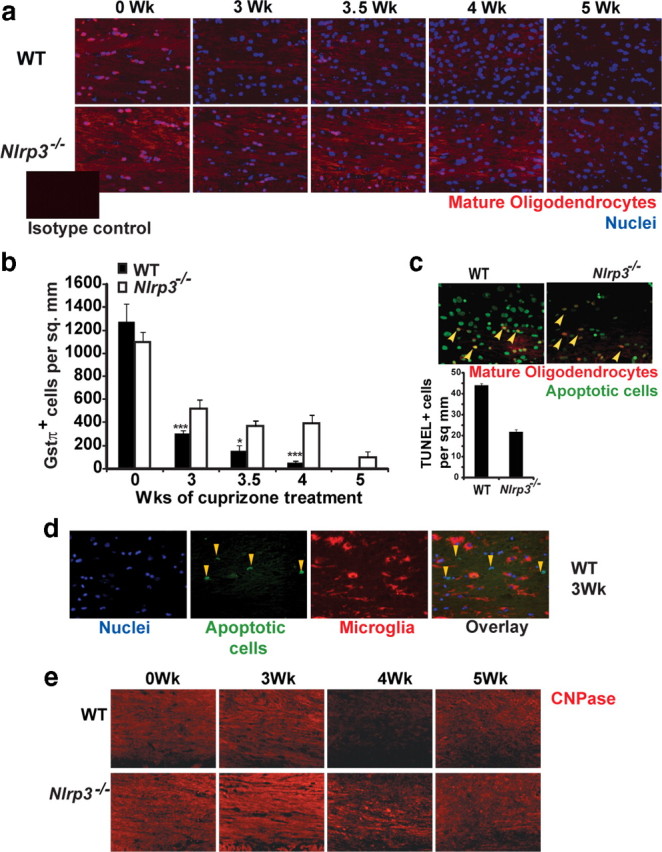
NLRP3 exacerbates mature oligodendrocyte depletion in the corpus callosum during cuprizone-induced demyelination. a, Mature myelinating oligodendrocytes in the corpus callosum were increased during cuprizone treatment of WT and Nlrp3−/− mice as identified with GSTπ staining (red). DAPI was used to label nuclei (blue). The inset shows isotype control. b, Nlrp3−/− mice (white bar) show a significant delay in the loss of mature oligodendrocytes compared with WT controls (black bars) (p = 0.004 at 3 weeks, p = 0.02 at 3.5 weeks, and p = 0.002 at 4 weeks). Mature oligodendrocyte depletion was quantitated by counting GSTπ+ cells that colocalized with a DAPI-positive nucleus. Each image was scored twice in a blind manner. Average counts with SE are depicted here. c, During demyelination, oligodendrocytes undergo apoptosis and are depleted from the corpus callosum. Mature oligodendrocytes undergoing apoptosis were detected by TUNEL (indicated by yellow arrows). The inset shows quantitation of TUNEL+ cells. Nlrp3−/− mice showed relatively less TUNEL+ cells compared with WT control mice. d, TUNEL+ cells are not microglia. Double staining for microglia by RCA and apoptotic cells (yellow arrows) by TUNEL showed no colocalization. e, CNPase staining of paraffin-embedded 5 μm brain sections from Nlrp3−/− and WT control mice during demyelination shows delayed loss of myelin in the corpus callosum of Nlrp3−/− mice. Representative images for 9–14 mice per time point are shown. In b and d, *p < 0.05, ***p < 0.005. Error bars indicate SEM.
Demyelination, mature oligodendrocyte death, astrogliosis, and microglial infiltration during cuprizone-induced demyelination are independent of IL-1β
The above results indicate the importance of NLRP3 in pathology associated with the cuprizone model; hence we examined the role of an inflammasome end product (i.e., IL-1β) in this model. A previous study of IL-1β−/− mice showed delayed remyelination but no difference in demyelination in the cuprizone model, although this latter issue was only peripherally addressed in that report (Mason et al., 2001). To elaborate on these results, we performed a more extensive analysis of cuprizone-induced demyelination in IL-1β−/− mice. IL-1β−/− mice showed no difference in demyelination (Fig. 5a) (p = 0.07 at 3 weeks, p = 0.55 at 4 weeks), loss of GSTπ+ mature oligodendrocyte (Fig. 5b) (p = 0.29 at 3 weeks, p = 0.16 at 4 weeks), accumulation of microglia (Fig. 5c) (p = 0.56 at 3 weeks, p = 0.15 at 4 weeks) or astrogliosis (Fig. 5d) (p = 0.80 at 3 weeks, p = 0.26 at 4 weeks) during demyelination. This indicates that all of the measured neuropathology observed in the cuprizone model is IL-1β independent.
Figure 5.

IL-1β does not appear to be required in demyelination, astrogliosis, microglial infiltration, and mature oligodendrocyte depletion. a, IL-1β−/− mice exhibit no difference in demyelination. Each circle represents the averaged observed LFB score from three readers for one mouse. Demyelination was quantitated by LFB-PAS staining. IL-1β−/− mice (open circles) show no difference in demyelination compared with WT controls (filled circles) (p = 0.07 at 3 weeks, p = 0.55 at 4 weeks). b, IL-1β−/− mice (white bars) exhibit no difference in mature oligodendrocyte number compared with age-matched WT controls (black bars) (p = 0.29 at 3 weeks, p = 0.16 at 4 weeks). Mature oligodendrocytes were measured by the GSTπ+ stain at the corpus callosum. c, IL-1β−/− mice (white bars) exhibit no difference in microglial infiltration compared with age-matched WT controls (black bars) (p = 0.56 at 3 weeks, p = 0.15 at 4 weeks). Microglia were measured by RCA+ staining at the corpus callosum after 3 and 4 weeks of cuprizone treatment. d, IL-1β−/− mice (white bars) exhibit no difference in astrogliosis when compared with age-matched WT controls (black bars) (p = 0.80 at 3 weeks, p = 0.26 at 4 weeks). Astrocytes were measured by the GFAP+ stain at the corpus callosum after 3 and 4 weeks of cuprizone treatment. GFAP was used to detect astrocyte accumulation in the corpus callosum. RCA+ or GFAP+ cells with an observable DAPI-stained nucleus were counted blindly twice. Cell counts for b–d are averages of between 6 and 10 mice per time point. Error bars indicate SEM.
Cuprizone-induced microglial accumulation, astrogliosis, and demyelination are partially dependent on caspase-1
NLRP3 is required for the processing of caspase-1, which in turn cleaves pro-IL-1β and IL-18 to their mature active forms (Sutterwala et al., 2006b). To establish whether NLRP3-dependent CNS inflammation and demyelination are caspase-1 dependent, we studied casp1−/− mice. Casp1−/− mice showed a statistically significant difference when compared with WT controls, in the extent of demyelination as measured by LFB (Fig. 6a) (p = 0.02 at 3 weeks, p = 0.07 at 4 weeks) and in the number of mature oligodendrocyte as detected by GSTπ immunostaining (p = 0.001 at 3 weeks, p = 0.45 at 4 weeks, and p = 0.39 at 5 weeks) after cuprizone treatment (Fig. 6b). Casp1−/− mice also showed delayed microglial infiltration (p = 0.008 at 3 weeks, p = 0.05 at 4 weeks, and p = 0.19 at 5 weeks) and astrogliosis (p = 0.02 at 3 weeks, p = 0.01 at 4 weeks, and p = 0.37 at 5 weeks) when compared with WT controls (Fig. 6c,d, respectively). These results suggest that the phenotype of casp1−/− mice is similar to mice deficient in the Nlrp3 gene.
Figure 6.

Roles of caspase-1 in demyelination, mature oligodendrocyte depletion, microglial infiltration, and astrogliosis. a, Casp1−/− (open circles) and age-matched WT mice (filled circles) exhibit a difference in the extent of demyelination (p = 0.02 at 3 weeks, p = 0.07 at 4 weeks). b, Casp1−/− mice (white bars) and age-matched WT controls (black bars) exhibit a difference in mature oligodendrocyte number (p = 0.001 at 3 weeks, p = 0.45 at 4 weeks, and p = 0.39 at 5 weeks). c, Casp1−/− mice (white bars) exhibit reduced microglial infiltration at the corpus callosum when compared with age-matched WT controls (black bars) (p = 0.008 at 3 weeks, p = 0.05 at 4 weeks, and p = 0.19 at 5 weeks). d, Casp1−/− mice (white bars) exhibit reduced astrogliosis at the corpus callosum when compared with age-matched WT controls (black bars) (p = 0.02 at 3 weeks, p = 0.01 at 4 weeks, and p = 0.37 at 5 weeks). Demyelination was quantitated by LFB-PAS staining, mature oligodendrocyte by GSTπ, microglia by RCA, and astrocytes by GFAP staining as described in Figure 3. *p < 0.05, **p < 0.01, ***p < 0.005. Error bars indicate SEM. Cell counts for b–d are averages of between 6 and 12 mice per time point.
Demyelination, mature oligodendrocyte death, astrogliosis, and microglial infiltration during cuprizone-induced demyelination are IL-18 dependent
IL-18 is one of the several caspase-1 cleavage substrates and since demyelination is IL-1β independent, we assessed its role as a candidate effector. To identify an effector molecule for caspase-1-induced demyelination in the cuprizone model, we used IL-18-deficient (IL-18−/−) mice. Microglial infiltration, astrogliosis, mature oligodendrocyte (ODG) depletion, and demyelination were studied as described previously. IL-18−/− and WT controls showed delayed demyelination (Fig. 7a) (p = 0.04 at 3 weeks) and mature oligodendrocyte depletion after 3 weeks of cuprizone treatment (Fig. 7b) (p = 0.00023 at 3 weeks). IL-18−/− mice showed significantly reduced microglial infiltration (Fig. 7c) (p = 0.019 at 3 weeks) and astrogliosis (Fig. 7d) (p = 0.004 at 3 weeks). These results indicate that IL-18 exacerbates demyelination and cellular infiltration in the cuprizone model and can partially attribute to the Nlrp3−/− phenotype.
Figure 7.

IL-18 exacerbates demyelination, mature oligodendrocyte depletion, microglial infiltration, and astrogliosis. a–d, Compared with WT controls (filled circles or bars), IL-18−/− mice (open circles or bars) showed reduced demyelination as determined by LFB (p = 0.04 at 3 weeks) (a), delayed mature oligodendrocyte depletion as determined by GSTπ+ cells (p = 0.00023 at 3 weeks) (b), reduced microglial infiltration (RCA+ cells) (p = 0.019 at 3 weeks) (c), and reduced astrogliosis (GFAP+ cells) at the corpus callosum (p = 0.004 at 3 weeks) (d). *p < 0.05, ***p < 0.005. Error bars indicate SEM. Cell counts for b–d are the averages of six to eight mice per time point.
Remyelination in the cuprizone model is IL-18 dependent
Regulation of oligodendrogenesis by progenitors is a potential therapeutic intervention for the functional loss after demyelination in MS. Previous studies in the cuprizone model have shown that remyelination is dramatically reduced in IL-1β−/− mice (Mason et al., 2001); thus, we investigated whether NLRP3 via its regulation of IL-1β processing may regulate oligodendrogenesis and remyelination. To assess the role of NLRP3 in remyelination, Nlrp3−/− mice along with age-matched C57BL/6 control (WT) mice were treated with cuprizone for 6 weeks and then returned to regular chow to allow for remyelination. Surprisingly, at 8, 10, and 12 weeks of this regimen when remyelination was allowed to occur for 2, 4, and 6 weeks, there was no difference in mature ODG repopulation (Fig. 8b) (p = 0.99 at 8 weeks, p = 0.74 at 10 weeks, and p = 0.56 at 12 weeks) and myelin staining (Fig. 8a) (p = 0.99 at 8 weeks, p = 0.36 at 10 weeks, and p = 0.89 at 12 weeks) of the corpus callosum in Nlrp3−/− mice relative to C57BL/6 controls. This suggests that remyelination does not appear to be influenced by the absence of Nlrp3. However, a more in-depth investigation showed that IL-18−/− mice exhibited a faster remyelination as measured by LFB-PAS staining (Fig. 8c) (p = 0.00015 at 8 weeks, p = 0.014 at 10 weeks) and CNPase staining (Fig. 8d) for myelin. These results reveal a previously unknown detrimental role for IL-18 in remyelination that requires more in-depth analysis that is beyond this report. We hypothesize that this harmful role of IL-18 during remyelination might negate the beneficial role of IL-1β described in the literature. This provides a feasible explanation for why Nlrp3−/− mice that lack both cytokines do not show a difference from wild-type controls.
Figure 8.

Overall remyelination in the cuprizone model is unaffected by NLRP3 but inhibited by IL-18. a, Nlrp3−/− (open circles) and WT mice (filled circles) show no difference in remyelination during the course of cuprizone treatment (p = 0.99 at 8 weeks, p = 0.36 at 10 weeks, p = 0.89 at 12 weeks). b, Nlrp3−/− (white bars) and WT (black bars) mice show no difference in mature oligodendrocyte repopulation during remyelination (p = 0.99 at 8 weeks, p = 0.74 at 10 weeks, p = 0.56 at 12 weeks). c, IL-18−/− (open circles) and WT mice (filled circles) show enhanced remyelination during the course of cuprizone treatment (p = 0.00015 at 8 weeks, p = 0.014 at 10 weeks). d, IL-18−/− mice show enhanced remyelination by CNPase staining compared with WT mice. **p < 0.01, ***p < 0.005. Error bars indicate SEM.
Discussion
The NLR genes, particularly those associated with the inflammasome function, have secured much interest as sensors of PAMPs (pathogen-associated molecular patterns) as well as DAMPs (damage-associated molecular patterns); however, their roles in CNS inflammatory disorders have not been extensively studied (Mariathasan et al., 2006; Martinon et al., 2006; Willingham et al., 2007). In this report, we provide evidence that NLRP3 has a role in regulating neuroinflammation and demyelination in a mouse model of cuprizone-induced demyelination. However, the outcome is much more complex than anticipated. We found that one of the end products of the inflammasome, namely IL-1β, has no role in any of the pathologic events that were investigated. Another inflammasome end product, IL-18, was found to exacerbate demyelination and inflammation. Conventional inflammasome components including NLRP3, caspase-1, and IL-18 exacerbated inflammatory readouts such as microglial accumulation and astrogliosis, whereas NLRP3, caspase-1, and IL-18 enhanced demyelination and accelerated the loss of mature oligodendrocytes. These data reveal a novel and important role for NLRP3 that has not been appreciated previously.
NLRP3 is known for its classic role in the formation of a multiprotein complex with ASC and procaspase-1 that is critical for caspase-1 cleavage and maturation, which in turn is important for the processing of pro-IL-1β/IL-18 to mature IL-1β/IL-18. In the CNS, IL-1β is released primarily by microglia and macrophages (Mason et al., 2001). IL-1β promotes leukocyte infiltration by inducing expression of many cytokines, chemokines, and adhesion molecules (Shaftel et al., 2007). The release of IL-1β also mobilizes neutrophils and other immune cells to aid in resolving infections and promoting wound healing. Chronic release of IL-1β is detrimental as it can contribute to skin rashes, inflammatory arthritis, and systemic fever (Goldbach-Mansky et al., 2006). For this reason, IL-1β production is tightly regulated at the levels of transcription, translation, and release. Previous studies examining the role of IL-1β in neuroinflammation have not yielded consistent findings. IL-1β levels in the CSF of MS patients correlate with disease susceptibility, severity, and progression (Kantarci et al., 2000; de Jong et al., 2002). IL-1β immunoreactivity has been found in activated microglia and macrophages during EAE in rats (Bauer et al., 1993). Treatment with either sIL-1R (soluble IL-1 receptor) or IL-1Ra (IL-1 receptor antagonist) reduces clinical signs of EAE in rats (Jacobs et al., 1991; Badovinac et al., 1998). IL-1β is cytotoxic to mature oligodendrocytes both in vivo and in vitro, while stimulating proliferation of both microglia and astrocytes (Takahashi et al., 2003). However, IL-1β−/− mice also display delayed remyelination, indicating a reparative role of IL-1β (Mason et al., 2001). Our study of IL-1β−/− mice in the cuprizone model showed no differences in demyelination, microglial infiltration, astrogliosis, or mature oligodendrocyte death after 0, 3, and 4 weeks of cuprizone treatment. This indicates that the demyelination observed in the cuprizone model is IL-1β independent even though IL-1β levels increase concurrent with inflammatory cell infiltration during demyelination (Mason et al., 2001).
Since IL-1β−/− and WT control mice showed no difference in demyelination, we explored the role of caspase-1 in demyelination. Caspase-1 has been implicated in both human and mouse neuroinflammation. Caspase-1 levels are significantly increased in peripheral blood mononuclear cells from MS patients (Huang et al., 2004). Moreover, caspase-1 is known to contribute to the pathology of EAE (Furlan et al., 1999; Ahmed et al., 2002). Our studies with casp1−/− mice showed delayed microglial infiltration, astrogliosis, a reduction in mature oligodendrocyte depletion, and a decrease in demyelination. This suggests a caspase-1/NLRP3-dependent but IL-1β-independent mechanism that leads to increased demyelination and the loss of mature oligodendrocytes. Several studies have shown that caspase-1 has numerous targets besides IL-1β and IL-18, with upward of 70 substrates (Shao et al., 2007; Keller et al., 2008). It is likely that the activation of these other substrates is responsible for the role of NLRP3 and caspase-1 in demyelination.
An alternate cytokine that might explain our data is IL-18. IL-18 is an 18 kDa member of the IL-1 family of cytokines. IL-18 is produced by several immune and nonimmune cells including monocytes, splenocytes, keratinocytes, microglia, macrophages, and astrocytes (Conti et al., 1999; Prinz and Hanisch, 1999; Sugama et al., 2002). In the CNS, IL-18 induces microglial production of proinflammatory cytokines such as IL-1β and TNFα and MMPs (matrix metalloproteinases). Extravasation of PMNs (polymorphonuclear leukocytes) and monocytes/macrophages is amplified by IL-18-dependent upregulation of ICAM-1 (intercellular adhesion molecule-1) on endothelial cells. IL-18 levels are elevated in demyelinating cerebral lesions of MS patients (Balashov et al., 1999; Losy and Niezgoda, 2001; Nicoletti et al., 2001). Moreover, expression of IL-18 and its receptor on oligodendrocytes is greater in brain tissue from patients with active MS than in patients with silent MS or from neuropathologically normal subjects (Cannella and Raine, 2004). In EAE, a murine model of MS in which the induction of MBP (myelin basic protein)-specific CD4+ T-cells secreting cytokines, particularly IFN-γ and TNF-α, results in limb paralysis, elevated IL-18 mRNA is seen in the CNS of EAE rats at onset and during the disease (Jander and Stoll, 1998; Wildbaum et al., 1998). However, EAE studies with mice deficient in IL-18 (IL-18−/−) have not always been consistent. Whereas Shi et al. (2000) reported that IL-18−/− mice are resistant to EAE, Gutcher et al. (2006) reported that IL-18−/− mice are susceptible to EAE but IL-18 receptor α-deficient mice (IL-18Rα−/−) are resistant to EAE indicating the role of a ligand other than IL-18 acting via IL-18Rα to cause EAE. In our hands, IL-18−/− mice replicated the data obtained with Nlrp3−/− mice at the 3 week time point of demyelination. IL-18-deficient mice showed delayed demyelination, indicating an exacerbatory role for IL-18 in demyelination. In addition, IL-18-deficient mice show enhanced remyelination, suggesting a role for IL-18 in inhibiting remyelination.
These results show that the neuroinflammatory component of the disease model is mediated by an NLRP3-, caspase-1-, and IL-18-dependent pathway. However, remyelination is NLRP3 independent and IL-18 dependent. Thus, the positive effects of IL-1β and the negative effects of IL-18 on remyelination might negate each other in Nlrp3−/− mice. Such a possibility is not without precedence. For example, another study has found that IL-18 negated the febrile effect associated with IL-1β (Gatti et al., 2002). In summary, the role of IL-18 in exacerbating neuroinflammation along with its inhibitory role in remyelination indicate that inhibition of IL-18 may prove to be a valuable therapeutic approach for demyelinating diseases such as MS. This finding has broad implications for inflammation in other acute and chronic neuroinflammatory diseases, including stroke and Alzheimer's disease.
Footnotes
This work was supported by the National Multiple Sclerosis Society Grants CA-1053-A-8 and RG-1785E. We thank Janice Weaver at the Lineberger Histopathology Core for assistance with histology, and Victoria Madden at the Microscopy Services Laboratory for assistance with microscopy.
References
- Ahmed Z, Doward AI, Pryce G, Taylor DL, Pocock JM, Leonard JP, Baker D, Cuzner ML. A role for caspase-1 and -3 in the pathology of experimental allergic encephalomyelitis: inflammation versus degeneration. Am J Pathol. 2002;161:1577–1586. doi: 10.1016/S0002-9440(10)64436-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen IC, Scull MA, Moore CB, Holl EK, McElvania-TeKippe E, Taxman DJ, Guthrie EH, Pickles RJ, Ting JP. The NLRP3 inflammasome mediates in vivo innate immunity to influenza A virus through recognition of viral RNA. Immunity. 2009;30:556–565. doi: 10.1016/j.immuni.2009.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnett HA, Mason J, Marino M, Suzuki K, Matsushima GK, Ting JP. TNF alpha promotes proliferation of oligodendrocyte progenitors and remyelination. Nat Neurosci. 2001;4:1116–1122. doi: 10.1038/nn738. [DOI] [PubMed] [Google Scholar]
- Arnett HA, Hellendall RP, Matsushima GK, Suzuki K, Laubach VE, Sherman P, Ting JP. The protective role of nitric oxide in a neurotoxicant-induced demyelinating model. J Immunol. 2002;168:427–433. doi: 10.4049/jimmunol.168.1.427. [DOI] [PubMed] [Google Scholar]
- Badovinac V, Mostarica-Stojković M, Dinarello CA, Stosić-Grujicić S. Interleukin-1 receptor antagonist suppresses experimental autoimmune encephalomyelitis (EAE) in rats by influencing the activation and proliferation of encephalitogenic cells. J Neuroimmunol. 1998;85:87–95. doi: 10.1016/s0165-5728(98)00020-4. [DOI] [PubMed] [Google Scholar]
- Balashov KE, Rottman JB, Weiner HL, Hancock WW. CCR5+ and CXCR3+ T cells are increased in multiple sclerosis and their ligands MIP-1α and IP-10 are expressed in demyelinating brain lesions. Proc Natl Acad Sci U S A. 1999;96:6873–6878. doi: 10.1073/pnas.96.12.6873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauer J, Berkenbosch F, Van Dam AM, Dijkstra CD. Demonstration of interleukin-1 beta in Lewis rat brain during experimental allergic encephalomyelitis by immunocytochemistry at the light and ultrastructural level. J Neuroimmunol. 1993;48:13–21. doi: 10.1016/0165-5728(93)90053-2. [DOI] [PubMed] [Google Scholar]
- Black RA, Kronheim SR, Cantrell M, Deeley MC, March CJ, Prickett KS, Wignall J, Conlon PJ, Cosman D, Hopp TP. Generation of biologically active interleukin-1 beta by proteolytic cleavage of the inactive precursor. J Biol Chem. 1988;263:9437–9442. [PubMed] [Google Scholar]
- Cannella B, Raine CS. Multiple sclerosis: cytokine receptors on oligodendrocytes predict innate regulation. Ann Neurol. 2004;55:46–57. doi: 10.1002/ana.10764. [DOI] [PubMed] [Google Scholar]
- Coeshott C, Ohnemus C, Pilyavskaya A, Ross S, Wieczorek M, Kroona H, Leimer AH, Cheronis J. Converting enzyme-independent release of tumor necrosis factor alpha and IL-1β from a stimulated human monocytic cell line in the presence of activated neutrophils or purified proteinase 3. Proc Natl Acad Sci U S A. 1999;96:6261–6266. doi: 10.1073/pnas.96.11.6261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Compeyrot-Lacassagne S, Tran TA, Guillaume-Czitrom S, Marie I, Koné-Paut I. Brain multiple sclerosis-like lesions in a patient with Muckle-Wells syndrome. Rheumatology (Oxford) 2009;48:1618–1619. doi: 10.1093/rheumatology/kep321. [DOI] [PubMed] [Google Scholar]
- Conti B, Park LC, Calingasan NY, Kim Y, Kim H, Bae Y, Gibson GE, Joh TH. Cultures of astrocytes and microglia express interleukin 18. Brain Res Mol Brain Res. 1999;67:46–52. doi: 10.1016/s0169-328x(99)00034-0. [DOI] [PubMed] [Google Scholar]
- de Jong BA, Huizinga TW, Bollen EL, Uitdehaag BM, Bosma GP, van Buchem MA, Remarque EJ, Burgmans AC, Kalkers NF, Polman CH, Westendorp RG. Production of IL-1beta and IL-1Ra as risk factors for susceptibility and progression of relapse-onset multiple sclerosis. J Neuroimmunol. 2002;126:172–179. doi: 10.1016/s0165-5728(02)00056-5. [DOI] [PubMed] [Google Scholar]
- Dong Y, Benveniste EN. Immune function of astrocytes. Glia. 2001;36:180–190. doi: 10.1002/glia.1107. [DOI] [PubMed] [Google Scholar]
- Dostert C, Pétrilli V, Van Bruggen R, Steele C, Mossman BT, Tschopp J. Innate immune activation through Nalp3 inflammasome sensing of asbestos and silica. Science. 2008;320:674–677. doi: 10.1126/science.1156995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franco-Pons N, Torrente M, Colomina MT, Vilella E. Behavioral deficits in the cuprizone-induced murine model of demyelination/remyelination. Toxicol Lett. 2007;169:205–213. doi: 10.1016/j.toxlet.2007.01.010. [DOI] [PubMed] [Google Scholar]
- Furlan R, Martino G, Galbiati F, Poliani PL, Smiroldo S, Bergami A, Desina G, Comi G, Flavell R, Su MS, Adorini L. Caspase-1 regulates the inflammatory process leading to autoimmune demyelination. J Immunol. 1999;163:2403–2409. [PubMed] [Google Scholar]
- Gatti S, Beck J, Fantuzzi G, Bartfai T, Dinarello CA. Effect of interleukin-18 on mouse core body temperature. Am J Physiol Regul Integr Comp Physiol. 2002;282:R702–R709. doi: 10.1152/ajpregu.00393.2001. [DOI] [PubMed] [Google Scholar]
- Goldbach-Mansky R, Dailey NJ, Canna SW, Gelabert A, Jones J, Rubin BI, Kim HJ, Brewer C, Zalewski C, Wiggs E, Hill S, Turner ML, Karp BI, Aksentijevich I, Pucino F, Penzak SR, Haverkamp MH, Stein L, Adams BS, Moore TL, et al. Neonatal-onset multisystem inflammatory disease responsive to interleukin-1beta inhibition. N Engl J Med. 2006;355:581–592. doi: 10.1056/NEJMoa055137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutcher I, Urich E, Wolter K, Prinz M, Becher B. Interleukin 18-independent engagement of interleukin 18 receptor-alpha is required for autoimmune inflammation. Nat Immunol. 2006;7:946–953. doi: 10.1038/ni1377. [DOI] [PubMed] [Google Scholar]
- Halle A, Hornung V, Petzold GC, Stewart CR, Monks BG, Reinheckel T, Fitzgerald KA, Latz E, Moore KJ, Golenbock DT. The NALP3 inflammasome is involved in the innate immune response to amyloid-beta. Nat Immunol. 2008;9:857–865. doi: 10.1038/ni.1636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanisch UK, Kettenmann H. Microglia: active sensor and versatile effector cells in the normal and pathologic brain. Nat Neurosci. 2007;10:1387–1394. doi: 10.1038/nn1997. [DOI] [PubMed] [Google Scholar]
- Harton JA, Linhoff MW, Zhang J, Ting JP. Cutting edge: CATERPILLER: a large family of mammalian genes containing CARD, pyrin, nucleotide-binding, and leucine-rich repeat domains. J Immunol. 2002;169:4088–4093. doi: 10.4049/jimmunol.169.8.4088. [DOI] [PubMed] [Google Scholar]
- Hazuda DJ, Strickler J, Kueppers F, Simon PL, Young PR. Processing of precursor interleukin 1 beta and inflammatory disease. J Biol Chem. 1990;265:6318–6322. [PubMed] [Google Scholar]
- Hornung V, Bauernfeind F, Halle A, Samstad EO, Kono H, Rock KL, Fitzgerald KA, Latz E. Silica crystals and aluminum salts activate the NALP3 inflammasome through phagosomal destabilization. Nat Immunol. 2008;9:847–856. doi: 10.1038/ni.1631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang WX, Huang P, Hillert J. Increased expression of caspase-1 and interleukin-18 in peripheral blood mononuclear cells in patients with multiple sclerosis. Mult Scler. 2004;10:482–487. doi: 10.1191/1352458504ms1071oa. [DOI] [PubMed] [Google Scholar]
- Jacobs CA, Baker PE, Roux ER, Picha KS, Toivola B, Waugh S, Kennedy MK. Experimental autoimmune encephalomyelitis is exacerbated by IL-1α and suppressed by soluble IL-1 receptor. J Immunol. 1991;146:2983–2989. [PubMed] [Google Scholar]
- Jander S, Stoll G. Differential induction of interleukin-12, interleukin-18, and interleukin-1beta converting enzyme mRNA in experimental autoimmune encephalomyelitis of the Lewis rat. J Neuroimmunol. 1998;91:93–99. doi: 10.1016/s0165-5728(98)00162-3. [DOI] [PubMed] [Google Scholar]
- Jha S, Ting JP. Inflammasome-associated nucleotide-binding domain, leucine-rich repeat proteins and inflammatory diseases. J Immunol. 2009;183:7623–7629. doi: 10.4049/jimmunol.0902425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanneganti TD, Ozören N, Body-Malapel M, Amer A, Park JH, Franchi L, Whitfield J, Barchet W, Colonna M, Vandenabeele P, Bertin J, Coyle A, Grant EP, Akira S, Núñez G. Bacterial RNA and small antiviral compounds activate caspase-1 through cryopyrin/Nalp3. Nature. 2006a;440:233–236. doi: 10.1038/nature04517. [DOI] [PubMed] [Google Scholar]
- Kanneganti TD, Body-Malapel M, Amer A, Park JH, Whitfield J, Franchi L, Taraporewala ZF, Miller D, Patton JT, Inohara N, Núñez G. Critical role for Cryopyrin/Nalp3 in activation of caspase-1 in response to viral infection and double-stranded RNA. J Biol Chem. 2006b;281:36560–36568. doi: 10.1074/jbc.M607594200. [DOI] [PubMed] [Google Scholar]
- Kantarci OH, Atkinson EJ, Hebrink DD, McMurray CT, Weinshenker BG. Association of two variants in IL-1beta and IL-1 receptor antagonist genes with multiple sclerosis. J Neuroimmunol. 2000;106:220–227. doi: 10.1016/s0165-5728(00)00202-2. [DOI] [PubMed] [Google Scholar]
- Keller M, Rüegg A, Werner S, Beer HD. Active caspase-1 is a regulator of unconventional protein secretion. Cell. 2008;132:818–831. doi: 10.1016/j.cell.2007.12.040. [DOI] [PubMed] [Google Scholar]
- Lequerre T, Vittecoq O, Saugier-Veber P, Goldenberg A, Patoz P, Frebourg T, Le Loet X. A cryopyrin-associated periodic syndrome with joint destruction. Rheumatology (Oxford) 2007;46:709–714. doi: 10.1093/rheumatology/kel399. [DOI] [PubMed] [Google Scholar]
- Li H, Willingham SB, Ting JP, Re F. Cutting edge: inflammasome activation by alum and alum's adjuvant effect are mediated by NLRP3. J Immunol. 2008;181:17–21. doi: 10.4049/jimmunol.181.1.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L, Belkadi A, Darnall L, Hu T, Drescher C, Cotleur AC, Padovani-Claudio D, He T, Choi K, Lane TE, Miller RH, Ransohoff RM. CXCR2-positive neutrophils are essential for cuprizone-induced demyelination: relevance to multiple sclerosis. Nat Neurosci. 2010;13:319–326. doi: 10.1038/nn.2491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Losy J, Niezgoda A. IL-18 in patients with multiple sclerosis. Acta Neurol Scand. 2001;104:171–173. doi: 10.1034/j.1600-0404.2001.00356.x. [DOI] [PubMed] [Google Scholar]
- Lucchinetti C, Brück W, Parisi J, Scheithauer B, Rodriguez M, Lassmann H. Heterogeneity of multiple sclerosis lesions: implications for the pathogenesis of demyelination. Ann Neurol. 2000;47:707–717. doi: 10.1002/1531-8249(200006)47:6<707::aid-ana3>3.0.co;2-q. [DOI] [PubMed] [Google Scholar]
- Mariathasan S, Weiss DS, Newton K, McBride J, O'Rourke K, Roose-Girma M, Lee WP, Weinrauch Y, Monack DM, Dixit VM. Cryopyrin activates the inflammasome in response to toxins and ATP. Nature. 2006;440:228–232. doi: 10.1038/nature04515. [DOI] [PubMed] [Google Scholar]
- Martinon F, Pétrilli V, Mayor A, Tardivel A, Tschopp J. Gout-associated uric acid crystals activate the NALP3 inflammasome. Nature. 2006;440:237–241. doi: 10.1038/nature04516. [DOI] [PubMed] [Google Scholar]
- Mason JL, Suzuki K, Chaplin DD, Matsushima GK. Interleukin-1β promotes repair of the CNS. J Neurosci. 2001;21:7046–7052. doi: 10.1523/JNEUROSCI.21-18-07046.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsushima GK, Morell P. The neurotoxicant, cuprizone, as a model to study demyelination and remyelination in the central nervous system. Brain Pathol. 2001;11:107–116. doi: 10.1111/j.1750-3639.2001.tb00385.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsushima K, Taguchi M, Kovacs EJ, Young HA, Oppenheim JJ. Intracellular localization of human monocyte associated interleukin 1 (IL 1) activity and release of biologically active IL 1 from monocytes by trypsin and plasmin. J Immunol. 1986;136:2883–2891. [PubMed] [Google Scholar]
- Mayer-Barber KD, Barber DL, Shenderov K, White SD, Wilson MS, Cheever A, Kugler D, Hieny S, Caspar P, Núñez G, Schlueter D, Flavell RA, Sutterwala FS, Sher A. Caspase-1 independent IL-1β production is critical for host resistance to Mycobacterium tuberculosis and does not require TLR signaling in vivo. J Immunol. 2010;184:3326–3330. doi: 10.4049/jimmunol.0904189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miwa K, Asano M, Horai R, Iwakura Y, Nagata S, Suda T. Caspase 1-independent IL-1beta release and inflammation induced by the apoptosis inducer Fas ligand. Nat Med. 1998;4:1287–1292. doi: 10.1038/3276. [DOI] [PubMed] [Google Scholar]
- Napoli I, Neumann H. Microglial clearance function in health and disease. Neuroscience. 2009;158:1030–1038. doi: 10.1016/j.neuroscience.2008.06.046. [DOI] [PubMed] [Google Scholar]
- Neven B, Callebaut I, Prieur AM, Feldmann J, Bodemer C, Lepore L, Derfalvi B, Benjaponpitak S, Vesely R, Sauvain MJ, Oertle S, Allen R, Morgan G, Borkhardt A, Hill C, Gardner-Medwin J, Fischer A, de Saint Basile G. Molecular basis of the spectral expression of CIAS1 mutations associated with phagocytic cell-mediated autoinflammatory disorders CINCA/NOMID, MWS, and FCU. Blood. 2004;103:2809–2815. doi: 10.1182/blood-2003-07-2531. [DOI] [PubMed] [Google Scholar]
- Nicoletti F, Di Marco R, Mangano K, Patti F, Reggio E, Nicoletti A, Bendtzen K, Reggio A. Increased serum levels of interleukin-18 in patients with multiple sclerosis. Neurology. 2001;57:342–344. doi: 10.1212/wnl.57.2.342. [DOI] [PubMed] [Google Scholar]
- Omoto Y, Tokime K, Yamanaka K, Habe K, Morioka T, Kurokawa I, Tsutsui H, Yamanishi K, Nakanishi K, Mizutani H. Human mast cell chymase cleaves pro-IL-18 and generates a novel and biologically active IL-18 fragment. J Immunol. 2006;177:8315–8319. doi: 10.4049/jimmunol.177.12.8315. [DOI] [PubMed] [Google Scholar]
- Pittock SJ, Lucchinetti CF. The pathology of MS: new insights and potential clinical applications. Neurologist. 2007;13:45–56. doi: 10.1097/01.nrl.0000253065.31662.37. [DOI] [PubMed] [Google Scholar]
- Plant SR, Iocca HA, Wang Y, Thrash JC, O'Connor BP, Arnett HA, Fu YX, Carson MJ, Ting JP. Lymphotoxin β receptor (Lt βR): dual roles in demyelination and remyelination and successful therapeutic intervention using Lt βR-Ig protein. J Neurosci. 2007;27:7429–7437. doi: 10.1523/JNEUROSCI.1307-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prinz M, Hanisch UK. Murine microglial cells produce and respond to interleukin-18. J Neurochem. 1999;72:2215–2218. doi: 10.1046/j.1471-4159.1999.0722215.x. [DOI] [PubMed] [Google Scholar]
- Rigante D, Ansuini V, Caldarelli M, Bertoni B, La Torraca I, Stabile A. Hydrocephalus in CINCA syndrome treated with anakinra. Childs Nerv Syst. 2006;22:334–337. doi: 10.1007/s00381-006-1280-3. [DOI] [PubMed] [Google Scholar]
- Schönbeck U, Mach F, Libby P. Generation of biologically active IL-1β by matrix metalloproteinases: a novel caspase-1-independent pathway of IL-1β processing. J Immunol. 1998;161:3340–3346. [PubMed] [Google Scholar]
- Shaftel SS, Carlson TJ, Olschowka JA, Kyrkanides S, Matousek SB, O'Banion MK. Chronic interleukin-1β expression in mouse brain leads to leukocyte infiltration and neutrophil-independent blood brain barrier permeability without overt neurodegeneration. J Neurosci. 2007;27:9301–9309. doi: 10.1523/JNEUROSCI.1418-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shao W, Yeretssian G, Doiron K, Hussain SN, Saleh M. The caspase-1 digestome identifies the glycolysis pathway as a target during infection and septic shock. J Biol Chem. 2007;282:36321–36329. doi: 10.1074/jbc.M708182200. [DOI] [PubMed] [Google Scholar]
- Shi FD, Takeda K, Akira S, Sarvetnick N, Ljunggren HG. IL-18 directs autoreactive T cells and promotes autodestruction in the central nervous system via induction of IFN-γ by NK cells. J Immunol. 2000;165:3099–3104. doi: 10.4049/jimmunol.165.6.3099. [DOI] [PubMed] [Google Scholar]
- Shornick LP, De Togni P, Mariathasan S, Goellner J, Strauss-Schoenberger J, Karr RW, Ferguson TA, Chaplin DD. Mice deficient in IL-1beta manifest impaired contact hypersensitivity to trinitrochlorobenzone. J Exp Med. 1996;183:1427–1436. doi: 10.1084/jem.183.4.1427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sospedra M, Martin R. Immunology of multiple sclerosis. Annu Rev Immunol. 2005;23:683–747. doi: 10.1146/annurev.immunol.23.021704.115707. [DOI] [PubMed] [Google Scholar]
- Sugama S, Cho BP, Baker H, Joh TH, Lucero J, Conti B. Neurons of the superior nucleus of the medial habenula and ependymal cells express IL-18 in rat CNS. Brain Res. 2002;958:1–9. doi: 10.1016/s0006-8993(02)03363-2. [DOI] [PubMed] [Google Scholar]
- Sutterwala FS, Ogura Y, Szczepanik M, Lara-Tejero M, Lichtenberger GS, Grant EP, Bertin J, Coyle AJ, Galán JE, Askenase PW, Flavell RA. Critical role for NALP3/CIAS1/Cryopyrin in innate and adaptive immunity through its regulation of caspase-1. Immunity. 2006a;24:317–327. doi: 10.1016/j.immuni.2006.02.004. [DOI] [PubMed] [Google Scholar]
- Sutterwala FS, Ogura Y, Zamboni DS, Roy CR, Flavell RA. NALP3: a key player in caspase-1 activation. J Endotoxin Res. 2006b;12:251–256. doi: 10.1177/09680519060120040701. [DOI] [PubMed] [Google Scholar]
- Takahashi JL, Giuliani F, Power C, Imai Y, Yong VW. Interleukin-1beta promotes oligodendrocyte death through glutamate excitotoxicity. Ann Neurol. 2003;53:588–595. doi: 10.1002/ana.10519. [DOI] [PubMed] [Google Scholar]
- Thomas PG, Dash P, Aldridge JR, Jr, Ellebedy AH, Reynolds C, Funk AJ, Martin WJ, Lamkanfi M, Webby RJ, Boyd KL, Doherty PC, Kanneganti TD. The intracellular sensor NLRP3 mediates key innate and healing responses to influenza A virus via the regulation of caspase-1. Immunity. 2009;30:566–575. doi: 10.1016/j.immuni.2009.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ting JP, Davis BK. CATERPILLER: a novel gene family important in immunity, cell death, and diseases. Annu Rev Immunol. 2005;23:387–414. doi: 10.1146/annurev.immunol.23.021704.115616. [DOI] [PubMed] [Google Scholar]
- Ting JP, Kastner DL, Hoffman HM. CATERPILLERs, pyrin and hereditary immunological disorders. Nat Rev Immunol. 2006;6:183–195. doi: 10.1038/nri1788. [DOI] [PubMed] [Google Scholar]
- Wildbaum G, Youssef S, Grabie N, Karin N. Neutralizing antibodies to IFN-γ-inducing factor prevent experimental autoimmune encephalomyelitis. J Immunol. 1998;161:6368–6374. [PubMed] [Google Scholar]
- Willingham SB, Bergstralh DT, O'Connor W, Morrison AC, Taxman DJ, Duncan JA, Barnoy S, Venkatesan MM, Flavell RA, Deshmukh M, Hoffman HM, Ting JP. Microbial pathogen-induced necrotic cell death mediated by the inflammasome components CIAS1/cryopyrin/NLRP3 and ASC. Cell Host Microbe. 2007;2:147–159. doi: 10.1016/j.chom.2007.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamasaki K, Muto J, Taylor KR, Cogen AL, Audish D, Bertin J, Grant EP, Coyle AJ, Misaghi A, Hoffman HM, Gallo RL. NLRP3/cryopyrin is necessary for interleukin-1β (IL-1β) release in response to hyaluronan, an endogenous trigger of inflammation in response to injury. J Biol Chem. 2009;284:12762–12771. doi: 10.1074/jbc.M806084200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Podojil JR, Chang J, Luo X, Miller SD. TGF-β-induced myelin peptide-specific regulatory T cells mediate antigen-specific suppression of induction of experimental autoimmune encephalomyelitis. J Immunol. 2010;184:6629–6636. doi: 10.4049/jimmunol.0904044. [DOI] [PMC free article] [PubMed] [Google Scholar]