

Abstract
Opitz G/BBB syndrome (OS) is a genetic disorder characterized by midline developmental defects. Male patients with the X-linked form of OS, caused by loss-of-function mutations in the MID1 gene, show high variability of the clinical signs. MID1 encodes a ubiquitin ligase that controls phosphatase 2A, but its role in the pathogenesis of the disease is still unclear. Here, we report a mouse line carrying a nonfunctional ortholog of the human MID1 gene, Mid1. Mid1-null mice show the brain anatomical defect observed in patients (i.e., hypoplasia of the anterior portion of the medial cerebellum, the vermis). We found that the presence of this defect correlates with motor coordination and procedural and nonassociative learning impairments. The defect is limited to the most anterior lobes of the vermis, the region of the developing cerebellum adjacent to the dorsal midbrain. Analyses at midgestation reveal that lack of Mid1 causes the shortening of the posterior dorsal midbrain, the rostralization of the midbrain/cerebellum boundary, and the downregulation of a key player in the development of this region, Fgf17. Thus, lack of Mid1 causes a misspecification of the midbrain/cerebellar boundary that results in an abnormal development of the most anterior cerebellar lobes. This animal model provides a tool for additional in vivo studies of the physiological and pathological role of the Mid1 gene and a system to investigate the development and function of anterior cerebellar domains.
Introduction
Opitz G/BBB syndrome (OS) is a congenital anomaly disorder characterized by developmental defects of midline structures (Opitz, 1987). OS patients present with hypertelorism, hypospadias, and laryngo-tracheo-esophageal (LTE) anomalies. They may also have cleft lip and palate, heart defects, and anal anomalies (Robin et al., 1996). In the X-linked OS form, male patients manifest the clinical signs with variable expressivity, whereas female carriers only show hypertelorism (Robin et al., 1995). A high percentage of X-linked OS patients present mental retardation and developmental delay, and approximately one-third of the patients subjected to magnetic resonance imaging (MRI) show anatomical brain abnormalities that mainly consist of hypoplasia of the anterior cerebellar vermis (Fontanella et al., 2008).
The cerebellum is the primary center for motor coordination and is essential for cognitive processing (Petrosini et al., 1998). In mammals, it starts developing early during embryogenesis from the dorsal most anterior portion of the hindbrain as two bilateral wings that eventually fuse along the midline to form the central vermis flanked by two lateral hemispheres. The neurons in the cerebellum originate from two germinal compartments that give rise, among others, to the two main cerebellar populations, granule cells that colonize an external layer [external granule layer (EGL)] and Purkinje cells (PCs) positioned just underneath. Around birth, the granule neurons start their inward migration through the PC to find their final location in the internal granule layer (IGL). In the meanwhile, the smooth surface of the cerebellum starts the process of foliation, a series of remodeling steps that confer the final species-specific lobular architecture (Wang and Zoghbi, 2001; Chizhikov and Millen, 2003; Sillitoe and Joyner, 2007).
The gene responsible for the X-linked form of OS is MID1 (Quaderi et al., 1997). MID1 encodes a ubiquitin ligase that belongs to the TRIM (tripartite motif) family (Meroni and Diez-Roux, 2005). MID1 is associated with the microtubules (MTs) and regulates the level of MT-associated phosphatase 2A (PP2A) by binding α4 (Cainarca et al., 1999; Schweiger et al., 1999; Liu et al., 2001; Trockenbacher et al., 2001). Recent data indicate that MID1 is involved in TNFα (tumor necrosis factor-α)-induced p38MAPK (p38 mitogen-activated protein kinase)-mediated apoptosis through the interaction with the α4/PP2A complex (Prickett and Brautigan, 2007). Mid1 expression during development was investigated in mouse, chicken, and human. Mid1 is almost ubiquitously expressed at early embryonic stages becoming more restricted to the tissues involved in the disease during organogenesis (Dal Zotto et al., 1998; Richman et al., 2002; Pinson et al., 2004). In early chick development, Mid1 is also expressed on the right side ectoderm of the Hensen's node (Granata and Quaderi, 2003). Despite the biochemical and expression data, the role of MID1 in development and in the pathogenesis of OS is still obscure. The mutations found in OS patients indicate loss of function as the mechanism underlying the disease, and we generated a Mid1-null mouse line to study its role in vivo. We report that, like OS patients, these mice display hypoplasia of the anterior cerebellum. We present data showing the involvement of Mid1 in the development of the vermal anterobasal lobe through the correct specification of the midbrain/cerebellum boundary.
Materials and Methods
Animals
All mouse were housed and handled in accordance with guidelines of the Institutional Animal Care and Use Committee of Cardarelli Hospital (Naples, Italy). Mid1 −/+ heterozygous females were generated by mating the male chimeras obtained upon injection of a Mid1 −/Y ES clone (supplemental Fig. S1, available at www.jneurosci.org as supplemental material) with C57BL/6 females. Mid1 −/Y mice and their Mid1 +/Y control littermates were generated by mating heterozygous female with wild-type males. Mice were bred for 10 generations with C57BL/6 mice to generate and analyze the mutant mice in a pure genetic background.
Tissue preparation
Mice were anesthetized and perfused transcardially with 4% paraformaldehyde. Adult and postnatal brains were harvested and processed using standard procedures either for embedding in OCT and cryosectioning (20 μm) or agarose embedding and vibratome sectioning (50 μm). For timed-pregnant mice, the day of the vaginal plug was designated embryonic day 0.5 (E0.5). Embryos were collected, fixed in 4% paraformaldehyde, embedded in OCT, and cryosectioned (20 μm).
RNA in situ hybridization
Digoxigenin-labeled antisense riboprobes were generated for Pax2, Otx2, En1 (V. Broccoli, DiBIT, Milan, Italy), and Fgf17 (D. Ornitz, Washington University School of Medicine, St. Louis, MO), and in situ hybridization was performed on cryosections as previously described (Surace et al., 2000).
Immunohistochemistry and histology
Immunohistochemistry was performed using standard protocols with rabbit anti-Calbindin (Swant; 1:20,000 on postnatal vibratome sections; 1:2000 on embryo cryosections) and mouse anti-NeuN (Millipore Bioscience Research Reagents; 1:1000) antibodies, and then detected using anti-rabbit or anti-mouse biotinylated secondary antibodies (1:200) and the ABC kit (Vector Laboratories). Nissl staining was performed using standard procedures.
Behavioral analyses
Subjects.
Adult male mice (40 Mid1 +/Y and 45 Mid1 −/Y) were housed in standard breeding cages at a constant temperature (22 ± 2°C) and on a 12 h light/dark cycle with food and water available ad libitum. Animals of the two groups were matched for age, and when possible they were littermates. Each experimental group included at least eight subjects.
Motor coordination tests.
For the walking wire task, mice were placed on 45° inclined 1 cm2 grid steel wire with the head directed toward the floor and observed for 120 s. Animals underwent two consecutive trials with an intertrial interval (ITI) of ∼30 min. The number of missteps was counted and the average of the two measurements was considered for statistical analyses. For the hanging wire test, mice were placed on a wire cage lid that was gently waved, so the mouse gripped the wire, and then turned upside down. Latency to fall off the grip was recorded with a cutoff time of 60 s.
Learning and memory tests.
To test motor learning, we used the rotarod task (Ugo Basile). Mice were placed on the accelerating rod reaching a final speed of 40 rpm in 5 min. They were tested for four trials a day for 5 consecutive days with 20 min ITI. The latency to fall off the rod was recorded (800 s cutoff time). Mean day latency was considered for statistical analyses. Long-term habituation of acoustic startle was measured using the SR-Lab System (San Diego Instruments). All animals were acclimatized to the Plexiglas cylinder for 5 min, during which time the background noise level (70 dB) was continually present. Individual mice were then exposed to six repeated presentations of 100 ms, 130 dB sound. The interstimulus interval was of 100 ms. The startle response was recorded for 65 ms measuring every 1 ms from the stimulus onset. The maximum startle amplitude recorded during the 65 ms sampling window was used as the dependent variable. The same procedure was repeated for 3 consecutive days. For statistical analysis, we calculated the percentage of startle response from day 1. The cross maze task consists of a transparent Plexiglas cross-shaped maze, with four sliding doors at the end of each arm, enclosed in a cue-controlled environment. Food-deprived animals (80–85% of their normal weight) were subjected to one session of habituation (10 min of free exploration to retrieve 10 food pellets scattered all over the maze). A mouse always had a choice between two arms to enter, with the third arm blocked off. Two start arms were used for both procedures. Animals received 15 trials a day, with an ITI of 40 s. In the egocentric procedure, no visual distal cues were visible and the position of the goal was changed according to the animal position. In the allocentric procedure, visual distal cues were visible and the position of the goal was fixed to the room cues but not to the animal position (Rinaldi et al., 2008). Entering the correct arm was scored as correct response and rewarded while entering the wrong arm was scored as incorrect. The percentage of correct responses/15 across days was considered for statistical analyses.
Statistical analysis
One-way ANOVA with a between-group factor genetic (two levels: wild type, Mid1 −/Y) was used to analyze the data of the walking wire and the hanging wire tasks. Two-way ANOVA for repeated measures with a between-group factor genetic (two levels: wild type, Mid1 −/Y) and testing days or training trials as repeated measures for horizontal activity (six levels: T1–T6), latency to fall off the rod (five levels: D1–D5), percentage of startle amplitude (three levels: D1–D3), and percentage of correct response in the cross maze tasks (five levels: D1–D5). Duncan's post hoc test was used when appropriate. Statistical significance was set at p < 0.05. Inferior colliculus (IC) length was determined using photographs of sagittal sections in ImageJ and evaluated by Student's t test analysis.
Results
Mid1 −/Y mice show cerebellar defects
To study Mid1 function in vivo, we generated a mouse line carrying a nonfunctional Mid1 gene by disruption of the first ATG-containing exon (supplemental Fig. S1A,B, available at www.jneurosci.org as supplemental material). Like in human, Mid1 is transcribed from the X chromosome, although in mouse it spans the pseudoautosomal boundary (Palmer et al., 1997). Northern blot analysis on total RNA from E11.5 embryos and reverse transcription-PCR on RNA from several adult tissues show the complete absence of the Mid1 transcript in male mutant mice (supplemental Fig. S1C,D, available at www.jneurosci.org as supplemental material). Consistently, immunoblot analysis with a specific antibody shows the complete absence of the Mid1 protein in Mid1 −/Y lysates (supplemental Fig. S1E, available at www.jneurosci.org as supplemental material). Mid1 −/Y males and Mid1 −/− females were indistinguishable from their wild-type and heterozygous littermates. We focused on male Mid1 −/Y as null mutants using Mid1 +/Y littermates as wild-type controls.
Direct observation of whole-mount adult brains suggested a somewhat abnormal midbrain/cerebellum junction region in Mid1 −/Y mice (data not shown). Histological analyses confirmed the presence of a malformed anterior cerebellum. Nissl staining of sagittal sections through the vermis showed hypoplasia and abnormalities of lobes I, II, and III. The defect is present, although with a certain degree of variability, in all the Mid1 −/Y mice analyzed (n = 9) and in none of their wild-type littermates (n = 7). In all null mice, lobe I, which is poorly pronounced in the C57/B6 mouse strain, is totally missing and lobe II is not completely formed. In many cases, the third lobe is also abnormal in shape (Fig. 1 A,B). Sagittal sections through the lateral hemispheres showed normal foliation (Fig. 1 C,D).
Figure 1.

Abnormal cerebellum in Mid1 −/Y mice. Sagittal sections through the cerebellar vermis (A, B) and hemisphere (C, D) of adult wild-type and null mice stained with Nissl. Anterior is to the left. The numbers of the vermal and names of lateral lobes are indicated. The arrow in B indicates the anterobasal defect in Mid1 −/Y mice.
To investigate the layer organization of the Mid1 −/Y adult cerebellar cortex, we analyzed the two main neuronal populations of the cerebellum, PCs and granule neurons. The Mid1 −/Y general cerebellar cortex layer architecture is normal, with PCs disposed in monolayer and their dendritic arbors directed toward the external surface to define the molecular layer (ML) and with the IGL positioned just inside the PC monolayer (Fig. 2). Although abnormal in shape, even the anterior vermal lobes maintain the correct layer organization and thickness (Fig. 2 A–D). The shape of the anterior lobes varies among mutant animals, and, as shown in the coronal section through the anterior part of the cerebellum, the defect is not uniform along the mediolateral axis where the presence of intermingled lobes and layers is observed (Fig. 2 E–H). The coronal sections confirm the normal structure of lobe IV and of the hemispheres (Fig. 2 E–H).
Figure 2.
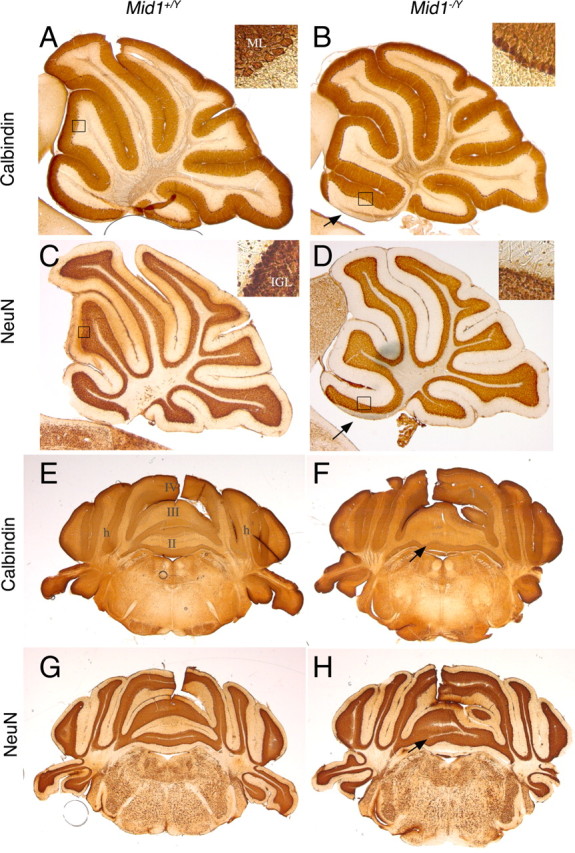
Normal ML and IGL organization in Mid1 −/Y adult mice. Sagittal sections through the vermis (A–D) and coronal sections at the level of the anterior cerebellum (E–H) are shown. The insets show magnification of the indicated areas. In null mice, PCs and their dendrites form a normal ML (detected by anti-Calbindin) and granule cells form a normal IGL (detected by anti-NeuN). The numbers of the vermal lobes are indicated in E. h, Hemisphere. The arrows indicate the defect in Mid1 −/Y brains.
Cerebellar defects in Mid1 knock-out originate prenatally
The development of the cerebellum is a long process that in the mouse begins as early as E9.5 and is completed by the second postnatal week (Chizhikov and Millen, 2003). Foliation begins around birth and reaches the definitive strain-specific architecture by postnatal day 15 (P15) (Sillitoe and Joyner, 2007). We analyzed sagittal sections of cerebella at P0, P2, and P7 (Fig. 3). At P0 and P2, the correct formation of the four principal fissures was observed in both wild-type and null mice (Fig. 3 A–D). The principal fissures determine the anchor points for the formation of the five cardinal lobes. The anterobasal cardinal lobe, which gives rise to the definitive lobes I, II, and III, is limited caudally by the preculminate fissure and rostrally by the isthmus; it appeared abnormal already in P0 and P2 Mid1 −/Y cerebella as shown by the upturning EGL in the most anterior part and by a more profound fissure delimiting future lobes II and III (Fig. 3 A–D). Consistent with the normal layer organization observed in adult mice, the granule cells at P0 form the EGL and progressively start their inward migration in both genotypes at P2 and P7 (Fig. 3 C–F).
Figure 3.
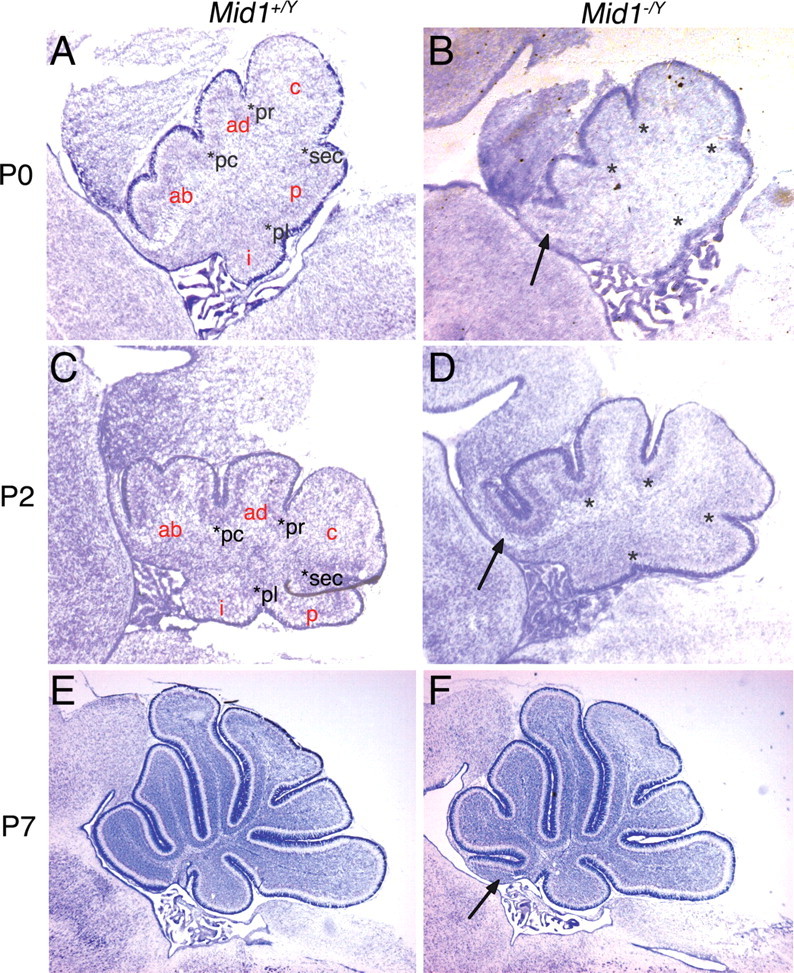
The anterobasal lobe defect in Mid1 −/Y mice is detected at birth. Sagittal sections through the vermis at birth (A, B), P2 (C, D), and P7 (E, F) stained with Nissl. The granule cells at P0 and P2 are still superficial and form the EGL, whereas at P7 they are starting to migrate inwards to eventually form the IGL. The four principal fissures (pc, preculminate; pr, primary; sec, secondary; pl, posterolateral) are normally formed in P0 and P2 Mid1 −/Y mice. The cardinal lobes generated by the principal fissures are correctly formed with the exception of the anterobasal lobe, rostral to the preculminate fissure, that shows the defect (ab, anterobasal; ad, anterodorsal; c, central; p, posterior; i, inferior). The development of the other cardinal lobes into the definitive foliation proceeds normally from P0 through P7. The asterisks indicate the principal fissures. The arrows indicate the defect.
Thus, Mid1 −/Y mice present developmental defects resulting in the postnatal abnormal formation of the anterobasal cerebellar vermis.
Mid1 −/Y vermal defect is caused by an incorrect definition of the tectum/cerebellum boundary
The defect is present as early as the formation of the primitive fissures around birth; we therefore analyzed the developing vermis before that stage. At E17.5, the surface of the cerebellum is smooth and the mutant is almost indistinguishable from the wild type. Disordered PCs are still disposed in multilayers underneath the EGL in both genotypes (Fig. 4). However, in the most anterior Mid1 −/Y cerebellum, some PCs are present in ectopic position within the isthmic region heading toward the tectum (Fig. 4 A,B). Analysis of the expression of Engrailed 1 (En1), a gene expressed in the tectum and the cerebellum, showed that PC migration in ectopic position coincides with a premature halt of the anterior EGL. The strong En1 expression in the IC also highlights a more rostral IC/cerebellar boundary and a slightly enlarged isthmic region in Mid1 −/Y embryos (Fig. 4 C–D′). These results suggest that the abnormal foliation presented by Mid1 −/Y mice might not be the result of a fissure formation anomaly, rather, the consequence of a poor definition of the boundary between the posterior tectum (i.e., the inferior colliculus) and the most anterior part of the medial cerebellum.
Figure 4.
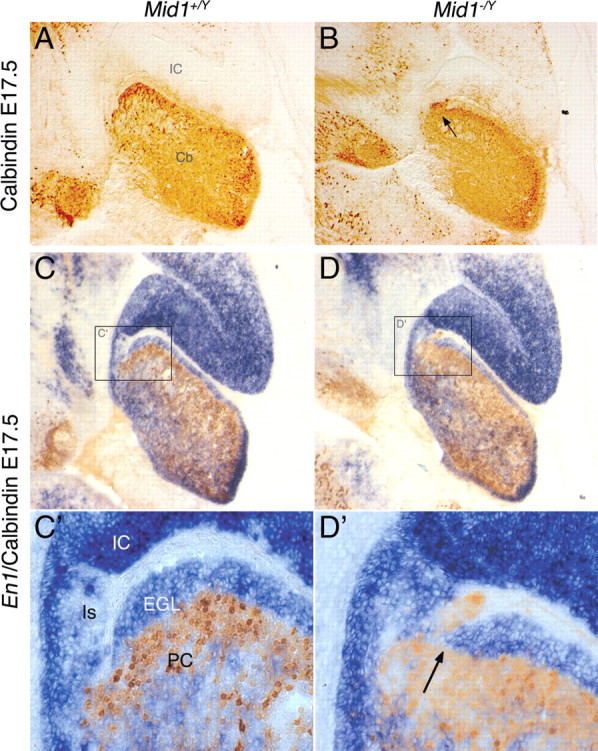
Inaccurate definition of the dorsal midbrain/cerebellum boundary in Mid1 −/Y mice. Immunohistochemistry with anti-Calbindin (A, B) and RNA in situ hybridization (En1) followed by anti-Calbindin (C–D′) on medial sagittal sections of E17.5 brains. C′, D′, Magnification of the delimited areas. Cb, Cerebellum; Is, isthmus. The arrows indicate ectopic PCs in Mid1 −/Y embryos.
At midgestation, Mid1 expression is observed in the proliferative compartments of both dorsal midbrain and in the cerebellum anlage (supplemental Fig. S2, available at www.jneurosci.org as supplemental material). By E13.5–E14.5, strong and specific expression in the tectum with a sharp caudal limit at the dorsal midbrain/hindbrain boundary (MHB) is observed (supplemental Fig. S2, available at www.jneurosci.org as supplemental material) (Dal Zotto et al., 1998). We analyzed markers expressed at the dorsal MHB at these stages (Wang and Zoghbi, 2001). We found no overt differences in the expression of several markers (e.g., Gbx2, Math1, En1, En2, Gli3) (data not shown), although they all suggested a mispositioning of the boundary. This observation was further confirmed by the expression patterns of Pax2 (at this stage limited to the cerebellum) and Otx2 (expressed in the dorsal midbrain) that show a more rostral position of the midbrain/cerebellum boundary in Mid1 −/Y embryos resulting in a shorter lower portion of the IC (Fig. 5 A,B). We measured the length of the IC [i.e., the distance between the posterior Otx2 expression limit and the IC physical bending on the ventricular side (Fig. 5 B, asterisk)] in Mid1 −/Y (n = 4) and Mid1 +/Y (n = 4) E14.5 embryos at three mediolateral positions of the presumptive vermal region. As shown in the graph, the null mice have a shorter IC with the difference more pronounced in the lateral sections of the vermis (Fig. 5 C). Although there is a trend toward a shorter IC, we do not observe statistically significant differences at E13.5. We tested programmed cell death occurrence in the IC at E12.5–E14.5. Few cells are undergoing apoptosis in this region at these stages, and they are not increased in null embryos (data not shown). Similarly, we did not find overt differences in the proliferation of the IC using the anti-Ki67 antibody (data not shown). Whatever the mechanism, the net effect of a shorter IC is a more anterior position of the dorsal midbrain/hindbrain boundary.
Figure 5.

Reduced IC length and rostralization of midbrain/cerebellar boundary in Mid1 −/Y embryos. Overlay of images of adjacent E14.5 sagittal sections hybridized with Pax2 (green) and Otx2 (purple) (A, B). The arrow indicates the mispositioning of the isthmus (B). C, E14.5 IC length (in millimeters) in wild-type (n = 4) and null (n = 4) mice. The length is the ventricular side distance between the two physical bending, indicated with the arrow and the asterisk in B, at three different positions of the vermal region: M, medial; M/L, mediolateral (∼120 μm from the midline); L, lateral (∼240 μm from the midline). t test, **p = 0.0042. Error bars indicate SEM.
Fgf17 downregulation in Mid1 −/Y midbrain/hindbrain boundary
Fibroblast growth factors (Fgfs) are key molecules in the establishment and development of the dorsal midbrain/cerebellum border. Early in development, Fgf8 is the main player in the isthmic organizer, the signaling center that coordinates the patterning of the cerebellum at the mesencephalon/rhombomere 1 (mes/r1) boundary (Liu et al., 1999; Martinez et al., 1999). At midgestation, Fgf8 is involved in the proper anteroposterior development of the mes/r1 region (Partanen, 2007). We analyzed Fgf8 at E10.5 and E12.5 and did not find alteration of its expression in null mice (data not shown). Another member of the Fgf8 subfamily, Fgf17, is expressed at the mes/r1 boundary but, differently from Fgf8, its expression extends temporally and from E13.5 to E14.5 Fgf17 is the only Fgf expressed in this region (Xu et al., 2000). At E12.5, we did not observe changes in the expression of Fgf17 (data not shown). However, at E13.5, we began to observe a decrease in the Fgf17 expression domain in Mid1 −/Y mice in both medial and mediolateral sagittal sections (Fig. 6 A–D). The downregulation of Fgf17 is striking at E14.5. At this stage in the mutant embryos, Fgf17 expression domain is maintained at the border between the IC and the cerebellar plate but heavily restricted along the anteroposterior axis (Fig. 6 E–H). Analysis of coronal sections at the same stage confirmed the strong reduction of Fgf17 signal (Fig. 6 I–L).
Figure 6.
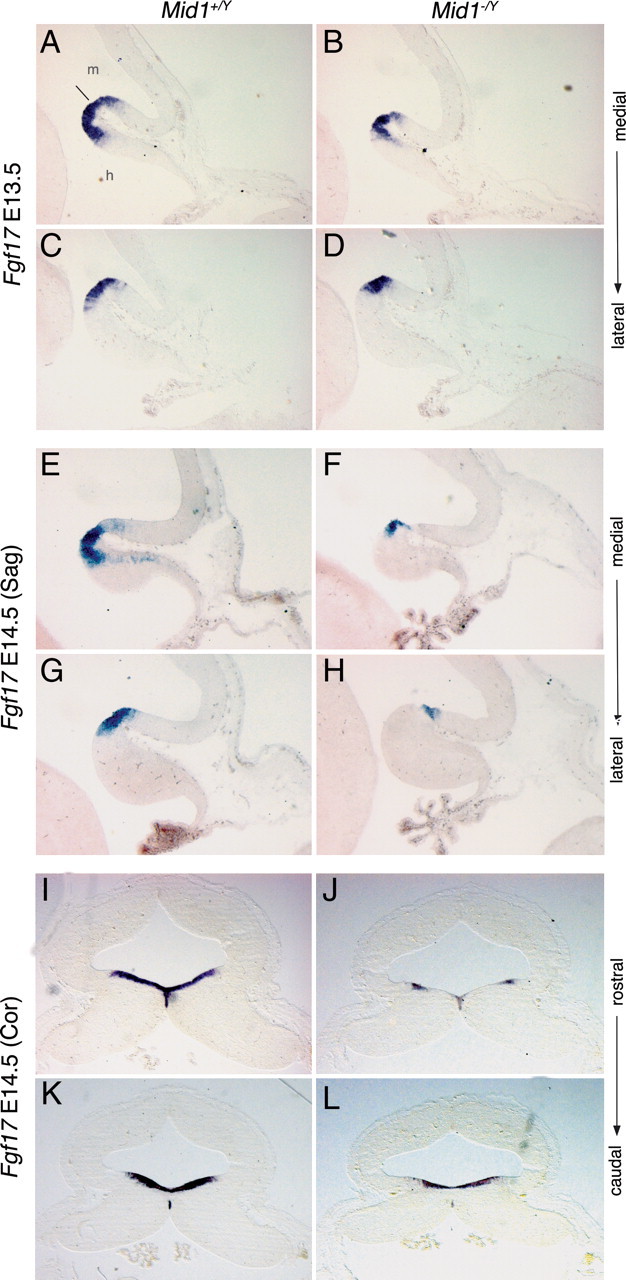
Fgf17 is downregulated in Mid1 −/Y embryos. RNA in situ hybridization of Fgf17 on E13.5 sagittal section [medial (A, B); mediolateral (C, D)], on E14.5 sagittal sections [medial (E, F); mediolateral (G, H)], and on E14.5 coronal sections [rostral (I, J); caudal (K, L)]. Downregulation of Fgf17 in Mid1 −/Y embryos starts at E13.5 and is more evident at E14.5 especially in the next to the midline region along the mediolateral axis and in the cerebellum along the anteroposterior axis. The line indicates the isthmus. m, Midbrain; h, hindbrain.
Together, these data indicate that Mid1 is important for the determination of the boundary between the tectum and the cerebellum and for the expression of a key regulator of this region, Fgf17.
Mid1 −/Y mice have motor coordination and motor learning impairment
We investigated whether the abnormalities observed in Mid1 −/Y mice correlates with cerebellum-dependent behavioral functions. To test motor coordination, we used the walking and the hanging wire tasks (Joyal et al., 1996). We found that Mid1 −/Y mice made more false steps walking on the inclined wire (wild type, 5.5 ± 0.97, vs Mid1 −/Y, 9.077 ± 0.92; F ( 1 , 26 ) = 7.181; p = 0.01) and were also impaired in hanging on the wire compared with wild-type animals (latency to fall off the grid for wild type, 61 ± 4.9 s; for Mid1 −/Y, 42 ± 5.7 s; F ( 1 , 25 ) = 6.07; p = 0.02). These effects were not attributable to muscular or primary sensorial deficits, since Mid1 −/Y mice were not impaired in any of these tasks (supplemental Fig. S3, available at www.jneurosci.org as supplemental material). We tested motor learning in the classic rotarod task. Although both groups improved their performance across days, Mid1 −/Y mice showed delayed motor learning (Fig. 7 A). The observed impairment is not simply attributable to general hypoactivity since Mid1 −/Y mice were hyperactive when compared with their wild-type littermates (supplemental Fig. S3, available at www.jneurosci.org as supplemental material).
Figure 7.

Mid1 −/Y mice show motor coordination and learning impairments. A, Mid1 −/Y mice demonstrate a worse performance in the rotarod task, showing a deficit in motor learning acquisition (F ( 1 , 48 ) = 5.042; p = 0.02). B, Control mice (p = 0.008), but not Mid1 −/Y mice, reduce the percentage of startle amplitude across days in the long-term habituation of acoustic startle task; the null mice do not habituate to the stimulus; the startle response between the two groups significantly differ on the third day (p = 0.02). C, D, Mid1 −/Y mice recorded fewer correct responses in the egocentric spatial version of the cross maze task (C) on the fourth (p = 0.01) and on the fifth day (p = 0.06), but not in the allocentric one (D). *p < 0.05, Mid1 −/Y versus wild type. Error bars indicate SEM.
We also tested long-term habituation of the acoustic startle response, a form of nonassociative learning selectively mediated by the cerebellar vermis and occurring when repeated exposure to an acoustic stimulus across days induces habituation of the startle response (Leaton and Supple, 1986). Whereas in the wild-type group the percentage of mean startle amplitude significantly decreased from day 1 to day 3, no significant changes across days were observed in Mid1 −/Y mice (Fig. 7 B). Indeed, the percentage of startle response between the two groups significantly differed on the third day. Thus, Mid1 −/Y vermal defect correlates with impairment in long-term habituation of acoustic startle.
The cerebellum has been also involved in other cognitive processes such as spatial learning, in particular in its egocentric form (Molinari et al., 1997a; Petrosini et al., 1998). We tested Mid1 −/Y mice in the egocentric version of the cross maze task: food-deprived animals are challenged to find food in a T-shaped maze in the absence of visual cues (Rinaldi et al., 2008). Mid1 −/Y mice, as their wild-type littermates, increased the percentage of correct responses across days (Fig. 7 C). However, the percentage of correct responses in the two groups differs on the fourth and fifth training day, suggesting a delayed learning curve of Mid1 −/Y animals. As control, we subjected different groups of animals to the allocentric version of the cross maze task, which has identical motor and motivational demand, but it is based on explicit learning and is dependent on the integrity of medial temporal lobe regions (Burguière et al., 2005). Allocentric spatial learning requires the animal to form a cognitive map of the environment and learn the position of the food relatively to visual cues. The performance of Mid1 −/Y mice in this nonprocedural version of the cross maze task was undistinguishable from wild-type animals (Fig. 7 D). Consistently, Mid1 −/Y mice were not impaired in a medial temporal lobe-dependent learning task, the passive avoidance (supplemental Fig. S3, available at www.jneurosci.org as supplemental material). These results clearly prove that learning impairment of Mid1 −/Y mice is not secondary to aspecific motor, sensorial, learning, memory, or motivational impairments and well correlate with a vermal defect.
Together, our results demonstrate that Mid1 is important for the specification of the most anterior medial part of the cerebellum and that, compatibly with a cerebellar defect, its deletion specifically affects motor coordination and nonassociative and procedural learning.
Discussion
Here, we report the generation and characterization of a mouse line null for Mid1, the ortholog of the X-linked Opitz syndrome gene, MID1. Like in human, the murine gene is transcribed from the X chromosome and the hemizygous Mid1 −/Y mice show a developmental phenotype (i.e., hypoplasia of the anterior region of the medial portion of the cerebellum, the vermis).
This mouse model perfectly recapitulates the anatomical CNS defect observed in OS male patients with MID1 mutations (i.e., cerebellar vermis hypoplasia) (Fontanella et al., 2008). Like many other clinical manifestations of OS, brain anatomical defects show variable expressivity and are not present in all patients. Indeed, there are cohorts with mild clinical presentations and in which brain defects were not observed (So et al., 2005). However, we recently reviewed the reports of OS patients with assessed MID1 mutations and found that more than one-third of the patients subjected to MRI examination exhibit brain abnormalities. Moreover, the common brain defect in OS patients with MID1 mutations is hypoplasia of the cerebellar vermis (Cox et al., 2000; De Falco et al., 2003; Pinson et al., 2004; Fontanella et al., 2008). As part of the neurological involvement, OS patients often also present intellectual impairment and psychomotor and speech delays. We found that lack of the murine Mid1 gene results in motor coordination defects as well as motor, nonassociative, and procedural learning impairments that may correlate with the developmental delays of OS patients. The cerebellum, traditionally associated with pure motor coordination functions, in the last decades has been linked more and more to cognitive responses (Molinari et al., 1997b; Petrosini et al., 1998; Tavano et al., 2007). These studies mainly rely on individuals with specific cerebellar lesions either as consequence of tumor surgery in human or experimentally induced in animal models. Consistent with behavioral data obtained on selective lesion of the vermis, Mid1 −/Y mice show hyperactivity, balance and motor coordination defects, motor and procedural learning deficits, and lack of long-term habituation to acoustic startle (Leaton and Supple, 1986; Joyal et al., 1996; Molinari et al., 1997a; Bobée et al., 2000; Callu et al., 2007). In the case of spatial egocentric learning, a major involvement of the hemispheres has been reported previously (Gaytán-Tocavén and Olvera-Cortés, 2004); however, our data suggest that the vermis may also contribute to this task. Together, behavioral deficits in Mid1 −/Y mice qualitatively recapitulate those observed after selective lesions of the cerebellar vermis. Few animal models with specific defects in the anterobasal vermis are reported and much still needs to be unraveled of the function and connectivity of this portion of the cerebellum. This animal model will be useful to further dissect the function of the different domains of the vermis.
As mentioned, in addition to the CNS defect, OS patients show craniofacial, tracheoesophageal, cardiac, and urogenital midline abnormalities that are not present in Mid1 knock-out mice. In fact, the Mid1 −/Y mice have a normal life span, and they are fertile and born at the expected Mendelian ratio, indicating that life-threatening defects such as LTE abnormalities, cleft palate, and major cardiac defects are not present in this mouse model. Moreover, histological analyses during prenatal development confirmed that (1) the LTE region and the palate develop normally; (2) no signs of hypospadias are observed, the urethra being well canalized in the phallic portion in E15.5 embryos; and (3) the major cardiac abnormalities presented by OS patients (ventricular and atrial septal defects) are not observed in E18.5 mice (supplemental Fig. S4, available at www.jneurosci.org as supplemental material). Although we cannot exclude that subtle defects of midline structures might be present in the Mid1 −/Y mice, it is a fact that manifest abnormalities, other than the cerebellar defect, are not present. The discrepancy between the human and the murine phenotype might be explained by the influence of the genetic background or by evolutionary developmental differences between the two species that may translate in different expressivity of the clinical signs. Indeed, Mid1 loss of function results in complete penetrance of developmental defects in both species, whereas the expressivity of the midline defects is different (i.e., all the Mid1 −/Y mice show the cerebellar defect and all OS patients with MID1 mutations show several midline signs, among which the cerebellar defect, with variable expressivity). The presence of a partially redundant gene that in the mouse is able to compensate the absence of Mid1 in non-neural compartments may underlie these differences. A natural candidate for this role is the close homolog of Mid1, Mid2 (Buchner et al., 1999). Mid2 belongs to the same family and interacts with Mid1 (Short et al., 2002). Functional redundancy between Mid1 and Mid2 has been demonstrated in chick. In fact, the effect of the downregulation of a cascade of genes in the Hensen's node in chick embryos exerted by an anti-Mid1/2 morpholino can be rescued by the expression of either of the two genes (Granata et al., 2005). The expression of Mid2 in Mid1 −/Y embryos observed by both RNA in situ hybridization and real-time PCR is not altered (data not shown) but redundancy may act through different mechanisms. Mid1/2 functional cooperation is an interesting issue to address also in the perspective of better understanding the high variability of the clinical manifestations in OS patients even carrying the same MID1 mutation.
We demonstrated that lack of Mid1 causes the abnormal development of the lobes that derive from the anterobasal cardinal lobe, the most anterior part of the developing vermis adjacent to the tectum. Absence of Mid1 causes a shortening of the E14.5 IC lower region with a consequent rostralization of the dorsal isthmus, the region at the boundary between the midbrain and hindbrain. This rostralization is concomitant with reduction in Fgf17 expression. As said, Fgf17 is a member of the subfamily of Fgfs expressed in the mes/rh1 boundary (Fgf8, 17, and 18) and is the only one expressed in this region from E13.5 to E14.5 when the other two have already been switched off (Xu et al., 1999, 2000). Consistent with our data, the complete lack of Fgf17 leads to malformation of the anterior vermis caused by an anticipated rostral differentiation of PCs (Xu et al., 2000). Moreover, in Fgf17-null brains, the inferior colliculus is highly hypoplastic (Xu et al., 2000). The IC phenotype in Mid1 −/Y mice is milder than in the Fgf17 mutant. This is very likely attributable to the fact that we do not have complete loss of Fgf17 expression but a strong reduction limited to a short window of time. On the contrary, the cerebellar phenotype appears more severe in the Mid1 −/Y than in the Fgf17 −/− mice, suggesting that Fgf signaling might not be the only mechanism implicated. However, we observed the presence of ectopic PCs in very rostral cerebellar/isthmic position consistent with the premature differentiation of PCs in the rostromedial part of the cerebellar plate proposed for the Fgf17 −/− mice. Although we still do not know whether the reduction of Fgf17 is caused by a transcriptional mechanism or by the absence of the midbrain/hindbrain boundary cells deputed to express it, the regulation of a member of the Fgf family by Mid1 is not unprecedented. Indeed, data in chick embryos indicated a positive downstream effect of Mid1/2 on the expression of Fgf8 in the ectoderm of the Hensen's node (Granata and Quaderi, 2003).
What is the mechanism through which Mid1 absence causes shortening of the IC, downregulation of Fgf17, and defects in the development of the anterobasal lobe is still to be determined. Major changes in proliferation and apoptosis do not seem to account for the reduction of the IC and rostralization of the isthmus, at least at E12.5–E13.5 stages. The cerebellum, the tectum, and the isthmic region are also affected in other mouse knock-out lines among which are the series of Gli3 mutants (Blaess et al., 2008). Interestingly, Gli3 is expressed, like Mid1, in the dorsal tectum at midgestation and GLI3 is implicated in three human pathological conditions sharing common features with Opitz syndrome (Biesecker, 2006). A fascinating hypothesis is that Mid1 might modulate Gli3 activity in the midbrain/cerebellar region by controlling PP2A levels (Krauss et al., 2008). Through this or other mechanisms, Mid1 may influence the migration, adhesion, or identity of the population of dorsal midbrain/hindbrain boundary cells that are crucial for the maintenance of the proper lineage separation between the two structures (Broccoli et al., 1999; Kala et al., 2008).
This mouse model will be crucial to determine the pathogenetic mechanisms and the physiological function of Mid1 during development, mechanisms that can be studied exploiting the cerebellar defect as a system but that can be relevant also in the occurrence of the other clinical manifestations of OS as well as in other human congenital disorders presenting the same signs.
Footnotes
This work was supported by Pierfranco e Luisa Mariani Foundation, Jérôme Lejeune Foundation, and the Italian Telethon Foundation (G.M.). We thank Edoardo Nusco, Ciro Bonetti, Ivana Peluso, Fabio Russo, Nicola Meola, and Christian Alfano for technical assistance, and Graciana Diez-Roux, Valeria Marigo, and Elena Rugarli for helpful discussion and critical reading of this manuscript.
References
- Biesecker LG. What you can learn from one gene: GLI3. J Med Genet. 2006;43:465–469. doi: 10.1136/jmg.2004.029181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blaess S, Stephen D, Joyner AL. Gli3 coordinates three-dimensional patterning and growth of the tectum and cerebellum by integrating Shh and Fgf8 signaling. Development. 2008;135:2093–2103. doi: 10.1242/dev.015990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bobée S, Mariette E, Tremblay-Leveau H, Caston J. Effects of early midline cerebellar lesion on cognitive and emotional functions in the rat. Behav Brain Res. 2000;112:107–117. doi: 10.1016/s0166-4328(00)00166-2. [DOI] [PubMed] [Google Scholar]
- Broccoli V, Boncinelli E, Wurst W. The caudal limit of Otx2 expression positions the isthmic organizer. Nature. 1999;401:164–168. doi: 10.1038/43670. [DOI] [PubMed] [Google Scholar]
- Buchner G, Montini E, Andolfi G, Quaderi N, Cainarca S, Messali S, Bassi MT, Ballabio A, Meroni G, Franco B. MID2, a homologue of the Opitz syndrome gene MID1: similarities in subcellular localization and differences in expression during development. Hum Mol Genet. 1999;8:1397–1407. doi: 10.1093/hmg/8.8.1397. [DOI] [PubMed] [Google Scholar]
- Burguière E, Arleo A, Hojjati M, Elgersma Y, De Zeeuw CI, Berthoz A, Rondi-Reig L. Spatial navigation impairment in mice lacking cerebellar LTD: a motor adaptation deficit? Nat Neurosci. 2005;8:1292–1294. doi: 10.1038/nn1532. [DOI] [PubMed] [Google Scholar]
- Cainarca S, Messali S, Ballabio A, Meroni G. Functional characterization of the Opitz syndrome gene product (midin): evidence for homodimerization and association with microtubules throughout the cell cycle. Hum Mol Genet. 1999;8:1387–1396. doi: 10.1093/hmg/8.8.1387. [DOI] [PubMed] [Google Scholar]
- Callu D, Puget S, Faure A, Guegan M, El Massioui N. Habit learning dissociation in rats with lesions to the vermis and the interpositus of the cerebellum. Neurobiol Dis. 2007;27:228–237. doi: 10.1016/j.nbd.2007.04.007. [DOI] [PubMed] [Google Scholar]
- Chizhikov V, Millen KJ. Development and malformations of the cerebellum in mice. Mol Genet Metab. 2003;80:54–65. doi: 10.1016/j.ymgme.2003.08.019. [DOI] [PubMed] [Google Scholar]
- Cox TC, Allen LR, Cox LL, Hopwood B, Goodwin B, Haan E, Suthers GK. New mutations in MID1 provide support for loss of function as the cause of X-linked Opitz syndrome. Hum Mol Genet. 2000;9:2553–2562. doi: 10.1093/hmg/9.17.2553. [DOI] [PubMed] [Google Scholar]
- Dal Zotto L, Quaderi NA, Elliott R, Lingerfelter PA, Carrel L, Valsecchi V, Montini E, Yen CH, Chapman V, Kalcheva I, Arrigo G, Zuffardi O, Thomas S, Willard HF, Ballabio A, Disteche CM, Rugarli EI. The mouse Mid1 gene: implications for the pathogenesis of Opitz syndrome and the evolution of the mammalian pseudoautosomal region. Hum Mol Genet. 1998;7:489–499. doi: 10.1093/hmg/7.3.489. [DOI] [PubMed] [Google Scholar]
- De Falco F, Cainarca S, Andolfi G, Ferrentino R, Berti C, Rodríguez Criado G, Rittinger O, Dennis N, Odent S, Rastogi A, Liebelt J, Chitayat D, Winter R, Jawanda H, Ballabio A, Franco B, Meroni G. X-linked Opitz syndrome: novel mutations in the MID1 gene and redefinition of the clinical spectrum. Am J Med Genet A. 2003;120A:222–228. doi: 10.1002/ajmg.a.10265. [DOI] [PubMed] [Google Scholar]
- Fontanella B, Russolillo G, Meroni G. MID1 mutations in patients with X-linked Opitz G/BBB syndrome. Hum Mutat. 2008;29:584–594. doi: 10.1002/humu.20706. [DOI] [PubMed] [Google Scholar]
- Gaytán-Tocavén L, Olvera-Cortés ME. Bilateral lesion of the cerebellar-dentate nucleus impairs egocentric sequential learning but not egocentric navigation in the rat. Neurobiol Learn Mem. 2004;82:120–127. doi: 10.1016/j.nlm.2004.05.006. [DOI] [PubMed] [Google Scholar]
- Granata A, Quaderi NA. The Opitz syndrome gene MID1 is essential for establishing asymmetric gene expression in Hensen's node. Dev Biol. 2003;258:397–405. doi: 10.1016/s0012-1606(03)00131-3. [DOI] [PubMed] [Google Scholar]
- Granata A, Savery D, Hazan J, Cheung BM, Lumsden A, Quaderi NA. Evidence of functional redundancy between MID proteins: implications for the presentation of Opitz syndrome. Dev Biol. 2005;277:417–424. doi: 10.1016/j.ydbio.2004.09.036. [DOI] [PubMed] [Google Scholar]
- Joyal CC, Meyer C, Jacquart G, Mahler P, Caston J, Lalonde R. Effects of midline and lateral cerebellar lesions on motor coordination and spatial orientation. Brain Res. 1996;739:1–11. doi: 10.1016/s0006-8993(96)00333-2. [DOI] [PubMed] [Google Scholar]
- Kala K, Jukkola T, Pata I, Partanen J. Analysis of the midbrain-hindbrain boundary cell fate using a boundary cell-specific Cre-mouse strain. Genesis. 2008;46:29–36. doi: 10.1002/dvg.20344. [DOI] [PubMed] [Google Scholar]
- Krauss S, Foerster J, Schneider R, Schweiger S. Protein phosphatase 2A and rapamycin regulate the nuclear localization and activity of the transcription factor GLI3. Cancer Res. 2008;68:4658–4665. doi: 10.1158/0008-5472.CAN-07-6174. [DOI] [PubMed] [Google Scholar]
- Leaton RN, Supple WF., Jr Cerebellar vermis: essential for long-term habituation of the acoustic startle response. Science. 1986;232:513–515. doi: 10.1126/science.3961494. [DOI] [PubMed] [Google Scholar]
- Liu A, Losos K, Joyner AL. FGF8 can activate Gbx2 and transform regions of the rostral mouse brain into a hindbrain fate. Development. 1999;126:4827–4838. doi: 10.1242/dev.126.21.4827. [DOI] [PubMed] [Google Scholar]
- Liu J, Prickett TD, Elliott E, Meroni G, Brautigan DL. Phosphorylation and microtubule association of the Opitz syndrome protein mid-1 is regulated by protein phosphatase 2A via binding to the regulatory subunit alpha 4. Proc Natl Acad Sci U S A. 2001;98:6650–6655. doi: 10.1073/pnas.111154698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez S, Crossley PH, Cobos I, Rubenstein JL, Martin GR. FGF8 induces formation of an ectopic isthmic organizer and isthmocerebellar development via a repressive effect on Otx2 expression. Development. 1999;126:1189–1200. doi: 10.1242/dev.126.6.1189. [DOI] [PubMed] [Google Scholar]
- Meroni G, Diez-Roux G. TRIM/RBCC, a novel class of “single protein RING finger” E3 ubiquitin ligases. Bioessays. 2005;27:1147–1157. doi: 10.1002/bies.20304. [DOI] [PubMed] [Google Scholar]
- Molinari M, Grammaldo LG, Petrosini L. Cerebellar contribution to spatial event processing: right/left discrimination abilities in rats. Eur J Neurosci. 1997a;9:1986–1992. doi: 10.1111/j.1460-9568.1997.tb00766.x. [DOI] [PubMed] [Google Scholar]
- Molinari M, Leggio MG, Solida A, Ciorra R, Misciagna S, Silveri MC, Petrosini L. Cerebellum and procedural learning: evidence from focal cerebellar lesions. Brain. 1997b;120:1753–1762. doi: 10.1093/brain/120.10.1753. [DOI] [PubMed] [Google Scholar]
- Opitz JM. G syndrome (hypertelorism with esophageal abnormality and hypospadias, or hypospadias-dysphagia, or “Opitz-Frias” or “Opitz-G” syndrome)—perspective in 1987 and bibliography. Am J Med Genet. 1987;28:275–285. doi: 10.1002/ajmg.1320280203. [DOI] [PubMed] [Google Scholar]
- Palmer S, Perry J, Kipling D, Ashworth A. A gene spans the pseudoautosomal boundary in mice. Proc Natl Acad Sci U S A. 1997;94:12030–12035. doi: 10.1073/pnas.94.22.12030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Partanen J. FGF signalling pathways in development of the midbrain and anterior hindbrain. J Neurochem. 2007;101:1185–1193. doi: 10.1111/j.1471-4159.2007.04463.x. [DOI] [PubMed] [Google Scholar]
- Petrosini L, Leggio MG, Molinari M. The cerebellum in the spatial problem solving: a co-star or a guest star? Prog Neurobiol. 1998;56:191–210. doi: 10.1016/s0301-0082(98)00036-7. [DOI] [PubMed] [Google Scholar]
- Pinson L, Augé J, Audollent S, Mattéi G, Etchevers H, Gigarel N, Razavi F, Lacombe D, Odent S, Le Merrer M, Amiel J, Munnich A, Meroni G, Lyonnet S, Vekemans M, Attié-Bitach T. Embryonic expression of the human MID1 gene and its mutations in Opitz syndrome. J Med Genet. 2004;41:381–386. doi: 10.1136/jmg.2003.014829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prickett TD, Brautigan DL. Cytokine activation of p38 mitogen-activated protein kinase and apoptosis is opposed by alpha-4 targeting of protein phosphatase 2A for site-specific dephosphorylation of MEK3. Mol Cell Biol. 2007;27:4217–4227. doi: 10.1128/MCB.00067-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quaderi NA, Schweiger S, Gaudenz K, Franco B, Rugarli EI, Berger W, Feldman GJ, Volta M, Andolfi G, Gilgenkrantz S, Marion RW, Hennekam RC, Opitz JM, Muenke M, Ropers HH, Ballabio A. Opitz G/BBB syndrome, a defect of midline development, is due to mutations in a new RING finger gene on Xp22. Nat Genet. 1997;17:285–291. doi: 10.1038/ng1197-285. [DOI] [PubMed] [Google Scholar]
- Richman JM, Fu KK, Cox LL, Sibbons JP, Cox TC. Isolation and characterisation of the chick orthologue of the Opitz syndrome gene, Mid1, supports a conserved role in vertebrate development. Int J Dev Biol. 2002;46:441–448. [PubMed] [Google Scholar]
- Rinaldi A, De Leonibus E, Cifra A, Minicocci E, Oliverio A, Mele A. Neural network activated during allocentric and egocentric spatial memory retrieval in mice: focus on striatal subregions. FENS Abstr. 2008;4 157.124. [Google Scholar]
- Robin NH, Feldman GJ, Aronson AL, Mitchell HF, Weksberg R, Leonard CO, Burton BK, Josephson KD, Laxová R, Aleck KA, Allanson JE, Guion-Almeida ML, Martin RA, Leichtman LG, Price RA, Opitz JM, Muenke M. Opitz syndrome is genetically heterogeneous, with one locus on Xp22, and a second locus on 22q11.2. Nat Genet. 1995;11:459–461. doi: 10.1038/ng1295-459. [DOI] [PubMed] [Google Scholar]
- Robin NH, Opitz JM, Muenke M. Opitz G/BBB syndrome: clinical comparisons of families linked to Xp22 and 22q, and a review of the literature. Am J Med Genet. 1996;62:305–317. doi: 10.1002/(SICI)1096-8628(19960329)62:3<305::AID-AJMG20>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- Schweiger S, Foerster J, Lehmann T, Suckow V, Muller YA, Walter G, Davies T, Porter H, van Bokhoven H, Lunt PW, Traub P, Ropers HH. The Opitz syndrome gene product, MID1, associates with microtubules. Proc Natl Acad Sci U S A. 1999;96:2794–2799. doi: 10.1073/pnas.96.6.2794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Short KM, Hopwood B, Yi Z, Cox TC. MID1 and MID2 homo- and heterodimerise to tether the rapamycin-sensitive PP2A regulatory subunit, Alpha 4, to microtubules: implications for the clinical variability of X-linked Opitz GBBB syndrome and other developmental disorders. BMC Cell Biol. 2002;3:1. doi: 10.1186/1471-2121-3-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sillitoe RV, Joyner AL. Morphology, molecular codes, and circuitry produce the three-dimensional complexity of the cerebellum. Annu Rev Cell Dev Biol. 2007;23:549–577. doi: 10.1146/annurev.cellbio.23.090506.123237. [DOI] [PubMed] [Google Scholar]
- So J, Suckow V, Kijas Z, Kalscheuer V, Moser B, Winter J, Baars M, Firth H, Lunt P, Hamel B, Meinecke P, Moraine C, Odent S, Schinzel A, van der Smagt JJ, Devriendt K, Albrecht B, Gillessen-Kaesbach G, van der Burgt I, Petrij F, et al. Mild phenotypes in a series of patients with Opitz GBBB syndrome with MID1 mutations. Am J Med Genet A. 2005;132:1–7. doi: 10.1002/ajmg.a.30407. [DOI] [PubMed] [Google Scholar]
- Surace EM, Angeletti B, Ballabio A, Marigo V. Expression pattern of the ocular albinism type 1 (Oa1) gene in the murine retinal pigment epithelium. Invest Ophthalmol Vis Sci. 2000;41:4333–4337. [PubMed] [Google Scholar]
- Tavano A, Grasso R, Gagliardi C, Triulzi F, Bresolin N, Fabbro F, Borgatti R. Disorders of cognitive and affective development in cerebellar malformations. Brain. 2007;130:2646–2660. doi: 10.1093/brain/awm201. [DOI] [PubMed] [Google Scholar]
- Trockenbacher A, Suckow V, Foerster J, Winter J, Krauss S, Ropers HH, Schneider R, Schweiger S. MID1, mutated in Opitz syndrome, encodes an ubiquitin ligase that targets phosphatase 2A for degradation. Nat Genet. 2001;29:287–294. doi: 10.1038/ng762. [DOI] [PubMed] [Google Scholar]
- Wang VY, Zoghbi HY. Genetic regulation of cerebellar development. Nat Rev Neurosci. 2001;2:484–491. doi: 10.1038/35081558. [DOI] [PubMed] [Google Scholar]
- Xu J, Lawshe A, MacArthur CA, Ornitz DM. Genomic structure, mapping, activity and expression of fibroblast growth factor 17. Mech Dev. 1999;83:165–178. doi: 10.1016/s0925-4773(99)00034-9. [DOI] [PubMed] [Google Scholar]
- Xu J, Liu Z, Ornitz DM. Temporal and spatial gradients of Fgf8 and Fgf17 regulate proliferation and differentiation of midline cerebellar structures. Development. 2000;127:1833–1843. doi: 10.1242/dev.127.9.1833. [DOI] [PubMed] [Google Scholar]